

Abstract
Background/Aim: Cisplatin combined with pemetrexed disodium heptahydrate (pemetrexed) is considered the standard treatment for patients with advanced, non-squamous, non-small-cell lung cancer. However, its growth-inhibitory effects on KRAS-dependent lung cancer as monotherapy and combination therapy are not well understood. The aim of this study was to compare the effects of cisplatin and pemetrexed on A549 cells as mono- and combination therapies and elucidate the underlying mechanisms. Materials and Methods: For in vitro studies, A549 cells were exposed to cisplatin with/without pemetrexed for 72 h. The results were then evaluated by cell viability, apoptosis, reactive oxygen species, terminal deoxynucleotidyl transferase dUTP nick-end labeling, and western blotting assays. Results: Our results revealed that cisplatin monotherapy was most cytotoxic to A549 cells, while cisplatin plus pemetrexed combination had an intermediate effect, and pemetrexed monotherapy induced a minimal cytotoxic effect on A549 cells. This effect was evidenced by cell viability results, inhibition of KRAS proto-oncogene, GTPase (KRAS)/Raf proto-oncogene, serine/ threonine kinase/mitogen-activated protein kinase kinase/ extracellular signal-regulated kinase pathways and apoptosis induction triggered by reactive oxygen species-mediated DNA damage. The immunoblotting result of conversion of microtubule-associated protein 1 light chain 3 alpha (LC3)-I to -II indicated that the greatest inducer of autophagy was combined treatment with cisplatin plus pemetrexed, while pemetrexed monotherapy had the lowest effect on autophagy induction, with cisplatin monotherapy having an intermediate effect. We found that the AKT serine/threonine kinase 1/mechanistic target of rapamycin kinase (mTOR) and AMP-activated protein kinase/mTOR signaling pathways were associated with autophagy activation. Interestingly, combination therapy with cisplatin plus pemetrexed was the primary eliminator of cellular senescence; cisplatin monotherapy had an intermediate effect, while pemetrexed monotherapy increased cellular senescence of A549 cells, as assessed by the expression of β-galactosidase protein. Conclusion: Cisplatin monotherapy may be more effective than pemetrexed monotherapy or cisplatin plus pemetrexed combination therapy against KRAS-dependent lung cancer.
Keywords: Cisplatin, pemetrexed, apoptosis, autophagy, senescence, KRAS
In terms of incidence and mortality, lung cancer is the most common cancer globally (1). Lung cancer is a leading cause of death, with non-small-cell lung cancer being the predominant form of the disease (2), accounting for nearly 80% of all lung cancer cases (3).
Mutations in the KRAS proto-oncogene, GTPase (KRAS) gene are frequently found in various types of human cancer, including of the lung, pancreas, and large intestine (4,5). Approximately 15-25% of patients with non-small cell lung cancer reportedly have KRAS mutations (6,7). These mutations alter the intrinsic GTPase activity of RAS and confer resistance to GTPase activators, which causes the accumulation of RAS in its active GTP union state, supporting the activation of KRAS (8,9). Constitutive activation of KRAS triggers stimulation of downstream signaling pathways, including the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/AKT serine/threonine kinase 1 (AKT)/mechanistic target of rapamycin kinase (mTOR) and Raf proto-oncogene, serine/threonine kinase (RAF)/ mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling cascades (10,11).
In multicellular organisms, apoptosis can occur in the form of programmed cell death (12). Apoptosis can be initiated via two crucial pathways: the extrinsic and the intrinsic (13). Various types of stress can induce programmed cell death (14). The generation of reactive oxygen species (ROS) is a crucial stressor that can trigger DNA damage (15). DNA damage can activate members of the caspase family, leading to cleavage of poly (ADP-ribose) polymerase (PARP), a hallmark of apoptosis (16). It has been reported that ROS-mediated DNA damage is related to cellular senescence (17). The production of DNA damage can lead to permanent arrest of the cell cycle (18). Under irreversible conditions, damaged cells remain alive but unable to proliferate, a phenomenon known as cellular senescence (19,20).
Several studies have reported the interaction between ROS generation and autophagy (21,22). Autophagy is considered a double-edged sword in cancer cells (23). Under nutrient starvation conditions, autophagy can promote cell survival by providing the energy required for cellular metabolism (24). On the other hand, autophagy can lead to cell death by consuming essential cellular components (25,26). Various studies have claimed that the PI3K/AKT/mTOR and AMP-activated protein kinase (AMPK)/mTOR pathways regulate autophagy to induce apoptosis (27,28).
Herein, we compared the anticancer effects of cisplatin and pemetrexed on KRAS-dependent A549 cells as mono- and combination therapies and clarified the underlying mechanisms.
Materials and Methods
Cell line and cell culture. A KRAS-mutated A549 cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). A549 cells were cultured in Roswell Park Memorial Institute 1640 medium containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and maintained at 37˚C in a humidified atmosphere containing 5% CO2.
Drug preparation. Cisplatin [PtCl2(NH3)2] and pemetrexed disodium heptahydrate [C20H19N5Na2O6·7H2O] were obtained from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). These drugs were dissolved in dimethyl sulfoxide for in vitro experiments.
Cell viability assay. The cytotoxicity of cisplatin and pemetrexed as single-drug or combined therapies in A549 cells was assessed using a water-soluble tetrazolium salt (WST-1) assay (Cell Proliferation Reagent WST-1; Roche, Tokyo, Japan). Into each well of a 96-well microtiter plate, 100 μl of a growing cell suspension (4×103 cells/well) was seeded, and 100 μl of cisplatin (200 nM) or pemetrexed (100 nM) solution as single-drug or combined treatments was added to each well (29). After incubation for 72 h at 37˚C in 5% CO2, 10 μl of WST-1 solution was added to each well, and the plates were incubated at 37˚C for an additional 4 h (29). The absorbance was then measured at 450 nm with a microplate enzyme-linked immunosorbent assay reader (Multiskan FC; Thermo Scientific, Tokyo, Japan). Data are presented as relative viability (%) by comparing drug-treated cells with DMSO-treated cells; the viability of DMSO-treated cells was assumed to be 100%.
