

Abstract
Changes in the nanopore ionic current during entry of a target molecule underlie the sensing capability and dominate the intensity and extent of applications of the nanopore approach. The volume exclusion model has been proposed and corrected to describe the nanopore current blockage. However, increasing evidence shows nonconformity with this model, suggesting that the ionic current within a nanopore should be entirely reconsidered. Here, we revisit the origin of nanopore current blockage from a theoretical perspective and propose that the noncovalent interactions between a nanopore and a target molecule affect the conductance of the solution inside the nanopore, leading to enhanced current blockage. Moreover, by considering the example of an aerolysin nanopore discriminating the cytosine DNA and methylcytosine DNA that differ by a single methyl group, we completely demonstrate, by nanopore experiments and molecular dynamics simulations, the essential nature of this noncovalent interaction for discrimination. Our conductance model suggests multiplicative effects of both volume exclusion and noncovalent interaction on the current blockage and provides a new strategy to achieve volume difference sensing at the atomic level with highly specific current events, which would promote the nanopore protein sequencing and its applications in real-life systems.
Keywords: Electrochemistry, Nanopore, Ionic current, Noncovalent interaction, Methylcytosine
Introduction
Measuring an ionic current through a single nanoscale channel is at the core of nanopore-based single-molecule sensing.1−3 When a single molecule enters a nanopore, it blocks the ionic current inside the channel, leading to an ionic current change, which is defined as current blockage. This phenomenon endows nanopore technology with a high sensitivity for reading molecules individually and has allowed this technique to be employed in single-molecule DNA sequencing,4,5 peptide/protein sensing,6−8 single-molecule reaction studies,9,10 enzymatic process measurement,11,12 and biomolecule interaction analysis.13 However, due to the lack of current response specificity and resolution, challenges arise when nanopore technology is applied in the detection of biomolecules in real-life samples and the discrimination of 20 amino acids for protein sequencing, respectively.14,15 Therefore, to expend and promote nanopore-based sensing, it is necessary to understand the effect and explore the origin of current blockage. Traditionally, current blockage has been considered to mainly result from the presence of a target molecule inside a nanopore, and this hypothesis led to the proposal of the volume exclusion model.16 Since the geometric structures and charge distributions of the nanopore and the target molecule affect the ion mobility inside the nanopore, further modulating the current blockage, correction terms were introduced to describe the changes in geometry and charge-induced current.17−20 However, it remains challenging to understand and elucidate the mechanism of the discrimination for ultrasmall volume differences and the opposite orders of current blockage and the weight/volume values for the same charged molecules. For example, dTMP, which has a smaller volume, produced an obviously larger current blockage than the dAMP, which has a larger volume, in β-CD-coupled α-hemolysin,4 while the current blockage of dAMP was larger than that of dTMP in MoS2 nanopore.21 Therefore, factors beyond volume, geometry, and charge must affect and even dominate the current blockage of the target molecule in some cases.
Here, based on the volume exclusion model, we discussed the possible electric conductivity change inside a nanopore during target molecule entry from a theoretical perspective. Moreover, we explored the factors affecting nanopore conductance based on the electrolytic conductance theories and further reveal the physical mechanism by which noncovalent interactions between the nanopore and the target molecule influence ion mobility inside the nanopore. Furthermore, taking the discrimination of methylcytosine and cytosine by an aerolysin nanopore as an example, we applied a combination of nanopore experiments and physical theories to exclude all other possible factors, such as volume, ion type, and ion strength, that may dominate the current difference and demonstrate the key role of the interactions between aerolysin and cytosine/methylcytosine. All-atom molecular dynamics (MD) simulations were applied to demonstrate the different interactions induced by cytosine and methylcytosine inside aerolysin and illustrate the interaction details. The results demonstrated a critical region defined as R1 for aerolysin and DNA interactions, and a mechanism by which aerolysin utilizes interaction differences to distinguish cytosine and methylcytosine was suggested, which was further directly verified by the experiments with a series mutant aerolysins.
Results and Discussion
Principle of the Conductance Change Induced by a Target Molecule Inside a Nanopore
A model of a target molecule inducing the current blockage via volume exclusion inside a nanopore has been well established. However, how the noncovalent interactions between the target molecule and amino acid residues within the nanopore lumen regulate the ionic current has been fully unexplored but is disclosed here for the very first time. Previous studies have shown that the amplitude of current blockage (ΔIb) is approximately proportional to the excluded atomic volume as ΔIb = −I0V/Vpore and have further proposed the following equation to estimate the current blockage of an electrically neutral target molecule:16
| 1 |
Where, σ is the solution conductivity inside the nanopore, ψ is the transmembrane potential across the nanopore, h is the effective sensing length of the nanopore beyond physical limitation, V is the volume of the target molecule, and f(dm/Dp, lm/h) corrects the error induced by the relative geometry of the target molecule and the nanopore, with dm, Dp, and lm representing the diameter of the molecule, the diameter of the nanopore, and the length of the molecule, respectively.
In the volume exclusion model, σ is normally treated as a constant. However, σ has been demonstrated to depend on factors such as the solution dielectric constant, solvent viscosity, field strength, and short-range ion related interactions.22 Therefore, the solution conductivity inside a nanopore changes during target molecule entry, suggesting the possibility of noncovalent interactions between the target molecule and amino acid residues within the nanopore lumen, including electrostatic interactions, vdW interactions, and hydrogen bonds, affecting the target molecule-induced current drop.
Since σ can be calculated from σ = Λcc, where Λc is the equivalent conductance and c represents the ion concentration, to further establish the correlation between the noncovalent interactions and current blockage, Λc is considered from the molecular perspective as
| 2 |
where J is the current density and is equal to ∑inieivi, X is the field strength, ni accounts for the ion number inside the effective sensing space of the nanopore, ei is the charge of an ion, and vi is the velocity component along the nanopore. Next, the ways in which electrostatic interactions, vdW interactions, and hydrogen bonds influence these coefficients are discussed individually.
