

SUMMARY
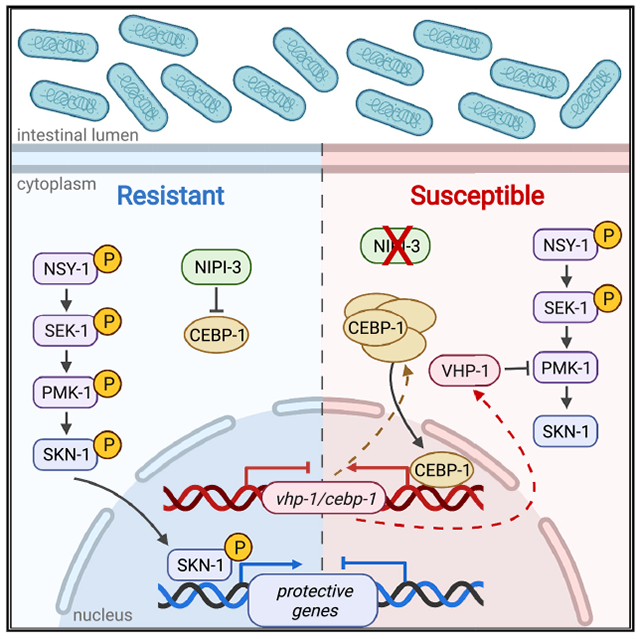
In Caenorhabditis elegans, ROS generated in response to intestinal infection induces SKN-1, a protective transcription factor homologous to nuclear factor erythroid 2-related factor 1 or 2 (NRF1/2) in mammals. Many factors regulate SKN-1, including the p38 mitogen-activated protein kinase (MAPK) cascade that activates SKN-1 by phosphorylation. In this work, another positive regulator of SKN-1 is identified: NIPI-3, a Tribbles pseudokinase. NIPI-3 has been reported to protect against intestinal infection by negatively regulating the CCAT enhancer binding protein (C/EBP) bZIP transcription factor CEBP-1. Here we demonstrate that CEBP-1 positively regulates the vhp-1 transcript, which encodes a phosphatase that dephosphorylates the p38 MAPK called PMK-1. The increased levels of VHP-1 caused by CEBP-1 transcriptional enhancement result in less PMK-1 phosphorylation, affecting SKN-1 activity and intestinal resistance to the pathogen. The data support a model in which NIPI-3’s negative regulation of CEBP-1 decreases VHP-1 phosphatase activity, allowing increased stimulation of SKN-1 activity by the p38 MAPK phosphorylation cascade in the intestine.
In brief
Both NIPI-3 and SKN-1 are known to play a role in intestinal innate immunity, and Wu et al. connect these two factors in a common pathway. NIPI-3 negatively regulates the enhancer binding protein CEBP-1, decreasing VHP-1 phosphatase activity and allowing increased stimulation of SKN-1 activity by p38 MAPK phosphorylation.
Graphical Abstract
INTRODUCTION
The inflammation that occurs as part of the innate immune response is necessary for the host to overcome many infectious agents. However, uncontrolled or inappropriate inflammation can be just as damaging as, or more so than, the pathogen. One group of factors that has been studied for their ability to counteract oxidative damage during the immune response is the Nrf (nuclear factor erythroid 2-related factor)/CNC (Cap’n’-collar) family of transcriptional regulators (Battino et al., 2018; Blackwell et al., 2015; Fuse and Kobayashi, 2017). Mammalian Nrf2 was discovered to be protective during infections as diverse as septic shock (Thimmulappa et al., 2006), pneumonia (Gomez et al., 2016), chronic granulomatous disease (CGD) (Segal et al., 2010), and periodontitis (Chiu et al., 2017).
Caenorhabditis elegans contains a functional ortholog of the Nrf proteins called SKN-1 (Blackwell et al., 2015). Like the Nrf proteins in mammals, SKN-1 is an important transcriptional regulator in a worm and was initially studied because of its contributions to development, oxidative stress resistance, and lifespan (An and Blackwell, 2003; Bowerman et al., 1992). We and others showed that like Nrf2, SKN-1 has a role in resistance to pathogens, including the human bacterial pathogens Pseudomonas aeruginosa and Enterococcus faecalis (Hoeven et al., 2011; van der Hoeven et al., 2012; Papp et al., 2012). Protection by SKN-1 from these pathogens was shown to depend on its activity in the intestine.
Because of SKN-1’s importance, much work has been done to identify components and regulatory mechanisms that control its activity (reviewed by Blackwell et al., 2015). Some of the more notable regulatory pathways include the p38 mitogen-activated protein kinase (MAPK) phosphorylation cascade that results in the phosphorylation of SKN-1 on activating residues (Inoue et al., 2005). The pathway is composed of NSY-1, a MAPKKK that phosphorylates the MAPKK called SEK-1, which in turn phosphorylates the p38 MAPK called PMK-1. Levels of phosphorylated PMK-1 can additionally be modulated by the phosphatase VHP-1 (Kim et al., 2004; Mizuno et al., 2004; Papp et al., 2012). In contrast, phosphorylation by GSK-3 on different residues is ultimately inhibitory (An et al., 2005). Phosphorylation by AKT1/2, of the insulin signaling pathway (Tullet et al., 2008), and SGK-1, of the mechanistic target of rapapmycin (mTOR) pathway, are also inhibitory (Mizunuma et al., 2014; Robida-Stubbs et al., 2012). There is also negative regulation by WDR-23, an E3 ubiquitin ligase that targets SKN-1 to the proteasome (Choe et al., 2009). Finally, reactive oxygen species (ROS) production by a nicotinamide adenine dinucleotidephosphate (NADPH) oxidase, called BLI-3, was shown to be necessary for SKN-1 activation on pathogens (Hoeven et al., 2011).
Many SKN-1 regulators were previously identified by screens, including screens under non-stress conditions to identify negative regulators (Choe et al., 2009; Kahn et al., 2008; Wang et al., 2010) and screens following exposure to chemical oxidants that induce SKN-1 activity to identify positive regulators (Wu et al., 2016, 2017). In contrast, we are carrying out a screen on human pathogens to identify positive regulators whose loss prevents SKN-1 activity. The screen is still in progress and will be reported in a later publication. However, while establishing the conditions for this screen, one gene of interest, nipi-3, was identified as a possible SKN-1 regulator.
NIPI-3 is a member of the Tribbles pseudokinase (TRB) family that regulates key signaling pathways by acting as scaffolds or adaptors. TRB function has been linked to diverse biological functions in eukaryotic cells, including cell differentiation, cell cycle, stress response, immune response, and cancer development, and has been studied and characterized in Drosophila melanogaster, Caenorhabditis elegans, and mammalian systems. TRBs have a central domain with scaffolding and/or adaptor functions thought to interact with components of major signaling pathways (reviewed by Eyers et al., 2017; Sakai et al., 2016). A genetic interaction of NIPI-3 with SKN-1 was of interest because previous work also identified a role for the NIPI-3 in pathogen resistance. NIPI-3 is required in the hypodermis of C. elegans for protection against the fungal pathogen Drechmeria coniospora and in the intestine for protection against P. aeruginosa (McEwan et al., 2016; Pujol et al., 2008a). However, a specific connection between TRBs/NIPI-3 and Nrf/SKN-1 activity has not been reported to our knowledge. Herein, we describe the mechanism by which NIPI-3 regulates SKN-1 activity in the intestine to affect pathogen resistance.
RESULTS
NIPI-3 is required for SKN-1 activation following pathogen exposure
To identify genes that affect SKN-1 activity on pathogens, two previously established SKN-1 reporter fusions were employed: Pgcs-1::gfp and Pgst-4::gfp (An and Blackwell, 2003; Leiers et al., 2003). Both reporters are strongly induced in the intestine of animals exposed to P. aeruginosa and E. faecalis in a SKN-1-dependent manner, as we reported previously. However, the induction of the Pgcs-1::gfp was observed to be stronger on P. aeruginosa, in contrast to the induction of Pgst-4::gfp, which was stronger on E. faecalis (Hoeven et al., 2011). Using these reporters, we observed that loss of nipi-3 by RNAi dramatically reduced induction of both reporters in the intestine to levels comparable to the skn-1 RNAi control (Figures 1A–1D). In addition to pathogen exposure, the reporter strains were exposed to an oxidant commonly used to induce SKN-1, 10 mM sodium arsenite (NaAsO2) (Inoue et al., 2005). It was determined that nipi-3 RNAi also reduced reporter activation in response to this stimulant (Figure S1). Therefore, the effect of nipi-3 RNAi on these SKN-1 reporters was not specific to a particular pathogen or to pathogen stress. However, because NIPI-3 and SKN-1 were independently reported in previous publications to affect intestinal immunity (Hoeven et al., 2011; McEwan et al., 2016), we decided to further investigate the nature of their interaction in the context of infection.
Figure 1. NIPI-3 is required for SKN-1 activation following pathogen exposure.
