

Abstract
The capacity of RNA viruses to adapt to new hosts and rapidly escape the host immune system is largely attributable to de novo genetic diversity that emerges through mutations in RNA. Although the molecular spectrum of de novo mutations—the relative rates at which various base substitutions occur—are widely recognized as informative toward understanding the evolution of a viral genome, little attention has been paid to the possibility of using molecular spectra to infer the host origins of a virus. Here, we characterize the molecular spectrum of de novo mutations for SARS-CoV-2 from transcriptomic data obtained from virus-infected cell lines, enabled by the use of sporadic junctions formed during discontinuous transcription as molecular barcodes. We find that de novo mutations are generated in a replication-independent manner, typically on the genomic strand, and highly dependent on mutagenic mechanisms specific to the host cellular environment. De novo mutations will then strongly influence the types of base substitutions accumulated during SARS-CoV-2 evolution, in an asymmetric manner favoring specific mutation types. Consequently, similarities between the mutation spectra of SARS-CoV-2 and the bat coronavirus RaTG13, which have accumulated since their divergence strongly suggest that SARS-CoV-2 evolved in a host cellular environment highly similar to that of bats before its zoonotic transfer into humans. Collectively, our findings provide data-driven support for the natural origin of SARS-CoV-2.
Keywords: SARS-CoV-2, molecular spectrum, de novo mutations, mutational signature, evolutionary origin, mRNA mutation
Graphical abstract
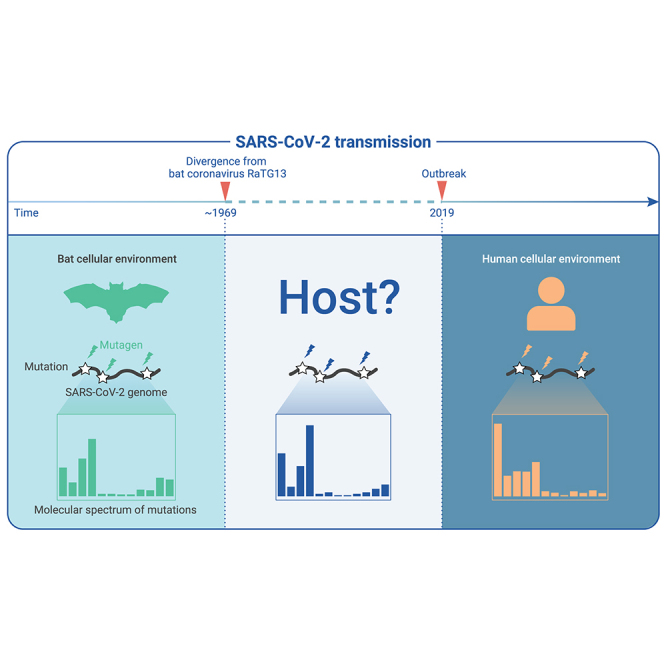
Public summary
-
•
The asymmetric de novo mutations in SARS-CoV-2 are induced by mutagenic mechanisms in the host cellular environment
-
•
De novo mutations determine the molecular spectrum of accumulated mutations during SARS-CoV-2 evolution
-
•
Molecular spectra of accumulated mutations in betacoronaviruses cluster according to the host species instead of the phylogenetic relationship
-
•
The mutations accumulated in SARS-CoV-2 prior to its transmission to humans are consistent with an evolutionary process in a bat host
Introduction
Since the first reports of coronavirus disease 2019 (COVID-19), controversies have persisted regarding the origin of its causative agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1 While many studies have proposed that a natural origin of SARS-CoV-2 provides the simplest explanation for its emergence,2, 3, 4, 5 counter arguments have speculated that an accidental laboratory escape of an engineered SARS-like coronavirus could not be excluded.6,7 One factor driving this prolonged controversy is a lack of empirical data that clearly support either possibility. Moreover, the search for related viruses in wild animals that are sufficiently genetically similar to SARS-CoV-2 has not yet shown fruitful results. Since its divergence from RaTG13—the genetically most similar virus identified to date—approximately 50 years ago,8, 9, 10, 11 SARS-CoV-2 had accumulated ∼500 mutations before its jump to human hosts (RaTG13 accumulated ∼600 mutations meanwhile).
As per the traditional aphorism, “the absence of evidence is not evidence of absence,” especially when considering the vast number of unexplored wild animals and the even greater number of viruses they harbor. While the considerable efforts of many research groups to search in nature for a closely related coronavirus may yet provide many insights into the origins of SARS-CoV-2, we instead turned our attention to the ∼500 base substitutions that have accumulated in SARS-CoV-2, because, we hypothesized, they could provide us with unprecedented statistical power to test if they accumulated through an evolutionary process that was consistent with those occurring in known, natural coronaviruses.
Virus evolution begins with a de novo mutation in its genome, thus providing new variations in the genetic material that is retained or lost under different selection pressures. On the one hand, mutations that confer a fitness advantage or disadvantage will increase or decrease in frequency through natural selection. For example, some mutations may affect transmission efficiency or capacity to escape from the host immune system. On the other hand, neutral mutations, which have little fitness effect, remain unaffected by natural selection, resulting in their genomic accumulation of an equal chance through random genetic drift.
Since de novo mutations are the starting point for genetic variation, their molecular spectrum—i.e., the relative rates at which all 12 possible types of base substitutions arise—has been widely recognized as an essential parameter for understanding genome evolution. Since this spectrum of mutation rates can be used to predict sequence changes under neutral processes, this metric can serve as a null model for optimizing phylogenetic reconstructions based on maximum likelihood or for detecting genomic signals of positive selection.12, 13, 14, 15, 16, 17, 18
In addition to its applications in evolutionary biology, the molecular spectrum has been used to describe somatic mutations accumulated in the genome of cancer cells and to identify etiological agents involved in tumorigenesis. Various mutational processes, such as exposure to mutagens and enzymatic modification, will each generate unique combinations of mutation types, termed “mutational signatures.” Therefore, the molecular spectrum can be used to infer the suite of operative mutational processes through which somatic mutations accumulated in the genome of a cancer cell.19, 20, 21 For example, excess C > A or G > T transversions, mainly caused by polycyclic aromatic hydrocarbons, have been identified as a mutational signature for tobacco smoking in the development of lung cancers.21,22
Following the same logic, we propose that the molecular spectrum of mutations that accumulated during the evolution of a viral genome may be informative for inferring the ancestral hosts of that virus, because viruses share the same sets of mutagens in the cellular environment as their hosts. However, we realize that this strategy heavily relies on the validity of three assumptions. First, the cellular environment is substantially variable among different hosts such that they can create mutational signatures sufficiently distinct in the viral genome for tracing its transmission history. Second, de novo mutations in the viral genome are predominantly introduced through processes specific to the host cellular environment, rather than through inherently viral mechanisms of mutagenesis. Third, the molecular spectrum of mutations accumulated in the evolution of a given virus is largely determined by de novo mutations rather than by natural selection, which in principle could blur any mutational signatures. We realize that the key to testing these assumptions is to characterize the molecular spectrum of de novo mutations in SARS-CoV-2, before natural selection has a chance to affect their apparent frequency.