First, the electrostatic interactions between the target molecule and the nanopore are considered. Relative to the charges on the target molecule, on the one hand, ions with the same charge affect the vi via the changed electrostatic potential ui as follows:
| 3 |
Where, ei is the charge of other ions, amino acid residues, and the target molecule, and rji quantifies the distance between ei and ej. The charges on the target molecule enhances the electrostatic potential of ions with the same charge, decreasing their vi. On the other hand, for oppositely charged ions, the electrostatic interactions would also change the quantity of the free ions as follows:
| 4 |
Where, nion-paired accounts for the paired anion/cation, and nion-bounded is the number of ions bound to the target molecule as a result of potentially strong electrostatic interactions between them. Assuming the target molecule can bind a total of a ions and the bonding of each ion is symmetric, nion-bounded can be calculated by
| 5 |
where nmolecule is the number of target molecules (normally one) and ΔG is the Gibbs free energy change. The electrostatic interactions between the target molecule and the nanopore lumen have a considerable effect on ΔG, leading to a change in the number of free ions confined within the nanopore. Therefore, strong electrostatic interactions would affect the current blockage by decreasing both vi and the free ion quantity ∑ini.
Second, the treatment of the dipole is the core of the vdW interactions between the target molecule and the nanopore.23 The vdW interactions includes dispersion forces between instantaneously induced dipoles, Debye forces between permanent dipoles and induced dipoles, and the Keesom force between permanent molecular dipoles. Here, the dipoles of the target molecule and amino acid residues in the nanopore lumen are considered as (+ep) and (−eq) separated by a distance rpq. The value of rpq changes with the formation of an induced dipole and the enhancement of the permanent dipole. Thus, the contribution of these interactions to the electrostatic potential, defined as the dipole-induced potential ui′, can be described as
| 6 |
where rpi and rqi are the distances between ei and ep and between ei and eq, respectively. A strong vdW interaction between the target molecule and the nanopore lumen can induce more instantaneous dipoles and even redistribute (+ep) and (−eq) and increase rpq. Both of these factors can enhance the total electrostatic potential, resulting in a smaller vi and deeper current blockage.
Third, hydrogen bonds between the target molecule and the nanopore are considered. Hydrogen bonds are unique noncovalent interactions that are stronger than vdW and electrostatic interactions but weaker than covalent bonds. Hydrogen bonds are responsible for many of the anomalous physical and chemical properties of compounds. Thus, hydrogen bonds have both covalent and noncovalent natures, making this factor complicated. On the one hand, since nanopores are full of water molecules in single-molecule sensing, the hydrogen-bond network changes upon formation of hydrogen bonds between the nanopore and the target molecule due to their directional control and saturability, thus affecting the ion mobility. On the other hand, the dielectric constant D is also affected by the hydrogen bonds. D actually represents the dipole conditions24 and can be described as
| 7 |
Here, T is the absolute temperature, kB is Boltzmann’s constant, ξ is the volume of the nanopore confinement, and the total dipole moment can be calculated by M(t) = ∑i=1NMi(t). The formation of hydrogen bonds between the target molecule and the nanopore lumen restrict part of the dipole moment and decrease D, further resulting in a higher electrostatic potential for the ions and a lower vi.
Therefore, in principle, the noncovalent interactions between the target molecule and the nanopore can indeed enhance the current blockage inside the nanopore.
Experimental Evidence for the Deeper Current Blockage Induced by Noncovalent Interactions
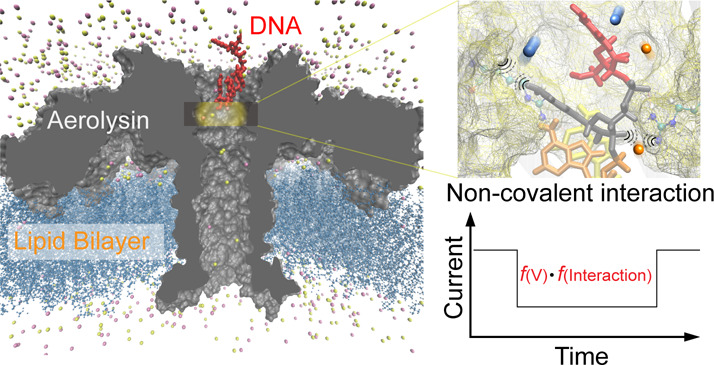
To demonstrate the correlation between noncovalent interactions and current drop enhancement, as shown in Figure 1a–b, the discrimination of methylcytosine by aerolysin nanopore was taken as an example due to the extremely long translocation duration for the ssDNA inside aerolysin (>1000-fold greater than that inside WT α-hemolysin). Notably, this long translocation duration suggests the strong interactions between the ssDNA and the aerolysin lumen.11 Aerolysin is a pore-forming toxin (PFT) from Aeromonas spp. that can self-assemble into a lipid bilayer and organize into circular heptameric pores.25 With many charged amino acid residues inside the lumen, aerolysin can form strong noncovalent interactions with oligonucleotides, which not only prolongs the translocation duration for them, but also results in single nucleotide difference. Furthermore, aerolysin can discriminate a single methyl group within an oligonucleotide.26 As shown in Figure 1c–e, the 4-nt oligonucleotide of mC-A3 (5′-mCAAA-3′) exhibits a 0.82 pA greater current blockage and a 1.36-fold longer translocation duration than that of C-A3 (5′-CAAA-3′) at +60 mV. However, since the volume of a single methyl group is approximately 0.03 nm3, according to eq 1, the estimated extra current drop it induces should be smaller than 0.01 pA in the aerolysin lumen with a volume of 29.52 nm3. Therefore, the solution conductivity σ must change obviously for methylated and normal ssDNA inside aerolysin, as we predicted. It should be noted that when the ssDNA is inside the nanopore, the σ of the position at which ssDNA remains plays the dominant role.
Figure 1.

