

Abstract
Evolving antimicrobial resistance has motivated the search for novel targets and alternative therapies. Caseinolytic protease (ClpP) has emerged as an enticing new target since its function is conserved and essential for bacterial fitness, and because its inhibition or dysregulation leads to bacterial cell death. ClpP protease function controls global protein homeostasis and is, therefore, crucial for the maintenance of the bacterial proteome during growth and infection. Previously, acyldepsipeptides (ADEPs) were discovered to dysregulate ClpP, leading to bactericidal activity against both actively growing and dormant Gram-positive pathogens. Unfortunately, these compounds had very low efficacy against Gram-negative bacteria. Hence, we sought to develop non-ADEP ClpP-targeting compounds with activity against Gram-negative species and called these activators of self-compartmentalizing proteases (ACPs). These ACPs bind and dysregulate ClpP in a manner similar to ADEPs, effectively digesting bacteria from the inside out. Here, we performed further ACP derivatization and testing to improve the efficacy and breadth of coverage of selected ACPs against Gram-negative bacteria. We observed that a diverse collection of Neisseria meningitidis and Neisseria gonorrhoeae clinical isolates were exquisitely sensitive to these ACP analogues. Furthermore, based on the ACP-ClpP cocrystal structure solved here, we demonstrate that ACPs could be designed to be species specific. This validates the feasibility of drug-based targeting of ClpP in Gram-negative bacteria.
Keywords: ClpP, antibiotics, dysregulation, Neisseria meningitidis, Neisseria gonorrhoeae, acyldepsipeptides (ADEPs), activators of self-compartmentalizing proteases (ACPs)
Graphical Abstract
Bacterial proteolytic systems are fascinating new targets for antibacterial therapies in the era of broad-spectrum antibiotic resistance. The caseinolytic protease P (ClpP) is one such main proteolytic complex.1 ClpP is a cylindrical serine protease comprised of two stacked heptameric rings; ClpP associates with and gets activated by AAA+ (ATPases associated with diverse cellular activities) hexameric chaperones such as ClpA or ClpX.2 Regulation of protein degradation is further specified with the help of certain adaptors that recognize and direct target proteins to the AAA+ chaperone, which unfolds and then translocates the substrates into the ClpP proteolytic chamber for degradation.3 In the absence of the chaperones, ClpP can only degrade small peptides of up to 30 amino acids and some unstructured proteins, however at a much lower efficiency compared to ClpP in complex with its AAA+ chaperones.4 ClpP-mediated degradation of proteins and peptides can influence important cellular functions and have broad implications on bacterial infectivity.5
Antibiotic development specific to ClpP6,7 has focused on several inhibitors that were efficient against Gram-positive bacteria, such as Staphylococcus aureus and Listeria mono-cytogenes, but less so against Gram-negative bacteria. These drugs included trans-β-lactones,8 phenyl esters,9 pyrimidines,10 boronates,11 and α-amino diphenyl phosphonates12 among others. Upon interacting with ClpP, these compounds would effectively inactivate its proteolytic function, but the antibacterial efficacy of these compounds was generally found to be low. In an alternative approach, acyldepsipeptides (ADEPs) were discovered to bind and activate ClpP independent of the cognate ATPase, thereby allowing dysregulated protein degradation that results in bacterial cell death.6,7,13,14 These analogues exhibited potent antibacterial activity against many Gram-positive bacteria13–16 but Gram-negative bacteria were generally more resistant, likely due to active efflux and/or limited penetration of the outer membrane.
Efforts in our laboratory have concentrated on the development of ClpP dysregulators (also termed activators) targeting Gram-negative bacteria. Using a high-throughput screening approach, four structurally distinct non-ADEP activators of ClpP were identified17 and were named activators of self-compartmentalizing proteases (ACPs). These ACPs were found to dysregulate ClpP and manifested the desired antibacterial activity against several Gram-negative bacteria, including Neisseria meningitidis.17
N. meningitidis and Neisseria gonorrhoeae are two closely related Gram-negative pathogens that cause very different diseases. N. meningitidis, commonly referred to as the meningococcus, resides asymptomatically in the posterior region of the nasopharynx of nearly 10% of adults. Occasionally, meningococci breach the nasopharyngeal barrier and cause rapidly progressing invasive meningococcal diseases including meningitis and septicemia, which, if left untreated, may result in mortality or debilitating sequelae.18,19 N. gonorrhoeae is a closely related but phenotypically distinct relative of meningococcus that is associated with gonorrhea, a widely prevalent sexually transmitted disease. The gonococcus can asymptomatically colonize the genital, rectal, and oropharyngeal mucosal tissues, and pathogenesis arises when an inappropriately intense inflammatory response inadvertently damages the surrounding tissues. Early and effective antibiotic treatment is, therefore, crucial for minimizing the risk of sequelae to Neisserial infection. Of particular challenge, both pathogenic Neisseria sp. have a highly plastic genome and natural competence for genetic transformation allowing them to avoid immune-mediated clearance and to efficiently exchange genetic material. For N. gonorrhoeae, this has led to the rapid rise in multidrug resistant strains which prompted the WHO and US Centers for Disease Control to consider this pathogen an urgent-level threat to public health, creating pressing need for identification and development of novel antibiotic therapy.20
In this study, we describe the generation and characterization of a large number of analogues of ACPs and ADEPs. We concentrated our efforts on targeting Neisserial ClpP. Several ACP1 analogues showed potent activity against N. meningitidis and N. gonorrhoeae. Furthermore, based on structural studies, this work validates that simpler non-ADEP ClpP activators can be further developed to target specific bacterial species.
RESULTS
Generation of ACP Analogues.
Through our initial high throughput screening of over 60 000 compounds in the Maybridge and Chembridge libraries, we previously reported four novel chemical entities that activated ClpP and called these ACP1–ACP5 (ACP4 and ACP5 have the same basic chemical structure and, therefore, these analogous molecules are referred to here as ACP4).17 In the search for ACPs specific for Gram-negative pathogens, we screened these ACPs against a panel of Gram-negative bacteria. We observed that the pathogenic Neisseria species, N. gonorrhoeae and N. meningitidis, were particularly susceptible to ACPs.17
Given the antibacterial potential of activating ClpP, we generated a total of 85 ACP1 and 46 ADEP analogues. The compound structures and activities against E. coli (EcClpP) and N. meningitidis (NmClpP) ClpP are given in Table S1, with compounds numbered according to their in vitro activity against purified NmClpP (see below). We also generated analogues of ACP3 and ACP4, but no significant activity was observed against EcClpP or NmClpP; hence, these two chemical scaffolds were not further optimized in the current study.
The syntheses of ADEP analogues were described before,14,21,22 while the synthetic approaches for the preparation of ACP1 analogues are given in Figure 1 and Figure S1. The vast majority, about 60% of the ACP1 analogues reported herein, were synthesized using a 4-step strategy outlined in Figure 1. In the first step, a series of aryl- or heteroarylthiols were alkylated with ethyl 2-bromoisobutyrate to afford the corresponding alkylated sulfide products (condition iii).23 In the second step, oxidation of the sulfides to the corresponding sulfones was accomplished with meta-chloroperbenzoic acid (mCPBA, condition iv).24 In the third step, the ethyl ester functionality was hydrolyzed to afford the corresponding carboxylic acids (condition v),25 which served as the penultimate intermediate to several ACP1 analogues. In the fourth and final step, the carboxylic acid precursors were coupled with a wide variety of amines/ammonium salts using the coupling agent (benzotriazol-1-yloxy)-tripyrrolidinophosphonium hexafluorophosphate (PyBOP) in the presence of Hünig’s base to afford the ACP1 analogues (condition ii).
Figure 1.
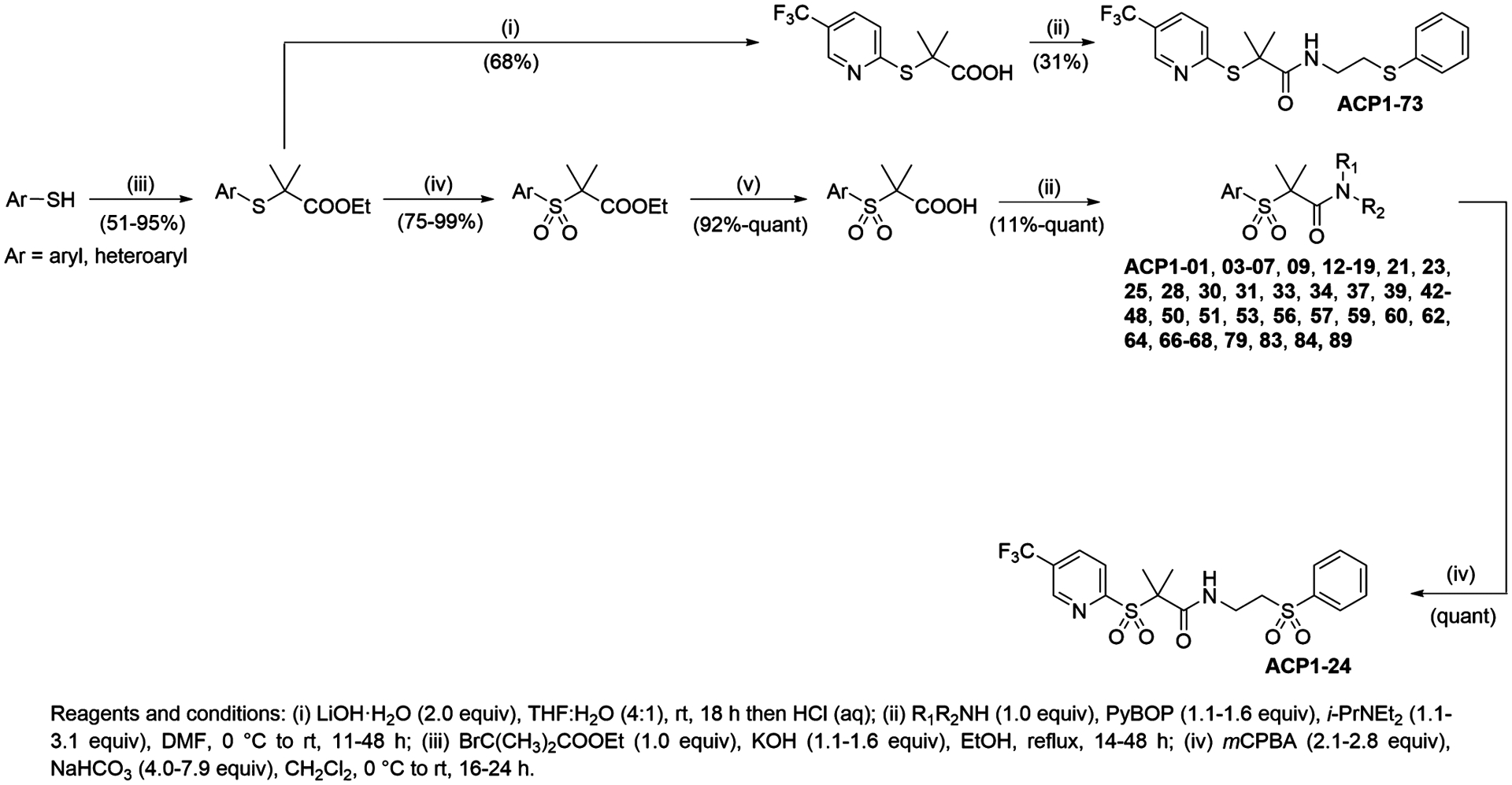
Synthesis of ACP1 analogues. Shown is the general synthetic approach used to obtain ACP1 analogues via late-stage amide coupling of the acid and amine components mediated by PyBOP.
