

Abstract
The cGAS–cyclic GMP–AMP (cGAMP)–stimulator of IFN genes (STING) pathway induces a powerful type I IFN (IFN-I) response and is a prime candidate for augmenting immunity in cancer immunotherapy and vaccines. IFN-I also has immune-regulatory functions manifested in several autoimmune diseases and is a first-line therapy for relapsing–remitting multiple sclerosis. However, it is only moderately effective and can induce adverse effects and neutralizing Abs in recipients. Targeting cGAMP in autoimmunity is unexplored and represents a challenge because of the intracellular location of its receptor, STING. We used microparticle (MP)–encapsulated cGAMP to increase cellular delivery, achieve dose sparing, and reduce potential toxicity. In the C57BL/6 experimental allergic encephalomyelitis (EAE) model, cGAMP encapsulated in MPs (cGAMP MPs) administered therapeutically protected mice from EAE in a STING-dependent fashion, whereas soluble cGAMP was ineffective. Protection was also observed in a relapsing–remitting model. Importantly, cGAMP MPs protected against EAE at the peak of disease and were more effective than rIFN-β. Mechanistically, cGAMP MPs showed both IFN-I–dependent and –independent immunosuppressive effects. Furthermore, it induced the immunosuppressive cytokine IL-27 without requiring IFN-I. This augmented IL-10 expression through activated ERK and CREB. IL-27 and subsequent IL-10 were the most important cytokines to mitigate autoreactivity. Critically, cGAMP MPs promoted IFN-I as well as the immunoregulatory cytokines IL-27 and IL-10 in PBMCs from relapsing–remitting multiple sclerosis patients. Collectively, this study reveals a previously unappreciated immune-regulatory effect of cGAMP that can be harnessed to restrain T cell autoreactivity.
A major discovery in innate immunity is that cyclic dinucleotides activate transcription factors required for type I IFN (IFN-I) expression. Among cyclic dinucleotides, 2′,3′ cyclic GMP–AMP (cGAMP) is produced by mammalian cells as a result of cytosolic DNA that binds and activates the enzyme cGAMP synthase (cGAS) (1). In addition to mammalian cells, certain bacteria such as Vibrio cholerae produce the analogue 3′,3′ cGAMP, which acts as a bacterial secondary messenger that also serves as a cytosolic pathogen-associated molecular pattern (2). Other bacteria also produce the cyclic dinucleotides cyclic-di-GMP and cyclic-di-AMP. These cyclic dinucleotides bind to the innate immune receptor stimulator of IFN genes (STING), which can activate the transcription factors, NF-κB and IRF3, to activate IFN-I gene expression (1, 3). IFN-Is represent host cell responses to viral or intracellular bacterial infections, but also play a critical role in the promotion of a Th1 and subsequent CTL response in adaptive immunity. This has rendered cGAMP and other STING activators as prime candidates to enhance T cell responses in both vaccine adjuvants and cancer immunotherapy and are currently explored for these purposes (4–7).
Despite its traditional role as a proinflammatory mediator, a critical role of IFN-I is in immune regulation (8). A prime example is the use of IFN-β for the treatment of multiple sclerosis (MS) (9), although the precise mechanism of IFN-mediated protection in this disease is unclear. MS remains a global health burden despite being one of the most recognized and studied autoimmune disorders. Currently, there is no cure for MS; however, many treatment options exist to slow the progression of the disease and alleviate the symptoms. MS patients are typically stratified into several clinical variants of the disease, the most frequent being relapsing–remitting MS (RRMS). Recombinant IFN-I therapy is one of the first-line therapies for RRMS. Compared with some newer RRMS medications, rIFN-I has been shown to have an excellent safety profile but is only moderately effective in comparison with newer disease modifying therapies (10). An alternative therapy that could trigger the production of a natural IFN-I and other related immunomodulatory factors may improve efficacy without sacrificing safety.
Although cyclic dinucleotides are currently actively pursued to augment immune response to cancer, their utility has not been tested clinically in the autoimmune diseases such as MS. cGAMP represents an attractive alternative to rIFN because certain forms, such as 2′,3′ cGAMP, is a self-molecule that is physiologically produced. However, moving cGAMP forward as a clinical therapy remains a challenge because of its hydrophilic nature and requirement for its delivery into the cytosol to bind to STING in innate immune cells. As a result, relatively high concentrations of cGAMP are required, which could raise toxicity. In many antitumor studies employing cGAMP, intratumoral delivery is required (6, 11). Site-specific injection is not a feasible route for the treatment of a systemic disease such as MS. To counter this problem, the encapsulation and delivery of cyclic dinucleotides in nanoparticles and microparticles (MPs) represent an attractive alternative (12–14). However, because of the limited linkages available for covalent binding to polymeric carriers, reliable delivery of cGAMP by particle technology has remained a major challenge.
In this study, we explore the use cGAMP encapsulated in MPs (cGAMP MPs) as a therapy in experimental autoimmune encephalomyelitis (EAE), a murine model of MS. The MPs we used efficiently encapsulate cGAMP, have a good safety profile, and result in much greater efficacy than soluble cGAMP in reducing severity and delaying the onset of chronic EAE. cGAMP MP therapy during peak of disease in the relapsing–remitting EAE model greatly reduced disease severity and prevented relapses more effectively than IFN-β. To explore the mechanism by which cGAMP attenuates EAE, we show that cGAMP MPs induce IL-27 and IL-10 secretion in a STING- and IFN-I–dependent fashion. Importantly, we extended the study to patients with RRMS and showed that the treatment of PBMCs from MS patients similarly caused an immunosuppressive phenotype marked by IL-27 and IL-10 induction. These results show a previously unrecognized impact of cGAMP in immune regulation, mediated by the induction of inhibitory cytokines, and provide the proof of principle that encapsulated STING agonists could be an alternative therapy for RRMS.
Materials and Methods
Mice
C57B6/J, SJL/J, Ifnar1−/−, Il10−/−, Tmem173gt/gt (Goldenticket), and IL-27 receptor α (Il27ra−/−) knockout mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were housed in specific pathogen–free conditions. Mouse care and experiments were subject to approved protocols under The University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee.
Induction of EAE
Nine-week-old female mice were induced to EAE by s.c. injection of an emulsion containing CFA (Sigma-Aldrich, St. Louis, MO), 200–300 μg myelin oligodendrocyte glycopeptide (MOG)35–55 (Bio-Synthesis, Lewisville, TX), and 200 μg tuberculosis toxin in the lower back. A total of 200–300 ng of pertussis toxin (List Biological Laboratories, Campbell, CA) was injected i.p. at day 0 and day 2. In SJL/J relapsing–remitting EAE experiments, 9-wk-old SJL/J female mice were induced to EAE as described above using 300 μg myelin proteolipid protein (PLP)139–151 (Bio-Synthesis). SJL/J mice showing EAE symptoms at day 15 (≥2.0) were stratified into groups with an equivalent average disease score before administration of therapy or control MP.
EAE scores were based on the development of progressive muscle weakness ranging from 0.5 to 5.0. This is described as the following: 0.5, tail weakness; 1.0, tail limp with no gait change; 1.5, tail limp with mild gait change or tail weakness with moderate gait change; 2.0, tail limp with moderate gait change and loss of balance; 2.5, tail limp and wobbly gait; 3.0, partial hind limb paralysis; 3.5, complete hind limb paralysis; 4.0, partial or total forelimb paralysis; 4.5, severe total body paralysis or rotational spinning; and 5.0, death. Mice were monitored daily until study completion or until a humane end point was reached (≥4.0). Mice achieving a peak disease score of ≥1.0 were considered positive for disease incidence. In the SJL/J model, mice achieving multiple-day increase of disease scores or increase above 0.5 after initial remission were considered positive for a relapse.
Histology
Mice received terminal doses of tribromoethanol prior to perfusion with PBS and subsequent 4% paraformaldehyde. Spinal columns were collected and maintained in 4% paraformaldehyde. Spinal cords were removed after 24 h and then embedded in paraffin. Spinal sections were sectioned transversely in 8-μm-thick sections using a microtome. H&E was used to determine inflammatory cell infiltration, and Luxol fast blue was used to determine degrees of demyelination in representative sections with periodic acid–Schiff counterstaining. Histology scoring was performed in a double-blinded fashion on thoracic and cervical sections. Increased histological scores were indicative of inflammatory cellular infiltrate.