In this study, we first tested each of these three assumptions using a computational strategy specifically developed for detecting de novo mutations in SARS-CoV-2 from the transcriptome of virus-infected cell lines. After validating the three assumptions, we constructed a phylogenetic tree for SARS-CoV-2 and related coronaviruses and identified hundreds of mutations that accumulated in its genome before jumping to human hosts. Finally, we investigated whether the accumulation of these mutations was compatible with other viruses in the phylogenetic tree that are reported to have a natural origin. Our data-driven investigation provides transparent and empirical support for the natural origin of SARS-CoV-2.
Results
The rationale for detecting de novo mutations in SARS-CoV-2
The identification of de novo mutations in SARS-CoV-2 has been technically challenging. For example, the molecular spectrum of de novo mutations cannot be inferred from within-individual polymorphisms in samples of bronchoalveolar lavage fluid23, 24, 25 or from mutations that accumulated among patients (i.e., among-patient polymorphisms),16,26,27 because we are specifically concerned with the extent to which the molecular spectrum of mutations that accumulated during virus evolution reflect the molecular spectrum of de novo mutations (refer to the third aforementioned assumption).
For evolutionary genomics studies, de novo mutations are ideally detected in newly synthesized virus genomes, before natural selection has a chance to act, for example, using RNA sequencing data from SARS-CoV-2-infected cells. However, de novo RNA mutations in SARS-CoV-2 cannot be directly inferred from mismatches between sequencing reads and the reference genome because of the high error rate inherent to high-throughput sequencing (10−3 to 10−4 errors per nucleotide), which is approximately two orders of magnitude higher than the average de novo RNA mutation rate (10−5 to 10−6 mutations per nucleotide).28 Furthermore, errors generated during library preparation—that is, in the procedures of reverse transcription and polymerase chain reaction (PCR)-based amplification—will also increase the complexity of identifying bona fide RNA mutations.29
Nevertheless, experimental strategies have been designed to detect rare de novo mutations in RNA viruses, such as for poliovirus30 and for Ebola virus.31 Acevedo et al. developed circular sequencing (CirSeq) in which RNA molecules are first circularized, then serve as the template for rolling circle reverse transcription; bona fide RNA mutations will appear periodically in the resultant complement DNA30 (Figure 1A, left panel). Different from CirSeq, replicated sequencing (Rep-seq), developed by Gout et al., could also be used to detect de novo mutations in RNA viruses, although this method was originally developed for detecting de novo mRNA mutations—the differences in sequence between an mRNA and its template DNA. In Rep-seq, each mRNA is barcoded with a unique oligonucleotide and is then reverse transcribed three times. Mismatches repeatedly observed in the sequencing reads that share the same barcode are considered to be bona fide RNA mutations that were extant in the mRNA29 (Figure 1A, right panel). In addition, a hybrid strategy named accurate RNA consensus sequencing (ARC-seq) was later developed, which uses a rolling circle strategy for multiple times of reverse transcription of an RNA molecule in conjunction with oligonucleotide barcodes for recognizing reads of the same RNA molecule.32
Figure 1.

The molecular spectrum of de novo SARS-CoV-2 mutations
(A) Schematic of two experimental approaches previously developed to detect RNA mutations. Bona fide RNA mutations (magenta stars) should be repeatedly detected, while errors generated during reverse transcription, PCR amplification, or high-throughput sequencing (green stars) should only be occasionally detected.
(B) Schematic of our junction-barcoding approach to detect RNA mutations for SARS-CoV-2. The genomic coordinates of a pair of upstream and downstream sites of sporadic junctions can serve as the molecular barcode to group sequencing reads derived from the same negative-sense subgenome into read families. Bona fide RNA mutations should be unanimously detected in a read family.
(C) Comparison of overall mismatch frequency between our junction-barcoding approach and the conventional computational approach.
(D) The numbers of de novo RNA mutations of 12 base-substitution types, with respect to the positive-sense SARS-CoV-2 genome. Two-tailed p values were calculated from binomial tests assuming an equal frequency for each type of base substitutions.
(E) The molecular spectrum of de novo SARS-CoV-2 mutations. Two-tailed p values were calculated from Fisher's exact tests.
Here, we sought to develop a strategy for detecting de novo mutations in SARS-CoV-2. SARS-CoV-2 is a positive-sense single-strand RNA virus,8,33,34 replicating its genome within host cells through two rounds of transcription using an RNA-dependent RNA polymerase (RdRp) encoded in the viral genome: the RdRp first transcribes the positive-sense genomic RNA to generate a few intermediate negative-sense genomic RNAs that can then serve as a template to transcribe several positive-sense RNA genomes. These positive-sense RNA genomes are then packed into individual virions (Figure S1A, left panel). This two-round transcription mechanism is also employed by SARS-CoV-2 to synthesize various positive-sense subgenomes that function as viral mRNAs for the translation of viral proteins (Figure S1A, right panel).
The SARS-CoV-2 subgenomes are nested within the genomic RNA and are produced by discontinuous transcription from positive-sense genomic RNA into intermediate negative-sense subgenomic RNA (i.e., via polymerase jumping, Figure S1B). In addition to canonical junctions generated by the leader-to-body fusion occurring between the leader and one of the eight body transcription-regulating sequences, a huge number of noncanonical fusions can be found at random sites in the viral genome.35 Most resultant noncanonical junctions are present at a low frequency, likely resulting from sporadic errors in discontinuous transcription.36 Nevertheless, the negative-sense subgenomic RNA bearing such sporadic junctions can serve as a template for transcription into multiple identical positive-sense subgenomic RNAs.37
We realized that sporadic junctions could serve as the molecular barcode for a negative-sense subgenomic RNA, since it is unlikely that two independently synthesized, positive-sense subgenomic RNAs will share identical genomic coordinates of a pair of upstream and downstream junction sites. Therefore, these “junction barcodes” can be used to group sequencing reads into families; each read family will include all sequencing reads derived from the positive-sense subgenomic RNAs that have been transcribed from the same negative-sense subgenomic RNA. Repeated detection of the same mismatch within a read family implies that an RNA mutation was present in the negative-sense subgenomes (Figure 1B). In contrast, errors generated during reverse transcription or sequencing can be excluded, as they will appear randomly (i.e., not at identical sites).
