

Abstract
In Alzheimer’s disease (AD), the molecular mechanisms involved in the neurodegeneration are still incompletely defined, though this aspect is crucial for a better understanding of the malady and for devising effective therapies. Mitochondrial dysfunctions and altered Ca2+ signaling have long been implicated in AD, though it is debated whether these events occur early in the course of the pathology, or whether they develop at late stages of the disease and represent consequences of different alterations. Mitochondria are central to many aspects of cellular metabolism providing energy, lipids, reactive oxygen species, signaling molecules for cellular quality control, and actively shaping intracellular Ca2+ signaling, modulating the intensity and duration of the signal itself. Abnormalities in the ability of mitochondria to take up and subsequently release Ca2+ could lead to changes in the metabolism of the organelle, and of the cell as a whole, that eventually result in cell death. We sought to investigate the role of mitochondria and Ca2+ signaling in a model of Familial Alzheimer’s disease and found early alterations in mitochondria physiology under stressful condition, namely, reduced maximal respiration, decreased ability to sustain membrane potential, and a slower return to basal matrix Ca2+ levels after a mild excitotoxic stimulus. Treatment with an inhibitor of the permeability transition pore attenuated some of these mitochondrial disfunctions and may represent a promising tool to ameliorate mitochondria and cellular functioning in AD and prevent or slow down cell loss in the disease.
Keywords: Alzheimer’s disease, hippocampus, mitochondria, membrane potential, Ca2+, excitotoxicity, Alisporivir
1. Introduction
Alzheimer’s disease is the most common form of neurodegenerative disease affecting millions of people worldwide (https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on June 2021). Biological hallmarks of the disease are extracellular deposition of amyloid plaques, composed mainly by fibrils of amyloid-ß (Aß) peptide, intracellular neurofibrillary tangles (NFTs) consisting of aggregated, hyperphosphorylated tau protein, and extensive neurodegeneration [1]. Clinical symptoms are cognitive impairment and dementia characterized by progressive, profound disability culminating with death [2,3,4]; it is proposed that subtle alterations in the physiology of the brain and the so-called preclinical phase might precede the overt manifestation of clinical symptoms by 2–3 decades [5]. The dominant hypothesis for explaining AD alterations postulates that the aggregation of Aß peptides initiate the cascade of events leading to inflammation, the formation of tau-tangles, synaptic loss, and neurodegeneration [6,7]. However, this hypothesis has been challenged, mainly because numerous clinical trials aimed at decreasing Aß burden have failed to deliver significant benefits to the patients, and because there is not a strong correlation between phases of amyloid deposition and degree of cognitive decline, reviewed in [8], or regional brain hypometabolism and amyloid plaque burden [9]. The majority of AD cases are sporadic (SAD) with no defined etiology other than age, while about 1% of AD cases are genetic (familial AD, FAD) caused by mutations in three genes responsible for the synthesis and processing of the ß-amyloid precursor protein (APP). These are APP, PSEN1, coding for presenilin-1, and PSEN2 coding for presenilin-2. The mutations are autosomal dominant and affected people present the common alterations of AD, often anticipating the onset of the disease by several years in comparison to the sporadic forms and showing a very rapid rate of progression. Although the familial cases are rare, their study has provided important clues that help the understanding of the pathology, both via the direct examination of affected individuals and also via the creation of animal models expressing the human mutated genes. We took advantage of a mouse model expressing both a mutated form of human PS2 and human APP (B6.152H transgenic line) [10,11] to investigate cellular alterations that might emerge early in the course of the disease and precede significant Aß deposition contributing to cellular impairment. Numerous mitochondria alterations have long been described in AD [12,13], ranging from a decrease in cytochrome c oxidase (COX) subunits described in the postmortem AD-brain [14], altered fission/fusion processes [15], and compromised mitophagy processes [16]. We evaluated mitochondria functions in primary hippocampal neurons derived from B6.152H newborn pups and observed a slight but consistent impairment in the capacity to sustain their primary functions during challenging circumstances i.e., in conditions of disablement of components of the respiratory chain, or when an extra workload imposes an increase in the respiratory capacity of the cells. In addition, treatment with low glutamate concentration induces rapid Ca2+ uptake in mitochondria, reflecting the cytosolic Ca2+ increase, which, compared to controls, in cells from the FAD mouse models, take longer to be cleared off from the matrix, and often is paired with sustained mitochondrial membrane depolarization. These subtle alterations could make B6.152H cells more vulnerable to toxic stimuli, or less capable to sustain intensive stimulations, and in the long run, this could compromise their fitness and survival.
2. Materials and Methods
2.1. Animal Handling and Care
The transgenic mouse line B6.152H was a kind gift from Dr. L. Ozmen (F. Hoffmann-La Roche Ltd., Basel, Switzerland). The line has the C57BL/6 strain background, and wild-type animals of this strain were used as controls. All procedures and experiments involving animals were conducted in accordance with the Italian and European Community Council Directive on Animal Care, and approved by the Italian Ministry of Health, protocol number: D2784.N.HEH.
2.2. Primary Neuronal Cultures
Primary neuronal cultures were isolated from the brain, the hippocampus region, of 0- to 1-day old newborn mice of either sex. Cells were seeded on poly-D-lysine (10 µg/mL), laminin (2 µg/mL)-coated coverslips at a density of 320,000 cells cm−2 in MEM based medium (Gibco 32360-026) supplemented with 20 mM Glucose, 1 mM Na-pyruvate; 1% N2 supplement (Thermo Fisher 17502048, Waltham, MA, USA), 9 µg/mL biotin, 0.5 mM L-glutamine, 0.5% B27 supplement (Thermo Fisher 17504044), 10% horse serum, 25 µg/mL penicillin, and 25 µg/mL streptomycin. The medium was replaced 48 h after seeding with the BME medium (Gibco 41010-026) containing 5 mM glucose and supplemented with 0.5 mM L-glutamine, 2% B27 supplement, and 1 mM Na-pyruvate. Experiments were performed after 14–17 days in vitro (DIV).
2.3. Adeno-Associated Virus (AAV) Production
Recombinant AAV vectors used in this study were prepared by the AAV Vector Unit at the International Center for Genetic Engineering and Biotechnology Trieste (http://www.icgeb.org/avucore-facility.html), as described previously [17,18]. Briefly, infectious, recombinant AAV vector particles were generated in HEK293T cell cultures grown in roller bottles, using a cross-packaging approach by which the vector genome is packaged into serotype-9 AAV capsid [19]. Viral stocks were obtained by PEG precipitation and subsequent cesium chloride (CsCl2) gradient centrifugation [20]. The titer of recombinant AAVs was determined by real-time PCR quantifying vector genomes (vg) against a standard curve of plasmid containing the specific vector genome [21], and the values obtained were in the range of 1 × 1012–1 × 1013 vg per milliliter.
2.4. Ca2+ Imaging
Cytosolic and mitochondrial Ca2+ measurements were performed using genetically encoded Ca2+ indicators (geci), jRGECO1 [22] (pAAV.Syn.NES-jRGECO1a.WPRE.SV40 Addgene plasmid #100854; http://n2t.net/addgene:100854; RRID:Addgene_100854) and 4mtD3cpv [23] (Addgene plasmid #36324; http://n2t.net/addgene:36324; RRID:Addgene_36324), respectively. A 4mtD3cpv probe was inserted into the pAAV.hSyn.eGFP.WPRE.bGH plasmid, at the sites Kpn1 and HindIII for virus production, after removing the eGFP coding sequence, (pAAV.hSyn.eGFP.WPRE.bGH Addgene plasmid #105539; http://n2t.net/addgene:105539; RRID: Addgene_105539) After 8–10 DIV, the cells were infected with approximately 1–2 × 109 viral particles mL−1 and cultivated for further 6–4 days to ensure appropriate expression and folding of the probes. Ca2+ levels were monitored using an inverted fluorescence microscope Nikon Eclipse Ti equipped with a Zyla-CMOS 4.2-P camera (Andor, Oxford Instruments, Abingdon, UK) and a 40× objective Nikon S Fluor Oil 1.30 DIC H/N2 WD 0.22). Excitation wavelengths were obtained with a 75 W Xenon Lamp (USHIO, UXLS50A) and a monochromator (Cairn Optoscan Monochromator, Cairn Research Ltd., Faversham, UK) controlled by NIS-E software (Nikon). Excitation wavelength for jRGECO1 was 535 nm, the beam passed first thorough an excitation filter 542/20 and a dichroic mirror FF-570-Di01, both from Semrock (Semrock Inc, IDEX Corporation, Lake Forest, IL, USA); the emitted light was collected through a Semrock BA 620/52 filter and the exposure time was 80 ms. 4mtD3cpv was excited at 425 nm, the beam passed first through a 438/20× filter (Semrock) and a dichroic mirror FF 458-Di02 (Semrock), and the emitted light passed first through a beam splitter (Optosplit T515LPXR-UF2, 301,699 Chroma) and was collected through two emission filters: FF01-479/40 for CFP and FF01-542/27 for YFP, (both filters from Semrock), and exposure time was 100 ms. The measurements were performed at 37 °C using a modified Krebs–Ringer buffer (mKRB) 140 mM NaCl, 2.8 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 5 mM glucose, pH 7.4 at 37 °C, to mimic the brain extracellular fluid composition, the coverslips were mounted into an open-topped chamber (RC-41LP, Warner Instruments).
