

Abstract

The potential anticancer effect of fluoroquinolone antibiotics has been recently unveiled and related to their ability to interfere with DNA topoisomerase II. We herein envisioned the design and synthesis of novel Ciprofloxacin and Norfloxacin nitric oxide (NO) photo-donor hybrids to explore the potential synergistic antitumor effect exerted by the fluoroquinolone scaffold and NO eventually produced upon light irradiation. Anticancer activity, evaluated on a panel of tumor cell lines, showed encouraging results with IC50 values in the low micromolar range. Some compounds displayed intense antiproliferative activity on triple-negative and doxorubicin-resistant breast cancer cell lines, paving the way for their potential use to treat aggressive, refractory and multidrug-resistant breast cancer. No significant additive effect was observed on PC3 and DU145 cells following NO release. Conversely, antimicrobial photodynamic experiments on both Gram-negative and Gram-positive microorganisms displayed a significant killing rate in Staphylococcus aureus, accounting for their potential effectiveness as selective antimicrobial photosensitizers.
Introduction
Since their early discovery as byproducts of chloroquine synthesis,1 quinolones have represented one of the most important classes of antibiotics for urinary and respiratory infection treatment.2−4 Quinolones exert their bactericidal activity by interfering with DNA gyrase in Gram-negative bacteria and topoisomerase IV in Gram-positive bacteria.5 Both enzymes belong to the topoisomerase family, which plays an essential role in the regulation of the DNA topological state, in DNA replication, and in the condensation and segregation of chromosomes.6,7 In the presence of quinolone, the enzyme forms a DNA/enzyme/drug ternary complex that perturbs DNA replication, leading to bacterial death or eukaryotic cell apoptosis.
Structural modifications of the first marketed compound of this class of molecules, nalidixic acid, generated compounds with greater potency, a broader spectrum of activity, improved pharmacokinetics, and lower frequency of acquired resistance. In particular, Norfloxacin (Nor) and Ciprofloxacin (Cip), belonging to the second-generation fluoroquinolones (Figure 1), displayed increased potency and affinity for Gram-negative bacteria due to the introduction of a fluorine atom at position 6. Subsequent structure modifications led to third- and fourth-generation fluoroquinolones, with improved efficacy against Gram-positive organisms.2,8,9 Also, structure–activity relationship (SAR) studies highlighted the importance of the N-1 substituent on the quinolone core together with the presence of either a carboxylic acid function in position 3 and a ketone function in position 4. Furthermore, to expand their spectrum of action and improve pharmacokinetics, a saturated heterocyclic ring containing an amine function was introduced at the 7-position from the second-generation fluoroquinolones.10
Figure 1.

Chemical structures of Ciprofloxacin, Norfloxacin, and Vosaroxin.
Of note, recent evidence indicates that higher doses of these drugs exert anticancer effects,11,12 and as expected, this effect is related to their ability to interfere with DNA topoisomerase II, the gyrase human counterpart, which is a well-known target of several anticancer drugs, such as epipodophyllotoxins (etoposide), anthracyclines (doxorubicin and daunorubicin), amsacrine, and mitoxantrone. In this view, we envisioned the design and synthesis of a novel class of 4-quinolone-based topoisomerase II inhibitors endowing the enhancement of their cytotoxic activity.
SAR studies on anticancer quinolones (Figure 2A,B)4,13−15 allowed identifying the types of structure modifications boosting anticancer activity, such as the reduction of their zwitterionic character by modifying the carboxylic group at the 3-position or by adding a proper substituent on the aliphatic heterocyclic amine at the 7-position. Out of these studies, Vosaroxin16 has reached phase III clinical trial investigation (Figure 1).17 Interestingly, Vosaroxin, such as other molecules of the same class, appears to be devoid of the typical side effects of topoisomerase II inhibitors, namely, significant cardiotoxicity and cross-resistance with other topoisomerase II inhibitors, while preserving the cytotoxic effect in multidrug-resistant (MDR) or inactivated p53 cell lines.13
Figure 2.

Antimicrobial (A) and antitumoral (B) structure–activity relationships of 4-quinolones.
The combination of different therapeutic functionalities with synergic or additive features within the same molecular scaffold represents a fascinating opportunity to improve the overall treatment efficacy. Furthermore, the possibility of administering a single drug bearing multiple biological “effectors” could allow higher control on pharmacokinetic and side effects while boosting the treatment outcome.18−22
In this context, the use of molecules able to release nitric oxide (NO) under the exclusive control of light, e.g., NO photo-donors, has been recently described for antibacterial and anticancer treatment.23,24 NO is physiologically produced by the NO synthase enzyme family from l-arginine and O225 and performs multiple physiological roles ranging from the control of the vascular tone to neurotransmission.26,27 Moreover, NO possesses antimicrobial properties28 and has been shown to be a key player in cancer biology, where its role seems to be regulated by several factors, such as the tumor cell subtype, NO cell sensitivity, exposure time, and cellular concentration.29 In fact, unlike pico- and nanomolar NO concentrations are known to boost cancer progression and invasiveness, micromolar NO concentrations promote cytotoxicity30 and interfere with P-glycoprotein activity,31 thus representing a promising option to tackle MDR phenomena. Due to the intrinsic difficulties posed by the direct administration of gaseous NO, specific NO donors,32 alone or included in nanomaterials, have been actively investigated.33,34 In particular, molecules able to release NO upon the application of an external stimulus, such as light, have attracted increasing attention due to the possibility of precisely controlling the NO production and release only at the site of interest.35−42
Based on the above, we herein report the design, synthesis, characterization, and molecular modeling studies of 12 Cip and Nor derivatives, endowed with a 4-nitro-3-trifluoromethyl-aniline moiety for the light-triggered release of NO. The new derivatives’ biological activity has also been evaluated, both in prokaryotic and tumor cells, along with the effect of NO release on cell viability and compared with Cip and Nor.
Results and Discussion
Design and Synthesis of Novel Ciprofloxacin and Norfloxacin Hybrids
The new Cip and Nor derivatives are characterized by a carboxylic or a methyl ester group at the 3-position and a NO photo-donor directly linked to the N-4 atom of the piperazine ring (compounds 1a, 1b, 3a, and 3b) or connected through an alkyl spacer made of two or three methylene units (compounds 6a–6d and 7a–7d) (Table 1).
Table 1. General Structure of Novel Ciprofloxacin and Norfloxacin NO Photo-Donor Hybrids.


The strategy developed for synthesizing final compounds 1a, 1b, 3a, and 3b is depicted in Scheme 1. Carboxylic acids 1a and 1b were prepared following a one-step procedure involving an aromatic nucleophilic substitution between 4-fluoro-1-nitro-2-(trifluoromethyl)benzene and Cip or Nor in DMSO at 120 °C for 1 h. Methyl esters 3a and 3b were synthesized in two steps, including a Fisher esterification between Cip or Nor with refluxing methanol, and p-toluenesulfonic acid (22 h) to afford esters 2a and 2b that were subsequently reacted with 4-fluoro-1-nitro-2-(trifluoromethyl)benzene in refluxing acetonitrile overnight to provide final compounds 3a and 3b.
Scheme 1. Synthetic Strategy for the Synthesis of Compounds 1–3a,b.

Reagents and conditions: (i) 4-fluoro-1-nitro-2-(trifluoromethyl)benzene, DMSO, 120 °C, 1 h; (ii) CH3OH, p-TsOH, 70 °C, 22 h; (iii) 4-fluoro-1-nitro-2-(trifluoromethyl)benzene, CH3CN, 80 °C, overnight.
Scheme 2 reports the synthetic pathway to achieve final compounds 6a–6d and 7a–7d. Starting from 4-fluoro-1-nitro-2-(trifluoromethyl)benzene, intermediates 4a and 4b were prepared through an aromatic nucleophilic substitution with 2-aminoethanol or 3-aminopropan-1-ol in acetonitrile at 60 °C overnight. Mesylation of the alcoholic function of compounds 4a and 4b provided the intermediates 5a and 5b that were reacted with derivatives 2a and 2b in refluxing acetonitrile overnight. The obtained methyl esters 6a–6d were hydrolyzed with a refluxing NaOH aqueous solution (2 M) for 24 h to afford the corresponding carboxylic acids 7a–7d.
Scheme 2. Synthetic Strategy for the Synthesis of Final Compounds 6a–6d and 7a–7d.

Reagents and conditions: (i) 1-aminoethanol or 1-aminopropan-3-ol, CH3CN, 60 °C, overnight; (ii) CH3SO2Cl, TEA, dry CH2Cl2, 0 °C, then room temperature, 1 h; (iii) 2a and 2b, CH3CN, reflux, overnight; (iv) aqueous NaOH 2 M, reflux, 24 h.
Spectroscopic and Photochemical Characterization of Novel Ciprofloxacin and Norfloxacin Derivatives
To investigate the spectroscopic behavior of the synthesized compounds, carboxylic acids 1a, 1b, and 7a–7d were selected for recording absorption and fluorescence spectra, showing the characteristic peaks at 400 and 450 nm, respectively (Figure S25). The amount of NO released from the selected compounds upon light irradiation was quantified using the Griess assay, in which NO2, generated upon the reaction of released NO with oxygen, reacts with the Griess reagent, generating a purple azo dye that can be spectroscopically monitored following its absorption peak at ∼540 nm.43 Therefore, aqueous solutions of compounds 1a, 1b, and 7a–7d (80 μM) were treated with the Griess reagent and irradiated with a white lamp for different time intervals (15 min, 1 h, and 2 h). All performed analyses showed the development of a light purple coloration, confirming a weak NO release, especially after 1 h irradiation time. Although the absorption peak of the analyte solution may weakly skew absorbance measurements at 540 nm, the obtained data indicate a trend in NO production; in particular, the amount of NO released seems to be in the order 7b > 1b > 7d > 1a = 7a > 7c. Furthermore, the nitrite concentration was quantified by measuring the absorbance at 540 nm with respect to a standard curve of NaNO2 in H2O. Data reported in Figure 3 confirmed a significant nitrite production by 7b, 1b, and 7d following 1 h irradiation, particularly evident for 7b ([NO2–] = 6.2 μM), while the extent of NO2 generated by other compounds was almost negligible. Except for compound 1b, these results suggest that longer spacers between the piperazine ring and the NO-donor moiety might favor the NO production yield.
Figure 3.
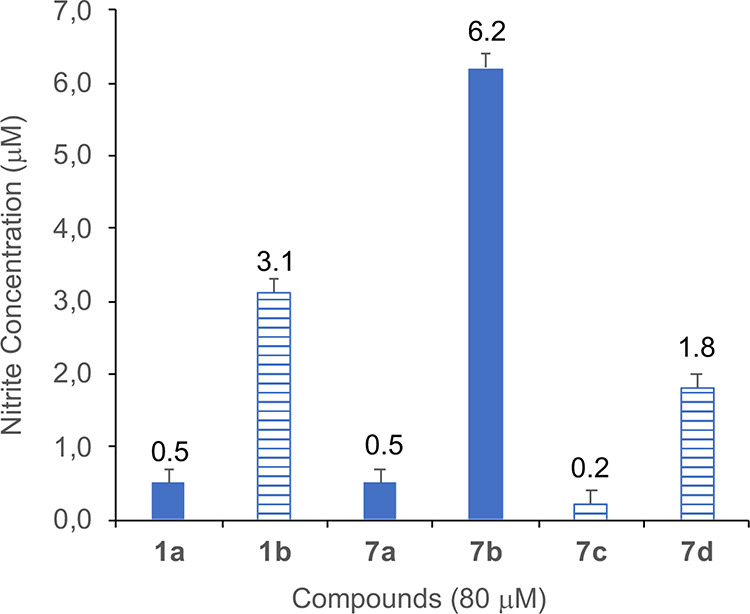
NO detection by a Griess test. Effects of 80 μM solution of 1a, 1b, and 7a–7d on NO production after 1 h of white light irradiation (mean ± SD of three independent experiments). Nitrite concentration was determined by comparing the test samples’ absorbance values to a standard curve generated by serial dilution of 50 μM NaNO2.
