

Abstract
Background
The immune and inflammatory responses play a considerable role in left ventricular remodeling after myocardial infarction (MI). Binding of AhR (aryl hydrocarbon receptor) to its ligands modulates immune and inflammatory responses; however, the effects of AhR in the context of MI are unknown. Therefore, we evaluated the potential association between AhR and MI by treating mice with a nontoxic endogenous AhR ligand, ITE (2‐[1’H‐indole‐3’‐carbonyl]‐thiazole‐4‐carboxylic acid methyl ester). We hypothesized that activation of AhR by ITE in MI mice would boost regulatory T‐cell differentiation, modulate macrophage activity, and facilitate infarct healing.
Methods and Results
Acute MI was induced in C57BL/6 mice by ligation of the left anterior descending coronary artery. Then, the mice were randomized to daily intraperitoneal injection of ITE (200 µg/mouse, n=19) or vehicle (n=16) to examine the therapeutic effects of ITE during the postinfarct healing process. Echocardiographic and histopathological analyses revealed that ITE‐treated mice exhibited significantly improved systolic function (P<0.001) and reduced infarct size compared with control mice (P<0.001). In addition, we found that ITE increased regulatory T cells in the mediastinal lymph node, spleen, and infarcted myocardium, and shifted the M1/M2 macrophage balance toward the M2 phenotype in vivo, which plays vital roles in the induction and resolution of inflammation after acute MI. In vitro, ITE expanded the Foxp3+ (forkhead box protein P3‐positive) regulatory T cells and tolerogenic dendritic cell populations.
Conclusions
Activation of AhR by a nontoxic endogenous ligand, ITE, improves cardiac function after MI. Post‐MI mice treated with ITE have a significantly lower risk of developing advanced left ventricular systolic dysfunction than nontreated mice. Thus, the results imply that ITE has a potential as a stimulator of cardiac repair after MI to prevent heart failure.
Keywords: 2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester, aryl hydrocarbon receptor, dendritic cells, myocardial infarction, regulatory T cells
Subject Categories: Heart Failure, Myocardial Infarction, Remodeling, Coronary Artery Disease
Nonstandard Abbreviations and Acronyms
- AhR
aryl hydrocarbon receptor
- DCs
dendritic cells
- Foxp3+
forkhead box protein P3‐positive
- ITE
2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester
- mDCs
mature dendritic cells
- mdLNs
mediastinal lymph nodes
- tDCs
tolerogenic dendritic cells
- Tregs
regulatory T cells
Clinical Perspective
What Is New?
We provide in vivo evidence of AhR (aryl hydrocarbon receptor) activation as a novel antiremodeling strategy in suppressing adverse remodeling of myocardial infarction.
Systemic administration of an endogenous AhR ligand induced regulatory T cell increases in the mediastinal lymph node, spleen, and postinfarct myocardium, which brought about earlier M1 to M2 macrophage transition and favorable remodeling in a preclinical model of myocardial infarction.
What Are the Clinical Implications?
Our findings suggest that AhR activation can successfully modulate postinfarct inflammation and improve left ventricular function via the control of regulatory T cells and subsequently a better immune environment for healing.
Short‐term administration and activation of AhR by a nontoxic endogenous ligand can be a novel therapeutic strategy for the prevention of heart failure in humans after myocardial infarction.
Acute myocardial infarction (MI) leads to cardiac dysfunction and, ultimately, ischemic heart failure (HF), which is associated with increased morbidity and mortality.1, 2 Although the probability of survival after MI has gradually increased in recent years because of advanced treatments, patients with acute MI have an increased risk of secondary events such as HF.3 Cardiac remodeling is important to prevent progression to HF after MI. Although structural and functional cardiac remodeling is multifactorial, inflammation and immune responses are critical components. Current clinical and experimental studies support an additional critical role for inflammation after MI.4, 5, 6 MI triggers a massive inflammatory response related to the infiltration of immune cells into the damaged myocardium. This phase involves reparative pathways necessary for cardiac healing.7 However, unbalanced or excessive inflammation contributes to the aggravation of adverse ventricular remodeling and impaired left ventricular function and structure. Progressive remodeling increases wall stress and loss of viable myocardium. Exposure of the heart to pathophysiological stress after MI induces cardiomyocyte hypertrophy and fibrosis.8 Cardiomyocyte hypertrophy during cardiac remodeling is characterized by alterations in cell size associated with sarcomeric and constitutive protein synthesis and changes in cardiac gene expression. These observations indicate that restriction of cardiac inflammation is important in repair of ischemic injury, and that persistent inflammatory signaling is also essential for healing. A recent approach with the interleukin‐1β inhibitor, canakinumab, as anti‐inflammatory therapy significantly diminished adverse serious cardiovascular events and hospitalization for HF in patients with prior MI, suggesting the potential of this inhibitor in anti‐inflammatory therapies as an antiremodeling remedy after MI.9, 10 We aimed to develop a novel therapeutic approach to prevent postinfarct HF.
AhR (aryl hydrocarbon receptor), which belongs to the subfamily of the bHLH/PAS (basic helix‐loop‐helix/Per‐ARNT‐Sim) family, is a transcription factor that regulates adaptive metabolism and environmental responses.11 AhR is activated by xenobiotic chemicals, such as 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, and leads to a broad spectrum of biological and immunotoxicological effects.12, 13 Several studies have shown that AhR plays a significant role in the immune system, leading to the mobilization and recruitment of regulatory T cells (Tregs) that promote tolerance in autoimmune and inflammatory diseases.14, 15, 16 In particular, AhR leads to upregulated Treg‐specific gene expression and inhibits the expression of specific genes associated with effector T‐cell function.17 An endogenous nontoxic ligand of AhR, ITE (2‐[1’H‐indole‐3’‐carbonyl]‐thiazole‐4‐carboxylic acid methyl ester), suppresses experimental autoimmune encephalomyelitis by promoting the production of functional Foxp3+ (forkhead box protein P3‐positive) Tregs and tolerogenic dendritic cells (tDCs).18 Dendritic cells (DCs), the most potent type of antigen‐presenting cell, regulate immunity and tolerance. In response to stimuli, DCs manipulate immunoregulatory functions, resulting in changes in their stage of differentiation, maturation, and functions.19 AhR may stimulate tDC‐supported differentiation of Tregs, thereby modulating inflammation during the postinfarct myocardial inflammation phase. In addition, because AhR can affect the control of monocyte differentiation and regulate the immune response, we examined the protective effect of DC activation in response to ITE. The physiological relevance of ITE during post‐MI remodeling and its subsequent effect on the immune system remain unclear.
Here, we examined whether ITE is a useful tool in promoting effective favorable cardiac remodeling after acute MI using a coronary artery ligated mouse model. We found that ITE improves cardiac remodeling and reduces the proinflammatory response through systemic activation of Treg differentiation and regulation of the M1/M2 macrophage profile in the injured myocardium.