C > U and G > U are over-represented in SARS-CoV-2 de novo mutations
Using the junction-barcoding approach (Figures S2 and S3A), we effectively distinguished reverse transcription or sequencing errors from the bona fide RNA mutations (Figure 1C, see supplemental materials and methods for details). From the RNA sequencing data for SARS-CoV-2-infected Vero cells,35 we identified a total of 197 de novo RNA point mutations in the SARS-CoV-2 genome. These mutations could be categorized into 12 distinct types with respect to the positive-sense genomic RNA (Figure 1D, with an example shown in Figure S4). To estimate the rate of each mutation type, we controlled for the nucleotide composition of the viral genome and the potential coverage bias generated during high-throughput sequencing. For this calculation, we estimated the coverage for each site by all read families (similar to RNA mutation calling in Figure S2, but no mismatch was required) and aggregated this coverage according to the nucleotide (A, C, G, or U) in the reference genome. We divided the number of mutations of each type by the total coverage of all sites with the nucleotide in the reference genome, and used this ratio to infer the molecular spectrum of mutations in SARS-CoV-2 in Vero cells (Figure 1E).
Among the 197 RNA mutations we identified in SARS-CoV-2, 122 were transitions (purine to purine or pyrimidine to pyrimidine interchanges) and 75 were transversions (interchanges of a purine to a pyrimidine or vice versa), which significantly deviated from the randomly expected ratio (4:8, p = 3 × 10−16, binomial test, Figure 1D). On average, transitions occurred three times more frequently (1.1 × 10−5 substitutions per site) than transversions (3.6 × 10−6 substitutions per site, Figure 1E), which was possibly attributable to the structural similarity between bases that are substituted in transitions.
In particular, C > U mutations appeared to be the most abundant transition (p = 6 × 10−5, binomial test with probability equal to ¼, Figure 1D). Among the eight types of RNA transversions, G > U mutations occurred much more frequently than the other seven mutation types (p = 8 × 10−6, binomial test with probability equal to ⅛, Figure 1D), reaching frequencies comparable with that of transitions. We then focused on these two major signatures of SARS-CoV-2 mutations, i.e., over-representation of G > U and C > U mutations, in subsequent analyses.
The molecular spectrum of de novo mutations shapes the spectrum of polymorphisms during the evolution of SARS-CoV-2 in human patients
Given that the de novo mutations provide the raw materials for virus evolution (Figure 2A), before the further investigation of the molecular mechanisms underlying the over-representation of G > U and C > U mutations, we first sought to determine the levels at which SARS-CoV-2 evolution in human patients was affected by the molecular spectrum of de novo mutations. To this end, we retrieved 34,853 high-quality sequences of SARS-CoV-2 variants isolated from patients worldwide (Table S1) from GISAID (global initiative on sharing all influenza data),38 and reconstructed the genomic sequence of their last common ancestor.
Figure 2.

The molecular spectrum of SARS-CoV-2 polymorphisms among patients
(A) The emergence of among-patient polymorphisms through the accumulation of de novo mutations. The frequency of a de novo mutation (the magenta star with an arrow pointing to it) may be increased by positive selection, decreased by negative selection, or changed through genetic drift due to chance events. If a mutation becomes predominant within a patient, it can be detected as an among-patient polymorphism.
(B) The molecular spectrum of among-patient polymorphisms at 4-fold degenerate sites in SARS-CoV-2.
(C) A scatterplot shows the molecular spectrum of de novo mutations versus among-patient polymorphisms at 4-fold degenerate sites in SARS-CoV-2. Each dot represents a base-substitution type, colored according to (B). Pearson’s correlation coefficient (r) and the corresponding p value are shown.
(D) The molecular spectrum of among-patient polymorphisms in the whole genome of SARS-CoV-2.
(E) Similar to (C), for all polymorphisms.
We then identified genetic differences between each variant and the ancestor and treated those differences observed in at least two patients as among-patient polymorphisms. This process enabled the removal of singletons that were potentially generated by sequencing errors. The molecular spectrum of SARS-CoV-2 among-patient polymorphisms (Figure 2B) highly resembled that of the de novo mutations at 4-fold degenerate sites (Figure 2C, r = 0.81, p = 0.001), as well as in the whole genome (Figures 2D and 2E, r = 0.86, p = 3 × 10−4). The signatures indicating over-representation of G > U and C > U among de novo mutations were also observed in among-patient polymorphisms. This finding indicated that the molecular spectrum of de novo mutations dominated the base-substitution types of polymorphisms in SARS-CoV-2 during its evolution in human patients.
It is worth noting that, despite their apparent similarity, the de novo mutation identified in this study is by definition different from the among-patient polymorphisms,16,26 because the latter has been influenced by natural selection related to the processes of infection, propagation, or release from infected cells.13,39 Only with the characterization of the molecular spectrum of de novo mutations can we thus examine the relative influence of mutation versus selection in driving the genomic evolution of SARS-CoV-2 (Figure 2A). The observation that the molecular spectrum of among-patient polymorphisms largely resembled that of de novo mutations indicated that the proportions of deleterious mutations were largely uniform among the 12 types of base substitutions.
Replication-independent asymmetric emergence of mutations on single-strand RNA
Presumably, the over-representation of G > U and C > U mutations could be consequences of transcriptional errors that produce RNAs carrying different sequences from that of the template (i.e., replication-dependent mutations), or these mismatches could result from exposure to environmental mutagens that can induce mutations in the absence of transcriptional machinery (i.e., replication-independent mutations).40 We realized that these two mechanisms could be distinguished by comparing the frequency of G > U (or C > U) mutations with that of its complement mutation, C > A (or G > A). For example, a mutation observed in the negative-sense genome (e.g., C > A, which will be transcribed into a G > U mutation in the positive-sense genome) that is generated during positive to negative strand transcription should occur at approximately equal frequency in negative to positive strand transcription (leading to a C > A mutation in the positive-sense genome), because the same polymerase performs both functions. Consequently, when the SARS-CoV-2 replication cycle (i.e., two rounds of transcription) is completed, the molecular spectrum of replication-dependent mutations should be “symmetric,” meaning that the frequencies of G > U and its complement mutation, C > A, should be similar (Figure 3A, left panel). Alternatively, if a mutation is generated in a replication-independent manner, for example, induced by mutagens specifically in the positive-sense (or negative-sense) single-strand RNA of SARS-CoV-2, then the frequency of complement mutations will not necessarily be symmetrical (Figure 3A, middle panel).
Figure 3.

Predictions and observations for various mutagenic mechanisms on the symmetry of mutations
(A) Predictions on the symmetry between a pair of complement base-substitution types for three potential mutagenic mechanisms. If de novo mutations are introduced during transcription by RdRp (left panel), or by a replication-independent mechanism in double-strand RNAs (right panel), mutations should be symmetric when a replication cycle is completed: a base-substitution type and its complement base-substitution type should arise at the same rate in the viral genome. On the contrary, if de novo mutations are introduced by a replication-independent mechanism specific to single-strand RNAs, mutations could be asymmetric (middle panel).
(B) The statistical assessment on the symmetry of mutations using Fisher's exact tests.
(C) Predictions for two potential mutagenic mechanisms in single-strand RNAs, positive-sense biased versus genomic-strand biased mutagenesis.