2.5. TMRM Experiment
The neuronal culture was incubated for 30 min at 37 °C with 10 nM TMRM, 1 μM cyclosporin H in a modified Krebs–Ringer buffer (mKRB) 140 mM NaCl, 2.8 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 5 mM glucose, pH 7.4 at 37 °C. The coverslip was then transferred onto a thermostated chamber on the microscope stage and incubated for a further 10 min to verify signal stability. A combination of respiratory chain and ATP-synthase inhibitors was used to verify their impact on the mitochondrial membrane potential. TMRM fluorescence was recorded using 40× objective (SFluor 40× N.A. 1.3, Nikon, Minato, Tokyo, Japan) on an inverted microscope (Nikon Ti-E). Fluorescence illumination used a 50–75 WLamp (USHIO UXLS50A) and 550 nm excitation wavelengths was obtained using a monochromator (Optoscan CAIRN-Research, Faversham, UK) controlled by NIS- ELEMENTS AR (Nikon) software. The emitted fluorescence was collected using an FF-570- Di01Dichroic (Semrock, Lake Forest, IL, USA) and a 620/52 nm (Semrock) filter. Images were acquired every 60 s, with 50 ms exposure times by a Zyla-CMOS 4.2-P camera (Andor, Oxford Instruments, Belfast, Northern Ireland). Where indicated, rotenone (1 µM) or antimycin-A (1 µM) were added. At the end of each experiment, 4 µM FCCP was added to assess the correct distribution of the dye. Images were exported as TIFF, analyzed with ImageJ (National Institutes of Health).
2.6. Superoxide Measurements
The cells (14–17 div) were incubated for 10 min in mKrebsRB with 200 nM MitoSOX and 1 μM cyclosporin H (CsH) to ensure proper loading of the cells, were subsequently washed two times with plain mKrebsRB and maintained in mKrebsRB 1 μM (CsH). The coverslip was then transferred on the thermostated chamber and kept on the microscope stage for 20 min before beginning with the acquisition. Fluorescence was recorded using 40× objective (SFluor 40× N.A. 1.3, Nikon, Minato, Tokyo, Japan) on an inverted microscope (Nikon Ti-E). Fluorescence illumination used a 50–75 WLamp (USHIO UXLS50A) and the sample was alternately illuminated with 405 and 490 nm excitation wavelengths, obtained using a monochromator (Optoscan CAIRN-Research, Faversham, UK) controlled by NIS-ELEMENTS AR (Nikon) software. Excitation light passed through a QUAD turret emission filter FFO l-390/482/563/640. The emitted fluorescence was collected using a DÌ03-R405/488/561/635 dichroic mirror and an FFO l-446/523/600/677 filter (all from (Semrock, Lake Forest, IL, USA). Multiple images were acquired for every coverslip, with 100 ms exposure times by a Zyla-CMOS 4.2-P camera (Andor, Oxford Instruments, Belfast, Northern Ireland).
2.7. H2O2 Measurements
WT and B6.152H 7 D.I.V. cultures were transduced with approximately 6.5 × 1013 U/mL viral particles carrying the gene for hsyn-mito roGFP2orp1. At day 14–17 in vitro, H2O2 production was measured using an inverted fluorescence microscope Nikon Eclipse Ti and a 40× objective Nikon S Fluor Oil 1.30 DIC H/N2 WD 0.22). Excitation wavelengths were obtained by a monochromator (Cairn Optoscan Monochromator, Cairn Research Ltd. UK). Cells were alternatively illuminated with 405 and 488 excitation wavelengths light passed through Semrock QUAD excitation filter FF01-390/482/563/640; emitted light first passed through the Semrock dichroic mirror Di03-R405/488/561/635, and 510 nm emitted light was collected through a Semrock emission filter FF01-446/523/600/677; the exposure time was 100 ms. The measurements were performed at 37 °C using a modified Krebs–Ringer buffer (mKRB) 140 mM NaCl, 2.8 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 5 mM glucose, pH 7.4 at 37 °C, to mimic the brain extracellular fluid composition; acquisitions were performed every minute. Images were next analyzed using the ImageJ software. Regions of interest (roi) were manually drawn on neuronal cell bodies where mitochondria expressing roGFP2orp1 were clearly visible, and the ratio between light emitted for the 405 nm excitation and the 488 nm excitation was calculated.
2.8. Protein Extraction and Western Blotting
Proteins were extracted from 14–17 DIV neuronal culture using an extraction buffer: 150 mM NaCl, 50 mM Tris-Cl (pH 8.0), 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS with a complete mini, EDTA-free protease inhibitors cocktail (Roche Applied Science). The extracts were quantified using the BCA Protein assay kit (QuantumProtein Assay, EuroClone) as per manufacturer instruction. The protein suspension was dissolved in reducing Laemli sample buffer and heated for 5 min at 50 °C. Thirty micrograms of the protein extract was loaded per lane and separated by SDS-PAGE (12.5% polyacrylamide gel) and subsequently transferred onto nitrocellulose membrane (GE Healthcare #10600001) and probed using the following antibodies: MitoProfile total OXPHOS rodent WB antibody cocktail (Abcam ab110413) 0.6 µg/mL; mouse anti Hsp90 1:1000 (BD Bioscience #610418). Species-specific, HRP-conjugated secondary antibodies (BioRad) have been used. Immuno-bands were visualized using the chemiluminescence reagent Westar Sun (Cyanogen) on an UviTech Mini HD9 system (Eppendorf). Band intensities were analyzed using the image processing software ImageJ (NIH, USA), using the HSP90 signal for normalization.
2.9. Immunofluorescence
Fourteen- to seventeen-day in vitro hippocampal neurons were fixed with a medium containing 4% PFA and 20% sucrose dissolved in phosphate buffered saline (PBS), for 10-min, all subsequent procedures were conducted at room temperature. The coverslips were then washed 3 times, for 5 min each, with PBS and incubated with quenching solution (0.24% NH4Cl in PBS) for 20 min and washed again at the end of incubation. Cells were then treated with 0.1% TRITON-X 100 in PBS (permeabilization solution) for 3-min, and subsequently incubated with 10% Goat serum, 2% BSA, 0.2% gelatin, in PBS for 30 min to block unspecific reaction sites and then incubated for 1 h with mouse anti-neurofilament 200 (1:100, Merck-Sigma N5389); rabbit anti-glial fibrillary acidic protein (1:100 Dako, Z0334). After rinsing, the cells were incubated for 45 min with Alexa Fluor secondary antibodies Fluor Plus 555 goat anti mouse (1:500 Thermo Fisher Scientific A32727), and Alexa Fluor Plus 488 goat anti rabbit (1:500 Thermo Fisher Scientific A32731. To mark the nuclei, the cells were incubated for 10 min with 5 µg/mL Hoechst 33,342 (Invitrogen H1399) and mounted on glass coverslips using Mowiol 4–88 (Millipore 475904-M). Images were collected with the Nikon Eclipse 80i ViCo fluorescence microscope system, equipped with a Qimaging Fast 1394 camera and subsequently elaborated with the image processing software ImageJ (NIH, USA).
3. Results
3.1. FAD Hippocampal Neurons Have Impaired Respiratory Capacity
Mitochondria dysfunction has long been implicated in the cellular alterations that characterize AD. To further address this issue, and with the aim of carefully probing mitochondria metabolism in AD, we used primary cortical neurons isolated from the hippocampi of newborn WT and B6.152H mice. This last genotype represents a model for Familial Alzheimer’s disease as it expresses the mutated human presenilin 2 (PS2-N141I) protein, together with the human mutated (Swedish variant K670N, M671L)Amyloid Precursor Protein (APP) [10,11]. Neuronal cells derived from the hippocampi of newborn pups, p0–p1, were grown for 14–17 days in vitro (div) to allow the expression of membrane channels and receptors, in a medium with a physiological glucose level (5 mM), to avoid potential toxic effects of the high glucose media commonly used [24], and to maintain a balance between aerobic and anaerobic metabolism in the cultured neurons. For all the following experiments with neurons, we seeded the same number of cells for both WT and B6.152H, checked the cells at regular intervals to control for possible contamination and to replenish consumed medium, and we did not observe differences in the growth rate or cell death between the two genotypes. The oxygen consumption rate (OCR) was measured in the cultures by means of the Seahorse XF Analyzer as described in [25]. Basal OCR and ATP synthesis-driven OCR (oligomycin-sensitive respiration) were comparable (not statistically different) in both WT and B6.152H neuronal cultures, while maximal OCR induced by the uncoupling of oxidative phosphorylation (achieved with the addition of the protonophore carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone, FCCP) (relevant substances used in this work are listed in Table 1, Section 2) was significantly reduced in B6.152H neurons compared to WT (Figure 1). Since sometimes the addition of oligomycin inhibits maximal respiration in the cells [26], the addition of FCCP was performed in separate wells from oligomycin, or, when added after oligomycin, only the wells at least doubling OCR with respect to basal OCR were considered.