For the synthesis of specific ACP1 analogues, alternative reagents and/or changes to synthetic strategy were required as described in Figure 1. For example, synthesis of ACP1-73 (a sulfide analogue) involved direct hydrolysis of the alkylated sulfidyl ester (condition i) in the second step to afford the corresponding acid which was subsequently coupled using PyBOP. Synthesis of ACP1-24 (a bis-sulfone analogue) involved oxidation of ACP1-06 with mCPBA. The synthesis of all other ACP1 analogues was accomplished using analogous approaches and is discussed in the Methods section and shown in Figure S1. All characterization data for listed ACP1 compounds are given in the Supplemental Experimental Procedures.
Compound Screening against NmClpP and EcClpP.
The generated ACP1 analogues were first screened by determining the relative degradation index using 25 μM of compound (RD25). This is a quantitative measure that we developed and used previously to assess the potency of compounds for activating ClpP degradation of fluorescently labeled casein (casein-FITC) as a model substrate.17 The closer the value of RD25 is to 1 (complete degradation), the better the compound is at activating ClpP to degrade casein-FITC, while a value of 0 indicates no detectable activation. It should be noted that NmClpP and N. gonorrhoeae ClpP (NgClpP) are basically identical proteins, differing by only 2 or 3 residues depending on the strains (e.g., compare UniProt sequences CLPP_NEIMB and A0A1D3FJB7_NEIGO).
The screen was done using both EcClpP and NmClpP. In Figure 2A,B and Table S1, the compounds are ranked and numbered according to their RD25 values with NmClpP. Table S2 correlates our current compound naming scheme with previously published compound names. Several of the ACP1 analogues (Figure 2A) and many of the ADEP analogues (Figure 2B) showed good activities against NmClpP and EcClpP. However, it is notable that the most active compounds were not the same for NmClpP and EcClpP. This raises the interesting prospect that these drugs can be developed to specifically target different bacteria. This issue is discussed further below.
Figure 2.
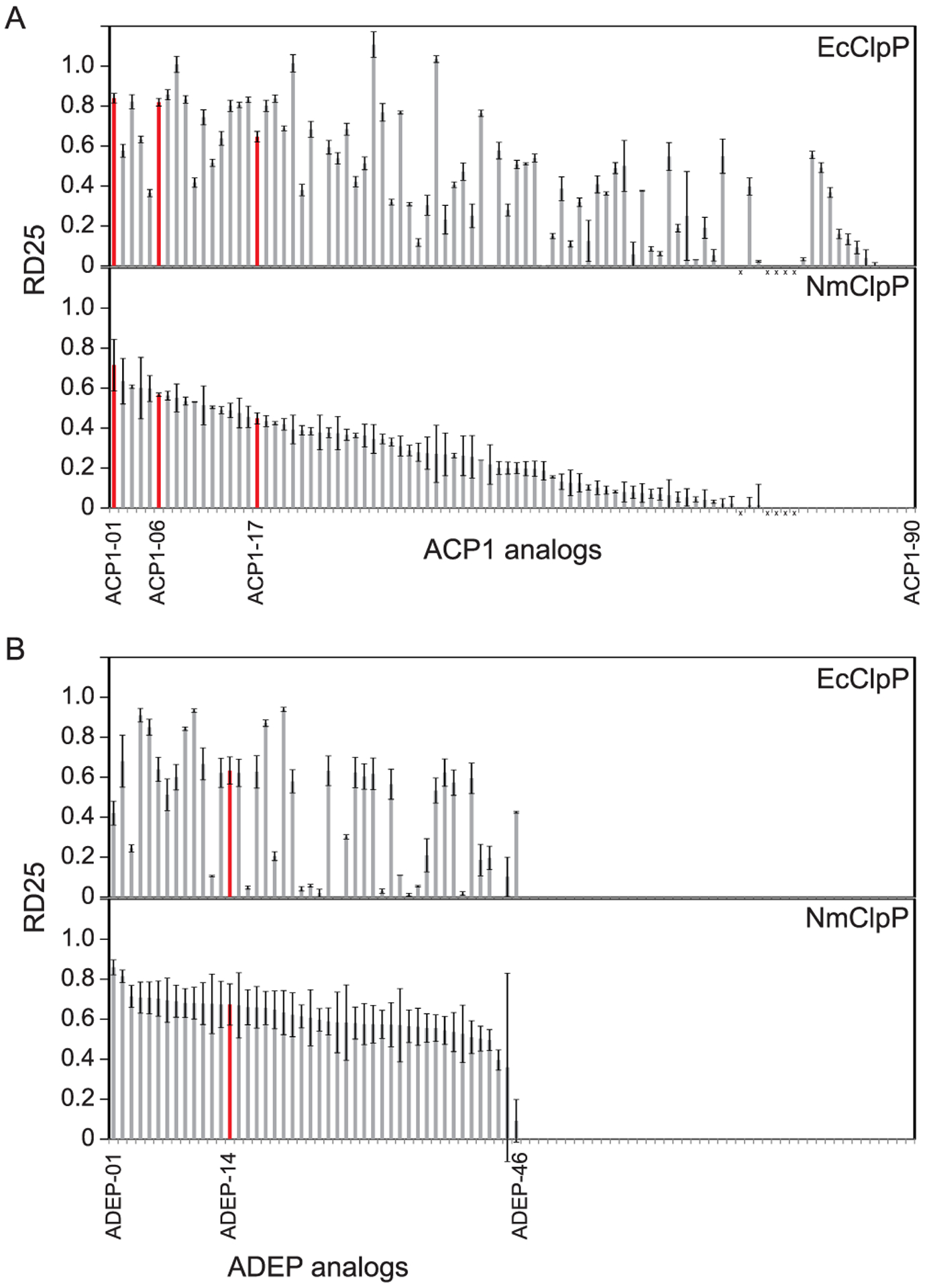
RD25 for EcClpP and NmClpP. (A) RD25 values were measured for EcClpP and NmClpP in the presence of 85 different ACP1 analogues. The “x” indicates five compounds that were not investigated due to compound purity issues. (B) RD25 values were measured for EcClpP and NmClpP in the presence of 46 different ADEP analogues. The compounds are numbered according to their relative activity on NmClpP, with ACP1-01 and ADEP-01 being the most active analogues and are listed in the same order in the top (EcClpP) and bottom (NmClpP) panels. Compounds highlighted in red are discussed in Figure 3.
We also tested these compounds against human ClpP protein (HsClpP) as a counter screen. For the ADEP compounds, the screen was done as part of prior study.26 Many of the ADEP compounds are active against HsClpP, but few of the ACP1 compounds showed good activity against HsClpP (Table S1). Furthermore, we had shown that some of the ADEPs have toxicity against mammalian cell lines by targeting the human mitochondrial ClpP,26 while that is not the case for the tested ACP1 analogues (data not shown). Hence, the ACP1 analogues could be more suitable for further development as antibiotics.
Antibacterial Activity.
ACP1 and ADEP analogues that showed good RD25 activity (typically RD25 > 0.4 against NmClpP) were further tested to determine minimum inhibitory concentrations (MICs) against wild type (WT) and ΔclpP N. meningitidis H44/76 strains. Analogues that specifically target the ClpP protease in bacteria with little off-target activity should inhibit the growth of the WT strain but not the growth of the ΔclpP strain. Among the ACP1 analogues, the best compounds which had good activity against the WT strain but reduced activity against the ΔclpP strain were ACP1-01 and ACP1-17 (Figure 3A,B). ACP1-06, labeled as ACP1b in our previous paper,17 has a weaker activity against WT N. meningitidis and no activity against the ΔclpP strain (Figure 3A,B). On the other hand, many of the ADEP analogues generated showed much higher specific activity toward the ClpP target and none of the ADEP analogues tested had any significant activity against the ΔclpP strain (Figure 3A,B and ref 14). The MIC values of the ADEP analogues were typically significantly lower, by a factor of about 10, than those of the ACP1s (Figure 3A). None of the ACP1 or ADEP compounds had a significant effect on the E. coli WT strain, MC4100 (data not shown).
Figure 3.