Intracellular ROS assay. The intracellular ROS level was determined using a Reactive Oxygen Species Detection Assay Kit (ab186029; Abcam, Tokyo, Japan). In brief, after treatment with cisplatin and pemetrexed as single-drug or combined therapy for 72 h, cells were harvested for staining with a working solution of deep red ROS dye. Subsequently, the cells were incubated at 37˚C for 60 min before flow cytometric (FCM) analysis.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay. For 72 h, A549 cells were treated with cisplatin or pemetrexed as single-drug therapy or in combination. An in situ Direct DNA Fragmentation (TUNEL) Assay Kit (ab66108; Abcam) was used to measure DNA fragmentation in apoptotic cells. In brief, cells were fixed with 1% paraformaldehyde in phosphate-buffered saline and placed on ice for 15 min. Subsequently, the samples were treated with a staining solution and incubated at 37˚C for 60 min. After the addition of rinse buffer, cells were resuspended in propidium iodide/RNase A solution and incubated at room temperature for 30 min for the FCM analysis.
Apoptosis assay. A549 cells were treated with cisplatin and pemetrexed as single-drug or combined therapy for 72 h. Apoptotic cell death was quantified by FCM using fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit with propidium iodide (PI) (BioLegend, San Diego, CA, USA).
Western blotting. For 72 h, A549 cells were treated with cisplatin and pemetrexed as single-drug or combined therapy. Whole protein lysates were isolated using M-PER Mammalian Protein Extraction Reagent (Thermo Scientific), which included a phosphatase inhibitor cocktail and a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Protein concentrations were assessed using BCA protein assay reagent (Thermo Scientific). Total cellular protein (40 μg) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). Milk-blocked blots were then incubated at 4˚C overnight with primary antibodies against the following proteins: KRAS, RAF, MEK, phospho (p)-MEK (Ser 217/221), ERK, p-ERK (Thr 202/Tyr 204), AKT, p-AKT (Ser 473), mTOR, p-mTOR (Ser 2448), AMPKα, p-AMPKα (Thr 172), microtubule-associated protein 1 light chain 3 alpha (LC3), β-galactosidase, β-actin, and cleaved PARP (Asp 214) (all from Cell Signaling Technology, Danvers MA, USA). They were then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology). Proteins of interest were revealed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, Rockford, IL, USA) and viewed using an Invitrogen iBright FL1000 Imaging System (Thermo Fisher Scientific). The bands were quantified with the densitometric program of iBright Imaging System and normalized against β-actin.
Statistical analysis. Statistical analysis was conducted using GraphPad PRISM software, v. 7.0 (GraphPad Software Inc., San Diego, CA, USA). Results are presented as the mean±standard deviation of three independent experiments and were analyzed by a one-way analysis of variance followed by Dunnett’s multiple comparison test. Values of p<0.05 were considered statistically significant.
Results
Effects of cisplatin and pemetrexed on the viability of A549 cells, alone and in combination. The effect of cisplatin and pemetrexed on the viability of A549 cells was determined using a WST-1 assay. As shown in Figure 1A, cisplatin monotherapy was most cytotoxic to A549 cells, whilst cisplatin and pemetrexed combination therapy had an intermediate effect; pemetrexed monotherapy had the least effect on A549 cells. As illustrated in Figure 1B, after treatment with cisplatin and pemetrexed, both as monotherapy and combination therapy, we noted that a proportion of A549 cells had grown round and become detached from the culture dish, features typical of apoptotic cells. Cisplatin monotherapy was more potent in inducing these apoptotic characteristics than pemetrexed monotherapy and the combination therapy.
Figure 1. Impact of cisplatin and pemetrexed on the viability of A549 cells. A: A549 cells were treated with cisplatin (200 nM) and pemetrexed (100 nM) alone and in combination for 72 h, and the cell survival rate was measured by the WST-1 assay. Data are presented as the mean±SD from three independent experiments. ***Significantly different at p<0.001 compared with the dimethyl sulfoxide (DMSO)-treated group using one-way analysis of variance with Dunnett’s multiple comparison test. B: After 72-h treatment with cisplatin (200 nM) and pemetrexed (100 nM) as singleagents and combined, the morphological changes of A549 cells were captured under optical microscopy.

Effects on inducing ROS-mediated DNA damage. Previous studies reported that many chemotherapy drugs induce cytotoxic effects via ROS-mediated DNA damage (30-32). Therefore, we hypothesized that cisplatin and pemetrexed might cause DNA damage through the generation of ROS. The intracellular ROS level was determined by FCM after treatment with cisplatin and pemetrexed alone and in combination to investigate ROS generation. As indicated in Figure 2, cisplatin monotherapy was more potent in increasing ROS production than pemetrexed monotherapy. In contrast, cisplatin plus pemetrexed combination therapy had an intermediate effect on increasing ROS generation.
Figure 2. The effect of cisplatin and pemetrexed on reactive oxygen species (ROS) production. A: A549 cells were incubated with cisplatin (200 nM) and pemetrexed (100 nM) alone and in combination for 72 h and then stained with ROS deep-red dye. The fluorescent signal was evaluated by flow cytometry, and the median fluorescence intensity (MFI) was determined. The histogram indicates the ROS levels after drug treatment. B: The bar diagram shows the quantification of ROS production. The results are presented as the mean±SD from three independent experiments. ***Significantly different at p<0.001 vs. control by one-way analysis of variance followed by Dunnett’s multiple comparison test.