Methods
Data of this study are available on request from the authors.
Induction of an Ml Model and Treatment With ITE
All animal experiments were approved by the Institutional Animal Care and Use Committee at the School of Medicine of the Catholic University of Korea (approval number: CUMC‐2018‐0035‐07). Male 7‐ to 8‐week‐old C57BL/6 mice were purchased from Orientbio Korea. Mice were anesthetized by intraperitoneal injection of tiletamine/zolazepam 12.5 mg/kg (Zoletil 50; Virbac Korea, Seoul, Korea) and xylazine 10 mg/kg (Rompun; Bayer, Seoul, Korea), intubated, and placed on a mechanical animal ventilator (Harvard Apparatus). MI was induced in C57BL/6 mice (20‒22 g) by ligation of the left anterior descending coronary artery. In the sham group, the chest was opened, but left anterior descending coronary artery ligation was not performed. For ITE‐treated mice, ITE (Tocris Bioscience, Bristol, UK) was prepared in DMSO (Sigma‐Aldrich, St. Louis, MO) at a concentration of 100 mmol/L and then diluted further with PBS before use. MI mice were randomly assigned and received daily intraperitoneal injection of vehicle (control group, n=16) or ITE (200 µg) for 1 week (ITE group, n=19). Mice were euthanized on day 5 or day 28 after MI under Rompun‐Zoletil anesthesia, followed by bilateral thoracotomy to assure euthanasia. For a survival analysis, mice treated with ITE (n=30) and controls (n=28) after MI or a sham‐operated group (n=5) were monitored to identify overall survival.
Histology
Hearts were rapidly excised, fixed in 4% paraformaldehyde solution, and embedded in paraffin. The paraffin blocks were cut into 5‐μm‐thick sections by using a microtome (Thermo Fisher Scientific, Waltham, MA). Serial sections were stained with Masson's trichrome, and infarct size was calculated as follows: total infarct circumference/total left ventricular circumference × 100.20 To visualize the whole infarct area, excised heart tissue was cut into 4 serial sections and stained with a 1% 2,‐3,‐5‐triphenyltetrazolium chloride (Sigma‐Aldrich) solution at 37°C for 20 minutes.
Echocardiography
Echocardiography was performed at 28 days after MI using an Affinity 50 imaging system (Philips, Amsterdam, the Netherlands). Briefly, mice were anesthetized with isoflurane (2.0%) in a mixture of oxygen and nitrous oxide (O2/N2O=3/7). The ejection fraction and fractional shortening were calculated by M‐mode tracings in short‐axis views at the papillary muscle level of the short‐axis views.
Generation of Bone Marrow‐Derived DCs
Bone marrow‐derived DCs were obtained from mice (C57BL/6, 7 weeks old, male) as previously described.21 Immature DCs were cultured with 1 µmol/L ITE or vehicle (control). To generate tDCs and mature DCs (mDCs), 10 ng/mL recombinant murine tumor necrosis factor alpha (BD Pharmingen, Mountain View, CA) and 1 µg/mL lipopolysaccharide (Sigma‐Aldrich) were added to immature DC cultures in the presence of ITE or PBS for 4 hours. Cells were harvested, and CD11c, CD40, CD80, CD86, and PD‐L1 surface marker expression was examined on a fluorescence‐activated cell sorter or in functional assays.
DC–T‐Cell Cocultures
DCs were cocultured with splenocytes from C57BL/6 mice in RPMI 1640 medium containing 10% fetal bovine serum (FBS) (DC:T‐cell ratio, 1:10) at 37°C for 72 hours. Next, the cells were harvested and analyzed by flow cytometry to identify the population of T cells. The cell culture supernatants were collected for cytokine measurements.
Analysis of Immunofluorescence Staining
Paraffin‐embedded cardiac tissue sections were deparaffinized and rehydrated. The sections were unmasked in antigen retrieval buffer (Abcam, Cambridge, UK) for 10 minutes at 95°C. After blocking, the sections were stained overnight at 4°C with 1 of the following antibodies: anti‐CD4 (GK1.5; Abcam), anti‐Foxp3 (D6O8R; Cell Signaling Technology, Danvers, MA), anti‐CD68 (FA‐11; Abcam), anti‐iNOS (inducible nitric oxide synthase) (Abcam), and anti‐ MR (mannose receptor; Abcam). After secondary antibody incubation, nuclei were counterstained with a DAPI solution (Dako, Carpinteria, CA). Stained slides were mounted in DAKO Fluorescence Mounting Medium, and fluorescence signals were imaged using fluorescence microscopy (LSM 510 Meta; Zeiss, Oberkochen, Germany). Images were acquired using ZEN 2012 software (Zeiss).
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA). The RNA purity and concentration of the purified RNA were determined by using a Thermo NanoDrop 2000 (Thermo Fisher), followed by cDNA synthesis with a cDNA synthesis kit (Roche, Basel, Switzerland), quantitative real‐time polymerase chain reaction (BioRad Laboratories, Hercules, CA), and real‐time polymerase chain reaction (BioRad). mRNA expression was normalized to GAPDH mRNA expression, and the lists of primers used for quantitative real‐time polymerase chain reaction are included in Table.
Table 1.
Primer Sequence for Quantitative Real‐Time Polymerase Chain Reaction
| Gene | Primer Sequence |
|---|---|
| CYP1A1 |
Forward 5′ TAA CCA TGA CCG GGA ACT GTG 3′ Reverse 5′ CTC CGA TGC ACT TTC GCT TG 3′ |
| TNFα |
Forward 5′ CAC AGA AAG CAT GAT CCG CGA CGT 3′ Reverse 5′ TGA GAG GGA GGC CAT TTG GGA 3′ |
| IL1β |
Forward 5′ GAG TGT GGA TCC CAA GCA AT 3′ Reverse 5′ ACG GAT TCC ATG GTG AAG TC 3′ |
| IL10 |
Forward 5′ GCT CTT ACT GAC TGG CAT GAG 3′ Reverse 5′ CGC AGC TCT AGG AGC ATG TG 3′ |
| TGFβ1 |
Forward 5′ TGA CGT CAC TGG AGT TGT ACG 3′ Reverse 5′ GGT TCA TGT CAT GGA TGG TGC 3′ |
| Foxp3 |
Forward 5′ ACCCAGGAAAGACAGCAACC 3′ Reverse 5′ GATCTGCTTGGCAGTGCTTG 3′ |
| GAPDH |
Forward 5′ AGA ACA TCA TCC CTG CAT CC 3′ Reverse 5′ CAC ATT GGG GGT AGG AAC AC 3′ |
Measurement of Cytokine Concentrations
To measure cytokine production, single‐cell suspensions were prepared from mouse spleens and cultured in RPMI 1640 medium containing 10% FBS and 1% antibiotics at a density of 1×106 cells/mL, followed by stimulation with 50 ng/mL PMA (Sigma‐Aldrich) and 1 μmol/L ionomycin (Sigma‐Aldrich). After 24 hours of incubation, the splenocyte culture supernatants were collected and analyzed to measure the concentrations of the cytokines interleukin (IL)‐10 and IL‐17 (ELISA kits; R&D Systems, Minneapolis, MN).