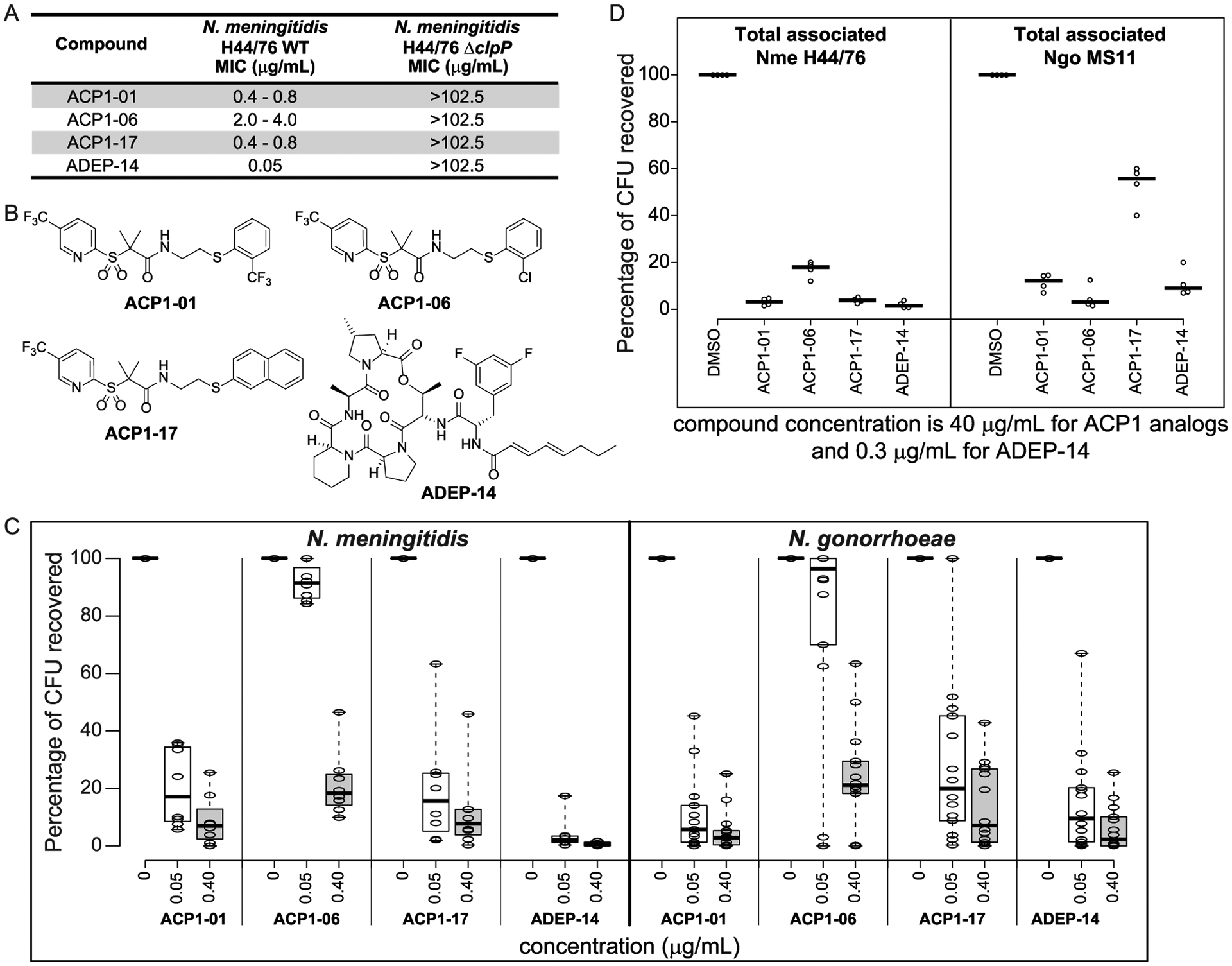
Antibacterial activity of ACP1-01, ACP1-06, ACP1-17, and ADEP-14. (A) Minimum inhibitory concentrations of selected analogues on the growth of WT N. meningitidis H44/76 compared to N. meningitidis H44/76 ΔclpP. Representative results from three independent experiments are shown. (B) Chemical structures of ACP1-01, ACP1-06, ACP1-17, and ADEP-14. (C) Shown are boxplots indicating the antibacterial susceptibility of various meningococcal (8 strains) and gonococcal (14 strains) reference strains to ACP1-01, ACP1-06, ACP1-17, and ADEP-14 after 20 h of treatment. Percent CFUs recovered was calculated as (CFUs recovered after treatment)/(CFUs of untreated bacteria) × 100. A total of three experiments were performed for each strain tested at each concentration of test compounds. The individual values were then averaged and converted to percentage CFU recovered. Treatment concentration resulting in less than 20% CFUs was considered as concentration at which strains were susceptible to the test compound. Boxplots were generated using BoxPlotR (http://shiny.chemgrid.org/boxplotr/). Thick horizontal lines represent the median. Whiskers extend to minimum and maximum values. (D) Effect of compounds on in vitro Neisserial infection. HeLa cells expressing human CEACAM1 or CEACAM5 were infected for 4 h with N. meningitidis H44/76 or N. gonorrhoeae MS11, respectively, and then treated with the indicated compounds at a final concentration of 40 μg/mL for ACP1 analogues and 0.3 μg/mL for ADEP-14 for 2 h, or with 1% DMSO for 2 h as a solvent control. CFUs recovered after treatment are reported as percent of CFUs observed after plating HeLa cell lysates obtained after treatment with saponin and pipetting to break the human cell membranes. Experiments were repeated four times and the thick horizontal lines represent the median.
ACP1-01, ACP1-06, ACP1-17, and ADEP-14 (Figure 3B) were used for further studies with Neisseria. In these studies, we sought to determine whether the drugs were active against a diverse collection of clinical isolates. Eight serogroup B meningococcal strains were selected from a well-characterized collection,27 and 14 gonococcal reference strains with diverse antibiotic susceptibilities were obtained from Public Health England.28 Details about the strains are provided in Table 1. We tested compound concentrations of 0.05 μg/mL and 0.4 μg/mL against the Neisserial strains (Figure 3C and Table S3) and found that ACP1-01 was generally more effective than ACP1-17, particularly with gonococci. ACP1-06 was the least effective of these analogues. ADEP-14 exhibited high activity against all meningococcal strains and most gonococcal strains.
Table 1.
Pathogenic Neisseria Strains Used to Test Susceptibility to ACPs and ADEP
| Neisseria meningitidis strains | |||
|---|---|---|---|
| number | name | source; country; year | classificationa |
| 2237 | n259/BZ 169 | Caugant Collection; Netherlands; 1985 | B:P1.5–2,16:F3–3:ST32 (cc32) |
| 2238 | n261/NG080 | Caugant Collection; Norway; 1981 | B:P1.7,16:F3–3:ST-32 (cc32) |
| 2239 | n262/NG144/82 | Caugant Collection; Norway; 1982 | B:P1.7,16:F3–3:ST-32 (cc32) |
| 2240 | n263/NG PB24 | Caugant Collection; Norway; 1985 | B:P1.7–2,16–7:F3–3:ST32 (cc32) |
| 2242 | n267/AK 50 | Caugant Collection; Greece; 1992 | B:P1.7–2,4:F1–5:ST41 (cc41/44) |
| 2243 | n274/88/03415 | Caugant Collection; Scotland; 1988 | B:P1.7–2,4:F1–5:ST46 (cc41/44) |
| 2249 | n280/SWZ107 | Caugant Collection; Switzerland; 1986 | B:1.ND,ND:F-ND:ST35 (cc35) |
| 2250 | n282/NG F26 | Caugant Collection; Norway; 1988 | B:1.ND,16:F-ND:ST14 (cc269) |
| Neisseria gonorrhoeae strains | |
|---|---|
| strain | country |
| N. gonorrhoeae; NCTC 13477 or WHO F | Isolated in Canada, 1991 |
| N. gonorrhoeae; NCTC 13478 or WHO G | Isolated in Thailand in 1997 |
| N. gonorrhoeae; NCTC 13479 or WHO K | Isolated by Dr. T. Muratani |
| N. gonorrhoeae; NCTC 13480 or WHO L | Isolated in Asia, 1996 |
| N. gonorrhoeae; NCTC 13481 or WHO M | Isolated in the Philippines, 1992 |
| N. gonorrhoeae; NCTC 13482 or WHO N | Isolated in Australia, 2001 |
| N. gonorrhoeae; NCTC 13483 or WHO O | Isolated in Canada, 1991 |
| N. gonorrhoeae; NCTC 13484 or WHO P | |
| N. gonorrhoeae; NCTC 13817 or WHO U | Isolated in Sweden, May 2011 |
| N. gonorrhoeae; NCTC 13818 or WHO V | Isolated in Karlstad, Sweden, 2012 |
| N. gonorrhoeae; NCTC 13819 or WHO W | Isolated in Hong Kong, 2007 |
| N. gonorrhoeae; NCTC 13820 or WHO X | Isolated in Kyoto, Japan, 2009 |
| N. gonorrhoeae; NCTC 13821 or WHO Y | Isolated in Quimper, France, June 2010 |
| N. gonorrhoeae; NCTC 13822 or WHO Z | Isolated in Australia 2013 |
B, Serogroup B; P1, porin type; ST, serotype; and cc, clonal complex. These are based on MLST classification method used for Neisseria meningitidis.
Most meningococcal strains treated in this study were susceptible to ACP1-01, ACP1-17, and ADEP-14 at the concentration of 0.4 μg/mL, with the exceptions being strains 2250 and 2242 (Figure 3C and Table S3). For example, 2250 (NG F26; Table 1) and 2242 (AK 50; Table 1) strains had reduced susceptibility with 25% and 18% survival, respectively, after treating with 0.40 μg/mL ACP1-01 (Figure 3C and Table S3). Meningococcal strain 2242 belongs to clonal complex 41/44, which is responsible for the majority of serogroup B mediated invasive meningococcal disease, whereas strain 2250 belongs to clonal complex 269 and was isolated from a carrier. Overall, these compounds have a broad coverage across various phylogenetically diverse Neisseria strains, and strains known to be resistant to other drugs are still susceptible to these ClpP activators.
One gonococcal strain, 13477 (WHO F; Table 1), was less susceptible to all test compounds (Figure 3C and Table S3). Curiously, this strain is not intrinsically drug resistant as it is susceptible to most known antibiotics.28 On the other hand, strain 13822 (WHO Z; Table 1) is known to be resistant to most antibiotics, including those currently in use (e.g., cefixime and ceftriaxone), but was moderately susceptible to ADEP-14 and highly sensitive to ACP1-01 at a concentration of 0.4 μg/mL (Figure 3C and Table S3). Interestingly, the multidrug resistant strain 13821 (WHO Y; Table 1) was susceptible to both ACP1s and ADEP-14 (Figure 3C and Table S3).
Given that antibiotics often have a reduced capacity to kill bacteria that are either tightly associated with and/or have been engulfed by mammalian cells, the drugs were next tested for their ability to clear Neisserial infection of human epithelial cells using a well-established in vitro model. The Neisserial Opa protein adhesins bind to several different cell surface glycoproteins of the CEACAM family.29 HeLa cells have been widely used as a model to understand this association because they do not normally express CEACAMs, but can be transfected so as to express individual receptor proteins.30 When Neisserial Opa proteins engage human CEACAMs, bacteria tightly adhere to the cell surface and then are engulfed by the epithelial cells. Neisseria adhering to the outside of these cells remain susceptible to the antibiotic gentamicin, while those that have been engulfed are resistant to this treatment because they are protected by the human cell membrane.30
To test the ability of ACP1-01, ACP1-06, ACP1-17, and ADEP-14 to clear a Neisserial infection of HeLa-CEACAM1 cells, Nme H44/76 (N. meningitidis) or Ngo MS11 (N. gonorrhoeae) were resuspended in serum-free growth media and human CEACAM-expressing HeLa cells were infected with these strains at an inoculum dose of 105 CFU/mL for 3 h. After which, the cells were washed and treated with the compounds for 2 h. This method determines the effect of drugs on total bacteria associated with human cells in vitro.