Table 1.
Key resources table.
| Reagent or Resource | Supplier | Identifier |
|---|---|---|
| Chemicals, peptides, cell culture media and supplements | ||
| Poly-D-Lysine | Merck | P6407 |
| Laminin | Merck | L2020 |
| Minimum Essential Medium, MEM | Gibco | 32360-026 |
| Basal Medium Eagle, BME | Gibco | 41010-026 |
| N2-supplement | Thermo-Fisher | 17502048 |
| B27-supplement | Thermo-Fisher | 17504044 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Merck | C2920 |
| Tetramethylrhodamine, methyl ester (TMRM) | Thermo Fisher | I34361 |
| MitoSOX™ Red | Thermo Fisher | M36008 |
| Cyclosporin H | Merck | SML1575 |
| Bicinchoninic Protein Assay Kit, Quantum Protein | Euroclone | EMP014500 |
| Chemiluminescent reagent Westar Sun | Cyagen | XLS063 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo-Fisher | H1399 |
| MOWIOL® 4-88 Reagent, Poly(vinyl alcohol) | Millipore | 475904-M |
| Antibodies | ||
| Mito profile Total Oxphos rodent WT | Abcam | ab110413 |
| anti HSP 90 | BD Bioscience | 610418 |
| anti-neurofilament | Merck | N5389 |
| anti glial fibrillary acidic protein | Dako | Z0334 |
| Alexa-Fluor Plus 555 goat anti-mouse IgG | Thermo-Fisher | A32727 |
| Alexa Fluor Plus 488 goat anti-rabbit IgG | Thermo-Fisher | A32731 |
| Recombinant proteins | ||
| jRGECO1 | Addgene | 100854 |
| 4mtD3cpv | Addgene | 36324 |
| mito roGFP2-Orp1 | Addgene | 65001 |
Figure 1.

Quantification of intact-cell respiratory rate, OCR, and of the extracellular acidification rate ECAR. Panel (A), Basal OCR, (expressed as pmols of O2 consumed per minute per 2 × 105 cells/well) combination of ATP synthesis-driven respiration plus the respiration caused by the leak of protons from the membrane (WT n = 50 wells, mean ranks = 57.88; B6.152H n = 50 wells mean ranks = 43.12; Mann–Whitney probability test Asymp. prob = 0.8); at the 0.05 level, the two distributions are not significantly different. Maximal OCR (uncoupled respiration), (WT n = 50 wells, mean ranks = 51.2; B6.152H n = 50 wells, mean ranks = 49.8; Mann Whitney probability test Asymp. prob = 0.011, at the 0.05 level the two distributions are significantly different. (WT, yellow; B6.152H, indigo). Panel (B), ECAR (expressed as mpH per minute per 2 × 105 cells/well) quantification: Basal values WT n = 47 wells, median: 16.97. B6.152H n = 42 wells, median: 10.82. At the 0.05 level. the two populations are significantly different, Mann–Whitney U test, asymp prob = 0.005. Maximal ECAR, after 2 μg/mL oligomycin addition, WT median: 28.5, B6.152H median: 23.77. At the 0.05 level, the two populations are significantly different, Mann–Whitney U test, asymp. prob. = 4.6 × 10−5. Box representing 25–75% of data, whiskers are 1–99% of data, white square represents the mean value; transverse line represents the median value. Cells were from at least 4 different culture preparations. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Simultaneously, in the same plates, we also quantified the extracellular acidification rate, ECAR, which is an indirect measurement of the glycolytic flux of the cells. The main contributor to ECAR is the lactic acid produced by glycolysis, but other cellular metabolic pathways can change the extracellular pH mostly by producing CO2, namely the TCA cycle. In our experimental conditions, B6.152H cultures showed a lower basal ECAR, compared to wild-type cells, which could be an indication of a glycolytic defect as already suggested for B6.152H cortical neurons [25]. Maximal ECAR is measured after the addition of oligomycin (2 μg/mL), which inhibits F1F0 ATP synthase activity, blocking mitochondrial ATP synthesis and shifting cellular energy production to glycolysis. B6.152H hippocampal neurons had significantly lower maximal ECAR than WT neurons, suggesting a possible diminished capacity for these cells to meet energy challenges.
3.2. FAD Hippocampal Neurons Are Less Able to Maintain a Stable Membrane Potential When Challenged with Inhibitors of the Respiratory Chain
On the day of experiment, the cells were incubated for 30 min at 37 °C with 10 nM TMRM supplemented with 1 μM Cyclosporin H (CSH) to ensure stable loading of the dye (Figure 2A). The coverslip was then transferred onto a thermostated chamber on the microscope stage and incubated for another 10 min to equilibrate the temperature and prevent focal drift due to thermal imbalance. As shown in (Figure 2B), the cultures are enriched in neurons but contain a mixed population of neurons and glial cells. For TMRM measurements, and for all subsequent imaging experiments, only neurons were examined, placing the region of interest (roi) on the soma of cells previously identified as neurons by their morphology in bright field images, round-shaped somata, and distinct processes [27], or by the specific targeting of genetically encoded sensors (see below).
Figure 2.

TMRM staining and immunofluorescence of mixed hippocampal mouse cultures. Panel (A), 15 div WT neuronal culture loaded with potentiometric dye TMRM 10 nM, imaged with the Nikon Eclipse Ti inverted fluorescence microscope, 40× magnification; fluorescence image superimposed on the bright field image used to select neurons. Panel (B), representative image of 15 div neuronal culture; paraformaldehyde fixed cells were stained with: 1:100 anti-neurofilament 200 (NF 200, Merck-Sigma # N5389) labeling neurons, red; and with 1:100 anti glial fibrillary acidic protein (GFAP, Dako # 20334) labeling glial cells, green, and counterstained with Hoechst 100 μg/mL, blue, to mark all nuclei. Images were collected with a Leica DMI 6000 inverted microscope, equipped with ORCA FLASH 4.0 camera (Hamamatsu), magnification 20×. Analysis of images and composition of panels performed with the software ImageJ.
The basal TMRM fluorescence levels between WT and B6.152H neurons were comparable (Figure 3 inset), indicating a similar loading of the dye, and excluding the possibility of conspicuous differences in plasma membrane or mitochondria membrane potential between the two genotypes, and allowing us to normalize the fluorescence data to the average of the initial 5 min acquisitions. Cultured neurons were then challenged with drugs known to inhibit the mitochondrial respiratory chain specifically at the level of complex I (rotenone) or complex III (antimycin), with the aim of identifying hidden defects in the capacity of mitochondrial ATP-synthase to maintain the inner membrane potential via hydrolysis of ATP molecules within the time frame of the experiment. As shown in Figure 3A, upon addition of rotenone mitochondria of B6.152H, cells lost at least part of their membrane potential much faster than WT mitochondria. This result could be attributed either to a defect in ATP-synthase activity, or/and to a reduced availability of ATP. Similar results were observed inhibiting complex III with antimycin. On the contrary, the cells responded in a similar way to the blockade of the catalytic activity of ATP-synthase with oligomycin (Figure 3B); we observed a slight initial depolarization and a small hyperpolarization in B6.152H cells, but overall, the cells maintained a stable mitochondrial membrane potential for the whole duration of the experiment, indicating that the respiratory complexes of the two genotypes are equally able to sustain analogous membrane potential within the experimental timeframe. The slight depolarization observed after oligomycin addition might be due to a modest inhibition of the plasma membrane Na+/K+ATPase by the drug [28,29]. Figure 3C compares the decrease of TMRM fluorescence at 20 min after the addition of either respiratory chain inhibitor, and prior to the addition of the uncoupler (FCCP). The protonophore FCCP is added to completely collapse the mitochondrial membrane potential (providing a minimal value for the normalization of the data), and it also allows for controlling the TMRM-specific signal.
Figure 3.

