

Abstract
Epigenetic mechanisms may contribute to idiopathic scoliosis (IS). We identified 8 monozygotic twin pairs with IS, 6 discordant (Cobb angle difference > 10°) and 2 concordant (Cobb angle difference ≤ 2°). Genome-wide methylation in blood was measured with the Infinium HumanMethylation EPIC Beadchip. We tested for differences in methylation and methylation variability between discordant twins and tested the association between methylation and curve severity in all twins. Differentially methylated region (DMR) analyses identified gene promoter regions. Methylation at cg12959265 (chr. 7 DPY19L1) was less variable in cases (false discovery rate (FDR) = 0.0791). We identified four probes (false discovery rate, FDR < 0.10); cg02477677 (chr. 17, RARA gene), cg12922161 (chr. 2 LOC150622 gene), cg08826461 (chr. 2), and cg16382077 (chr. 7) associated with curve severity. We identified 57 DMRs where hyper- or hypo-methylation was consistent across the region and 28 DMRs with a consistent association with curve severity. Among DMRs, 21 were correlated with bone methylation. Prioritization of regions based on methylation concordance in bone identified promoter regions for WNT10A (WNT signaling), NPY (regulator of bone and energy homeostasis), and others predicted to be relevant for bone formation/remodeling. These regions may aid in understanding the complex interplay between genetics, environment, and IS.
Keywords: idiopathic scoliosis, monozygotic twin, epigenome-wide association study, DNA methylation, bone, discordant, curve severity, differentially methylated region
1. Introduction
Adolescent idiopathic scoliosis (IS) is a three-dimensional spinal deformity affecting 1–3% of otherwise normal prepubescent and adolescent individuals [1,2]. Screening programs, conservative treatment, and surgical care in the case of progressive curvatures impose significant personal, familial, financial, and societal costs across the lifetime of affected individuals. The etiology of IS remains unknown. However, it has been shown to have a strong familial component [3] with a sibling recurrence risk of 18%, and heritability estimates of approximately 87.5% [4,5,6].
Traditional genetic association methods including familial linkage studies [7,8,9,10,11,12,13,14,15,16,17], exome sequencing [18,19,20,21,22,23,24,25,26,27], and genome wide association studies (GWAS) [28,29,30,31,32,33,34,35,36] have resulted in a number of positive associations with IS, of which only a few loci, notably those in or near ADGRG6 [31,37,38,39,40,41,42,43] and LBX1 [21,29,30,44,45,46,47,48,49,50,51,52,53,54,55,56,57], have been replicated across multiple independent study populations [58,59]. While familial and case–control designs have added to our understanding of IS, the complex heterogenic nature of IS [58,59,60,61,62] has limited our understanding of the genetic underpinnings of this particular disorder. The combination of our inability to relate specific genetic variants to the biology of IS, the low reproducibility of results, increased prevalence of more severe curves among females [63], and the wide variation in phenotype has increased interest in the potential role of environmental and/or epigenetic factors in the etiology of IS [59,64,65].
One frequently studied mechanism of epigenetic regulation is DNA methylation in which a methyl group is added to the cytosine nucleotide within a DNA sequence. Typically occurring in a cytosine phosphate guanine (CpG) dinucleotide pair, methylation has the capacity to change chromatin structure and alter transcription factor binding [66]. This is a reversible event that may provide a link between genetic variation, environment, and disease [67,68]. Although tissue-specific, up to 80% of the variation in the epigenome may be due to genotype [69], therefore a challenge among epigenome wide association studies (EWAS) is determining whether the observed epigenetic phenotype association is due to environmental or genetic effects. Studying monozygotic (MZ) twins, is one way to minimize this concern. MZ twins discordant for the phenotype of interest are near perfect genetic matches, therefore their DNA methylation levels can be compared to shed light on the phenotypic expression of the disease.
Previous epigenome-wide association studies have provided evidence supporting the role of DNA methylation in numerous complex musculoskeletal diseases including osteoarthritis [70], osteoporosis [70], cerebral palsy [71], and Paget’s disease of bone [72]. The role of DNA methylation in IS has not been well studied. Targeted studies have reported associations between methylation and IS near the COMP [73] and PITX1 [74] genes. Two recent studies [75,76] of ESR1 and ESR2 methylation from paravertebral muscle tissue in females with IS supports potential interrelationship between sex hormone levels, methylation, and the clinical manifestation of IS. ESR1 methylation levels from paravertebral muscle tissue on the concave side of curve were associated with curve severity [75], and furthermore, ESR2 promoter methylation levels differed between concave and convex sides of the curves [76]. Epigenome-wide discovery analyses in MZ twins are limited to studies including only one [77] and two [78] MZ twin pairs discordant for IS. There is a strong need for additional epigenome wide analyses to understand the potential role of DNA methylation in IS. Therefore, the aim of this EWAS was to identify differences in DNA methylation levels between monozygotic twin pairs discordant for IS. Within twin pairs, we also aimed to determine if differences in methylation were associated with differences in curve severity.
2. Materials and Methods
2.1. Study Population
Peripheral whole blood samples were obtained from 8 female monozygotic twin pairs (n = 16 individuals) diagnosed with idiopathic scoliosis (IS). Participants were identified from an existing registry, the Genetics of Idiopathic Scoliosis project (the GenesIS project), that has been described by Baschal et al. [19]. A diagnosis of IS required that subjects had no congenital deformities or other co-existing genetic disorders and a standing anteroposterior radiograph showing a curvature of at least 10° by the Cobb method [79]. There were 6 twin discordant twin pairs (difference in primary curve Cobb angle >10°) and 2 concordant twin pairs in our study population. The difference in the primary curvature among the two concordant twin pairs was <1 and 2°, respectively (Table 1). Written informed consent and assent, when appropriate, was obtained from all study participants and/or their legal guardians in accordance with protocols approved through the Johns Hopkins School of Medicine Institutional Review Board and the University of Colorado Anschutz Medical Campus Institutional Review Board.
Table 1.
Demographics and Clinical Characteristics.
| Twin Pair | ID | Case Status | Curve Degree † | Age * |
|---|---|---|---|---|
| Discordant | ||||
| 1 | 15505 | Case | 48 | 44.8 |
| 15501 | Control | 41/37 | 44.8 | |
| 2 | 15643 | Case | 75 | 81.4 |
| 15642 | Control | 35 | 81.9 | |
| 3 | 16012 | Case | 50 | 16.3 |
| 16009 | Control | 22 | 16.3 | |
| 4 | 16615 | Case | 52/48 | 33.4 |
| 16611 | Control | 32/28 | 33.3 | |
| 5 | 18453 | Case | 34 | 5.6 |
| 18454 | Control | 23 | 5.6 | |
| 6 | 19294 | Case | 56/43 | 48.7 |
| 19292 | Control | 12 | 48.7 | |
| Concordant | ||||
| 7 | 16037 | NA | 45 | 42.8 |
| 16038 | NA | 45 | 42.8 | |
| 8 | 18721 | NA | 26/33 | 25.3 |
| 18722 | NA | 29/31 | 25.5 | |
† Major curve(s), * Age at sample collection.
2.2. DNA Methylation Processing
Genomic DNA was extracted from fresh whole blood using a standard phenol-chloroform purification procedure [80]. DNA was further purified using the Zymo DNA Clean & Concentrator kit, followed by Nanodrop quantification. Approximately 1 µg DNA was bisulfite converted using the Zymo EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA). The precipitated DNA was dispensed onto the Infinium MethylationEPIC 850 K BeadChip (Illumina). The EPIC chip provides methylation measurements across the genome. All sequencing was performed at the University of Colorado Genomics and Microarray Shared Resource. Twin pairs were consistently arranged in sequential order on the plate to minimize within and between batch effects.
The 850 K Infinium platform includes 866,836 annotated probes representing individual cytosine phosphate guanine (CpG) probe sites. Data were normalized using the SWAN normalization method implemented within the minfi R package [81]. Standard quality control checks were performed at both the sample and probe level using the minfi R package [81]. Probes or samples that failed any of the following standard filtering procedures were treated as missing data: probe was not detectable above background noise (n = 776 [<0.1%]), probes with a low bead count (>5% of samples with a beadcount <3) (n = 17,272 [2%]), cross reactive probes (n = 17,028 [2%]), dropped during initial quality control processing (n = 528 [<0.1%]). Probes located on sex chromosomes (n = 15,648 [1.8%]) and probes that included known SNPs (n = 165,678 [19.1%]) were also excluded from the analysis. Consistent with recommendations of Logue et al. [82], we filtered low variability probes known to be associated with poor reproducibility. We filtered out all probes with β range value < 0.05%, n = 145,163 (16.7%). In total, 504,743 probes met the inclusion criteria and were included in subsequent steps.
The microarray methylation measurements were performed in two batches. The ComBat function implemented in the sva R package [83] was used to adjust the SWAN normalized M values for potential batch effects. The batch adjusted M-values were used in all statistical analyses. The current Illumina annotation for the 850 K platform is on hg19. Only probes which match 100% to a single location were used for further analyses.
We used three analytical approaches to identify individual CpG sites (Figure 1): (A) we performed a discordant differentially methylated position (DMP) analysis, testing for differences in methylation (batch adjusted M-values) between six twin pairs where the difference in the primary spinal curvature Cobb angle was >10°, (B) Using all twins, we tested the association between within twin pair differences in methylation ( methylation levels in affected/more severe twin–methylation levels unaffected/less severe twin) and differences in curve severity within twin pairs ( primary curve magnitude in affected/more severe twin–primary curve magnitude is unaffected/less severe twin), (C) We tested for differences in methylation variability between the discordant twin pairs (Differentially Variable Position [DVP] analysis). The methods workflow is outlined in Figure 1.
Figure 1.

Study Methodologic Workflow. Differentially methylated position (DMP) analyses were used to identify single probes. We used two DMP strategies (1) Discordant DMP Analysis (A), differences in methylation between cases (twin with more severe IS) relative to controls (twin with less sever IS) (2) Severity DMP Analysis (B) looked at association between difference in curve severity and methylation , where methylation levels in affected/more severe twin–methylation levels unaffected/less severe twin and primary curve magnitude in affected/more severe twin–primary curve magnitude is unaffected/less severe twin. We also looked at differences in variability at single probes (differentially variable position analysis, DVP) among cases compared to controls (C). Region analyses based on single probes from (A,B) were used to identify regions of consistent methylation effects within promoter regions. We only considered regions with 5 or more probes where direction of effect was consistent across all probes. DVP probes (C) were not considered in the region analysis due to challenges interpreting a ‘consistent’ direction of effect based on variability.
Methylation is tissue specific. Confounding due to differences in cell composition between cases and controls is a potential concern in epigenome-wide association studies [84]. Cell proportions were estimated from methylation values using the minifi [81] R package. The distribution of CD8T, CD4T, B Cells, natural killer Cells, monocytes, and neutrophils was similar in cases compared to controls (Appendix A, Table A1). Due to the small sample size and similar distribution of cell proportions across all individuals, we did not adjust for cell type in subsequent analyses.