In Vitro Antitumor Activity Evaluation of Novel Fluoroquinolone Derivatives
The effect of the novel hybrid derivatives on cellular viability of a panel of tumor cell lines of different tissue origins (DU145 and PC3: prostate; MCF-7, MCF7/ADR, and MDA-MB231: breast; HCT116: colon) was investigated through the MTT assay following 3 days treatment with the compounds. Histograms reported in Figures 4 and 5 represent the IC50 values extrapolated from the corresponding concentration–response curves. In each graph, IC50 values of new derivatives were compared to the reference compounds, e.g., Cip or Nor. All compounds showed effects at micromolar concentrations (Tables S1 and S2).
Figure 4.

IC50 values obtained following 72 h treatment with Ciprofloxacin (C) and its derivatives in the MTT assay (mean ± ES 4/5 independent experiments; *p < 0.05, **p < 0.01, ***p < 0.001 vs Cip).
Figure 5.

IC50 values obtained following 72 h treatment with Norfloxacin (N) and its derivatives in the MTT assay (mean ± ES 4/5 independent experiments; °p < 0.05, °°p < 0.01, °°°p < 0.001 vs Nor).
Figure 4 shows that all Cip derivatives were significantly more potent than the parent compound in DU145 and PC3 prostate cancer cells in HCT116 colorectal cancer cells (the more potent being compound 7b, with an IC50 = 1.83 μM) and in MDA-MB231 breast cancer cells (IC50 values lower than 2.50 μM for compounds 7a and 7b) while only compounds 7a and 7b were more potent than Cip in MCF7 cells (Table S1). In general, carboxylic acids 7a and 7b, where the alkyl linker connects the fluoroquinolone scaffold to the NO photo-donor moiety, showed lower IC50 values when compared to their corresponding methyl esters 6a and 6b. Specifically, this trend was more pronounced for compound 7a, which was 2- to 7-fold more potent than 6a in the tested cell lines. In contrast, the difference in potency was lower when comparing 6b and 7b. These data indicate that a shorter alkyl bridge improves the cytotoxic effect (7avs7b). On the other hand, results obtained for methyl esters 6a and 6b suggest that a longer alkyl chain provides better cytotoxic properties. Finally, compounds lacking the alkyl bridge (1a and 3a) displayed an ambiguous trend on the tested cell lines. Indeed, DU145 and HCT116 cell lines were more sensitive to the methyl ester 3a, whereas the PC3 and MDA-MB231 cell lines displayed a slight susceptibility toward the carboxylic acid 1a.
A similar trend was observed for the SAR of the novel Nor hybrids, except for the better activity of carboxylic acid 1b in all the tested cell lines when compared to its methyl ester analog 3b. In particular, results obtained in DU145 and PC3 cells showed that all Nor derivatives were significantly more potent than the reference compound, with best results obtained for 7d in the DU145 cell line (IC50 = 1.56 μM) and 7c in the PC3 cell line (IC50 = 2.33 μM). In MCF7 and MDA-MB231 cells, compounds 1b and 7d showed a significantly lower IC50 than Nor, while 7c was the most potent among all the novel hybrids in both breast cancer cell lines (IC50 = 2.27 and 1.52 μM for MCF7 and MDA-MB231, respectively). Interestingly, in HCT116 cells, only 7c and 7d resulted to be more potent than Nor (Figure 5 and Table S2).
Last, the effects of all the newly synthesized compounds on cell viability were not limited to cancer cell lines; indeed, as shown on Table 2, which report the IC50 values obtained by the MTT assay following 72 h treatment with the compounds, also the non-tumorigenic HBL100 and the WH1 fibroblast cell lines responded to Cip, Nor, and to their derivatives at similar extent compared to cancer cell lines (Table 2).
Table 2. IC50 Values Obtained with the MTT Assay following 72 h Treatment with Ciprofloxacin (Cip), Norfloxacin (Nor), and Their Derivatives.
| IC50 (μM) ± ESa |
IC50 (μM) ± ESa |
||||
|---|---|---|---|---|---|
| Compd | WH1 | HBL100 | Compd | WH1 | HBL100 |
| 1a | 9.17 ± 0.60 | 7.79 ± 0.66 | 1b | 5.35 ± 1.14b | 9.26 ± 1.79 |
| 3a | 8.38 ± 1.20 | 10.57 ± 2.40 | 3b | 9.00 ± 1.91 | 11.95 ± 1.70 |
| 6a | 10.5 ± 2.50 | 9.76 ± 0.90 | 6c | 10.63 ± 1.90 | 10.85 ± 0.60 |
| 6b | 13.2 ± 3.40 | 12.21 ± 2.00 | 6d | 9.67 ± 1.65 | 3.68 ± 0.20b |
| 7a | 5.70 ± 0.92 | 9.14 ± 0.44 | 7c | 3.51 ± 0.40b | 11.09 ± 0.20 |
| 7b | 7.05 ± 1.20 | 9.19 ± 0.66 | 7d | 8.94 ± 0.91 | 10.40 ± 0.90 |
| Cip | 14.2 ± 2.10 | 10.28 ± 1.00 | Nor | 14.07 ± 2.10 | 12.43 ± 0.80 |
Mean ± ES 4/5 independent experiments.
p < 0.05 vs Nor.
To evaluate if NO released upon light irradiation could affect cell viability, the MTT assay was performed on DU145 and PC3 cells following treatment with two compounds that showed interesting potency against the two prostate cancer cell lines and different degrees of NO release. Specifically, the Cip and the Nor derivative that showed to develop the highest and the lowest NO levels by the Griess assay, namely, 7b and 7c, respectively, were selected for this experiment. Treatment was followed by 1 h irradiation with white or blue light and 48 h incubation in a drug-free medium. As shown in Figure 6, no differences in IC50 values were observed in cells treated with 7b and 7c and irradiated (both with white or blue light) with respect to cells treated and kept in the dark. However, compound 7c seems to be more active on DU145 cells when irradiated under blue light with respect to white light.
Figure 6.

IC50 values obtained following 24 h treatment with compound 7b or 7c with 1 h irradiation and 48 h incubation in the drug-free medium and MTT assay (mean ± ES 3/4 independent experiments. *p < 0.05 vs white light).
Overall, these results might indicate that the amount of NO released could be inadequate to induce cell death, thus confirming that the NO release does not play an additive role in the new fluoroquinolone derivatives’ toxic effect.
A significant obstacle to the successful chemotherapeutic treatment of tumors is their inadequate response to anticancer drugs due to intrinsic or acquired resistance phenomena. Tumor cells can be insensitive to drug treatment at the therapy onset (intrinsic resistance), or they can initially respond to anticancer agents, becoming refractory on subsequent treatment cycles (acquired resistance). For instance, the development of MDR following an initial drug response severely limits the success of doxorubicin (DOX) treatment of breast cancers.44,45 DOX is an anthracycline antibiotic targeting topoisomerase II and represents a mainstay in clinical management of early-stage and metastatic breast cancer. MDR to DOX involves few biochemical alterations, such as reduced drug accumulation, increased detoxification, increased DNA repair, topoisomerase II alterations, or in cell cycle regulation. In this scenario, identifying MDR-modulating agents or drugs able to escape MDR phenomena is of utmost importance in anticancer research.
As expected, the DOX-resistant MCF7/ADR cell line, obtained by selecting MCF7 cells exposed to increasing DOX concentrations, is significantly less sensitive to anthracycline treatment with respect to the wild-type cell line, with a resistance index (R.I.) of 22 (Figure 7). Interestingly, MCF7/ADR cells showed similar sensitivity to several Cip and Nor derivatives (Table 3) as observed for the MCF7 cell line, with the best IC50 values obtained for carboxylic acids 1a and 1b (8.72 and 5.63 μM, respectively).
Figure 7.
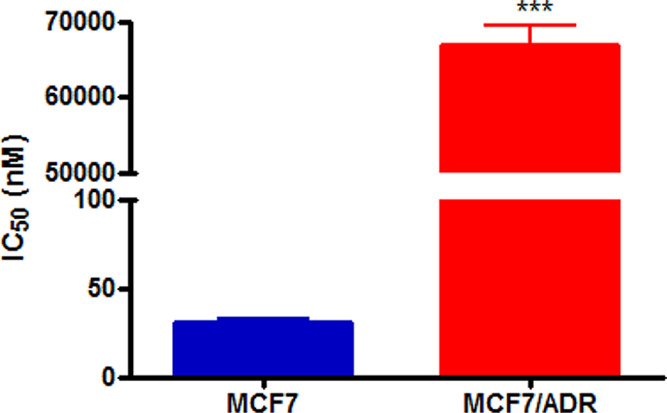
IC50 values obtained following 72 h treatment with DOX and the MTT assay (mean ± ES 4/5 independent experiments; ***p < 0.001 vs MCF7).
Table 3. IC50 Values Obtained with the MTT Assay following 72 h Treatment with DOX and Ciprofloxacin (Cip)/Norfloxacin (Nor) Derivatives.
| IC50 (μM) ± ESa |
IC50 (μM) ± ESa |
||||||
|---|---|---|---|---|---|---|---|
| Compd | MCF7 | MCF7/ADR | R.I. | Compd | MCF7 | MCF7/ADR | R.I. |
| 1a | 8.80 ± 1.59 | 8.72 ± 1.25 | 1.0 | 1b | 5.24b ± 0.70 | 5.63b ± 0.07 | 1.1 |
| 3a | 15.45 ± 1.20 | 19.55 ± 3.20 | 1.3 | 3b | 11.68 ± 1.40 | 15.04 ± 2.10 | 1.3 |
| 6a | 16.84 ± 2.40 | 15.12 ± 2.30 | 0.9 | 6c | 15.04 ± 0.10 | 17.15 ± 1.00 | 1.1 |
| 6b | 12.12 ± 1.10 | 18.76 ± 0.10 | 1.5 | 6d | 10.53 ± 1.20 | 28.04 ± 1.10 | 2.7 |
| 7a | 3.12 ± 0.62 | 14.69 ± 4.20 | 4.7 | 7c | 2.27b ± 0.31 | 18.31 ± 1.40 | 8.1 |
| 7b | 5.23 ± 0.82 | 23.55 ± 3.60 | 4.5 | 7d | 5.83 ± 0.82 | 20.02 ± 0.30 | 3.4 |
| Cip | 8.85 ± 0.09 | 19.12 ± 0.50 | 2.2 | Nor | 19.37 ± 0.70 | 20.06 ± 1.40 | 1.0 |
| DOX | 0.031 ± 0.001 | 66.90 ± 2.56 | 22 | ||||
Mean ± ES 4/5 independent experiments.
p < 0.05 vs reference compound.
In summary, the in vitro experiments showed that derivatives 7a and 7b are the most effective Cip analogs against all considered cell lines and with respect to the reference compound, indicating that regardless of the linker length, the carboxylic acid moiety outperforms the ester function (Figure 8). In line with these results, Nor derivatives 7c and 7d displayed a similar trend, and compound 7c was the best performing of the series (Figure 8). Cytotoxicity studies performed on MCF7/ADR resistant cell lines confirmed that the carboxylic moiety is essential for the anticancer activity and that only derivatives 1a and 1b, where the p-nitro-trifluoromethyl aniline is directly linked to the fluoroquinolone scaffold, can provide IC50 values in the low micromolar range (Figure 8).
Figure 8.

Summary of the most potent compounds synthesized in this work with their IC50 values on the tested cancer cell lines.