As shown in Figure 3D, there was no significant effect of 1% DMSO, the solvent used for the drug stocks, on cells or bacteria. We tested the effect of a range of ACP1 and ADEP concentrations, from 0.3 μg/mL to 40 μg/mL, on HeLa-CEACAM cells and found that cell viability was over 80% after 2 h treatment with compound concentrations up to 40 μg/mL. Since Neisseria is internalized by CEACAM-expressing HeLa cells, it was pertinent to use doses higher than the MIC values to overcome potential uptake issues by host cells. Hence, a compound concentration of 40 μg/mL for ACP1s and 0.3 μg/mL for ADEP-14 was selected for the infection experiments. Treatment with either of the selected ACP1s or ADEP, except for ACP1-06, resulted in nearly 95% reduction of viable Nme H44/76 recovered, while ACP1-01, ACP1-06, and ADEP-14 caused >80% reduction in the Ngo MS11 (Figure 3D). These results suggest that ACP1-01 and ADEP-14 can effectively clear infection caused by both meningococci and gonococci pathogens in this model.
Crystal Structure of NmClpP-ACP1-06 Complex.
In order to investigate how these ACP1 compounds bind to Neisserial ClpP, and whether ACP1s can be further modified to specifically target certain bacterial species but not others, we determined the X-ray crystal structure of NmClpP with ACP1-06. We had previously solved the structure of ACP1-06 with EcClpP,31 thus allowing direct structural comparisons that can guide future binding and specificity optimization of ACP1.
Addition of ACP1-06 dissolved in DMSO to the protein solution at high concentration often caused precipitation that prevented crystallization of the complex. Exposing NmClpP crystals to solid ACP1-06 in their crystallization drops allowed determination of the structure of the NmClpP-ACP1-06 complex (Table 2 and Figure 4). The asymmetric unit of the NmClpP-ACP1-06 complex crystals contains a single heptameric ring, two of which in the crystal lattice assemble into the biologically relevant tetradecamer in the extended conformation. While strong electron density for the sulfone group of ACP1-06 is found at identical locations in all hydrophobic sites of NmClpP, only one site, the interface between chains E and F (Figure 4A), showed sufficiently clear electron density to trace the entire chemical structure of ACP1-06 with confidence. However, the average density for the ligand is lower than that of the surrounding protein residues, which is expected given the dual configuration and some residual mobility, e.g., probable rotation of the ring portion of the (trifluoromethyl)pyridyl group of ACP1-06 (Figure 4B,C).
Table 2.
Crystallographic Data Collection and Refinement Statistics
| data collection | APS-14ID-B |
|---|---|
| λ (Å) | 1.000 |
| Space group | P22121 |
| Cell dimensions | |
| a, b, c (Å) | 97.3, 119.2, 127.8 |
| α, β, γ (deg) | 90.0, 90.0 90.0 |
| Resolution (Å)a | 97.33–1.64 (1.69–1.64) |
| R meas a,b | 0.116 (0.506) |
| R pim a,c | 0.077 (0.431) |
| I/σ(I)a | 12.1 (1.0) |
| CC1/2 (%) | 98.2 (75.1) |
| Completeness | 98.8 (91.4) |
| Total/unique reflections | 1 012 426/179 842 |
| Multiplicity | 5.6 (2.1) |
| Z (molecules/ASU)d | 7 |
| Refinement | |
| Rwork/Rfreee,f | 23.7/28.2 |
| No. atoms (non-hydrogens) | 10 544 |
| Protein | 9775 |
| Ligand | 65 |
| Water | 704 |
| Average B (Å2) | 31.8 |
| Protein (Å2) | 31.2 |
| ACP1-06 (Å2) | 62.6 |
| Waters (Å2) | 36.7 |
| RMSD Bonds (Å) | 0.012 |
| RMSD Angles (deg) | 1.28 |
| Ramachandran favored (%) | 98.0 |
| Ramachandran allowed (%) | 1.7 |
| Ramachandran outliers (%) | 0.3 |
| No. of TLS groups | 1 |
| Clashscore | 6.76 |
| PDB code | 6W9T |
Values in parentheses are for highest-resolution shell.
Rmeas: Multiplicity-independent R factor = Σhkl [Nhkl/(Nhkl − 1)]1/2 Σi | Ii(hkl) – 〈I(hkl)〉|/Σhkl Σi Ii(hkl).
Rpim: Precision indicating merging R factor = Σhkl [1/(Nhkl − 1)]1/2 Σi |Ii(hkl) – 〈I(hkl)〉|/Σhkl Σi Ii(hkl).
Za: Number of ClpP subunits in the asymmetric unit.
Rwork: = Σ ‖Fobs| – |Fcalc‖/Σ |Fobs|, where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively.
Rfree: Statistic is the same as Rwork except calculated on 5% of the total reflections chosen randomly and omitted from the refinement.
Figure 4.
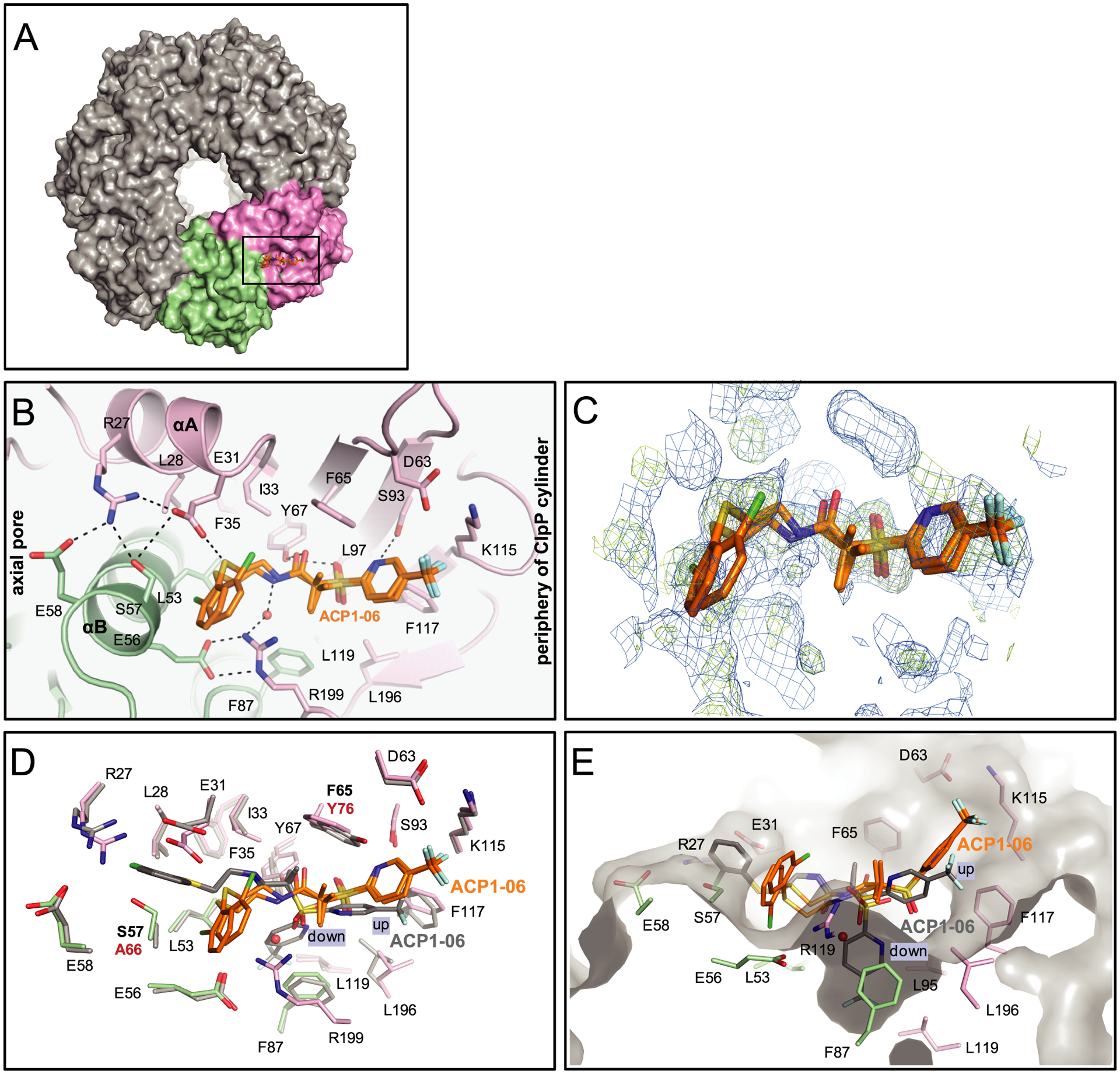
Binding modes of ACP1-06 on NmClpP. (A) Surface representation of the NmClpP tetradecamer viewed from top along the 7-fold symmetry axis, with the two adjacent subunits with bound ACP1-06 colored pink and green. A single ACP1-06 molecule is modeled in each heptameric ring (shown inside box). Panels B–E are enlarged views of this section. (B) The two binding modes of ACP1-06 in the NmClpP hydrophobic site. ACP1-06 binds to the hydrophobic site formed by two adjacent subunits. Hydrogen bonding interactions are shown as black dashed lines. A water molecule is shown as a red sphere. Residues that form the van der Waals binding surface are represented as sticks. A water-mediated hydrogen bond between the peptidic amino group of ACP1-06 and R199 of NmClpP stabilizes both configurations. (C) Simulated annealing composite omit maps showing electron density for the bound ACP1-06 and for protein residues and water molecules within 2.0 Å from the ligand. For clarity, only the ACP1-06 molecule is shown in the electron density maps. The 2Fo–Fc omit map (blue mesh) is contoured at 0.5 σ, while the Fo–Fc omit map (green mesh) is contoured at 2.0 σ. (D) Comparison of the hydrophobic sites and ACP1-06 binding configurations in NmClpP and EcClpP. Residues of EcClpP are shown in gray wires, while those for NmClpP are in light green and pink as in (B). The two binding configurations (up and down) of EcClpP are also shown as dark gray sticks superimposed with those found in NmClpP. In EcClpP, the residues that gate the narrow binding channel where the fragment of ACP1-06 closer to the axial pore inserts are A66 (labeled in bold red) and E40. In NmClpP, the equivalent pair is S57 and E31, preventing the formation of the channel and redirecting the western fragment to an alternate site bounded by the side chains of L53, E56, S57, and R199. (E) Alternate view of (D) showing only the site residues of NmClpP as sticks and the binding surface in gray. The small narrow pocket that accommodates the down configuration of ACP1-06 in EcClpP is shown in the center. The fragment of ACP1-06 nearest the axial pore in NmClpP binds an alternate pocket and causes a shift of the binding mode for the rest of the molecule from that found in EcClpP.