2.3. Statistical Analysis
We used descriptive statistics to summarize the demographic and clinical characteristics of all subjects included in the study. Wilcoxon signed rank tests were used to compare the distribution of the five cell types between case and control twins (Table 1 and Appendix A, Table A1). Normalized M-values were used in all analyses. β-values and percent methylation were also reported to facilitate biological interpretation. In the discordant DMP analysis, paired t-tests were used to test for differences in M values between discordant twins (n = 6 pairs, n = 12 individuals). The individual with the more severe curvature was designated as the “case”, and the corresponding twin with the less severe curvature was designated as the “control.” For the DVP discordant analysis, a regularized version of Bartlett’s test was used to identify differentially variable probes between discordant twin pairs. For the curve severity DMP analysis involving all twin pairs, linear regression models were used to test the association between differences in methylation () and differences in curve severity within twin pairs (). To account for multiple testing, false-discovery rate (FDR) adjusted p values were estimated using the algorithm described by Benjamini and Hochberg [85].
An exploratory analysis was used to identify differentially methylated regions (DMR) using the mCSEA [86] R package among promoter regions with a minimum of 5 probes. Significance was assessed based on 100,000 permutations. The DMR analysis was implemented for the discordant DMP and the curve severity DMP analyses. Based on the exploratory nature of the DMR analysis, only regions where 100% of probes were in the same direction of effect and the FDR adjusted p value was < 0.05 were considered significant.
Functional gene overrepresentation analysis was performed based on the results of the discordant DMP and curves severity DMP analyses. The top DMRs, nominal p value <0.001, were included in the DMR overrepresentation analysis. Overrepresentation analyses of the severity and discordant gene lists were conducted using PANTHER v16.0, test release 20,210,224 (PantherDB.org) [87,88,89]. Custom gene background inputs were used in accordance with gene promoter region DMRs analyzed within the Infinium HumanMethylation EPIC Beadchip platform. The Annotation Data Sets Gene Ontology (GO) Cellular Component Complete, GO Molecular Function Complete, and GO Biological Process Complete were analyzed separately, each using a Fisher’s Exact Test and Bonferroni correction. We report overrepresented GO terms with a Bonferroni-adjusted p < 0.05. Significant terms were reviewed; parent terms were manually removed and REVIGO [90] was used to eliminate redundant terms.
2.4. Prioritization of Candidates: Blood Methylation Correlation
While the tissue of origin for IS has not been determined, the primary clinical manifestation is that of the bony spinal column. Ebrahimi et al. [91] previously reported on methylation levels in whole blood versus trabecular bone. We utilized these data to prioritize our methylation candidates as ones that may have a functional role in bone. We reviewed the distribution of correlation coefficients, representing the strength of association between methylation levels in blood versus bone, among all probes evaluated by Ebrahimi et al. [91] Probes were considered strongly correlated if the correlation coefficients exceeded the 75th percentile among all probes evaluated by Ebrahimi et al. [91] (ρ = 0.49). We then reviewed the correlation coefficients for probes identified as candidates in our DMP and DMR analyses. For the DMR analysis, we reported the maximum correlation coefficient among all probes in the DMR as well as the percentage of strongly, positively correlated probes across the entire region.
3. Results
3.1. Demographics
The study population included 6 discordant and 2 concordant female monozygotic twin pairs with idiopathic scoliosis (IS, see Table 1). The average age among all individuals at the time of blood acquisition was 37.3 years (±22.5). The average Cobb angle of the primary curve was 39.6° (±15.3). The average difference in age between twin pairs at the time of sample acquisition was 1.2 months (range: 0 to 6 months). The average difference in curve severity among all twins was 19° (range: 0 to 44°). Among discordant twins, the average difference in curve severity was 25° (range: 11 to 44°).
3.2. Discordant Curvature Analysis
In the discordant analysis, none of the individual CpG probes were significant at the FDR adjusted p = 0.10. Differentially methylated region (DMR) analyses identified 200 promoter regions (containing 5–14 CpG sites) that were significant at the FDR adjusted p value of 0.05. Among these regions, 58 DMRs included probes/sites where the direction of effect (hypermethylation or hypomethylation) was consistent across 100% of the probes (Appendix A, Table A2). The most significant DMR represented a region on chr. 14 in the promoter region for the BCL2L2-PABPN1 gene (FDR adjusted p = 0.0113, see Figure 2). Using the Panther enrichment algorithm with these 58 DMRs, we identified 1 significantly enriched gene ontology (GO) term (Appendix A, Table A3).
Figure 2.
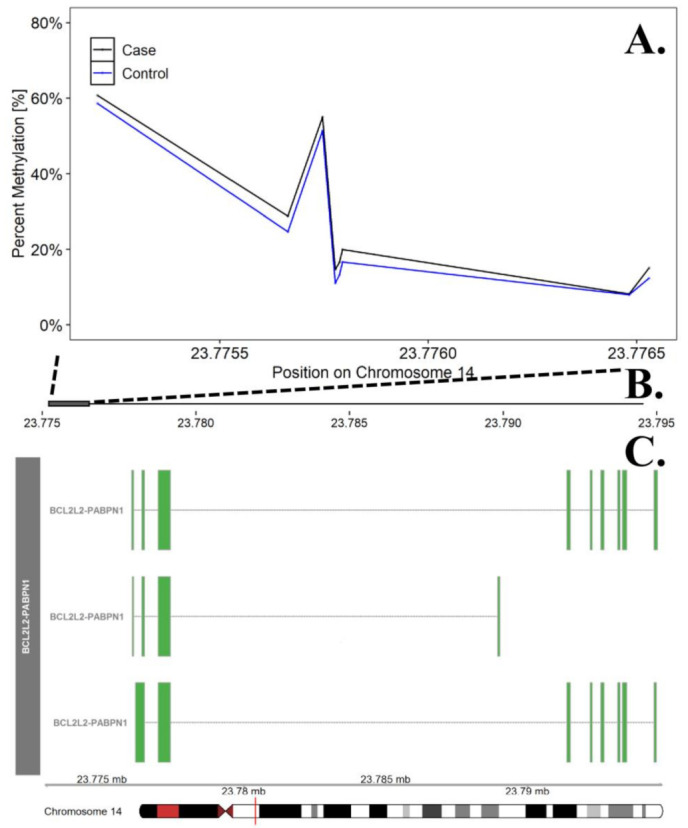
Differentially Methylated Region in the BCL2L2-PABN1 Promoter Region on Chromosome 14. The top panel (A) describes differences in percent methylation between cases and controls at each probe included in the promoter region for BCL2L2-PABN1. This region was the most significant DMR in the discordant twin analysis. The X axis represents the position (mb) of the probes. The middle panel (B) represents the location of promoter region (solid square) relative to the entire gene, represented in the bottom panel. Multiple known isoforms of BCL2L2-PABN1 are represented in the bottom panel (C), boxes represent exons and lines represent introns. The red line on the ideogram, bottom of the figure, represents location of the region within the chromosome.
In addition to differences in methylation levels, we looked for differences in methylation variability, which may provide valuable information about the heterogenous environmental exposures that contribute to disease etiology. In the differentially variable position (DVP) analysis, methylation variability at cg02477677 was significantly lower (FDR adjusted p value = 0.0791) in cases with a more severe curve compared to unaffected or less severely affected controls (Figure 3). The cg02477677 CpG probe is an open sea probe on chr. 7 near DPY19L1.
Figure 3.

Scatter Plot of Differential Variability Between Case and Control Twins. Methylation levels (% methylation or β values) at the cg12959265 probe, an open sea probe near the DPY19L1 gene on chromosome 7 in IS cases and IS controls. The triangle represents the mean % methylation and error bars represent +/− 1 standard deviation. The plot illustrates the large difference in variability at cg12959265 in IS cases vs. IS controls.
3.3. Curve Severity Analysis
We also tested whether the difference in methylation between cases and controls was associated with the difference in curve severity between cases and controls. We identified 4 CpG sites where the difference in methylation was significantly associated (FDR adjusted p value = 0.0753) with the difference in curve severity (Figure 4). At each of these open sea probes, increasing disparity in curve severity between cases and controls was associated with a pattern of hypomethylation.
Figure 4.

Volcano Plot: Curve Severity Analysis The volcano plot describes the effect size and p value for every probe tested in the curve severity analysis. The Y axis represents the −log10(p values) and the X axis represents the change in M value for every one-degree difference in curve severity between the twin pairs for each of the respective probes tested. Increasing curve disparity was more often associated with hypomethylation (decreased M values, left or negative side of the plot) than hypermethylation. The four FDR significant probes (FDR adj p = 0.0753) are highlighted in red, cg08826461 (nominal p value = 3.37 × 10−7), cg16382077 (nominal p value = 3.85 × 10−7), cg12922161 (nominal p value = 5.32 × 10−7), and cg02477677 (nominal p value = 5.97 × 10−7).
For every 1 degree increase in the difference in curve severity in cases compared to controls, batch adjusted M-values decreased by an average of between 0.012 to 0.027 units. Significant probes included cg02477677 (slope: 0.015 units, near the RARA gene on chr. 17, nominal p value = 5.97 × 10−7); cg08826461 (slope: 0.027 units chr. 2, does not map to a known gene, nominal p value = 3.37 × 10−7), cg12922161 (slope: −0.012 chr. 2, maps to LOC150622, nominal p value = 5.32 × 10−7), and cg16382077 (slope: 0.021 units, chr. 7, does not map to a known gene, nominal p value = 3.85 × 10−7).
The differentially methylated region analyses identified n = 197 promoter regions (ranging from 5 to 34 CpG sites) significant at the FDR adjusted p value of 0.05. Among these, 28 regions included probes where the direction of effect (difference in curve severity was either positively or negatively associated with the difference in methylation between twin pairs) was consistent across 100% of the probes (Appendix A, Table A4). The top DMR consisted of 34 probes on chr. 20 within the promoter region for the NNAT gene (FDR adjusted p value = 0.0237, Figure 5). Using Panther, we identified 15 significantly enriched ontologies (Appendix A, Table A5). The top biological process terms included pituitary gland development (GO:0021983) and anterior/posterior pattern specification (GO:0009952).
Figure 5.

Differentially Methylation Region in the NNAT Promoter Region on Chromosome 20. The top panel (A) presents the slope estimates from the curve severity analysis that represent the change in methylation between cases and controls per one-degree change in curve severity at each of the 34 probes included in the promoter region for the NNAT gene. This region was the most significant DMR in the curve severity analysis. The X axis represents the position (mb) of the probes. The middle panel (B) represents the location of promoter region (solid square) relative to the entire gene, represented in the bottom panel. Multiple known isoforms of the NNAT gene are represented in the bottom panel (C), boxes represent exons and lines represent introns. The red line on the ideogram, bottom of the figure, represents location of the region within the chromosome.
3.4. Candidate Prioritization
Ebrahimi et al. [91] conducted an epigenome-wide analysis to measure the correlation between methylation levels in whole blood and trabecular bone. We used these correlation coefficients to prioritize the methylation candidates identified in our study. Among the four probes identified as candidates in our DMP analysis (cg02477677, cg12922161, cg08826461, and cg16382077), only one probe, cg08826461, was strongly correlated with bone tissue (ρ = 0.494, FDR adjusted p value = 0.41329). Among DMRs, we prioritized candidate regions where either one or more probes within the DMR was significantly correlated with bone (FDR adjusted p value of less than 0.10), or, greater than 50% of probes included in the region were strongly correlated with bone. We identified 13 priority regions based on the discordant DMR analysis and 8 priority candidate regions based on the severity DMR analysis (Table 2).
Table 2.