Flow Cytometry Analysis
Spleens and mediastinal lymph nodes (mdLNs) were harvested and mechanically processed by passage through a 70‐μm cell strainer. Cell‐surface markers were stained with PerCP‐Cy5.5‐conjugated anti‐CD4 (RM4‐5; BioLegend, San Diego, CA) and Allophycocyanin‐conjugated anti‐CD25 (PC61; BioLegend) antibodies for Treg analysis. Intracellular staining was preformed after fixation and permeabilization with a Foxp3 fix/perm buffer set (BioLegend) by incubating cells with a Phycoerythrin‐conjugated anti‐FOXP3 antibody (FJK‐16s; Invitrogen). DCs were stained with fluorescein isothiocyanate–conjugated anti‐CD11c (N418; BioLegend), Allophycocyanin‐conjugated anti‐CD40 (3/23; BD Biosciences, Franklin Lakes, NJ), Alexa Fluor‐488‐conjugated anti‐CD80 (2D10, BioLegend), Alexa Fluor‐647‐conjugated anti‐CD86 (GL‐1; BioLegend), and Brilliant Violet421‐conjugated anti‐CD274 (10F.9G2; BioLegend) antibodies. Flow cytometric analysis was conducted by using a FACSCanto II flow cytometer (BD Biosciences), and the results were analyzed with FlowJo software (Tree Star, San Carlos, CA).
Statistical Analysis
Quantitative data and statistical analyses were conducted using Prism version 7.0 (GraphPad Software, San Diego, CA). Based on an unpaired Student t test or 1‐way ANOVA followed by Bonferroni's multiple comparison test, a P<0.05 was considered statistically significant. Survival analysis were studied by the Kaplan‐Meier curve and compared with a log‐rank (Mantel‐Cox) test.
Results
ITE Treatment Improves Cardiac Structure and Function After MI
We performed histological analysis of cardiac tissue sections to examine the effects of ITE on cardiac remodeling and function in mice after MI. Figure 1A shows a schematic depicting the animal experiments. At 28 days after MI, Masson's trichrome and 2,3,5‐triphenyltetrazolium chloride staining showed that the infarct size in mice injected with ITE once a day for 1 week was significantly smaller compared with control mice (P<0.0001; Figure 1B and 1C). Serial echocardiographic monitoring to assess cardiac function and morphology showed no significant differences in cardiac function including left ventricular ejection fraction and fractional shortening up to 2 weeks (Figure S1), but ITE treatment significantly increased the fractional shortening (28.9%–34.8%, P<0.001) and ejection fraction (10.6%–14.0%, P<0.01) compared with control at 28 days after MI (Figure 1D and 1E). Mice treated with ITE exhibited an increased survival rate compared with control mice (Figure 1F; log‐rank P=0.3981). The results suggest that ITE therapy induces improvements in cardiac structure and function, thereby resulting in increased survival after acute MI.
Figure 1. ITE improves cardiac function after MI.
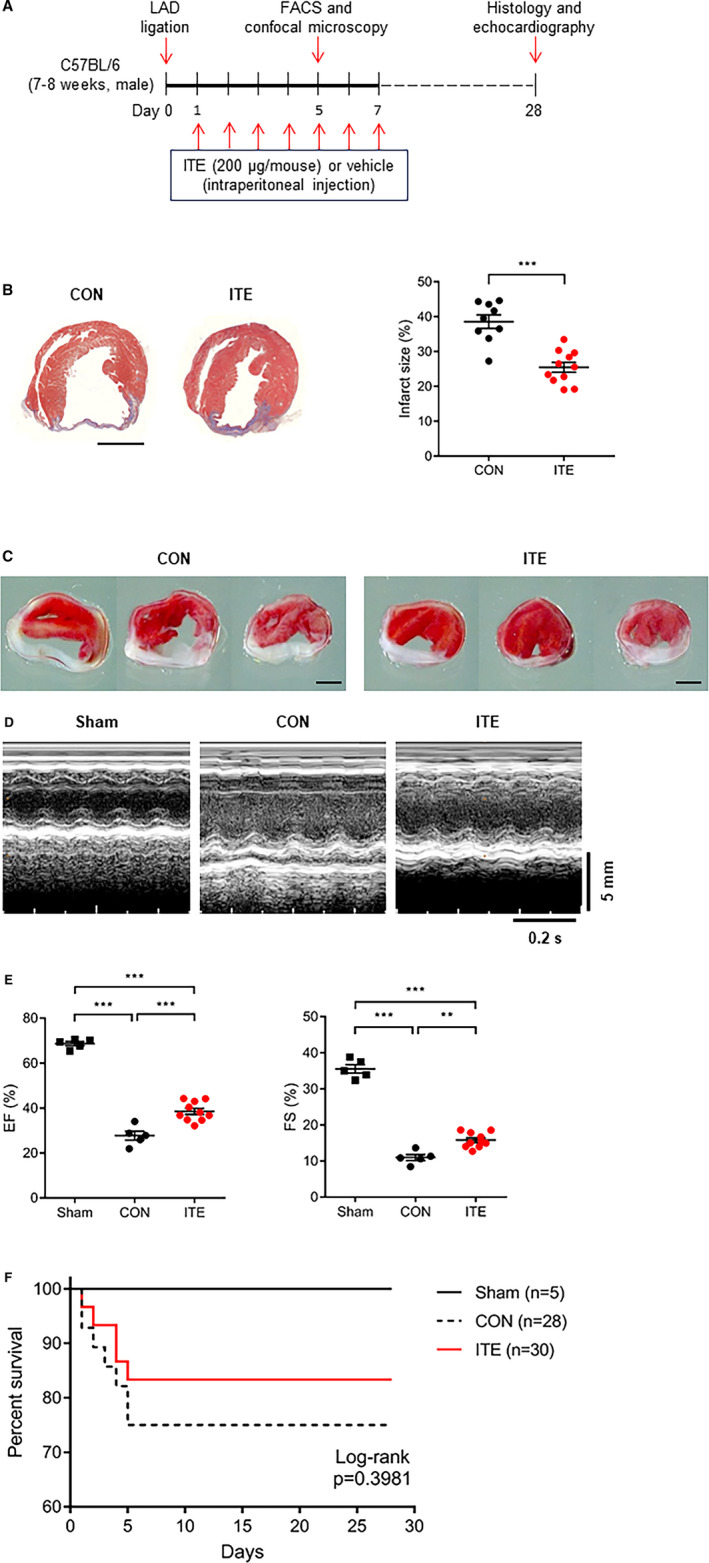
A, Schematic showing the experimental timeline. B, Representative images showing Masson’s trichrome staining of cardiac tissues from the control (n=9) and ITE‐treated (n=12) groups at 28 days after MI (left panel) and quantitative analysis of infarct size (right panel). Scale bar, 2 mm. The results were compared using an unpaired t test. ***P<0.001. C, Representative images showing 2,3,5‐triphenyltetrazolium chloride staining of cardiac tissues from the control (left) and ITE‐treated (right) groups at 28 days after MI (n=7 per group). Scale bar, 2 mm. D, Representative echocardiography M‐mode images of sham, control, and ITE‐treated mice. E, Echocardiography data showing the left ventricular EF and FS in the control and ITE‐treated groups at 28 days after MI (n=5: sham and control, n=10: ITE). **P<0.01. ***P<0.001. F, Kaplan‐Meier 28‐day survival analysis after acute MI in vehicle control (n=28) and ITE‐treated mice (n=30) or sham operation (n=5). The results were compared using 1‐way ANOVA. Data are presented as mean±SEM. CON indicates control; EF, ejection fraction; FACS, fluorescence‐activated cell sorter; FS, fractional shortening; ITE, 2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; LAD, left anterior descending coronary artery; and MI, myocardial infarction.