In Vitro Antimicrobial Activity on Pseudomonas aeruginosa
Considering that fluoroquinolones, such as Cip and Nor, are among the few antibiotics used to control the growth of P. aeruginosa, an opportunistic pathogen representing one of the most relevant agents of nosocomial infections, we preliminarily determined whether the novel derivatives could also be effective against the model strain PAO1. Our results indicate that, while Cip and Nor had minimal inhibitory concentration (MIC) values of 1.82 ± 1.19 and 4.16 ± 1.80 μg/mL, respectively, none of our newly designed derivatives showed antimicrobial activity comparable or better than reference compounds (MIC ∼25 μg/mL). These results might indicate that the fluoroquinolone scaffold’s structural changes compromise the antimicrobial activity, probably due to an impaired interaction with the microbial cell wall and/or with the bacterial target, e.g., the DNA gyrase. To rule out the first hypothesis, we investigated the ability of our novel compounds to bind the PAO1 cell wall. Binding experiments showed that upon incubation of P. aeruginosa PAO1 cells with our fluoroquinolones, a decrease in absorbance was observed in the supernatants (Figures S26 and S27), indicating good binding rates with the outer layer of the cell wall. However, it should be underlined that a good interaction between the molecules and the bacterial cell wall is not predictive of an effective uptake.
Since most compounds showed an absorption peak in the 390–395 nm region, we next investigated their activity upon irradiation with blue light. For a preliminary evaluation, we selected compound 7c, showing the highest absorption peak in the violet-blue range and some degree of activity against DU145 cancer cells. Upon irradiation with a light-emitting diode at 405 ± 10 nm and a fluence not toxic to cells (20 J/cm2), compound 7c did not show any antimicrobial activity against P. aeruginosa PAO1 (Figure 9a), indicating that despite its interaction with the cell wall, this is not sufficient to induce killing by photo-oxidative stress.
Figure 9.
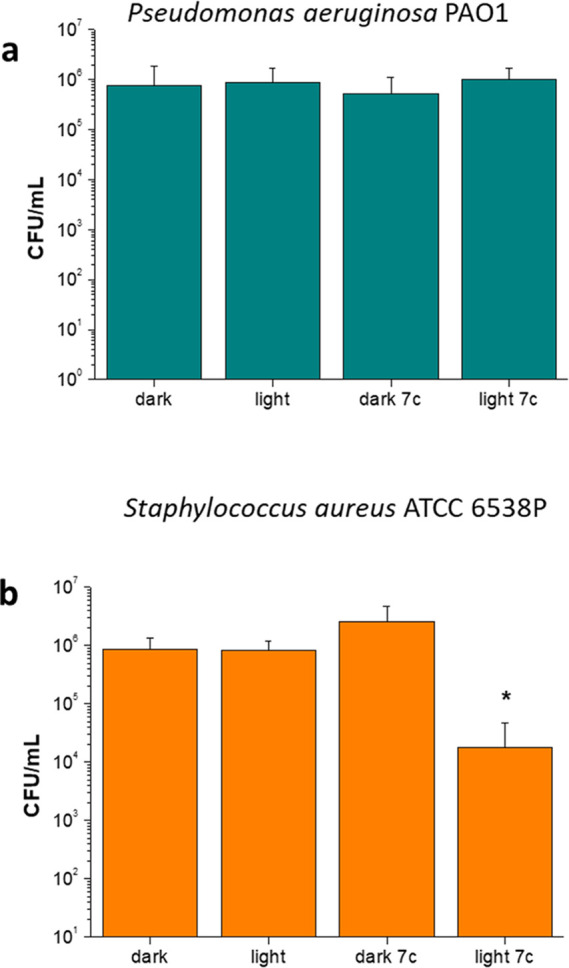
Photodynamic treatment of P. aeruginosa PAO1 (a) and S. aureus ATCC 6538P (b). Bacteria were incubated in the dark with or without 7c (10 μM) for 10 min and then irradiated under blue light (20 J/cm2). Dark controls were not irradiated. Viable cells are expressed as CFU/mL. Values represent the mean of at least three independent experiments, *p < 0.05.
It is well established that one of the major challenges in treating Gram-negative bacteria, with respect to the Gram-positive ones, is the difficulty of antimicrobial agents to strongly bind and cross their cell wall, which significantly differs in composition as compared to the latter. In order to shed light on the specific behavior of our fluoroquinolones, we then evaluated the effectiveness of compound 7c in killing Gram-positive Staphylococcus aureus. Interestingly, the photoactivation of 7c caused a significant (p < 0.05) 2 log unit decrease in S. aureus with respect to the dark incubated control (Figure 9b). This observation might support the hypothesis that compound 7c is able to efficiently bind the cell wall and probably cross the cytoplasmic membrane of S. aureus, ultimately inducing a photo-oxidative stress at the cell-wall level and/or at the cytoplasmic level, thus causing cellular death upon light irradiation. This result indirectly further demonstrates that despite the fact that our fluoroquinolones bind the cell wall of P. aeruginosa, this is not sufficient to induce bacterial killing by photoactivation. The selective activity of derivative 7c toward Gram-positive bacteria indicates that our derivatives act as neutral antimicrobial photosensitizers (aPSs). In fact, it has been reported that neutral photosensitizers are usually active toward Gram-positive microorganisms and not in the Gram-negative ones, being able to overcome the murein barrier of the first, but not the outer membrane of the cell wall of the latter.46 Based on the observed selectivity of 7c, it could be hypothesized that despite the fact that molecular modeling studies (see the following paragraph) account for the effective binding of fluoroquinolones with both topoisomerase II and gyrase, their biological activity against P. aeruginosa is hampered by an ineffective uptake. In the future, this could be overcome by incorporating these molecules within suitably designed carriers.
Computational Studies
Molecular modeling studies were carried out to investigate the interactions with the reference cellular and bacterial targets. The calculated free energies of binding (ΔG) to the catalytic site of the human topoisomerase IIα (Topo IIα), bacterial topoisomerase IIA (Topo IIA), and bacterial gyrase (Gyr) for the novel compounds are reported in Table 4. First, the crystal structure of the Topo IIα isoform (PDB ID: 5GWK) was used for docking studies.47
Table 4. Calculated Free Energies of Binding (ΔG, in kcal/mol) of the Novel Compounds to the Catalytic Sites of Topo IIα, Topo IIA, and Gyr.
| Compd | calcd. ΔG human Topo IIα | calcd. ΔG bacterial Topo IIA | calcd. ΔG bacterial Gyr |
|---|---|---|---|
| Cip | –9.1 | –11.1 | –7.9 |
| Nor | –8.5 | –10.6 | –7.0 |
| 1a | –11.6 | –11.8 | –8.4 |
| 1b | –10.8 | –10.8 | –8.1 |
| 3a | –10.8 | –9.68 | –8.1 |
| 3b | –10.7 | –12.1 | –9.3 |
| 6a | –10.7 | –13.8 | –9.8 |
| 6b | –10.1 | –10.7 | –8.1 |
| 6c | –10.5 | –11.9 | –9.2 |
| 6d | –10.0 | –13.4 | –9.4 |
| 7a | –11.3 | –13.8 | –9.7 |
| 7b | –11.4 | –14.1 | –8.9 |
| 7c | –10.6 | –12.9 | –9.2 |
| 7d | –13.2 | –15.1 | –10.5 |
| DOX | –13.5 | –12.5 | –12.5 |
All compounds displayed better in silico affinity toward Topo IIα than Cip and Nor, while DOX showed greater affinity, as demonstrated by experimental data. The Cip derivatives have lower free energies of binding than the Nor derivatives, except for compounds 6d and 7d. The 1a carboxyl group coordinates Mg2+ via a salt bond of 1.73 Å (Figure 10A). Furthermore, the complex is stabilized, within the catalytic site, by the H-bond with the Gly760 and Asp541 backbone, while the nitro group establishes electrostatic interactions with Arg487 (Figure 10B).
Figure 10.

3D superposition of the best-docked pose for 1a bound to the Topo IIα (A) and binding site interactions (B). RMSD superposing on the receptor (C) and starting structure (D).
The protein aids the anchoring of the ligand within the pocket by a cation-π link formed between Arg487 and the nitro group portion of the ligand. The neighboring DNA bases also contribute to the stabilization of the complex.
Once the general interaction model was established, equilibrium molecular dynamics (MD) simulations (100 ns) were performed to analyze the Topo IIα/DNA/1a ternary model system’s evolution and stability. The RMSDs of the tertiary structures (Topo IIα/DNA/1a) compared to the first ones at time 0 were analyzed and plotted during the 100 ns MD simulation (Figure 10C). The overall RMSD for the protein system appeared to have reached the equilibrium after 10 ns (Figure 10D), and the stabilization of the protein–ligand complex was reached after 7 ns, keeping the complex’s extensive hydrogen-bonding network constant. The energies of binding, calculated by the Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA) methodology (see the Experimental Section), including the time average, along MD simulation trajectories were employed to assess the strength of the interactions between the ligand and the binding pocket in the dynamic environment. Compound 1a shows a stable fluctuation that settles after the first 10 ns and records an average value of −17.4 kcal/mol in the remaining 90 ns (Figure S28).
The docked laying of 1b is very similar to that of 1a despite a loss of 0.8 kcal/mol, probably due to the ethyl group’s worse stacking. It is interesting to note that compounds 7a, 7b, and 7d have lower calculated free energies of binding than other compounds against human Topo IIα. Indeed, these compounds have shown excellent results in the tested cancer cell lines.
All the novel compounds were anchored in the active sites of Topo IIA (PDB ID: 2XCT) of S. aureus and DNA gyrase B (PBD ID: 6MS1) of P. aeruginosa. Anchored poses with the lowest bond energy, hydrogen bonds, noncovalent interactions, such as the π–π interactions, and the details of the π-cationic interactions were recorded and validated.
All the compounds analyzed showed better in silico affinity on bacterial topoisomerase than Cip and Nor, except compound 3a, while all compounds showed lower in silico activity against DNA gyrase. Despite the good in silico results, the compounds have a worse antimicrobial activity in vitro than the reference drugs (Cip and Nor). It seems that the novel compounds bind to the outer membrane of the Gram-negative cell wall, but the crossing of the cytoplasmic membrane to reach the cytoplasmic environment and then the molecular target could represent the issue to overcome. Thus, further investigations are needed to shed light on the observed impairment of novel derivatives’ antimicrobial activity.
Conclusions
In this work, we successfully managed to design and synthesize twelve novel Cip and Nor derivatives endowed with a NO photo-donor moiety. The light-triggered release of NO has been demonstrated by spectroscopic and photochemical studies, showing the release of this gasotransmitter in the micromolar range, especially for compound 7b. Docking studies confirmed that these novel chemical entities effectively bind to both bacterial and human topoisomerases, with better calculated free binding energies with respect to the parent compounds. P. aeruginosa PAO1 was not sensitive to the novel derivatives, and photoactivation experiments support the hypothesis that this could be ascribable to an inefficient uptake.
As far as anticancer activity is concerned, all novel fluoroquinolone derivatives displayed strong anticancer potency on a panel of different cancer cell lines, which was especially remarkable for compounds 7a–7d. On the other hand, the light-triggered release of NO from compounds 7b and 7c did not grant an additional cytotoxic effect on PC3 and DU145 prostate cancer cell lines, although a better response to the compounds was observed following blue light irradiation with respect to white light irradiation. Further studies focused on the precise mechanism of action of these compounds and on the role of NO in these cell lines are in progress.
Importantly, our data showed that some of the tested compounds, including compounds 1a, 1b, and 7a–7d, exhibit cytotoxic effects on MDA-MB231 cells, which are representative of triple-negative breast cancer (TNBC), one of the most aggressive and refractory forms of breast cancer. TBNC does not respond to endocrine therapy or other currently available targeted agents;48−50 thus, alternative therapeutic options that can selectively address this tumor subset are urgently needed. In this view, the promising results obtained with our novel derivatives on MDA-MB231 cells represent an encouraging starting point for developing and optimizing more effective treatment. Furthermore, compounds 1a and 1b also displayed a strong cytotoxic effect on DOX-resistant MCF7/ADR breast cancer cells, making these hybrids promising candidates for MDR breast cancer treatment. As expected, most of our fluoroquinolone derivatives displayed a certain extent of toxicity on healthy cells; this issue could be overcome in the future by encapsulating those molecules within suitably designed delivery systems.