In vitro primary cell culture
Bone marrow dendritic cells (BMDCs) and bone marrow–derived macrophages (BMDMs) were derived using established protocols (15, 16). Cells from both male and female mice were used. In brief, progenitor cells were flushed from bone marrow of 8–12-wk-old mice and made into a single-cell suspension, and RBCs were lysed with ammonium–chloride–potassium. BMDCs were cultured for 8 d in 10% serum supplemented RPMI 1640 (Life Technologies) containing 40 ng/ml GM-CSF (PeproTech, Rocky Hill, NJ). BMDMs were cultured for 7 d in DMEM (Life Technologies) containing 20% serum and 30% conditioned L929 media supernatant. Cells were plated overnight in 10% serum media prior to experimentation the next day. Cells were primed with 100 ng/ml LPS (MilliporeSigma) unless otherwise noted. For mixed splenocytes studies, spleens were taken from 8–12-wk-old mice, processed into a single-cell suspension, and depleted of CD8+ cells by positive selection using CD8α MACS beads (Miltenyi Biotec). For coculture experiments, BMDCs were cultured as described, then treated with particles for 24 h before the addition of sex-matched isolated naive CD4+ cells (Miltenyi Biotec) at a 1:5 ratio in the presence of αCD3 mAb for an additional 24 h. rIL-12p70, rIL-6, and rTGF-β were purchased from BioLegend. ERK (1049738–54-6) and CREB (666–15) inhibitors were purchased from MilliporeSigma (Burlington, MA).
Immunoblotting
Cells were treated as indicated, then washed with cold PBS prior to lysing with radioimmunoprecipitation assay buffer containing phosphatase and protease inhibitors (Roche Diagnostics). Equal amounts of protein were determined by BCA assay (Thermo Fisher Scientific) and combined with SDS-PAGE loading dye. Samples were resolved on 4–12% NuPAGE gels (Thermo Fisher Scientific) and transferred to nitrocellulose membranes. Blots were blocked in 5% reconstituted powdered milk and incubated with a primary Ab before washing and adding secondary Abs conjugated to HRP. Blots were washed of excess secondary Ab, developed using Super-Signal substrate (Thermo Fisher Scientific) and imaged by autoradiography. Primary Abs included the following: p-STAT1 (9167), STAT1 (9176), p-STING (72971), STING (50494), p-CREB (9198), CREB (9197), p-ERK (4370), and ERK (9107). All primary Abs were from Cell Signaling Technology (Danvers, MA). HRP-conjugated β-actin (sc-47778) was from Santa Cruz Biotechnology (Dallas, TX).
Flow cytometry
Cells were stimulated with 50 ng/ml PMA and 1 μg/ml ionomycin or with 5 μg/ml αCD3 mAb as specified in figure legends in the presence of brefeldin A (BioLegend). Cells were collected, washed twice in FACS buffer, and stained for surface markers on ice for 20 min. Cells were washed twice of excess Ab and fixed with FOXP3 Fix/Perm Solution (BioLegend) at room temperature for 20 min. Cells were washed in FACS buffer prior to washing and suspension in FOXP3 Perm Buffer for 15 min. Permeabilized cells were incubated with intracellular Abs for 30 min. Excess Ab was washed off twice before suspension in FACS buffer. Surface markers include mouse α-mouse CD3-PeCy7 (145–2C11), α-mouse CD25-APC/Cy7 (PC61), α-mouse CD4-PerCP/Cy5.5 (GK1.5), α-mouse CD11b–Brilliant Violet 510 (M1/70), α-mouse CD11c–Pacific Blue (N418), α-mouse CD45.2– Brilliant Violet 605 (104), α-mouse F4/80-APC (BM8), α-mouse Ly-6C–Alexa Fluor 700 (HK1.4), and α-mouse CD115-PerCP/Cy5.5 (AFS98). Intracellular marker included mouse α-mouse IL-10–APC (JES5–16E3). All mouse Abs were purchased from BioLegend. Mouse samples were run on a BD LSR II or BD LSRFortessa (BD Biosciences, Franklin Lakes, NJ) and analyzed using FlowJo (Ashland, OR).
In vivo biodistribution and local cytokine production
Nine-week-old female wild-type (WT) mice were induced to EAE as described. Texas Red–conjugated acetalated dextran (Ace-DEX) MPs loaded with or without cGAMP were injected i.m. at days 9, 11, and 13. WT mice without EAE were also injected at corresponding timepoints. All mice were euthanized at day 16. Injected leg, lungs, heart, brain, kidneys, liver, spleen, and inguinal lymph nodes (LNs) were evaluated by fluorescence imaging using an IVIS Lumina III (PerkinElmer, Waltham, MA). Spleens and LNs were processed, stained for cell surface markers, and analyzed by flow cytometry. Additional mice were induced to EAE and injected with PBS, soluble cGAMP, or cGAMP MPs every other day for three injections. Mice were sacrificed at indicated time points after final injection. Muscle from injected leg was weighed and homogenized in PBS containing protease inhibitor. Supernatants were collected and used for cytokine measurements.
Mouse ELISA
Supernatants were collected and assayed for quantitative cytokine measurements by ELISA. For mouse samples, IL-10 and IL-17 ELISA kits were purchased from BioLegend, IFN-γ from BD Biosciences, and IL-27 ELISA kit from Thermo Fisher Scientific. All assays were run in accordance with the manufacturers’ protocols. Murine IFN-β ELISA was developed in-house using primary Abs from Santa Cruz Biotechnology (sc-57201) and R&D Systems (32400–1; Minneapolis, MN), rIFN-β standard from R&D Systems (12401–1), and secondary α Rabbit IgG HRP-conjugate from Cell Signaling Technology (707). Human cell supernatants were analyzed by ELISA for IFN-β (PBL Assay Science), IL-27 (R&D Systems), and IL-10 (BD Biosciences).
Patient samples and analysis
Peripheral blood samples were collected from seven RRMS patients not treated with immunomodulatory therapies prior to blood sample collection. For these in vitro studies, fresh PBMCs were separated using Ficoll-Paque (GE Healthcare). Isolated PBMCs were suspended in complete RPMI 1640 and plated with cGAMP MPs, blank MPs, or soluble cGAMP for 24 h. Surface markers were stained with CD14 PE-Cy5 (SK3; BD Biosciences). Intracellular staining was performed after stimulation with LPS (L2654; Sigma-Aldrich) and incubated at 37°C, 5% CO2 for 1 h. Brefeldin A (eBioscience) was added for an additional 3 h. Cells were fixed, permeabilized, and stained with fluorescein-conjugated Abs against human IFN-β (MMHB-3, PBL Assay Science, Piscataway, NJ), αIL-10–APC (BD Biosciences), or αIL-27–APC (BD Biosciences). The percentages of cells expressing each cytokine in CD14+-gated monocytes were determined using a BD FACSCalibur and CellQuest software (BD Biosciences).
Quantitative RT-PCR
CD14+ monocytes were separated from the PBMCs of four RRMS patients using CD14 MicroBeads (Miltenyi Biotec). The purity of separated cells (>98%) was confirmed by FACS. The monocytes were treated with MPs for 8 h. The total RNA was extracted, and cDNA was synthesized using a High-Capacity cDNA Archive Kit (Applied Biosystems). The primers for MX1, IFN-β, and 18S mRNA were purchased from Applied Biosystems, and gene expression was measured by RT-PCR using TaqMan Gene Expression Assays (Applied Biosystems) in triplicates. The results are expressed as the average relative gene expression, normalized against the 18S mRNA expression for each subject.
Human ELISA
The effect of cGAMP MPs on the monocyte and cocultured CD4 T cells cytokine secretion was examined in five RRMS patients using ELISA for the cytokine measurements in the supernatants. Separated CD14+ monocytes (1 × 106 cells per condition) were cultured for 24 h with cGAMP MPs after LPS priming. In coculture conditions, CD14+ monocytes were primed with LPS then treated with cGAMP MPs for 12 h. The cells were washed with PBS after 12 h and were cocultured (1:2) with the CD4+ T cells for another 24 h. CD14+ monocytes or CD4 T cells were separated from the peripheral blood of untreated RRMS patients using negative magnetic bead separation (130–050-201,130–096-533; Miltenyi Biotec). Supernatants were collected for cytokine secretion measurements using IFN-β (PBL Assay Science), IL-27 (R&D Systems), and IL-10 (BD Pharmingen) ELISA kits. Assays were performed using manufacturers’ protocol.
Study approval
Human samples were collected upon obtaining The University of North Carolina at Chapel Hill Institutional Review Board–approved informed consent, protocol UNC-14–0705. Mouse care and experiments were subject to approved protocols under The University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee.
Statistics
All studies were evaluated for statistical significance by either two-tailed Student t test, one-way ANOVA, or Mann–Whitney U tests using GraphPad PRISM, as indicated in the corresponding figure legend. All experiments were repeated at least twice and run in biological replicates as indicated in the figure legends. Graphical results are shown as mean ± SEM. Significance was determined with a p value <0.05, as indicated in the figure legends.