Effect of the inhibition of the ETC or of the F1F0ATP-synthase on the mitochondrial membrane potential. Panel (A) shows traces of TMRM fluorescence for WT, yellow, and B6.152H, indigo, neurons. Where indicated, the cells were challenged with 2 μM rotenone and 4 μM FCCP. Values are normalized over the mean fluorescence of the first five acquisitions; fluorescence intensity was acquired every min. Inset: Absolute TMRM initial values for WT, (yellow) n = 51 cells, and B6.152H (indigo) neurons n = 67 cells; test t p = 0.11. Box representing 25–75% of data, whiskers: 1–99% of data, white square: Mean; transverse line: Median. Panel (B) shows mean traces of TMRM fluorescence of WT and B6.152H neurons challenged with 1 μg/mL oligomycin and 4 μM FCCP. Panel (C) shows the quantification of the effects produced on the mitochondrial membrane potential by the above-mentioned inhibitors expressed as the normalized difference between initial fluorescence F0, minus the fluorescence measured at 25 min,. Rotenone experiment WT n = 22, B6.152H n = 34 p = 1.5 × 10−4 test t (normal distribution); antimycin experiments: WT n = 16, B6.152H n = 24 p = 0.003 Mann–Whitney test. Oligomycin experiments: WT n = 11, B6.152H n = 14 p = 0.48, test t (normal distribution). Cells were derived from at least three independent preparations, both for WT and the B6.152H genotype. Columns represent mean value plus/minus standard deviation; red line represents the median of the values. ** p ≤ 0.01, *** p ≤ 0.001.
The ATP-synthase complex has been shown to be reduced in several post-mortem brain samples of Alzheimer’s disease patients [30], and defects in the enzyme have also been described in an AD mouse model [31]; a decreased expression of several nuclear encoded ATP-synthase genes have been found in brain areas early affected by AD [32]. To investigate possible ATP-synthase defects in our experimental model, we measured the total ATP content of the culture using a commercial kit, as per manufacturer instructions (Abcam Luminescent ATP detection assay kit ab113849) and did not find an appreciable difference between WT and B6.152H cells (results not shown). It needs stressing, however, that not only we have evaluated total ATP contributed by all the cell types present in the culture, and not just neurons [32], but also, and most relevant, that part of the cellular ATP is produced by glycolysis. To more directly evaluate whether the functioning of the mitochondrial ATP-synthase is impaired in the AD transgenic model, we measured ATP-synthase reverse activity in mitochondria derived from the brain of 3- and 6-month old WT and B6.152H mice, a protocol adapted from [33]. No appreciable differences were observed in this case between the two genotypes (Figure 4), but also in this case, the mitochondria are derived from whole brain tissue (mainly from cortical and hippocampal areas).
Figure 4.

Measurements of the F1F0 ATP-synthase activity in isolated brain mitochondria. Quantification of the amount of ATP hydrolyzed per minute per mg of mitochondrial protein extracted from the forebrain of WT and B6.152H mice of 3 and 6 months of age. Mean of multiple probing from at least three independent mitochondria preparation for each genotype and for each age. For p ≤ 0.05, the differences among phenotypes were not statistically significant. Columns represent mean value plus/minus SEM.
3.3. Mitochondrial Reactive Oxygen Species Production
A large body of scientific literature points to the effect of reactive oxygen species (ROS) in the progression and possibly the onset of neurodegenerative disorders, and mitochondria dysfunctions have been examined as possible early events in the pathogenesis [34,35], with mitochondria being important ROS producers within the cells. It has been shown that the malfunctioning of the mitochondrial respiratory chain, impairing the flux of electrons (e−) from complex I to final O2 reduction to H2O at the level of complex IV, often results in the production of reactive oxygen species due to the leak of e− from reduced cofactors. The anion radical superoxide (O2−•) is produced by direct reaction of a single e− with molecular O2, and the excess of matrix superoxide can initiate oxidative damage. The dye hydroethidine and its mitochondrial derivative MitoSOX™ are commonly used to measure O2−• in living cells. MitoSOX™ accumulates within the mitochondrial matrix driven by the inner membrane potential. Given that in resting conditions the membrane potential is comparable between WT and B6.152H neurons, the MitoSOX™ signal appears adequate to measure basal O2−• production in mitochondria of WT and B6.152H neuronal cultures. No significant differences were observed however (Figure 5). Often, inhibitors of the respiratory chain are added to increase ROS production and to better visualize it in imaging experiments We could not exploit MitoSOX™ to assess O2−• in conjunction with rotenone and antimycin inhibition of the respiratory chain, since such treatments cause the depolarization of mitochondria, which in the end lead to exit of the dye from the matrix and to its redistribution in the cytoplasm.
Figure 5.
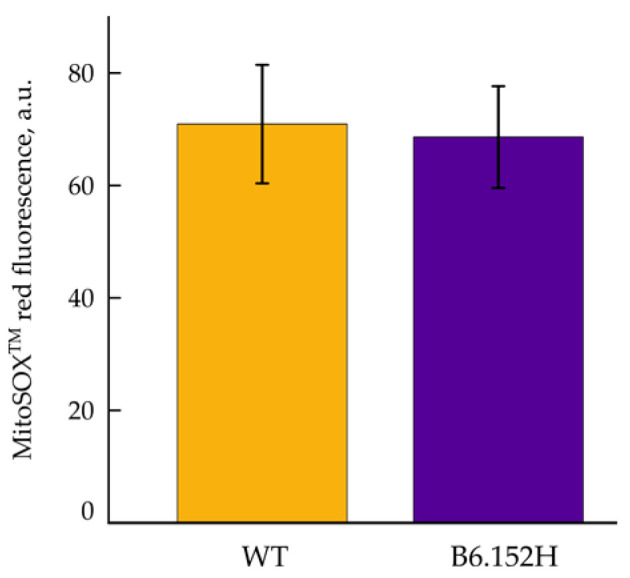
Measurements of superoxide production in primary hippocampal neurons. Basal MitoSOX fluorescence. WT, yellow, and B5.152H, purple, hippocampal neurons loaded with 200 nM MitoSOX™. The result is expressed as the average fluorescence for 8 WT and 6 B6.152H coverslips, respectively, with multiple neurons, derived from at least three independent preparations. For p ≤ 0.05, the differences between the two genotypes were not statistically significant. Columns represent mean value plus/minus SD.
Mitochondrial O2−• is rapidly detoxified to the less reactive hydrogen peroxide (H2O2) by matrix Manganese Superoxide dismutase (Mn-SOD). To measure H2O2 production, we used a genetically encoded, mitochondrially targeted probe named mito-ro-GFP2-orp1 (Addgene #65001) [36] modified by adding the human synapsin 1 promoter to achieve specific expression in neurons; nine div neurons were infected with adeno-associated virus (AAV) carrying the mitochondria targeted ROS probe. In our experimental settings, we could not detect any increase of H2O2 upon inhibition of the respiratory chain with rotenone or antimycin (results not shown); in addition, the basal ratio fluorescence of the probe was comparable between WT and B6.152H neurons (Figure 6), as seen with MitoSOX™ probe, meaning that either in our cultures, ROS production is quite low and efficiently counteracted by the intracellular antioxidative mechanisms, or that the sensitivity of the system is too low to detect subtle differences.
Figure 6.

Measurements of H2O2 production in primary hippocampal neuronal cultures. Panel (A): Representative image of hippocampal neurons expressing the mitochondrial targeted probe for H2O2, mito roGFP2-orp1. Fluorescence image recorded illuminating the cells with a 488 nm excitation wavelength and superimposed to the corresponding bright field image. Panel (B), quantification of the basal fluorescence of the probe expressed as the ratio between 405 and 488 nm illuminations. Symbols represent individual cells obtained from at least three independent preparations for each genotype, WT, n = 34 cells, median = 0.18; B6.152H n = 32 cells, median = 0.14. Box representing 25–75% of data, whiskers: 1–99% of data, white square: Mean; transverse line: Median. For p ≤ 0.05, the difference between the two genotypes is not statistically significant, Mann–Whitney probability test, asymp. prob. = 0.17.