Transporting C-A3 and mC-A3 through a wild-type aerolysin membrane channel. (a) All-atom model of the full-length aerolysin nanopore system. Aerolysin (gray) was inserted into a lipid bilayer membrane (dark blue), while nucleotides (red) were placed at the entrance of the pore. (b) Structure of methylated and unmethylated oligonucleotides containing methylcytosine and cytosine, respectively. The only difference between them was the addition of a methyl group which is marked in red. (c) Raw single-channel recording traces of ionic current and corresponding typical events of mC-A3 (red) and C-A3 (blue) at +60 mV, respectively. (d) Current histograms of the two individual pure samples at +60 mV. The residual current histograms were fitted to Gaussian function. (e) Translocation duration histograms reflecting the different interactions between pure samples and the pore. The histograms were fitted by a single exponential function with values of 5.5 ± 0.2 ms (mC-A3) and 4.7 ± 0.1 ms (C-A3) at +60 mV. (f) Voltage dependence of the translocation durations (left y-axis) and event frequency (right y-axis) for mC-A3 (red line) and C-A3 (blue line), respectively (for details, see Supplementary Tables S1 and S2). The event frequencies of the pure C-A3 and mC-A3 increased linearly with the applied voltage. All data were performed in 1.0 M KCl, 10 mM Tris, and 1.0 mM EDTA, pH 8.0, at 20 ± 2 °C in the presence of 2.0 μM oligonucleotides.
To further investigate the factors that may be crucial for modulating the conductance σ or the equivalent conductance Λc, the theory of electrolytic conductance was explored. Based on the Debye–Huckel–Onsager theory,22 the theoretical Λc for dilute solutions can be described as
| 8 |
where Λ0 is the limiting conductance at c = 0, ΔX/X is the relaxation term, and ΔΛe measures the electrophoretic countercurrent with
| 9 |
| 10 |
The numerical values of α0 and β0 were predicted to depend only on the solvent properties (dielectric constant D and viscosity η), absolute temperature, and valence type of the electrolyte. Since the valence type of the electrolyte and the absolute temperature inside the nanopore did not change during the measurement, the solution conductivity change could be ascribed to solvent properties and/or ion concentration inside the aerolysin lumen.
The possible change in ion concentration inside the aerolysin lumen for mC-A3 compared to C-A3 is related to the methylation-induced decrease in the solvent volume. Considering the tiny volume of methyl group described above, no obvious change should be induced by the solvent volume. Nevertheless, to exclude this factor from the experimental perspective, Li+, with a smaller volume than K+, was used to achieve a better response to the solvent volume decrease. It should be noted that although the hydrated radius of Li+ (3.82 Å) is larger than that of K+ (3.31 Å), their hydrated radii are quite similar to the radius of the aerolysin lumen, especially in the critical R1 region, which is smaller than 4.0 Å (Figure S1), necessitating the dehydration of these ions to traverse the region when DNA is inside aerolysin. The results show that methyl-DNA induced a smaller extra current drop in LiCl (Figure S2a and Figure S3). This smaller difference in the blockage current between methyl-DNA and DNA in LiCl seems to be caused only by the lower conductivity of LiCl (Figure S4 and Figure S5). To quantify this effect, the methyl group induced current blockage percentage is defined as
| 11 |
where Wc and Wmc are the peak widths of C-A3 and mC-A3, respectively. The results show that the value of R in LiCl (85.7%) is almost equal to that in KCl (87.5%), demonstrating that the change in solvent volume induced by the methyl group is not the key factor for the decrease in σ. In addition, this observation demonstrated that the decrease in σ is independent of the change in the ion-bound volume, considering that Li+ can bind more efficiently to DNA than K+.27 Moreover, even the high ion strengths lead to comparable R values in both LiCl and KCl (Figure S2b–d), which further supports the observation above. Therefore, the stronger noncovalent interactions between mC-A3 and the aerolysin lumen that would modulate solvent properties are expected account for the extra current blockage compared to C-A3.
The longer translocation duration of mC-A3 has been ascribed to the stronger interactions between mC-A3 and aerolysin than between C-A3 and aerolysin. To further prove this, we carried out voltage-dependent experiments (Figure 1f and Tables S1–S2). As shown in Figure 1f, the frequencies of both mC-A3 and C-A3 show a linear voltage dependence. According to a previous study on voltage-dependent frequency,11 this relationship suggests that the capture of mC-A3 and C-A3 by aerolysin is limited by the biased diffusion, suggesting that the most of the translocation duration is inside the aerolysin lumen rather than at the cap. More importantly, mC-A3 experiences an anomalous translocation duration at +60 mV, which indicates a higher translocation energy barrier that requires a larger driving force.28 This high energy barrier only for mC-A3 further supports the stronger interactions between mC-A3 and the aerolysin lumen. Moreover, the fraction of enhanced interaction between aerolysin and mC-A3 contributing to current change was estimated. Since the volume excluded by the target molecule could centralize the ionic current sensing from the whole nanopore to the region where the molecule remains, it would increase the effects of interaction-induced conductance change. Based on eq 1, the blockage current is correlated to the product of the volume exclusion effect and the interaction effect. According to the residual current for mC-A3 and C-A3 as presented in Figure 1c, an extra single methyl group that accounts for approximately 3.16% of the volume of C-A3 induced approximately 6.43% current blockage enhancement. Therefore, compared to C-A3, the enhanced interactions of mC-A3 results in a ∼ 5.63% decrease in conductance (for details, see Experimental Methods). Thus, the methylcytosine-enhanced noncovalent effect corresponds to a current difference of approximately 64.1% between mC-A3 and C-A3, while the extra volume of the methyl group is approximately 35.9%. It should be noted that this fraction is a relative value which represents the relative conductance change induced by the enhancement of noncovalent interactions of mC-A3 compared to C-A3.