Results
MPs target dendritic cells in mice with EAE
cGAMP is a small hydrophilic molecule that requires cytosolic delivery to stimulate STING. Efficient delivery of this agonist has been challenging, hindering its therapeutic potential because of the requirement for high concentrations that may cause vascular toxicity. Our group and others have successfully encapsulated cGAMP into nanoparticles or MPs, and a comparison of different particle compositions showed that particles composed of biodegradable and acid-labile Ace-DEX are superior to others in the terms of encapsulation efficiency of cGAMP (12, 13, 17). In the current study, we show that multiple batches of cGAMP MPs showed highly reproducible characteristics, with small variations in size, surface charge, and drug loading (Supplemental Fig. 1A, 1B), reflecting its consistency, which is a major advantage of this fabrication process. We delivered fluorescently labeled (Texas Red dye), empty Ace-DEX MPs (blank MPs), or labeled cGAMP MP by the clinically feasible route of i.m. injection and examined their tissue distribution using in vivo fluorescence imaging. The majority of particles were found at the injection site (Fig. 1A) but also disseminate proximally to the inguinal LN (Fig. 1B) in healthy mice as well as in mice with EAE. Interestingly, despite loss of muscle tissue, associated with the lower limb weakness in EAE, particles readily trafficked to the proximal LN after injection (Fig. 1B). Particles were not detected by in vivo imaging in any other peripheral nonimmune organs (Supplemental Fig. 2A). LNs and spleens were evaluated for cell-specific uptake of MPs. Flow cytometry analysis showed a significant increase in the percentage and total number of CD11c+ dendritic cells (DCs), CD11b+F4/80+ macrophages, and CD11b+CD115+Ly-6Clow+ patrolling monocytes in MP-treated mice compared with controls. Texas Red MPs were detected in the LNs of both healthy and mice with EAE. The inclusion of cGAMP in MP (indicated as a plus sign [+] for both cGAMP and MP) did not cause a difference in MP uptake (Fig. 1C–E). An increase in MP-positive APCs was also detected in the spleen (Supplemental Fig. 2B). More cGAMP particles were detected in DCs within the spleen than blank MPs by both percentage and total number (Supplemental Fig. 2B), and these differences were more significant in the EAE group. This suggests that cGAMP and EAE might affect particle trafficking, retention, and/or processing in the spleen. The active interest in STING stems from its capacity to induce the IFN-I. Fig. 1F indicates that administration of cGAMP in mice with EAE can increase IFN-β production in a time-dependent fashion at the injection site. However, only cGAMP MPs maintained this significant increase compared with control-treated mice past 6 h.
FIGURE 1.
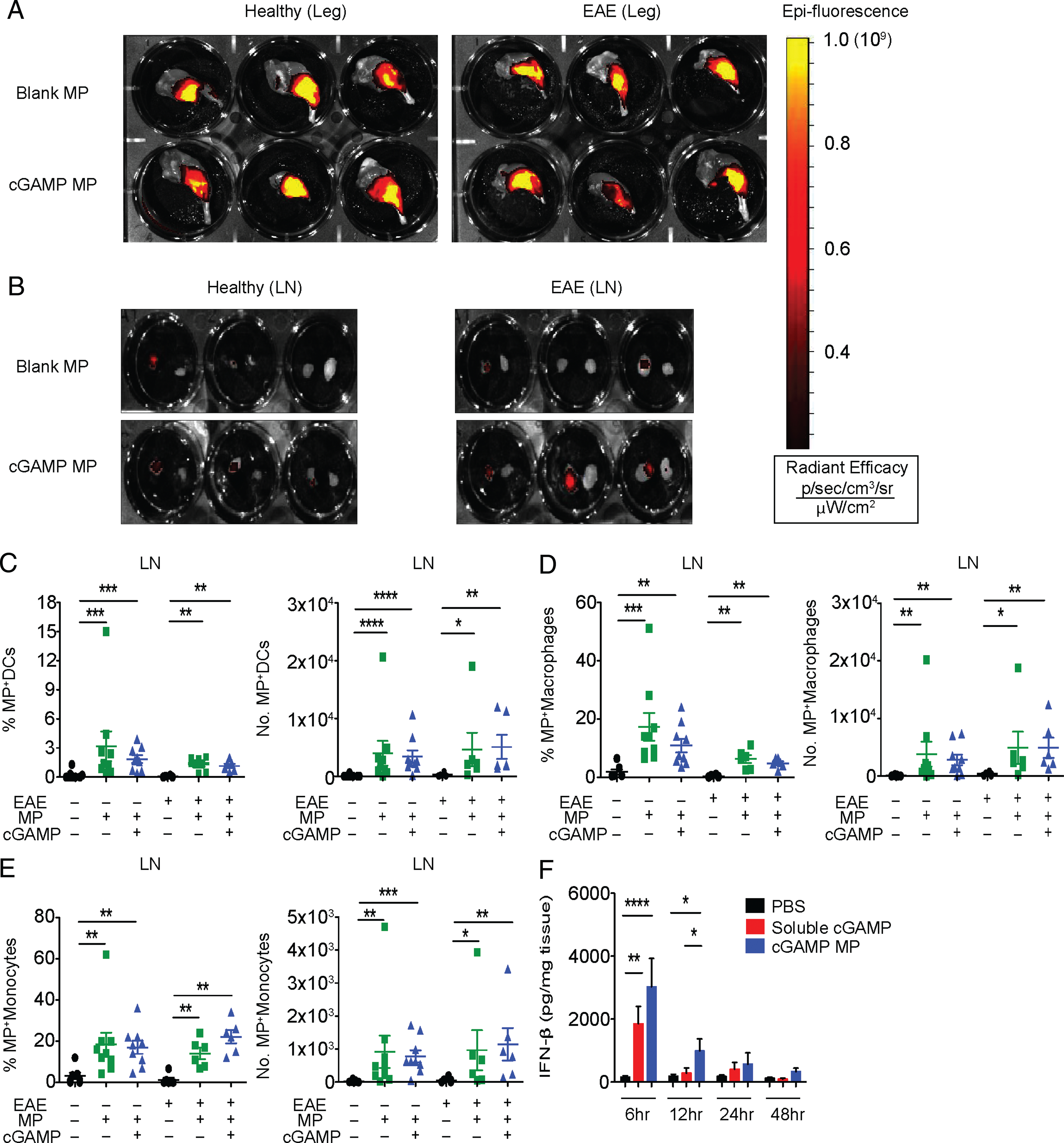
cGAMP MP biodistribution and local cytokine induction. (A–D) Fluorescently labeled cGAMP MP (5 μg of cGAMP) or equivalent blank MPs were injected into the hind leg of healthy or EAE-induced mice at days 9, 11, and 13 postimmunization (p.i.). Mice were euthanized 3 d after final injection. (A and B) Representative fluorescent images of injected leg muscle and LNs after MPs treatment, respectively. (C–E) LNs from mice were processed and stained for DC, macrophage, and monocyte surface markers and evaluated for the presence of fluorescent MPs by flow cytometry (n = 6–9). (F) Five micrograms of cGAMP were injected into mice at days 9, 11, and 13 after EAE induction. Mice were sacrificed at 6, 12, 24, and 48 h after final injection and evaluated for IFN-β in the muscle (n = 6). Combined data of two experiments, all data between treatment groups were tested by Mann–Whitney U tests. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
cGAMP MPs mitigate acute EAE in a STING- and IFN-I–dependent manner
Given that cGAMP MPs can induce the production of IFN-β in mice with EAE, we postulated that cGAMP MPs could induce a therapeutic effect in this disease model (18). To test this, we immunized mice with the autoantigen MOG35–55 with pertussis toxin to induce EAE. After disease onset (day 9), soluble cGAMP or cGAMP MP (each at 5 μg cGAMP) were delivered i.m. every other day for a total of five injections (Fig. 2A). cGAMP MPs reduced the disease score and delayed disease onset, whereas an equivalent dose of soluble cGAMP was ineffective, indicating the advantage of intracellular particle delivery (Fig. 2B). cGAMP MPs effectively prevented disease in 50% of treated mice, whereas soluble cGAMP prevented disease manifestation in only one mouse (10%) (Fig. 2B). Moreover, this i.m. therapy was effective at a 20-fold lower dose than what has been reported for i.v. cyclic di-GMP to reduce EAE scores (19). To explore the molecular pathways by which cGAMP MPs mediate their effect, mice bearing a nonfunctional mutation in the gene encoding STING (Tmem173gt/gt) were tested, and the in vivo protective effect of cGAMP MPs was abolished (Fig. 2C), indicating that cGAMP MPs protect mice from EAE by targeting its receptor STING. To determine if cGAMP MPs are effective during the peak of disease and if this can be achieved in an relapsing–remitting model of MS, we treated SJL/J mice with relapsing–remitting EAE at the peak of disease (day 15) (Fig. 2D). cGAMP MPs effectively reduced disease scores and prevented relapses that were observed in this mouse model. Furthermore, a comparative study indicated that cGAMP MPs were superior to rIFN-β in reducing disease scores and relapse incidence during the treatment (Fig. 2D). This suggests that cGAMP MP may attenuate relapsing–remitting EAE via mechanisms that are beyond IFN-I induction. Representative histology of spinal cord showed less cellular infiltrate and demyelination in the cGAMP MP–treated mice compared with PBS-treated controls (Fig. 2E, 2F).
FIGURE 2.