3.4. FAD Hippocampal Neurons Are More Sensitive to Glutamate Excitotoxicity
3.4.1. Effect of Glutamate on Cytosolic Ca2+ Transients
Glutamate is considered the principal excitatory neurotransmitter in the brain [37], important for the fundamental memory and learning processes [38,39]. Its concentration has to be maintained within tight boundaries for proper signal transmission, and to avoid toxic effects for the cells. Moreover, excitotoxicity has been proposed as a concurrent mechanism for the development of neurodegenerative disorders together with soluble amyloid beta (Aβ) oligomers [40] and tau protein hyperphosphorylation [41]. In order to induce a mild excitotoxic stress to the neurons and to evaluate their responses, we treated 14–17 div WT and B6.152H hippocampal neurons with a low concentration of glutamate (2 μM), which is one order of magnitude above the basal extracellular neurotransmitter concentration [42]. Glutamate is known to induce an increase in intracellular [Ca2+] due to a complex series of events: (i) Activation of AMPA receptors with depolarization and Ca2+ influx through voltage gated channels; (ii) opening of ionotropic NMDA receptors; and (iii) activation of Group I mGlu receptors. To evaluate the variations of intracellular Ca2+ content in cultured neurons after the toxic stimulus, WT and B6.152H neurons, 9 div, were infected with AA-virus expressing the cytosol targeted Ca2+ probe jRGECO [22] under the human synapsin 1 promoter. The cells were then examined 5 days post-infection to allow complete expression and folding of the probe. Basal fluorescence was comparable between WT and B6.152H neurons, indicating a similar expression of the probe between the two genotypes; as shown in Figure 7, we could sometimes observe spontaneous, synchronous Ca2+ oscillations in the resting cells of both genotypes. jRGECO is not a ratiometric probe, and accordingly, the intensity of the signal does not allow one to directly correlate it to [Ca2+]. Although a ratiometric dye would be easier to calibrate in terms of [Ca2+], we chose jRGECO for these experiments for its fluorescence intensity and suitability to detect the Ca2+ changes due to neuronal action potentials [22]. Information on the amplitude of the [Ca2+] changes with a non ratiometric probe can, however, be obtained by the measurements of the ΔF/F0 values. Being non ratiometric, in addition, means the probe is potentially affected by the pitfalls or artefacts due to movements of the focal plane or of the cells during the acquisitions, so extra care was taken to ensure stability of the focal plane, and movements of the cells were controlled for during offline analysis. The cells were challenged with the addition of 2 µM glutamate to mimic a sustained excitatory signal that elicited a large cytosolic Ca2+ increase. The rate of fluorescence increases, and the peak amplitude reached were comparable between the two genotypes, with no significant difference, indicating analogous endowment of cellular receptors and channels. After the rapid Ca2+ rise, its concentration progressively decreased to a lower plateau level; we then measured the jrGECO ΔF/F0 ratio 20 min post glutamate addition to investigate whether the cells were equally able to extrude excess Ca2+ from the cytosol and return toward the basal level. In the time frame of the experiment, we observed that both cell types reached a comparable cytosolic [Ca2+]. As discussed above, this probe does not allow the quantification of the absolute values of cytosolic [Ca2+] in the neurons, but the calculation of the ΔF/F0 permits one to compare the variation in [Ca2+] produced by the stimulus in the two cell types. Potassium chloride (KCl) 30 mM was added at minute 25, towards the end of acquisition, to further depolarize the plasma membrane, promote the opening of Voltage-Operated Channels (VOCs), and evaluate the residual capacity of the cells to accumulate Ca2+.
Figure 7.

Cytosolic Ca2+ transient in primary hippocampal neurons induced by glutamate. Panel (A), representative image of 14 div hippocampal neurons expressing cytoplasm-targeted jrGECO. Panel (B), representative traces of jrGECO fluorescence expressed as the normalized values of the difference between time point F minus the average of the initial 5 min of acquisition (F0); where indicated the cells were challenged with 2 μM glutamate and 30 mM KCl. Panel (C), quantification of the jrGECO fluorescence measured at the maximum peak reached after glutamate addition and expressed as normalized difference with initial fluorescence F0. WT n = 144 cells, median value = 4.13; B6.152H n = 121 cells, median value = 4.2. Two sample t-test prob. = 0.9; at the 0.05 level, the difference between the means is not significantly different from 0. Panel (D), jrGECO fluorescence measured 20 min after glutamate addition and expressed as before. WT n = 144, median value = 2.33; B6.152H n = 121 cells, median = 2.24; Mann–Whitney test Asymp prob = 0.1. At the 0.05 level, the two distributions are not significantly different. Box plot: Box height, 25–75%; whiskers, 1–99%; white square: Mean; transverse line: Median; curve: Normal distribution.
3.4.2. Effect of Glutamate on Mitochondrial Ca2+ Transients
Mitochondria actively participate in the control of cellular Ca2+ content, helping in finely shaping Ca2+ signals [43,44]; in addition, the activity of the respiratory chain is modulated by matrix Ca2+ due to its effects on key mitochondrial dehydrogenases. In the next series of experiments, we investigated the [Ca2+] changes within the mitochondrial matrix brought about by the glutamate challenge. For these experiments, WT and B6.152H neurons were infected with AAV expressing the mitochondrial Ca2+ probe 4mtD3cpv [23] under the control of the human synapsin 1 promoter. To ensure complete expression and folding of the probe, the cells were infected at least 5–7 days prior to the experiment. In the case of 4mtD3cpv, the ratio between the fluorescence emitted at 520 and 480 nm is directly proportional to the [Ca2+], independently of the amount of probe expression level [23,45]. The cells of the two genotypes have comparable mitochondrial resting Ca2+ levels (Figure 8 panel C). As before, the cells were then challenged with 2 μM glutamate in the presence of 2 mM external Ca2+. As expected, a rapid and large rise of matrix Ca2+ was observed upon glutamate addition. The first part of the response was generally homogeneous, with the majority of cells of both genotypes reaching a very similar Ca2+ peak with the same rate, suggesting that the functioning of the mitochondrial calcium uniporter (MCU) is comparable between the genotypes. The fluorescence of 4mtD3cpv was then monitored for about 25 min after glutamate addition and it was observed that after the first fast mitochondrial Ca2+ uptake, the behavior of the cells was quite variable. In some cells, the Ca2+ concentration steadily decreased toward basal, while in others, the Ca2+ inside the mitochondria begun to oscillate, giving rise to waves of Ca2+ that were very rarely observed in resting conditions. These oscillations were more frequently observed in mitochondria of B6.152H cells, 27.3% of the total, compared to 20% of all observed WT cells, and were clearly not attributable to changes in the focal plane, or movements of the cells, since 4mtD3cpv is a ratiometric probe that allows one to correct for these artifacts. Finally, the cells were challenged with the addition of 30 mM KCl, to produce a sustained plasma-membrane depolarization and prolonged opening of voltage activated Ca2+ channels. Mitochondria responded to KCl with a large Ca2+ peak that was comparable to that obtained with the addition of glutamate. We calculated the ratio between YFP and CFP fluorescence for 4mtd3cpv after 20 min from glutamate addition in order to measure the residual Ca2+ concentration in mitochondria after the stimulation. An appreciable difference between the Ca2+ level of WT and B6.152H cells was observed, with transgenic mitochondria showing a significantly higher level of Ca2+ (Figure 8 panel F). The inability of mitochondria to efficiently clear matrix [Ca2+] could be detrimental for the organelles, since a prolonged increase in matrix Ca2+ concentration could keep some of the Ca2+-dependent mitochondrial dehydrogenases active, and could also sensitize/prime the mitochondrial permeability transition pore (PTP) and promote its opening in the presence of a second cue (e.g., oxidation, changes in matrix pH, presence of toxic oligomeric species) [46,47].
Figure 8.

Mitochondrial Ca2+ transients in hippocampal primary cultures induced by glutamate. Panel (A), representative image of 14 div hippocampal neurons expressing mitochondria-targeted cameleon probe 4mtD3cpv. Panel (B), representative traces of mitochondrial Ca2+ uptake in hippocampal neurons expressing 4mtD3cpv, left WT; right B6.152H cells. Where indicated the cells were challenged with 2 μM glutamate and 30 mM KCl addition. Panels (C–E), quantifications of 4mtD3cpv ratio fluorescence for, respectively: C basal values (WT n = 111 cells, median = 2.83; B6.152H n = 89 cells, median = 2.66, Mann–Whitney probability test, exact prob. = 0.063: at the 0.05 level, the two distributions are not significantly different); (D), peak glutamate (WT n = 111 cells, median = 6.5; B6.152H n = 89 cells, median = 5.6, Mann–Whitney probability test, exact prob. = 0.858: at the 0.05 level, the two distributions are not significantly different); (E) peak KCl (WT n = 95 cells, median = 6.76; B6.152H n = 71 cells, median = 6.54, Mann–Whitney probability test, exact prob. = 0.65: at the 0.05 level, the distributions are not significantly different). Panel (F), quantification of the 4mtD3cpv fluorescence ratio measured 20 min after the addition of 2 μM glutamate (WT n = 111, median = 3.07; B6.152H n = 85 median = 3.79, Mann–Whitney probability test, exact prob. = 0.029: at the 0.05 level, the distributions are significantly different). Box plot: Box height, 25–75%; whiskers, 1–99%; white square, mean; transverse line median; curve, normal distribution equation. The spontaneous movements of mitochondria during the acquisition were controlled for in off-line analysis to ensure the evaluated organelles rested within the boundary of the outlined roi. * p ≤ 0.05.