In the complex structure, ACP1-06 was modeled in two binding configurations and refined to almost equal occupancies of 0.51 for configuration A and 0.49 for configuration B, respectively (Figure 4C). In these configurations, the backbones of the compound fragments closest to the periphery have nearly identical orientations up to the gem-dimethyl groups, while those of the fragments closest to the axial pore differ through C–S bond rotation, placing the Cl atoms of the ortho-chlorophenyl moieties at 180° relative to each other. In one configuration, the ortho-Cl group is solvent exposed, while in the other configuration, it occupies a pocket formed by L53, E56, S57 of one subunit and R199 at the C-terminus of the adjacent subunit (Figure 4B). Interestingly, unlike in the EcClpP-ACP1-06 complex, where the fragment closest to the axial pore is fully extended and inserts the chlorophenyl group in a very narrow channel between the αA helix of one subunit and the αB helix of the adjacent subunit, this fragment is kinked in the NmClpP-ACP1-06 complex (Figure 4B,D). This is due to the presence of the S57-E31 hydrogen bond that blocks the channel in NmClpP. This hydrogen bond is not present in EcClpP as the equivalent residues are A66 and E40, which results in ample space for the insertion of the planar chlorophenyl group (Figure 4D,E). This orientation of the chlorophenyl group in NmClpP is further stabilized by the interaction between the carboxylate group of E31 and the S atom of the compound (Figure 4B).
Placement in the narrow pocket of the hydrophobic site of the ACP1-06 fragment closest to the axial pore causes the fragment at the periphery to assume a single configuration in NmClpP (Figure 4E) distinct from its up and down configurations observed in the EcClpP-ACP1-06 structure,31 despite the structural conservation of the binding site in NmClpP and EcClpP. In general, there is a slight translation and rotation of this fragment, resulting in new interactions with hydrophobic site residues, and in its inability to assume the down configuration found in the EcClpP-ACP1-06 complex where it occupies a small cavity (Figure 4E). The new interactions include a solvent-mediated hydrogen bond between R199 and the peptidic amino group of ACP1-06, and reorganized hydrogen bonding interactions between Y67 and the peptidic carbonyl and sulfonyl oxygens of ACP1-06 (Figure 4B). In the EcClpP-ACP1-06 structure, R199 interacts with the sulfone group of ACP1-06, and no solvent-mediated interaction with the small molecule exists. In addition, S93 in NmClpP forms a hydrogen bond with the N atom of the pyridyl group of ACP1-06; an interaction not observed in the EcClpP-ACP1-06 complex despite the serine residue being conserved (Figure 4B,D). Finally, the trifluoromethyl group is solvent exposed, with the electronegative fluorine atoms potentially interacting with solvent molecules and the positively charged amino group of K115 (Figure 4B,E). The planar pyridine ring is further stabilized by van der Waals contacts with nearby hydrophobic residues F65, F117, and L196.
To measure the binding affinity of ACP1-06 to NmClpP and EcClpP, casein-FITC degradation assays were carried out in the presence of ClpP and a range of ACP1-06 concentrations (Figure 5A,B). Initial velocities (υ0) were determined from the data collected over three independent experiments and nonlinear regression analysis was then performed on the initial velocities using the Hill equation (see Methods). The apparent K0.5s (equivalent to EC50) obtained were 6.1 (0.3) μM and 16.1 (0.7) μM for the binding of ACP1-06 to EcClpP and NmClpP, respectively, with similar Hill coefficients (h) of about 2 (Figure 5C,D). Since the Hill coefficients are greater than 1, this indicates that ACP1-06 binds cooperatively to both proteases. We had previously obtained a Kd of 3.2 (0.5) μM for the binding of ACP1-06 to EcClpP using isothermal titration calorimetry (ITC),17 which is close but not the same as the currently obtained value. Note that Kd measured by ITC is different from K0.5 (EC50) obtained from enzymatic assays since the latter is measured in the presence of substrate. Unfortunately, despite repeated attempts, no usable data could be obtained for the binding of ACP1-06 to NmClpP using ITC. Nevertheless, the apparent K0.5s obtained from the activity assays (Figure 5) clearly show that ACP1-06 binds with weaker affinity to NmClpP compared to EcClpP, which is consistent with the obtained structures (Figure 4).
Figure 5.
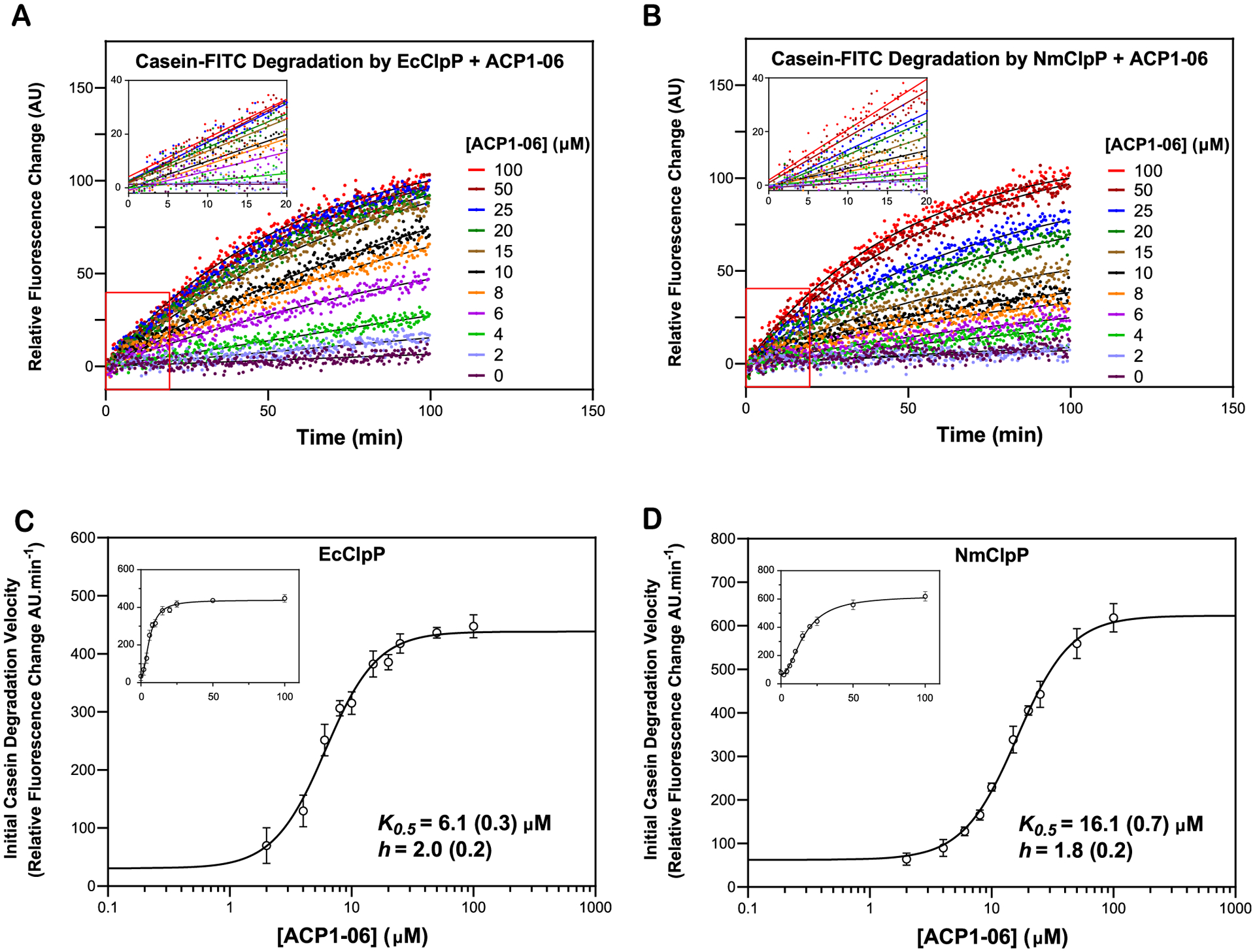
Measurement of the binding of ACP1-06 to EcClpP and NmClpP. (A,B) Shown are representative curves of casein-FITC degradation by EcClpP or NmClpP in the presence of different concentrations of ACP1-06. Data were normalized to the 100 μM ACP1-06 reaction. Experiments were repeated three times. Solid black lines show fits to single exponentials. Insets show the linear fits from 0.66 to 20 min. (C,D) K0.5 (equivalent to EC50) and h (Hill coefficient) for ACP1-06 binding to EcClpP and NmClpP were determined by fitting the change in initial degradation velocity of casein-FITC by compound-activated ClpP as a function of compound concentration to a Hill equation (see Methods). The data and fits are shown as semilog plots in the main figures and as regular plots in the insets. The data points in the absence of ACP1-06 are not shown on the semilog plots. Error bars represent the standard deviations from the average of three repeats.
On the basis of the above structural observations and affinity measurements, it is possible to consider that the differences in the binding of ACP1-06 to EcClpP versus NmClpP can be further exploited for the design of species-specific ClpP activators. Such compounds could then be further developed as antibiotics targeting specific bacterial species.
DISCUSSION
In this study, we have tested the activity of 85 ACP1 and 46 ADEP derivatives. These two classes of drugs have previously been shown to have antibacterial activity by virtue of their ability to dysregulate the cylindrical protease, ClpP, so that it actively degrades cytoplasmic proteins unchecked by the regulatory chaperones, such as ClpX and ClpA, with which it typically forms complexes.6,7,13,17 Our prior work revealed that the pathogenic Neisseria sp. were particularly sensitive to a variety of ClpP-targeting drugs, prompting us to use them as models with which to perform structure–activity relationship (SAR) studies to reveal features that may promote ClpP dysregulation.