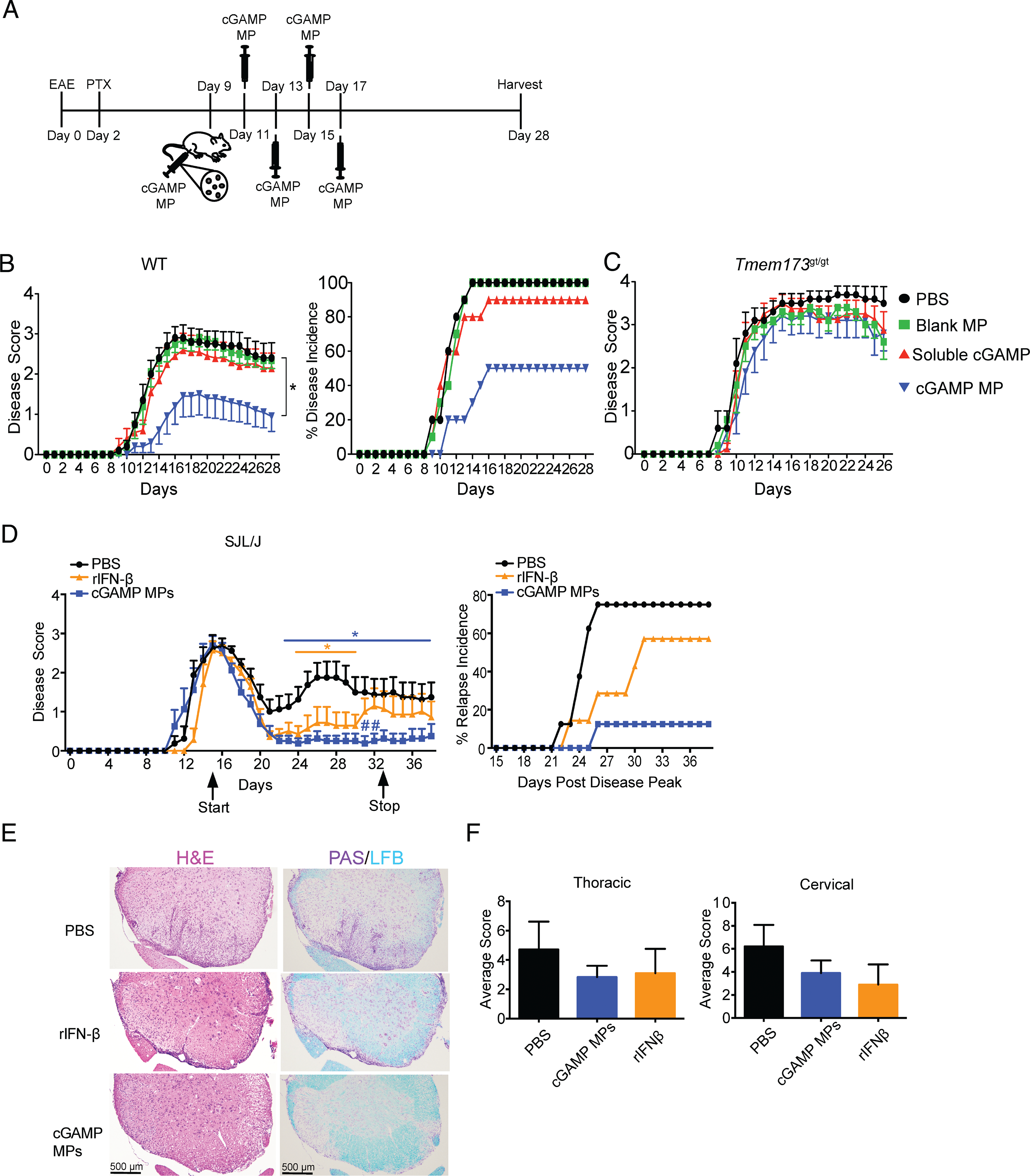
cGAMP MPs tamper EAE in WT mice. (A) Treatment schematic of acute EAE model by soluble cGAMP or cGAMP MP. Mice received 5 μg cGAMP in MPs or equivalent controls. (B) Mice as indicated in (A) were monitored for disease severity and disease incidence for 28 d (n = 10). (C) Tmem173gt/gt mice were treated as indicated in (A) and monitored for severity of disease (n = 5–10). (D) SJL/J mice with EAE were treated with cGAMP MP (5 μg cGAMP) or rIFN-β (10,000 U/mouse) beginning at peak of disease (day 15) every other day until day 33. Mice were monitored for disease severity and incidences of relapse after initial remission. (E) Representative histological images of the cervical spinal cord (D) by H&E and Luxol fast blue (LFB) + periodic acid–Schiff (PAS) stain and average histological scores (F) at day 44. Area under the curve (AUC) was determined for mouse disease scores and tested by Mann–Whitney U test in (B) and (C). Two-way ANOVA was used to determine significance in (D). Data are presented as mean ± SEM. *p < 0.05, #p < 0.05, significance between cGAMP MP and rIFN-β.
Mice with EAE given chronic doses of cGAMP MP show no signs of in vivo toxicity
IFN-I therapy can induce proinflammatory cytokines (20), leading to side effects that may cause its discontinued use (21). Thus, it is important to determine if cGAMP MPs cause toxicity. We evaluated for toxicity EAE mice given three doses of cGAMP MPs (5, 7.5, and 10 μg) upon disease onset and every other day up to day 17 for a total of five doses. Mice treated with cGAMP MPs did not exhibit weight loss, a measure of toxicity, but instead mitigated some of the weight loss in untreated mice, indicating that the treatment was well tolerated (Supplemental Fig. 3A). Sera were screened for changes in liver enzymes (alanine aminotransferase, hemoglobin, platelets, and WBC counts were all within normal limits) (Supplemental Fig. 3B, 3C). Sera from cGAMP MP–treated mice did not have elevated IL-6, TNF-α, or IL-1β (Supplemental Fig. 3D). Thus, cGAMP MPs did not cause detectable toxicity.
cGAMP MPs induce STING-dependent IL-27 and IL-10 production in cultured APCs
IFN-I directly or indirectly exert anti-inflammatory effects on several immune cell types critical in EAE and MS (22–24), perhaps leading to its therapeutic efficacy. Based on our in vivo results, we hypothesized cGAMP MPs may induce anti-inflammatory cytokine production from APCs. Low to medium doses of cGAMP MPs induced increased IL-10 and IL-27 compared with soluble cGAMP in WT LPS-primed BMDCs (Fig. 3A, 3B). cGAMP MP–induced increases of these cytokines were lost in Tmem173gt/gt cells, confirming that cGAMP MP is working through STING activation (Fig. 3A, 3B). There was a partial decrease in the magnitude of IL-10 and IL-27 production in cGAMP MP–treated Ifnar−/− BMDCs relative to WT, indicating that IFN-I induced IL-10 and IL-27 secretion. However, significant amounts of IL-10 and IL-27 were still detectable in Ifnar−/− cells above the negative control, suggesting that an IFN-independent pathway also contributed to the induction of these cytokines. A similar experiment was conducted with BMDM cells with similar results (Fig. 3C, 3D). As described earlier, reproducibility of MP therapies is a critical hurdle to overcome prior to clinical use. In addition to reproducible chemical and physical properties, we demonstrate the biological reproducibility of IFN-β, IL-27, and IL-10 induction by three independent batches of cGAMP MPs (Supplemental Fig. 4). Taken together, these data suggest that cGAMP MPs activate STING to induce an anti-inflammatory IL-27 and IL-10, which are dependent on STING and predominantly mediated by IFN-I interaction with IFN-αβR. However, an IFN-independent effect was also detected.
FIGURE 3.