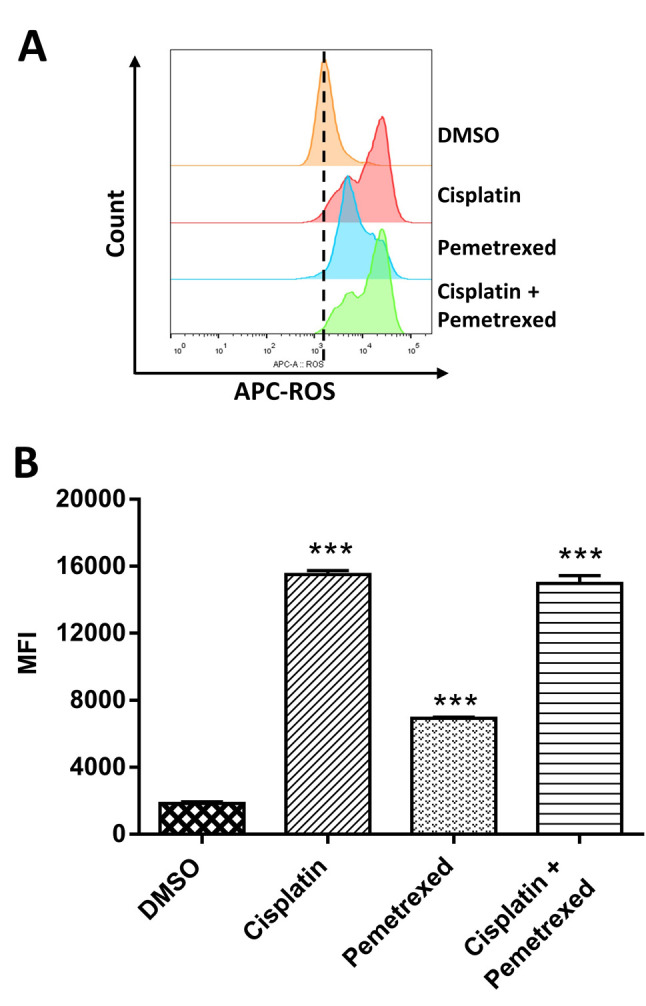
To determine whether cell death caused by cisplatin and pemetrexed was due to DNA fragmentation, we performed a TUNEL assay. As shown in Figure 3A, after 72 h of cisplatin monotherapy, the proportion of cells with fragmented DNA increased from 0.18% to 78%. In contrast, the combination therapy had an intermediate effect, resulting in a rate of 71%, while pemetrexed monotherapy showed a fragmented cell rate of 20.4%. These results are consistent with the findings of the ROS-generation assay.
Figure 3. The effect of cisplatin and pemetrexed on DNA fragmentation. A: A549 cells were stained with fluorescein isothiocyanate-dUTP dye after incubation with cisplatin (200 nM) and pemetrexed (100 nM) as single-agent therapy and combined therapy for 72 h. The fluorescence signal was measured by flow cytometry. (B) The bar diagram shows the quantification of DNA fragmentation. The results were expressed as the mean±SD from three independent experiments. ***Significantly different at p<0.001 vs. control by one-way analysis of variance followed by Dunnett’s multiple comparison test.

Effect on apoptosis of A549 cells. Previous studies reported that ROS-mediated DNA damage triggered growth inhibition via activation of apoptosis signaling (32-34). To investigate whether or not growth inhibition due to apoptosis occurred, we conducted annexin V-FITC and propidium iodide fluorescence staining. As illustrated in Figure 4A and B, combined treatment with cisplatin and pemetrexed resulted in an intermediate increase in the apoptotic population, while cisplatin monotherapy was the most potent apoptosis-inducing agent. By contrast, pemetrexed monotherapy induced the lowest proportion of apoptotic cells.
Figure 4. Effect of cisplatin and pemetrexed on the induction of apoptosis in A549 cells using an annexin V-fluorescein isothiocyanate/propidium iodide (PI) apoptosis detection kit. A: A549 cells were treated with cisplatin (200 nM), and pemetrexed (100 nM) alone and in combination for 72 h and then flow cytometry was used to evaluate apoptosis. Quadrant 1 shows necrotic cells; quadrant 2 shows late apoptotic cells; quadrant 3 shows early apoptotic cells; quadrant 4 shows viable cells. B: Quantification of apoptotic cells. The results were expressed as the mean±SD from three independent experiments. Significance was determined by one-way analysis of variance followed by Dunnett’s multiple comparison test at: *p<0.05, **p<0.01 and ***p<0.001 when compared with the control. C: To evaluate the expression of cleaved poly (ADP-ribose) polymerase (PARP) (Asp 214), western blotting was performed. β-Actin served as a loading control. Representative immunoblots with quantification relative to β-actin from three independent experiments are shown.

Several studies have claimed that ROS-mediated DNA damage can activate a caspase cascade (33,35,36). The cleavage of PARP, a DNA-repair enzyme, by activated caspases (16,37) is a hallmark of apoptosis (38,39). To examine the expression of cleaved PARP, we performed immunoblotting. As shown in Figure 4C, the strongest inducer of cleaved PARP was cisplatin monotherapy, while combination therapy with cisplatin and pemetrexed had an intermediate effect on increasing cleaved PARP. Pemetrexed monotherapy induced the lowest amount of cleaved PARP. This result is consistent with the findings from the apoptosis assay.