Priority DMRs Based on High Correlation with Bone.
| Nearest Gene | Chr. | Start Position | End Position | Number of Probes | DMR Nominal p Value | DMR FDR p Value | Maximum Bone Correlation | FDR Adj. p Value for Maximum Bone Correlation | Percent Strongly Positively Correlated Probes Across DMR |
|---|---|---|---|---|---|---|---|---|---|
| Discordant DMR Analysis | |||||||||
| WNT10A | chr2 | 219,744,145 | 219,745,748 | 9 | 2.17 × 10−5 | 0.0113 | 0.83 | 0.0307 | 33.3% |
| CRISP2 | chr6 | 49,681,178 | 49,681,774 | 11 | 2.19 × 10−5 | 0.0113 | 0.89 | 0.0128 | 100.0% |
| RBPJL | chr20 | 43,934,854 | 43,935,551 | 12 | 2.20 × 10−5 | 0.0113 | 0.72 | 0.1048 | 66.7% |
| KDM2B | chr12 | 122,018,574 | 122,020,205 | 14 | 2.21 × 10−5 | 0.0113 | 0.83 | 0.0336 | 50.0% |
| IL27 | chr16 | 28,518,114 | 28,519,597 | 9 | 4.34 × 10−5 | 0.0156 | 0.75 | 0.0844 | 33.3% |
| CA14 | chr1 | 150,229,143 | 150,230,345 | 9 | 6.51 × 10−5 | 0.0196 | 0.78 | 0.0585 | 33.3% |
| C9orf47 | chr9 | 91,604,473 | 91,606,140 | 12 | 2.64 × 10−4 | 0.0318 | 0.79 | 0.0518 | 18.2% |
| STAB1 | chr3 | 52,528,714 | 52,529,393 | 8 | 3.04 × 10−4 | 0.0329 | 0.79 | 0.0534 | 12.5% |
| ACY3 | chr11 | 67,415,183 | 67,418,365 | 8 | 3.26 × 10−4 | 0.0329 | 0.72 | 0.1045 | 62.5% |
| MPG | chr16 | 125,896 | 128,009 | 11 | 3.29 × 10−4 | 0.0329 | 0.82 | 0.0368 | 10.0% |
| ESM1 | chr5 | 54,281,198 | 54,282,459 | 13 | 3.97 × 10−4 | 0.0360 | 0.79 | 0.0526 | 61.5% |
| TMEM219 | chr16 | 29,972,752 | 29,974,294 | 6 | 5.57 × 10−4 | 0.0431 | 0.77 | 0.0667 | 66.7% |
| CREBBP | chr16 | 3,930,112 | 3,931,489 | 5 | 6.16 × 10−4 | 0.0457 | 0.76 | 0.0725 | 80.0% |
| Severity DMR Analysis | |||||||||
| GANC | chr15 | 42,565,522 | 42,566,390 | 7 | 3.38 × 10−4 | 0.0357 | 0.88 | 0.0153 | 28.6% |
| NME3 | chr16 | 1,821,559 | 1,822,346 | 8 | 3.59 × 10−4 | 0.0366 | 0.76 | 0.0729 | 37.5% |
| SLC6A5 | chr11 | 20,619,598 | 20,621,109 | 8 | 3.59 × 10−4 | 0.0366 | 0.80 | 0.0489 | 28.6% |
| RAB22A | chr20 | 56,883,532 | 56,885,003 | 8 | 4.38 × 10−4 | 0.0391 | 0.78 | 0.0594 | 25.0% |
| ACTN4 | chr19 | 39,137,911 | 39,138,334 | 7 | 4.89 × 10−4 | 0.0407 | 0.84 | 0.0298 | 33.3% |
| NPY | chr7 | 24,322,873 | 24,324,570 | 8 | 5.58 × 10−4 | 0.0421 | 0.84 | 0.0276 | 37.5% |
| RAB38 | chr11 | 87,908,558 | 87,909,729 | 9 | 6.33 × 10−4 | 0.045 | 0.73 | 0.0995 | 44.4% |
| COPB1 | chr11 | 14,521,639 | 14,522,617 | 6 | 7.79 × 10−4 | 0.0495 | 0.88 | 0.0149 | 50.0% |
Maximum Bone Correlation = maximum correlation coefficient representing strength of correlation between blood and bone CpG sites across all sites included in the DMR (from Ebrahimi et al.), FDR p Value for Maximum Bone Correlation = FDR adjusted p value for maximum correlation coefficient across all sites included in the DMR (from Ebrahimi et al.)., Percent Strongly Correlated Probes Across DMR = Percentage of probes across the entire region where the correlation coefficient representing strength of correlation between blood and bone CpG sites is greater than 75th percentile among all probes tested in Ebrahimi et al.
4. Discussions
We utilized an epigenome-wide association study (EWAS) in monozygotic (MZ) twins to identify individual methylation sites and regions across the genome relevant to idiopathic scoliosis (IS). We identified a single CpG site where methylation variability was different between discordant MZ twins and identified CpG sites where increasing curve severity was more often associated with hypomethylation. Differentially methylated region (DMR) analyses identified multiple regions potentially indicative of unique methylation changes within twin pairs discordant for IS as well as unique methylation patterns associated with curve severity. Integration of a peripheral blood/bone methylation dataset allowed us to prioritize regions and sites based on their potential relevance to the IS disease process in bone. Collectively, these results highlight both new and previously reported pathways related to IS curve progression including those involved in neurogenesis and body segmentation.
Differential variability in methylation represents large shifts in methylation that may reflect differential epigenetic and/or environmental effects in cases relative to controls. Differential variability analyses in disease-discordant monozygotic twins have been used to identify DNA methylation signatures associated with Type 1 Diabetes [92] and rheumatoid arthritis [93]. In our analysis, variability at cg0247767, chr. 7 near DPY19L1, was significantly different between discordant twin pairs. DPY19L1 is a transmembrane protein localized in the endoplasmic reticulum that regulates neuronal migration and extension during development [94,95]. Zebrafish with mutations within this gene demonstrate spinal axial curvatures [96]; however, the phenotype has not been studied in detail.
We also identified four individual CpG sites that were associated with curve severity (cg02477677, cg12922161, cg08826461, and cg1638077). At these sites, an increase in curve severity tended to be associated with a decrease in methylation (hypomethylation) within the twin pairs. One of the probes, cg12922161, maps to a location near LOC150622/SILC1, a non-coding RNA gene. Although the function of this non-coding RNA is not well known, it has been shown to regulate neuron outgrowth and neuroregeneration via cis-acting activation of the transcription factor SOX11 [97]. To date, select non-coding RNAs as non-protein coding regulatory transcripts within the genome have been hypothesized to functionally participate in the initiation and progression of IS [98]. The cg02477677 probe was also associated with curve severity. This probe maps to a region near RARA on chr. 17, which encodes a transcription factor for the retinoic acid receptor protein During development, RA signaling plays an essential role in embryonic body axis extension, left-right somite synchronization, and limb development [99]. It is a central mechanism underlying bilateral symmetry during development of the mouse embryo [100]. Right-left asymmetries have previously been hypothesized as a potential contributing factor to IS based on the increased prevalence of IS among individuals demonstrating vestibular and posterior basicranial morphological asymmetries in MRI cross-sectional studies [101,102,103].
Regions of differentially methylated probes (DMRs) may have more important functional implications than methylation levels at a single CpG site, particularly in promoter regions which are areas of the genome where methylation levels tend to be negatively correlated with gene expression [67,68]. Based on the discordant analysis, we identified 58 significant DMRs in known promoter regions, the most significant region included methylation sites with the promoter region for the BCL2L2-PABPN1 gene on chr. 14. BCL2L2-PABPN1 is a paralog of PABPN1, which is associated with the development of oculopharyngeal muscular dystrophy, a disease characterized by muscular weakness in eyelids, pharyngeal musculature, and limbs [104]. In the curve severity analysis, we identified 28 significant DMRs in known promoter regions, the most significant included probes with the promoter region for the NNAT gene on chr. 20. This gene is important for brain development and implicated in neurodegenerative diseases including anterior horn disease [105]. The paternal copy of the NNAT gene is exclusively expressed due to imprinting [106,107]. This is potentially relevant to IS given the sex bias of progressive curvatures (females > males), and the higher percentage of affected offspring from paternal IS cases compared to maternal IS cases (80% vs. 56%) [63].
Enrichment analyses of the top DMRs from both the discordant and curve severity results revealed both broad, non-specific ontologies and select ontologies related to neurogenesis, axon guidance and neuron differentiation, all of which can be supported by current literature from both family-based exome sequencing and genome-wide association (GWAS) studies [49,50]. Combined with results in the current study, these data suggests a potential role of neuropathological processes underlying the development and progression of IS.
The complex genetic architecture underlying IS is further complicated by the lack of a clear tissue target. Despite the major clinical manifestation and therapeutic dilemma of the spinal curvature, IS ultimately affects multiple tissue types, one of which is bone. To understand the potential functional relevance of our methylation results in osseous tissue, we reviewed overlap between our identified CpG sites and those reported by Ebrahimi et al. [91] correlating the same methylation markers in matched blood and trabecular bone samples. We identified 21 regions where the DNA methylation in our dataset was correlated with methylation in bone tissue, and therefore, could potentially be considered biologically relevant (Table 2). Annotation of two of the regions implicated the NPY gene on chr. 7 and the WNT10A gene on chr. 2. The NPY gene encodes a neuropeptide expressed throughout the central and peripheral nervous systems [108] and is an essential regulator of bone homeostasis and metabolism [109]. NPY is also a local regulator of osteoblastic lineage and is responsive to mechanical stimuli with potential roles in fracture healing and osteoarthritis [109]. The WNT10A gene is a member of the WNT gene class and functions within the WNT10A/β-catenin signaling pathway in regulation of adult epithelial proliferation [110,111], mesenchymal stem cell regulation by stimulating osteoblastogenesis [112], coordination of vertebrate segmentation, and motile cilia function [113].
The role of DNA methylation in IS has not been well studied in current literature outside of two targeted studies and extremely small genome-wide discovery analyses. Mao et al. [73] reported IS cases were associated with increased methylation near the promotor region for the COMP gene on chr. 19 and more importantly, decreased COMP expression. Shi et al. [74] identified significantly higher levels of methylation in IS cases versus controls in a region near the pituitary homeobox-1 (PITX1) gene, a homeobox transcriptional regulator that plays a role in maintenance of side-to-side musculoskeletal symmetry during development [114]. In our study, methylation levels in the promoter regions for the PITX1 and/or the COMP gene were not differentially methylated in the discordant and/or curve severity analysis. However, pituitary gland development (GO:0021983) and the anterior/posterior pattern specification (GO:0009952) ontologies represented the top 2 most enriched terms in the curve severity DMR analysis (see Appendix A, Table A5).
Meng et al. [78] and Liu et al. [77] conducted the only other IS EWAS studies in the current literature. They used a similar strategy, testing for methylation differences in peripheral blood samples in a discovery cohort (1 and 2 MZ twin pairs, respectively) discordant for curve progression. A second cohort consisting of individuals with IS versus controls was used to confirm methylation sites or regions identified in the discovery cohort. Meng et al. [78] identified a single probe cg01374129 (near the HSA2 gene) that was significantly hypomethylated in the progressive group compared to the non-progressive group. Liu et al. [77] identified a DMR near the promoter region for the NDN gene that was significantly associated with IS. These studies were limited in that the discovery EWAS was performed in a very limited number of individuals. Our study builds on these initial findings with added methylation data from MZ twins both concordant and discordant in their spinal curves. Our complementary analyses provide a list of candidate sites and regions across the genome that may assist in the development of prognostic tools capable of identifying individuals at risk for curve progression. Methylation is tissue specific, we were also able to prioritize hits identified in our analysis based on known correlation with methylation in bone tissue. Additional validation in larger cohorts is needed to confirm these methylation markers as relevant and explore their utility in the clinical setting.