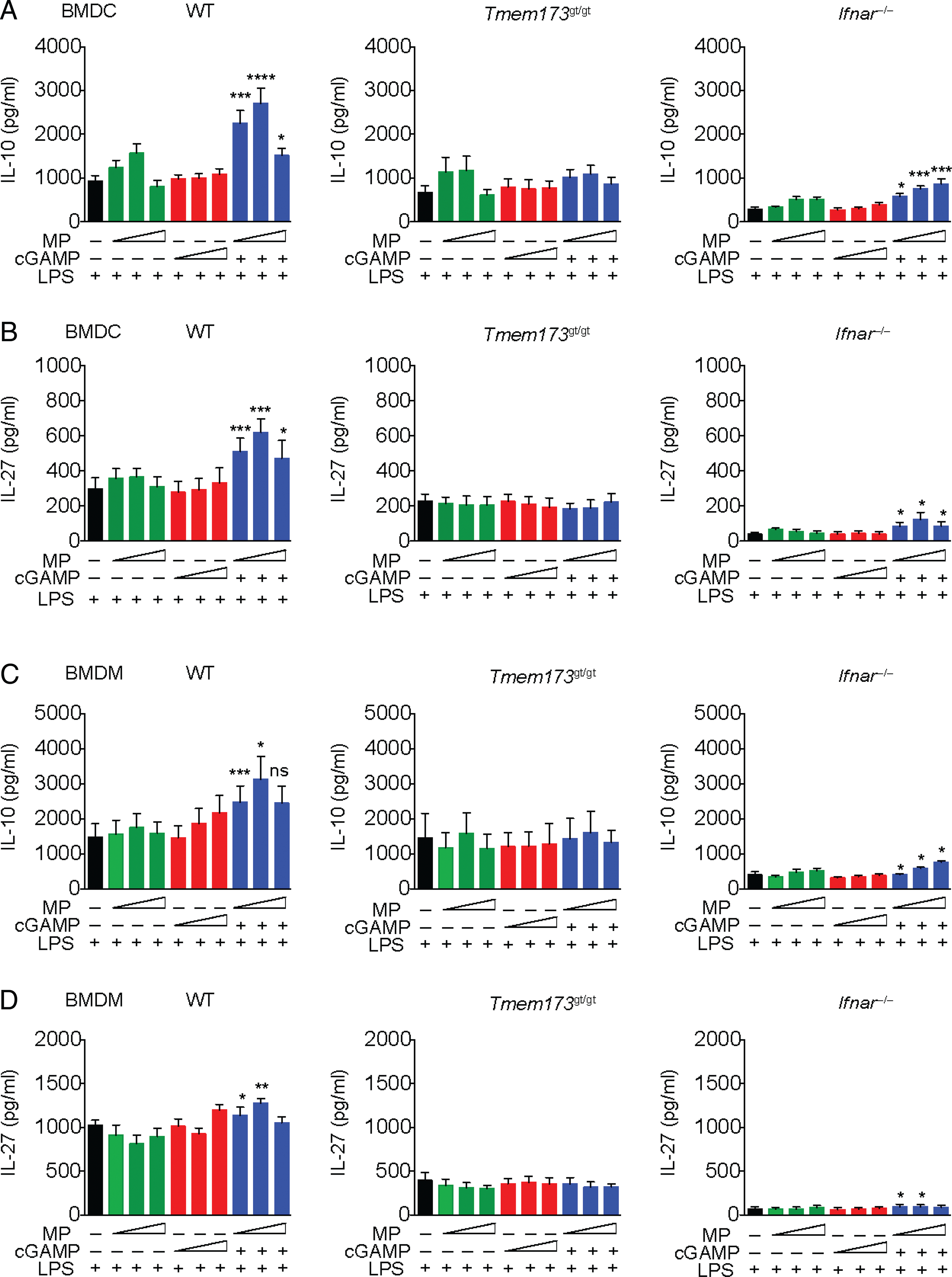
cGAMP MPs induce STING-dependent IL-10 and IL-27 production in APCs in vitro. (A and B) BMDCs from WT, Tmem173gt/gt, or Ifnar−/− mice were primed with LPS for 3 h prior to the addition of increasing concentrations of MP-encapsulated cGAMP (0.2–5 μg/ml), equivalent soluble cGAMP, or blank MP controls. IL-10 and IL-27 were measured in supernatants. (C and D) BMDMs from WT, Tmem173gt/gt, or Ifnar−/− mice were conditioned as described above and assayed for IL-10 and IL-27. All supernatants were collected after 24 h and assayed by ELISA. Combined data of two experiments, six biological replicates (n = 6). Student t test was used for cGAMP MP concentrations and their equivalent soluble dose. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
cGAMP MPs require ERK and CREB to induce IL-10 in vitro
The signaling required for the production of IL-10 is complex, involving several signaling pathways and autocrine feedback. We next examined the pathways by which cGAMP MP induces IL-10 production in BMDC because this has not been investigated. cGAMP MPs robustly promote STING phosphorylation by 1 h, whereas soluble cGAMP did not have a significant effect (Fig. 4A, 4B). Soluble cGAMP induced a detectable but nonsignificant increase in pSTAT1 compared with media control, indicative of IFN-I signaling. In contrast, cGAMP MPs significantly increased STAT1 signaling, reflecting a strong IFN induction (Fig. 4A, 4B). ERK is an important mediator of cell proliferation and survival and an upstream mediator of IL-10 production through downstream CREB signaling (25, 26). We observed an increase in the ratio of phosphorylated to total ERK as early as 30 min, which increased over time after cGAMP MP treatment (Fig. 4A, 4B). CREB phosphorylation was also increased in cGAMP MP–treated group (Fig. 4A, 4B). To test if these pathways were critical for the induction of IL-10 by cGAMP MP, BMDCs were LPS-stimulated as before but given either an ERK or CREB inhibitor prior to cGAMP MP treatment. Increased IL-10 induced by cGAMP MPs was reduced to basal control levels with increasing ERK or CREB inhibitor concentrations (Fig. 4C). These data support the link between ERK and downstream CREB and IL-10 (27), suggesting that cGAMP MP promotes ERK and CREB activation to generate IL-10 in BMDCs.
FIGURE 4.
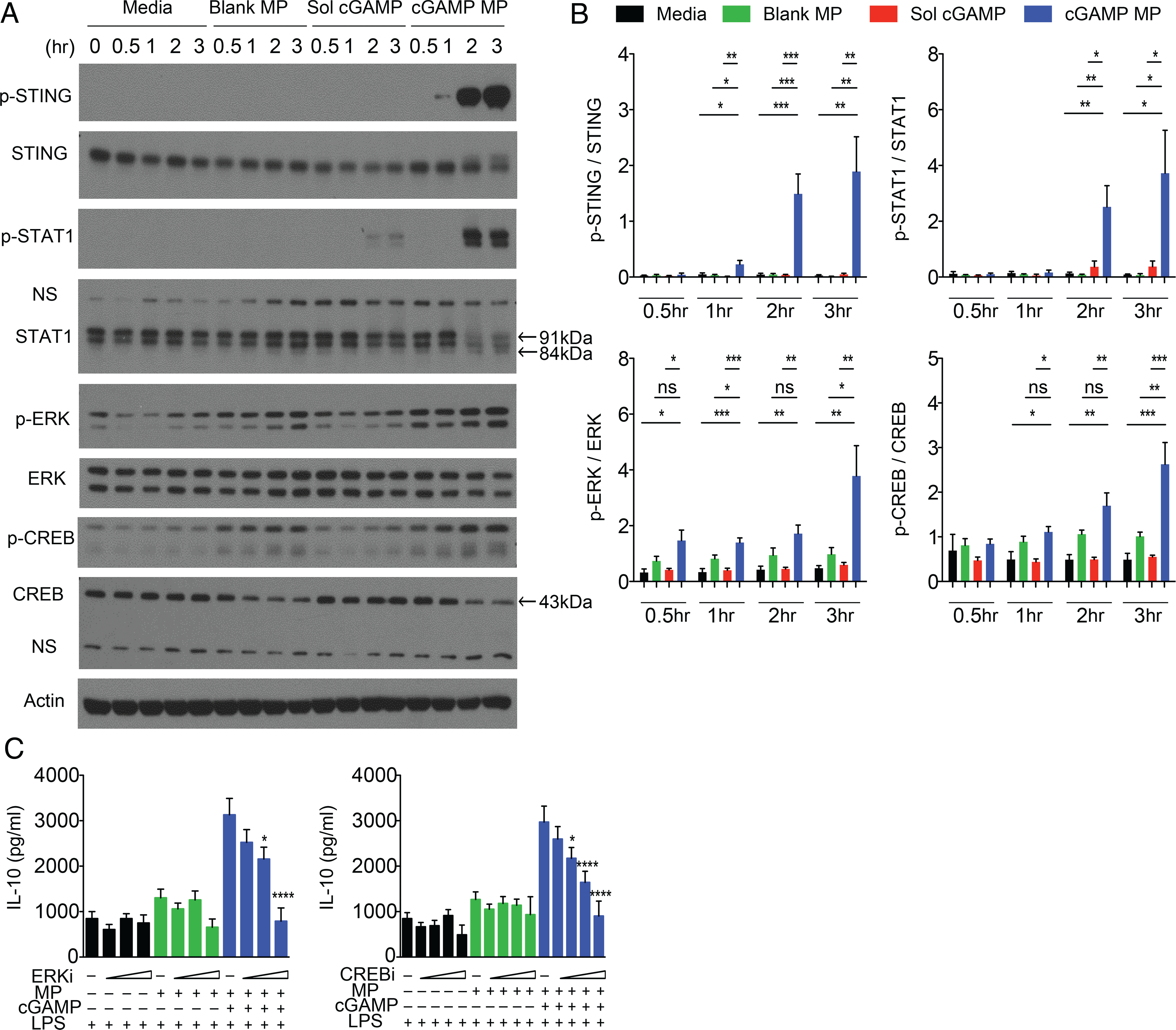
BMDC production of IL-10 induced by cGAMP MPs requires ERK/CREB signaling. (A and B) BMDC were treated with 1 μg/ml cGAMP MPs or equivalent controls for up to 3 h. Cells were evaluated for phosphorylated and total protein expression by Western blot at the indicated timepoints. (A) Representative Western blots of two experiments, four total mice. Arrows indicate expected size for interrogated protein with nonspecific bands indicated on blots. Increases in phosphorylated protein–to–total protein ratio was determined by densitometry. (B). Samples were determined for significance by one-way ANOVA. (C) BMDC were primed with LPS for 3 h and treated with increasing concentrations of ERK inhibitor (ERKi; 10–25 μM) or CREB inhibitor (CREBi; 2.5–15 μM) for 30 min prior to the addition of MPs for 24 h. All samples in (C) were evaluated for significance when compared with their noninhibitor control by Student t test (n = 4–6). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
cGAMP MPs induce anti-inflammatory cytokines in splenocytes, even in the presence of activated and polarized T cells
The acute phase of EAE and MS is driven by adaptive immune cells, whereas IFN-I therapy is an inhibitor of this response (28). Next, we determined if cGAMP MPs also inhibit adaptive immunity through the induction of IL-10. cGAMP MPs treatment of splenocyte cultures caused a significant increase in IFN-β over soluble cGAMP, but this waned over 3 d. IL-10 secretion lagged behind IFN-I and rose from days 1 to 3 (Fig. 5A). These results demonstrate that cGAMP MPs induce a robust IFN-β response and IL-10 production in splenocytes. To assess the effect of cGAMP MPs when CD4 T cells are activated, as during autoimmunity, we added soluble αCD3 mAb to activate T cells in the splenocyte culture. IFN-β was induced by the highest dose of cGAMP MP in the presence of αCD3 mAb–activated T cells. IL-27 and IL-10 increased nearly 20-fold compared with untreated cultures at this dose of cGAMP (Fig. 5B). These increases in IL-27 and IL-10 were accompanied by a decrease in IL-17 and IFN-γ, cytokines associated with Th17 and Th1 cells, respectively, which are pathogenic in the context of autoimmunity. As with BMDC and BMDM monocultures, we observed that cGAMP MPs can induce STING-dependent, but IFN-independent, IL-27 secretion (Fig. 5C). Additionally, we studied the effect of cGAMP MPs under the Th1- and Th17-polarizing conditions, and cGAMP MPs remained effective in the induction of IL-10 (Supplemental Fig. 5). These data suggest that cGAMP MP can cause the induction of anti-inflammatory cytokines even in the presence of activated Th1 and Th17 cells.