While E. coli and Neisserial ClpP sequences are highly conserved (~70% identity),31 there are clear differences in the ability of some ACP1 and ADEP analogues to activate target protein degradation by these two protein variants. This suggests the possibility of developing new analogues with the desired specificity for pathogenic Neisseria but with minimal effect on other bacteria in the microbiome. The crystal structures of NmClpP and EcClpP in complex with ACP1-06, both of which our group determined,31 show that ACP1s bind these targets differently. The binding modes arise from different binding surfaces, especially near the axial pores. In particular, the A66 (EcClpP) to S57 (NmClpP) residue change (Figure 4D) eliminates the possibility of a narrow axial channel in NmClpP, whose equivalent location in EcClpP serves as a docking site for the ortho-chlorophenyl moiety of ACP1-06 (Figure 4E). However, an adjacent binding pocket exists in NmClpP where the said moiety binds in the crystal structure. The net effect is the significant reorganization of stabilizing protein–ligand interactions and the species-specific ACP1-06 binding modes. It was previously assumed that the “down configuration” of ACP1-06 in EcClpP, where the (trifluoromethyl)pyridine moiety binds deep into a narrow hydrophobic pocket,31 is the principal binding configuration responsible for its ClpP activating effects. Apparently, this binding configuration is not observed in NmClpP as the change in binding mode configuration prevents the (trifluoromethyl)pyridine moiety from reaching into the tunnel. Therefore, it is reasonable to propose that the ACP1-06 fragment proximal to the axial pore can be modified into aliphatic moieties similar to those found in ADEPs that bind NmClpP.14,31 This modification can potentially reorganize the electrostatic interaction networks near the axial pores and lead to greater activation of NmClpP.
Antibiotics are the only viable treatment option against Neisserial infection and yet resistance to classical antibiotics is unavoidable. The gonococci exemplify this point, since strains that are resistant to clinically relevant doses of antibiotics have begun to emerge.28 The novelty of ClpP as an antimicrobial target stems from the fact that it stimulates the unchecked proteolytic degradation of bacterial proteins, effectively digesting the bacterium from the inside out. It has been shown that the presence of a second antibiotic acting on a different pathway allows ClpP-activating compounds to kill even slow growing and persister strains.32 By rapidly destroying, rather than impeding the growth of bacteria in the tissues, this new class of ClpP activating compounds provides the exciting prospect of synergizing with the immune effector mechanisms to clear infection by otherwise difficult to treat pathogens.
MATERIALS AND METHODS
Synthesis of Additional ACP1 Analogues.
During the course of our investigations, it was observed that the synthesis of certain ACP1 analogues, particularly those which contained different heterocyclic frameworks on the western portion of the molecule, were not easily accessible using the aforementioned 4-step strategy (Figure 1) and alternate routes were developed. For example, for the ACP1 analogues synthesized in Figure S1A, bromoisobutyryl bromide was coupled with 3-phenylpropylamine to afford a brominated intermediate (condition i) that could be used to alkylate a variety of aryl- and heteroarylthiols (condition ii). The corresponding sulfides were oxidized late-stage to afford the desired ACP1 analogues (condition iii). This strategy provided access to a variety of analogues containing medicinally relevant heterocyclic moieties such as 2-pyrimidinyl (ACP1-58), 4-pyridyl (ACP1-70), 1,3,4-thiadiazolyl (ACP1-87), and 1-methyl-1H-imidazolyl (ACP1-88). Synthesis of the benzothiazole-containing analogue (ACP1-78) required an alternate synthetic approach to obtain the desired acid precursor (starting from 2-mercaptobenzothiazole using conditions iv and v), which was coupled with 3-phenylpropylamine (condition vi) and oxidized (condition iii) to afford the desired product.
A series of ACP1 analogues which replaced the gemdimethyl group in the central portion of the molecule with a variety of cycloalkyl groups (ring size = 3–6) was accomplished as outlined in Figure S1B. For example, 5-(trifluoromethyl)pyridine-2-thiol or 4-(trifluoromethyl)-benzenethiol were alkylated with benzyl bromoacetate to afford the corresponding sulfide products (condition i), which were then oxidized to the corresponding sulfones (condition ii).23 Using a known set of reaction conditions, bis-alkylation of the α-sulfonyl benzyl ester intermediates with various dibromoalkanes proceeded smoothly to afford the desired cycloalkylated products (condition iii).33 Deprotection of the benzyl ester groups using Pd-catalyzed hydrogenolysis afforded the desired acid precursors (condition iv), which were then coupled with a variety of amines (condition v) to yield the desired cycloalkylated analogues.
A series of functionalized ACP1 analogues, which contained polar functional groups on the eastern aryl portion of the molecule, were synthesized as outlined in Figure S1C. For example, 3-bromo-4-chlorobenzoic acid was alkylated with benzyl bromide under basic conditions to afford the benzyl ester product (condition i). The resulting aryl bromide was coupled with N-Boc-propargylamine using a Pd-catalyzed Sonogashira cross-coupling reaction to afford the desired coupled product, which was then deprotected with HCl to afford the corresponding ammonium salt (conditions ii and iii, respectively). Amide coupling of the HCl salt with acids using PyBOP afforded the desired amide products (condition iv), which were then subjected to Pd-catalyzed reduction; this led to reduction of the alkyne moiety as well as concomitant deprotection of the benzyl ester group to afford the desired acid precursors ACP1-38 and ACP1-41 (condition v). ACP1-41 was subsequently coupled with different amines using PyBOP to yield the desired analogues ACP1-26, 29, 36, and 40.
In a similar late-stage functionalization strategy, additional ACP1 analogues with polar functional groups on the eastern aryl portion of the molecule were prepared starting from aryl nitro analogues ACP1-21 and ACP1-37 as shown in Figure S1D. These were reduced to the corresponding anilines ACP1-08 and ACP1-11 using stannous chloride dihydrate (condition i). ACP1-11 was subsequently coupled with various nucleophiles (e.g., amines, phenol and thiophenol) in the presence of CDI (1,1′-carbonyldiimidazole, condition (ii) to afford the desired ACP1 analogues (e.g., ureas, carbamate and S-thiocarbamate, respectively). Alternatively, ACP1-11 was reacted with trimethylsilyl isocyanate or phenyl isocyanate to afford the corresponding urea analogues ACP1-80 or ACP1-81 (condition iii).
An ACP1 analogue containing an aryl-sulfonamide group on the eastern portion of the molecule (ACP1-02) was prepared starting from 4-bromo-3-(trifluoromethyl)benzenesulfonamide as outlined in Figure S1E. The aryl bromide was coupled with N-Boc-propargylamine using a Pd-catalyzed Sonogashira cross-coupling reaction (condition i) to afford the desired coupled product, which was then deprotected with trifluoroacetic acid (TFA) to afford the corresponding ammonium salt (conditions ii and iii, respectively). The final product was obtained following a Pd-catalyzed reduction (condition iv). Lastly, the synthesis of N-sulfonyl urea analogue ACP1-86 was prepared using an alternative approach involving trapping the isocyanate (derived from the corresponding N-sulfonyl carbamate) with 2-((2-chlorophenyl)thio)ethan-1-amine34 (Figure S1F).
It should be mentioned that ACP1 analogues which contained the 5-(trifluoromethyl)pyridyl functionality in the western portion of the molecule were prone to undergo an intramolecular Smiles-type rearrangement35 under the reaction conditions (forming a minor side product through the loss of SO2); for this reason, we recommend that reaction times for amide couplings be kept to a minimum and closely monitored by TLC and/or crude 1H NMR analysis to avoid side product formation. ACP1-71, ACP1-74, ACP1-75, ACP1-76, and ACP1-77 were isolated as mixtures of rearranged side-products and were not investigated further.
The characterization of all compounds is given in supplemental experimental procedures. HPLC chromatograms are given for ACP1-01, ACP1-06, ACP1-17, and ADEP-14.
ADEP natural products and related analogues were prepared according to known solution-phase14–16,22,36 and solid-phase21 methods.
Protein Expression and Purification.
All proteins were expressed from IPTG-inducible promoters. ClpP constructs were expressed in a BL21(DE3) 1146D strain, which lacks the gene for endogenous E. coli ClpP. EcClpP and NmClpP were expressed and purified generally as previously described for EcClpP.31,37 Protein concentrations were determined by absorbance at 280 nm with extinction coefficients calculated using ProtParam (http://ca.expasy.org/tools/protparam.html) or by Bradford assay (Bio-Rad Laboratories).
Briefly, EcClpP was expressed from a plasmid containing an IPTG inducible promoter in BL21(DE3) 1146 strain. Cells were grown to an OD600 of 0.6 and induced with 1 mM IPTG for 3 h at 37 °C. Harvested cells were resuspended in 50 mM TrisHCl pH 7.5, 150 mM KCl, 10% glycerol and lysed by French Press. Cell debris was removed by centrifugation at 15 000 rpm using a Sorval SS34 rotor at 4 °C. Solid ammonium sulfate was added slowly to the clarified supernatant at 4 °C with gentle stirring until the solution was 40% ammonium sulfate. Stirring continued for 60 min before precipitated proteins were removed by centrifugation at 15 000 rpm using a Sorval SS34 rotor at 4 °C for 15 min. Solid ammonium sulfate was added to the supernatant again at 4 °C with gentle stirring until the solution was 60% ammonium sulfate. The ClpP containing precipitate was collected by centrifugation at 15 000 rpm using a Sorval SS34 rotor at 4 °C for 30 min. The supernatant was discarded, and the pellet was dissolved in a small volume of 50 mM TrisHCl, pH 7.5, 150 mM KCl, 1 mM DTT, 10% glycerol (buffer A). The dissolved pellet solution was dialyzed extensively to remove residual ammonium sulfate before loading onto a Q-Sepharose column (all columns from GE Healthcare Life Sciences) equilibrated with buffer A. ClpP was eluted from the column with a linear gradient from 0 to 500 mM KCl in buffer A. ClpP eluted at approximately 250 mM. Elution fractions containing ClpP were pooled and dialyzed against 50 mM MES pH 6.0, 10% glycerol (buffer B). ClpP protein was loaded onto an SP-Sepharose column equilibrated with buffer B and eluted with a linear gradient from 0 to 250 mM KCl in buffer B. ClpP containing fractions were pooled and dialyzed against ClpP storage buffer (50 mM TrisHCl, pH 7.5, 200 mM KCl, 10% glycerol) before being snap frozen in liquid nitrogen and stored at −80 °C.