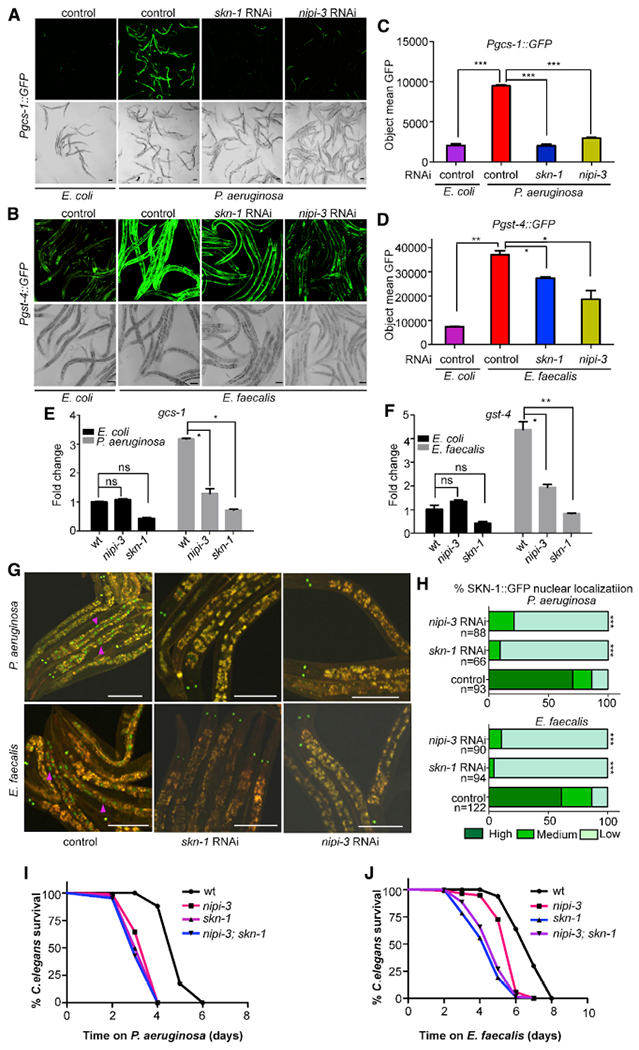
(A) Expression pattern of Pgcs-1::gfp in worms exposed to P. aeruginosa PA14 and E. coli HT115 for 7 h.
(B) Pgst-4::gfp expression pattern in worms exposed to E. faecalis OG1RF and E.coli HT115 for 16 h. Scale bars are 100 μm.
(C and D) Quantification of GFP fluorescence from animals containing the Pgcs-1::gfp and Pgst-4::gfp expression reporters in (A) and (B) with a Cytation 5 image reader as described in STAR Methods. The y axis denotes the average number of pixels measured in nematodes after pathogen exposure. Three biological replicates with at least 50 worms/replicate were analyzed. Error bars represent the standard error the mean (SEM). *p < 0.05, **p < 0.01, ***p < 0.001.
(E and F) qRT-PCR analysis of SKN-1-dependent genes gcs-1 and gst-4 induced in wild-type (N2), skn-1, and nipi-3 animals when exposed to P. aeruginosa PA14 and E. coli HT115 for 7 h and E. faecalis OG1RF and E. coli HT115 for 16h. Gene expression values are relative to the act-1 housekeeping gene, with the wild-type animals exposed to E. coli set to 1. Error bars represent the SEM. Significantly lower levels of gcs-1 and gst-4 expression were observed in skn-1, nipi-3, and wild-type animals exposed to E. coli compared with wild-type animals exposed to pathogens. *p < 0.05; **p < 0.01; ns, not significant.
(G) Worms containing the integrated SKN-1B/C::GFP transgene were exposed to P. aeruginosa PA14 for 7 h (upper panel) and to E. faecalis OG1RF for 16 h (lower panel). SKN-1B/C::GFP localization was observed by fluorescence microscopy, and a few intestinal nuclei displaying the nuclear localization are marked with purple arrows as examples.
(H) Degree of nuclear localization was scored as described in STAR Methods and is given as a percentage for each category. The number of worms used in scoring each experimental condition is indicated (n). Significantly higher levels of SKN-1B/C::GFP nuclear localization were observed in wild-type worms exposed to the pathogens compared with skn-1 and nipi-3 animals. Scale bars are 100 μm. ***p < 0.001.
(I and J) Survival over time of C. elegans of the indicated genotypes on lawns of P. aeruginosa (I) or E. faecalis (J). p < 0.0001 for wild type compared with all mutants. p < 0.05 for nipi-3 compared with skn-1 and nipi-3; skn-1. p = ns for skn-1 compared with nipi-3; skn-1. The data are representative of three independent trials performed in triplicate. Sample sizes, median survival, and p values of all trials are shown in Table S1. Related data in Figure S1.
To confirm the results obtained by RNAi and GFP reporters, we examined endogenous gene expression of the SKN-1-regulated genes using qRT-PCR and a nipi-3 mutant strain containing a hypomorphic allele, because animals with loss-of-function mutations in nipi-3 are not viable past mid-larval development (Kim et al., 2016; Pujol et al., 2008a). As observed previously, gcs-1 (Figure 1E) and gst-4 (Figure 1F) expression was increased when animals were exposed to a pathogen (Hoeven et al., 2011). However, the induction failed to occur in nipi-3 mutant animals, consistent with the GFP reporters. To look at SKN-1 activation more directly, we used a translational reporter, SKN-1(B/C)::GFP, which fluorescently labels the transcription factor (An and Blackwell, 2003). Under inducing conditions, such as pathogen exposure, SKN-1(B/C)::GFP localizes to the nuclei of the intestinal cells (An and Blackwell, 2003; Hoeven et al., 2011; McCallum et al., 2016). As shown in representative pictures following exposure to a pathogen, this nuclear localization is clearly visible in the RNAi control animals, but not in those that have been treated with nipi-3 and skn-1 RNAi (Figure 1G). Categorical scoring revealed that the difference was statistically significant (Figure 1H).
Because loss of skn-1 and nipi-3 was previously reported to result in pathogen sensitivity (Hoeven et al., 2011; McEwan et al., 2016), epistasis analysis was performed to determine whether the pathogen sensitivity phenotypes were consistent with SKN-1 and NIPI-3 operating in the same pathway or different pathways. Using a loss-of-function allele of skn-1, the sensitivity of the skn-1; nipi-3 double mutant was not significantly different from that of the skn-1 single mutant, suggesting that SKN-1 is epistatic to NIPI-3 (Figures 1I and 1J). Moreover, a strain of C. elegans containing a gain-of-function allele, skn-1(lax120) was observed to be impervious to loss of nipi-3 (Figures S2A and S2B). Suppression of nipi-3 by RNAi in skn-1(lax120) animals also did not affect the constitutive expression of the Pgst-4::gfp reporter (Figures S2C and S2D). Altogether, the data are consistent with a model in which NIPI-3 is required for SKN-1 activation.
NIPI-3’s effects on SKN-1 activation require CEBP-1
In the previous work that originally identified a role for NIPI-3 in intestinal immunity, a suppressor screen revealed that loss of the gene encoding a CCAT enhancer binding protein (C/EBP) bZIP transcription factor, CEBP-1, restored resistance in a nipi-3 background (McEwan et al., 2016). Based on these data, NIPI-3 was proposed to normally suppress CEBP-1 activity, which in turn suppresses defense mechanisms in the intestine. Based on the findings in Figure 1 that are consistent with NIPI-3 acting in a pathway with SKN-1, we propose the model depicted in Figure 2A, in which SKN-1 activity is suppressed by CEBP-1, which in turn is suppressed by NIPI-3.
Figure 2. NIPI-3’s effect on SKN-1 activation requires CEBP-1.