Experimental Section
General Remarks
Reagent-grade chemicals were purchased from Sigma-Aldrich or Fluorochem and were used without further purification. All reactions were monitored by thin-layer chromatography (TLC) performed on silica gel Merck 60 F254 plates; the spots were visualized by UV light (λ = 254 and 366 nm) and an iodine chamber. Melting points were determined on a Büchi B-450 apparatus in capillary glass tubes and are uncorrected. Flash chromatography purification was performed on a Merck silica gel 60, 0.040–0.063 mm (230–400 mesh) stationary phase using glass columns with a diameter between 1 and 4 cm. Nuclear magnetic resonance spectra (1H NMR and 13C NMR recorded at 500 and 125 MHz, respectively) were obtained on Varian INOVA spectrometers using CDCl3, acetone-d6, CD3OD, and DMSO-d6 with a 0.03% of TMS as an internal standard. Coupling constants (J) are reported in hertz. Signal multiplicities are characterized as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), and app (apparent). Purities of all compounds were ≥95% as determined by microanalysis (C, H, and N) that was performed on a Carlo Erba instrument model E1110; all the results agreed within ±0.4% of the theoretical values.
General Procedure for the Synthesis of Carboxylic Acids 1a and 1b
To a suspension of Cip or Nor (0.100 g, 0.3 mmol) in 5 mL of DMSO was added 4-fluoro-1-nitro-2-(trifluoromethyl)benzene (0.063 g, 0.3 mmol) under stirring. The color of the suspension immediately turned yellow. The reaction was carried out in a sealed Pyrex vial at 120 °C for 60 min. After cooling to room temperature, the product was precipitated with deionized water and decanted. The solid yellow residue was repeatedly washed in sequence with deionized water, isopropanol, and diethyl ether. The obtained solid was dried under N2 and did not require any further purification. According to this procedure, the following products have been obtained.
1-Cyclopropyl-6-fluoro-7-(4-(4-nitro-3-(trifluoromethyl)phenyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (1a)
Yellow solid (89%): mp 296–298 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.67 (s, 1H), 8.13 (d, J = 9.5 Hz, 1H), 7.94 (d, J = 13.5 Hz, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.37 (d, J = 2.5 Hz, 1H), 7.31 (dd, J = 9.5, 2.5 Hz, 1H), 3.86–3.82 (m, 1H), 3.78 (t, J = 5.0 Hz, 4H), 3.54 (t, J = 4.8 Hz, 4H), 1.32 (q, J = 7.0 Hz, 2H), 1.22–1.19 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 176.35, 165.92, 152.89, 148.10, 144.59 (d, JCF = 9.9 Hz), 139.19, 135.59, 128.85, 124.33, 121.36, 118.59, 115.29, 111.67 (d, JCF = 5.1 Hz), 111.08 (d, JCF = 22.3 Hz), 106.76, 106.18, 48.53, 45.96, 35.87, 7.60. Anal. Calcd. for C24H20F4N4O5: C, 55.39; H, 3.87; N, 10.77. Found: C, 55.42; H, 3.89; N, 10.74.
1-Ethyl-6-fluoro-7-(4-(4-nitro-3-(trifluoromethyl)phenyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (1b)
Yellow solid (74%): mp 310–312 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.97 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.97 (d, J = 13.0 Hz, 1H), 7.36 (s, 1H), 7.31 (d, J = 9.0 Hz, 1H), 7.22 (d, J = 7.0 Hz, 1H), 4.61 (q, J = 7.0 Hz, 2H), 3.77 (br s, 4H), 3.53 (br s, 4H), 1.43 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 176.25, 166.06, 153.06, 148.58, 144.86, 137.19, 135.77, 128.80, 124.35, 119.81, 119.32, 117.47, 115.32, 111.68 (d, JCF = 6.25 Hz), 111.42 (d, JCF = 28.7 Hz), 105.78, 48.64, 46.06, 41.00, 14.29. Anal. Calcd. for C23H20F4N4O5: C, 54.33; H, 3.96; N, 11.02. Found: C, 54.50; H, 3.94; N, 10.99.
General Procedure for the Synthesis of Ciprofloxacin and Norfloxacin Methyl Esters 2a and 2b
To a warmed suspension of the starting fluoroquinolone (1.00 g, 3.00 mmol) in 100 mL of CH3OH was dropped p-toluenesulfonic acid (3.00 g, 15.7 mmol) dissolved in 5 mL of CH3OH via a syringe. The resulting solution was refluxed for 22 h. The reaction mixture was then cooled to room temperature, and the reaction solvent was removed under vacuum. To the resulting yellow oil was added a saturated solution of Na2CO3 (50 mL), and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated at reduced pressure. The crude was purified by flash chromatography using a CH2Cl2/CH3OH gradient eluting system. According to this procedure, the following products have been obtained.
Methyl 1-Cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (2a)
White solid (91%): mp 230–233 °C; 1H NMR (500 MHz, CDCl3): δ 8.54 (s, 1H), 8.02 (d, J = 13.3 Hz, 1H), 7.29–7.27 (m, 1H), 3.91 (s, 3H), 3.46–3.42 (m, 1H), 3.29–3.27 (m, 4H), 3.15–3.13 (m, 4H), 2.40 (s, 2H), 1.33 (t, J = 6.5 Hz, 2H), 1.14 (q, J = 6.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 173.24, 166.61, 153.61 (d, JCF = 247.5 Hz), 148.46, 145.15 (d, JCF = 10.0 Hz), 138.18, 123.14 (d, JCF = 7.5 Hz), 113.46 (d, JCF = 22.5 Hz), 110.21, 104.87, 52.20, 51.19, 46.06, 34.65, 8.28. Anal. Calcd for C18H20FN3O3: C, 62.60; H, 5.84; N, 12.17. Found: C, 62.78; H, 5.86; N, 12.19.
Methyl 1-Ethyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (2b)
White solid (92%): mp 189–190 °C; 1H NMR (500 MHz, CDCl3): δ 8.42 (s, 1H), 8.06 (d, J = 13.0 Hz, 1H), 6.73 (d, J = 7.0 Hz, 1H), 4.20 (q, J = 7.5 Hz, 2H), 3.92 (s, 3H), 3.22–3.21 (m, 4H), 3.10–3.08 (m, 4H), 1.92 (s, 2H), 1.54 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 173.15, 166.72, 153.39 (d, JCF = 238.0 Hz), 148.30, 145.36 (d, JCF = 10.5 Hz), 136.22, 123.87 (d, JCF = 6.6 Hz), 113.80 (d, JCF = 22.9 Hz), 110.26, 103.85, 52.17, 51.36, 49.10, 46.09, 14.50. Anal. Calcd for C17H20FN3O3: C, 61.25; H, 6.05; N, 12.61. Found: C, 61.32; H, 6.06; N, 12.58.
General Procedure for the Synthesis of Methyl Esters 3a and 3b
To a solution of the appropriate fluoroquinolone methyl ester (2a or 2b) in anhydrous CH3CN (10 mL) at 40 °C was added 4-fluoro-1-nitro-2-(trifluoromethyl)benzene under stirring. The color of the solution immediately turned yellow. The temperature was raised up to 80 °C, and the reaction mixture was left under stirring overnight. The solvent was evaporated, and the crude was diluted with CH2Cl2 (50 mL) and washed with deionized water (3 × 25 mL). The organic phase was dried over Na2SO4, filtered, and reduced under vacuum. The residue was purified by flash chromatography using a CH2Cl2/CH3OH gradient eluting system. According to this procedure, the following products have been obtained.
Methyl 1-Cyclopropyl-6-fluoro-7-(4-(4-nitro-3-(trifluoromethyl)phenyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (3a)
The title compound was obtained using 0.265 g (0.77 mmol) of 2a and 0.172 g (0.82 mmol) of 4-fluoro-1-nitro-2-(trifluoromethyl)benzene. Yellow solid (61%): mp 265–267 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.44 (s, 1H), 8.12 (d, J = 9.5 Hz, 1H), 7.78 (d, J = 13.5 Hz, 1H), 7.47 (d, J = 7.5 Hz, 1H), 7.37 (d, J = 2.5 Hz, 1H), 7.31 (dd, J = 9.5, 2.5 Hz, 1H), 3.77–3.74 (m, 4H), 3.73 (s, 3H), 3.68–3.63 (m, 1H), 3.44–3.42 (m, 4H), 1.27–1.23 (m, 2H), 1.13–1.10 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 173.12, 166.52, 153.44 (d, JCF = 247.5 Hz), 153.15, 148.65, 143.88 (d, JCF = 12.5 Hz), 138.15 (d, JCF = 10.0 Hz), 128.70, 126.46 (d, JCF = 33.8 Hz), 123.9 (d, JCF = 7.5 Hz), 123.47, 121.29, 115.41, 113.91 (d, JCF = 22.5 Hz), 112.72 (d, JCF = 6.3 Hz), 110.56, 105.03, 52.30, 49.54, 47.29, 34.67, 8.36. Anal. Calcd. for C25H22F4N4O5: C, 56.18; H, 4.15; N, 10.48. Found: C, 56.31; H, 4.13; N, 10.51.
Methyl 1-Ethyl-6-fluoro-7-(4-(4-nitro-3-(trifluoromethyl)phenyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (3b)
The title compound was obtained using 0.500 g (1.5 mmol) of 2b and 0.314 g (1.5 mmol) of 4-fluoro-1-nitro-2-(trifluoromethyl)benzene. Yellow solid (52%): mp 236–238 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.65 (s, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.83 (d, J = 13.0 Hz, 1H), 7.37 (d, J = 2.5 Hz, 1H), 7.31 (dd, J = 9.5, 3.0 Hz, 1H), 7.08 (d, J = 7.0 Hz, 1H), 4.42 (q, J = 7.0 Hz, 2H), 3.76–3.74 (m, 7H), 3.43–3.41 (m, 4H), 1.38 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 171.46, 165.10, 153.28, 152.93, 151.31, 148.85, 143.63 (d, JCF = 10.0 Hz), 136.16, 135.54, 128.83, 124.59, 123.54, 122.74 (d, JCF = 6.3 Hz), 115.34, 111.92 (d, JCF = 22.5 Hz), 111.72 (d, JCF = 6.3 Hz), 109.15, 105.88, 51.16, 48.86, 48.05, 46.17, 14.23. Anal. Calcd. for C24H22F4N4O5: C, 55.17; H, 4.24; N, 10.72. Found: C, 55.28; H, 4.25; N, 10.75.
2-((4-Nitro-3-(trifluoromethyl)phenyl)amino)ethanol (4a)
To a solution of 2-aminoethanol (1.00 g, 16.4 mmol) in anhydrous CH3CN (10 mL) was added 4-fluoro-1-nitro-2-(trifluoromethyl)benzene (2.32 g, 11.1 mmol). The reaction was left under stirring at 60 °C overnight. Then, the reaction solvent was removed under reduced pressure and the residue was dissolved in EtOAc and washed with a saturated solution of NaHCO3 (3 × 25 mL). The organic phase was dried over Na2SO4, filtered, and concentrated under vacuum. The crude was purified by flash chromatography eluting with a 20% of Cy in EtOAc to give the desired product. Yellow solid (72%): 1H NMR (500 MHz, CD3OD): δ 8.01 (d, J = 9.0 Hz, 1H), 7.04 (d, J = 2.5 Hz, 1H), 6.81 (dd, J = 9.0, 2.5 Hz, 1H), 3.74 (t, J = 5.5 Hz, 2H), 3.35 (t, J = 5.5 Hz, 2H); 13C NMR (125 MHz, CD3OD): δ 154.65, 136.41, 130.34, 127.27 (t, JCF = 18.4 Hz), 125.08, 122.92, 113.05, 112.26, 61.15, 46.26. Anal. Calcd. for C9H9F3N2O3: C, 43.21; H, 3.63; N, 11.20. Found: C, 43.12; H, 3.64; N, 11.22.