Limitations
Our study includes several limitations. First, the samples were obtained after disease onset. We cannot exclude the possibility that differences in methylation were caused by changes in curve severity. Although samples within each twin pair were obtained no more than six months apart and thus age at sample acquisition was balanced across the twin pairs, there was substantial heterogeneity in age at sample acquisition across the twin pairs. This is potentially problematic if age modifies the effect of methylation on curve progression. Similarly, we used a case–control design. Cases and controls were defined based on curve pattern information available at the time of sample acquisition. Misclassification of controls is possible if spinal progression occurred over the lifetime of the individual.
5. Conclusions
A better understanding of the genetic, epigenetic, and environmental factors underlying IS onset and/or curve progression has significant clinical implications [115,116]. DNA methylation markers may provide value as a prognostic tool for predicting both the initiation and progression of this disorder and furthermore, may also aid in the identification of homogenous subgroups of individuals allowing for more personalized treatment algorithms. In the current study, we identified methylation at specific sites across the genome. Differentially methylation region (DMR) promoter enrichment analyses identified several biologically relevant ontologies related to pituitary gland development, body segmentation and neuronal differentiation. We prioritized the DMR candidates based on known correlation between methylation in blood versus bone. Priority candidates include DMRs in promoter regions related to the WNT signaling pathway (WNT10A), a signaling pathway that is relevant to bone formation and remodeling [117], and neuropeptide Y (NPY), a regulator of bone and energy homeostasis [109]. This information allows for further targeted studies aimed at understanding the functional relevance of these findings in relation to IS and axial spinal development, alignment, and side-to-side symmetry.
Appendix A
Table A1.
Proportion of Blood Cell Populations in IS Cases and Controls.
| Cases | Control | ||||||
|---|---|---|---|---|---|---|---|
| Median | Min | Max | Median | Min | Max | p Value | |
| CD8T Cell (%) | 11.5% | 8.3% | 33.6% | 12.2% | 6.9% | 18.7% | 0.1953 |
| CD4T Cell (%) | 18.5% | 7.7% | 28.3% | 14.3% | 9.3% | 18.0% | 0.1953 |
| Natural Killer Cell Count (%) | 5.3% | 2.2% | 8.8% | 5.7% | 1.9% | 8.4% | 0.7422 |
| Bcell Count (%) | 5.8% | 3.5% | 10.3% | 6.2% | 2.9% | 10.3% | 0.6406 |
| Monocyte Cell Count (%) | 7.5% | 3.4% | 9.5% | 8.0% | 5.1% | 10.9% | 0.6406 |
| Neutrophil Cell Count (%) | 55.3% | 25.7% | 67.2% | 58.4% | 52.0% | 72.9% | 0.1953 |
Table A2.
Analysis of Discordant Curve Severity in Twin Pairs Identified 57 Differentially Methylated Regions where Hyper- or Hypo-methylation was Consistent Across all Probes in the Region.
| Feature/Gene | Chr. | Start Position | End Position | Number of Probes | Nominal p Value | FDR p Value | Maximum Bone Correlation | FDR p Value for Maximum Bone Correlation | Percent Strongly Positively Correlated Probes Across DMR |
|---|---|---|---|---|---|---|---|---|---|
| WNT10A | chr2 | 219,744,145 | 219,745,748 | 9 | 2.17 × 10−5 | 0.0113 | 0.83 | 0.0307 | 33.3% |
| BCL2L2 * | chr14 | 23,775,206 | 23,776,530 | 8 | 2.17 × 10−5 | 0.0113 | 0.50 | 0.4069 | 14.3% |
| CRISP2 | chr6 | 49,681,178 | 49,681,774 | 11 | 2.19 × 10−5 | 0.0113 | 0.89 | 0.0128 | 100.0% |
| SLFN13 | chr17 | 33,773,921 | 33,777,219 | 11 | 2.19 × 10−5 | 0.0113 | −0.27 | 0.7380 | 0.0% |
| RBPJL | chr20 | 43,934,854 | 43,935,551 | 12 | 2.20 × 10−5 | 0.0113 | 0.72 | 0.1048 | 66.7% |
| KDM2B | chr12 | 122,018,574 | 122,020,205 | 14 | 2.21 × 10−5 | 0.0113 | 0.83 | 0.0336 | 50.0% |
| MS4A3 | chr11 | 59,822,727 | 59,828,426 | 13 | 2.21 × 10−5 | 0.0113 | −0.57 | 0.2917 | 7.7% |
| GPR21 | chr9 | 125,794,756 | 125,797,284 | 14 | 2.21 × 10−5 | 0.0113 | 0.38 | 0.5940 | 0.0% |
| IL27 | chr16 | 28,518,114 | 28,519,597 | 9 | 4.34 × 10−5 | 0.0156 | 0.75 | 0.0844 | 33.3% |
| AZU1 | chr19 | 826,359 | 827,821 | 9 | 4.34 × 10−5 | 0.0156 | −0.28 | 0.7263 | 0.0% |
| CA14 | chr1 | 150,229,143 | 150,230,345 | 9 | 6.51 × 10−5 | 0.0196 | 0.78 | 0.0585 | 33.3% |
| HTRA4 | chr8 | 38,830,814 | 38,831,857 | 11 | 6.58 × 10−5 | 0.0196 | 0.29 | 0.7117 | 0.0% |
| ESRP2 | chr16 | 68,269,763 | 68,271,177 | 13 | 6.62 × 10−5 | 0.0196 | 0.35 | 0.6385 | 0.0% |
| ELANE | chr19 | 850,975 | 852,311 | 8 | 8.68 × 10−5 | 0.0199 | 0.45 | 0.4788 | 0.0% |
| CLDN15 | chr7 | 100,880,751 | 100,882,286 | 10 | 8.74 × 10−5 | 0.0199 | 0.68 | 0.1542 | 20.0% |
| RUNX1 | chr21 | 36,259,179 | 36,422,112 | 14 | 8.85 × 10−5 | 0.0199 | 0.60 | 0.2518 | 21.4% |
| NFE2 | chr12 | 54,689,278 | 54,696,210 | 14 | 8.85 × 10−5 | 0.0199 | −0.36 | 0.6204 | 0.0% |
| C6orf229 | chr6 | 24,799,059 | 24,799,757 | 5 | 9.45 × 10−5 | 0.0199 | 0.70 | 0.1293 | 20.0% |
| SLC25A16 | chr10 | 70,287,181 | 70,287,493 | 6 | 1.07 × 10−5 | 0.0216 | −0.45 | 0.4813 | 0.0% |
| EPHA2 | chr1 | 16,482,553 | 16,483,528 | 8 | 1.09 × 10−4 | 0.0216 | 0.47 | 0.4470 | 0.0% |
| TNFSF13 | chr17 | 7,460,690 | 7,462,249 | 12 | 1.32 × 10−4 | 0.0223 | 0.65 | 0.1888 | 8.3% |
| UBASH3A | chr21 | 43,822,540 | 43,823,863 | 6 | 1.69 × 10−4 | 0.0252 | −0.64 | 0.1916 | 0.0% |
| C7orf49 | chr7 | 134,852,662 | 134,855,381 | 9 | 1.74 × 10−4 | 0.0256 | 0.59 | 0.2703 | 11.1% |
| ADAP2 | chr17 | 29,247,612 | 29,248,848 | 9 | 1.95 × 10−4 | 0.0266 | −0.39 | 0.5675 | 0.0% |
| AMT | chr3 | 49,459,855 | 49,461,563 | 11 | 1.97 × 10−4 | 0.0266 | 0.37 | 0.5997 | 0.0% |
| C9orf47 | chr9 | 91,604,473 | 91,606,140 | 12 | 2.64 × 10−4 | 0.0318 | 0.79 | 0.0518 | 18.2% |
| CD3D | chr11 | 118,213,272 | 118,214,927 | 8 | 2.78 × 10−4 | 0.0318 | −0.20 | 0.8262 | 0.0% |
| HSPB6 | chr19 | 36,247,867 | 36,248,907 | 8 | 2.82 × 10−4 | 0.0318 | 0.24 | 0.7811 | 0.0% |
| STAB1 | chr3 | 52,528,714 | 52,529,393 | 8 | 3.04 × 10−4 | 0.0329 | 0.79 | 0.0534 | 12.5% |
| ACY3 | chr11 | 67,415,183 | 67,418,365 | 8 | 3.26 × 10−4 | 0.0329 | 0.72 | 0.1045 | 62.5% |
| MPG | chr16 | 125,896 | 128,009 | 11 | 3.29 × 10−4 | 0.0329 | 0.82 | 0.0368 | 10.0% |
| KLRD1 | chr12 | 10,455,788 | 10,460,639 | 8 | 3.34 × 10−4 | 0.0329 | −0.73 | 0.1030 | 12.5% |
| FES | chr15 | 91,427,184 | 91,428,456 | 10 | 3.50 × 10−4 | 0.033 | 0.39 | 0.5737 | 0.0% |
| ESM1 | chr5 | 54,281,198 | 54,282,459 | 13 | 3.97 × 10−4 | 0.036 | 0.79 | 0.0526 | 61.5% |
| CTSG | chr14 | 25,045,625 | 25,046,267 | 6 | 4.28 × 10−4 | 0.0382 | −0.36 | 0.6151 | 0.0% |
| HMGN2 | chr1 | 26,797,576 | 267,987,40 | 6 | 4.71 × 10−4 | 0.0403 | −0.45 | 0.4827 | 0.0% |
| LRG1 | chr19 | 4,540,003 | 4,540,782 | 6 | 4.71 × 10−4 | 0.0403 | −0.42 | 0.5212 | 0.0% |
| LOC100130933 | chr17 | 73,641,809 | 73,642,991 | 10 | 4.81 × 10−4 | 0.0405 | 0.44 | 0.5036 | 0.0% |
| HMGCR | chr5 | 74,632,477 | 74,637,028 | 5 | 5.10 × 10−4 | 0.0419 | 0.71 | 0.1165 | 20.0% |
| RPSAP52 | chr12 | 66,220,754 | 66,221,950 | 6 | 5.14 × 10−4 | 0.0419 | −0.77 | 0.0683 | 0.0% |
| MIR145 | chr5 | 148,808,721 | 148,810,180 | 7 | 5.18 × 10−4 | 0.0419 | −0.38 | 0.5867 | 0.0% |
| PILRA | chr7 | 99,970,448 | 99,971,016 | 5 | 5.31 × 10−4 | 0.0419 | 0.37 | 0.6031 | 0.0% |
| TMEM219 | chr16 | 29,972,752 | 29,974,294 | 6 | 5.57 × 10−4 | 0.0431 | 0.77 | 0.0667 | 66.7% |
| GIMAP1 | chr7 | 150,412,503 | 150,415,143 | 5 | 5.86 × 10−4 | 0.0451 | 0.33 | 0.6550 | 0.0% |
| CREBBP | chr16 | 3,930,112 | 3,931,489 | 5 | 6.16 × 10−4 | 0.0457 | 0.76 | 0.0725 | 80.0% |
| MAST2 | chr1 | 46,268,158 | 46,269,120 | 6 | 6.21 × 10−4 | 0.0457 | 0.70 | 0.1248 | 16.7% |
| TAGLN3 | chr3 | 111,717,534 | 111,718,245 | 8 | 6.29 × 10−4 | 0.0457 | 0.68 | 0.1531 | 12.5% |
| S100P | chr4 | 6,694,923 | 6,695,698 | 9 | 6.30 × 10−4 | 0.0457 | −0.51 | 0.3861 | 0.0% |
| FAM53C | chr5 | 137,672,901 | 137,675,418 | 7 | 6.48 × 10−4 | 0.0458 | −0.25 | 0.7705 | 0.0% |
| GAMT | chr19 | 1,401,372 | 1,402,626 | 5 | 6.58 × 10−4 | 0.0459 | −0.35 | 0.6314 | 0.0% |
| H6PD | chr1 | 9,293,583 | 9,303,499 | 10 | 6.77 × 10−4 | 0.0462 | 0.39 | 0.5714 | 0.0% |
| ITGAE | chr17 | 3,704,471 | 37,058,75 | 10 | 6.77 × 10−4 | 0.0462 | −0.45 | 0.4790 | 0.0% |
| CDH9 | chr5 | 27,036,352 | 27,040,099 | 10 | 6.77 × 10−4 | 0.0462 | −0.42 | 0.5366 | 0.0% |
| ELP2 | chr18 | 33,709,151 | 33,709,799 | 7 | 6.91 × 10−4 | 0.0462 | −0.22 | 0.8067 | 0.0% |
| LRP11 | chr6 | 150,185,188 | 150,186,488 | 8 | 7.16 × 10−4 | 0.0462 | 0.54 | 0.3423 | 12.5% |
| FIGLA | chr2 | 71,017,541 | 71,018,823 | 11 | 7.46 × 10−4 | 0.0473 | 0.61 | 0.2324 | 18.2% |
| CAPN13 | chr2 | 31,020,802 | 31,031,755 | 6 | 8.07 × 10−4 | 0.0496 | 0.48 | 0.4419 | 0.0% |
* BCL2L2-PABPN1, Maximum Bone Correlation = maximum correlation coefficient representing strength of correlation between blood and bone CpG sites across all sites within the DMR (from Ebrahimi et al.), FDR p Value For Maximum Bone Correlation = FDR adjusted p value for maximum correlation coefficient across all sites included in the DMR (from Ebrahimi et al.), Percent Positively Strongly Correlated Probes Across DMR = Percentage of probes across the entire region where the correlation coefficient representing strength of correlation between blood and bone CpG sites is greater than 75th percentile among all probes tested in Ebrahimi et al.