3.5. Glutamate Excitotoxicity Impairs FAD Hippocampal Neurons Membrane Potential, Effect of Alisporivir
To evaluate the ability of the cells to maintain the membrane potential in the presence of a large and sustained Ca2+ influx, the neuronal cultures were challenged with 2 µM glutamate, in the presence of physiologic extracellular Ca2+. Figure 9A shows representative traces of TMRM fluorescence measured in WT and B6.152H hippocampal neurons treated with glutamate; fluorescence measurements (arbitrary units) were normalized to the difference between the average of initial fluorescence, minus the average of fluorescence signal measured after the addition of the protonophore FCCP. We observed that WT neurons can better sustain the excitotoxic challenge posed by glutamate, while double transgenic neurons seem to be more sensitive to the treatment, and a large proportion of cells lost their membrane potential faster than WT ones, as a consequence of the treatment.
Figure 9.

Effect of glutamate treatment on the mitochondrial membrane potential of primary hippocampal neurons. Panel (A), representative traces of WT (left), B6.152H (middle), and B6.152H cells treated with 1.5 μM Alisporivir (right), loaded with the potentiometric probe TMRM. Where indicated 2 μM glutamate and 4 μM FCCP were added. Time point TMRM acquisitions are normalized to the difference between the average of initial fluorescence (first 5 min acquisitions), minus the average of fluorescence signal measured after the addition of the uncoupler FCCP (last 3 min acquisitions). Panel (B), violin plot, quantification of basal TMRM fluorescence expressed in arbitrary units; respectively for WT, yellow n = 167 cells; B6.152H, indigo, n = 89 cells; B6.152H plus Alisporivir, purple, n = 130 cells. ANOVA one-way test, at the 0.05 level, the population means are not significantly different. Box height, 25–75%; whiskers, 1–99%; white circle, median; transverse line, median. Panel (C), quantification of TMRM fluorescence 20 min after 2 μM glutamate addition, expressed as % of initial (mean of 5 acquisitions) fluorescence. Color code as in previous panel. WT, n = 11 coverslip (157 cells); B6.152H, n = 11 coverslip (87 cells); B6.152H plus 1.5 μM Alisporivir n = 8 coverslip (129 cells). ANOVA one-way test: α = 0.05 Tukey and Fisher test, the difference between WT and B6.152H is statistically significant, p = 0.02 as well as the difference between B6.152H and B6.152H plus Alisporivir, p = 0.048; while the difference between WT and B6.152H plus Alisporivir is not significant. * p ≤ 0.05.
The loss of membrane potential caused by the addition of glutamate and the increase of matrix [Ca2+] could be due to the incapacity of the respiratory chain to work at a sufficient rate when challenged with a sustained ion entry. However, it could also be caused by the opening of the permeability transition pore. We checked whether the PTP was involved in the depolarization caused by the glutamate excitotoxicity by treating B6.152H neurons with a low concentration (1.5 μM) of the inhibitor Alisporivir (formerly known as Debio 025), which inhibits PTP opening via cyclophilin D binding, but, differently from its parent compound, cyclosporin A, does not bind to calcineurin [48,49]. Cells were treated with 1.5 μM Alisporivir for 5 min prior the additions of glutamate. We controlled for the fact that Alisporivir per se does not change the basal membrane potential, and as seen in Figure 9, panel B, we did not observe any difference in basal fluorescence between WT, B6.152H neurons, and B6.152H neurons treated with the drug. On the other hand, Alisporivir treatment delayed, and often significantly reduced, the drop of membrane potential in B6.152H neurons challenged with glutamate (Figure 9 panel C). Alisporivir also had a mild effect on WT neurons, slightly increasing basal TMRM fluorescence, compared to untreated cells.
4. Discussion
In this study, we reported subtle alterations in mitochondrial functionality in cultured hippocampal neurons derived from a mouse model of AD, which could make neurons more susceptible to challenging conditions, or contribute to the accumulation of stress-induced modification that can make them more vulnerable. We observed a decrease in maximal respiratory rate (OCR) in intact hippocampal neurons isolated from newborn B6.152H pups carrying the human proteins APP and PS2 bearing mutations responsible for familial AD. Basal and ATP-linked OCR were comparable between WT and B6.152H cells, suggesting that in normal conditions, these cells carry out regular metabolic functions. The defect instead became apparent when the cells were challenged with a stimulus that increases the rate of the respiratory chain, which could mimic the necessity to increase the rate of ATP production [50,51]. Parallel to this, a decrease in basal and maximal glycolytic flux was also measured, which could exacerbate the consequences for the cell of coping with a stimulus increasing the cellular workload and the need for additional ATP production [25].
Defects in mitochondrial functionality or molecular endowment have been consistently reported for AD patients and also AD animal models [52,53,54], while mitochondrial F1Fo ATP synthase and single components of the respiratory chain have been found defective in both AD patients and AD models [30,31,32,55]. We did not observe a reduction in single respiratory components of the ETC or ATP-synthase (Supplementary Figure S1): Protein extract obtained from four independent WT and B6.152H neuronal cultures were probed with antibodies against individual components of the ETC and the F1FO ATP-synthase and normalized against the HSP90 protein, employed here as a housekeeper and as loading control. Nevertheless, the defect could lay in the organization of mitochondrial supra-molecular complexes, which are thought to be fundamental in the efficient e− transport and H+ translocation, and for the maintenance of the mitochondria cristae structure [56,57,58]. In addition, if the defect lays specifically in one cell type it would be difficult to spot it with this approach since the cultures used are enriched in neuronal cells but also bear other cell types.
The potential small defects at the level of the electron transport chain (ETC) or the ATP-synthase were confirmed by monitoring the mitochondrial membrane potential of the neurons challenged with different, specific inhibitors. A faster dissipation of mitochondrial membrane potential was indeed observed in B6.152H neurons compared to controls when the electron flux was blocked either at the level of complex I or complex III. This effect would be indicative of a potential defect in the working of the ATP-synthase functioning in “reverse mode”, hydrolyzing ATP to maintain the membrane potential [59], or to an insufficient supply of ATP to the enzyme. Measurements of ATP-synthase activity or total ATP content did not highlight any difference between WT and B6.152H genotypes, but this could be due to the limits of the experimental approach.
The latent defect observed in this experimental model could become detrimental in conditions of increased energy demand i.e., to restore ion gradients across the plasma membrane during periods of intense signaling, or to cope with toxic stimuli. Excess glutamate is known to cause excitotoxic damage to the brain, which can end up in terminal cellular disfunction and cell death, and has been implicated in the neurodegeneration characteristic of AD [60,61,62]. In our experimental model, a low micromolar glutamate addition produced a rapid and transient Ca2+ increase in the cytosol and in the mitochondrial matrix of both WT and B6.152H neurons, followed by a recovery towards resting values. B6.152H mitochondria took longer to extrude excessive matrix Ca2+. Excessive [Ca2+] could be detrimental for mitochondria [63], ending up reducing ATP synthesis [64,65] and inhibiting complex I activity [66]. In addition, a large and persistent load of matrix Ca2+ could sensitize the permeability transition pore (PTP) favoring its open state [67].
A physiological increase in cytosolic Ca2+ produces a depolarization of the mitochondrial membrane potential, likely caused by Ca2+ cycling across the mitochondrial inner membrane [68,69], but a toxic extracellular glutamate increase provokes the decrease in mitochondrial membrane potential and also a perturbation of Ca2+ homeostasis [70]. Moreover, a large and persistent decrease of mitochondrial membrane potential would make mitochondria unable to provide sufficient ATP for the cellular needs, making it more vulnerable to internal and external stressors and potentially setting in motion the series of event leading to cell death [71]. Upon the addition of glutamate, we observed different responses from the cultured neurons with some cells keeping their basal membrane potential unchanged and others experiencing different degrees of depolarization. On average, the B6:152H cells experienced a larger decrease in membrane potential than WT neurons, 20 min after glutamate addition. Paradoxically, the average [Ca2+] within mitochondria is higher in B6.152H cells than in controls, despite no significant average difference in cytosolic [Ca2+] and a larger depolarization of the organelles in the transgenic cells. It needs to be stressed that these results are the average of several tens of cells, while it has been impossible to monitor Ca2+ levels (either cytoplasmic or mitochondrial) and mitochondrial membrane potential in the same cells. Accordingly, in cells with dramatic drops in mitochondrial membrane potential, the matrix [Ca2+] will not decrease to the basal level, but it will simply equilibrate with the cytoplasmic [Ca2+] i.e., it will be close to that in cells with normal mitochondrial membrane potential. On the contrary, in cells with only partial depolarization, the efficacy of Ca2+ efflux from the mitochondria via the Na+/Ca2+ exchanger (NCLX, exchanging 3 Na+ for 1 Ca2+) would be reduced [72], and the matrix Ca2+ would be higher compared to controls The average [Ca2+] in the mitochondrial matrix will thus depend on the percentage of B6.152H cells with the membrane potential identical, lower, or even completely depolarized compared to the controls. Most relevant, however, preventive incubation of the cells with a micromolar concentration of an inhibitor of the PTP delayed, and in some cases completely avoided, the decrease in the mitochondrial membrane potential suggesting that the PTP might be involved, and inferring that in the AD model, it could be more sensitive to stimuli promoting its opening. We judged the potentially protective effect of Alisporivir very interesting as it could be developed into a therapeutic tool for AD, and in vivo experiments on the FAD mouse model are presently being started by our group.