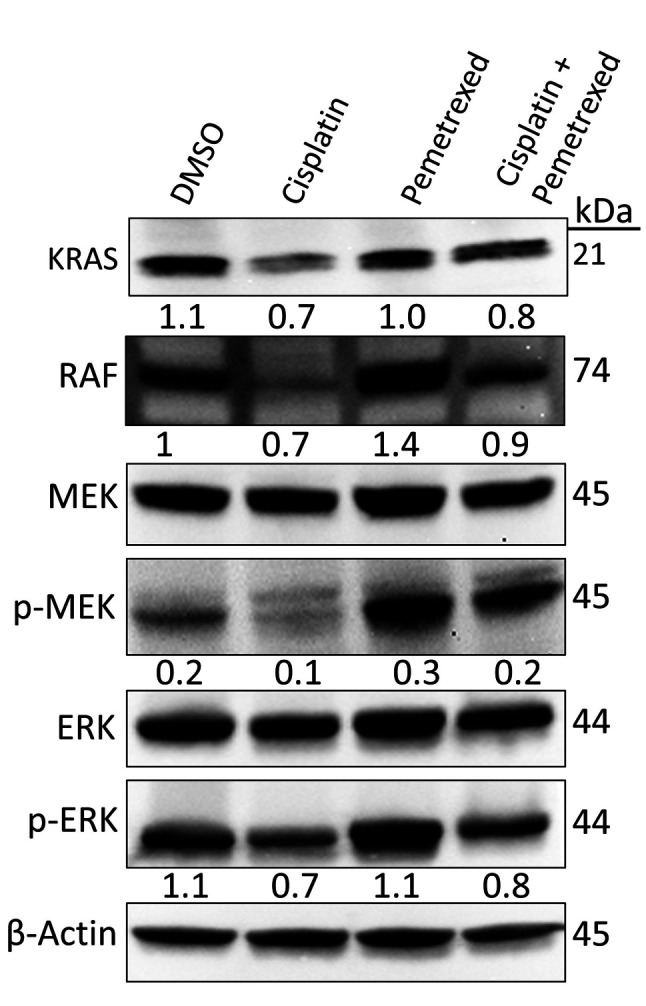
Effects on the RAF/MEK/ERK signaling pathway. Previous studies have revealed that the RAF/MEK/ERK signaling pathway plays a vital role in cell growth, cell-cycle arrest, and in prevention of apoptosis and drug resistance in various cancer cell lines (40-42). The effects of cisplatin and pemetrexed on RAF/MEK/ERK signaling were evaluated by immunoblotting. As indicated in Figure 5, combined treatment with cisplatin and pemetrexed had an intermediate inhibitory effect on the RAF/MEK/ERK signaling cascade, whereas cisplatin monotherapy induced a maximum inhibitory effect. In contrast, when A549 cells were treated with pemetrexed alone, we observed activation of the RAF/MEK/ERK signaling cascade. These results suggest that cisplatin monotherapy is more effective at inhibiting RAF/MEK/ERK signaling in A549 cells than pemetrexed monotherapy and cisplatin plus pemetrexed combination therapy.
Figure 5. Effect of cisplatin and pemetrexed on KRAS proto-oncogene, GTPase (KRAS) signaling in KRAS-dependent A549 cells. Representative western blot data of the levels of KRAS proto-oncogene, GTPase (KRAS), Raf proto-oncogene, serine/threonine kinase (RAF), mitogen-activated protein kinase kinase (MEK), phospho (p)-MEK (Ser 217/221), ERK and p-ERK (Thr 202/Tyr 204) after 72-h treatment with cisplatin (200 nM) and pemetrexed (100 nM) alone and in combination. β-Actin was used as a loading control. Representative immunoblots with quantification relative to β-actin or non-phosphorylated forms from three independent experiments are shown.
Effects on autophagy. mTOR is a signaling molecule of the PI3K/AKT/mTOR signaling pathway closely connected with the inhibition of autophagy (43,44). Several studies have reported that inhibition of the PI3K/AKT/mTOR signaling pathway can induce autophagy (45,46). To determine whether or not cisplatin and pemetrexed induce autophagy, A549 cells were treated for 72 h. The levels of total and phosphorylated mTOR and AKT were examined by immunoblotting. As indicated in Figure 6, cisplatin and pemetrexed exerted a distinctive inhibitory effect on the AKT/mTOR signaling pathway both as monotherapies and in combination.
Figure 6. Effect of cisplatin and pemetrexed on autophagy induction in A549 cells. Representative western blot data of the levels of AKT serine/threonine kinase 1 (AKT), phospho (p)-AKT (Ser 473), mechanistic target of rapamycin kinase (mTOR), p-mTOR (Ser 2448), protein kinase AMP-activated catalytic subunit alpha (AMPKα), p-AMPKα (Thr 172) and microtubule-associated protein 1 light chain 3 alpha (LC3) after 72-h treatment with cisplatin (200 nM) and pemetrexed (100 nM) alone and in combination. β-Actin served as a loading control. Representative immunoblots with quantification relative to β-actin, non-phosphorylated forms, or LC3-I from three independent experiments are shown.