Table A3.
Based on 58 DMRs for Discordant Curve Severity, one Significantly Enriched Ontology was Identified. Analysis of Discordant Curve Severity in Twin Pairs Identified n = 57 Differentially Methylated Regions where Hyper- or Hypomethylation was Consistent Across all Probes in the Region.
| Category | Fold Enrichment | Bonferroni Adjusted p Value |
|---|---|---|
| Gene Ontology Cellular Component | ||
| secretory granule lumen (GO:0034774) | 3.8 | 7.29 × 10−3 |
Table A4.
Analysis of Curve Severity in Twin Pairs Identified 28 Differentially Methylated Regions where Slope Representing Association between Difference in Methylation and Difference in Curve Severity within each Twin Pair was Consistent Across all Probes in the Region.
| Feature/Gen | Chr. | Start Position | End Position | Number of Probes | Nominal p Value | FDR p Value | Maximum Bone Correlation | FDR p Value for Maximum Bone Correlation | Percent Strongly Positively Correlated Probes Across DMR |
|---|---|---|---|---|---|---|---|---|---|
| NNAT | chr20 | 36,148,133 | 36,149,750 | 34 | 1.11 × 10−5 | 0.0237 | 0.57 | 0.2898 | 12.1% |
| TMEM232 | chr5 | 110,021,543 | 110,062,837 | 11 | 5.09 × 10−5 | 0.0237 | 0.68 | 0.1441 | 30.0% |
| SLC22A20 | chr11 | 64,979,837 | 64,981,596 | 9 | 8.43 × 10−5 | 0.0252 | 0.66 | 0.1680 | 33.3% |
| PDE12 | chr3 | 57,541,377 | 57,543,243 | 5 | 9.89 × 10−5 | 0.0252 | 0.69 | 0.1347 | 40.0% |
| SUV420H2 | chr19 | 55,850,082 | 55,852,507 | 5 | 1.98 × 10−4 | 0.0299 | 0.70 | 0.1324 | 40.0% |
| CYR61 | chr1 | 86,045,347 | 86,046,661 | 10 | 2.22 × 10−4 | 0.0299 | 0.71 | 0.1148 | 40.0% |
| ZNF440 | chr19 | 11,924,860 | 11,925,219 | 7 | 2.63 × 10−4 | 0.0333 | 0.63 | 0.2110 | 14.3% |
| LOC150622 | chr2 | 6,072,139 | 6,072,801 | 5 | 3.16 × 10−4 | 0.0348 | 0.61 | 0.2383 | 40.0% |
| GANC | chr15 | 42,565,522 | 42,566,390 | 7 | 3.38 × 10−4 | 0.0357 | 0.88 | 0.0153 | 28.6% |
| TMEM87A | chr15 | 42,565,522 | 42,566,390 | 7 | 3.38 × 10−4 | 0.0357 | NA | NA | NA |
| NME3 | chr16 | 1,821,559 | 1,822,346 | 8 | 3.59 × 10−4 | 0.0366 | 0.76 | 0.0729 | 37.5% |
| SLC6A5 | chr11 | 20,619,598 | 20,621,109 | 8 | 3.59 × 10−4 | 0.0366 | 0.80 | 0.0489 | 28.6% |
| HSPB6 | chr19 | 36,247,867 | 36,248,907 | 8 | 3.98 × 10−4 | 0.0378 | 0.24 | 0.7811 | 0.0% |
| RAB22A | chr20 | 56,883,532 | 56,885,003 | 8 | 4.38 × 10−4 | 0.0391 | 0.78 | 0.0594 | 25.0% |
| SLC1A1 | chr9 | 4,489,544 | 4,490,288 | 6 | 4.60 × 10−4 | 0.0394 | 0.60 | 0.2551 | 16.7% |
| ACTN4 | chr19 | 39,137,911 | 39,138,334 | 7 | 4.89 × 10−4 | 0.0407 | 0.84 | 0.0298 | 33.3% |
| PRKD1 | chr14 | 30,396,845 | 30,397,763 | 6 | 4.96 × 10−4 | 0.0407 | 0.49 | 0.4133 | 16.7% |
| STL | chr6 | 125,284,212 | 125,284,659 | 6 | 4.96 × 10−4 | 0.0407 | 0.22 | 0.8030 | 0.0% |
| CPXM1 | chr20 | 2,781,122 | 2,782,348 | 9 | 5.06 × 10−4 | 0.0411 | 0.43 | 0.5202 | 0.0% |
| INSR | chr19 | 7,294,087 | 7,295,192 | 5 | 5.27 × 10−4 | 0.0413 | 0.61 | 0.2438 | 20.0% |
| NPY | chr7 | 24,322,873 | 24,324,570 | 8 | 5.58 × 10−4 | 0.0421 | 0.84 | 0.0276 | 37.5% |
| RAB38 | chr11 | 87,908,558 | 87,909,729 | 9 | 6.33 × 10−4 | 0.045 | 0.73 | 0.0995 | 44.4% |
| CNTNAP5 | chr2 | 124,782,117 | 124,783,254 | 9 | 6.33 × 10−4 | 0.045 | 0.63 | 0.2146 | 11.1% |
| AKR7L | chr1 | 19,600,471 | 19,601,069 | 7 | 7.52 × 10−4 | 0.0495 | 0.49 | 0.4219 | 0.0% |
| COPB1 | chr11 | 14,521,639 | 14,522,617 | 6 | 7.79 × 10−4 | 0.0495 | 0.88 | 0.0149 | 50.0% |
| MEI1 | chr22 | 42,095,347 | 42,095,536 | 5 | 7.91 × 10−4 | 0.0495 | 0.65 | 0.1891 | 40.0% |
| PPP2R1B | chr11 | 111,637,044 | 111,638,422 | 5 | 7.91 × 10−4 | 0.0495 | 0.69 | 0.1381 | 40.0% |
| KCNB1 | chr20 | 48,098,642 | 48,100,238 | 8 | 7.97 × 10−4 | 0.0495 | 0.55 | 0.3315 | 12.5% |
Maximum Bone Correlation = maximum correlation coefficient representing strength of correlation between blood and bone CpG sites across all sites included in the DMR (from Ebrahimi et al.), FDR p Value for Maximum Bone Correlation = FDR adjusted p value for maximum correlation coefficient across all sites included in the DMR (from Ebrahimi et al.), Percent Strongly Positively Correlated Probes Across DMR = Percentage of probes across the entire region where the correlation coefficient representing strength of correlation between blood and bone CpG sites is greater than 75th percentile among all probes tested in Ebrahimi et al., NA = probes unavailable.
Table A5.
Significantly Enriched Ontologies Based on Curve Severity DMR Promoter Analysis.
| Category | Fold Enrichment | Bonferroni Adjusted p Value |
|---|---|---|
| Gene Ontology Molecular Function | ||
| RNA polymerase II cis-regulatory region sequence-specific DNA binding (GO:0000978) | 2.4 | 1.27 × 10−3 |
| DNA-binding transcription factor activity, RNA polymerase II-specific (GO:0000981) | 2.3 | 3.65 × 10−4 |
| regulatory region nucleic acid binding (GO:0001067) | 2.2 | 1.04 × 10−3 |
| transcription regulator activity (GO:0140110) | 1.9 | 2.04 × 10−2 |
| Gene Ontology Cellular Component | ||
| chromatin (GO:0000785) | 2.3 | 7.60 × 10−3 |
| Gene Ontology Biologic Process | ||
| pituitary gland development (GO:0021983) | 12.0 | 8.09 × 10−3 |
| anterior/posterior pattern specification (GO:0009952) | 5.2 | 3.91 × 10−3 |
| mesenchyme development (GO:0060485) | 4.8 | 8.84 × 10−3 |
| heart morphogenesis (GO:0003007) | 4.4 | 2.44 × 10−3 |
| embryonic organ development (GO:0048568) | 3.3 | 4.70 × 10−3 |
| negative regulation of cell differentiation (GO:0045596) | 3.0 | 6.46 × 10−3 |
| head development (GO:0060322) | 2.7 | 2.37 × 10−3 |
| tube development (GO:0035295) | 2.7 | 3.04 × 10−3 |
| neuron differentiation (GO:0030182) | 2.5 | 5.09 × 10−3 |
| anatomical structure morphogenesis (GO:0009653) | 2.0 | 3.30 × 10−3 |
Author Contributions
Conceptualization: P.M.C. and N.H.-M. Methodology: P.M.C., L.A.V. and N.H.-M. Software: P.M.C., L.A.V., and G.D.T. Validation: N/A. Formal analysis: P.M.C. Investigation: N.H.-M., E.A.T. and C.I.W. Resources: N.H.-M., P.E., F.M. and K.Å. Data curation: P.M.C. and E.A.T. Writing—original draft preparation: P.M.C. Writing—review and editing: P.M.C., E.A.T., G.D.T., L.A.V., C.I.W., P.E., F.M., K.Å. and N.H.-M. Visualization: P.M.C. Supervision: N.H.-M. Project administration: E.A.T. and N.H.-M. Funding acquisition: N.H.-M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially funded by NIH NIAMS R01 AR068292.