ITE Promotes the Differentiation of Foxp3+ Tregs on Day 5 After MI
To confirm ITE‐induced changes in Treg populations and cytokine production in vivo, mice were euthanized 5 days after MI. Cells were isolated from the heart‐draining mdLNs and spleen. Flow cytometry revealed that the Foxp3+ Treg population in the mdLNs and spleens from mice treated with ITE was larger than that in corresponding tissues from control mice (Figure 2A and 2B). Next, we examined splenocyte cytokine profiles. ITE treatment increased the expression of IL‐10 and decreased the expression of IL‐17 in the spleen of treated mice compared with that of control mice (Figure 2C). Next, we examined cardiac Treg infiltration of the healing infarct after MI. An increased number of Tregs in the infarcted myocardium was observed in ITE‐treated mice compared with control mice at 5 days after MI (Figure 2D). Collectively, the results show that ITE treatment increases the Foxp3+ Treg populations in the lymph nodes, spleen, and infarcted myocardium.
Figure 2. ITE increased Foxp3+ Treg populations.
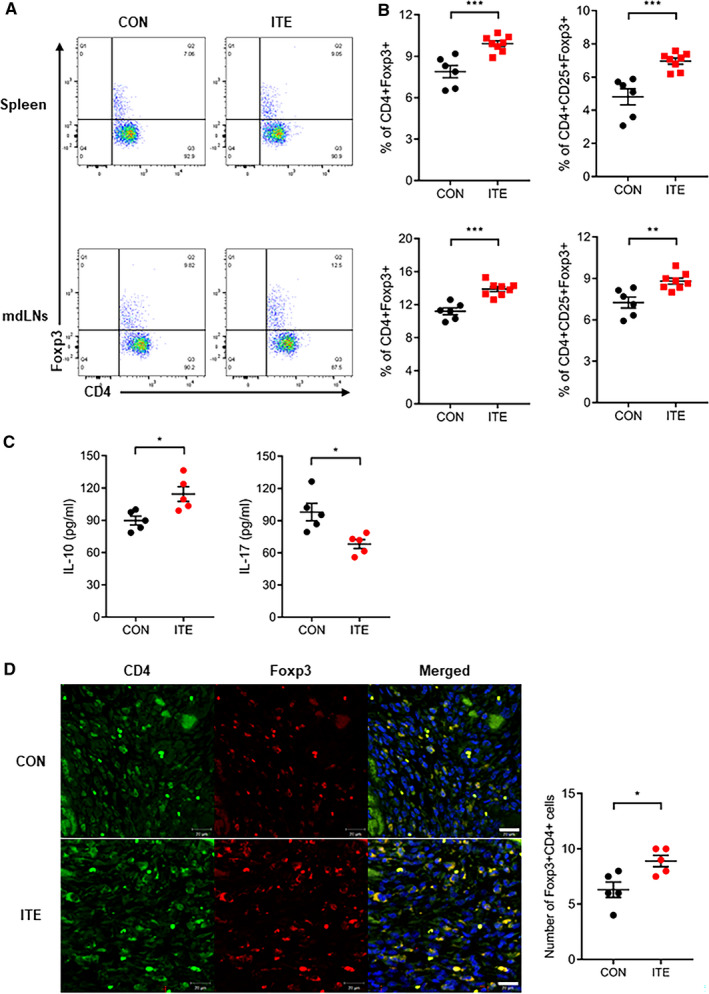
A, Representative fluorescence‐activated cell sorter plots showing the CD4+Foxp3+ Treg populations in the spleen and mdLNs of mice in the control and ITE‐treated groups at 5 days after MI. B, Dot plots showing the CD4+Foxp3+ Tregs and CD4+CD25+Foxp3+ Tregs (n=6: control, n=8: ITE). C, Levels of cytokines (picograms per milliliter) produced by splenocytes in the control and ITE‐treated groups at 5 days after MI (n=5 for per group). D, Confocal immunofluorescence images of Tregs in the infarcted myocardium at 5 days after MI. Cells were stained for CD4 (green) and Foxp3 (red) (left). A merged image is shown on the right (n=5 for per group). Scale bars=20 µm. The control and ITE groups were compared with an unpaired t test. Data are presented as mean±SEM. CON indicates control; Foxp3+, forkhead box protein P3‐positive; IL, interleukin; ITE, 2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; mdLNs, mediastinal lymph nodes; MI, myocardial infarction; and Tregs, regulatory T cells. *P<0.05. **P<0.01. ***P<0.001.
ITE Modulates a Shift in Macrophage Subsets in the Myocardium After MI
To determine whether ITE alters macrophage polarization, immunofluorescence staining was performed to evaluate myocardial infiltration by M1 and M2 macrophages. ITE promoted polarization to an M2 phenotype in the myocardium at post‐MI day 5. In contrast, nontreated mice showed accumulation of M1 macrophages at post‐MI day 5. The CD68+iNOS+ M1 macrophage population in infarcted heart tissue was far lower in ITE‐treated mice than in control mice on day 5 (Figure 3A), whereas the CD68+MR+ M2 macrophage population was increased in the ITE‐treated mice (Figure 3B). To identify ITE‐induced AhR activation in the infarcted myocardium, we confirmed the mRNA expression of Cyp1a1 (cytochrome P450 1A1), which has been used as a biomarker for AhR activation. The ITE group showed increased Cyp1a1 expression compared with the sham and control groups (Figure 4A). Additionally, to determine the expression of cytokines in the infarcted myocardium, we examined cytokine profiles by quantitative real‐time polymerase chain reaction. The expression of mRNA transcripts encoding tumor necrosis factor alpha and IL‐1β after MI was significantly increased in the control group compared with the sham and ITE groups. The expression of IL‐10 and transforming growth factor beta 1 in post‐MI infarcted tissue from the ITE group was much higher than that in corresponding tissue from the sham and control groups (Figure 4B), suggesting that ITE modulates immune responses in the infarcted myocardium.