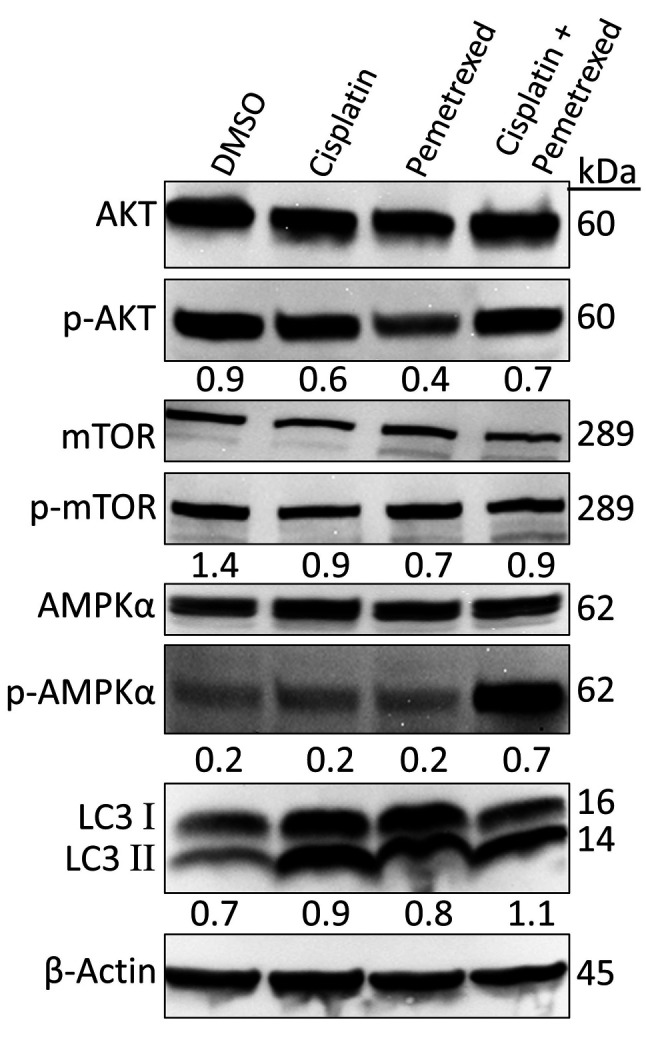
In contrast, AMPK is a key regulator of cellular metabolism and energy balance (47). Previous studies have reported that mTOR is a sensor of changes in the cellular energy state through AMPK (48,49). Activation of AMPK can inhibit mTOR-dependent signaling, which can trigger protein synthesis inhibition (49,50). Several studies have reported that AMPK/mTOR signaling is associated with autophagy, and AMPK can enhance autophagy initiation (49,51). As shown in Figure 6, combined treatment with cisplatin plus pemetrexed increased the expression of p-AMPKα (Thr 172), triggering inhibition of mTOR-dependent signaling. Importantly, when A549 cells were treated with cisplatin and pemetrexed as monotherapies, AMPK activation was not observed.
LC3 is currently broadly used to study autophagy (52,53). During autophagy, conversion of LC3 type II from LC3 type I occurs; therefore, an increase in the ratio of LC3-II/LC3-I expression is considered an autophagy marker (54,55). As shown in Figure 6, cisplatin plus pemetrexed treatment was most potent for increasing the LC3-II/LC3-I ratio, whereas cisplatin monotherapy had an intermediate effect and pemetrexed monotherapy had the least autophagy induction activity.
Effects on cellular senescence. Previous studies have reported that chemotherapy drugs can induce cellular senescence of cancer cells (56-59). Some claim that some chemotherapy drugs can remove cellular senescence (60,61). Therefore, it is crucial to characterize the senescence status correctly when managing patients with cancer.
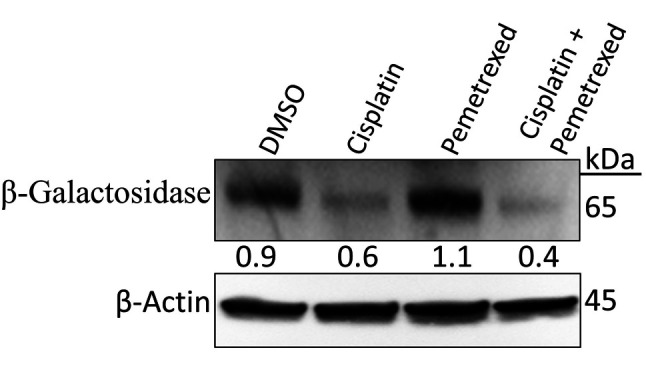
To examine the effect of cisplatin and pemetrexed treatment on cellular senescence in A549 cells, the level of β-galactosidase, a biomarker for senescent cells, was determined by immunoblotting. A549 cells were exposed to drugs for 72 h. As shown in Figure 7, cisplatin plus pemetrexed combined treatment most potently reduced expression of β-galactosidase, whereas cisplatin monotherapy had a milder effect. In contrast, in A549 cells that were exposed to pemetrexed monotherapy, β-galactosidase expression was induced, suggesting that pemetrexed monotherapy may promote adverse effects (62).
Figure 7. Effect of cisplatin and pemetrexed on cellular senescence in A549 cells. A549 cells were treated with cisplatin (200 nM) and pemetrexed (100 nM) alone and in combination for 72 h. The expression level of cellular senescence marker β-galactosidase was determined by western blotting. β-Actin was used as a loading control. Representative immunoblots with quantification relative to β-actin from three independent experiments are shown.
Discussion
Cisplatin and pemetrexed are well-known anticancer drugs but to our knowledge, this is the first study to elucidate the anticancer effects of cisplatin and pemetrexed as monotherapy or combination therapy and to propose potential mechanisms of action in A549 cells.
ROS have been reported to promote or slow the progression of cancer cells (63,64). Furthermore, ROS play a fundamental role in pathological and physiological processes (65,66). Many studies have claimed that excessive ROS generation by chemotherapy drugs can trigger DNA damage and lead to the onset of apoptosis (63,67). In the present study, cisplatin monotherapy most potently increased ROS-mediated DNA damage, causing apoptosis of A549 cells, as evidenced by PARP cleavage (Figure 2-4). In contrast, we found that pemetrexed monotherapy induced greater expression of β-galactosidase, a biomarker for senescent cells, than cisplatin monotherapy or its combination treatment (Figure 7).
The RAS/RAF/MEK/ERK pathway (also known as the mitogen-activated protein kinase signal transmission pathway) has been reported to be crucial in the regulation of several physiological processes, including cell division, growth, development, and death (68,69). This pathway is the core of the signaling network involved in cell division, growth, and development (70,71). RAS acts as a key upstream molecule of the RAF/MEK/ERK signaling pathway (71,72). In the present study, we found that cisplatin monotherapy was the most effective agent at impeding RAF/MEK/ERK signaling in KRAS-dependent A549 cells, suggesting cell growth inhibition (Figure 5).