Molecular Insight into the Enhanced Noncovalent Interaction Induced by a Methyl Group
To further support the enhanced interactions between mC-A3 and aerolysin during the translocation, an all-atom MD simulation of the aerolysin nanopore system was performed using the program NAMD29 to gain insights into the noncovalent interaction between aerolysin and C-A3/mC-A3. After the equilibration of the aerolysin system (for details, see the Supporting Information, Methods), steered molecular dynamics (SMD) simulations were then performed to pull the oligonucleotides through the pore at a constant velocity of 0.25 Å/ps along the z-axis (Figure S6). The backbone of the oligonucleotides, regarded as SMD atoms, was attached to a dummy atom via a virtual spring. During the whole translocating process, the steering force (SMD force) along with the traverse orientation increased to overcome the dynamic interactions between the oligonucleotide and residues in the aerolysin lumen. As expected, the SMD force increased rapidly when the ssDNA entered the aerolysin pore (Figure 2a), especially in the z-axis regions from 29.8 to 47.4 Å and from −19.4 to −7.6 Å, which are denoted as the R1 region and the R2 region, respectively. Note that both C-A3 and mC-A3 needed a stronger SMD force to be threaded through R1 and R2, which is consistent with our previous finding that the aerolysin has two sensing regions.30 Furthermore, the SMD force applied to mC-A3 became nearly 1.52-fold stronger than that applied to C-A3 when the oligonucleotides traversed the R1 region, while those applied to C-A3 and mC-A3 in the R2 region were comparable. This result reveals a much stronger noncovalent interactions between mC-A3 and the R1 region of aerolysin than that between C-A3 and the R1 region. Furthermore, the slower constant velocity for ssDNA through aerolysin and the different orientations of ssDNA at the entrance of the pore also reveal obviously a stronger interaction than C-A3 experiences when it traverses the R1 region (Figure S7–S8), supporting the importance of the R1 region.
Figure 2.

Nonbonded interaction between mC-A3 (red line) or C-A3 (blue line) and the aerolysin protein. (a) SMD simulations for the translocation of mC-A3 (red line) and C-A3 (blue line) through the pore with a constant velocity of 0.25 Å/ps. (b left) Frames of mC-A3 (red box) and C-A3 (blue box) located near R1. The amino acid residues in R1 are marked in yellow. (right) vdW force (upper) and electrostatic force (lower) exerted on DNA by the aerolysin protein. (c) Decomposed vdW force (upper) and electrostatic force (lower) along the transport orientation of oligomers. Both forces on mC-A3 (red) and C-A3 (blue) affected all amino acid residues in the aerolysin lumen.
To rationalize the importance of the R1 region, two frames shown in Figure 2b with mC-A3 and C-A3 located in the R1 region from the SMD simulation were further simulated. In these two frames, the centers of mass for mC-A3 and C-A3 are both at position 35.2 Å in the R1 region. Then, the MD simulations were carried out in the NPT ensemble for 20 ns with applying a harmonics constraint to restrain the backbone of C-A3 or mC-A3. Nonbonded interactions were calculated to describe the effects of the R1 region on mC-A3 and C-A3, respectively. Nonbonded interactions are generally divided into two components: vdW interactions and electrostatic interactions. Figure 2c shows that mC-A3 experiences an ∼9.0 kcal mol–1 Å–1 stronger vdW force with the R1 region than does C-A3, while the mean value of the electrostatic force on mC-A3 is comparable to that on C-A3. Moreover, the nonbonded force, the resultant of vdW and electrostatic forces on mC-A3, is indeed 1.35-fold stronger than those on C-A3 (Figure S9), which suggests a potential correlation with the 1.36-fold longer duration for mC-A3. To further investigate the effects of nonbonded interaction on translocation duration, we consider the direction of the forces (Figure 2d). The vdW force exerted on mC-A3 is prone to repel its translocation, while the electrostatic force is apt to repel the translocation of C-A3, demonstrating that the nonbonded interaction indeed prolongs the translocation duration. In addition to the nonbonded interactions, hydrogen bonds have also been considered. The geometry criterion was used to define the hydrogen bonds based on the coordinates of all the atoms in the frames from the above simulation. As shown in Figure S10, mC-A3 exhibits an obvious tendency to form hydrogen bonds with the R1 region during the whole simulation, and mC-A3 is prone to form more than one hydrogen bond with the R1 region at one time. These above results strongly confirm that mC-A3 exhibits a strong noncovalent interaction with the R1 region, leading to a long duration and high blockage amplitude. These distinct noncovalent interactions for mC-A3 and C-A3 would affect the ion pathway inside the aerolysin, no matter for K+ or Cl– (Figure S11). The heterogeneous ion density along the aerolysin lumen during mC-A3 and C-A3 inside further leads to the z-axis dependent electrostatic potential (Figure S12), which shows a higher potential gradient in R1 region for mC-A3 occupied aerolysin, demonstrating the more ionic flow blocked by mC-A3.
Considering that every amino acid residue participates in unique interactions with DNA, we sought insight into the interaction within the R1 region. In terms of the double β-barrel structure of the aerolysin transmembrane region (Figure 3a), the contributions of three pairs of individual sites of amino acid residues in the R1 region were studied: (I) T218 and S278 (Figure 3b); (II) R220 and S276 (Figure 3c); and (III) D222 and T274 (Figure 3d). As shown in Figure 3b, Pair I near the z-axis position of 43.8 Å is inclined to interact with C-A3 or mC-A3 through vdW forces rather than electrostatic forces. Moreover, the strengths of the vdW force and electrostatic force between T218 and mC-A3 are valued as high as 7.6 and 5.6 kcal mol–1 Å–1, respectively, while the strengths of these forces between T218 and C-A3 remain lower than 2.0 kcal mol–1 Å–1 during the whole simulation. Therefore, mC-A3 experiences a stronger interaction with T218. For S278, its vdW forces and electrostatic forces for both C-A3 and mC-A3 could reach strengths greater than 9.0 kcal mol–1 Å–1. This result suggests that S278 experiences relatively strong nonbonded interactions with C-A3 and mC-A3, but this force is not responsible for the distinct current blockage. Surprisingly, in Pair II, the MD results for C-A3 and mC-A3 show the strongest nonbonded interactions with R220 among all the sites in R1 (Figure 3c). Compared to the entire R1 region, R220 possesses comparable values and fluctuations for both vdW and electrostatic forces. Therefore, R220 dominates the nonbonded interaction between R1 and oligonucleotides. This result supports the key role of R220 in aerolysin nanopore sensing as observed experimentally.11 More importantly, it indicates that the sensing capability of aerolysin is assisted by other amino acid residues in R1 rather than being determined only by the spot R220. In contrast, S276 in Pair II, which is located near the z-axis position of 37.0 Å, hardly interacts with C-A3 and mC-A3 by either vdW or electrostatic forces (Figure 3c). At the bottom of the R1 region, D222 and T274 (Pair III), the electrostatic force experienced by D222 (∼18 kcal mol–1 Å–1) is stronger than the vdW force with the oligonucleotides (Figure 3d). However, T274, as another site in Pair III, experiences a much stronger vdW force and electrostatic force with mC-A3 than with C-A3.