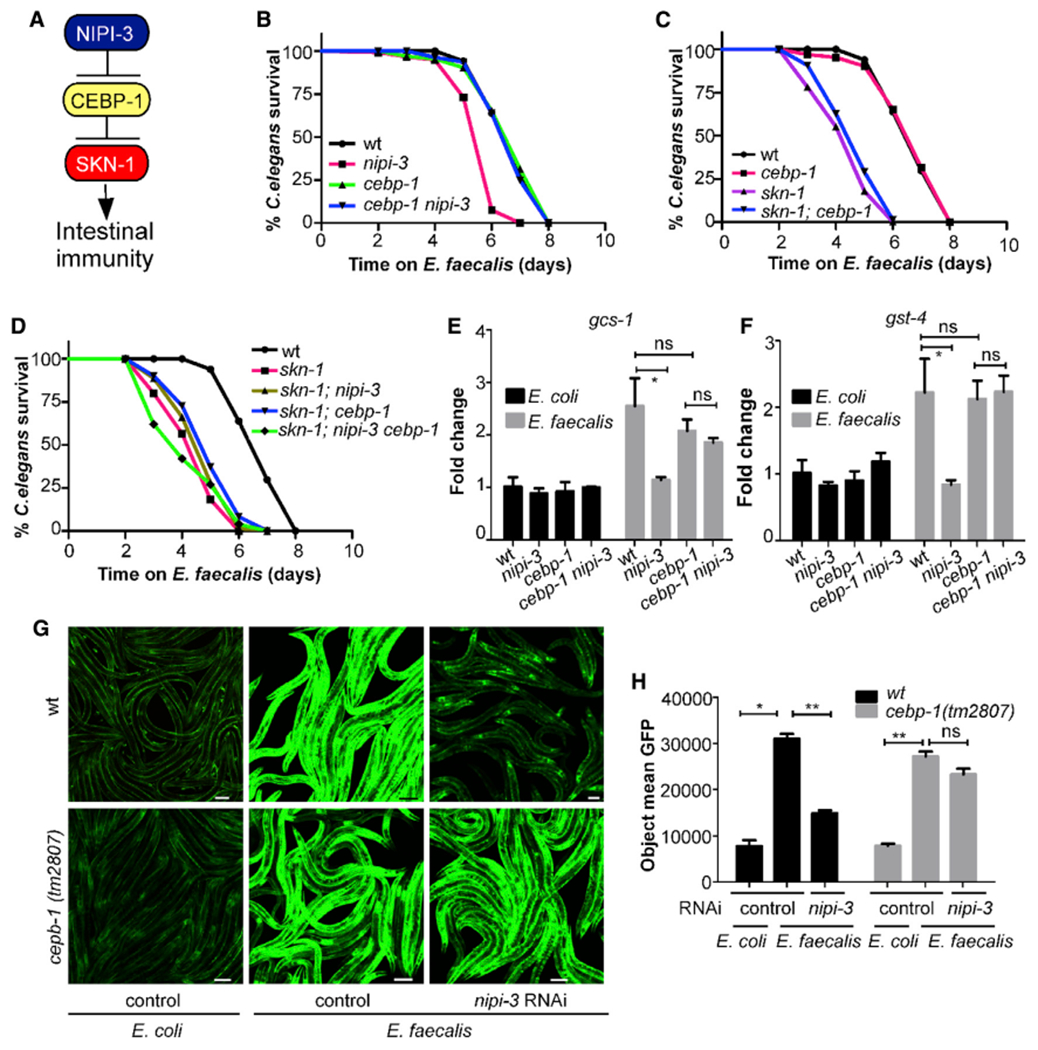
(A) Proposed model for the connection between NIPI-3 and SKN-1.
(B) Survival curves of wild type, nipi-3(fr4), cebp-1(tm2807), and nipi-3(fr4) cebp-1(tm2807) on E. faecalis OG1RF. p < 0.0001 for nipi-3 compared with all other strains. p = ns for wild type, cebp-1, and cebp-1 nipi-3 compared with one another.
(C) Survival curves ofwild type, cebp-1(tm2807), skn-1(zu135), and skn-1(zu135); cebp-1(tm2807) on E. faecalis OG1RF. p < 0.0001 forwild type compared with skn-1 and skn-1; cebp-1 and for cebp-1 compared with skn-1 and skn-1; cebp-1. p = ns for wild type compared with cebp-1 and for skn-1 compared with skn-1; cebp-1.
(D) Survival curves of wild type, skn-1, skn-1; nipi-3, skn-1; cebp-1, and skn-1; nipi-3 cebp-1 on E. faecalis OG1RF. p < 0.0001 forwild type compared with all strains. p = ns for comparisons between all strains except wild type. For the survival experiments, animals were exposed to E. faecalis after treatment with cdc-25.1 RNAi. Killing assays were performed in triplicate and repeated independently three times. Sample sizes, median survival, and p values of all trials are shown in Table S1.
(E and F) Defect in gcs-1 (E) and gst-4 (F) expression levels in the nipi-3(fr4) background are suppressed by the loss-of-function allele of cebp-1(tm2807) as revealed by qRT-PCR. Wild-type (N2), nipi-3(fr4), cebp-1(tm2807), and nipi-3(fr4) cebp-1(2807) animals were collected following exposure to E. faecalis or E. coli for 16 h. Gene expression values are relative to the act-1 housekeeping gene, with the N2 animals exposed to E. coli set to 1. The data shown are the mean ± SEM of the three independent experiments, each of which was performed in triplicate. *p < 0.05; ns, not significant.
(G) Confocal microscopy of nipi-3 RNAi-treated or control RNAi-treated Pgst-4::gfp animals of the indicated genotypes after exposure to E. faecalis or E. coli for 16 h. The images are representative of three independent experiments (n > 150 adult nematodes per strain). Scale bars are 100 μm.
(H) Quantification of mean GFP fluorescence for animals in (G) with a Cytation 5 image reader as described in STAR Methods. Three biological replicates with at least 50 worms/replicate were analyzed. Error bars represent the SEM. *p < 0.05; **p < 0.01; ns, not significant. Related data in Figure S2.
The relationship between NIPI-3 and CEBP-1 on intestinal immunity was previously explored using animals exposed to P. aeruginosa or E. coli expressing the P. aeruginosa toxin ToxA (McEwan et al., 2016). To observe whether the effects were the same on E. faecalis, we tested the sensitivity of the nipi-3 and cebp-1 single and double mutants following exposure to this pathogen (Figure 2B). Loss of cebp-1 restored pathogen sensitivity to wild-type levels in the nipi-3 background, as had been previously observed for animals feeding on P. aeruginosa (Figure S3A) (McEwan et al., 2016). These data, along with the data presented in Figure 1, verify that the NIPI-3 influence on intestinal immunity is not confined to a particular pathogen. Consistent with the model (Figure 2A), a mutant containing a deletion in both cebp-1 and skn-1 displayed the same pathogen sensitivity phenotype as the skn-1 single mutant (Figures 2C and S3B). A nipi-3 cebp-1; skn-1 triple mutant also was as sensitive to the pathogen as the skn-1 single-mutant animals (Figures 2D and S3C). Looking at the expression of genes induced by the pathogen in a SKN-1-dependent manner, we observed that the addition of the cebp-1 loss-of-function allele to the nipi-3 background largely restored gst-4 and gcs-1 expression levels (Figures 2E, 2F, S3D, and S3E). The same finding was observed using the Pgst-4::gfp reporter strain; loss of cebp-1 function rescued induction in animals exposed to nipi-3 RNAi (Figures 2G and 2H). Altogether, these data are consistent with a model in which CEBP-1 inhibits SKN-1 activation during pathogen stress but is normally prevented from doing so by NIPI-3.
CEBP-1 affects vhp-1, cebp-1, and nipi-3 transcript levels
As an enhancer binding protein, CEBP-1 binds to a conserved DNA motif in the promoters of its target genes to activate their expression. To understand the mechanism by which CEBP-1 affects SKN-1 activation, we examined a previous chromatin immunoprecipitation sequencing (ChIP-seq) analysis that identified potential CEBP-1 binding sites. Interestingly, the skn-1 promoter was reported as having a CEBP-1 binding site (Kim et al., 2016). Additional notable genes were vhp-1 and sek-1, encoding components of the p38 MAPK pathway. As mentioned, the p38 MAPK pathway is central to C. elegans innate immune regulation, because it regulates several key transcription factors, including SKN-1, by a phosphorylation cascade (Figure 3A) (Hoeven et al., 2011; Papp et al., 2012). We performed qRT-PCR on wild-type (N2), nipi-3, cebp-1, and nipi-3 cebp-1 animals exposed to E. coli and pathogen. Figure 3 shows the assays performed with E. faecalis, whereas Figure S4 shows those with P. aeruginosa. The expression of genes resulting from exposure to the E. coli control differ somewhat between Figure 3 and Figure S4 because of the difference in time points, 16 h compared with 8 h, which were optimized for the respective pathogens. Although a slight, sometimes significant, rise in sek-1 expression levels was evident in non-infected nipi-3 animals, there was no difference in pathogen-infected animals between the different backgrounds (Figures 3B and S4A). A similar pattern was observed for the skn-1 transcript, with elevated levels in the non-infected, but not in the infected, nipi-3 animals (Figures 3C and S4B). In contrast, vhp-1 transcript levels were found to be significantly elevated in both E. faecalis- and P. aeruginosa-infected nipi-3 mutant animals (Figures 3D and S4C).
Figure 3. NIPI-3 affects vhp-1 transcript levels.