FIGURE 5.
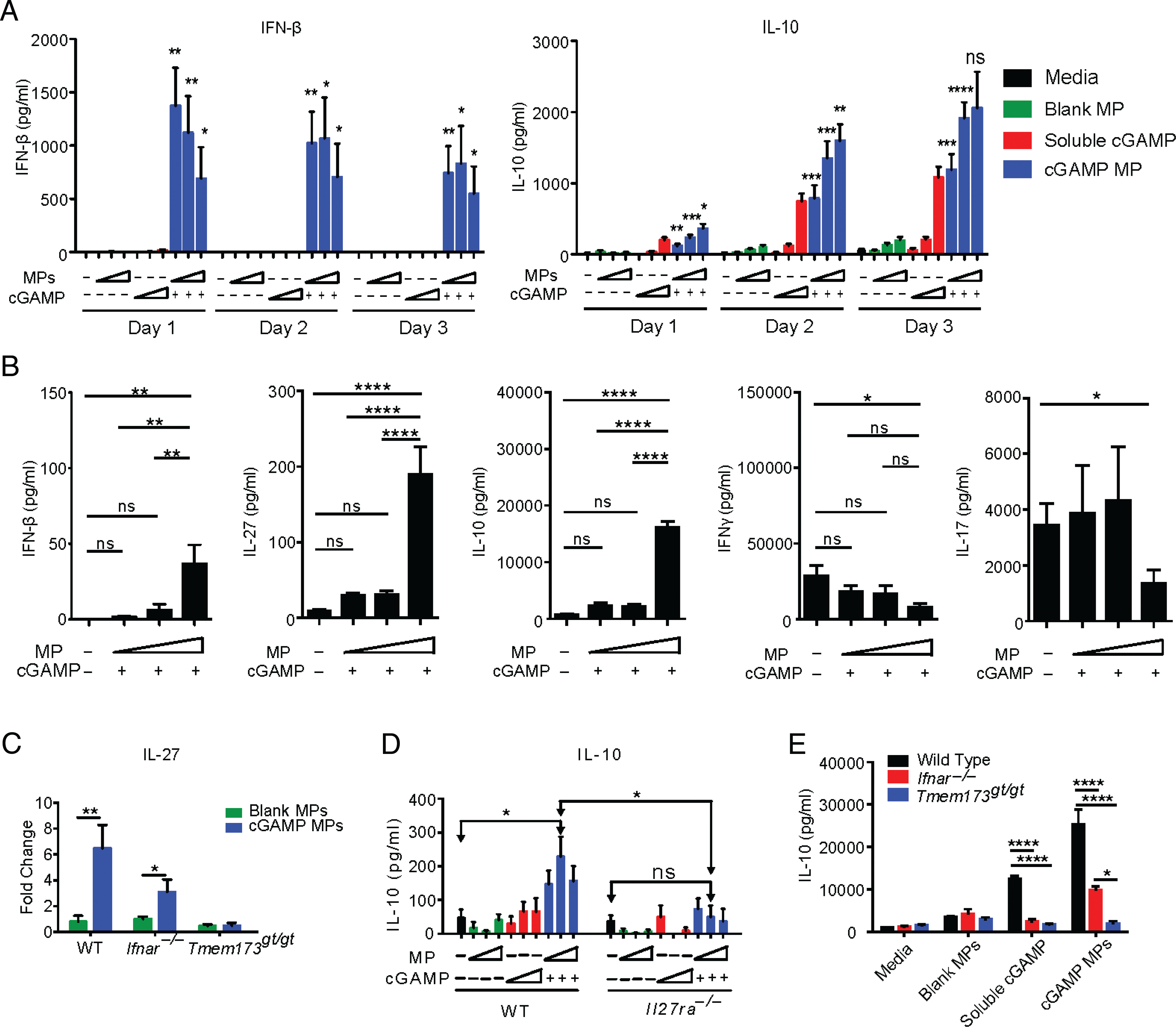
cGAMP MPs induce anti-inflammatory signatures in splenocytes. (A) CD8-depleted splenocytes were treated with increasing concentrations of cGAMP MPs (0.2–5 μg/ml) for up to 3 d, and cytokines were measured supernatants. cGAMP concentrations in cGAMP MP were compared with equivalent soluble cGAMP controls by Student t test. (B) CD8-depleted splenocytes were stimulated by soluble αCD3 mAb and treated for 3 d with increasing concentrations of cGAMP MPs. Groups were compared by one-way ANOVA. (C) CD8-depleted splenocytes from WT, Ifnar−/−, and Tmem173gt/gt mice were stimulated with soluble αCD3 mAb and treated for 3 d with cGAMP MPs (5 μg/ml cGAMP) or blank MPs. IL-27 was measured in supernatants and expressed as fold change over soluble αCD3 mAb controls. Significance was determined by Student t test. (D) WT BMDC were treated for 24 h with increasing concentrations of cGAMP MPs or controls. WT or Il27ra−/− naive CD4+ T cells were added to each condition for an additional 24 h with αCD3 mAb stimulation. Groups with each genotype were compared by one-way ANOVA. cGAMP MP doses between genotype were compared by Student t test. (E) WT, Ifnar−/−, and Tmem173gt/gt CD8-depleted splenocytes were treated with cGAMP MPs (5 μg/ml cGAMP) or equivalent control dose during 3 d of αCD3 mAb stimulation. Treatment groups were compared between genotypes by one-way ANOVA. Data represent the combination of at least two experiments (n = 6–12). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
IL-27 signaling in CD4+ T cells promote IL-10 (29). To determine if IL-27 signaling in T cells was critical for the induction of IL-10 by cGAMP MPs, we cocultured BMDCs with WT and Il27ra−/− CD4 T cells after pretreating the BMDCs with cGAMP MP. Cultures with Il27ra−/− T cells lost IL-10 induction from cGAMP MPs (Fig. 5D). Tmem173gt/gt splenocytes also lacked this cGAMP MP–mediated IL-10 induction (Fig. 5E). However, similar to experiments with BMDM and BMDC shown earlier in Fig. 3, splenocytes lacking Ifnar continued to produce a significant amount of IL-10 under cGAMP MP stimulation, although reduced in comparison with that produced by WT cells (Fig. 5E). Taken together, these in vitro data indicate that cGAMP MPs engages the STING pathway to promote IL-27, which is then required for downstream IL-10 induction, but induction of both IL-27 and IL-10 occurs through IFNAR-dependent and -independent pathways.
cGAMP MPs mediate EAE protection by IL-27 and IL-10
Our in vitro data indicate that cGAMP MPs induce immunoregulatory cytokines IL-27 (30) and IL-10 (31). We hypothesized that they are the key cytokines contributing to the protection observed in cGAMP MP–treated mice with EAE. To assess if cGAMP MP can induce IL-27 and IL-10 in vivo, we injected mice with cGAMP MPs or soluble cGAMP i.m. and measured IL-27 and IL-10 levels at the injection site. cGAMP MPs significantly increased IL-27 6 and 12 h postinjection and increased IL-10 at the 12-h timepoint (Fig. 6A).
FIGURE 6.