NmClpP was expressed and cells lysed as for EcClpP. Solid ammonium sulfate was added slowly to clarified lysate containing NmClpP at 4 °C with gentle stirring until the solution was 60% ammonium sulfate. Stirring continued for 60 min before precipitated proteins were removed by centrifugation at 15 000 rpm using a Sorval SS34 rotor at 4 °C for 30 min. The NmClpP-containing supernatant was dialyzed extensively against buffer A with several changes of buffer. NmClpP protein was loaded onto a Q-sepharose column equilibrated with buffer A. The protein was eluted with a linear gradient from 0 to 500 mM KCl in buffer A. Protein containing fractions were pooled and dialyzed back into buffer A. The protein was loaded onto a MonoQ 10/100 column equilibrated with buffer A and eluted with a linear gradient from 0 to 500 mM KCl in buffer A. The fractions containing NmClpP were pooled and dialyzed against ClpP storage buffer, snap frozen and stored at −80 °C.
Degradation Assays and Compound Screen.
For the RD25 index17 determination, each reaction mixture contained 3.6 μM ClpP, 2.5% DMSO and 25 μM of compound in buffer of 25 mM TrisHCl, pH 7.5, and 100 mM KCl in a total volume of 200 μL. The background control reaction consisted of the same reagents but lacking the compound. Reactions were carried out using black 96-well plates and were preincubated for 10 min at 37 °C before 4.5 μM casein-FITC and 15.5 μM unlabeled casein were added. EcClpAP-dependent degradation of the same casein-FITC substrate was used as a control. Each EcClpAP reaction contained 3.6 μM EcClpP, 3 μM EcClpA, and 0.3 mM ATP in PD+ buffer (25 mM HEPES, pH 7.5, 20 mM MgCl2, 30 mM KCl, 0.03% Tween 20, and 10% glycerol), and an ATP-regeneration system (13 units/mL of creatine kinase and 16 mM creatine phosphate). Change in fluorescence (485 nm excitation, 535 nm emission) was monitored over 6 h on an EnSpire (PerkinElmer) plate reader equipped with a fluorescence module. Casein-FITC degradation by compound-activated ClpP after 6 h at 37 °C was compared to the EcClpAP-dependent casein-FITC degradation after 6 h at 37 °C using the equation for RD25 that we previously established.17
Measurement of the Apparent Binding Constant of ACP1-06 for EcClpP and NmClpP.
EcClpP and NmClpP protease activities were assessed by measuring the degradation rate of bovine milk casein labeled with fluorescein isothiocyanate (casein-FITC, Sigma-Aldrich) in the presence of ACP1-06. Reactions were conducted in buffer containing 25 mM HEPES, pH 8.0, 100 mM KCl, 0.02% Triton X-100 (v/v), 1 mM DTT and 1% DMSO. ClpP was held at a final monomeric concentration of 3.6 μM, and ACP1-06 at 0–100 μM. Reactions were preincubated for 10 min at 37 °C before initiating the reaction with addition of 19.5 μM casein and 4.5 μM casein-FITC in a 96-well black flat-bottom plate. Fluorescence (485 nm excitation, 535 nm emission) was monitored for 100 min at 1 min intervals on an EnSpire Multilabel Plate Reader (PerkinElmer). Kinetic analysis was then performed by determining the initial velocities (0.66–20 min) from data collected over three independent experiments. Nonlinear regression analysis was performed on initial velocity data using the Hill equation:
υ0 is the initial velocity, Vmax is the maximal velocity, h is the Hill coefficient, and K0.5 is the microscopic apparent dissociation constant (equivalent to an EC50). The resulting K0.5 and h are given in Figure 5.
Neisseria Susceptibility Assay.
To determine the susceptibility concentration, Neisseria meningitidis H44/76 or Neisseria gonorrhoeae MS11 was grown as a lawn on GC-agar (BD Biosciences) with 1% IsoVitaleX (BD Biosciences) at 37 °C in a moist atmosphere with 5% CO2 for 16 to 18 h. Cells from the agar-media plates were collected with a sterile cotton swab and resuspended in Brain Heart Infusion (BHI, from Difco) growth media with 1% IsoVitaleX (Becton Dickinson, Sparks, USA) to about 106 CFU/mL. Each compound was diluted in growth media to 2× working concentration. 50 μL of the cell suspension was mixed with 50 μL of the diluted compound in a single well of a 96-well plate. These plates were sealed with Breathe-Easy sealing membrane (Sigma-Aldrich) and were then incubated at 37 °C in a moist atmosphere with 5% CO2. After 16 to 18 h, minimum inhibitory concentrations were determined optically as the lowest concentration of compound that completely inhibited growth. Treated cultures were then serially diluted in PBS+2 (PBS supplemented with MgCl2 and CaCl2) and spotted on GC agar plates supplemented with 1% IsoVitaleX and incubated for 18–20 h, following which, CFUs were counted. Treatment concentrations resulting in hazy or no growth were considered as inhibitory concentrations. For analysis, the percentage of CFUs recovered post-treatment to pretreatment are reported.
Antibacterial Activity.
The effects of four compounds on bacterial infectivity were tested using a previously described in vitro method30 with slight modifications. Briefly, HeLa cells stably expressing human CEACAM1 or CEACAM5, which allow meningococcal or gonococcal Opa-mediated adhesion and engulfment, were cultured in RPMI media supplemented with heat-inactivated FBS and glutaMAX to a confluency of 70%. Cells were then seeded overnight on a 24-well plate at a concentration of 105/mL. These cells were infected with meningococcal strain H44/76 or gonococcal strain MS11 at a calculated MOI of 10 for 4 h at 37 °C with 5% CO2 to allow bacterial adherence. Infected cells were washed twice with PBS+2 to remove any nonadherent bacteria, and treated with ACP1-01 (40 μg/mL), ACP1-06 (40 μg/mL), ACP1-17 (40 μg/mL), and ADEP-14 (0.3 μg/mL) or 1% DMSO (the solvent for the ACP1s and ADEPs) for 2 h, after which the infected cells were washed again with PBS+2. Total associated meningococci were quantified by determining CFUs.30 Infected and uninfected HeLa cells were analyzed microscopically to assess effects of treatment on mammalian cells, verifying that effects seen were not due to loss of the human cells.
Crystallization of NmClpP and Structure Determination of the NmClpP-ACP1-06 Complex.
Crystals of apo-NmClpP were grown at 23 °C by mixing at a 1:1 ratio a protein solution (11 mg/mL) with crystallization solution (0.2 M ammonium acetate, 40% 2-methyl-2,4-pentanediol (MPD)) on sitting drop plates. Large crystals grew within 2 weeks. To generate the NmClpP-ACP1-06 complex, solid ACP1-06 was added to the crystallization drops. The crystals were transferred to cryoprotectant solution containing 20% glycerol and immediately flash-frozen in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source-14-ID-B beamline (BioCARS), then indexed, integrated using DIALS,38 and scaled in the CCP4 package.39 The structure of the NmClpP-ACP1-06 complex was solved by molecular replacement using the apo-NmClpP structure as a search model.31 Initial refinement was performed in Phenix40 with simulated annealing and coordinate shaking to remove model bias, and subsequent modeling and refinement were performed in Coot41 and Phenix using translation-libration-screw (TLS) parameters with individual coordinate, B-factor and occupancy refinement. After complete modeling of protein and solvent, the ligand ACP1-06 was modeled into the electron density and refined as above. Initially, the occupancies of ACP1-06 modeled into the difference electron density map in two configurations were set equally at 0.5. Subsequent refinement as described above resulted in occupancies of 0.51 and 0.49 for the two configurations, suggesting that they appear in equal proportions in the crystal lattice. Atomic coordinates for the NmClpP-ACP1-06 complex have been deposited with PDB ID 6W9T (see also Table 2).
Supplementary Material
Supplementary Experimental Procedures; Characterization Data of ACP1 Analogues (PDF)
Figure S1: Detailed synthetic scheme for the remainder of the ACP1 analogues; (A–E) show detailed synthetic routes for the remainder of the ACP1 analogues; Table S2: Naming of ACP1 and ADEP analogues (PDF)
Antibacterial susceptibility of various meningococcal (8 strains) and gonococcal (14 strains) reference strains to ACP1-01, ACP1-06, ACP1-17, and ADEP-14; Values listed refer to percentage of CFU recovered in the presence of different compound concentrations; This table provides the data shown in Figure 3C (XLSX)
RD25 values of ACP1 and ADEP analogues in EcClpP, NmClpP, and HsClpP activation (XLSX)
ACKNOWLEDGMENTS
MFM was supported by the Precision Medicine Initiative (PRiME) at the University of Toronto internal fellowship number PMRF2019-007. JDG held the Natural Science and Engineering Research Council of Canada’s (NSERC) Postgraduate Scholarship-Doctoral (PGS-D) award. This work was supported by a CIHR Project grant (PJT-148564) to EFP, SDG, RAB, and WAH, and the Canada Research Chairs program (EFP). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DEAC02-06CH11357. Use of BioCARS was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number P41 GM118217. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.0c00599
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00599.
The authors declare no competing financial interest.
Contributor Information
Jordan D. Goodreid, Department of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada.
Vitor Hugo Balasco Serrão, Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Emil F. Pai, Department of Biochemistry and Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 1M1, Canada; Ontario Cancer Institute/Princess Margaret Hospital, Toronto, Ontario M5G 1L7, Canada;.
Robert A. Batey, Department of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada.
Scott D. Gray-Owen, Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Walid A. Houry, Department of Biochemistry and Department of Chemistry, University of Toronto, Toronto, Ontario M5G 1M1, Canada;.