Previous studies have reported many vital molecules and signaling pathways responsible for regulating autophagy (73-75). The PI3K/AKT/mTOR signaling pathway is well-studied, and critical in regulating the cell cycle, apoptosis, and autophagy (43,76). Drugs that suppress the PI3K/AKT/mTOR signaling pathway have been reported to induce autophagy (77,78). In our study, we found that cisplatin and pemetrexed inhibited AKT/mTOR signaling, triggered activation of autophagy (Figure 6). Various studies have reported that AMPK positively regulates autophagy and inhibits the mTOR-dependent signaling pathway (28,79,80). The AKT/mTOR signaling pathway is involved in regulating autophagy (81). AKT controls autophagy mainly through the alteration of mTOR activity (82-84). Our results showed that cisplatin plus pemetrexed therapy induced AMPK activation, thereby stimulating autophagy through the blockade of the mTOR-dependent signaling pathway (Figure 6). In addition, several studies have claimed that LC3 is an autophagosome marker and can be used for monitoring autophagy (85-87). In actuality, the LC3-II/LC3-I ratio is a hallmark of the degree of activation of autophagy (88,89). In the present study, our approach to detect the conversion of LC3 (LC3-I to LC3-II) by western blotting revealed cisplatin plus pemetrexed combination therapy to be more potent than both cisplatin and pemetrexed monotherapies for inducing autophagy in A549 cells (Figure 6).
Finally, this study revealed that cisplatin and pemetrexed induced growth inhibition in A549 cells in four explicit systems (Figure 8). Firstly, these drugs enhance ROS-mediated DNA damage, which triggers apoptosis. Secondly, they induced autophagy by regulating the AMPK/mTOR and PI3K/AKT/ mTOR signaling pathways. Thirdly, the generation of fragmented DNA may regulate cellular senescence in A549 cells. Fourthly, these drugs impede the RAS/RAF/MEK/ERK signaling pathway, thereby inhibiting the growth of A549 cells. Overall, our findings suggest that cisplatin monotherapy is the most potent against A549 cells, as evidenced by the cell viability outcome, RAS/RAF/MEK/ERK pathway inhibition, and induction of apoptosis triggered by excessive generated ROS.
Figure 8. The hypothetical mechanism underlying cisplatin- and pemetrexed-induced autophagy, cellular senescence, and apoptosis in A549 cells. A: Drug treatment enhances reactive oxygen species (ROS)-mediated DNA damage to trigger apoptosis and induce cellular senescence. B: Drug treatment suppresses the KRAS proto-oncogene, GTPase (KRAS)/Raf proto-oncogene, serine/threonine kinase (RAF)/ mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway to induce apoptosis. C: Cisplatin and pemetrexed induce autophagy in A549 cells through the regulation of the AKT serine/threonine kinase 1 (AKT)/mechanistic target of rapamycin kinase (mTOR) and AMP-activated protein kinase (AMPK)/mTOR signaling pathways.

Conclusion
Our results revealed the mechanisms of action of cisplatin and pemetrexed alone and in combination against A549 cells. We conclude that cisplatin monotherapy may be more efficient than pemetrexed monotherapy or cisplatin plus pemetrexed combination therapy in eliminating KRAS-dependent cells.
Conflicts of Interest
The Authors declare no competing financial interests.
Authors’ Contributions
Md Mohiuddin and Kazuo Kasahara conceived this study; Md Mohiuddin carried out the experiments; Md Mohiuddin and Kazuo Kasahara discussed and interpreted the results; Md Mohiuddin wrote the article; Kazuo Kasahara supervised the experiments and project.
Acknowledgements
The Authors would like to thank Ms. Miki Kashiwano (Department of Respiratory Medicine, Graduate School of Medical Sciences, Kanazawa University) for her technical assistance. This work was supported by a Grant-in-Aid for Scientific Research (C) (JSPS KAKENHI Grant Number 17K09606) to K.K. The funders had no role in the study design, data collection or interpretation, or decision to submit the work for publication.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300. doi: 10.21037/tlcr.2016.06.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jancík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010:150960. doi: 10.1155/2010/150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westcott PM, To MD. The genetics and biology of KRAS in lung cancer. Chin J Cancer. 2013;32(2):63–70. doi: 10.5732/cjc.012.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, Nafa K, Riedel ER, Hsu M, Pao W, Miller VA, Ladanyi M. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14(18):5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Del Re M, Rofi E, Restante G, Crucitta S, Arrigoni E, Fogli S, Di Maio M, Petrini I, Danesi R. Implications of KRAS mutations in acquired resistance to treatment in NSCLC. Oncotarget. 2017;9(5):6630–6643. doi: 10.18632/oncotarget.23553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang K, DeClue JE, Vass WC, Papageorge AG, McCormick F, Lowy DR. Suppression of c-ras transformation by GTPase-activating protein. Nature. 1990;346(6286):754–756. doi: 10.1038/346754a0. [DOI] [PubMed] [Google Scholar]
- 10.Castellano E, Downward J. RAS interaction with PI3K: More than just another effector pathway. Genes Cancer. 2011;2(3):261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, Evangelisti C, Martelli AM, Montalto G, Cervello M, McCubrey JA. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011;3(3):192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandey SS, Singh S, Pathak C, Tiwari BS. “Programmed cell death: A process of death for survival” – How far terminology pertinent for cell death in unicellular organisms. J Cell Death. 2018;11:1179066018790259. doi: 10.1177/1179066018790259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisner V, Picard M, Hajnóczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018;20(7):755–765. doi: 10.1038/s41556-018-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14. doi: 10.1186/1477-3163-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, Herceg Z, Wang ZQ, Schulze-Osthoff K. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13(3):978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JH, Hales CN, Ozanne SE. DNA damage, cellular senescence and organismal ageing: causal or correlative. Nucleic Acids Res. 2007;35(22):7417–7428. doi: 10.1093/nar/gkm681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509(7501):439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24(22):2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015;4:184–192. doi: 10.1016/j.redox.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388. doi: 10.1038/cdd.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li F, Guo H, Yang Y, Feng M, Liu B, Ren X, Zhou H. Autophagy modulation in bladder cancer development and treatment (Review) Oncol Rep. 2019;42(5):1647–1655. doi: 10.3892/or.2019.7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26(4):605–616. doi: 10.1038/s41418-018-0252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Liu Y, Wang D, Xu Y, Dong R, Yang Y, Lv Q, Chen X, Zhang Z. The upstream pathway of mTOR-mediated autophagy in liver diseases. Cells. 2019;8(12):1597. doi: 10.3390/cells8121597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang P, Jiang B, Li Y, Liu Z, Zhang P, Zhang M, Huang X, Xiao X. Autophagy promotes angiogenesis via AMPK/Akt/mTOR signaling during the recovery of heat-denatured endothelial cells. Cell Death Dis. 2018;9(12):1152. doi: 10.1038/s41419-018-1194-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakai A, Kasahara K, Ohmori T, Kimura H, Sone T, Fujimura M, Nakao S. MET increases the sensitivity of gefitinib-resistant cells to SN-38, an active metabolite of irinotecan, by up-regulating the topoisomerase I activity. J Thorac Oncol. 2012;7(9):1337–1344. doi: 10.1097/JTO.0b013e31825cca4c. [DOI] [PubMed] [Google Scholar]
- 30.Huang G, Pan ST. ROS-mediated therapeutic strategy in chemo-/radiotherapy of head and neck cancer. Oxid Med Cell Longev. 2020;2020:5047987. doi: 10.1155/2020/5047987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SJ, Kim HS, Seo YR. Understanding of ROS-inducing strategy in anticancer therapy. Oxid Med Cell Longev. 2019;2019:5381692. doi: 10.1155/2019/5381692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davalli P, Marverti G, Lauriola A, D’Arca D. Targeting oxidatively induced DNA damage response in cancer: Opportunities for novel cancer therapies. Oxid Med Cell Longev. 2018;2018:2389523. doi: 10.1155/2018/2389523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863(12):2977–2992. doi: 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Zhao X, Tang M, Li L, Lei Y, Cheng P, Guo W, Zheng Y, Wang W, Luo N, Peng Y, Tong A, Wei Y, Nie C, Yuan Z. The role of ROS and subsequent DNA-damage response in PUMA-induced apoptosis of ovarian cancer cells. Oncotarget. 2017;8(14):23492–23506. doi: 10.18632/oncotarget.15626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuo Y, Xiang B, Yang J, Sun X, Wang Y, Cang H, Yi J. Oxidative modification of caspase-9 facilitates its activation via disulfide-mediated interaction with Apaf-1. Cell Res. 2009;19(4):449–457. doi: 10.1038/cr.2009.19. [DOI] [PubMed] [Google Scholar]
- 36.Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32(33):3789–3797. doi: 10.1038/onc.2012.556. [DOI] [PubMed] [Google Scholar]
- 37.Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Darzynkiewicz Z. Cleavage of Poly(ADP-ribose) polymerase measured in situ in individual cells: relationship to DNA fragmentation and cell cycle position during apoptosis. Exp Cell Res. 2000;255(1):125–132. doi: 10.1006/excr.1999.4796. [DOI] [PubMed] [Google Scholar]
- 39.He J, Whitacre CM, Xue LY, Berger NA, Oleinick NL. Protease activation and cleavage of poly(ADP-ribose) polymerase: an integral part of apoptosis in response to photodynamic treatment. Cancer Res. 1998;58(5):940–946. [PubMed] [Google Scholar]
- 40.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16(3):368–377. doi: 10.1038/cdd.2008.148. [DOI] [PubMed] [Google Scholar]
- 42.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18(2):189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 43.Xu F, Na L, Li Y, Chen L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020;10:54. doi: 10.1186/s13578-020-00416-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Zhai C, Cheng J, Mujahid H, Wang H, Kong J, Yin Y, Li J, Zhang Y, Ji X, Chen W. Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PLoS One. 2014;9(3):e90563. doi: 10.1371/journal.pone.0090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Pi C, Wang G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed Pharmacother. 2018;103:699–707. doi: 10.1016/j.biopha.2018.04.072. [DOI] [PubMed] [Google Scholar]
- 46.Wang N, He J, Pan C, Wang J, Ma M, Shi X, Xu Z. Resveratrol activates autophagy via the AKT/mTOR signaling pathway to improve cognitive dysfunction in rats with chronic cerebral hypoperfusion. Front Neurosci. 2019;13:859. doi: 10.3389/fnins.2019.00859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamargo-Gómez I, Mariño G. AMPK: Regulation of metabolic dynamics in the context of autophagy. Int J Mol Sci. 2018;19(12):3812. doi: 10.3390/ijms19123812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garza-Lombó C, Schroder A, Reyes-Reyes EM, Franco R. mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Curr Opin Toxicol. 2018;8:102–110. doi: 10.1016/j.cotox.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin SC, Hardie DG. AMPK: Sensing glucose as well as cellular energy status. Cell Metab. 2018;27(2):299–313. doi: 10.1016/j.cmet.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12(16):1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- 51.Yuan J, Zhao X, Hu Y, Sun H, Gong G, Huang X, Chen X, Xia M, Sun C, Huang Q, Sun Y, Kong W, Kong W. Autophagy regulates the degeneration of the auditory cortex through the AMPK-mTOR-ULK1 signaling pathway. Int J Mol Med. 2018;41(4):2086–2098. doi: 10.3892/ijmm.2018.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Runwal G, Stamatakou E, Siddiqi FH, Puri C, Zhu Y, Rubinsztein DC. LC3-positive structures are prominent in autophagy-deficient cells. Sci Rep. 2019;9(1):10147. doi: 10.1038/s41598-019-46657-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dhingra A, Alexander D, Reyes-Reveles J, Sharp R, Boesze-Battaglia K. Microtubule-associated protein 1 light chain 3 (LC3) isoforms in RPE and retina. Adv Exp Med Biol. 2018;1074:609–616. doi: 10.1007/978-3-319-75402-4_74. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Zhang X, Chu ESH, Chen X, Kang W, Wu F, To KF, Wong VWS, Chan HLY, Chan MTV, Sung JJY, Wu WKK, Yu J. Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J. 2018;32(1):37–51. doi: 10.1096/fj.201601393R. [DOI] [PubMed] [Google Scholar]