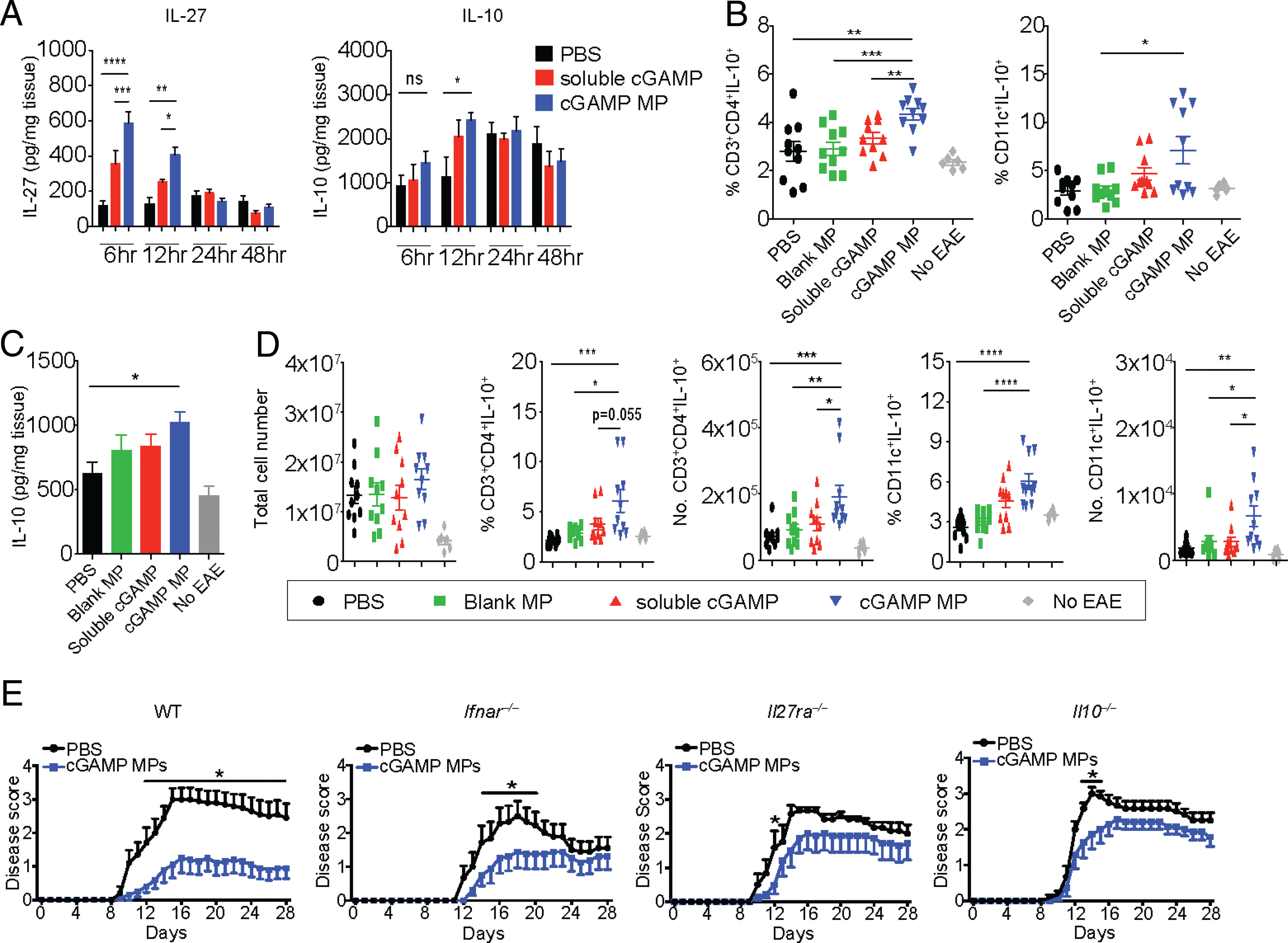
cGAMP MPs protect WT mice from EAE by IL-27–dependent IL-10 production. (A) Soluble (5 μg) or equivalent cGAMP MPs were injected i.m. into mice at days 9, 11, and 13 after EAE induction. Samples from mice were obtained at 6, 12, 24, and 48 h after final injection and evaluated for local cytokine production (n = 6). (B–D) WT mice were induced to EAE and injected i.m. with 5 μg cGAMP in MPs or equivalent levels of blank MP or cGAMP controls at days 9 and 11. Samples were collected on day 12. No EAE represents healthy control mice. Percentage and total number flow cytometry analysis of IL-10–producing CD4+T cells and CD11c+ DCs after PMA/ionomycin stimulation. Data from two experiments (n = 6–10). (E) WT, Ifnar−/−, Il27ra−/−, or Il10−/− mice were induced to EAE and injected i.m. with 5 μg cGAMP MPs on days 9, 11, 13, 15, and 17 and sacrificed at day 28. (E) Disease scores of two combined experiments per genotype (n = 7–10). Significance between groups was determined by two-way ANOVA. All other statistics were evaluated by Mann–Whitney U tests. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We next analyzed the impact of cGAMP MP on cells that produced IL-10 in mice with EAE. Analysis of spleen at day 12 postimmunization indicated that cGAMP MP–treated mice had a higher percentage of both IL-10+CD4 T cells and IL-10+CD11c+ DCs than untreated, MP-treated, or soluble cGAMP–treated mice (Fig. 6B). Furthermore, increased IL-10 production was observed in total splenocytes upon ex vivo stimulation with PMA/ionomycin as measured by an ELISA (Fig. 6C). There seems to be two distinct populations of CD11c+IL-10+ cells in the cGAMP MP–treated splenocytes. We surmised that it may be due to the limited percentage of splenic DCs that took up cGAMP MP compared with the higher uptake in DCs in LNs that are adjacent to the injection site. As a result, some splenic DCs were unaffected by cGAMP MP. In the draining inguinal LNs of mice with EAE, there was no difference in the total cell counts among untreated and blank MP–, soluble cGAMP–, and cGAMP MP–treated groups, indicating that the overall immune cell numbers were the same. However, cGAMP MP caused a significant increase in the percentage and total number of IL-10+CD4+ T cells and IL-10+CD11c+ cells in the draining LNs compared with mice injected with soluble cGAMP or blank particles (Fig. 6D).
To determine if the anti-inflammatory cytokines IL-27 and IL-10 were essential for cGAMP MP–mediated protection against EAE, Il27ra−/− mice were treated with cGAMP MPs upon disease onset. As before, cGAMP MPs protected WT mice from EAE when compared with a PBS-treated control (Fig. 6E, left). cGAMP MPs also protected Ifnar−/− mice from day 14 to 20 during peak of disease (Fig. 6E, left center). In contrast, cGAMP MPs did not significantly protect Il27ra−/− (Fig. 6E, right center) and Il10−/− (Fig. 6E, right) mice except at day 12 and days 14 to 16, respectively, indicating that the protective effect is primarily mediated through IL-27 and subsequent IL-10 and that the protection occurs through both IFN-I–dependent and –independent pathways. Taken together, these studies support the conclusion that cGAMP MPs protect mice from EAE through IL-10, which requires IL-27 for its optimal induction.
cGAMP MPs promote anti-inflammatory cytokines in immune cells from RRMS patients
Our data indicate that cGAMP MPs induce IFN-I and related anti-inflammatory cytokines to protect mice in the EAE model. To demonstrate clinical relevance, we next tested if this effect can also be observed in cells from RRMS patients. We obtained PBMC and isolated monocytes from recently diagnosed RRMS patients who had not yet begun treatment and treated magnetic bead-separated human CD14+ monocytes with cGAMP MPs (5 μg/ml cGAMP) (Fig. 7A). cGAMP MP, but not soluble cGAMP, promoted the upregulation of IFN-I– and IFN-induced Mx1 gene expression as tested by quantitative RT-PCR. Flow cytometry also showed a dramatic and significant increase in IFN-β and strong IL-27 and IL-10 responses in monocytes treated with cGAMP MP, but not with soluble cGAMP (Fig. 7B). Human monocytes were highly sensitive to cGAMP MP stimulation compared with mouse APCs and produced ample IL-27 after this treatment and additionally produced IL-10 (Fig. 7C). Finally, we cocultured patients’ isolated monocytes with autologous CD4 T cells and confirmed in a coculture that cGAMP MPs still induced IL-27 and IL-10 production (Fig. 7D). These results show that cGAMP MP induced IFN-β and the anti-inflammatory IL-27 and IL-10 in monocytes isolated from RRMS patients.
FIGURE 7.