Institutional Review Board Statement
Written informed consent was obtained from study subjects who were enrolled in accordance with protocols approved by the Johns Hopkins School of Medicine Institutional Review Board and the University of Colorado Anschutz Medical Campus Institutional Review Board (Colorado Multiple Institutional Review Board, Studies #06-1161 and 07-0417). All procedures involving human participants were performed in accordance with the ethical standards of these institutional review boards, the 1964 Declaration of Helsinki and its later amendments, or comparable ethical standards.
Informed Consent Statement
Written informed consent was obtained for all study participants in accordance with protocols approved through the Johns Hopkins School of Medicine Institutional Review Board and the University of Colorado Anschutz Medical Campus Institutional Review Board.
Data Availability Statement
Data is available from the author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Altaf F., Drinkwater J., Phan K., Cree A.K. Systematic Review of School Scoliosis Screening. Spine Deform. 2017;5:303–309. doi: 10.1016/j.jspd.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein S.L., Dolan L.A., Cheng J.C., Danielsson A., Morcuende J.A. Adolescent idiopathic scoliosis. Lancet. 2008;371:1527–1537. doi: 10.1016/S0140-6736(08)60658-3. [DOI] [PubMed] [Google Scholar]
- 3.Miller N.H. Genetics of familial idiopathic scoliosis. Clin. Orthop. Relat. Res. 2007;462:6–10. doi: 10.1097/BLO.0b013e318126c062. [DOI] [PubMed] [Google Scholar]
- 4.Khanshour A.M., Wise C.A. Pathogenesis of Idiopathic Scoliosis. Springer; Berlin, Germany: 2018. The Genetic Architecture of Adolescent Idiopathic Scoliosis; pp. 51–74. [Google Scholar]
- 5.Tang N.L., Yeung H.Y., Hung V.W., Di Liao C., Lam T.P., Yeung H.M., Lee K.M., Ng B.K., Cheng J.C. Genetic epidemiology and heritability of AIS: A study of 415 Chinese female patients. J. Orthop Res. 2012;30:1464–1469. doi: 10.1002/jor.22090. [DOI] [PubMed] [Google Scholar]
- 6.Ward K., Ogilvie J., Argyle V., Nelson L., Meade M., Braun J., Chettier R. Polygenic inheritance of adolescent idiopathic scoliosis: A study of extended families in Utah. Am. J. Med. Genet. A. 2010;152:1178–1188. doi: 10.1002/ajmg.a.33145. [DOI] [PubMed] [Google Scholar]
- 7.Miller N.H., Mims B., Child A., Milewicz D.M., Sponseller P., Blanton S.H. Genetic analysis of structural elastic fiber and collagen genes in familial adolescent idiopathic scoliosis. J. Orthop. Res. 1996;14:994–999. doi: 10.1002/jor.1100140621. [DOI] [PubMed] [Google Scholar]
- 8.Miller N.H., Schwab D.L., Sponseller P., Shugert E., Bell J., Maestri N. Genomic Search for X-Linkage in Familial Adolescent Idiopathic Scoliosis. In: Stokes I.A., editor. Research into Spinal Deformities 2. IOS Press; Amsterdam, The Netherlands: 1998. pp. 209–213. [Google Scholar]
- 9.Wise C.A., Barnes R., Gillum J., Herring J.A., Bowcock A.M., Lovett M. Localization of susceptibility to familial idiopathic scoliosis. Spine (Phila Pa 1976) 2000;25:2372–2380. doi: 10.1097/00007632-200009150-00017. [DOI] [PubMed] [Google Scholar]
- 10.Chan V., Fong G.C., Luk K.D., Yip B., Lee M.K., Wong M.S., Lu D.D., Chan T.K. A genetic locus for adolescent idiopathic scoliosis linked to chromosome 19p13.3. Am. J. Hum. Genet. 2002;71:401–406. doi: 10.1086/341607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Justice C.M., Miller N.H., Marosy B., Zhang J., Wilson A.F. Familial idiopathic scoliosis: Evidence of an X-linked susceptibility locus. Spine (Phila Pa 1976) 2003;28:589–594. doi: 10.1097/01.BRS.0000049940.39801.E6. [DOI] [PubMed] [Google Scholar]
- 12.Miller N.H., Justice C.M., Marosy B., Doheny K.F., Pugh E., Zhang J., Dietz H.C., 3rd, Wilson A.F. Identification of candidate regions for familial idiopathic scoliosis. Spine (Phila Pa 1976) 2005;30:1181–1187. doi: 10.1097/01.brs.0000162282.46160.0a. [DOI] [PubMed] [Google Scholar]
- 13.Alden K.J., Marosy B., Nzegwu N., Justice C.M., Wilson A.F., Miller N.H. Idiopathic scoliosis: Identification of candidate regions on chromosome 19p13. Spine (Phila Pa 1976) 2006;31:1815–1819. doi: 10.1097/01.brs.0000227264.23603.dc. [DOI] [PubMed] [Google Scholar]
- 14.Miller N.H., Marosy B., Justice C.M., Novak S.M., Tang E.Y., Boyce P., Pettengil J., Doheny K.F., Pugh E.W., Wilson A.F. Linkage analysis of genetic loci for kyphoscoliosis on chromosomes 5p13, 13q13.3, and 13q32. Am. J. Med. Genet. A. 2006;140:1059–1068. doi: 10.1002/ajmg.a.31211. [DOI] [PubMed] [Google Scholar]
- 15.Montanaro L., Parisini P., Greggi T., Di Silvestre M., Campoccia D., Rizzi S., Arciola C.R. Evidence of a linkage between matrilin-1 gene (MATN1) and idiopathic scoliosis. Scoliosis. 2006;1:21. doi: 10.1186/1748-7161-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gurnett C.A., Alaee F., Bowcock A., Kruse L., Lenke L.G., Bridwell K.H., Kuklo T., Luhmann S.J., Dobbs M.B. Genetic linkage localizes an adolescent idiopathic scoliosis and pectus excavatum gene to chromosome 18 q. Spine (Phila Pa 1976) 2009;34:E94–E100. doi: 10.1097/BRS.0b013e31818b88a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marosy B., Justice C.M., Vu C., Zorn A., Nzegwu N., Wilson A.F., Miller N.H. Identification of susceptibility loci for scoliosis in FIS families with triple curves. Am. J. Med. Genet. A. 2010;152:846–855. doi: 10.1002/ajmg.a.33222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buchan J.G., Alvarado D.M., Haller G.E., Cruchaga C., Harms M.B., Zhang T., Willing M.C., Grange D.K., Braverman A.C., Miller N.H., et al. Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum. Mol. Genet. 2014;23:5271–5282. doi: 10.1093/hmg/ddu224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baschal E.E., Wethey C.I., Swindle K., Baschal R.M., Gowan K., Tang N.L., Alvarado D.M., Haller G.E., Dobbs M.B., Taylor M.R., et al. Exome sequencing identifies a rare HSPG2 variant associated with familial idiopathic scoliosis. G3. 2014;5:167–174. doi: 10.1534/g3.114.015669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patten S.A., Margaritte-Jeannin P., Bernard J.C., Alix E., Labalme A., Besson A., Girard S.L., Fendri K., Fraisse N., Biot B., et al. Functional variants of POC5 identified in patients with idiopathic scoliosis. J. Clin. Investig. 2015;125:1124–1128. doi: 10.1172/JCI77262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grauers A., Wang J., Einarsdottir E., Simony A., Danielsson A., Akesson K., Ohlin A., Halldin K., Grabowski P., Tenne M., et al. Candidate gene analysis and exome sequencing confirm LBX1 as a susceptibility gene for idiopathic scoliosis. Spine J. 2015;15:2239–2246. doi: 10.1016/j.spinee.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Li W., Li Y., Zhang L., Guo H., Tian D., Li Y., Peng Y., Zheng Y., Dai Y., Xia K., et al. AKAP2 identified as a novel gene mutated in a Chinese family with adolescent idiopathic scoliosis. J. Med. Genet. 2016;53:488–493. doi: 10.1136/jmedgenet-2015-103684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haller G., Alvarado D., McCall K., Yang P., Cruchaga C., Harms M., Goate A., Willing M., Morcuende J.A., Baschal E., et al. A polygenic burden of rare variants across extracellular matrix genes among individuals with adolescent idiopathic scoliosis. Hum. Mol. Genet. 2016;25:202–209. doi: 10.1093/hmg/ddv463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao W., Chen C., Zhou T., Yang S., Gao B., Zhou H., Lian C., Wu Z., Qiu X., Yang X., et al. Rare coding variants in MAPK7 predispose to adolescent idiopathic scoliosis. Hum. Mutat. 2017;38:1500–1510. doi: 10.1002/humu.23296. [DOI] [PubMed] [Google Scholar]
- 25.Einarsdottir E., Grauers A., Wang J., Jiao H., Escher S.A., Danielsson A., Simony A., Andersen M., Christensen S.B., Akesson K., et al. CELSR2 is a candidate susceptibility gene in idiopathic scoliosis. PLoS ONE. 2017;12:e0189591. doi: 10.1371/journal.pone.0189591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baschal E.E., Terhune E.A., Wethey C.I., Baschal R.M., Robinson K.D., Cuevas M.T., Pradhan S., Sutphin B.S., Taylor M.R.G., Gowan K., et al. Idiopathic Scoliosis Families Highlight Actin-Based and Microtubule-Based Cellular Projections and Extracellular Matrix in Disease Etiology. G3. 2018;8:2663–2672. doi: 10.1534/g3.118.200290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terhune E.A., Cuevas M.T., Monley A.M., Wethey C.I., Chen X., Cattell M.V., Bayrak M.N., Bland M.R., Sutphin B., Trahan G.D., et al. Mutations in KIF7 implicated in idiopathic scoliosis in humans and axial curvatures in zebrafish. Hum. Mutat. 2021;42:392–407. doi: 10.1002/humu.24162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S., Gao X., Londono D., Devroy S.E., Mauldin K.N., Frankel J.T., Brandon J.M., Zhang D., Li Q.Z., Dobbs M.B., et al. Genome-wide association studies of adolescent idiopathic scoliosis suggest candidate susceptibility genes. Hum. Mol. Genet. 2011;20:1456–1466. doi: 10.1093/hmg/ddq571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi Y., Kou I., Takahashi A., Johnson T.A., Kono K., Kawakami N., Uno K., Ito M., Minami S., Yanagida H., et al. A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 2011;43:1237–1240. doi: 10.1038/ng.974. [DOI] [PubMed] [Google Scholar]
- 30.Nelson L.M., Chettier R., Ogilvie J.W., Ward K. Candidate Genes for Susceptibility of Adolescent Idiopathic Scoliosis Identified Through a Large Genome-Wide Association Study; Proceedings of the Scoliosis Research Society 46th Annual Meeting & Course; Louisville, KY, USA. 14–17 September 2011. [Google Scholar]
- 31.Kou I., Takahashi Y., Johnson T.A., Takahashi A., Guo L., Dai J., Qiu X., Sharma S., Takimoto A., Ogura Y., et al. Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet. 2013;45:676–679. doi: 10.1038/ng.2639. [DOI] [PubMed] [Google Scholar]