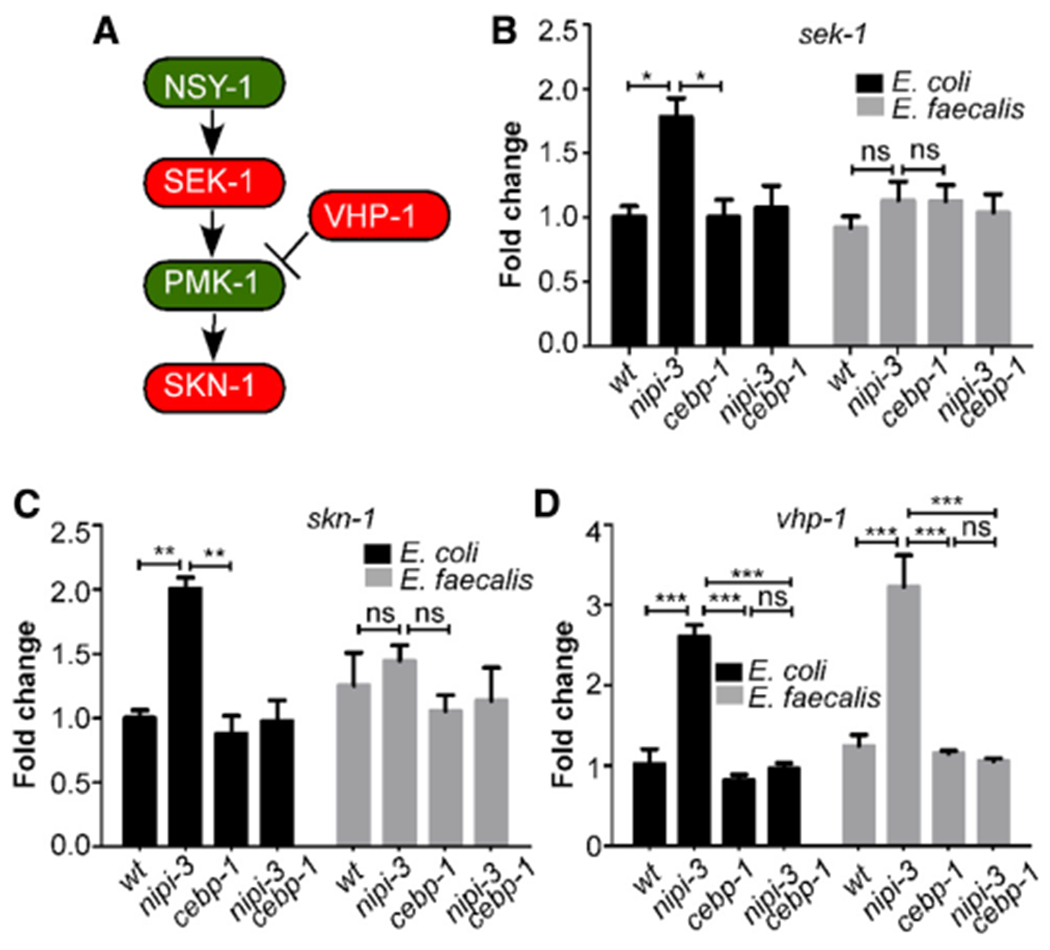
(A) Model forthe regulation of SKN-1 by the p38 MAPK pathway as established in previous work.
(B-D) Transcript levels of sek-1 (B), skn-1 (C), and vhp-1 (D) of the indicated genotypes measured by qRT-PCR are presented. Gene expression values are relative to the act-1 housekeeping gene, with the wild-type (N2) animals exposed to E. coli set to 1. Data are the average of three independent replicates. Error bars represent the SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. Related data in Figure S3.
The rise in vhp-1 transcript levels is consistent with a model in which CEBP-1 enhances transcription of this gene when uninhibited by the presence of NIPI-3. By what mechanism does CEBP-1 have greater activity when NIPI-3 is not present? A previous study examining the relationship between CEBP-1 and NIPI-3 during development observed that both transcript and protein levels of CEBP-1 were higher in larval stage 1 (L1) nipi-3 mutant animals (Kim et al., 2016). We measured cebp-1 transcript levels in adult animals and documented a significant increase on E. coli, as well as on pathogens (Figures 4A and S4D). In addition, we observed that the cebp-1 transcript was significantly downregulated in the cebp-1 background. Because the primers used amplify the cebp-1 mutant and the wild-type alleles equally well, the data suggest that CEBP-1 positively regulates the expression of its own gene. We also examined nipi-3 gene expression in the nipi-3 mutant using primers that amplify the nipi-3 mutant allele as efficiently as the wild type. Interestingly, nipi-3 transcript levels were increased on both E. coli and pathogen in the nipi-3 mutant animals (Figures 4B and S4E). These data are consistent with a model in which CEBP-1 positively regulates its own and nipi-3]’s gene expression (Figure 4C). The regulation of CEBP-1’s own gene is predicted to generate a positive feedback loop. In contrast, the positive regulation of the nipi-3 transcript by CEBP-1, coupled to the negative regulation of CEBP-1 by the resulting protein NIPI-3, predicts a negative feedback loop (Figure 4C).
Figure 4. CEBP-1 and NIPI-3 are in a regulatory feedback loop that is both transcriptional and post-translational.
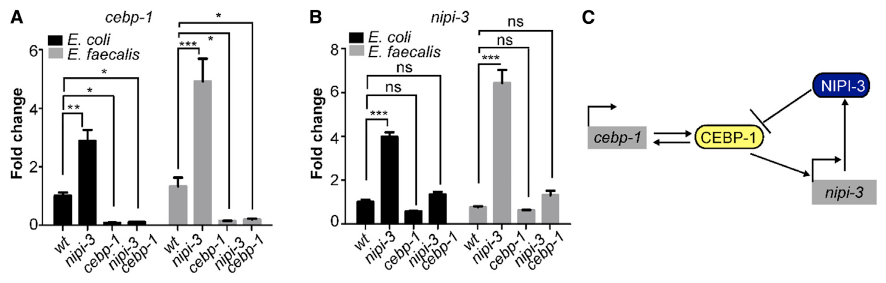
(A and B) Gene expression levels as measured by qRT-PCR of cebp-1 (A) and nipi-3 (B) in the indicated worm genotypes (wild-type (N2), nipi-3(fr4), cebp-1(tm2807), and nipi-3(fr4) cebp-1(tm2807) animals) following exposure to E. faecalis or E. coli for 16 h. That data shown are the mean ± SEM of three independent experiments, each of which was performed in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
(C) Schematic of the CEBP-1/NIPI-3 regulatory feedback loops. See main text for details. Related data in Figure S4.
NIPI-3 regulates the PMK-1 p38 MAPK pathway via the phosphatase VHP-1
The finding that vhp-1 transcript levels are higher in the nipi-3 mutant on pathogens provides a possible explanation for how loss of NIPI-3 results in less SKN-1 activity and increased pathogen susceptibility (Figure 5A). It is well established that VHP-1 negatively regulates the p38 MAPK pathway by dephosphorylating PMK-1 (Kim et al., 2004; Mizuno et al., 2004). Increased VHP-1 levels result in less PMK-1 activity and therefore less SKN-1 activity (Papp et al., 2012). If loss of nipi-3 increases VHP-1 levels, we predict less PMK-1 phosphorylation. To test this hypothesis, we examined total and phosphorylated PMK-1 (p-PMK-1) levels following exposure to a pathogen (Figure 5B) and a non-pathogen (Figure S5A). As observed previously (Kim et al., 2004; Mizuno et al., 2004), a control consisting of vhp-1 RNAi in the wild-type background resulted in a significant increase in the amount of phosphorylated, but not total, PMK-1 detected. In contrast, the total and phosphorylated levels of PMK-1 were significantly decreased following pmk-1 RNAi. In keeping with our expectations, the nipi-3 mutant background caused a significant decrease in the amount of phosphorylated PMK-1 without significantly affecting total levels, and these PMK-1 phosphorylation levels were restored in the nipi-3 cebp-1 double mutant (Figures 5B and S5A).
Figure 5. NIPI-3 regulates the PMK-1 p38 MAPK pathway via the phosphatase VHP-1.