Figure 3. AhR activation by ITE drives macrophage polarization in the infarcted myocardium.
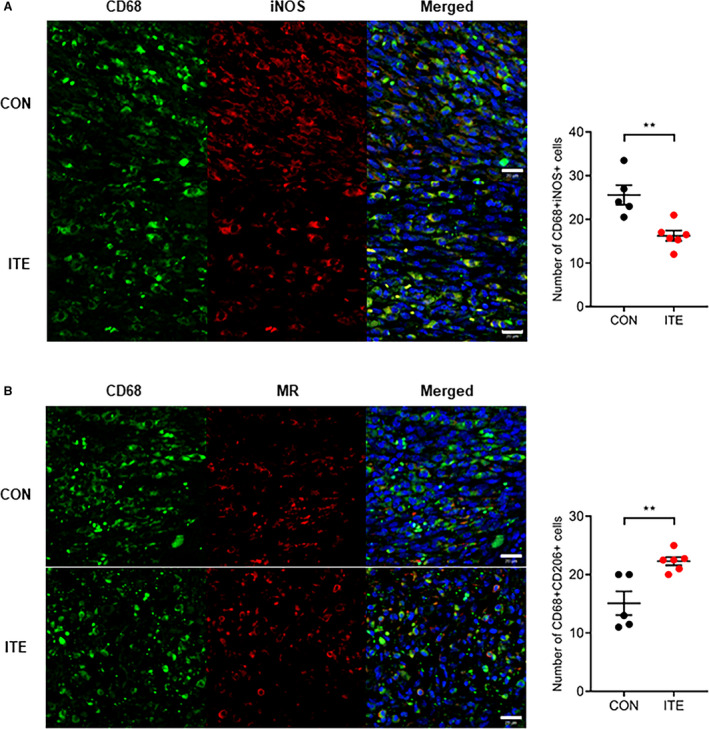
A, Representative confocal IF images of M1 macrophages in infarcted hearts at 5 days after MI. Sections were stained for CD68 (green) and iNOS (red) (left). The number of CD68+iNOS+ cells in control and ITE‐injected mice was counted (right). B, Representative confocal IF images of M2 macrophages in infarcted hearts at 5 days after MI. Cells were stained for CD68 (green) and MR (red) (left). The number of CD68+MR+ cells in control and ITE‐injected mice was counted (right) (n=5: control, n=6: ITE). Scale bars=20 µm. Data are presented as mean±SEM and were analyzed using an unpaired t test. AhR indicates aryl hydrocarbon receptor; CON, control; IF, immunofluorescence; iNOS, inducible nitric oxide synthase; ITE, 2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; MI, myocardial infarction; and MR, mannose receptor. **P<0.01.
Figure 4. ITE regulates the production of anti‐inflammatory and proinflammatory cytokines.
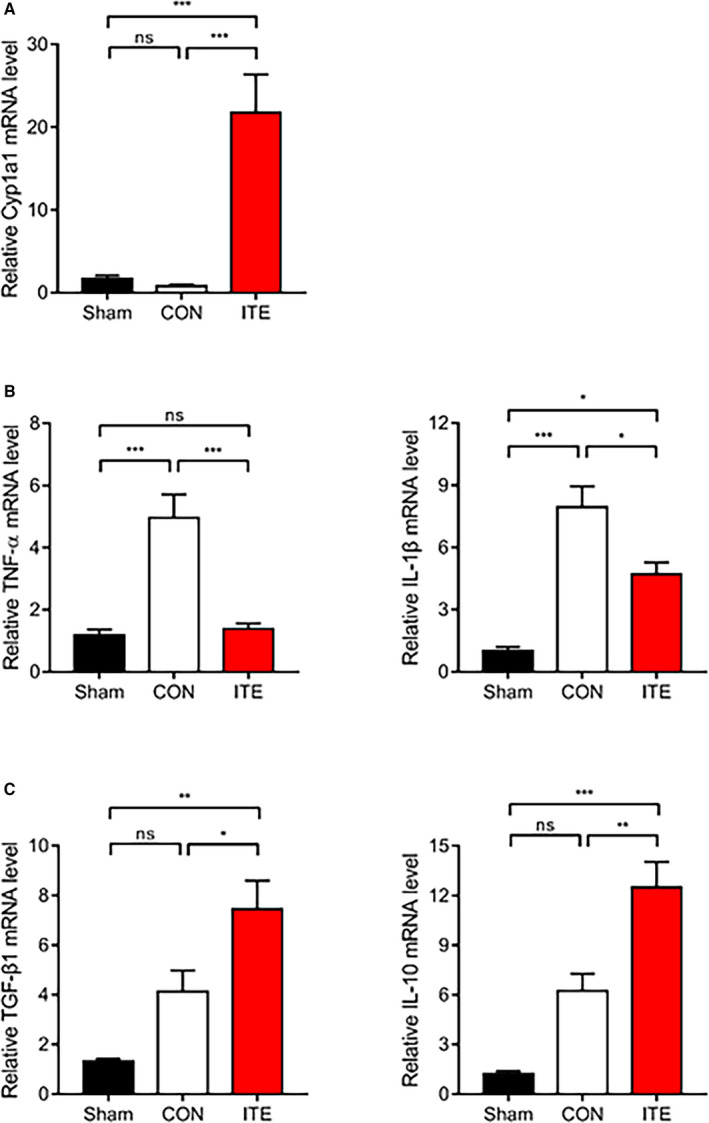
A, Relative mRNA expression of Cyp1a1 in infarcted tissues determined by quantitative real‐time polymerase chain reaction. Expression of mRNA transcripts encoding the proinflammatory cytokines TNF‐α and IL‐1β (B) and the anti‐inflammatory cytokines TGF‐β1 and IL‐10 (C) in infarcted tissues at 5 days after MI (n=5‒7 per group). The values are normalized to GAPDH and expressed as the fold‐change from sham values. The results were compared using 1‐way ANOVA. Data are presented as mean±SEM. CON indicates control; Cyp1a1, cytochrome P450 1A1; IL, interleukin; ITE, 2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; MI, myocardial infarction; ns, not significant; TGF‐β1, transforming growth factor beta 1; and TNF‐α, tumor necrosis factor alpha. *P<0.05. **P<0.01. ***P<0.001.