- 32.Miyake A., Kou I., Takahashi Y., Johnson T.A., Ogura Y., Dai J., Qiu X., Takahashi A., Jiang H., Yan H., et al. Identification of a susceptibility locus for severe adolescent idiopathic scoliosis on chromosome 17q24.3. PLoS ONE. 2013;8:e72802. doi: 10.1371/journal.pone.0072802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Z., Tang N.L., Xu L., Qin X., Mao S., Song Y., Liu L., Li F., Liu P., Yi L., et al. Genome-wide association study identifies new susceptibility loci for adolescent idiopathic scoliosis in Chinese girls. Nat. Commun. 2015;6:8355. doi: 10.1038/ncomms9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogura Y., Kou I., Miura S., Takahashi A., Xu L., Takeda K., Takahashi Y., Kono K., Kawakami N., Uno K., et al. A Functional SNP in BNC2 Is Associated with Adolescent Idiopathic Scoliosis. Am. J. Hum. Genet. 2015;97:337–342. doi: 10.1016/j.ajhg.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma S., Londono D., Eckalbar W.L., Gao X., Zhang D., Mauldin K., Kou I., Takahashi A., Matsumoto M., Kamiya N., et al. A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females. Nat. Commun. 2015;6:6452. doi: 10.1038/ncomms7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kou I., Otomo N., Takeda K., Momozawa Y., Lu H.F., Kubo M., Kamatani Y., Ogura Y., Takahashi Y., Nakajima M., et al. Genome-wide association study identifies 14 previously unreported susceptibility loci for adolescent idiopathic scoliosis in Japanese. Nat. Commun. 2019;10:3685. doi: 10.1038/s41467-019-11596-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mogha A., Benesh A.E., Patra C., Engel F.B., Schoneberg T., Liebscher I., Monk K.R. Gpr126 functions in Schwann cells to control differentiation and myelination via G-protein activation. J. Neurosci. 2013;33:17976–17985. doi: 10.1523/JNEUROSCI.1809-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J.F., Yang G.H., Pan X.H., Zhang S.J., Zhao C., Qiu B.S., Gu H.F., Hong J.F., Cao L., Chen Y., et al. Association of GPR126 gene polymorphism with adolescent idiopathic scoliosis in Chinese populations. Genomics. 2015;105:101–107. doi: 10.1016/j.ygeno.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 39.Karner C.M., Long F., Solnica-Krezel L., Monk K.R., Gray R.S. Gpr126/Adgrg6 deletion in cartilage models idiopathic scoliosis and pectus excavatum in mice. Hum. Mol. Genet. 2015;24:4365–4373. doi: 10.1093/hmg/ddv170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qin X., Xu L., Xia C., Zhu W., Sun W., Liu Z., Qiu Y., Zhu Z. Genetic Variant of GPR126 Gene is Functionally Associated With Adolescent Idiopathic Scoliosis in Chinese Population. Spine (Phila Pa 1976) 2017;42:E1098–E1103. doi: 10.1097/BRS.0000000000002123. [DOI] [PubMed] [Google Scholar]
- 41.Man G.C., Tang N.L., Chan T.F., Lam T.P., Li J.W., Ng B.K., Zhu Z., Qiu Y., Cheng J.C. Replication Study for the Association of GWAS-associated Loci with Adolescent Idiopathic Scoliosis Susceptibility and Curve Progression in a Chinese Population. Spine (Phila Pa 1976) 2019;44:464–471. doi: 10.1097/BRS.0000000000002866. [DOI] [PubMed] [Google Scholar]
- 42.Kou I., Watanabe K., Takahashi Y., Momozawa Y., Khanshour A., Grauers A., Zhou H., Liu G., Fan Y.H., Takeda K., et al. A multi-ethnic meta-analysis confirms the association of rs6570507 with adolescent idiopathic scoliosis. Sci. Rep. 2018;8:11575. doi: 10.1038/s41598-018-29011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu G., Liu S., Lin M., Li X., Chen W., Zuo Y., Liu J., Niu Y., Zhao S., Long B., et al. Genetic polymorphisms of GPR126 are functionally associated with PUMC classifications of adolescent idiopathic scoliosis in a Northern Han population. J. Cell Mol. Med. 2018;22:1964–1971. doi: 10.1111/jcmm.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao W., Peng Y., Liang G., Liang A., Ye W., Zhang L., Sharma S., Su P., Huang D. Association between common variants near LBX1 and adolescent idiopathic scoliosis replicated in the Chinese Han population. PLoS ONE. 2013;8:e53234. doi: 10.1371/journal.pone.0053234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang H., Yang Q., Liu Y., Guan Y., Zhan X., Xiao Z., Wei Q. Association between ladybird homeobox 1 gene polymorphisms and adolescent idiopathic scoliosis: A MOOSE-compliant meta-analysis. Medicine (Baltimore) 2019;98:e16314. doi: 10.1097/MD.0000000000016314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang J., Xing D., Li Z., Chua S., Li S. Association Between rs11190870 Polymorphism Near LBX1 and Susceptibility to Adolescent Idiopathic Scoliosis in East Asian Population: A Genetic Meta-Analysis. Spine (Phila Pa 1976) 2014;39:862–869. doi: 10.1097/BRS.0000000000000303. [DOI] [PubMed] [Google Scholar]
- 47.Chen S., Zhao L., Roffey D.M., Phan P., Wai E.K. Association of rs11190870 near LBX1 with adolescent idiopathic scoliosis in East Asians: A systematic review and meta-analysis. Spine J. 2014;14:2968–2975. doi: 10.1016/j.spinee.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 48.Jiang H., Qiu X., Dai J., Yan H., Zhu Z., Qian B., Qiu Y. Association of rs11190870 near LBX1 with adolescent idiopathic scoliosis susceptibility in a Han Chinese population. Eur. Spine J. 2013;22:282–286. doi: 10.1007/s00586-012-2532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y.L., Gao S.J., Xu H., Liu Y., Li H.L., Chen X.Y., Ning G.Z., Feng S.Q. The association of rs11190870 near LBX1 with the susceptibility and severity of AIS, a meta-analysis. Int. J. Surg. 2018;54:193–200. doi: 10.1016/j.ijsu.2018.01.051. [DOI] [PubMed] [Google Scholar]
- 50.Cao Y., Min J., Zhang Q., Li H., Li H. Associations of LBX1 gene and adolescent idiopathic scoliosis susceptibility: A meta-analysis based on 34,626 subjects. BMC Musculoskelet Disord. 2016;17:309. doi: 10.1186/s12891-016-1139-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo L., Yamashita H., Kou I., Takimoto A., Meguro-Horike M., Horike S., Sakuma T., Miura S., Adachi T., Yamamoto T., et al. Functional Investigation of a Non-coding Variant Associated with Adolescent Idiopathic Scoliosis in Zebrafish: Elevated Expression of the Ladybird Homeobox Gene Causes Body Axis Deformation. PLoS Genet. 2016;12:e1005802. doi: 10.1371/journal.pgen.1005802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu S., Wu N., Zuo Y., Zhou Y., Liu J., Liu Z., Chen W., Liu G., Chen Y., Chen J., et al. Genetic Polymorphism of LBX1 Is Associated with Adolescent Idiopathic Scoliosis in Northern Chinese Han Population. Spine (Phila Pa 1976) 2017;42:1125–1129. doi: 10.1097/BRS.0000000000002111. [DOI] [PubMed] [Google Scholar]
- 53.Xu L., Wu Z., Xia C., Tang N., Cheng J.C.Y., Qiu Y., Zhu Z. A Genetic Predictive Model Estimating the Risk of Developing Adolescent Idiopathic Scoliosis. Curr. Genom. 2019;20:246–251. doi: 10.2174/1389202920666190730132411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chettier R., Nelson L., Ogilvie J.W., Albertsen H.M., Ward K. Haplotypes at LBX1 have distinct inheritance patterns with opposite effects in adolescent idiopathic scoliosis. PLoS ONE. 2015;10:e0117708. doi: 10.1371/journal.pone.0117708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Londono D., Kou I., Johnson T.A., Sharma S., Ogura Y., Tsunoda T., Takahashi A., Matsumoto M., Herring J.A., Lam T.P., et al. A meta-analysis identifies adolescent idiopathic scoliosis association with LBX1 locus in multiple ethnic groups. J. Med. Genet. 2014;51:401–406. doi: 10.1136/jmedgenet-2013-102067. [DOI] [PubMed] [Google Scholar]
- 56.Nada D., Julien C., Samuels M.E., Moreau A. A Replication Study for Association of LBX1 Locus with Adolescent Idiopathic Scoliosis in French-Canadian Population. Spine (Phila Pa 1976) 2018;43:172–178. doi: 10.1097/BRS.0000000000002280. [DOI] [PubMed] [Google Scholar]
- 57.Fan Y.H., Song Y.Q., Chan D., Takahashi Y., Ikegawa S., Matsumoto M., Kou I., Cheah K.S., Sham P., Cheung K.M., et al. SNP rs11190870 near LBX1 is associated with adolescent idiopathic scoliosis in southern Chinese. J. Hum. Genet. 2012;57:244–246. doi: 10.1038/jhg.2012.11. [DOI] [PubMed] [Google Scholar]
- 58.Edery P., Margaritte-Jeannin P., Biot B., Labalme A., Bernard J.C., Chastang J., Kassai B., Plais M.H., Moldovan F., Clerget-Darpoux F. New disease gene location and high genetic heterogeneity in idiopathic scoliosis. Eur. J. Hum. Genet. 2011;19:865–869. doi: 10.1038/ejhg.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorman K.F., Julien C., Moreau A. The genetic epidemiology of idiopathic scoliosis. Eur. Spine J. 2012;21:1905–1919. doi: 10.1007/s00586-012-2389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lowe T.G., Edgar M., Margulies J.Y., Miller N.H., Raso V.J., Reinker K.A., Rivard C.H. Etiology of idiopathic scoliosis: Current trends in research. J. Bone Jt. Surg. Am. 2000;82:1157–1168. doi: 10.2106/00004623-200008000-00014. [DOI] [PubMed] [Google Scholar]
- 61.Dunwoodie S.L., Kusumi K. The Genetics and Development of Scoliosis. 2nd ed. Springer International Publishing; Cham, Switzerland: 2018. p. 1. [DOI] [Google Scholar]
- 62.Heary R.F., Madhavan K. Genetics of scoliosis. Neurosurgery. 2008;63:222–227. doi: 10.1227/01.NEU.0000320384.93384.28. [DOI] [PubMed] [Google Scholar]
- 63.Kruse L.M., Buchan J.G., Gurnett C.A., Dobbs M.B. Polygenic threshold model with sex dimorphism in adolescent idiopathic scoliosis: The Carter effect. J. Bone Jt. Surg. Am. 2012;94:1485–1491. doi: 10.2106/JBJS.K.01450. [DOI] [PubMed] [Google Scholar]
- 64.Arpon A., Milagro F.I., Ramos-Lopez O., Mansego M.L., Riezu-Boj J.I., Martinez J.A., Project M. Methylome-Wide Association Study in Peripheral White Blood Cells Focusing on Central Obesity and Inflammation. Genes. 2019;10:444. doi: 10.3390/genes10060444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grauers A., Rahman I., Gerdhem P. Heritability of scoliosis. Eur. Spine J. 2012;21:1069–1074. doi: 10.1007/s00586-011-2074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Domcke S., Bardet A.F., Adrian Ginno P., Hartl D., Burger L., Schubeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–579. doi: 10.1038/nature16462. [DOI] [PubMed] [Google Scholar]
- 67.Gutierrez-Arcelus M., Lappalainen T., Montgomery S.B., Buil A., Ongen H., Yurovsky A., Bryois J., Giger T., Romano L., Planchon A., et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife. 2013;2:e00523. doi: 10.7554/eLife.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lappalainen T., Greally J.M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 2017;18:441–451. doi: 10.1038/nrg.2017.32. [DOI] [PubMed] [Google Scholar]
- 69.Gertz J., Varley K.E., Reddy T.E., Bowling K.M., Pauli F., Parker S.L., Kucera K.S., Willard H.F., Myers R.M. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7:e1002228. doi: 10.1371/journal.pgen.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Delgado-Calle J., Fernandez A.F., Sainz J., Zarrabeitia M.T., Sanudo C., Garcia-Renedo R., Perez-Nunez M.I., Garcia-Ibarbia C., Fraga M.F., Riancho J.A. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2013;65:197–205. doi: 10.1002/art.37753. [DOI] [PubMed] [Google Scholar]