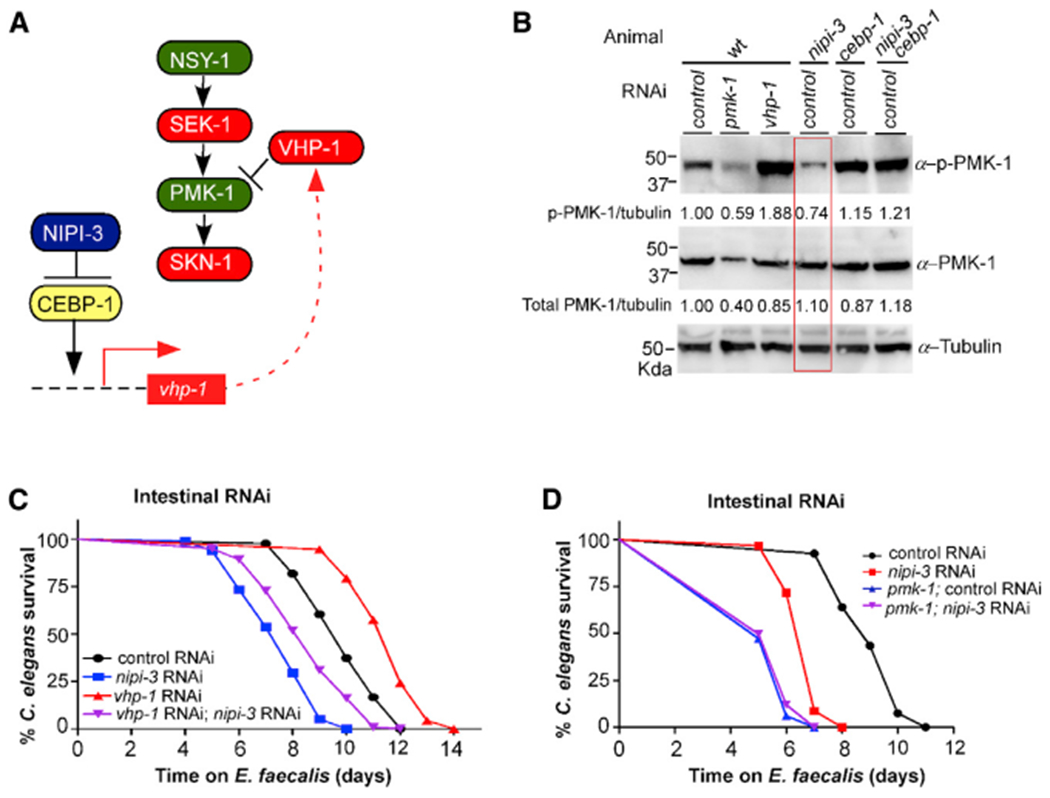
(A) Model for NIPI-3 regulating intestinal innate immunity in C. elegans by controlling SKN-1 activity through the VHP-1 phosphatase.
(B) Loss of nipi-3 results in lower levels of PMK-1 phosphorylation. Immunoblot analysis using an α-phospho-p38 antibody, an α-p38 antibody, and an α-tubulin antibody (loading control) from lysates of the indicated animals exposed to E. faecalis for 16 h. Band intensities relative to α-tubulin are indicated below each blot, with the control set to 1. The lane containing the nipi-3 lysate is boxed.
(C) Epistasis analysis of intestine-only RNAi animals, either wild type or also containing the pmk-1(km25) allele, exposed to nipi-3 and vhp-1 RNAi. p < 0.0001 for all strain comparisons.
(D) Epistasis analysis of intestine-only RNAi animals exposed to nipi-3 RNAi in both wild type and pmk-1(km25) mutant backgrounds. p < 0.0001 for all strain comparisons except for pmk-1; control RNAi to pmk-1; nipi-3 RNAi, which is not significant. The VP303 background was used for all strains to enable intestine-specific RNAi, and C. elegans survival on E. faecalis was tracked over time. Data are representative of three trials. Sample sizes, median survival, and p values of all trials are shown in Table S1. Related data in Figure S5.
The model presented in Figure 5A leads to the prediction that the pathogen sensitivity phenotype evident in nipi-3 animals will be relieved if vhp-1 expression is lowered. However, animals containing the nipi-3 hypomorphic allele were unable to develop properly when exposed to vhp-1 RNAi (McEwan et al., 2016). Overall, there is significant evidence for NIPI-3 and VHP-1 being required during development and being regulated differently in different tissues (see Discussion) (Kim et al., 2016; McEwan et al., 2016; Pujol et al., 2008a). To eliminate these complications, we used an intestinal-specific RNAi strain and exposed animals to nipi-3 and vhp-1 RNAi before placing them on E. faecalis. Knockdown of nipi-3, just in the intestine, caused a pathogen sensitivity phenotype, whereas knockdown of vhp-1 resulted in increased resistance, as previously reported (Kim et al., 2004; McEwan et al., 2016). As predicted, knockdown of both genes simultaneously resulted in partial rescue of the nipi-3 pathogen sensitivity phenotype (Figure 5C). The same patterns of sensitivity and resistance were observed when the RNAi-exposed animals were infected with P. aeruginosa (Figure S5B).
Overall, our data are consistent with the model presented in Figure 5A in which NIPI-3 functions upstream of the p38 MAPK, PMK-1, to affect SKN-1 activity. However, prior experiments using a pmk-1 null mutant found that the addition of nipi-3 RNAi caused an additive effect on the pathogen sensitivity phenotype, indicative of the two components operating in different pathways. In addition, RNA sequencing (RNA-seq) analysis did not show a clear overlap among the genes regulated by PMK-1 and NIPI-3, as would be expected (McEwan et al., 2016). Because PMK-1 and NIPI-3 are present in several tissues of C. elegans and appear to have different regulatory relationships with each other depending on the context (see Discussion), we reasoned that knocking down nipi-3 only in the intestine of pmk-1 animals might clarify an epistatic relationship. Therefore, the strain containing the pmk-1 null allele was crossed with the intestinal-specific RNAi strain VP303 and exposed to control or nipi-3 RNAi, followed by exposure to E. faecalis (Figure 5D) or P. aeruginosa (Figure S5C). nipi-3 RNAi, just in the intestine, did not increase sensitivity to either pathogen beyond what was observed with the pmk-1 null. In conclusion, the data are consistent with the model presented in Figure 5A in which NIPI-3 and CEBP-1 affect the levels of VHP-1, which controls the amount of phosphorylated PMK-1 present to activate SKN-1 during intestinal infection.
DISCUSSION
The data presented herein support a connection between two important biological regulators: SKN-1/Nrf and NIPI-3/TRB. By several different assays, reduced SKN-1 activity was observed under inducing conditions without NIPI-3. Most experiments employed intestinal pathogen exposure as the inducing condition, but the dependence of SKN-1 activation on NIPI-3 was also apparent following challenge with oxidant, suggesting a universal mechanism of SKN-1 regulation to be studied in other contexts. Regulation of SKN-1 by NIPI-3 depended on CEBP-1, consistent with a previous report of NIPI-3 controlling intestinal immunity by negatively regulating CEBP-1 (McEwan et al., 2016). As an enhancer binding protein, it is logical that CEBP-1 might exert its effects on SKN-1 activity by enhancing the transcription of a negative SKN-1 regulator. In the absence of NIPI-3, unfettered CEBP-1 activity led to the enhanced transcription of vhp-1, encoding a phosphatase known to negatively regulate the p38 MAPK pathway that activates SKN-1 (Kim et al., 2004; Mizuno et al., 2004; Papp et al., 2012). CEBP-1 also enhanced transcription of its gene and of nipi-3, creating positive and negative feedback loops, respectively. Loss of nipi-3 led to less phosphorylation of PMK-1, and knockdown of vhp-1 in the intestine rescued the sensitivity caused by loss of nipi-3, consistent with NIPI-3 exerting its effects on the p38 MAPK pathway through VHP-1. Epistasis analysis of intestinal pathogen resistance was also consistent with NIPI-3 acting on the p38 MAPK pathway to regulate SKN-1, as depicted in Figure 5A. However, it is important to realize that NIPI-3 appears to regulate immunity and other processes differently depending on the context.
As mentioned, C. elegans can suffer hypodermal infections by pathogens such as D. coniospora, in addition to the intestinal infections used in this study (Powell and Ausubel, 2008; Pujol et al., 2008b). It was in this context that the tribbles pseudokinase NIPI-3 was first discovered (Pujol et al., 2008a). Although nipi-3 is expressed in several tissues, including the hypodermis, pharynx, intestine, and subsets of neurons, protection from D. coniospora infection only required expression in the hypodermal cells (Pujol et al., 2008a). This is in direct contrast to what was observed following P. aeruginosa infection, in which intestinal nipi-3 expression was crucial, and suggests that NIPI-3 carries out its effects cell autonomously (McEwan et al., 2016). In addition, genetic analysis of how NIPI-3 regulated protection against D. coniospora infection in the hypodermis defined a pathway different from that elucidated for intestinal infection here and in previous work (McEwan et al., 2016). Although NIPI-3 acts upstream of the p38 MAPK pathway for hypodermal infection, a protein kinase C delta homolog (TPA-1) and a BiP/GRP78 chaperone homolog (HSP-3) were demonstrated to be directly downstream of NIPI-3 and to feed into the p38 MAPK pathway via TIR-1 to NSY-1; CEBP-1 is not involved (Pujol et al., 2008a; Ziegler et al., 2009).
In addition to infection resistance, NIPI-3 regulates development. When studying a complete deletion of nipi-3 (nipi-3(0)), rather than a hypomorphic allele, it was discovered that total loss of NIPI-3 causes developmental arrest between the L2 and the L3 larval stages (Kim et al., 2016). The requirement for NIPI-3 in development was not specific to any tissue. By screening for suppressors, loss of cebp-1 was discovered to suppress the lethal phenotype of the nipi-3(0) mutation, confirming a genetic connection between nipi-3 and cebp-1. Moreover, the p38 MAPK signaling pathway, specifically SEK-1, was shown to be downstream of NIPI-3 and CEBP-1 in this context. When L1 animals were examined, loss of nipi-3 resulted in more phosphorylation of PMK-1, a sign of more p38 MAPK signaling activity (Kim et al., 2016). This is the opposite of what was observed in adult animals during intestinal infection in this work and during hypodermal infection in the D. coniospora study (Pujol et al., 2008a). Comparing the current study to these past investigations yields three key takeaways: (1) NIPI-3 is an important, cell-autonomous component of pathogen resistance to both hypodermal and intestinal infections, (2) NIPI-3 is an important regulator of development in multiple tissues, and (3) NIPI-3 regulation of the p38 MAPK signaling pathway is context dependent.