Figure 3.

Nonbonded pair interaction between oligonucleotides and amino acid residues in the R1 region of the aerolysin lumen. (a) Double β-barrel structure of the one chain in the aerolysin R1 region. (b–d) vdW force (left) and electrostatic force (right) between mC-A3 (red line) or C-A3 (blue line) and Pair I (T218 and S278) (b), Pair II (R220 and S276) (c), and Pair III (D222 and T274) (d).
The individual amino acid residues in the R1 region play a similar role in the hydrogen bonds (Table S3) as in the nonbonded interactions described above. R220 is the major residue involved in the hydrogen bonds with C-A3, with an occupancy of 61.0%, and with mC-A3, with an occupancy of 115.3%, where more than one hydrogen bond could be formed. More interestingly, the hydrogen bonds formed by T274 can only be observed in mC-A3 simulation with an occupancy of 37.1%, which agrees with the result that T274 forms a much stronger nonbonded interaction with mC-A3 than C-A3. In contrast, D222, which could obviously interact with C-A3 and mC-A3 via nonbonded interactions, hardly formed hydrogen bonds with them during the simulation. Therefore, the simulation results demonstrate the stronger noncovalent interactions between the aerolysin lumen and mC-A3 and further suggest the critical role of the R1 region of aerolysin in interaction-enhanced methylcytosine sensing. The results support the following detailed interaction-enhanced mechanism in the R1 region: (1) R220 and D222 participate in strong noncovalent interactions with both mC-A3 and C-A3, holding them in the R1 region with an efficient long resident time; (2) T218 and T274 in the R1 region interact with the functional groups of mC-A3 and C-A3 and exert an obviously stronger noncovalent force on mC-A3 than on C-A3, leading to the enhanced current blockages of mC-A3.
To further verify this interaction-enhanced mechanism of aerolysin, mutations of the critical amino acids were performed to modify the noncovalent interactions with the oligonucleotides. T274 was demonstrated to form the most distinct noncovalent interactions with mC-A3 and C-A3 either via nonbonded interactions or hydrogen bonds among the amino acids in R1, as presented in Figure 3b–d and Table S3, and is considered as the key factor for mC-A3 discrimination. Therefore, T274 is taken as an example to evaluate the importance of nonbonded interactions and hydrogen bonds by mutating to serine (S) or valine (V), which replaces the methyl group and hydroxyl group of the threonine residue, respectively (Figure S15a). As presented in Figures S13 and S14, the T274S and T274 V could be expressed and purified, resulting in a stable open pore current. As shown in Figures S15b and S15c, both of these two mutant aerolysins could also distinguish mC-A3 from C-A3; however, the 34.1% and 57.1% separation achieved in T274S and T274V mutant aerolysin, respectively, was obviously lower than that in WT aerolysin (87.5%) (Figure S16). These results demonstrated that the nonbonded interactions and hydrogen bonds formed by T274 in the R1 region of aerolysin are both essential for methylcytosine determination. Moreover, to further evaluate the volume exclusion factor, we replaced the threonine with tryptophan (W), which has the largest volume of the 20 natural amino acids. As we expected, T274W aerolysin showed the smallest open pore current among WT aerolysin and T274 mutations (Figure S14); however, it could not distinguish mC-A3 from C-A3 as shown in Figure S15d. This observation further supports the results described above that the volume exclusion is not the key factor for methylcytosine discrimination in aerolysin.
Conclusion
In summary, while the blockage current inside a nanopore is mainly predicted by the volume exclusion model, we revisited the origin of the blockage current induced by the target molecule. Focusing on the solution conductance change inside the nanopore, we first demonstrated the ability of the interaction between the target molecule and the amino acid residues within the nanopore lumen to modulate the nanopore ionic current. In the theoretical model, the noncovalent interactions could modulate the ionic current mainly in the following three ways: (1) the electrostatic interactions affect the ion mobility and the free ion quantity inside the lumen; (2) the vdW interactions introduce dipole induced electrostatic potential to influence ion mobility; and (3) the hydrogen bonds have an enormous influence on the dipole moment and further lead to the change in dielectric constant inside the nanopore. Actually, the volume excluded by the target molecule could centralize the ionic current sensing from the whole nanopore to the region in which the molecule remains, which would help to increase the effects of interaction-induced changes in conductance. Therefore, the blockage current is correlated to the product of the volume exclusion effect and the interaction effect. Using an aerolysin nanopore that can discriminate methylcytosine from cytosine as a model, we explored all potential factors for inducing methyl group enhanced current blockage. The experimental results suggested that the enhanced current blockage could be ascribed to the interaction between methylcytosine and the amino acid residues in the aerolysin lumen, while the volume, ion concentration, and ion type were excluded. Moreover, the separation of mC-A3 and C-A3 remained at 85.7% ∼ 105.3% in 1.0 to 3.0 M KCl and LiCl solutions, further demonstrating that the current blockage difference is also independent of the ion-bound volume change. Furthermore, the MD simulation results confirmed the enhanced interaction between mC-A3 and aerolysin, and further illustrated the mechanism of the critical sensing region interacting with C-A3 and mC-A3. Currently, many studies are devoted to exploring the nanopores with narrower sensing points to achieve higher spatial resolution based on the volume exclusion model. However, the origin of current blockage revealed here indicates a new sensing principle and strategy to utilize interactions to realize high temporal and spatial resolution, which could be used for ultrasmall volume differences and enantiomer and isomer discrimination, such as post-translational modification and chiral molecules.