3-((4-Nitro-3-(trifluoromethyl)phenyl)amino)propan-1-ol (4b)
To a solution of 3-aminopropan-1-ol (0.143 g, 1.90 mmol) in anhydrous CH3CN (5 mL) was added 4-fluoro-1-nitro-2-(trifluoromethyl)benzene (0.200 g, 0.95 mmol). The reaction was left under stirring at 60 °C for 12 h in a closed glass Pyrex vial. After the reaction was complete, the reaction solvent was removed under reduced pressure and the residue was repeatedly triturated with n-hexane till the formation of a yellow solid. The solid was decanted, solubilized in EtOAc, and washed with a saturated solution of NaHCO3 (3 × 25 mL). The organic phase was dried over Na2SO4, filtered, and reduced under pressure, affording the pure desired product that did not require any further purification. Yellow solid (quantitative): 1H NMR (500 MHz, acetone-d6): δ 8.03 (d, J = 8.5 Hz, 1H), 7.09 (d, J = 3.0 Hz, 1H), 6.88 (dd, J = 6.5, 3.0 Hz, 1H), 6.67 (br s, 1H), 3.82–3.68 (m, 3H), 3.40 (q, J = 6.5 Hz, 2H), 1.90–1.85 (m, 2H); 13C NMR (125 MHz, acetone-d6): δ 154.19, 136.02, 130.26, 126.74 (t, JCF = 38.3 Hz), 123.68 (d, JCF = 270.8 Hz), 112.00 (d, JCF = 145.5 Hz), 60.01, 40.97, 29.45. Anal. Calcd. for C10H11F3N2O3: C, 45.46; H, 4.20; N, 10.60. Found: 45.33; H, 4.21; N, 10.63.
General Procedure for the Synthesis of Methanesulfonates 5a and 5b
To a solution of the appropriate alcohol (4a or 4b) in anhydrous CH2Cl2 was added TEA at 0 °C. The reaction mixture was left under stirring for 30 min, and then methanesulfonyl chloride was added dropwise using a dropping funnel. The reaction was left under stirring at room temperature for 60 min. After this time, the solvent was removed under reduced pressure, the resulting residue was dissolved in CH2Cl2 and washed in sequence with a saturated solution of NH4Cl (25 mL) and a saturated solution of NaHCO3. The organic phase was dried over Na2SO4, filtered, and evaporated. The crude products were directly used in the next step with no further purification or characterization. According to this procedure, the following products have been synthesized.
2-((4-Nitro-3-(trifluoromethyl)phenyl)amino)ethyl Methanesulfonate (5a)
The title compound was obtained using 0.100 g (0.4 mmol) of 4a in 5 mL of anhydrous CH2Cl2, 166 μL (1.2 mmol) of TEA, and 62 μL (0.8 mmol) of methanesulfonyl chloride. Yellow oil (quantitative).
3-((4-Nitro-3-(trifluoromethyl)phenyl)amino)propyl Methanesulfonate (5b)
The title compound was obtained using 0.300 g (1.13 mmol) of 4b in 10 mL of anhydrous CH2Cl2, 0.345 g (3.40 mmol) of TEA, and 0.328 g (2.26 mmol) of methanesulfonyl chloride. Yellow oil (quantitative).
General Procedure for the Synthesis of Methyl Esters 6a–6d
In a two-neck round-bottom flask, the appropriate fluoroquinolone methyl ester (2a or 2b) was added to the appropriate methanesulfonate solution (5a or 5b) in anhydrous CH3CN under a N2 atmosphere. The reaction mixture was refluxed under stirring overnight. Then, the solvent was removed under reduced pressure, and a saturated solution of NaHCO3 was added to the residue. The aqueous phase was extracted with CH2Cl2 (3 × 25 mL), and then the organic phases were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by flash chromatography using a CH2Cl2/CH3OH gradient eluting system. According to this procedure, the following products have been obtained.
Methyl 1-Cyclopropyl-6-fluoro-7-(4-(2-((4-nitro-3-(trifluoromethyl)phenyl)amino)ethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (6a)
The title compound was synthesized using 0.276 g (0.8 mmol) of 2a in 15 mL of anhydrous CH3CN and 0.129 g (0.39 mmol) of 5a. Yellow solid (69%): mp 231–233 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.44 (s, 1H), 8.08 (d, J = 9.5 Hz, 1H), 7.75 (d, J = 13.0 Hz, 1H), 7.47 (t, J = 5.0 Hz, 1H), 7.43 (d, J = 7.5 Hz, 1H), 7.16 (s, 1H), 6.88 (dd, J = 9.0, 2.5 Hz, 1H), 3.73 (s, 3H), 3.66–3.62 (m, 1H), 3.37 (app. q, J = 6.0 Hz, 2H), 3.28–3.24 (m, 4H), 2.70–2.65 (m, 4H), 2.63 (t, J = 6.0 Hz, 2H), 1.26–1.24 (m, 2H), 1.11–1.08 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 171.52, 164.95, 153.57, 152.38 (d, JCF = 193.0 Hz), 148.22, 143.85 (d, JCF = 9.6 Hz), 138.05, 133.55, 129.70, 124.82 (d, JCF = 32.9 Hz), 123.63, 121.79 (d, JCF = 6.1 Hz), 121.46, 111.50 (d, JCF = 22.6 Hz), 108.98, 106.18, 55.97, 52.42, 51.23, 49.52, 34.71, 7.51. Anal. Calcd. for C27H27F4N5O5: C, 56.15; H, 4.71; N, 12.13. Found: C, 55.94; H, 4.70; N, 12.17.
Methyl 1-Cyclopropyl-6-fluoro-7-(4-(3-((4-nitro-3-(trifluoromethyl)phenyl)amino)propyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (6b)
The title compound was synthesized using 0.350 g (1.01 mmol) of 2a in 15 mL of anhydrous CH3CN and 0.195 g (0.57 mmol) of 5b. Yellow solid (50%): mp 202–204 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.44 (s, 1H), 8.08 (d, J = 9.5 Hz, 1H), 7.75 (d, J = 13.5 Hz, 1H), 7.59 (t, J = 5.5 Hz, 1H), 7.43 (d, J = 7.5 Hz, 1H), 7.07 (s, 1H), 6.85 (dd, J = 9.5, 2.5 Hz, 1H), 3.73 (s, 3H), 3.67–3.62 (m, 1H), 3.29–3.23 (m, 6H), 2.61–2.56 (m, 4H), 2.46 (t, J = 7.0 Hz, 2H), 1.77 (m, 2H), 1.27–1.23 (m, 2H), 1.11–1.08 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 171.53, 164.96, 152.42 (d, JCF = 203.8 Hz), 148.22, 143.88 (d, JCF = 10.0 Hz), 138.06, 133.38, 129.77, 125.02, 123.62, 121.77 (d, JCF = 6.3 Hz), 121.45, 111.50 (d, JCF = 22.5 Hz), 108.99, 106.16, 54.87, 52.48, 51.24, 49.57, 40.42, 34.70, 25.40, 7.51. Anal. Calcd. for C28H29F4N5O5: C, 56.85; H, 4.94; N, 11.84. Found: C, 57.01; H, 4.95; N, 11.81.
Methyl 1-Ethyl-6-fluoro-7-(4-(2-((4-nitro-3-(trifluoromethyl)phenyl)amino)ethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (6c)
The title compound was synthesized using 0.400 g (1.2 mmol) of 2b in 15 mL of anhydrous CH3CN and 0.197 g (0.6 mmol) of 5a. Yellow solid (61%): mp 245–247 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.64 (s, 1H), 8.08 (d, J = 9.5 Hz, 1H), 7.79 (d, J = 13.5 Hz, 1H), 7.47 (t, J = 5.0 Hz, 1H), 7.15 (s, 1H), 7.03 (d, J = 7.0 Hz, 1H), 6.88 (dd, J = 9.5, 2.5 Hz, 1H), 4.40 (q, J = 7.0 Hz, 2H), 3.73 (s, 3H), 3.37 (q, J = 6.0 Hz, 2H), 3.27–3.25 (m, 4H), 2.67–2.65 (m, 4H), 2.62 (t, J = 6.0 Hz, 2H), 1.37 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 171.46, 165.11, 153.40, 152.29 (d, JCF = 213.5 Hz), 148.78, 144.16 (d, JCF = 10.3 Hz), 136.16, 133.53, 129.71, 124.81 (d, JCF = 31.9 Hz), 123.62, 122.54 (d, JCF = 6.3 Hz), 121.45, 111.81 (d, JCF = 22.4 Hz), 109.10, 105.69, 55.96, 52.45, 51.14, 49.54, 48.02, 14.19. Anal. Calcd. for C26H27F4N5O5: C, 55.22; H, 4.81; N, 12.38. Found: C, 55.40; H, 4.80; N, 12.40.
Methyl 1-Ethyl-6-fluoro-7-(4-(3-((4-nitro-3-(trifluoromethyl)phenyl)amino)propyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (6d)
The title compound was synthesized using 0.465 g (1.4 mmol) of 2b in 15 mL of anhydrous CH3CN and 0.240 g (0.7 mmol) of 5b. Yellow solid (50%): mp 216–218 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.64 (s, 1H), 8.08 (d, J = 9.5 Hz, 1H), 7.78 (d, J = 13.5 Hz, 1H), 7.59 (t, J = 5.5 Hz, 1H), 7.07 (s, 1H), 7.03 (d, J = 7.0 Hz), 6.84 (dd, J = 9.0, 2.0 Hz, 1H), 4.40 (q, J = 7.0 Hz, 2H), 3.73 (s, 3H), 3.28–3.20 (m, 6H), 2.60–2.54 (br m, 4H), 2.45 (t, J = 7.0 Hz, 2H), 1.77 (m, 2H), 1.37 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 171.47, 165.11, 153.40, 152.33 (d, JCF = 223.4 Hz), 148.78, 144.18 (d, JCF = 10.3 Hz), 136.16, 133.37, 129.81, 125.00, 123.61, 122.52 (d, JCF = 6.3 Hz), 121.44, 111.79 (d, JCF = 22.5 Hz), 109.10, 105.66, 54.88, 52.50, 51.14, 49.60, 48.01, 40.43, 25.39, 14.19. Anal. Calcd. for C27H29F4N5O5: C, 55.96; H, 5.04; N, 12.08. Found: C, 56.08; H, 5.05; N, 12.06.
General Procedure for the Synthesis of Carboxylic Acids 7a–7d
In a round-bottom flask, the appropriate methyl ester (6a–6d) and a solution of 2 M NaOH were refluxed for 24 h under vigorous stirring. After this time, the reaction mixture was cooled, and a solution of 2 M HCl was added up to the isoelectric point. The obtained precipitate was filtered under vacuum and washed in sequence with deionized water, isopropanol, and diethyl ether. The solid was then purified by flash chromatography using a CH2Cl2/CH3OH gradient eluting system. According to this procedure, the following products have been obtained.