We postulate that the context dependence of NIPI-3 regulation in terms of tissue, developmental stage, and pathogen exposure explain the discrepancies between the current and the previous investigation into NIPI-3’s regulation of intestinal immunity. The previous study looked at interactions with the p38 MAPK pathway, specifically PMK-1, in the context of the whole animal using epistasis and gene expression analysis (McEwan et al., 2016). It was concluded that NIPI-3 was likely affecting intestinal immunity in a pathway parallel to the p38 MAPK pathway, because loss of nipi-3 and pmk-1 in the whole animal resulted in an additive effect on pathogen resistance and transcriptome analysis did not reveal significant overlap in gene expression changes between pmk-1 and nipi-3 mutant animals. However, when the interaction was explored by reducing nipi-3 expression only in the intestine, the results were more consistent with these components being in the same pathway (Figures 5D and S5C) (Figures 5D and S4B). Tissue-specific gene expression studies in the worm would clarify the question and are just starting to become possible (Kaletsky et al., 2016, 2018). We predict that transcriptome analysis of infected intestinal tissue only will display significant overlap in gene expression changes among nipi-3, pmk-1, and skn-1 animals.
All NIPI-3-regulated events elucidated in C. elegans thus far involve the p38 MAPK pathway. An important, outstanding question in the field is how NIPI-3 achieves context-dependent regulation of this pathway (Andrusiak and Jin, 2016). TRBs like TRB1, TRB2, and TRB3 in humans and NIPI-3 in C. elegans have a central pseudokinase domain that lacks key residues necessary for phosphorylation but is thought to function as a scaffold and/or adaptor to interact with components of major signaling pathways. In addition, the C-terminal domain contains an E3 ubiquitin ligase binding site and a MAPK/ERK binding site (reviewed by Sakai et al., 2016; Eyers et al., 2017). Interestingly, in the context of certain mammalian systems, TRB2 was shown to ubiquitinate the CEBP-1 homolog C/EBPα by pairing with an E3 ubiquitin ligase, COP-1, leading to C/EBPα degradation (Keeshan et al., 2010). Loss of nipi-3 in C. elegans also resulted in high levels of cebp-1 transcript in the context of intestinal infection (Figure 4A) and during development, in which levels of CEBP-1 protein were also measured as higher (Kim et al., 2016). Whether NIPI-3 also employs an E3 ubiquitin ligase to facilitate the degradation of CEBP-1 is an interesting subject for future investigation; C. elegans does not encode an obvious COP-1 homolog. In the context of hypodermal immunity, in which CEBP-1 is not involved, it will also be of interest to understand the protein domains and mechanism or mechanisms employed by NIPI-3 to regulate pathogen resistance. Identification of additional components and elucidation of functional mechanisms involved in NIPI-3 activity will lead to better understanding of how it achieves context-dependent regulation. Because NIPI-3 is a member of the tribbles pseudokinase family, which has multiple roles in eukaryotic development, cell differentiation, cell cycle, stress response, and cancer development (Sakai et al., 2016; Eyers et al., 2017), further study may affect understanding of this significant group of proteins.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests regarding resources and reagents should be directed to and will be fulfilled by the lead contact, Danielle A. Garsin (danielle.a.garsin@uth.tmc.edu).
Materials availability
The C. elegans strains generated in this study will be shared upon request, but we may require a payment to cover shipping and a completed Materials Transfer Agreement if there is potential for commercial application.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report any datasets requiring accession numbers, DOIs, or unique identifiers. Nor does this paper report original code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For general maintenance, C. elegans strains were grown at 20°C on E. coli OP50 (Brenner, 1974) that was spotted onto Nematode Growth (NG) agar plates as previously described (Hope, 1999). New strains were constructed using standard procedures, and all genotypes were confirmed by PCR or sequencing. The nipi-3 hypomorphic mutation strain IG544 nipi-3 (fr4), the cebp-1 mutant strain CZ8920 cebp-1(tm2807), and the vhp-1 mutant strain JT366 vhp-1(sa366) II were obtained from the Caenorhabditis Genetic Center and each of them was backcrossed with N2 Bristol six, seven and four times, respectively. C. elegans and bacterial strains used in this study are listed in the Key resources table. For experiments requiring synchronized animals, L1 stage worms on non-starved plates were washed off, filtered through a 10 μm filter (pluriSelect, pluriStrainer 10 μm), harvested by centrifugation, transferred to seeded plates, and grown to the desired stage.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-p38 | Peterson et al., 2019 | N/A |
| anti-phospho-p38 | Cell Signaling | Cat# 9211; RRID: AB_331641 |
| anti-α-tubulin | Sigma-Aldrich | Cat# T9026; RRID: AB_477579 |
| anti-mouse lgG, HRP-linked conjugate | Promega | Cat# W4021; RRID: AB_430834 |
| anti-rabbit lgG, HRP-linked conjugate | Promega | Cat# W4011; RRID: AB_430833 |
| Bacterial strains and virus strains | ||
| Enterococcus faecalis OG1RF | Dunny et al., 1978 | WB Cat# WBStrain00041967; RRID:WB-STRAIN: WBStrain00041967 |
| Pseudomonas aeruginosa PA14 | Rahme et al., 1995 | WB Cat# WBStrain00041978; RRID:WB-STRAIN: WBStrain00041978 |
| Escherichia coli OP50 | Brenner, 1974 | WB Cat# WBStrain00041969; RRID:WB-STRAIN: WBStrain00041969 |
| Escherichia coli HT115(DE3) | Timmons et al., 2001 | WB Cat# WBStrain00041080; RRID:WB-STRAIN: WBStrain00041080 |
| Chemicals, peptides, and recombinant proteins | ||
| Agarose | Thermo Fisher | Cat# 207569 |
| Red-Taq polymerase 2x Master mix | As One | Cat# AO180303 |
| Palmitic acid | Sigma-Aldrich | Cat# P0500 |
| SuperSignal™ West Atto Ultimate Sensitivity Substrate | Thermo Fisher | Cat# A38555 |
| tetramisole hydrochloride | Sigma-Aldrich | Cat# 152119 |
| Experimental models: Organisms/strains | ||
| C. elegans: Strain: N2 (Bristol) | Caenorhabditis Genetics Center | WB Cat# WBStrain00000001; RRID:WB-STRAIN:WBStrain00000001 |
| C. elegans: Strain: GF181 nipi-3(fr4) X; C. elegans: Parent strain: IG544 nipi-3(fr4) X | This study; Pujol et al., 2008a) | N/A; WB Cat# WBStrain00021982; RRID:WB-STRAIN: WBStrain00021982 |
| C. elegans: Strain: GF173 cebp-1(tm2807) X; C. elegans: Parent strain: CZ8920 cebp-1(tm2807) X | This study; Yan et al., 2009 | N/A; WB Cat# WBStrain00005416; RRID:WB-STRAIN: WBStrain00005416 |
| C. elegans: Strain: GF190 cebp-1(tm2807) nipi-3 (fr4) X | This study | N/A |
| C. elegans: Strain: EU31 skn-1(zu135)IV/nT1 [unc-?(n754) let-?] (IV;V) | Caenorhabditis Genetics Center | WB Cat# WBStrain00007251; RRID:WB-STRAIN:WBStrain00007251 |
| C. elegans: Strain: GF193 skn-1(zu135) IV/nT1 [unc-?(n754) let-?] (IV;V); nipi-3 (fr4) X | This study | N/A |
| C. elegans: Strain: GF197 skn-1(zu135) IV/nT1 [unc-?(n754) let-?] (IV;V); cebp-1(tm2807) X | This study | N/A |
| C. elegans: Strain: GF201 skn-1(zu135) IV/nT1 [unc-?(n754) let-?] (IV;V); cebp-1(tm2807) nipi-3(fr4) X | This study | N/A |
| C. elegans: Strain: VP303 rde-1(ne219) V; kbIs7 [nhx-2p::rde-1 + rol-6(su1006)] | Caenorhabditis Genetics Center | WB Cat# WBStrain00040175; RRID:WB-STRAIN:WBStrain00040175 |
| C. elegans: Strain: LD001 ldIs7[skn-1b/c::GFP + rol-6(su1006)] | Caenorhabditis Genetics Center | WB Cat# WBStrain00024124; RRID:WB-STRAIN:WBStrain00024124 |
| C. elegans: Strain: LD1171 IdIs3 [gcs-1p::gfp+pRF4(rol-6(su1006))] | Caenorhabditis Genetics Center | WB Cat# WBStrain00024128; RRID:WB-STRAIN: WBStrain00024128 |