(D) The molecular spectrum of among-patient polymorphisms in a negative-sense, single-strand RNA virus, Influenza A virus (subtype H1N1). Two-tailed p values were calculated from Fisher's exact tests.
(E) The molecular spectrum of de novo mutations in a negative-sense, single-strand RNA virus, Ebola virus. De novo mutations were identified from isolated virions, at which time replication cycles have completed. Error bars represent standard errors (N = 21) of the average mutation rates of each base-substitution type. Two-tailed p values were calculated using the t tests.
We reasoned that among-patient polymorphisms in SARS-CoV-2 could be used to investigate whether the mutagenic mechanisms underlying the over-representation of some mutation types were replication dependent or independent because polymorphisms were generated by complete replication cycles. The polymorphism data revealed that G > U transversions occurred with greater frequency than C > A (p = 2 × 10−116, Fisher's exact test) and that C > U transitions occurred with greater frequency than G > A (p = 1 × 10−199, Figure 3B). This asymmetric distribution indicates that the observed over-representation of G > U and C > U mutations unlikely results from a replication-dependent process.
Presumably, replication-independent mutations can occur in either double- or single-strand RNA. We reasoned that mutations arising in double-strand RNA should also lead to a symmetric molecular spectrum (Figure 3A, right panel). This possibility was excluded by the observation that, among SARS-CoV-2 isolates from human patients, G > U and C > U polymorphisms were distributed in asymmetrically greater numbers (Figure 3B), supporting the likelihood that the mechanism responsible for introducing these mutations involved single-strand RNA. Furthermore, since the negative-sense RNA of SARS-CoV-2 is mainly present in the double-strand RNA (i.e., paired with positive-sense RNA; Figure 3A), we therefore proposed that the observed G > U and C > U mutations were most likely introduced to the single-strand positive-sense RNA.
Asymmetric emergence of de novo mutations on the genomic-strand RNA
Thus far, our results indicated that the disproportionate abundance of G > U and C > U mutations in SARS-CoV-2 likely arose in positive-sense single-strand RNAs. There are two possible mechanisms that could account for this outcome. First, the positive-sense RNA is more vulnerable to mutagens—for example, due to destruction of the RNA secondary structure by translating ribosomes, which subsequently exposes the single-strand RNAs to mutagens. Second, the viral genetic information spends the majority of its life cycle as a positive-sense RNA. We thus reasoned that the molecular spectrum of mutations in negative-sense single-strand RNA viruses could be used to investigate which of the two mechanisms underlay the emergence of single-strand RNA mutations, since the negative-sense RNA was predominant in these viruses (Figure 3C).
We first assessed this mechanism by analyzing the genetic polymorphisms among 1,839 confirmed variants (Table S2) of the negative-sense single-strand RNA virus, Influenza A virus, collected during the 2009 pandemic.38 The asymmetric frequency of the among-patient polymorphisms that we observed in SARS-CoV-2, a positive-sense RNA virus (i.e., over-representation of G > U and C > U on the positive strand), was reversed in Influenza A virus: G > U and C > U polymorphisms were more abundant in the negative-sense genome (p = 2 × 10−10 and 0.05, respectively, Fisher's exact tests, Figure 3D). This result allowed us to exclude the possibility that a positive-sense-specific mechanism was responsible for the over-representation of G > U and C > U mutations.
Furthermore, the molecular spectrum of mutations in the negative-sense single-strand RNA virus, Ebola (Zaire ebolavirus), was previously characterized using CirSeq with virions isolated from 293T cells,31 which have completed cycles of replication. Asymmetric accumulation of G > U and C > U mutations were observed in the negative-sense genomic RNA of the Ebola virus (Figure 3E), which further supported a replication-independent mutation mechanism on single-strand RNAs that acts on the strand carrying the genetic information.
Asymmetric emergence of de novo RNA mutations in host cellular environment
In light of these findings, we next sought to determine whether these replication-independent mutations were introduced to the genomic-strand RNA in the extracellular virion environment, where the viral RNA is protected by the capsid, or if they occurred in the cellular environment following host invasion (Figure 4A). To address this issue, we reasoned that positive-sense single-strand, persistent yeast RNA viruses (e.g., Saccharomyces 20S RNA narnavirus and Saccharomyces 23S RNA narnavirus) could be used to test if the eukaryotic cellular environment was able to induce G > U or C > U mutations in single-strand RNAs (Figure 4A). These viruses represented a strong experimental model for this question because they persist in yeast cells as naked RNA, without a capsid.41 Therefore, the introduction of G > U and C > U mutations is dependent on mutagenic mechanisms within the yeast cellular environment, and without which the asymmetric accumulation of mutations will not be detectable (Figure 4A).
Figure 4.

Predictions and observations for mutagenic processes in virions versus in host cells
(A) Predictions on the symmetry of mutations for mutagenic processes in virions versus in host cells.
(B) The molecular spectrum of de novo mutations that we detected in 20S RNA narnavirus from previously published ARC-seq data. Two-tailed p values were calculated from Fisher's exact tests.
(C) The molecular spectrum of yeast mRNA mutations that we detected from previously published ARC-seq data. Two-tailed p values were calculated from Fisher's exact tests.
(D) A scatterplot shows the molecular spectrum of de novo mutations in 20S RNA narnavirus versus in yeast endogenous mRNAs. Each dot represents a base-substitution type, colored according to Figure 1E. Pearson's correlation coefficient (r) and the corresponding p value are shown.
Both CirSeq42 and ARC-seq32 experiments were previously conducted in budding yeast, although the aims of the previous studies were to identify mutations in endogenous RNAs. We reasoned that some reads might be derived from the persistent yeast RNA viruses, which can be used to calculate the symmetry of mutations in the virus genomes. While the yeast strain used in the CirSeq generated minimal reads from either 20S or 23S RNA narnaviruses, the ARC-seq data contained 43,034 sequencing reads from the 20S RNA narnavirus genome (but no reads from the 23S RNA narnavirus). Among the reads that mapped to the 20S RNA narnavirus genome, we identified a significantly higher abundance of G > U mutations than C > A mutations (46 versus 22, p = 0.003, Fisher's exact test) and more C > U than G > A mutations (157 versus 69, p = 1 × 10−8, Fisher's exact test, Figure 4B). These results indicated that the yeast cellular environment was sufficient to induce the asymmetric emergence of G > U and C > U mutations in a positive-sense single-strand RNA viral genome.
Postulating that mutagens in the cellular environment cannot discriminate endogenous and viral RNAs, we further predicted that G > U and C > U mutations should also be over-represented in yeast endogenous mRNAs. To test this prediction, we characterized the molecular spectrum of mRNA mutations in the same ARC-seq dataset for the budding yeast.32 The results showed that the molecular spectrum of mutations was highly similar between the 20S RNA narnavirus and that of the yeast-derived mRNAs (r = 0.93, p = 2 × 10−5, Figures 4C and 4D). Furthermore, G > U and C > U mutations occurred more frequently in the endogenous mRNAs than C > A and G > A mutations, respectively (p < 10−2311 and 10−9126, respectively, Fisher's exact tests, Figure 4C). A similar molecular spectrum of mRNA mutations was also observed in the CirSeq data for the budding yeast42 (r = 0.84, p = 1 × 10−3, Figure S5). The similarities between these molecular spectra indicated the possibility that mutations were induced by the same mutagens in the cellular environment of yeast.