- 55.Kaludercic N, Maiuri MC, Kaushik S, Fernández ÁF, de Bruijn J, Castoldi F, Chen Y, Ito J, Mukai R, Murakawa T, Nah J, Pietrocola F, Saito T, Sebti S, Semenzato M, Tsansizi L, Sciarretta S, Madrigal-Matute J. Comprehensive autophagy evaluation in cardiac disease models. Cardiovasc Res. 2020;116(3):483–504. doi: 10.1093/cvr/cvz233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102(20):1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mikuła-Pietrasik J, Niklas A, Uruski P, Tykarski A, Książek K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell Mol Life Sci. 2020;77(2):213–229. doi: 10.1007/s00018-019-03261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang L, Fang J, Chen J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017;3:17049. doi: 10.1038/cddiscovery.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mohiuddin M, Kimura H, Sone T, Matsuoka H, Saeki K, Terada N, Nguyen MAT, Kasahara K. Pemetrexed disodium heptahydrate suppressed apoptosis by pRB hyperphosphorylation and CDKs overexpression in pc9 cells. Cancer Science. 2021;112:327–327. [Google Scholar]
- 60.Hansel C, Jendrossek V, Klein D. Cellular senescence in the lung: The central role of senescent epithelial cells. Int J Mol Sci. 2020;21(9):3279. doi: 10.3390/ijms21093279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu M, Meng P, Ling X, Zhou L. Advancements in therapeutic drugs targeting of senescence. Ther Adv Chronic Dis. 2020;11:2040622320964125. doi: 10.1177/2040622320964125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H, Campisi J. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7(2):165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44(5):479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Khan MA, Sethi G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules. 2019;9(11):735. doi: 10.3390/biom9110735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bardaweel SK, Gul M, Alzweiri M, Ishaqat A, ALSalamat HA, Bashatwah RM. Reactive oxygen species: the dual role in physiological and pathological conditions of the human body. Eurasian J Med. 2018;50(3):193–201. doi: 10.5152/eurasianjmed.2018.17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zarkovic N. Roles and functions of ROS and RNS in cellular physiology and pathology. Cells. 2020;9(3):767. doi: 10.3390/cells9030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ivanova D, Zhelev Z, Aoki I, Bakalova R, Higashi T. Overproduction of reactive oxygen species - obligatory or not for induction of apoptosis by anticancer drugs. Chin J Cancer Res. 2016;28(4):383–396. doi: 10.21147/j.issn.1000-9604.2016.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 69.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol Lett. 2016;12(5):3045–3050. doi: 10.3892/ol.2016.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007. doi: 10.3892/etm.2020.8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773(8):1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 73.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khandia R, Dadar M, Munjal A, Dhama K, Karthik K, Tiwari R, Yatoo MI, Iqbal HMN, Singh KP, Joshi SK, Chaicumpa W. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: Current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. 2019;8(7):674. doi: 10.3390/cells8070674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, Li W, Hu J, Lu C, Liu Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020;11(9):797. doi: 10.1038/s41419-020-02998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim KY, Park KI, Kim SH, Yu SN, Park SG, Kim YW, Seo YK, Ma JY, Ahn SC. Inhibition of autophagy promotes salinomycin-induced apoptosis via reactive oxygen species-mediated PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human prostate cancer cells. Int J Mol Sci. 2017;18(5):1088. doi: 10.3390/ijms18051088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xia J, Dai L, Wang L, Zhu J. Ganoderic acid DM induces autophagic apoptosis in non-small cell lung cancer cells by inhibiting the PI3K/Akt/mTOR activity. Chem Biol Interact. 2020;316:108932. doi: 10.1016/j.cbi.2019.108932. [DOI] [PubMed] [Google Scholar]
- 79.Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sridharan S, Jain K, Basu A. Regulation of autophagy by kinases. Cancers (Basel) 2011;3(2):2630–2654. doi: 10.3390/cancers3022630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castets P, Rion N, Théodore M, Falcetta D, Lin S, Reischl M, Wild F, Guérard L, Eickhorst C, Brockhoff M, Guridi M, Ibebunjo C, Cruz J, Sinnreich M, Rudolf R, Glass DJ, Rüegg MA. mTORC1 and PKB/Akt control the muscle response to denervation by regulating autophagy and HDAC4. Nat Commun. 2019;10(1):3187. doi: 10.1038/s41467-019-11227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Altomare DA, Khaled AR. Homeostasis and the importance for a balance between AKT/mTOR activity and intracellular signaling. Curr Med Chem. 2012;19(22):3748–3762. doi: 10.2174/092986712801661130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA. 2010;107(44):18880–18885. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Toledo M, Batista-Gonzalez A, Merheb E, Aoun ML, Tarabra E, Feng D, Sarparanta J, Merlo P, Botrè F, Schwartz GJ, Pessin JE, Singh R. Autophagy regulates the liver clock and glucose metabolism by degrading CRY1. Cell Metab. 2018;28(2):268–281.e4. doi: 10.1016/j.cmet.2018.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sarkar C, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, Lipinski MM. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10(12):2208–2222. doi: 10.4161/15548627.2014.981787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bhutia SK, Praharaj PP, Bhol CS, Panigrahi DP, Mahapatra KK, Patra S, Saha S, Das DN, Mukhopadhyay S, Sinha N, Panda PK, Naik PP. Monitoring and measuring mammalian autophagy. Methods Mol Biol. 2019;1854:209–222. doi: 10.1007/7651_2018_159. [DOI] [PubMed] [Google Scholar]
- 89.Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010;221(2):117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]