| C. elegans: Strain: CL2166 dvIs19 [(pAF15) gst-4p::GFP::NLS] III | Caenorhabditis Genetics Center | WB Cat# WBStrain00005102; RRID:WB-STRAIN: WBStrain00005102 |
| C. elegans: Strain: SPC167 dvIs19 III; skn-1(lax120) IV | Caenorhabditis Genetics Center | WB Cat# WBStrain00034419; RRID:WB-STRAIN: WBStrain00034419 |
| C. elegans: Strain: KU25 pmk-1(km25) IV | Caenorhabditis Genetics Center | WB Cat# WBStrain00024040; RRID:WB-STRAIN: WBStrain00024040 |
| C. elegans: Strain: GF225 vhp-1(sa366) II; C. elegans: Parent strain: JT366 vhp-1(sa366) II | This study; Nix et al., 2011 | N/A; WB Cat# WBStrain00022797; RRID:WB-STRAIN: WBStrain00022797 |
| C. elegans: Strain: GF169 IdIs3 cebp-1(tm2807) X | This study | N/A |
| C. elegans: Strain: GF166 dvIs19 III; cebp-1(tm2807) X | This study | N/A |
| C. elegans: Strain: GF229 rde-1(ne219)V; kbIs 7 pmk-1(km25) IV | This study | N/A |
| C. elegans: Strain: GF233 (can’t grow to L3) pmk-1(km25) IV; nipi-3(fr4) X | This study | N/A |
| C. elegans: Strain: GF235 (can’t grow to L3) vhp-1(sa366) II; nipi-3(fr4) X | This study | N/A |
| Oligonucleotides | ||
| See Table S2 | N/A | |
| Software | ||
| GraphPad Prism 9.0 | GraphPad Prism Software, Inc | https://www.graphpad.com/ |
| Image Lab | Bio-rad | https://www.bio-rad.com/ |
METHOD DETAILS
RNAi Interference
RNAi clones were from the C. elegans library (Geneservices, UK), with the same bacterial strain (HT115(DE3)) containing the empty vector L4440 as a control. All clones were verified by sequencing. RNAi experiments were conducted by feeding worms from the L1- to L4-stage at 16°C on agar plates with bacteria expressing the double stranded RNA corresponding to the gene to be knocked down. In general, RNAi bacteria were cultured to stationary phase, 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added, and then the culture was incubated for another hour. The culture was then concentrated 5-fold prior to being seeded onto NGM plates containing 100 μg/ml carbenicillin and 1 mM IPTG for 5 hours to induce dsRNA expression at 37°C. Double RNAi knockdowns were obtained by mixing the two bacterial RNAi cultures in a 1:1 ratio. To render the worms sterile prior to killing assays, larvae were exposed to cdc-25.1 RNAi during development than L4 or early adult worms were transferred to P. aeruginosa or E. faecalis plates for pathogen exposure experiments.
Killing assays
Killing assays were conducted as previously described (Garsin et al., 2001; Tan et al., 1999). Briefly, E. faecalis OG1RF grown in Brain Heart Infusion (BHI) to late log phase was seeded on BHI agar plates with 10 μg/ml gentamycin and 10 μg/ml nystatin and incubated at 37°C for 24 hours. For P. aeruginosa killing assays, P. aeruginosa PA14 was cultured in Luria broth (LB), seeded on slow-killing plates, and incubated, first, for 24 hours at 37°C, and then for 14 hours at 25°C. The killing assay plates contained a ring of palmitic acid (10 μg/ml in ethanol), which precipitates out of solution and forms a physical barrier that helps prevent the worms from escaping. A total of 90~180 L4 larvae were transferred to three replica plates. Worms were scored as live and dead at 24-hour intervals.
Arsenite Assay
To examine SKN-1-dependent gene expression, we used two reporter strains: Pgcs-1::gfp and Pgst-4::gfp. To investigate the effect of arsenite stress on the gcs-1 promoter-driven GFP expression, N2 worms carrying the Pgcs-1::gfp transgene were exposed to vector control, skn-1 and nipi-3 RNAi during the L1 to L4 larval stages and then animals were transferred to NGM plates with 10 mM arsenite for 5 hours at 20°C prior to microscopic examination. To investigate the effect of arsenite stress on gst-4 promoter-driven GFP expression, N2 worms carrying the Pgst-4::gfp transgene were treated with vector control, skn-1 or nipi-3 RNAi. L4 stage animals were collected and transferred to wells of a 24-well plates with 200 μl M9 buffer containing 10 mM arsenite. After 1 hour at 20°C, the animals were collected in a 1.5 mL tubes and washed once with M9 buffer and recovered on NGM plates pre-seeded with E. coli OP50 for 4 hours. The animals were paralyzed with 25 mM levamisole for 15 min. Anesthetized worms were mounted on 2% agarose pads and imaged using an Olympus FLUOVIEW FV 3000 confocal microscope equipped with Fluoview FV315-SW software.
RNA isolation and qRT-PCR analysis
RNA was extracted from about 1,000 L4 larvae exposed to E. faecalis OG1RF for 16 hours or P. aeruginosa PA14 for 8 hours. RNA from animals exposed to E. coli HT115 was extracted as a control at the same time points. The RNA was extracted with Direct-zol™ RNA Miniprep plus (Zymoresearch) and cDNA synthesis was executed using Primer Script RT Master Mix (TakaRa cat# RR036A). qRT-PCR was performed on an CFX 96™ Real-Time system (BioRad) using SYBR Green Master mix. The primer sequences are listed in the Key resources table. All values were normalized to act-1. One-tailed t tests were performed with GraphPad Prism 9.0. A P value less than or equal to 0.05 was considered significant.
Fluorescence Microscopy
To investigate the expression of gst-4 and gcs-1, animals containing Pgst-4::gfp, Pgcs-1::gfp were exposed to E. faecalis, P. aeruginosa and E. coli strains for 16 or 7 hours at 25°C and paralyzed with 25 mM levamisole. Anesthetized worms were mounted on 2% agarose pads and imaged using an Olympus FLUOVIEW FV 3000 confocal microscope equipped with Fluoview FV315-SW software. The mean level of GFP fluorescence intensity per animal was quantified using a CYTATION 5 image reader (BioTeK). Briefly, approximately 50~80 worms in 100 μl M9 buffer with 25 mM levamisole were transferred to a 96-well plate (Corning Incorporated Costar, 3603). GFP quantifications were performed by using Gen5 3.08 software and three biological replicates were used for each experiment.
SKN-1B/C::GFP expression was analyzed by the above-mentioned fluorescent microscopy of worms exposed to E. faecalis for 16 hours and P. aeruginosa for 7 hours. To discriminate intestinal autofluorescence from SKN-1B/C::GFP fluorescence, we used a combination of the EGFP and mCherry filter sets. This combination allowed autofluorescence to be detected as a yellow to red signal. The degree of nuclear localization in the intestinal cells was scored as previously described (An and Blackwell, 2003; Inoue et al., 2005). Briefly, no nuclear localization of SKN-1B/C::GFP, nuclear localization in the anterior or posterior of the worm, and nuclear localization in all intestinal cells are categorically indicated by low, medium, and high, respectively. All fluorescence microcopy experiments shown were independently repeated at least three times.
Western blot analysis
Worms of each genotype (2000~3000 individuals of L4 stage worms after pathogen exposure) were collected and washed with M9 buffer and boiled in home-made sample buffer. Samples were boiled, spun down, and supernatants were flash frozen in dry ice and then heated at 80°C for 10 min. Proteins were resolved on a 12% SDS-polyacrylamid gel by electrophoresis, transferred to a PVDF membrane, and incubated with rabbit anti-phospho-p38 MAPK (Cell Signaling, #9211) at a 1:2,000 dilution, mouse anti-tubulin (Sigma, #T9026) at a 1:4,000 dilution, or anti-p38 MAPK at a 1:1000 dilution (obtained from Dr. Read Pukkila-Worley at UMass Medical School). Following washing, the blots were incubated with 1:2,000 secondary HRP conjugated anti-mouse (for anti-tubulin), or anti-rabbit (for anti-phospho-p38 and anti-p38 MAPK) antibody for 1 hour and subsequently washed 3 times for 3 min intervals. Blots were developed using SuperSignal™ West Atto Ultimate Sensitivity Substrate (ThermoFisher, A38555) and visualized using a ChemiDoc™ MP Imaging System (BIO-RAD). Image lab (software) was used to quantify the intensity of the immunoblot bands.