Variation among host cells in providing the cellular environment for asymmetric mutations
Although the particular mutagenic mechanisms that caused the observed asymmetric G > U mutations in the cellular environment remain unknown, we hypothesized that reactive oxygen species (ROS) could serve as a strong candidate,43 particularly considering that oxidative stress is associated with the infection of some respiratory viruses.44 Specifically, some ROS can oxidize guanine to 8-oxoguanine and thereby induce G > U transversions after an additional round of transcription.45,46 Alternatively, we also suspected that chemicals with similar property to polycyclic aromatic hydrocarbons, which are also well known to induce G > T somatic mutations in the lung cancer samples among tobacco smokers,47 could serve as potential cytosolic mutagens of viral RNA. For C > U mutation, potential candidates that could induce its accumulation included RNA-editing activity by cytidine deaminases.48,49
Given the broad array of potential mechanisms, we reasoned that, regardless of the exact nature of the mutagens that caused asymmetric accumulation of G > U or C > U RNA mutations in the cellular environment, these mutagens were unlikely to discriminate between RNA and DNA.50 Consequently, somatic mutations in DNA would also arise,51, 52, 53 particularly in the coding strand, which is exposed to the cellular environment in the single-strand state during transcription54, 55, 56 (illustrated in Figure 5A). Based on this assumption, we investigated the capacity of the cellular environment to generate G > T and C > T somatic mutations in the coding strand of genomic DNA to subsequently infer its capacity to induce G > U and C > U mutations in RNA viruses. To this end, we retrieved the somatic mutations identified for each of the 36 human tissues55 from publicly available Genotype-Tissue Expression (GTEx) data.57
Figure 5.

Variation among 36 human tissues in providing the cellular environment for asymmetric mutations in RNA viruses
(A) The rationale underlying assessment of cellular environments in generating asymmetric mutations in RNA based on somatic mutations in the coding strand of DNA.
(B) A scatterplot shows the asymmetric accumulation of two types of somatic mutations among 36 human tissues. 1, adipose subcutaneous; 2, adipose visceral omentum; 3, adrenal gland; 4, artery aorta; 5, artery coronary; 6, artery tibial; 7, brain caudate basal ganglia; 8, brain cortex; 9, brain frontal cortex BA9; 10, brain hippocampus; 11, brain hypothalamus; 12, brain nucleus accumbens basal ganglia; 13, brain putamen basal ganglia; 14, breast mammary tissue; 15, colon sigmoid; 16, colon transverse; 17, esophagus gastroesophageal junction; 18, esophagus mucosa; 19, esophagus muscularis; 20, heart atrial appendage; 21, heart left ventricle; 22, liver; 23, lung; 24, muscle skeletal; 25, nerve tibial; 26, ovary; 27, pancreas; 28, pituitary; 29, prostate; 30, skin not sun-exposed suprapubic; 31, skin sun-exposed lower leg; 32, small intestine terminal ileum; 33, spleen; 34, stomach; 35, thyroid; 36, whole blood. Odds ratios and two-tailed p values were calculated with Fisher's exact tests. Dots were colored according to the false discovery rates (Q values).
We characterized the molecular spectra of somatic mutations that emerged in these 36 human tissues (Figure S6) and projected them into a two-dimensional space based on the levels of asymmetry in G > T (versus C > A) and C > T (versus G > A) base substitutions (Figure 5B). Among them, 20 tissues, including the lung, showed asymmetry in both G > T and C > T mutations, while 10 tissues showed asymmetry only in G > T mutations. The cellular environments of the remaining six tissues could not induce detectable asymmetry in either G > T or C > T. These results indicated that human tissues exhibit wide-ranging differences in their capacity to induce various types of de novo mutations.
To further investigate the cellular environment that likely drove SARS-CoV-2 evolution in human patients, we plotted the asymmetries of G > U and C > U for SARS-CoV-2 among-patient polymorphisms (abbreviated as pSCV2 in the figure) into the two-dimensional space and found that the cellular environment where SARS-CoV-2 propagated in patients was most similar to that of the lung (Figure 5B). This finding is in agreement with numerous reports that showed the airborne transmission58 of SARS-CoV-2 and supports a cellular environment-dependent genomic evolution of SARS-CoV-2.
The molecular spectrum of 529 accumulated mutations in SARS-CoV-2 prior to its transmission to humans resembled that of coronaviruses evolved in bats
Thus far, our results showed that the molecular spectrum of mutations that accumulated during SARS-CoV-2 evolution is reflective of the asymmetric emergence of de novo mutations (Figure 2) caused by host cellular environments (Figures 3 and 4). Given that different types of base substitutions are disproportionately induced in various cell environments (Figure 5), we reasoned that the ancestral cellular environment where SARS-CoV-2 propagated prior to its transmission to humans could, in principle, be inferred from the mutations in the SARS-CoV-2 genome that accumulated during that period. These mutations could be identified from a phylogenetic tree including SARS-CoV-2 and related coronaviruses.
We built an evolutionary tree (Figure 6A), including the last common ancestor of SARS-CoV-2 isolated from patients and its closely related coronaviruses isolated from Rhinolophus bats (RaTG13, RshSTT200, and ZC45) and pangolins (GD-1 and GX-P5L), using Rc-o319 from bats as an outgroup.8,59, 60, 61, 62, 63 We then reconstructed the ancestral sequence for each internal node (N1–N5) and determined which mutations accumulated in the evolutionary history represented by each branch in the phylogenetic tree (B1–B9, Figure 6A). Based on the parsimony principle we labeled seven branches (B1–B4 and B6–B8) that represented the evolutionary history exclusively in the cellular environments of bats and two that represented a mixed evolutionary history in bats and pangolins (B5 and B9, Figure 6A).
Figure 6.

The molecular spectra of mutations accumulated in SARS-CoV-2 and related viruses
(A) The maximum likelihood phylogenetic, tree including SARS-CoV-2 and related coronaviruses, using Rc-o319 as an outgroup. Internal nodes are labeled as N1–N5, and the icon on the side of a tip indicates the host species from which a SARS-CoV-2-related virus was isolated. The branches are labeled as B0–B9, among which the red branch (B0) represents the evolutionary history in which the host organism is to be determined. The molecular spectrum of accumulated mutations is shown on the top of each branch, and the icon inside shows the inferred host species for the branch according to the parsimony principle.
(B) A heatmap shows Pearson's correlation coefficient (r) between a pair of molecular spectra. Two scatterplots are shown to exemplify the similarity in the molecular spectrum.