ITE‐Treated DCs Have a Tolerogenic Function
Several approaches have shown that the regulatory capacity of in vivo expansion of the Foxp3+ Treg compartment depends on AhR activation.18, 22, 23 DCs can also modulate T‐cell activation and polarization, contributing to Foxp3+ Treg induction. To gain insight into the effect of AhR activation in response to ITE in vitro, we investigated the expression of costimulatory molecules, including CD40, CD80, CD86, and PD‐L1 in DCs. We treated immature DCs in vitro with 1 µmol/L ITE and found that the expression of costimulatory molecules by ITE‐treated mDCs was slightly lower than that by untreated control cells (Figure 5A and 5B). DCs can induce the differentiation of CD4+ T cells into Tregs; therefore, splenocytes were cocultured with DCs and analyzed. ITE‐treated mDCs increased the Foxp3+ Treg population to a greater extent than nontreated mDCs (Figure 6A and 6B). These data were confirmed by measuring Foxp3 mRNA levels (Figure 6C). We showed that ITE could induce tDCs and inhibit their differentiation and maturation. Next, we examined the functional effects on Treg expansion in response to ITE. Compared with mDCs, ITE‐mDCs promoted the Foxp3+CD4+CD25+ Treg population and increased the mRNA expression of Foxp3 (Figure 6A through 6C). These results suggest that ITE upregulates the immunotolerogenic effect of DCs when cocultured with splenocytes.
Figure 5. ITE inhibits the expression of costimulatory molecules by LPS‐induced DCs.
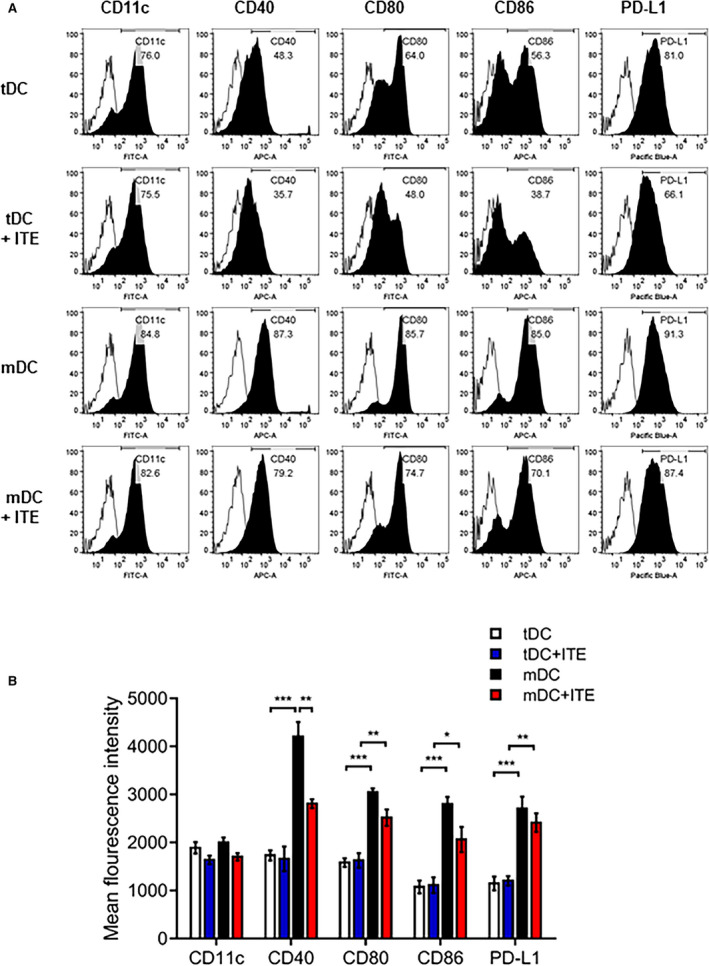
A, DC subsets and ITE‐treated cells were stained with specific antibodies and analyzed by flow cytometry. B, Bar graph showing the mean fluorescence intensity, expressed as the mean±SEM (n=5 independent DC preparations). *P<0.05. **P<0.01. ***P<0.001. APC, allophycocyanin; DC, dendritic cells; FITC, fluorescein isothiocyanate; ITE, 2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; LPS, lipopolysaccharide; mDC, mature dendritic cells; and tDC, tolerogenic dendritic cells.
Figure 6. Immunosuppressive characteristics of ITE‐primed tDCs.
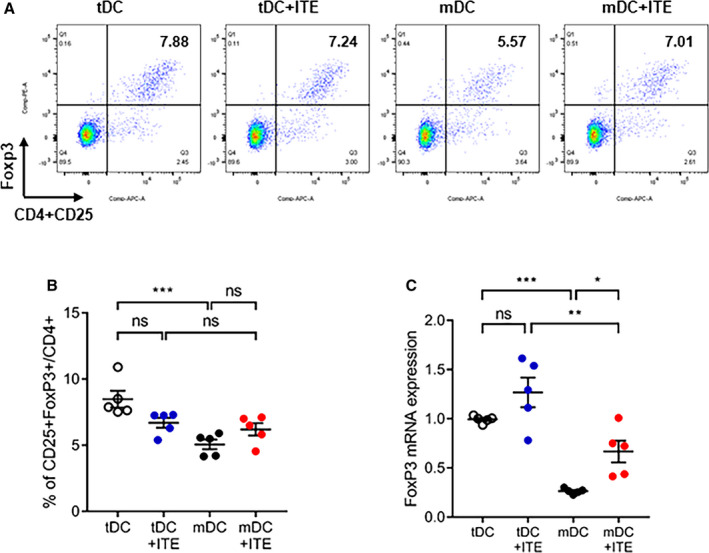
A, Splenocytes were cocultured with tDCs or mDCs and stimulated with or without ITE for 72 hours at 37°C. B, The bar graphs show the mean percentage of CD4+CD25+Foxp3+ cells. C, Relative Foxp3 mRNA expression was measured by quantitative real‐time polymerase chain reaction. Foxp3 mRNA expression was quantified relative to that of GAPDH. Data are presented as mean±SEM and were compared using 1‐way ANOVA (n=5 per group). APC indicates allophycocyanin; Foxp3 indicates forkhead box protein P3; ITE, 2‐(1’H‐indole‐3’‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; mDCs, mature dendritic cells; ns, not significant; and tDCs, tolerogenic dendritic cells. *P<0.05. **P<0.01. ***P<0.001.
Discussion
Our study revealed the effects of AhR stimulation by ITE (a nontoxic endogenous AhR ligand) on functional and structural improvements after MI in mice. ITE treatment for 1 week significantly reduced infarct size measured by histology and markedly improved the left ventricular ejection fraction and fractional shortening observed by echocardiography in MI mice. These antiremodeling effects could be derived from increases in the Foxp3+ Treg populations of the spleen and lymph nodes and subsequent M2 macrophage predominance in infarcted hearts, creating a better environment for healing and favorable remodeling. These findings support the conclusion that ITE, a nontoxic AhR stimulator, can be clinically translated into a novel antiremodeling therapy for the prevention of post‐MI HF in humans.