QUANTIFICATION AND STATISTICAL ANALYSIS
Following categorical scoring of SKN-1B/C::GFP animals (see above), statistical differences were determined by Chi Square and Fischer’s exact tests. For gene expression quantification using fluorescent measures or qRT-PCR, each experimental condition was compared pairwise to the control condition. Significance was determined using unpaired t tests for experiments with only two samples or one-way ANOVA followed by Tukey’s multiple comparison tests for experiments with multiple samples. Statistically significant differences are indicted in the figure with asterisks and brackets mark the compared conditions. Mantel-Cox log rank analysis was used to compare survival curves and to calculate the median survival. The median survival and comparison values for all survival experiments and their replicates can be found in Table S1. For all statistical tests, P values < 0.05 were considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Supplementary Material
Highlights.
NIPI-3 and SKN-1 both affect C. elegans intestinal innate immunity
NIPI-3 negatively regulates CEBP-1, an enhancer of vhp-1 and cebp-1 expression
VHP-1 dephosphorylates the MAPK called PMK-1, reducing activation of SKN-1
Thus, NIPI-3 and SKN-1 are in a common genetic pathway affecting immunity
ACKNOWLEDGMENTS
We thank Dr. Read Pukkila-Worley at University of Massachusetts for providing us with the antibody against PMK-1. Thanks to Melissa R. Cruz for excellent technical support. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Research reported in this publication was supported by the National Institutes of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI076406 and R01AI150045 (to D.A.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109529.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- An JH, and Blackwell TK (2003). SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 17, 1882–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JH, Vranas K, Lucke M, Inoue H, Hisamoto N, Matsumoto K, and Blackwell TK (2005). Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proc. Natl. Acad. Sci. USA 102, 16275–16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrusiak MG, and Jin Y (2016). Context Specificity of Stress-activated Mitogen-activated Protein (MAP) Kinase Signaling: The Story as Told by Caenorhabditis elegans. J. Biol. Chem 291, 7796–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battino M, Giampieri F, Pistollato F, Sureda A, de Oliveira MR, Pittalà V, Fallarino F, Nabavi SF, Atanasov AG, and Nabavi SM (2018). Nrf2 as regulator of innate immunity: A molecular Swiss army knife!. Biotechnol. Adv 36, 358–370. [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY, and Isik M (2015). SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med 88 (Pt B), 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman B, Eaton BA, and Priess JR (1992). skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell 68, 1061–1075. [DOI] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AV, Saigh MA, McCulloch CA, and Glogauer M (2017). The Role of NrF2 in the Regulation of Periodontal Health and Disease. J. Dent. Res 96, 975–983. [DOI] [PubMed] [Google Scholar]
- Choe KP, Przybysz AJ, and Strange K (2009). The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol. Cell. Biol 29, 2704–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny GM, Brown BL, and Clewell DB (1978). Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc. Natl. Acad Sci. U S A 75, 3479–3483. 10.1073/pnas.75.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers PA, Keeshan K, and Kannan N (2017). Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 27, 284–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuse Y, and Kobayashi M (2017). Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 22, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA, Sifri CD, Mylonakis E, Qin X, Singh KV, Murray BE, Calderwood SB, and Ausubel FM (2001). A simple model host for identifying Gram-positive virulence factors. Proc. Natl. Acad. Sci. USA 98, 10892–10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez JC, Dang H, Martin JR, and Doerschuk CM (2016). Nrf2 Modulates Host Defense during Streptococcus pneumoniae Pneumonia in Mice. J. Immunol 797, 2864–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeven Rv., McCallum KC, Cruz MR, and Garsin DA (2011). Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog. 7, e1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope IA (1999). C. elegans A Practical Approach (Oxford University Press; ). [Google Scholar]
- Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, and Matsumoto K (2005). The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 19, 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn NW, Rea SL, Moyle S, Kell A, and Johnson TE (2008). Proteasomal dysfunction activates the transcription factor SKN-1 and produces a selective oxidative-stress response in Caenorhabditis elegans. Biochem. J 409, 205–213. [DOI] [PubMed] [Google Scholar]
- Kaletsky R, Lakhina V, Arey R, Williams A, Landis J, Ashraf J, and Murphy CT (2016). The C. elegans adult neuronal IIS/FOXO transcriptome reveals adult phenotype regulators. Nature 529, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletsky R, Yao V, Williams A, Runnels AM, Tadych A, Zhou S, Troyanskaya OG, and Murphy CT (2018). Transcriptome analysis of adult Caenorhabditis elegans cells reveals tissue-specific gene and isoform expression. PLoS Genet. 14, e1007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan K, Bailis W, Dedhia PH, Vega ME, Shestova O, Xu L, Toscano K, Uljon SN, Blacklow SC, and Pear WS (2010). Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood 116, 4948–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K, and Ausubel FM (2004). Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc. Natl. Acad. Sci. USA 101, 10990–10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Thakur N, Piggott CA, Omi S, Polanowska J, Jin Y, and Pujol N (2016). Coordinated inhibition of C/EBP by Tribbles in multiple tissues is essential for Caenorhabditis elegans development. BMC Biol. 14, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiers B, Kampkötter A, Grevelding CG, Link CD, Johnson TE, and Henkle-Dührsen K (2003). A stress-responsive glutathione S-transferase confers resistance to oxidative stress in Caenorhabditis elegans. Free Radic. Biol. Med 34, 1405–1415. [DOI] [PubMed] [Google Scholar]
- McCallum KC, Liu B, Fierro-Gonzàlez JC, Swoboda P, Arur S, Miranda-Vizuete A, and Garsin DA (2016). TRX-1 Regulates SKN-1 Nuclear Localization Cell Non-autonomously in Caenorhabditis elegans. Genetics 203, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DL, Feinbaum RL, Stroustrup N, Haas W, Conery AL, Anselmo A, Sadreyev R, and Ausubel FM (2016). Tribbles ortholog NIPI-3 and bZIP transcription factor CEBP-1 regulate a Caenorhabditis elegans intestinal immune surveillance pathway. BMC Biol. 14, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, Kim DH, Ausubel FM, and Matsumoto K (2004). The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 23, 2226–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizunuma M, Neumann-Haefelin E, Moroz N, Li Y, and Blackwell TK (2014). mTORC2-SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging Cell 18, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix P, Hisamoto N, Matsumoto K, and Bastiani M (2011). Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc. Natl. Acad Sci. U S A 108, 10738–10743. 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp D, Csermely P, and Sőti C (2012). A role for SKN-1/Nrf in pathogen resistance and immunosenescence in Caenorhabditis elegans. PLoS Pathog. 8, e1002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson ND, Cheesman HK, Liu P, Anderson SM, Foster KJ, Chhaya R, Perrat P, Thekkiniath J, Yang Q, Haynes CM, and Pukkila-Worley R (2019). The nuclear hormone receptor NHR-86 controls anti-pathogen responses in C. elegans. PLoS Genet. 15, e1007935. 10.1371/journal.pgen.1007935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR, and Ausubel FM (2008). Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol. Biol 415, 403–427. [DOI] [PubMed] [Google Scholar]
- Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, Goncharov A, Jin Y, Chisholm AD, and Ewbank JJ (2008a). Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr. Biol 18, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, and Ewbank JJ (2008b). Anti-fungal innate immunityin C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 4, e1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, and Ausubel FM (1995). Common virulence factors for bacterial pathogenicity in plants and animals. Science 268, 1899–1902. 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, and Blackwell TK (2012). TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 15, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S, Miyajima C, Uchida C, Itoh Y, Hayashi H, and Inoue Y (2016). Tribbles-Related Protein Family Members as Regulators or Substrates of the Ubiquitin-Proteasome System in Cancer Development. Curr. Cancer Drug Targets 16, 147–156. [DOI] [PubMed] [Google Scholar]
- Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, et al. (2010). NADPH oxidase limits innate immune responses in the lungs in mice. PLoS ONE 5, e9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MW, Mahajan-Miklos S, and Ausubel FM (1999). Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. USA 96, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, and Biswal S (2006). Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest 116, 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, and Fire A (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112. 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, and Blackwell TK (2008). Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 182, 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoeven R, McCallum KC, and Garsin DA (2012). Speculations on the activation of ROS generation in C. elegans innate immune signaling. Worm 1, 160–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Robida-Stubbs S, Tullet JM, Rual JF, Vidal M, and Blackwell TK (2010). RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 6, e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CW, Deonarine A, Przybysz A, Strange K, and Choe KP (2016). The Skp1 Homologs SKR-1/2 Are Required for the Caenorhabditis elegans SKN-1 Antioxidant/Detoxification Response Independently of p38 MAPK. PLoS Genet. 12, e1006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CW, Wang Y, and Choe KP (2017). F-Box Protein XREP-4 Is a New Regulator of the Oxidative Stress Response in Caenorhabditis elegans. Genetics 206, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, and Jin Y (2009). The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 138, 1005–1018. 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler K, Kurz CL, Cypowyj S, Couillault C, Pophillat M, Pujol N, and Ewbank JJ (2009). Antifungal innate immunity in C. elegans: PKCdelta links G protein signaling and a conserved p38 MAPK cascade. Cell Host Microbe 5, 341–352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report any datasets requiring accession numbers, DOIs, or unique identifiers. Nor does this paper report original code.