(C) The distribution of r for the bootstrapped mutation spectra. In all 10,000 paired bootstrapped observations, r(B0, B1) was greater than r(B0, pSCV2), meaning that the p value was smaller than 0.0001. Numbers in the brackets represent the 95% confident intervals (CI) of r.
The results showed that the molecular spectra of the nine branches appeared similar (Figure 6A, inset on top of each branch), and were highly correlated with each other (Figure 6B). However, these spectra were only moderately correlated with the spectrum of mutations that accumulated during SARS-CoV-2 evolution in human patients, the spectrum of de novo viral mutations in Vero cells, or the spectrum of somatic mutations in the lung (Figure 6B). For example, the asymmetric emergence and accumulation of G > U mutations in the viral genome, which was observed in Vero cells (Figure 1D) and among human patients (Figure 2C), respectively, was no longer detectable in the seven bat-exclusive or two bat-pangolin branches (p > 0.5 in all one-sided Fisher's exact tests). This finding is in agreement with previous reports of low production of ROS and high concentrations of endogenous antioxidants in bat cells.64 These observations indicated that bats (probably also pangolins) provided a cellular environment for the genomic evolution of RNA viruses that substantially differed from that of humans.
We determined the 529 base substitutions that apparently accumulated in the SARS-CoV-2 genome since its divergence from RaTG13 (represented by branch B0 in Figure 6A). The molecular spectrum of this branch (Figure 6A, inset in the top left corner) was highly correlated with the bat-related branches (B1–B9), but showed a much lower correlation with the spectrum of mutations that accumulated in human patients (pSCV2, Figures 6B and 6C). The patterns held when only fragments of the SARS-CoV-2 genome (e.g., the coding sequence of the spike protein) were used for the comparison (Figure S7). These observations suggested that, after its divergence from RaTG13, SARS-CoV-2 likely evolved in a host cellular environment similar to that of bats prior to its zoonotic transfer into humans.
The apparent similarity in the molecular spectra between the mutations accumulated in branch B0 and those in the bat-related branches (B1–B9) could be either attributable to their propagation in a common cellular environment, or a common inherent mutational bias caused by shared genetic variation in the genes that modulate the relative rates of various de novo base substitutions among closely related viruses. To distinguish these two possibilities, we included the genetically more distant betacoronaviruses, SARS-CoV and MERS-CoV and their related viruses, in the analysis (Figure 7A). Bats were reported to be the natural host for both SARS-CoV and MERS-CoV, and zoonotic transfers from their putative intermediate hosts (market civets and dromedary camels) into humans led to SARS and MERS outbreaks in 2003 and 2012, respectively.65, 66, 67
Figure 7.

The similarity in mutation spectrum among genetically diverse coronaviruses isolated from various hosts
(A) The maximum likelihood phylogenetic trees constructed separately for SARS-CoV-related and MERS-CoV-related viruses, using BM48-31 and HKU4 as outgroups, respectively. The known phylogenetic relationship among SARS-CoV-2-related, SARS-CoV-related, and MERS-CoV-related viruses is depicted by dashed lines, which only reflect the tree topology and give no meaning to branch lengths.
(B) The principal-component analysis plot depicts similarity in molecular spectrum. Dots were colored according to the inferred host species. Green, orange, and cyan ellipses represent the 95% confidence intervals for bat, Rhinolophus bats, and human cellular environment, respectively.
(C) Similar to (B), dots were colored according to the phylogenetic lineage.
We constructed phylogenetic trees separately for SARS-CoV-related and MERS-CoV-related viruses, and labeled putative host species for each branch according to the parsimony principle (Figure 7A). If the molecular spectrum was shaped largely by the host cellular environment, we predicted that all branches representative of evolutionary history in the same host species would exhibit similar molecular spectra. On the contrary, if the molecular spectrum was an inherent feature encoded in the virus genome that becomes more distinct as genetic distance increases, we predicted that the lineages of SARS-CoV-2, SARS-CoV, and MERS-CoV would each exhibit their own mutational signatures.
To test these two predictions, we estimated the proportions of each base-substitution type for each branch (Figure 7A). To visualize the similarity across molecular spectra, we performed principal-component analysis, projecting these branches into a two-dimensional space (Figures 7B and 7C). The results showed that 17 branches with a reported evolutionary history exclusive to bats clustered together (Figure 7B), while the viruses from three distinct lineages of betacoronaviruses did not (Figure 7C). This observation indicated that the molecular spectrum of virus genome evolution mainly reflected the cellular environment in which viruses propagated, rather than their phylogenetic relationship. We consequently used the 95% confidence ellipse estimated from these 17 bat-exclusive branches (Figure 7B) to define the borderline of the bat cellular environment in the two-dimensional space.
Branches that represented the host history in camels (B18 and B19) fell outside of the 95% confidence ellipse (Figure 7B), indicating that camels had a distinct cellular environment from that of bats. Branches that represented host history entirely in humans (pSCV2), or a host history partly in humans (B10 for a SARS patient and pMERS for among-patient polymorphisms in MERS), also fell outside of the 95% confidence ellipse (Figure 7B). Notably, they appeared to cluster together with the spectra of de novo mutations detected in SARS-CoV-2 (mSCV2), Ebola virus (mEbola), or poliovirus (mPV), which were cultivated in primate cell lines. Furthermore, the 95% confidence ellipse of these six human-related molecular spectra was not overlapped with that estimated from the spectra of the 17 bat-exclusive branches (Figure 7B), highlighting the potential application of our approach for detecting a jumping event from bats to a new host.
The branch leading to SARS-CoV-2 (B0) was located within the 95% confidence ellipse defined by the 17 bat-exclusive branches (Figure 7B) and, in particular, was within the 95% confidence ellipse defined by the 13 Rhinolophus-exclusive branches. Considering the consistency of this approach in identifying well-established host jumping events (e.g., from bats to camels for MERS-CoV as shown in branches B18 and B19, and from bats to humans for SARS-CoV as shown in branch B10, Figure 7B), we concluded that since its divergence from RaTG13, SARS-CoV-2 most likely propagated primarily in a cellular environment highly similar to bats, particularly Rhinolophus bats, prior to its zoonotic spillover into humans.
Discussion
The central dogma of molecular biology asserts that the genetic information of cellular organisms is stored in DNA, which must be transcribed into mRNA for transmission of the genetic information into functional proteins. Although the presence of mRNA mutations has been confirmed in a few cellular organisms,29,32,42,68,69 they affect only a limited number of proteins due to the transient nature of mRNA. However, for RNA viruses whose genomic information is stored in RNA, mutations in RNA can have a long-term influence because such mutations have a chance of being inherited. In this study, we exploit this influence to infer the evolutionary history for RNA viruses, in particular SARS-CoV-2.