Experimental Methods
Nanopore Experiments
Materials and Reagents
1,2-Diphytanoyl-sn-glycero-3-phosphocholine was obtained from Avanti Polar Lipids (Alabaster, AL, USA). DNA was synthesized and purified by Sangon Biotech (Shanghai, China). WT proaerolysin was purified and activated at Nanjing University (Nanjing, China). The Decane and trypsin-EDTA were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Single-Molecule Assays
A Delrin bilayer cup (Warner Instruments, Hamden, CT, USA) was used to perform the experiments. DMPC solution was spread across a 50 μm orifice in the Delrin partition. WT or mutant aerolysin monomers were added into the grounded cis chamber to form a heptameric pore. A pair of Ag/AgCl electrodes were used to apply appropriate potentials across the membrane. Oligonucleotides (mC-A3 or C-A3) were added into the cis chamber for a final concentration of 5 μM which for the purpose of a high-speed collection for a large amount of data. All of the experiments were conducted by premixing DNA samples and electrolyte solution before the formation of a single aerolysin pore. Currents were recorded using an Axopatch 200B patch-clamp amplifier (Molecular Devices, Sunnyvale, CA, USA) coupled with a Digidata 1440A A/D converter (Molecular Devices, USA). The signals were filtered at a frequency of 5 kHz and acquired with Clampex 10.4 software (Molecular Devices, USA) at a sampling frequency of 100 kHz.
Data Analysis
To remove the collision events, a reduction of events in durations <0.2 ms was required. Data were analyzed with MOSAIC31,32 and OriginLab 8.0 (OriginLab Corporation, Northampton, MA, USA).
Mutation, Expression, and Purification of Proaerolysin
The construction of recombinant proaerolysin plasmids refers to our previous work.33−35 The primers used for plasmid mutation are as follows: T274S, 5′- GGTAGTACCAGTACCAGTCTGAGTCAGAGCG-3′ (forward) and 5′-CAGACTGGTACTGGTACTACCACCGTTTTG-3′ (reverse); T274V, 5′-GGTAGTACCGTCACCAGTCTGAGTCAGAGCG-3′ (forward) and 5′-CAGACTGGTGACGGTACTACCACCGTTTTG-3′ (reverse); T274W, 5′-GGTAGTACCTGGACCAGTCTGAGTCAGAGCG-3′ (forward) and 5′-CAGACTGGTCCAGGTACTACCACCGTTTTG-3′ (reverse), respectively. The mutant expression vectors were constructed by the site-directed mutagenesis with high-fidelity PCR enzymes (Invitrogen, 12368010). The PCR cycling conditions and the expression and purification processes of proaerolysin refer to our previous studies.33−36
Molecular Dynamics Simulations
Model Construction
The wild-type aerolysin cryo-EM structure coordinates were obtained from Protein Data Bank entry 5JZT.37 The aerolysin was first solvated by the SOLVATE program38 to produces a 3-Å-think shell of water around the aerolysin using 8 Gaussians to approximate the protein surface. The resulting structure was merged with a 200 Å x 200 Å path of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) lipid bilayer, aligned to the xy-plane with the hydrophobic β-barrel of aerolysin embedded in the bilayer while its hydrophilic cap protruded above the membrane. All the lipid and water molecules overlapping with the protein nanopore were removed. The nanopore–lipid system was solvated with pre-equilibrated TIP3P water molecules39 using VMD.40 K+ and Cl– ions were added to the solution to neutralize the aerolysin system and achieve a concentration of 1.0 M. A ssDNA of 5′-CAAA-3′ was derived from a model of double-stranded DNA, created with the 3D-DART web server,41 while 5′-mCAAA-3′ was generated by patching a methyl group to cytosine of 5′-CAAA-3′ described by the CHARMM36 force field.42
System Equilibrium
All MD simulations were performed using the program NAMD29 with the visualization and analysis by VMD.40 The aerolysin protein and oligonucleotide molecule were described by the CHARMM36 force field.42 After a 5000-step minimization, the aerolysin nanopore system was equilibrated in the NPT ensemble at 295 K and 1 atm for 2 ns with the heavy atoms (non-hydrogen) of protein restrained and allowing the relaxation of lipid, water, ions, and protein hydration atoms. Afterward, the alpha carbons of the protein were still restrained, and the equilibration simulation lasted 10 ns. Next, all the restraints were removed, and the system was relaxed for 60 ns. The 5′-CAAA-3′ and 5′-mCAAA-3′ were also equilibrated in NPT ensemble for 20 ns at 295 K in a 40 Å × 40 Å × 40 Å water box at 1.0 M KCl. The integration time step chosen was 2 fs. All NPT ensemble simulations were carried out with a Langevin piston for pressure control and Langevin dynamics for temperature control.43 Periodic boundary conditions were employed in all directions. van der Waals energies were calculated using a 12 Å cutoff and the particle-mesh-Ewald (PME) method was used to treat long-range electrostatics.44
Constant-Velocity SMD and MD Simulations
Steered molecule dynamic simulation was used to pull ssDNA through the aerolysin nanopore. The oligonucleotide was put at the entrance of aerolysin with a certain position (−10.0, 0.0, 65.0) for the mass center of 5′-CAAA-3′ and 5′-mCAAA-3′. The initial conformations of oligonucleotides were determined by the equilibrium of NPT ensemble and the translocation was initiated by its 3′ end. The SMD atoms, backbone of oligomers, are attached to a dummy atom via a virtual spring with a spring constant of 7.0 kcal mol–1 Å–2. The dummy atom is moved to traverse the pore from cis to trans with a constant velocity of 0.25 Å ps–1.
For the study of noncovalent interaction between oligonucleotides and aerolysin protein, two frames, selected from the trajectories of C-A3 and mC-A3 in SMD simulation with the center of mass for the two oligonucleotides both at position 35.2 Å, were used to perform MD simulation for 20 ns. Harmonic constraints were imposed on the backbone of the oligomers to restrain the oligonucleotides around R1 region. Based on the trajectories from MD simulation, the NAMD energy plugin in VMD was used to calculate the nonbonded interaction between oligonucleotides with the whole aerolysin protein or individual amino acid residues in the R1 region using a 12 Å cutoff for each snapshot of the MD trajectory. Hydrogen bonds between the oligonucleotides and aerolysin protein were calculated with the donor–acceptor distance (H···A distance) shorter than 3.0 Å and the H–D···A angle lower than 20°. The hydrogen bonds formed between the two oligonucleotides and amino acid residues in the R1 region are counted in each frame from the last 10 ns MD simulation.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (22027806, 22090050), the China Postdoctoral Science Foundation (2020M671429), the Excellent Research Program of Nanjing University (ZYJH004), and the Fundamental Research Funds for the Central Universities (14380239). We thanked Prof. Hu Qiu for the discussions about the MD simulations and Prof. Todd C. Sutherland for polishing the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00109.