Wound healing after MI requires an orchestrated inflammatory response. Induction of Foxp3+ Tregs provides important benefits in reducing infarct size and preventing HF following acute MI.24, 25 Several methods can be used for in vitro Foxp3+ Treg expansion, but the capacity of these methods to lead to prolonged activation of Foxp3+ Tregs is limited.26, 27, 28 Some studies have reported improved wound healing after MI by assessing the regulatory capacity of Tregs; unfortunately, these treatments have not translated to the clinic.29 Strategies aimed at manipulating functional Foxp3+ Tregs in vivo would be more applicable to clinical practice. Potential approaches to generate functional Foxp3+ Tregs responding to AhR activation have demonstrated the ability to establish anti‐inflammatory immune responses in settings of experimental autoimmunity and transplant rejection.14, 30, 31, 32 In this regard, AhR activation in acute MI could have the potential to modulate the immune response and reduce infarct size. The massive release of cytokines and inflammatory responses during ischemic cardiac injury play important roles in postischemic healing. Therefore, understanding the pathogenesis and pharmacodynamics of ischemia is essential to attenuate ischemic injury in heart tissues by regulating myocardial inflammation. AhR is an endogenous ligand‐dependent transcription factor that is intrinsically linked to mediating immune disorders and regulating inflammation.33 MI is associated with an intensive inflammatory response and spontaneous repression, which results in an important physiological process for effective recovery. Thus, AhR is an attractive target in MI and inflammatory disease.
Myocardial ischemic injury following acute MI promotes profound inflammation, and timely suppression of inflammatory responses via expansion of Foxp3+ Treg populations attenuates myocardial necrosis and adverse remodeling. After MI, macrophage subsets play a central role in cardiac remodeling.34, 35 Tregs coordinate a switch in macrophage polarity, which helps to remove dead tissue and repair the wound after ischemic injury.21 Cardiac‐resident macrophages mostly disappear within the first 24 hours, and monocytes replenish with the cardiac macrophage pool after acute MI.36 The infiltrating monocytes then differentiate into M1 macrophages responsible for the proinflammatory response. Subsequently, cytokines, chemokines, and growth factors influence the reparative phase coordinated by M2 macrophages. In this process, we hypothesized that ITE could further induce macrophage polarization from M1 to M2, thereby leading to better antiremodeling effect. Here, we showed that ITE abrogated excessive inflammation and promoted Treg populations in the mdLNs and spleen, which could induce a rapid shift from an M1 macrophage phenotype to an M2 macrophage phenotype in the infarcted myocardium (Figure 3A and 3B), achieving timely suppression and spatial containment of the postinfarction inflammatory reaction at post‐MI day 5. Although significant pathophysiological differences at the initial phase was not observed between ITE treatment and control, therapeutic Treg activation or expansion by ITE treatment enhanced M2‐like monocyte differentiation within 5 days, which induced better healing at a later phase (28 days after MI). Activated M2‐like macrophages are well known to mediate anti‐inflammatory processes and exhibit wound‐healing properties.37 Our studies clearly demonstrated that ITE could stimulate local M2‐like macrophage proliferation (Figure 3B), producing transforming growth factor beta 1 and IL‐10 in the myocardium in post‐MI mice (Figure 4B).
Upregulated AhR expression has been shown to accelerate DC‐induced anti‐inflammatory activities.32, 38 DCs serve as potent antigen‐presenting cells and the first line of defense against pathological infections. Many studies have attempted to regulate tDCs to prevent autoimmune disease.39, 40 Here, we found that ITE conferred tolerogenic functions on DCs by inhibiting DC maturation. In particular, ITE inhibited the expression of costimulatory molecules, including CD40, CD80, CD86, and PD‐L1, in lipopolysaccharide‐treated DCs derived from murine bone marrow‐derived DCs (Figure 5A and 5B). In addition to the limited lipopolysaccharide‐mediated inflammatory response of DCs, ITE expanded the Foxp3+ Treg population in vitro (Figure 6A). These results strongly support that ITE treatment after MI downregulates the immune response in the infarcted myocardium.
In the present study, we focused on the proof of concept of immune modulatory potential of ITE after acute MI; however, it is limited for understanding the detailed mechanisms underlying the immune responses after ITE treatment. For instance, the in vivo absorption, metabolism, distribution, and excretion rates of ITE remain unclear.41 Therefore, more intensive studies, including the detailed pharmacokinetics, will need to be performed to define the biodistribution of ITE and its primary effects on a tissue‐specific manner in future work.
In conclusion, an earlier M1 to M2 macrophage transition and rapid Treg expansion induced by ITE contribute to potent immune modulation, which improves the systolic function of the heart in MI mice. Treg populations are increased in the mdLNs, spleen, and infarcted myocardium. This altered immune environment within the infarcted heart promotes wound remodeling and preserves left ventricular function after myocardial tissue damage. Thus, ITE, an endogenous AhR ligand, may serve as a highly potent new compound for the antiremodeling treatment of MI.
Sources of Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF‐2018R1D1A1B07049375 and 2019R1A2C2085516).
Disclosures
None.
Supporting information
Figure S1
(J Am Heart Assoc. 2021;10:e020502. DOI: 10.1161/JAHA.120.020502.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.020502
For Sources of Funding and Disclosures, see page 13.