1-Cyclopropyl-6-fluoro-7-(4-(2-((4-nitro-3-(trifluoromethyl)phenyl)amino)ethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (7a)
The title compound was obtained starting from 0.159 g (0.28 mmol) of 6a and 25 mL of 2 M NaOH. Yellow solid (75%): mp 229–231 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.59 (s, 1H), 8.08 (d, J = 9.0 Hz, 1H), 7.83 (d, J = 13.5 Hz, 1H), 7.52–7.47 (m, 2H), 7.16 (s, 1H), 6.89 (d, J = 8.5 Hz, 1H), 3.70 (br s, 1H), 3.29 (m, 6H), 2.70–2.65 (br s, 4H), 2.63 (t, J = 5.5 Hz, 2H), 1.28–1.27 (br m, 2H), 1.09 (br s, 2H); 13C NMR (125 MHz, DMSO-d6): δ 175.45, 166.42, 153.18, 147.48, 146.44, 138.66, 133.53, 129.73, 124.96, 124.71, 123.65, 119.32, 111.11 (d, JCF = 21.3 Hz), 105.87, 55.97, 53.84, 52.43, 49.53, 40.11, 7.53. Anal. Calcd. for C26H25F4N5O5: C, 55.42; H, 4.47; N, 12.43. Found: C, 55.61; H, 4.48; N, 12.40.
1-Cyclopropyl-6-fluoro-7-(4-(3-((4-nitro-3-(trifluoromethyl)phenyl)amino)propyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (7b)
The title compound was obtained starting from 0.112 g (0.19 mmol) of 6b and 25 mL of 2 M NaOH. Yellow solid (74%): mp 235–237 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.66 (s, 1H), 8.08 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 13.0 Hz, 1H), 7.63 (s, 1H), 7.57 (d, J = 7.0 Hz, 1H), 7.08 (s, 1H), 6.86 (dd, J = 9.0, 2.0 Hz, 1H), 3.86–3.80 (br m, 1H), 3.41–3.33 (br s, 4H), 3.29–3.25 (m, 2H), 2.61 (br s, 4H), 2.48–2.42 (br m overlapped with DMSO, 2H), 1.79 (br s, 2H), 1.33–1.30 (m, 2H), 1.20–1.17 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 176.32, 165.88, 153.96, 152.59 (d, JCF = 152.5 Hz), 147.99, 139.15, 133.40, 129.79, 124.8 (d, JCF = 32.5 Hz), 123.61, 121.44, 118.57, 110.94 (d, JCF = 23.8 Hz), 106.73, 106.33, 54.88, 52.25, 50.63, 40.34, 35.84, 28.99, 7.55. Anal. Calcd. for C27H27F4N5O5: C, 56.15; H, 4.71; N, 12.13. Found: C, 56.02; H, 4.70; N, 12.15.
1-Ethyl-6-fluoro-7-(4-(2-((4-nitro-3-(trifluoromethyl)phenyl)amino)ethyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (7c)
The title compound was obtained starting from 0.177 g (0.31 mmol) of 6c and 25 mL of 2 M NaOH. Yellow solid (62%): mp 225–227 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.94 (s, 1H), 8.07 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 13.5 Hz, 1H), 7.47 (t, J = 5.5 Hz, 1H), 7.21–7.14 (m, 2H), 6.88 (dd, J = 9.5, 2.0 Hz, 1H), 4.58 (q, J = 7.0 Hz, 2H), 3.39–3.33 (m, 6H), 2.69–2.65 (m, 4H), 2.63 (t, J = 6.0 Hz, 2H), 1.41 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 176.12, 166.08, 153.84, 152.50 (d, JCF = 160.0 Hz), 148.47, 145.42 (d, JCF = 10.0 Hz), 137.18, 133.54, 129.71, 124.82 (d, JCF = 32.5 Hz), 123.63, 121.46, 119.19 (d, JCF = 7.5 Hz), 111.15 (d, JCF = 22.5 Hz), 107.06, 105.76, 55.93, 52.37, 49.45, 49.42, 49.03, 14.29. Anal. Calcd. for C25H25F4N5O5: C, 54.45; H, 4.57; N, 12.70. Found: C, 54.31; H, 4.56; N, 12.66.
1-Ethyl-6-fluoro-7-(4-(3-((4-nitro-3-(trifluoromethyl)phenyl)amino)propyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (7d)
The title compound was obtained starting from 0.077 g (0.13 mmol) of 6d and 25 mL of 2 M NaOH. Yellow solid (36%): mp 249–251 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.94 (s, 1H), 8.07 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 13.5 Hz, 1H), 7.59 (s, 1H), 7.17 (s, 1H), 7.06 (s, 1H), 6.84 (br m,1H), 4.59 (app s, 2H), 3.29–3.21 (br m, 6H), 2.62–2.53 (br m, 4H), 2.46 (t, J = 6.0 Hz, 2H), 1.81–1.73 (br m, 2H), 1.42 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 176.14, 166.04, 153.77, 152.44 (d, JCF = 163.8 Hz), 148.59, 137.16, 133.55, 129.76, 126.58, 125.47, 124.85 (d, JCF = 31.3 Hz), 123.60, 121.43, 119.26, 111.27 (d, JCF = 22.5 Hz), 107.12, 69.77, 49.07, 33.65, 31.26, 28.98, 22.06, 14.38. Anal. Calcd. for C26H27F4N5O5: C, 55.22; H, 4.81; N, 12.38. Found: C, 55.07; H, 4.82; N, 12.35.
Griess Test
The stock solution of the Griess reagent (purchased by Sigma Aldrich Srl) was obtained by dissolving 200 mg of the powder in 5 mL of H2O MilliQ. For nitrite quantification, each selected compound was dissolved in anhydrous DMSO to obtain a 1 mM solution (A). Subsequently, 160 μL of each solution A was diluted with 840 μL of H2O MilliQ to obtain solution B (160 μM). A total of 500 μL of B was then added to a quartz cuvette containing 500 μL of Griess reagent solution (80 μM). The resulting solutions were then irradiated with a 300 W tungsten lamp at a distance of 40 cm for 15 min, 1 h, and 2 h, recording the absorbance spectrum of each sample at each time point and reading the peak increase at 540 nm. The relationship of absorbance and concentrations of nitrite was constructed by drawing a standard curve using the known concentrations of NaNO2 (5, 10, 20, 30, 40, and 50 μM).
Cell Lines and In Vitro Culture Conditions
The cell lines DU145 (HTB-81, human prostate carcinoma), PC3 (CRL-1435, human prostate adenocarcinoma), MCF7 (HTB-22, human breast adenocarcinoma), MDA-MB231 (HTB-26, human breast adenocarcinoma), and HCT116 (CCL-247, human colorectal carcinoma) were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA); WH1 human fibroblasts were kindly provided by Dr. Guven.51 All the cells were maintained under standard culture conditions (37 °C; 5% CO2) in the RMPI1640 medium (PC3, MCF7, and MDA-MB231 cells), while DU145 and HCT116 cells were in the DMEM and Iscove’s medium (WH1 cells) supplemented with 10% fetal calf serum, 1% glutamine, and 1% antibiotics mixture; for HCT116 and DU145 cells, 1% sodium pyruvate and 1% non-essential amino acids were also added to the culture medium.
Cell Viability Assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed on all the cell lines tested as previously described52 with minor modifications. Briefly, according to the growth profiles previously defined for each cell line, adequate numbers of cells were plated in each well of a 96-well plate in 0.1 mL of complete culture medium. Cells were allowed to attach for 24 h before the treatment at 37 °C for 72 h with the compounds at concentrations ranging between 0.1 and 75 μM, bringing the final volume to 0.2 mL/well. Each experiment included eight replications per concentration tested; control samples were run with 0.2% DMSO. At the end of the incubation period, MTT (0.05 mL of a 2 mg/mL stock solution in PBS) was added to each well for 3 h at 37 °C. Cell supernatants were then carefully removed, the blue formazan crystals formed through MTT reduction by metabolically active cells were dissolved in 0.120 mL of DMSO, and the corresponding optical densities were measured at 570 nm using a Universal Microplate Reader EL800 (Bio-Tek, Winooski, VT).
To evaluate the contribution of NO release on cell viability, the MTT assay was performed on DU145 and PC3 cells following treatment with 7b and 7c with 1 h irradiation and 48 h incubation in a drug-free medium.
IC50 values were estimated from the resulting concentration–response curves by nonlinear regression analysis using GraphPad Prism software, v. 5.0 (GraphPad, San Diego, CA, USA). Differences between IC50 values were evaluated statistically by analysis of variance with a Bonferroni post-test for multiple comparisons.
Minimum Inhibitory Concentration Determination
P. aeruginosa PAO1 was chosen as a model microorganism53 and was grown overnight in a Luria Bertani (LB) medium at 37 °C on an orbital shaker at 200 rpm. Minimum inhibitory concentrations (MICs) of quinolone and quinolone derivatives were determined against P. aeruginosa PAO1 by a broth dilution method. Overnight cultures were diluted 100-fold to give a cellular concentration of 107 CFU/mL. Decreasing concentrations of compounds, from 200 to 0.1 μg/mL, were added to bacterial samples in a two-fold dilution series. Upon 24 h of incubation at 37 °C, the bacterial samples were observed for microbial growth, and MIC values were determined as minimal concentrations of drugs at which no turbidity was detectable. The assays were performed at least three times.
Bacterial Binding Assay
The relative efficiency of fluoroquinolone and derivatives in binding P. aeruginosa cells was determined by an indirect method.54P. aeruginosa PAO1 overnight cultures were centrifuged (5000 rpm for 10 min), and the supernatant was removed. Pellets were suspended and 10-fold diluted in PBS. Fluoroquinolone and fluoroquinolone derivatives were administered at 10 μM and incubated for 1 h at 37 °C in the dark to allow the interaction between the compounds and cells. After dark incubation, samples were centrifuged (10,000 rpm for 10 min), and the visible spectra of the supernatant were recorded (k = 380–700 nm) and compared with the corresponding visible spectrum of each compound.
Antimicrobial Photo-Inactivation Test
Upon the overnight growth of P. aeruginosa PAO1 and S. aureus ATCC6538P (methicillin susceptible S. aureus, MSSA), cultures were diluted in phosphate buffer (PBS–KH2PO4/K2HPO4, 10 mM, pH 7.4) to reach a concentration of ∼106 CFU/mL. Compound 7c was added to a cell suspension at a final concentration of 10 μM. Cells were incubated in the dark for 10 min and then irradiated under light at 410 ± 10 nm (20 J/cm2) or incubated in the dark as a control. Soon after irradiation, the bacterial concentration was evaluated by the viability count technique. Briefly, an aliquot of each sample was 10-fold serially diluted in PBS and a volume of 10 μL of each diluted and undiluted sample was inoculated on LB agar. After overnight incubation at 37 °C, the corresponding cellular concentration was calculated and expressed as CFU/mL.
Molecular Docking
Flexible ligand docking experiments were performed employing AutoDock 4.2.6 software implemented in YASARA (v. 19.5.5, YASARA Biosciences GmbH, Vienna, Austria)55,56 using the crystal structure of the human Topo IIα (PDB ID: 5GWK), bacterial Topo IIA (PDB ID: 2XCT), and bacterial DNA gyrase (PDB ID: 6M1S) retrieved from the PDB Data Bank as a fully optimized one and the Lamarckian genetic algorithm (LGA). The maps were generated by the program AutoGrid (4.2.6) with a spacing of 0.375 Å and dimensions that encompass all atoms extending 5 Å from the surface of the structure of the crystallized ligands. All the parameters were inserted at their default settings as previously reported.57 In the docking tab, the macromolecule and ligand are selected, and GA parameters are set as ga_runs = 100, ga_pop_size = 150, ga_num_evals = 25,000,000, ga_num_generations = 27,000, ga_elitism = 1, ga_mutation_rate = 0.02, ga_crossover_rate = 0.8, ga_crossover_mode = two points, ga_cauchy_alpha = 0.0, ga_cauchy_beta = 1.0, and number of generations for picking worst individual = 10. Since no water molecules are directly involved in complex stabilization, they were not considered in the docking process (although in the crystallized structure of the bacterial DNA gyrase there are three structural water molecules that form hydrogen bonds between the ligand and some of the amino acids present on the enzymatic site, this network of H-bonds is intrinsic with the structure of the crystallized ligand. Docking calculations performed with the crystallized water molecules led to unsatisfactory results; therefore, the removal of water molecules in the peripheral regions of the binding site does not influence the calculated free binding energies in any way). All protein amino acid residues were kept rigid, whereas all single bonds of ligands were treated as fully flexible. The values of the energies of docking, in kcal/mol, have been calculated employing the “hybrid” force field implemented in AutoDock that contains terms based on molecular mechanics as well as empirical. Although the prediction of absolute binding energies may be less accurate compared to more computationally expensive, purely force field-based methods, this semi-empirical approach is considered as well-suited for the relative rankings.58
Molecular Optimization
The semi-empirical calculations were performed using the parameterized model number 6 Hamiltonian59 as implemented in the MOPAC package (MOPAC2016 v. 18.151, Stewart Computational Chemistry, Colorado Springs, Colorado, USA). All molecules were fully optimized employing the eigenvector following the algorithm and a gradient minimization of 0.01 together with the precise and ddmin = 0 keywords.