REFERENCES
- (1).Baker TA, and Sauer RT (2006) ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem. Sci 31 (12), 647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Olivares AO, Baker TA, and Sauer RT (2016) Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol 14 (1), 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mahmoud SA, and Chien P (2018) Regulated Proteolysis in Bacteria. Annu. Rev. Biochem 87, 677–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bewley MC, Graziano V, Griffin K, and Flanagan JM (2009) Turned on for degradation: ATPase-independent degradation by ClpP. J. Struct. Biol 165 (2), 118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gottesman S (2019) Trouble is coming: Signaling pathways that regulate general stress responses in bacteria. J. Biol. Chem 294 (31), 11685–11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, and Houry WA (2018) The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol 13 (6), 1413–1425. [DOI] [PubMed] [Google Scholar]
- (7).Lin F, Mabanglo MF, Houry WA, and Batey RA (2019) Development of Small Molecules to Modulate the Activity of the ATP-Dependent ClpP Protease as a Novel Antibacterial and Anticancer Drug Target. In 2019 Medicinal Chemistry Reviews (Bronson JJ, Ed.) Vol. 54, pp 379–405. [Google Scholar]
- (8).Bottcher T, and Sieber SA (2009) Structurally refined beta-lactones as potent inhibitors of devastating bacterial virulence factors. ChemBioChem 10 (4), 663–6. [DOI] [PubMed] [Google Scholar]
- (9).Hackl MW, Lakemeyer M, Dahmen M, Glaser M, Pahl A, Lorenz-Baath K, Menzel T, Sievers S, Bottcher T, Antes I, Waldmann H, and Sieber SA (2015) Phenyl Esters Are Potent Inhibitors of Caseinolytic Protease P and Reveal a Stereogenic Switch for Deoligomerization. J. Am. Chem. Soc 137 (26), 8475–83. [DOI] [PubMed] [Google Scholar]
- (10).Mundra S, Thakur V, Bello AM, Rathore S, Asad M, Wei L, Yang J, Chakka SK, Mahesh R, Malhotra P, Mohmmed A, and Kotra LP (2017) A novel class of Plasmodial ClpP protease inhibitors as potential antimalarial agents. Bioorg. Med. Chem 25 (20), 5662–5677. [DOI] [PubMed] [Google Scholar]
- (11).Moreira W, Ngan GJ, Low JL, Poulsen A, Chia BC, Ang MJ, Yap A, Fulwood J, Lakshmanan U, Lim J, Khoo AY, Flotow H, Hill J, Raju RM, Rubin EJ, and Dick T (2015) Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio 6 (3), No. e00253–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Moreno-Cinos C, Sassetti E, Salado IG, Witt G, Benramdane S, Reinhardt L, Cruz CD, Joossens J, Van der Veken P, Brotz-Oesterhelt H, Tammela P, Winterhalter M, Gribbon P, Windshugel B, and Augustyns K (2019) alpha-Amino Diphenyl Phosphonates as Novel Inhibitors of Escherichia coli ClpP Protease. J. Med. Chem 62 (2), 774–797. [DOI] [PubMed] [Google Scholar]
- (13).Brotz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, and Labischinski H (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med 11 (10), 1082–7. [DOI] [PubMed] [Google Scholar]
- (14).Goodreid JD, Janetzko J, Santa Maria JP Jr., Wong KS, Leung E, Eger BT, Bryson S, Pai EF, Gray-Owen SD, Walker S, Houry WA, and Batey RA (2016) Development and Characterization of Potent Cyclic Acyldepsipeptide Analogues with Increased Antimicrobial Activity. J. Med. Chem 59 (2), 624–46. [DOI] [PubMed] [Google Scholar]
- (15).Socha AM, Tan NY, LaPlante KL, and Sello JK (2010) Diversity-oriented synthesis of cyclic acyldepsipeptides leads to the discovery of a potent antibacterial agent. Bioorg. Med. Chem 18 (20), 7193–202. [DOI] [PubMed] [Google Scholar]
- (16).Carney DW, Schmitz KR, Truong JV, Sauer RT, and Sello JK (2014) Restriction of the conformational dynamics of the cyclic acyldepsipeptide antibiotics improves their antibacterial activity. J. Am. Chem. Soc 136 (5), 1922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Leung E, Datti A, Cossette M, Goodreid J, McCaw SE, Mah M, Nakhamchik A, Ogata K, El Bakkouri M, Cheng YQ, Wodak SJ, Eger BT, Pai EF, Liu J, Gray-Owen S, Batey RA, and Houry WA (2011) Activators of cylindrical proteases as antimicrobials: identification and development of small molecule activators of ClpP protease. Chem. Biol 18 (9), 1167–78. [DOI] [PubMed] [Google Scholar]
- (18).Virji M (2009) Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol 7 (4), 274–86. [DOI] [PubMed] [Google Scholar]
- (19).Read RC (2014) Neisseria meningitidis; clones, carriage, and disease. Clin. Microbiol. Infect 20 (5), 391–5. [DOI] [PubMed] [Google Scholar]
- (20).Wi T, Lahra MM, Ndowa F, Bala M, Dillon JR, Ramon-Pardo P, Eremin SR, Bolan G, and Unemo M (2017) Antimicrobial resistance in Neisseria gonorrhoeae: Global surveillance and a call for international collaborative action. PLoS medicine 14 (7), No. e1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Goodreid JD, dos Santos ES, and Batey RA (2015) A Lanthanide(III) Triflate Mediated Macrolactonization/Solid-Phase Synthesis Approach for Depsipeptide Synthesis. Org. Lett 17 (9), 2182–2185. [DOI] [PubMed] [Google Scholar]
- (22).Goodreid JD, Wong K, Leung E, McCaw SE, Gray-Owen SD, Lough A, Houry WA, and Batey RA (2014) Total synthesis and antibacterial testing of the A54556 cyclic acyldepsipeptides isolated from Streptomyces hawaiiensis. J. Nat. Prod 77 (10), 2170–81. [DOI] [PubMed] [Google Scholar]
- (23).Brown PJ, Winegar DA, Plunket KD, Moore LB, Lewis MC, Wilson JG, Sundseth SS, Koble CS, Wu Z, Chapman JM, Lehmann JM, Kliewer SA, and Willson TM (1999) A ureido-thioisobutyric acid (GW9578) is a subtype-selective PPARalpha agonist with potent lipid-lowering activity. J. Med. Chem 42 (19), 3785–8. [DOI] [PubMed] [Google Scholar]
- (24).Faucher AM, White PW, Brochu C, Grand-Maitre C, Rancourt J, and Fazal G (2004) Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase. J. Med. Chem 47 (1), 18–21. [DOI] [PubMed] [Google Scholar]
- (25).Berry A, Cirillo PF, Hickey ER, Riether D, Thomson DS, Ermann M, Jenkins JE, Mushi I, Taylor M, Chowdhury C, Palmer CF, and Blumire N (2008) Compounds which modulate the CB2 receptor, CA2657247A1.
- (26).Wong KS, Mabanglo MF, Seraphim TV, Mollica A, Mao YQ, Rizzolo K, Leung E, Moutaoufik MT, Hoell L, Phanse S, Goodreid J, Barbosa LRS, Ramos CHI, Babu M, Mennella V, Batey RA, Schimmer AD, and Houry WA (2018) Acyldepsipeptide Analogs Dysregulate Human Mitochondrial ClpP Protease Activity and Cause Apoptotic Cell Death. Cell chemical biology 25 (8), 1017–1030. [DOI] [PubMed] [Google Scholar]
- (27).Brehony C, Wilson DJ, and Maiden MCJ (2009) Variation of the factor H-binding protein of Neisseria meningitidis. Microbiology 155 (12), 4155–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Unemo M, Del Rio C, and Shafer WM (2016) Antimicrobial Resistance Expressed by Neisseria gonorrhoeae: A Major Global Public Health Problem in the 21st Century. Microbiol Spectr 4 (3), 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sadarangani M, Pollard AJ, and Gray-Owen SD (2011) Opa proteins and CEACAMs: pathways of immune engagement for pathogenic Neisseria. FEMS microbiology reviews 35 (3), 498–514. [DOI] [PubMed] [Google Scholar]
- (30).Gray-Owen SD, Dehio C, Haude A, Grunert F, and Meyer TF (1997) CD66 carcinoembryonic antigens mediate interactions between Opa-expressing Neisseria gonorrhoeae and human polymorphonuclear phagocytes. EMBO J 16 (12), 3435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mabanglo MF, Leung E, Vahidi S, Seraphim TV, Eger BT, Bryson S, Bhandari V, Zhou JL, Mao YQ, Rizzolo K, Barghash MM, Goodreid JD, Phanse S, Babu M, Barbosa LRS, Ramos CHI, Batey RA, Kay LE, Pai EF, and Houry WA (2019) ClpP protease activation results from the reorganization of the electrostatic interaction networks at the entrance pores. Commun. Biol 2, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, and Lewis K (2013) Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503 (7476), 365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Alonso DA, Nájera C, and Varea M (2002) Synthesis and Reactivity of π-Electron-Deficient (Arylsulfonyl)acetates. Helv. Chim. Acta 85 (12), 4287–4305. [Google Scholar]
- (34).Neustadt BR (1994) Facile preparation of N-(sulfonyl)-carbamates. Tetrahedron Lett 35 (3), 379–380. [Google Scholar]
- (35).Holden CM, and Greaney MF (2017) Modern Aspects of the Smiles Rearrangement. Chem. - Eur. J 23 (38), 8992–9008. [DOI] [PubMed] [Google Scholar]
- (36).Hinzen B, Raddatz S, Paulsen H, Lampe T, Schumacher A, Habich D, Hellwig V, Benet-Buchholz J, Endermann R, Labischinski H, and Brotz-Oesterhelt H (2006) Medicinal chemistry optimization of acyldepsipeptides of the enopeptin class antibiotics. ChemMedChem 1 (7), 689–93. [DOI] [PubMed] [Google Scholar]
- (37).Wojtyra UA, Thibault G, Tuite A, and Houry WA (2003) The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J. Biol. Chem 278 (49), 48981–90. [DOI] [PubMed] [Google Scholar]
- (38).Winter G, Waterman DG, Parkhurst JM, Brewster AS, Gildea RJ, Gerstel M, Fuentes-Montero L, Vollmar M, Michels-Clark T, Young ID, Sauter NK, and Evans G (2018) DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. Sec. D, Struct. Biol 74 (2), 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Potterton L, Agirre J, Ballard C, Cowtan K, Dodson E, Evans PR, Jenkins HT, Keegan R, Krissinel E, Stevenson K, Lebedev A, McNicholas SJ, Nicholls RA, Noble M, Pannu NS, Roth C, Sheldrick G, Skubak P, Turkenburg J, Uski V, von Delft F, Waterman D, Wilson K, Winn M, and Wojdyr M (2018) CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr. Sec. D, Struct. Biol 74 (2), 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Experimental Procedures; Characterization Data of ACP1 Analogues (PDF)
Figure S1: Detailed synthetic scheme for the remainder of the ACP1 analogues; (A–E) show detailed synthetic routes for the remainder of the ACP1 analogues; Table S2: Naming of ACP1 and ADEP analogues (PDF)
Antibacterial susceptibility of various meningococcal (8 strains) and gonococcal (14 strains) reference strains to ACP1-01, ACP1-06, ACP1-17, and ADEP-14; Values listed refer to percentage of CFU recovered in the presence of different compound concentrations; This table provides the data shown in Figure 3C (XLSX)
RD25 values of ACP1 and ADEP analogues in EcClpP, NmClpP, and HsClpP activation (XLSX)