References
- 1.Torabi A, Cleland JG, Khan NK, Loh PH, Clark AL, Alamgir F, Caplin JL, Rigby AS, Goode K. The timing of development and subsequent clinical course of heart failure after a myocardial infarction. Eur Heart J. 2008;29:859–870. DOI: 10.1093/eurheartj/ehn096. [DOI] [PubMed] [Google Scholar]
- 2.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD; Joint ESCAAHAWHFTFftUDoMI , Katus HA, Lindahl B, Morrow DA, Clemmensen PM, et al. Third universal definition of myocardial infarction. Circulation. 2012;126:2020–2035. DOI: 10.1161/CIR.0b013e31826e1058. [DOI] [PubMed] [Google Scholar]
- 3.Chow SL, Maisel AS, Anand I, Bozkurt B, de Boer RA, Felker GM, Fonarow GC, Greenberg B, Januzzi JL Jr, Kiernan MS, et al. Role of biomarkers for the prevention, assessment, and management of heart failure: a scientific statement from the American Heart Association. Circulation. 2017;135:e1054–e1091. DOI: 10.1161/CIR.0000000000000490. [DOI] [PubMed] [Google Scholar]
- 4.Frantz S, Bauersachs J, Ertl G. Post‐infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. 2009;81:474–481. DOI: 10.1093/cvr/cvn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. DOI: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 6.Tardif J‐C, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, et al. Efficacy and safety of low‐dose colchicine after myocardial infarction. N Engl J Med. 2019;381:2497–2505. DOI: 10.1056/NEJMoa1912388. [DOI] [PubMed] [Google Scholar]
- 7.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. DOI: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. DOI: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 9.Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, Ridker PM. Anti‐inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139:1289–1299. DOI: 10.1161/CIRCULATIONAHA.118.038010. [DOI] [PubMed] [Google Scholar]
- 10.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. DOI: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 11.Nebert DW. Aryl hydrocarbon receptor (AHR):“Pioneer member” of the basic‐helix/loop/helix per‐Arnt‐sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog Lipid Res. 2017;67:38–57. DOI: 10.1016/j.plipres.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larigot L, Juricek L, Dairou J, Coumoul X. AhR signaling pathways and regulatory functions. Biochim Open. 2018;7:1–9. DOI: 10.1016/j.biopen.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14:801–814. DOI: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(h)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. DOI: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 15.Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. 2019;19:184–197. DOI: 10.1038/s41577-019-0125-8. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Ye Z, Kijlstra A, Zhou Y, Yang P. Activation of the aryl hydrocarbon receptor affects activation and function of human monocyte‐derived dendritic cells. Clin Exp Immunol. 2014;177:521–530. DOI: 10.1111/cei.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quintana FJ, Sherr DH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev. 2013;65:1148–1161. DOI: 10.1124/pr.113.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah A‐M, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2010;107:20768–20773. DOI: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. DOI: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 20.Takagawa J, Zhang Y, Wong ML, Sievers RE, Kapasi NK, Wang Y, Yeghiazarians Y, Lee RJ, Grossman W, Springer ML. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area‐ and length‐based approaches. J Appl Physiol. 2007;102:2104–2111. DOI: 10.1152/japplphysiol.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choo EH, Lee J‐H, Park E‐H, Park HE, Jung N‐C, Kim T‐H, Koh Y‐S, Kim E, Seung K‐B, Park C, et al. Infarcted myocardium‐primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation. 2017;135:1444–1457. DOI: 10.1161/CIRCULATIONAHA.116.023106. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Ma J, Takeuchi M, Usui Y, Hattori T, Okunuki Y, Yamakawa N, Kezuka T, Kuroda M, Goto H. Suppression of experimental autoimmune uveoretinitis by inducing differentiation of regulatory T cells via activation of aryl hydrocarbon receptor. Invest Ophthalmol Vis Sci. 2010;51:2109–2117. DOI: 10.1167/iovs.09-3993. [DOI] [PubMed] [Google Scholar]
- 23.Goettel J, Gandhi R, Kenison J, Yeste A, Murugaiyan G, Sambanthamoorthy S, Griffith A, Patel B, Shouval D, Weiner H, et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep. 2016;17:1318–1329. DOI: 10.1016/j.celrep.2016.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zacchigna S, Martinelli V, Moimas S, Colliva A, Anzini M, Nordio A, Costa A, Rehman M, Vodret S, Pierro C, et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat Commun. 2018;9:2432. DOI: 10.1038/s41467-018-04908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nahrendorf M, Swirski FK. Regulating repair: regulatory T cells in myocardial infarction. Circ Res. 2014;115:7–9. DOI: 10.1161/CIRCRESAHA.114.304295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL, June CH, Miller JS, Wagner JE, Blazar BR. Generation and large‐scale expansion of human inducible regulatory T cells that suppress graft‐versus‐host disease. Am J Transplant. 2011;11:1148–1157. DOI: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Huizinga TW, Toes RE. De novo generation and enhanced suppression of human CD4+CD25+ regulatory T cells by retinoic acid. J Immunol. 2009;183:4119–4126. doi: 10.4049/jimmunol.0901065. [DOI] [PubMed] [Google Scholar]
- 28.Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, McKenna DH, Bromberg JS, Levine BL, Riley JL, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. 2011;3:83ra41. DOI: 10.1126/scitranslmed.3001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gajarsa JJ, Kloner RA. Left ventricular remodeling in the post‐infarction heart: a review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail Rev. 2011;16:13–21. DOI: 10.1007/s10741-010-9181-7. [DOI] [PubMed] [Google Scholar]
- 30.Abron JD, Singh NP, Mishra MK, Price RL, Nagarkatti M, Nagarkatti PS, Singh UP. An endogenous aryl hydrocarbon receptor ligand, ITE, induces regulatory T cells and ameliorates experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2018;315:G220–G230. DOI: 10.1152/ajpgi.00413.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nugent LF, Shi G, Vistica BP, Ogbeifun O, Hinshaw SJ, Gery I. ITE, a novel endogenous nontoxic aryl hydrocarbon receptor ligand, efficiently suppresses EAU and T‐cell‐mediated immunity. Invest Ophthalmol Vis Sci. 2013;54:7463–7469. DOI: 10.1167/iovs.12-11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschläger M, Roncarolo MG. Activation of the aryl hydrocarbon receptor promotes allograft‐specific tolerance through direct and dendritic cell–mediated effects on regulatory T cells. Blood. 2008;112:1214–1222. DOI: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. Int Immunol. 2013;25:335–343. DOI: 10.1093/intimm/dxt011. [DOI] [PubMed] [Google Scholar]
- 34.Gombozhapova A, Rogovskaya Y, Shurupov V, Rebenkova M, Kzhyshkowska J, Popov SV, Karpov RS, Ryabov V. Macrophage activation and polarization in post‐infarction cardiac remodeling. J Biomed Sci. 2017;24:1–11. DOI: 10.1186/s12929-017-0322-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 36.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, et al. Differential contribution of monocytes to heart macrophages in steady‐state and after myocardial infarction. Circ Res. 2014;115:284–295. DOI: 10.1161/CIRCRESAHA.115.303567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. DOI: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Platzer B, Richter S, Kneidinger D, Waltenberger D, Woisetschläger M, Strobl H. Aryl hydrocarbon receptor activation inhibits in vitro differentiation of human monocytes and langerhans dendritic cells. J Immunol. 2009;183:66–74. DOI: 10.4049/jimmunol.0802997. [DOI] [PubMed] [Google Scholar]
- 39.Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis. 2010;69:2042–2050. DOI: 10.1136/ard.2009.126383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomson A, Robbins PD. Tolerogenic dendritic cells for autoimmune disease and transplantation. Ann Rheum Dis. 2008;67:iii90–iii96. DOI: 10.1136/ard.2008.099176. [DOI] [PubMed] [Google Scholar]
- 41.Boule LA, Burke CG, Jin GB, Lawrence BP. Aryl hydrocarbon receptor signaling modulates antiviral immune responses: ligand metabolism rather than chemical source is the stronger predictor of outcome. Sci Rep. 2018;8:1826. DOI: 10.1038/s41598-018-20197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1