Acknowledgments
We would like to thank Cristina Fasolato, Paola Pizzo, and Paulo Magalhaes (University of Padua) for helpful discussion. B6.152H mouse lines used for the authors’ research were kindly donated by L. Ozmen and F. Hoffmann—La Roche Ltd. (Basel, Switzerland).
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/cells10082046/s1, Figure S1: Expression of proteins of the ETC and ATP-synthase is comparable between Wt and B6.152H primary cells. Figure S2 Effect of glutamate treatment on WT neuronal culture in the presence of Alisporivir
Author Contributions
Conceptualization, E.B.; methodology, G.R., L.Z. and E.B.; validation, E.B.; formal analysis, G.R. and E.B.; investigation, G.R. and E.B.; data curation, E.B.; writing—original draft preparation, E.B.; writing—review and editing, E.B. and T.P.; visualization, E.B.; supervision, E.B.; project administration, E.B.; funding acquisition, T.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondazione Cassa di Risparmio di Padua e Rovigo (CARIPARO Foundation) Excellence project2017 (2018/113); Euro Bioimaging Project Roadmap/ESFRI from the European Commission, and Telethon Italy Grant GGP16029A to T.P.
Institutional Review Board Statement
All procedures were conducted in accordance with the Italian and European Communities Council Directive on Animal Care 2010/63/EU on the protection of animals used for scientific purposes and were approved by the Italian Ministry of Health (Project code: D2784.N.HEH).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are reported within the article and in the Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jack C.R., Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B., Holtzman D.M., Jagust W., Jessen F., Karlawish J., et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albert M.S., DeKosky S.T., Dickson D., Dubois B., Feldman H.H., Fox N.C., Gamst A., Holtzman D.M., Jagust W.J., Petersen R.C., et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R., Kawas C.H., Klunk W.E., Koroshetz W.J., Manly J.J., Mayeux R., et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperling R.A., Aisen P.S., Beckett L.A., Bennett D.A., Craft S., Fagan A.M., Iwatsubo T., Jack C.R., Kaye J., Montine T.J., et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vermunt L., Sikkes S.A.M., Hout A., Handels R., Bos I., Flier W.M., Kern S., Ousset P., Maruff P., Skoog I., et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019;15:888–898. doi: 10.1016/j.jalz.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardy J.A., Higgins G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson P.T., Alafuzoff I., Bigio E.H., Bouras C., Braak H., Cairns N.J., Castellani R.J., Crain B.J., Davies P., Del Tredici K., et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altmann A., Ng B., Landau S.M., Jagust W.J., Greicius M.D. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain. 2015;138:3734–3746. doi: 10.1093/brain/awv278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards J.G., Higgins G.A., Ouagazzal A.M., Ozmen L., Kew J.N.C., Bohrmann B., Malherbe P., Brockhaus M., Loetscher H., Czech C., et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003;23:8989–9003. doi: 10.1523/JNEUROSCI.23-26-08989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozmen L., Albientz A., Czech C., Jacobsen H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener. Dis. 2008;6:29–36. doi: 10.1159/000170884. [DOI] [PubMed] [Google Scholar]
- 12.Weidling I.W., Swerdlow R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020;330:113321. doi: 10.1016/j.expneurol.2020.113321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bubber P., Haroutunian V., Fisch G., Blass J.P., Gibson G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 14.Kish S.J., Mastrogiacomo F., Guttman M., Furukawa Y., Taanman J.-W., DožicDožic S., Pandolfo M., Lamarche J., DiStefano L., Chang L.-J. Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer’s disease and in hereditary spinocerebellar ataxia disorders: A nonspecific change? J. Neurochem. 1999;72:700–707. doi: 10.1046/j.1471-4159.1999.0720700.x. [DOI] [PubMed] [Google Scholar]
- 15.Manczak M., Calkins M.J., Reddy P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang E.F., Hou Y., Palikaras K., Adriaanse B.A., Kerr J.S., Yang B., Lautrup S., Hasan-Olive M.M., Caponio D., Dan X., et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019;22:401–412. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arsic N., Zacchigna S., Zentilin L., Ramirez-Correa G., Pattarini L., Salvi A., Sinagra G., Giacca M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 2004;10:844–854. doi: 10.1016/j.ymthe.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Rossi A., Galla L., Gomiero C., Zentilin L., Giacca M., Giorgio V., Calì T., Pozzan T., Greotti E., Pizzo P. Calcium signaling and mitochondrial function in presenilin 2 knock-out mice: Looking for any loss-of-function phenotype related to Alzheimer’s disease. Cells. 2021;10:204. doi: 10.3390/cells10020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inagaki K., Fuess S., Storm T.A., Gibson G.A., Mctiernan C.F., Kay M.A., Nakai H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayuso E., Mingozzi F., Montane J., Leon X., Anguela X.M., Haurigot V., Edmonson S.A., Africa L., Zhou S., High K.A., et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- 21.Zentilin L., Giacca M. Competitive PCR for precise nucleic acid quantification. Nat. Protoc. 2007;2:2092–2104. doi: 10.1038/nprot.2007.299. [DOI] [PubMed] [Google Scholar]
- 22.Dana H., Mohar B., Sun Y., Narayan S., Gordus A., Hasseman J.P., Tsegaye G., Holt G.T., Hu A., Walpita D., et al. Sensitive red protein calcium indicators for imaging neural activity. eLife. 2016;5:e12727. doi: 10.7554/eLife.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer A.E., Giacomello M., Kortemme T., Hires S.A., Lev-Ram V., Baker D., Tsien R.Y. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem. Biol. 2006;13:521–530. doi: 10.1016/j.chembiol.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Tomlinson D.R., Gardiner N.J. Glucose neurotoxicity. Nat. Rev. Neurosci. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- 25.Theurey P., Connolly N.M.C., Fortunati I., Basso E., Lauwen S., Ferrante C., Moreira Pinho C., Joselin A., Gioran A., Bano D., et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell. 2019;18:e12924. doi: 10.1111/acel.12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruas J.S., Siqueira-Santos E.S., Amigo I., Rodrigues-Silva E., Kowaltowski A.J., Castilho R.F. Underestimation of the maximal capacity of the mitochondrial electron transport system in oligomycin-treated cells. PLoS ONE. 2016;11:e0150967. doi: 10.1371/journal.pone.0150967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benson D.L., Watkins F.H., Steward O., Banker G. Characterization of GABAergic neurons in hippocampal cell cultures. J. Neurocytol. 1994;23:279–295. doi: 10.1007/BF01188497. [DOI] [PubMed] [Google Scholar]
- 28.Pedersen P.L., Carafoli E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem. Sci. 1987;12:146–150. doi: 10.1016/0968-0004(87)90071-5. [DOI] [Google Scholar]
- 29.Arato-Oshima T., Matsui H., Wakizaka A., Homareda H. Mechanism Responsible for Oligomycin-Induced Occlusion of Na+ within Na/K-ATPase*. [(accessed on 22 June 2021)]; doi: 10.1074/jbc.271.41.25604. Available online: https://reader.elsevier.com/reader/sd/pii/S0021925819783331?token=0F8A6B0F5E2E9CEB6893CBD67E210FC4F611C83A1B80DDDE8E14A8FB5CB21DD6179CA16812213BB9590235C387978DE7&originRegion=eu-west-1&originCreation=20210622170219. [DOI] [PubMed]
- 30.Schägger H., Ohm T.G. Human diseases with defects in oxidative phosphorylation: 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur. J. Biochem. 1995;227:916–921. doi: 10.1111/j.1432-1033.1995.tb20219.x. [DOI] [PubMed] [Google Scholar]
- 31.Beck S.J., Guo L., Phensy A., Tian J., Wang L., Tandon N., Gauba E., Lu L., Pascual J.M., Kroener S., et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016;7:11483. doi: 10.1038/ncomms11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang W.S., Reiman E.M., Valla J., Dunckley T., Beach T.G., Grover A., Niedzielko T.L., Schneider L.E., Mastroeni D., Caselli R., et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA. 2008;105:4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luvisetto S., Pietrobon D., Azzone G.F. Uncoupling of oxidative phosphorylation. 1. Protonophoric effects account only partially for uncoupling. Biochemistry. 1987;26:7332–7338. doi: 10.1021/bi00397a021. [DOI] [PubMed] [Google Scholar]