Supporting methods. Figures S1–S16: supplementary schemes and experimental results. Table S1: mean values of event frequency for mC-A3 and C-A3 at different applied potentials. Table S2: mean values of duration for mC-A3 and C-A3 at different applied potentials. Table S3: hydrogen bonds between the two oligonucleotides and amino acid residues in the R1 region of aerolysin (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kasianowicz J. J.; Brandin E.; Branton D.; Deamer D. W. Characterization of Individual Polynucleotide Molecules Using a Membrane Channel. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (24), 13770–13773. 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deamer D.; Akeson M.; Branton D. Three Decades of Nanopore Sequencing. Nat. Biotechnol. 2016, 34 (5), 518–524. 10.1038/nbt.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W.; Friedman A. K.; Baker L. A. Nanopore Sensing. Anal. Chem. 2017, 89 (1), 157–188. 10.1021/acs.analchem.6b04260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J.; Wu H.-C.; Jayasinghe L.; Patel A.; Reid S.; Bayley H. Continuous Base Identification for Single-Molecule Nanopore DNA Sequencing. Nat. Nanotechnol. 2009, 4 (4), 265–270. 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- Manrao E. A.; Derrington I. M.; Laszlo A. H.; Langford K. W.; Hopper M. K.; Gillgren N.; Pavlenok M.; Niederweis M.; Gundlach J. H. Reading DNA at Single-Nucleotide Resolution with a Mutant MspA Nanopore and Phi29 DNA Polymerase. Nat. Biotechnol. 2012, 30 (4), 349–353. 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland T. C.; Long Y.-T.; Stefureac R.-I.; Bediako-Amoa I.; Kraatz H.-B.; Lee J. S. Structure of Peptides Investigated by Nanopore Analysis. Nano Lett. 2004, 4 (7), 1273–1277. 10.1021/nl049413e. [DOI] [Google Scholar]
- Rosen C. B.; Rodriguez-Larrea D.; Bayley H. Single-Molecule Site-Specific Detection of Protein Phosphorylation with a Nanopore. Nat. Biotechnol. 2014, 32 (2), 179–181. 10.1038/nbt.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galenkamp N. S.; Biesemans A.; Maglia G. Directional Conformer Exchange in Dihydrofolate Reductase Revealed by Single-Molecule Nanopore Recordings. Nat. Chem. 2020, 12 (5), 481–488. 10.1038/s41557-020-0437-0. [DOI] [PubMed] [Google Scholar]
- Luchian T.; Shin S.; Bayley H. Single-Molecule Covalent Chemistry with Spatially Separated Reactants. Angew. Chem., Int. Ed. 2003, 42 (32), 3766–3771. 10.1002/anie.200351313. [DOI] [PubMed] [Google Scholar]
- Qing Y.; Ionescu S. A.; Pulcu G. S.; Bayley H. Directional Control of a Processive Molecular Hopper. Science 2018, 361 (6405), 908–912. 10.1126/science.aat3872. [DOI] [PubMed] [Google Scholar]
- Cao C.; Ying Y.-L.; Hu Z.-L.; Liao D.-F.; Tian H.; Long Y.-T. Discrimination of Oligonucleotides of Different Lengths with a Wild-Type Aerolysin Nanopore. Nat. Nanotechnol. 2016, 11 (8), 713–718. 10.1038/nnano.2016.66. [DOI] [PubMed] [Google Scholar]
- Tan C. S.; Riedl J.; Fleming A. M.; Burrows C. J.; White H. S. Kinetics of T3-DNA Ligase-Catalyzed Phosphodiester Bond Formation Measured Using the α-Hemolysin Nanopore. ACS Nano 2016, 10 (12), 11127–11135. 10.1021/acsnano.6b05995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornblower B.; Coombs A.; Whitaker R. D.; Kolomeisky A.; Picone S. J.; Meller A.; Akeson M. Single-Molecule Analysis of DNA-Protein Complexes Using Nanopores. Nat. Methods 2007, 4 (4), 315–317. 10.1038/nmeth1021. [DOI] [PubMed] [Google Scholar]
- Thakur A. K.; Movileanu L. Real-Time Measurement of Protein-Protein Interactions at Single-Molecule Resolution Using a Biological Nanopore. Nat. Biotechnol. 2019, 37 (1), 96–101. 10.1038/nbt.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressiot B.; Bacri L.; Pelta J. The Promise of Nanopore Technology: Advances in the Discrimination of Protein Sequences and Chemical Modifications. Small Methods 2020, 4, 2000090. 10.1002/smtd.202000090. [DOI] [Google Scholar]
- Talaga D. S.; Li J. Single-Molecule Protein Unfolding in Solid State Nanopores. J. Am. Chem. Soc. 2009, 131 (26), 9287–9297. 10.1021/ja901088b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner J. E.; Kasianowicz J. J.; Nablo B. J.; Robertson J. W. F. Theory for Polymer Analysis Using Nanopore-Based Single-Molecule Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (27), 12080–12085. 10.1073/pnas.1002194107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. C.; Kannam S. K.; Harrer S.; Downton M. T.; Moore S.; Wagner J. M. Geometric Dependence of the Conductance Drop in a Nanopore Due to a Particle. Phys. Rev. E 2014, 89 (4), 042702. 10.1103/PhysRevE.89.042702. [DOI] [PubMed] [Google Scholar]