- 71.Mohandas N., Bass-Stringer S., Maksimovic J., Crompton K., Loke Y.J., Walstab J., Reid S.M., Amor D.J., Reddihough D., Craig J.M. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin. Epigenet. 2018;10:25. doi: 10.1186/s13148-018-0457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diboun I., Wani S., Ralston S.H., Albagha O.M. Epigenetic analysis of Paget’s disease of bone identifies differentially methylated loci that predict disease status. eLife. 2021;10:e65715. doi: 10.7554/eLife.65715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mao S.H., Qian B.P., Shi B., Zhu Z.Z., Qiu Y. Quantitative evaluation of the relationship between COMP promoter methylation and the susceptibility and curve progression of adolescent idiopathic scoliosis. Eur. Spine J. 2018;27:272–277. doi: 10.1007/s00586-017-5309-y. [DOI] [PubMed] [Google Scholar]
- 74.Shi B., Xu L., Mao S., Xu L., Liu Z., Sun X., Zhu Z., Qiu Y. Abnormal PITX1 gene methylation in adolescent idiopathic scoliosis: A pilot study. BMC Musculoskelet Disord. 2018;19:138. doi: 10.1186/s12891-018-2054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Janusz P., Chmielewska M., Andrusiewicz M., Kotwicka M., Kotwicki T. Methylation of Estrogen Receptor 1 Gene in the Paraspinal Muscles of Girls with Idiopathic Scoliosis and Its Association with Disease Severity. Genes. 2021;12:790. doi: 10.3390/genes12060790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chmielewska M., Janusz P., Andrusiewicz M., Kotwicki T., Kotwicka M. Methylation of estrogen receptor 2 (ESR2) in deep paravertebral muscles and its association with idiopathic scoliosis. Sci. Rep. 2020;10:22331. doi: 10.1038/s41598-020-78454-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu G., Wang L., Wang X., Yan Z., Yang X., Lin M., Liu S., Zuo Y., Niu Y., Zhao S., et al. Whole-Genome Methylation Analysis of Phenotype Discordant Monozygotic Twins Reveals Novel Epigenetic Perturbation Contributing to the Pathogenesis of Adolescent Idiopathic Scoliosis. Front. Bioeng. Biotechnol. 2019;7:364. doi: 10.3389/fbioe.2019.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meng Y., Lin T., Liang S., Gao R., Jiang H., Shao W., Yang F., Zhou X. Value of DNA methylation in predicting curve progression in patients with adolescent idiopathic scoliosis. EBioMedicine. 2018;36:489–496. doi: 10.1016/j.ebiom.2018.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shands A.R.J., Eisberg H.B. The incidence of scoliosis in the state of Delaware; a study of 50,000 minifilms of the chest made during a survey for tuberculosis. J. Bone Jt. Surg. Am. 1955;37:1243–1249. doi: 10.2106/00004623-195537060-00012. [DOI] [PubMed] [Google Scholar]
- 80.Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY, USA: 1989. [Google Scholar]
- 81.Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Logue M.W., Smith A.K., Wolf E.J., Maniates H., Stone A., Schichman S.A., McGlinchey R.E., Milberg W., Miller M.W. The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics. 2017;9:1363–1371. doi: 10.2217/epi-2017-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jaffe A.E., Irizarry R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genom. Biol. 2014;15:R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological) 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 86.Martorell-Marugan J., Gonzalez-Rumayor V., Carmona-Saez P. mCSEA: Detecting subtle differentially methylated regions. Bioinformatics. 2019;35:3257–3262. doi: 10.1093/bioinformatics/btz096. [DOI] [PubMed] [Google Scholar]
- 87.Mi H., Muruganujan A., Casagrande J.T., Thomas P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mi H., Thomas P. PANTHER pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009;563:123–140. doi: 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Supek F., Bosnjak M., Skunca N., Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ebrahimi P., Luthman H., McGuigan F.E., Akesson K.E. Epigenome-wide cross-tissue correlation of human bone and blood DNA methylation—can blood be used as a surrogate for bone? Epigenetics. 2020;16:92–105. doi: 10.1080/15592294.2020.1788325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paul D.S., Teschendorff A.E., Dang M.A., Lowe R., Hawa M.I., Ecker S., Beyan H., Cunningham S., Fouts A.R., Ramelius A., et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016;7:13555. doi: 10.1038/ncomms13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Webster A.P., Plant D., Ecker S., Zufferey F., Bell J.T., Feber A., Paul D.S., Beck S., Barton A., Williams F.M.K., et al. Increased DNA methylation variability in rheumatoid arthritis-discordant monozygotic twins. Genom. Med. 2018;10:64. doi: 10.1186/s13073-018-0575-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Watanabe K., Takebayashi H., Bepari A.K., Esumi S., Yanagawa Y., Tamamaki N. Dpy19l1, a multi-transmembrane protein, regulates the radial migration of glutamatergic neurons in the developing cerebral cortex. Development. 2011;138:4979–4990. doi: 10.1242/dev.068155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Watanabe K., Bizen N., Sato N., Takebayashi H. Endoplasmic Reticulum-Localized Transmembrane Protein Dpy19L1 Is Required for Neurite Outgrowth. PLoS ONE. 2016;11:e0167985. doi: 10.1371/journal.pone.0167985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Data Z.H. Phenotype Annotation. [(accessed on 17 May 2021)];1994–2006 Available online: https://zfin.org/ZDB-FIG-070117-942.
- 97.Perry R.B., Hezroni H., Goldrich M.J., Ulitsky I. Regulation of Neuroregeneration by Long Noncoding RNAs. Mol. Cell. 2018;72:553–567. doi: 10.1016/j.molcel.2018.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Z., Li X., Shen J., Zhang L., Chan M.T.V., Wu W.K.K. Emerging roles of non-coding RNAs in scoliosis. Cell Prolif. 2020;53:e12736. doi: 10.1111/cpr.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cunningham T.J., Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015;16:110–123. doi: 10.1038/nrm3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sirbu I.O., Duester G. Retinoic-acid signalling in node ectoderm and posterior neural plate directs left-right patterning of somitic mesoderm. Nat. Cell Biol. 2006;8:271–277. doi: 10.1038/ncb1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hitier M., Hamon M., Denise P., Lacoudre J., Thenint M.A., Mallet J.F., Moreau S., Quarck G. Lateral Semicircular Canal Asymmetry in Idiopathic Scoliosis: An Early Link between Biomechanical, Hormonal and Neurosensory Theories? PLoS ONE. 2015;10:e0131120. doi: 10.1371/journal.pone.0131120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rousie D., Joly O., Berthoz A. Posterior Basicranium asymmetry and idiopathic scoliosis. arXiv. 20090911.5042 [Google Scholar]
- 103.Carry P.M., Duke V.R., Brazell C.J., Stence N., Scholes M., Rousie D.L., Hadley Miller N. Lateral semi-circular canal asymmetry in females with idiopathic scoliosis. PLoS ONE. 2020;15:e0232417. doi: 10.1371/journal.pone.0232417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hino H., Araki K., Uyama E., Takeya M., Araki M., Yoshinobu K., Miike K., Kawazoe Y., Maeda Y., Uchino M., et al. Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2004;13:181–190. doi: 10.1093/hmg/ddh017. [DOI] [PubMed] [Google Scholar]
- 105.Joseph R.M. Neuronatin gene: Imprinted and misfolded: Studies in Lafora disease, diabetes and cancer may implicate NNAT-aggregates as a common downstream participant in neuronal loss. Genomics. 2014;103:183–188. doi: 10.1016/j.ygeno.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 106.Kikyo N., Williamson C.M., John R.M., Barton S.C., Beechey C.V., Ball S.T., Cattanach B.M., Surani M.A., Peters J. Genetic and functional analysis of neuronatin in mice with maternal or paternal duplication of distal Chr 2. Dev. Biol. 1997;190:66–77. doi: 10.1006/dbio.1997.8681. [DOI] [PubMed] [Google Scholar]
- 107.Evans H.K., Wylie A.A., Murphy S.K., Jirtle R.L. The neuronatin gene resides in a “micro-imprinted” domain on human chromosome 20q11. 2. Genomics. 2001;77:99–104. doi: 10.1006/geno.2001.6612. [DOI] [PubMed] [Google Scholar]
- 108.Lee N.J., Herzog H. NPY regulation of bone remodelling. Neuropeptides. 2009;43:457–463. doi: 10.1016/j.npep.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 109.Wu J.Q., Jiang N., Yu B. Mechanisms of action of neuropeptide Y on stem cells and its potential applications in orthopaedic disorders. World J. Stem. Cells. 2020;12:986–1000. doi: 10.4252/wjsc.v12.i9.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu M., Horrell J., Snitow M., Cui J., Gochnauer H., Syrett C.M., Kallish S., Seykora J.T., Liu F., Gaillard D., et al. WNT10A mutation causes ectodermal dysplasia by impairing progenitor cell proliferation and KLF4-mediated differentiation. Nat. Commun. 2017;8:15397. doi: 10.1038/ncomms15397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vink C.P., Ockeloen C.W., ten Kate S., Koolen D.A., Ploos van Amstel J.K., Kuijpers-Jagtman A.M., van Heumen C.C., Kleefstra T., Carels C.E. Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur. J. Hum. Genet. 2014;22:1063–1070. doi: 10.1038/ejhg.2013.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cawthorn W.P., Bree A.J., Yao Y., Du B., Hemati N., Martinez-Santibanez G., MacDougald O.A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone. 2012;50:477–489. doi: 10.1016/j.bone.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grimes D.T., Boswell C.W., Morante N.F., Henkelman R.M., Burdine R.D., Ciruna B. Zebrafish models of idiopathic scoliosis link cerebrospinal fluid flow defects to spine curvature. Science. 2016;352:1341–1344. doi: 10.1126/science.aaf6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gurnett C.A., Alaee F., Kruse L.M., Desruisseau D.M., Hecht J.T., Wise C.A., Bowcock A.M., Dobbs M.B. Asymmetric lower-limb malformations in individuals with homeobox PITX1 gene mutation. Am. J. Hum. Genet. 2008;83:616–622. doi: 10.1016/j.ajhg.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Feinberg A.P. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 116.Hamilton J.P. Epigenetics: Principles and practice. Dig. Dis. 2011;29:130–135. doi: 10.1159/000323874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Baron R., Kneissel M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data is available from the author upon reasonable request.