- 34.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 35.Nunomura A., Perry G., Aliev G., Hirai K., Takeda A., Balraj E.K., Jones P.K., Ghanbari H., Wataya T., Shimohama S., et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 36.Albrecht S.C., Barata A.G., Großhans J., Teleman A.A., Dick T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011;14:819–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Y., Danbolt N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014;121:799–817. doi: 10.1007/s00702-014-1180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes J.R., Mukherjee B., Rogers B.C., Nafar F., Gosse M., Parsons M.P. The relationship between glutamate dynamics and activity-dependent synaptic plasticity. J. Neurosci. 2020;40:2793–2807. doi: 10.1523/JNEUROSCI.1655-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penn A.C., Zhang C.L., Georges F., Royer L., Breillat C., Hosy E., Petersen J.D., Humeau Y., Choquet D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature. 2017;549:384–388. doi: 10.1038/nature23658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Findley C.A., Bartke A., Hascup K.N., Hascup E.R. Amyloid beta-related alterations to glutamate signaling dynamics during Alzheimer’s disease progression. ASN Neuro. 2019;11:1759091419855541. doi: 10.1177/1759091419855541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang Z., Liu F., Iqbal K., Grundke-Iqbal I., Gong C.-X. Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. J. Alzheimer’s Dis. 2009;17:531–539. doi: 10.3233/JAD-2009-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herman M.A., Jahr C.E. Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walsh C., Barrow S., Voronina S., Chvanov M., Petersen O.H., Tepikin A. Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta. 2009;1787:1374–1382. doi: 10.1016/j.bbabio.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Pizzo P., Drago I., Filadi R., Pozzan T. Mitochondrial Ca2+ homeostasis: Mechanism, role, and tissue specificities. Pflug. Arch. 2012;464:3–17. doi: 10.1007/s00424-012-1122-y. [DOI] [PubMed] [Google Scholar]
- 45.Greotti E., Fortunati I., Pendin D., Ferrante C., Galla L., Zentilin L., Giacca M., Kaludercic N., Di Sante M., Mariotti L., et al. mCerulean3-based cameleon sensor to explore mitochondrial Ca2+ dynamics in vivo. iScience. 2019;16:340–355. doi: 10.1016/j.isci.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petronilli V., Cola C., Bernardi P. Modulation of the Mitochondrial Cyclosporin A-Sensitive Permeability Transition Pore. II. The Minimal Requirements for Pore Induction Underscore a Key Role for Transmembrane Electrical Potential, Matrix pH, and Matrix Ca2+—PubMed. [(accessed on 23 June 2021)]; Available online: https://pubmed.ncbi.nlm.nih.gov/7678245/ [PubMed]
- 47.Szabo I., Bernardi P., Zoratti M. Modulation of the mitochondrial megachannel by divalent cations and protons. J. Biol. Chem. 1992;267:2940–2946. doi: 10.1016/S0021-9258(19)50677-9. [DOI] [PubMed] [Google Scholar]
- 48.Sweeney Z.K., Fu J., Wiedmann B. From chemical tools to clinical medicines: Nonimmunosuppressive cyclophilin inhibitors derived from the cyclosporin and sanglifehrin scaffolds. J. Med. Chem. 2014;57:7145–7159. doi: 10.1021/jm500223x. [DOI] [PubMed] [Google Scholar]
- 49.Hansson M.J., Mattiasson G., Månsson R., Karlsson J., Keep M.F., Waldmeier P., Ruegg U.T., Dumont J.-M., Besseghir K., Elmér E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004;36:407–413. doi: 10.1023/B:JOBB.0000041776.31885.45. [DOI] [PubMed] [Google Scholar]
- 50.Yadava N., Nicholls D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchetti P., Fovez Q., Germain N., Khamari R., Kluza J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020;34:13106–13124. doi: 10.1096/fj.202000767R. [DOI] [PubMed] [Google Scholar]
- 52.Maurer I., Zierz S., Möller H.-J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging. 2000;21:455–462. doi: 10.1016/S0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 53.Bosetti F., Brizzi F., Barogi S., Mancuso M., Siciliano G., Tendi E.A., Murri L., Rapoport S.I., Solaini G. Cytochrome c oxidase and mitochondrial F 1 F 0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging. 2002;23:371–376. doi: 10.1016/S0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 54.Rhein V., Song X., Wiesner A., Ittner L.M., Baysang G., Meier F., Ozmen L., Bluethmann H., Dröse S., Brandt U., et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA. 2009;106:20057–20062. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emmerzaal T.L., Rodenburg R.J., Tanila H., Verweij V., Kiliaan A.J., Kozicz T. Age-dependent decrease of mitochondrial complex II activity in a familial mouse model for Alzheimer’s disease. J. Alzheimer’s Dis. 2018;66:75–82. doi: 10.3233/JAD-180337. [DOI] [PubMed] [Google Scholar]
- 56.Strauss M., Hofhaus G., Schröder R.R., Kühlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27:1154–1160. doi: 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lenaz G., Baracca A., Barbero G., Bergamini C., Dalmonte M.E., del Sole M., Faccioli M., Falasca A., Fato R., Genova M.L., et al. Mitochondrial Respiratory Chain Super-Complex I–III in Physiology and Pathology|Elsevier Enhanced Reader. [(accessed on 26 June 2021)]; doi: 10.1016/j.bbabio.2010.01.025. Available online: https://reader.elsevier.com/reader/sd/pii/S0005272810000368?token=B01B254C181842A8A9B92B51BC0C5C1BDF3F23D059E5DD736A23C5023209AAD183CD01E52F954B3E848A9ECA700EB856&originRegion=eu-west-1&originCreation=20210626143920. [DOI] [PubMed]
- 58.Davies K.M., Strauss M., Daum B., Kief J.H., Osiewacz H.D., Rycovska A., Zickermann V., Kuhlbrandt W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. USA. 2011;108:14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chinopoulos C. Mitochondrial consumption of cytosolic ATP: Not so fast. FEBS Lett. 2011;585:1255–1259. doi: 10.1016/j.febslet.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 60.Ong W.-Y., Tanaka K., Dawe G.S., Ittner L.M., Farooqui A.A. Slow excitotoxicity in Alzheimer’s disease. J. Alzheimer’s Dis. 2013;35:643–668. doi: 10.3233/JAD-121990. [DOI] [PubMed] [Google Scholar]
- 61.Lewerenz J., Maher P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 2015;9:469. doi: 10.3389/fnins.2015.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zádori D., Veres G., Szalárdy L., Klivényi P., Vécsei L. Alzheimer’s disease: Recent concepts on the relation of mitochondrial disturbances, excitotoxicity, neuroinflammation, and kynurenines. J. Alzheimer’s Dis. 2018;62:523–547. doi: 10.3233/JAD-170929. [DOI] [PubMed] [Google Scholar]
- 63.Pandya J.D., Nukala V.N., Sullivan P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics. 2013;5:10. doi: 10.3389/fnene.2013.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fagian M.M., da Silva L.P., Vercesi A.E. Inhibition of oxidative phosphorylation by Ca2+ or Sr2+: A competition with Mg2+ for the formation of adenine nucleotide complexes. Biochim. Biophys. Acta Bioenerg. 1986;852:262–268. doi: 10.1016/0005-2728(86)90231-8. [DOI] [PubMed] [Google Scholar]
- 65.Fink B.D., Bai F., Yu L., Sivitz W.I. Regulation of ATP production: Dependence on calcium concentration and respiratory state. Am. J. Physiol. Physiol. 2017;313:C146–C153. doi: 10.1152/ajpcell.00086.2017. [DOI] [PubMed] [Google Scholar]
- 66.Malyala S., Zhang Y., Strubbe J.O., Bazil J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019;15:e1006719. doi: 10.1371/journal.pcbi.1006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernardi P., Rasola A., Forte M., Lippe G. The mitochondrial permeability transition pore: Channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 2015;95:1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duchen M.R. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berndt N., Kann O., Holzhütter H.-G. Physiology-based kinetic modeling of neuronal energy metabolism unravels the molecular basis of NAD(P)H fluorescence transients. J. Cereb. Blood Flow Metab. 2015;35:1494–1506. doi: 10.1038/jcbfm.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vergun O., Keelan J., Khodorov B.I., Duchen M.R. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J. Physiol. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Plotegher N., Filadi R., Pizzo P., Duchen M.R. Excitotoxicity revisited: Mitochondria on the verge of a nervous breakdown. Trends Neurosci. 2021;44:342–351. doi: 10.1016/j.tins.2021.01.001. [DOI] [PubMed] [Google Scholar]
- 72.Boyman L., Williams G.S., Khananshvili D., Sekler I., Lederer W.J. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013;59:205–213. doi: 10.1016/j.yjmcc.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are reported within the article and in the Supplementary Materials.