Molecular Dynamics Simulations
The MD simulations of the human Topo IIα/DNA/1a ternary model system were performed with the YASARA Structure package (19.11.5).55 A periodic simulation cell with boundaries extending 8 Å from the surface of the complex was employed. The box was filled with water, with a maximum sum of all bump water of 1.0 Å and a density of 0.997 g/mL with an explicit solvent. YASARA’s pKa utility was used to assign pKa values at pH 7.4,60 and system charges were neutralized with NaCl (0.9% by mass). Water molecules were deleted to readjust the solvent density to 0.997 g/mL. The final system dimensions were approximately 122 × 122 × 122 Å3. The ligand force-field parameters were generated with the AutoSMILES utility,57 which employs semi-empirical AM1 geometry optimization. Moreover, the assignment of charges, by the assignment of the AM1BCC atom and bond types with refinement was performed using the RESP charges, and finally the assignments of general AMBER force field atom types. Optimization of the hydrogen bond network of the various enzyme–ligand complexes was obtained using the method established by Hooft et al.61 This model allowed addressing ambiguities arising from multiple side-chain conformations and protonation states that are not well resolved in the electron density. The protein was treated with an AMBER ff14SB force field and62 the ligand with GAFF2,63 and the TIP3P model was used for water. The cutoff was 8 Å for van der Waals forces (the default used by AMBER),64 and no cutoff was applied to electrostatic forces (using the Particle Mesh Ewald algorithm).65 A 100 ps MD simulation was run on the solvent only. The entire system was then energy-minimized using first the steepest descent minimization to remove conformational stress followed by a simulated annealing minimization until convergence (<0.01 kcal/mol Å). The equations of motions were integrated with multiple timesteps of 1.25 fs for bonded interactions and 2.5 fs for nonbonded interactions using the NPT ensemble at a temperature of 298 K and a pressure of 1 atm. The temperature was controlled using the Berendsen thermostat,66 and the pressure was controlled using the solvent-probe pressure control mode barostat.67 The MD simulation was then initiated with an equilibration period of 10 ns for the assessment of the ligand’s correct pose, and a classical production MD simulation of 100 ns was performed analogously to other experiments reported by us.68,69 The MD trajectories were recorded every 100 ps.
MM/PBSA Calculation of the Energies of Binding during the MD Simulation
To this purpose, we used the iPBSA script, according to the procedure reported in detail in the original publication,70 with the algorithm for MM/PBSA implemented in the freely available AmberTools21 suite,71 to analyze the ligand/enzyme complex coordinates recorded during the MD simulation.
Acknowledgments
All the authors gratefully acknowledge the Italian MUR for supporting grant PRIN 2017 (Code 201744BN5T) and PON R&I funds 2014–2020 (CUP: E66C18001320007, AIM1872330, activity 1).
Glossary
ABBREVIATIONS
- aPSs
antimicrobial photosensitizers
- CFU
colony forming unit
- Cip
Ciprofloxacin
- DOX
doxorubicin
- Gyr
gyrase
- MD
molecular dynamics
- MDR
multidrug resistance
- MIC
minimal inhibitory concentration
- NO
nitric oxide
- Nor
Norfloxacin
- PBS
phosphate buffered saline
- p-TsOH
para-toluenesulfonic acid
- R.I.
resistance index
- TEA
triethylamine
- TLC
thin-layer chromatography
- TMS
tetramethylsilane
- Topo
topoisomerase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00917.
Author Contributions
All authors contributed to the present paper and have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Emmerson A. M.; Jones A. M. The quinolones: decades of development and use. J. Antimicrob. Chemother. 2003, 51, 13–20. 10.1093/jac/dkg208. [DOI] [PubMed] [Google Scholar]
- Andersson M. I.; MacGowan A. P. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51, 1–11. 10.1093/jac/dkg212. [DOI] [PubMed] [Google Scholar]
- Novelli A.; Rosi E. Pharmacological properties of oral antibiotics for the treatment of uncomplicated urinary tract infections. Aust. J. Chem. 2017, 29, 10–18. 10.1080/1120009X.2017.1380357. [DOI] [PubMed] [Google Scholar]
- Mohammed H. H. H.; Abuo-Rahma G. E.-D. A. A.; Abbas S. H.; Abdelhafez E.-S. M. N. Current trends and future directions of fluoroquinolones. Curr. Med. Chem. 2019, 26, 3132–3149. 10.2174/0929867325666180214122944. [DOI] [PubMed] [Google Scholar]
- Weigel L. M.; Steward C. D.; Tenover F. C. GyrA mutations associated with fluoroquinolone resistance in eight species of Enterobacteriaceae. Antimicrob. Agents Chemother. 1998, 42, 2661–2667. 10.1128/AAC.42.10.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux J. J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Gubaev A.; Klostermeier D. DNA-induced narrowing of the gyrase N-gate coordinates T-segment capture and strand passage. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14085–14090. 10.1073/pnas.1102100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau J. M. Fluoroquinolones: mechanism of action, classification, and development of resistance. Surv. Ophthalmol. 2004, 49, S73–S78. 10.1016/j.survophthal.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Zhang M. Q.; Haemers A. Quinolone antimicrobial agents: structure-activity relationships. Pharmazie 1991, 46, 687–700. [PubMed] [Google Scholar]
- Zhang G. F.; Zhang S.; Pan B.; Liu X.; Feng L. S. 4-Quinolone derivatives and their activities against Gram positive pathogens. Eur. J. Med. Chem. 2018, 143, 710–723. 10.1016/j.ejmech.2017.11.082. [DOI] [PubMed] [Google Scholar]
- Yadav V.; Varshney P.; Sultana S.; Yadav J.; Saini N. Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC cancer 2015, 15, 581. 10.1186/s12885-015-1560-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold C.; Ocker M.; Ganslmayer M.; Gerauer H.; Hahn E. G.; Schuppan D. Ciprofloxacin induces apoptosis and inhibits proliferation of human colorectal carcinoma cells. Br. J. Cancer 2002, 86, 443–448. 10.1038/sj.bjc.6600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisacchi G. S.; Hale M. R. A ″Double-edged″ scaffold: antitumor power within the antibacterial quinolone. Curr. Med. Chem. 2016, 23, 520–577. 10.2174/0929867323666151223095839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Aal M. A. A.; Abdel-Aziz S. A.; Shaykoon M. S. A.; Abuo-Rahma G. E.-D. A. Towards anticancer fluoroquinolones: A review article. Arch. Pharm. 2019, 352, 1800376. 10.1002/ardp.201800376. [DOI] [PubMed] [Google Scholar]
- Sissi C.; Palumbo M. The quinolone family: from antibacterial to anticancer agents. Curr. Med. Chem.: Anti-Cancer Agents 2003, 3, 439–450. 10.2174/1568011033482279. [DOI] [PubMed] [Google Scholar]
- Hawtin R. E.; Stockett D. E.; Byl J. A. W.; McDowell R. S.; Tan N.; Arkin M. R.; Conroy A.; Yang W.; Osheroff N.; Fox J. A. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS One 2010, 5, e10186 10.1371/journal.pone.0010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravandi F.; Ritchie E. K.; Sayar H.; Lancet J. E.; Craig M. D.; Vey N.; Strickland S. A.; Schiller G. J.; Jabbour E.; Pigneux A.; Horst H. A.; Récher C.; Klimek V. M.; Cortes J. E.; Carella A. M.; Egyed M.; Krug U.; Fox J. A.; Craig A. R.; Ward R.; Smith J. A.; Acton G.; Kantarjian H. M.; Stuart R. K. Phase 3 results for vosaroxin/cytarabine in the subset of patients ≥60 years old with refractory/early relapsed acute myeloid leukemia. Haematologica 2018, 103, e514–e518. 10.3324/haematol.2018.191361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amata E.; Dichiara M.; Arena E.; Pittalà V.; Pistarà V.; Cardile V.; Graziano A. C. E.; Fraix A.; Marrazzo A.; Sortino S.; Prezzavento O. Novel sigma receptor ligand-nitric oxide photodonors: molecular hybrids for double-targeted antiproliferative effect. J. Med. Chem. 2017, 60, 9531–9544. 10.1021/acs.jmedchem.7b00791. [DOI] [PubMed] [Google Scholar]
- Amata E.; Dichiara M.; Gentile D.; Marrazzo A.; Turnaturi R.; Arena E.; La Mantia A.; Tomasello B. R.; Acquaviva R.; Di Giacomo C.; Rescifina A.; Prezzavento O. Sigma receptor ligands carrying a nitric oxide donor nitrate moiety: synthesis, in silico, and biological evaluation. ACS Med. Chem. Lett. 2020, 11, 889–894. 10.1021/acsmedchemlett.9b00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giacomo V.; Di Valerio V.; Rapino M.; Bosco D.; Cacciatore I.; Ciulla M.; Marrazzo A.; Fiorito J.; Di Stefano A.; Cataldi A. MRJF4, a novel histone deacetylase inhibitor, induces p21 mediated autophagy in PC3 prostate cancer cells. Cell. Mol. Biol. 2015, 61, 17–23. [PubMed] [Google Scholar]
- Olivieri M.; Amata E.; Vinciguerra S.; Fiorito J.; Giurdanella G.; Drago F.; Caporarello N.; Prezzavento O.; Arena E.; Salerno L.; Rescifina A.; Lupo G.; Anfuso C. D.; Marrazzo A. Antiangiogenic effect of (±)-haloperidol metabolite II valproate ester [(±)-MRJF22] in human microvascular retinal endothelial cells. J. Med. Chem. 2016, 59, 9960–9966. 10.1021/acs.jmedchem.6b01039. [DOI] [PubMed] [Google Scholar]
- Sozio P.; Fiorito J.; Di Giacomo V.; Di Stefano A.; Marinelli L.; Cacciatore I.; Cataldi A.; Pacella S.; Turkez H.; Parenti C.; Rescifina A.; Marrazzo A. Haloperidol metabolite II prodrug: asymmetric synthesis and biological evaluation on rat C6 glioma cells. Eur. J. Med. Chem. 2015, 90, 1–9. 10.1016/j.ejmech.2014.11.012. [DOI] [PubMed] [Google Scholar]
- Bonavida B. Sensitizing activities of nitric oxide donors for cancer resistance to anticancer therapeutic drugs. Biochem. Pharmacol. 2020, 176, 113913. 10.1016/j.bcp.2020.113913. [DOI] [PubMed] [Google Scholar]
- Hays E.; Bonavida B. Nitric oxide-mediated enhancement and reversal of resistance of anticancer therapies. Antioxidants 2019, 8, 407. 10.3390/antiox8090407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer R. M. J. The l-arginine: nitric oxide pathway. Curr Opin Nephrol Hypertens. 1993, 2, 122–128. 10.1097/00041552-199301000-00018. [DOI] [PubMed] [Google Scholar]
- Moncada S. Nitric oxide. J. Hypertens. Suppl. 1994, 12, S35–S39. [PubMed] [Google Scholar]