It is worth noting that four out of five branches that represented a mixed host history in bats and in a non-human organism (pangolins in B5 and B9, civets in B11, camels in B20, and hedgehogs in B25) fell within the 95% confidence ellipse estimated from the 17 bat-exclusive branches (Figure 7B). A plausible explanation is that the majority of the base substitutions present in these four bat-mixed branches were accumulated in bat cellular environments, in light of the unique cellular features reported in bats compared with other mammals, i.e., high concentrations of endogenous antioxidants.64 In other words, our approach can identify a host jumping event from molecular spectra only if sufficient mutations have accumulated in the new host. In the future, characterization of molecular spectra of RNA mutations in additional species (especially in bats) using CirSeq or Rep-seq will be promising for tracing the transmission route of SARS-CoV-2. Some additional caveats to the conclusions drawn from our results are discussed in the supplemental information.
Although this study focuses on the relative rates among base-substitution types (i.e., molecular spectrum), we estimate an average rate of 1.73 × 10−5 de novo mutations per nucleotide in SARS-CoV-2 (Figure 1E). Despite the proofreading mechanism70 provided by its nonstructural protein 14, SARS-CoV-2 mutations still occur at a rate three to four orders of magnitude higher than DNA mutations,29 which enables rapid immunological escape, while leaving genomic integrity maintained by natural selection for infectivity.
In addition, our analyses focused on the detection of point mutations. Although not highlighted in the results, we also estimated the molecular spectrum of indels with a similar computational strategy that used junctions as molecular barcodes for the intermediate negative-sense subgenomic RNA (see Figure S2 and supplemental materials and methods). This work identified 2 small insertions and 96 deletions (Figures S3B and S3C), the majority of which were shorter than 6 nucleotides.
Although the significant illness and death caused by the SARS-CoV-2-induced coronavirus disease 2019 pandemic has led to a multitude of studies of this virus, basic understanding is still lacking for several of its key features, such as its origin.1, 2, 3, 4, 5, 6, 7 We show that the mutations accumulated in SARS-CoV-2 prior to its transmission to humans are fully consistent with a natural evolutionary process in a Rhinolophus bat host. In addition to this theoretical purport, our methods will also be useful to identify the natural hosts of other RNA viruses and could be potentially applied toward the prevention of future outbreaks.
Acknowledgments
We thank Dr. Xionglei He from Sun Yat-sen University, Dr. Lucas Carey from Peking University, and Dr. Taolan Zhao from Institute of Genetics and Developmental Biology CAS for discussion. We acknowledge the authors and laboratories for generating and submitting the sequences to the GISAID Database on which this research is based. The list is detailed in Tables S1 and S2. This work was supported by grants from the National Natural Science Foundation of China (31922014).
Author contributions
W.Q. designed the study. K.-J.S., C.W., Y.W., and Q.H. performed data analyses. K.-J.S., C.W., Q.H., and W.Q. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published Online: August 12, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xinn.2021.100159.
Contributor Information
Qing Huan, Email: qhuan@genetics.ac.cn.
Wenfeng Qian, Email: wfqian@genetics.ac.cn.
Lead contact website
Supplemental information
Document S1. Supplemental material and methods and Figures S1–S7
References
- 1.Rasmussen A.L. On the origins of SARS-CoV-2. Nat. Med. 2021;27:9. doi: 10.1038/s41591-020-01205-5. [DOI] [PubMed] [Google Scholar]
- 2.Andersen K.G., Rambaut A., Lipkin W.I., et al. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu S.L., Saif L.J., Weiss S.R., Su L. No credible evidence supporting claims of the laboratory engineering of SARS-CoV-2. Emerg. Microbes Infect. 2020;9:505–507. doi: 10.1080/22221751.2020.1733440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi Z.L. Origins of SARS-CoV-2: focusing on science. Infect. Dis. Immun. 2021;1:3–4. doi: 10.1097/ID9.0000000000000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu C.I., Wen H., Lu J., et al. On the origin of SARS-CoV-2—the blind watchmaker argument. Sci. China Life Sci. 2021 doi: 10.1007/s11427-021-1972-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloom J.D., Chan Y.A., Baric R.S., et al. Investigate the origins of COVID-19. Science. 2021;372:694. doi: 10.1126/science.abj0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calisher C.H., Carroll D., Colwell R., et al. Science, not speculation, is essential to determine how SARS-CoV-2 reached humans. Lancet. 2021;398:209–211. doi: 10.1016/S0140-6736(21)01419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou P., Yang X.L., Wang X.G., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lytras S., Hughes J., Martin D., et al. Exploring the natural origins of SARS-CoV-2 in the light of recombination. bioRxiv. 2021 doi: 10.1101/2021.01.22.427830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boni M.F., Lemey P., Jiang X., et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020;5:1408–1417. doi: 10.1038/s41564-020-0771-4. [DOI] [PubMed] [Google Scholar]
- 11.Kar S., Leszczynski J. From animal to human: interspecies analysis provides a novel way of ascertaining and fighting COVID-19. Innovation. 2020;1:100021. doi: 10.1016/j.xinn.2020.100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z. Paml 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 13.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. U S A. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M.B., Dowsett I.T., Carr D.T., et al. Defining the impact of mutation accumulation on replicative lifespan in yeast using cancer-associated mutator phenotypes. Proc. Natl. Acad. Sci. U S A. 2019;116:3062–3071. doi: 10.1073/pnas.1815966116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu Y.O., Siegal M.L., Hall D.W., Petrov D.A. Precise estimates of mutation rate and spectrum in yeast. Proc. Natl. Acad. Sci. U S A. 2014;111:E2310–E2318. doi: 10.1073/pnas.1323011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Maio N., Walker C.R., Turakhia Y., et al. Mutation rates and selection on synonymous mutations in SARS-CoV-2. Genome Biol. Evol. 2021;13 doi: 10.1093/gbe/evab087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Z. Estimating the pattern of nucleotide substitution. J. Mol. Evol. 1994;39:105–111. doi: 10.1007/BF00178256. [DOI] [PubMed] [Google Scholar]