- Si W.; Aksimentiev A. Nanopore Sensing of Protein Folding. ACS Nano 2017, 11 (7), 7091–7100. 10.1021/acsnano.7b02718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J.; Sarthak K.; Si W.; Gao L.; Aksimentiev A. Rapid and Accurate Determination of Nanopore Ionic Current Using a Steric Exclusion Model. Acs Sensors 2019, 4 (3), 634–644. 10.1021/acssensors.8b01375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Liu K.; Bulushev R. D.; Khlybov S.; Dumcenco D.; Kis A.; Radenovic A. Identification of Single Nucleotides in MoS2 Nanopores. Nat. Nanotechnol. 2015, 10 (12), 1070–1076. 10.1038/nnano.2015.219. [DOI] [PubMed] [Google Scholar]
- Fuoss R. M. Review of the Theory of Electrolytic Conductance. J. Solution Chem. 1978, 7 (10), 771–782. 10.1007/BF00643581. [DOI] [Google Scholar]
- Tschumper G. S. Reviews in Computational Chemistry, Volume 26. Rev. Comp Ch 2009, 39–90. 10.1002/9780470399545.ch2. [DOI] [Google Scholar]
- Neumann M. The Dielectric Constant of Water. Computer Simulations with the MCY Potential. J. Chem. Phys. 1985, 82 (12), 5663–5672. 10.1063/1.448553. [DOI] [Google Scholar]
- Parker M. W.; Buckley J. T.; Postma J. P. M.; Tucker A. D.; Leonard K.; Pattus F.; Tsernoglou D. Structure of the Aeromonas Toxin Proaerolysin in Its Water-Soluble and Membrane-Channel States. Nature 1994, 367 (6460), 292–295. 10.1038/367292a0. [DOI] [PubMed] [Google Scholar]
- Yu J.; Cao C.; Long Y.-T. Selective and Sensitive Detection of Methylcytosine by Aerolysin Nanopore under Serum Condition. Anal. Chem. 2017, 89 (21), 11685–11689. 10.1021/acs.analchem.7b03133. [DOI] [PubMed] [Google Scholar]
- Kowalczyk S. W.; Wells D. B.; Aksimentiev A.; Dekker C. Slowing down DNA Translocation through a Nanopore in Lithium Chloride. Nano Lett. 2012, 12 (2), 1038–1044. 10.1021/nl204273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton M. F.; Discala F.; Bacri L.; Foster D.; Pelta J.; Oukhaled A. Exploration of Neutral Versus Polyelectrolyte Behavior of Poly(Ethylene Glycol)s in Alkali Ion Solutions Using Single-Nanopore Recording. J. Phys. Chem. Lett. 2013, 4 (13), 2202–2208. 10.1021/jz400938q. [DOI] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C.; Li M.-Y.; Cirauqui N.; Wang Y.-Q.; Dal Peraro M.; Tian H.; Long Y.-T. Mapping the Sensing Spots of Aerolysin for Single Oligonucleotides Analysis. Nat. Commun. 2018, 9 (1), 2823. 10.1038/s41467-018-05108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balijepalli A.; Ettedgui J.; Cornio A. T.; Robertson J. W. F.; Cheung K. P.; Kasianowicz J. J.; Vaz C. Quantifying Short-Lived Events in Multistate Ionic Current Measurements. ACS Nano 2014, 8 (2), 1547–1553. 10.1021/nn405761y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstater J. H.; Briggs K.; Robertson J. W. F.; Ettedgui J.; Marie-Rose O.; Vaz C.; Kasianowicz J. J.; Tabard-Cossa V.; Balijepalli A. MOSAIC: A Modular Single-Molecule Analysis Interface for Decoding Multistate Nanopore Data. Anal. Chem. 2016, 88 (23), 11900–11907. 10.1021/acs.analchem.6b03725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-Q.; Cao C.; Ying Y.-L.; Li S.; Wang M.-B.; Huang J.; Long Y.-T. Rationally Designed Sensing Selectivity and Sensitivity of an Aerolysin Nanopore via Site-Directed Mutagenesis. Acs Sensors 2018, 3 (4), 779–783. 10.1021/acssensors.8b00021. [DOI] [PubMed] [Google Scholar]
- Wang Y.-Q.; Li M.-Y.; Qiu H.; Cao C.; Wang M.-B.; Wu X.-Y.; Huang J.; Ying Y.-L.; Long Y.-T. Identification of Essential Sensitive Regions of the Aerolysin Nanopore for Single Oligonucleotide Analysis. Anal. Chem. 2018, 90 (13), 7790–7794. 10.1021/acs.analchem.8b01473. [DOI] [PubMed] [Google Scholar]
- Wu X.-Y.; Wang M.-B.; Wang Y.-Q.; Li M.-Y.; Ying Y.-L.; Huang J.; Long Y.-T. Precise Construction and Tuning of an Aerolysin Single-Biomolecule Interface for Single-Molecule Sensing. CCS Chem. 2019, 1 (3), 304–412. 10.31635/ccschem.019.20180025. [DOI] [Google Scholar]
- Lu S.; Wu X.; Li M.; Ying Y.; Long Y. Diversified Exploitation of Aerolysin Nanopore in Single-molecule Sensing and Protein Sequencing. View 2020, 1 (4), 20200006. 10.1002/VIW.20200006. [DOI] [Google Scholar]
- Iacovache I.; De Carlo S.; Cirauqui N.; Dal Peraro M.; van der Goot F. G.; Zuber B. Cryo-EM Structure of Aerolysin Variants Reveals a Novel Protein Fold and the Pore-Formation Process. Nat. Commun. 2016, 7 (1), 12062. 10.1038/ncomms12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmüller H.; Groll V.. Solvate 1.0.1; 1996. –2010; http://www.mpibpc.mpg.de/grubmueller/solvate.
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- van Dijk M.; Bonvin A. M. J. J. 3D-DART: A DNA Structure Modelling Server. Nucleic Acids Res. 2009, 37 (suppl_2), W235–W239. 10.1093/nar/gkp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103 (11), 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- Batcho P. F.; Case D. A.; Schlick T. Optimized Particle-Mesh Ewald/Multiple-Time Step Integration for Molecular Dynamics Simulations. J. Chem. Phys. 2001, 115 (9), 4003–4018. 10.1063/1.1389854. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.