- Star R. A. Nitric oxide. Am. J. Med. Sci. 1993, 306, 348–358. 10.1097/00000441-199311000-00015. [DOI] [PubMed] [Google Scholar]
- Orlandi V. T.; Bolognese F.; Rolando B.; Guglielmo S.; Lazzarato L.; Fruttero R. Anti-Pseudomonas activity of 3-nitro-4-phenylfuroxan. Microbiology 2018, 164, 1557–1566. 10.1099/mic.0.000730. [DOI] [PubMed] [Google Scholar]
- Thomas D. D.; Ridnour L. A.; Isenberg J. S.; Flores-Santana W.; Switzer C. H.; Donzelli S.; Hussain P.; Vecoli C.; Paolocci N.; Ambs S.; Colton C. A.; Harris C. C.; Roberts D. D.; Wink D. A. The chemical biology of nitric oxide: implications in cellular signaling. Free Radical Biol. Med. 2008, 45, 18–31. 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2016, 15, 185–203. 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riganti C.; Miraglia E.; Viarisio D.; Costamagna C.; Pescarmona G.; Ghigo D.; Bosia A. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005, 65, 516–525. [PubMed] [Google Scholar]
- Huang Z.; Fu J.; Zhang Y. Nitric oxide donor-based cancer therapy: advances and prospects. J. Med. Chem. 2017, 60, 7617–7635. 10.1021/acs.jmedchem.6b01672. [DOI] [PubMed] [Google Scholar]
- Pieretti J. C.; Pelegrino M. T.; Nascimento M. H. M.; Tortella G. R.; Rubilar O.; Seabra A. B. Small molecules for great solutions: Can nitric oxide-releasing nanomaterials overcome drug resistance in chemotherapy?. Biochem. Pharmacol. 2020, 176, 113740. 10.1016/j.bcp.2019.113740. [DOI] [PubMed] [Google Scholar]
- Alimoradi H.; Greish K.; Gamble A. B.; Giles G. I. Controlled delivery of nitric oxide for cancer therapy. Pharm. Nanotechnol. 2019, 7, 279–303. 10.2174/2211738507666190429111306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferroni C.; Del Rio A.; Martini C.; Manoni E.; Varchi G. Light-induced therapies for prostate cancer treatment. Front. Chem. 2019, 7, 719. 10.3389/fchem.2019.00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapozzi V.; Ragno D.; Guerrini A.; Ferroni C.; Pietra E. d.; Cesselli D.; Castoria G.; Di Donato M.; Saracino E.; Benfenati V.; Varchi G. Androgen receptor targeted conjugate for bimodal photodynamic therapy of prostate cancer in vitro. Bioconjugate Chem. 2015, 26, 1662–1671. 10.1021/acs.bioconjchem.5b00261. [DOI] [PubMed] [Google Scholar]
- Rapozzi V.; Varchi G.; Della Pietra E.; Ferroni C.; Xodo L. E. A photodynamic bifunctional conjugate for prostate cancer: an in vitro mechanistic study. Invest. New Drugs 2017, 35, 115–123. 10.1007/s10637-016-0396-x. [DOI] [PubMed] [Google Scholar]
- Ballestri M.; Caruso E.; Guerrini A.; Ferroni C.; Banfi S.; Gariboldi M.; Monti E.; Sotgiu G.; Varchi G. Core-shell poly-methyl methacrylate nanoparticles covalently functionalized with a non-symmetric porphyrin for anticancer photodynamic therapy. J. Photochem. Photobiol. B 2018, 186, 169–177. 10.1016/j.jphotobiol.2018.07.013. [DOI] [PubMed] [Google Scholar]
- Caruso E.; Cerbara M.; Malacarne M. C.; Marras E.; Monti E.; Gariboldi M. B. Synthesis and photodynamic activity of novel non-symmetrical diaryl porphyrins against cancer cell lines. J. Photochem. Photobiol. B 2019, 195, 39–50. 10.1016/j.jphotobiol.2019.04.010. [DOI] [PubMed] [Google Scholar]
- Ferroni C.; Sotgiu G.; Sagnella A.; Varchi G.; Guerrini A.; Giuri D.; Polo E.; Orlandi V. T.; Marras E.; Gariboldi M.; Monti E.; Aluigi A. Wool Keratin 3D scaffolds with light-triggered antimicrobial activity. Biomacromolecules 2016, 17, 2882–2890. 10.1021/acs.biomac.6b00697. [DOI] [PubMed] [Google Scholar]
- Caruso E.; Malacarne M. C.; Banfi S.; Gariboldi M. B.; Orlandi V. T. Cationic diarylporphyrins: In vitro versatile anticancer and antibacterial photosensitizers. J. Photochem. Photobiol. B 2019, 197, 111548. 10.1016/j.jphotobiol.2019.111548. [DOI] [PubMed] [Google Scholar]
- Sorrenti V.; Pittalà V.; Romeo G.; Amata E.; Dichiara M.; Marrazzo A.; Turnaturi R.; Prezzavento O.; Barbagallo I.; Vanella L.; Rescifina A.; Floresta G.; Tibullo D.; Di Raimondo F.; Intagliata S.; Salerno L. Targeting heme oxygenase-1 with hybrid compounds to overcome Imatinib resistance in chronic myeloid leukemia cell lines. Eur. J. Med. Chem. 2018, 158, 937–950. 10.1016/j.ejmech.2018.09.048. [DOI] [PubMed] [Google Scholar]
- Moorcroft M. J.; Davis J.; Compton R. G. Detection and determination of nitrate and nitrite: a review. Talanta 2001, 54, 785–803. 10.1016/S0039-9140(01)00323-X. [DOI] [PubMed] [Google Scholar]
- Wang J.; Seebacher N.; Shi H.; Kan Q.; Duan Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571. 10.18632/oncotarget.19187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J.; Qiu L.; Ge L.; Zhou J.; Ji Q.; Yang Y.; Long M.; Wang D.; Teng L.; Chen J. Overcoming multidrug resistance by intracellular drug release and inhibiting p-glycoprotein efflux in breast cancer. Biomed. Pharmacother. 2021, 134, 111108. 10.1016/j.biopha.2020.111108. [DOI] [PubMed] [Google Scholar]
- George S.; Hamblin M. R.; Kishen A. Uptake pathways of anionic and cationic photosensitizers into bacteria. Photochem. Photobiol. Sci. 2009, 8, 788–795. 10.1039/b809624d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. C.; Li T. K.; Farh L.; Lin L. Y.; Lin T. S.; Yu Y. J.; Yen T. J.; Chiang C. W.; Chan N. L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- Lee A.; Djamgoz M. B. A. Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat. Rev. 2018, 62, 110–122. 10.1016/j.ctrv.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Bosch A.; Eroles P.; Zaragoza R.; Vin̈a J. R.; Lluch A. Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer Treat. Rev. 2010, 36, 206–215. 10.1016/j.ctrv.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Yu K. D.; Zhu R.; Zhan M.; Rodriguez A. A.; Yang W.; Wong S.; Makris A.; Lehmann B. D.; Chen X.; Mayer I.; Pietenpol J. A.; Shao Z. M.; Symmans W. F.; Chang J. C. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin. Cancer Res. 2013, 19, 2723–2733. 10.1158/1078-0432.CCR-12-2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover C. K.; Pham X. Q.; Erwin A. L.; Mizoguchi S. D.; Warrener P.; Hickey M. J.; Brinkman F. S. L.; Hufnagle W. O.; Kowalik D. J.; Lagrou M.; Garber R. L.; Goltry L.; Tolentino E.; Westbrock-Wadman S.; Yuan Y.; Brody L. L.; Coulter S. N.; Folger K. R.; Kas A.; Larbig K.; Lim R.; Smith K.; Spencer D.; Wong G. K.-S.; Wu Z.; Paulsen I. T.; Reizer J.; Saier M. H.; Hancock R. E. W.; Lory S.; Olson M. V. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Orlandi V. T.; Martegani E.; Bolognese F.; Trivellin N.; Mat’átková O.; Paldrychová M.; Baj A.; Caruso E. Photodynamic therapy by diaryl-porphyrins to control the growth of Candida albicans. Cosmetics 2020, 7, 31. 10.3390/cosmetics7020031. [DOI] [Google Scholar]
- Flaberg E.; Guven H.; Savchenko A.; Pavlova T.; Kashuba V.; Szekely L.; Klein G. The architecture of fibroblast monolayers of different origin differentially influences tumor cell growth. Int. J. Cancer 2012, 131, 2274–2283. 10.1002/ijc.27521. [DOI] [PubMed] [Google Scholar]
- Gariboldi M. B.; Taiana E.; Bonzi M. C.; Craparotta I.; Giovannardi S.; Mancini M.; Monti E. The BH3-mimetic obatoclax reduces HIF-1α levels and HIF-1 transcriptional activity and sensitizes hypoxic colon adenocarcinoma cells to 5-fluorouracil. Cancer Lett. 2015, 364, 156–164. 10.1016/j.canlet.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Krieger E.; Koraimann G.; Vriend G. Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins 2002, 47, 393–402. 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- Floresta G.; Gentile D.; Perrini G.; Patamia V.; Rescifina A. Computational tools in the discovery of FABP4 Ligands: A statistical and molecular modeling approach. Mar. drugs. 2019, 17, 624. 10.3390/md17110624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile D.; Fuochi V.; Rescifina A.; Furneri P. M. New anti SARS-Cov-2 targets for quinoline derivatives chloroquine and hydroxychloroquine. Int. J. Mol. Sci. 2020, 21, 5856. 10.3390/ijms21165856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huey R.; Morris G. M.; Olson A. J.; Goodsell D. S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E.; Nielsen J. E.; Spronk C. A. E. M.; Vriend G.. Fast empirical pKa prediction by Ewald summation. Computational Drug Discovery and Design; Springer: New York, NY, 2006, 25, 481–486, 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Krieger E.; Dunbrack R.; Hooft R.; Krieger B. Assignment of protonation states in proteins and ligands: combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Bio. 2012, 819, 405–421. 10.1007/978-1-61779-465-0_25. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Krieger E.; Vriend G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresta G.; Dichiara M.; Gentile D.; Prezzavento O.; Marrazzo A.; Rescifina A.; Amata E. Morphing of Ibogaine: A successful attempt into the search for sigma-2 receptor ligands. Int. J. Mol. Sci. 2019, 20, 488. 10.3390/ijms20030488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresta G.; Patamia V.; Gentile D.; Molteni F.; Santamato A.; Rescifina A.; Vecchio M. Repurposing of FDA-approved drugs for treating iatrogenic botulism: A paired 3D-QSAR/docking approach†. ChemMedChem 2020, 15, 256–262. 10.1002/cmdc.201900594. [DOI] [PubMed] [Google Scholar]
- Sahakyan H. Improving virtual screening results with MM/GBSA and MM/PBSA rescoring. J. Comput.-Aided Mol. Des. 2021, 35, 731–736. 10.1007/s10822-021-00389-3. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Aktulga H. M.; Belfon K.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Giambasu G.; Gilson M. K.; Gohlke H.; Goetz A.W.; Harris R.; Izadi S.; Izmailov S. A.; Jin C.; Kasavajhala K.; Kaymak M. C.; King E.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Machado M.; Man V.; Manathunga M.; Merz K. M.; Miao Y.; Mikhailovskii O.; Monard G.; Nguyen H.; O’Hearn K. A.; Onufriev A.; Pan F.; Pantano S.; Qi R.; Rahnamoun A.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Skrynnikov N. R.; Smith J.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xue Y.; York D. M.; Zhao S.; Kollman P. A. (2021), Amber 2021; University of California, San Francisco. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.