- 18.Siepel A., Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 2004;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- 19.Tate J.G., Bamford S., Jubb H.C., et al. COSMIC: the catalogue of somatic mutations. Cancer Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexandrov L.B., Kim J., Haradhvala N.J., et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexandrov L.B., Nik-Zainal S., Wedge D.C., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hainaut P., Pfeifer G.P. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–374. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 23.Di Giorgio S., Martignano F., Torcia M.G., et al. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020;6:eabb5813. doi: 10.1126/sciadv.abb5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L., Liu W., Zhang Q., et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 2020;9:313–319. doi: 10.1080/22221751.2020.1725399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen Z., Xiao Y., Kang L., et al. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect Dis. 2020;71:713–720. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panchin A.Y., Panchin Y.V. Excessive G-U transversions in novel allele variants in SARS-CoV-2 genomes. PeerJ. 2020;8:e9648. doi: 10.7717/peerj.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teng X., Li Q., Li Z., et al. Compositional variability and mutation spectra of monophyletic SARS-CoV-2 clades. Genomics Proteomics Bioinformatics. 2020;18:648–663. doi: 10.1016/j.gpb.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanjuan R., Nebot M.R., Chirico N., et al. Viral mutation rates. J. Virol. 2010;84:9733–9748. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gout J.F., Thomas W.K., Smith Z., et al. Large-scale detection of in vivo transcription errors. Proc. Natl. Acad. Sci. U S A. 2013;110:18584–18589. doi: 10.1073/pnas.1309843110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acevedo A., Brodsky L., Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitfield Z.J., Prasad A.N., Ronk A.J., et al. Species-specific evolution of Ebola virus during replication in human and bat cells. Cell Rep. 2020;32:108028. doi: 10.1016/j.celrep.2020.108028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reid-Bayliss K.S., Loeb L.A. Accurate RNA consensus sequencing for high-fidelity detection of transcriptional mutagenesis-induced epimutations. Proc. Natl. Acad. Sci. U S A. 2017;114:9415–9420. doi: 10.1073/pnas.1709166114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu F., Zhao S., Yu B., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D., Lee J.Y., Yang J.S., et al. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y., Sun J., Li Y., et al. The strand-biased transcription of SARS-CoV-2 and unbalanced inhibition by remdesivir. iScience. 2021;24:102857. doi: 10.1016/j.isci.2021.102857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sola I., Almazan F., Zuniga S., Enjuanes L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2015;2:265–288. doi: 10.1146/annurev-virology-100114-055218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shu Y., McCauley J. GISAID: global initiative on sharing all influenza data—from vision to reality. Euro Surveill. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei C., Chen Y.M., Chen Y., Qian W. The missing expression level-evolutionary rate anticorrelation in viruses does not support protein function as a main constraint on sequence evolution. Genome Biol. Evol. 2021;13:evab049. doi: 10.1093/gbe/evab049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanjuan R., Domingo-Calap P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016;73:4433–4448. doi: 10.1007/s00018-016-2299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickner R.B., Fujimura T., Esteban R. Viruses and prions of Saccharomyces cerevisiae. Adv. Virus Res. 2013;86:1–36. doi: 10.1016/B978-0-12-394315-6.00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gout J.F., Li W., Fritsch C., et al. The landscape of transcription errors in eukaryotic cells. Sci. Adv. 2017;3:e1701484. doi: 10.1126/sciadv.1701484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dai D.P., Gan W., Hayakawa H., et al. Transcriptional mutagenesis mediated by 8-oxoG induces translational errors in mammalian cells. Proc. Natl. Acad. Sci. U S A. 2018;115:4218–4222. doi: 10.1073/pnas.1718363115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delgado-Roche L., Mesta F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020;51:384–387. doi: 10.1016/j.arcmed.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong Q., Lin C.L. Oxidative damage to RNA: mechanisms, consequences, and diseases. Cell Mol. Life Sci. 2010;67:1817–1829. doi: 10.1007/s00018-010-0277-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z., Wu J., Deleo C.J. RNA damage and surveillance under oxidative stress. IUBMB Life. 2006;58:581–588. doi: 10.1080/15216540600946456. [DOI] [PubMed] [Google Scholar]
- 47.Kucab J.E., Zou X., Morganella S., et al. A compendium of mutational signatures of environmental agents. Cell. 2019;177:821–836.e16. doi: 10.1016/j.cell.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris R.S., Dudley J.P. APOBECs and virus restriction. Virology. 2015;479-480:131–145. doi: 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blanc V., Davidson N.O. APOBEC-1-mediated RNA editing. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010;2:594–602. doi: 10.1002/wsbm.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohno M., Sakumi K., Fukumura R., et al. 8-Oxoguanine causes spontaneous de novo germline mutations in mice. Sci. Rep. 2014;4:4689. doi: 10.1038/srep04689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hofer T., Badouard C., Bajak E., et al. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol. Chem. 2005;386:333–337. doi: 10.1515/BC.2005.040. [DOI] [PubMed] [Google Scholar]
- 52.Hofer T., Seo A.Y., Prudencio M., Leeuwenburgh C. A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol. Chem. 2006;387:103–111. doi: 10.1515/BC.2006.014. [DOI] [PubMed] [Google Scholar]
- 53.Yan L.L., Zaher H.S. How do cells cope with RNA damage and its consequences? J. Biol. Chem. 2019;294:15158–15171. doi: 10.1074/jbc.REV119.006513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duan C., Huan Q., Chen X., et al. Reduced intrinsic DNA curvature leads to increased mutation rate. Genome Biol. 2018;19:132. doi: 10.1186/s13059-018-1525-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garcia-Nieto P.E., Morrison A.J., Fraser H.B. The somatic mutation landscape of the human body. Genome Biol. 2019;20:298. doi: 10.1186/s13059-019-1919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haradhvala N.J., Polak P., Stojanov P., et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016;164:538–549. doi: 10.1016/j.cell.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.GTEx Consortium Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lam T.T., Jia N., Zhang Y.W., et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020;583:282–285. doi: 10.1038/s41586-020-2169-0. [DOI] [PubMed] [Google Scholar]
- 60.Xiao K., Zhai J., Feng Y., et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. 2020;583:286–289. doi: 10.1038/s41586-020-2313-x. [DOI] [PubMed] [Google Scholar]
- 61.Hu D., Zhu C., Ai L., et al. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg. Microbes Infect. 2018;7:154. doi: 10.1038/s41426-018-0155-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murakami S., Kitamura T., Suzuki J., et al. Detection and characterization of bat sarbecovirus phylogenetically related to SARS-CoV-2. Jpn. Emerg Infect. Dis. 2020;26:3025–3029. doi: 10.3201/eid2612.203386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hul V., Delaune D., Karlsson E.A., et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. bioRxiv. 2021 doi: 10.1101/2021.01.26.428212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanadhita D., Satyaningtijas A.S., Agungpriyono S. Bats oxidative stress defense. J. Riset Veteriner Indonesia (Journal Indonesian Vet. Research) 2019;3 doi: 10.20956/jrvi.v20953i20951.26035. [DOI] [Google Scholar]
- 65.Guan Y., Zheng B.J., He Y.Q., et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 66.Alagaili A.N., Briese T., Mishra N., et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. mBio. 2014;5 doi: 10.1128/mBio.00884-14. e00884–00814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gordon A.J., Satory D., Halliday J.A., Herman C. Lost in transcription: transient errors in information transfer. Curr. Opin. Microbiol. 2015;24:80–87. doi: 10.1016/j.mib.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carey L.B. RNA polymerase errors cause splicing defects and can be regulated by differential expression of RNA polymerase subunits. eLife. 2015;4:e09945. doi: 10.7554/eLife.09945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.V'Kovski P., Kratzel A., Steiner S., et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Supplemental material and methods and Figures S1–S7