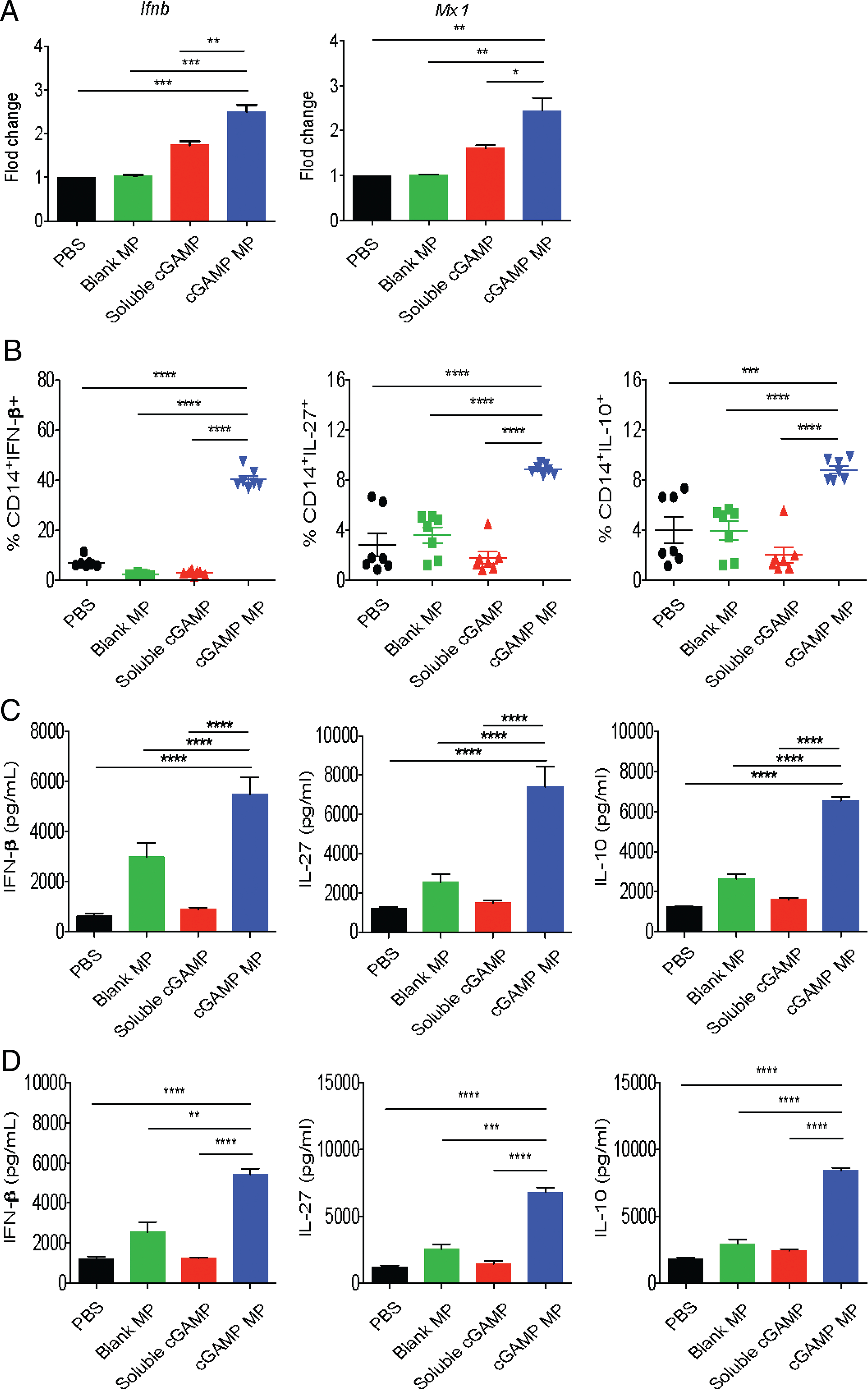
Human PBMCs express anti-inflammatory signatures in response to cGAMP MPs. (A) Monocytes isolated from PBMCs from RRMS patients were treated with 5 ug/ml cGAMP MPs for 8 h and evaluated for IFN-I gene expression by quantitative RT-PCR (qRT-PCR). (B) RRMS patient PBMCs were treated with cGAMP MPs for 24 h and stimulated with LPS prior to monocyte staining for flow cytometry. (C) Monocytes from RRMS patients were cultured with cGAMP MPs for 24 h, and supernatants were collected for ELISA. (D) RRMS patient monocytes were stimulated with LPS and pretreated with cGAMP MPs for 12 h. Washed monocytes were cocultured with autologous CD4+T cells for 12 h. Treatment groups were compared by a one-way ANOVA. Data are combined of at least two independent experiments (n = 4–7). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
The variety of applications for which IFN-I is used highlights the complex function of this cytokine family. In host immunity, IFN-I is an important factor in the initial antiviral response (32) and is an inducer of Th1-biased immunity (33). This property has made it useful for promoting balanced Th1/Th2 responses in vaccine design against pathogens. In tumor immunity, IFN-I can cause DC cross-presentation of Ag (34), making it a robust activator of cytotoxic CD8 T cells (35). Along with other antitumor properties, this has made it an important cancer immunotherapeutic, particularly in melanoma (36). However, this proinflammatory response is dampened with chronic IFN-I. In persistent viral infections, continuous IFN-I signaling has been shown to stunt the generation of new Th1 responses to the virus (37). This ties with the use of IFN-I in RRMS, in which its immunoregulatory properties has made it a mainstay of treatment for this condition for decades. However, use of rIFN-I has more recently been described as only a moderately effective therapy compared with newer medications and faces challenges such as the elicitation of neutralizing Abs against the recombinant protein that can further reduce efficacy.
The technology presented in this study was designed to build on the success of rIFN-I technology by using cGAMP to induce endogenous IFN-I and circumvent some of drawbacks associated with rIFN therapy. Our findings suggest that cGAMP could be a unique therapeutic, harnessing both IFN-dependent and -independent effects to support an immunoregulatory environment. STING is one of several innate immune sensors, including TLR3, TLR7, TLR9, MAVS, and RIG-I that are proficient in inducing IFN-I. Unlike IFN-promoting TLRs, STING signaling is independent of TRIF and MyD88 (38). STING is activated by cyclic dinucleotides as opposed to MAVS and RIG-I, which are activated by RNA that is highly sensitive to RNase. The complication of using small cyclic dinucleotide as a therapeutic agent arises when trying to target STING directly, as it is located in the cytosol and can be endoplasmic reticulum associated (39). For this reason, delivery of free STING agonists, such as cyclic dinucleotides, is often inefficient and requires high concentrations that could cause toxicity. A solution to rectify this problem is by using a particulate carrier to deliver STING agonists to appropriate cells. This has been shown to be an improvement to soluble delivery in applications such as vaccines and cancer (12–14). So far, this approach has not been used to elicit an immune-regulatory and anti-inflammatory response, which may be beneficial for RRMS. This study uses an encapsulation platform and showed superiority of cGAMP MP over soluble cGAMP in IFN-β induction and reduction of clinical scores in mice with EAE, but also in the elicitation of disease-attenuating cytokines IL-27 and IL-10, which are partially independent of IFN signaling.
Our previous study suggests that the cGAMP MP are taken up by APCs in vitro (12). The present study confirms in vivo uptake of particles by both macrophages and DCs. DCs and macrophages are some of the most prolific producers of IFN-I (8). Furthermore, in EAE and MS, they have crucial roles in priming autoreactive T cells but also can directly suppress these cells and promote homeostasis (40, 41). The used particles are in the range of 1–2 μm, which are in an ideal size range for uptake by DCs (42).
Prior to this work, the effect of cGAMP on EAE was unknown, although DNA activation of the STING pathway has been studied (19). For the use of IFN-I to treat RRMS, patient compliance and robust safety profile have been continual selling points. However, IFN-I in cancer immunotherapy was associated with severe side effects in patients (43). Given that we have shown STING signaling is inherently different from direct IFN-I signaling, it was important to determine if cGAMP MPs elicited systemic toxicity. Although high concentrations of STING agonists have been reported to be toxic in vitro (44), a formal evaluation of in vivo toxicity following administration has not been reported. The use of MPs to deliver cGAMP allowed dose sparing, leading to the use of low doses of i.m. cGAMP MPs as a clinically feasible way of alleviating disease. Safety testing showed that chronic administration of cGAMP MPs does not have significant side effects in EAE mice based on known indicators of systemic toxicity and actually improved some measured outcomes, suggesting that this could be a safe treatment.
To explore the mechanism by which cGAMP MP might be mitigating EAE, we examined several potential pathways. Of its immunomodulating properties, IL-10 induction has gained attention as a critical downstream mediator of IFN-I protection because of its production directly or indirectly in many MS-relevant cell types (22, 23). In contrast to IL-6, IL-10 causes sustained STAT3 signaling (45), which promotes SOCS3, causing inhibition of cytokine production (46). This STAT3 signaling is impaired in some MS patients, resulting in poor IL-10 response; thus, an exogenous agonist to activate this pathway may provide a remedy for this dysfunction (46). In regard to IFN, IL-10 production is markedly reduced with or without TLR agonists in primary Ifnar−/− myeloid cells, showing the importance of IFN-I signaling to IL-10 production by innate immune cells (47). IFN-I can directly signal IL-10 production in some cells (22) but appears to rely partially on IL-27 as an intermediary. IL-27 is a critical factor in the differentiation of IL-10–producing Tr1 cells (29) and promotes IL-10 in Th1, Th17, and regulatory T cells (30, 48). Regulatory T cell subsets further benefit from IL-27 by the suppression of FasL (49) and upregulation of survival factors such as cFLIP, BcL-6, and ICOS (50, 51). Thus, IL-27 can promote their survival through these different mechanisms. IL-27 has also been reported to directly inhibit IL-2 production (52) and the differentiation of Th17 cells (53). Indeed, an inverse correlation of low IL-27 and high Th17 and a direct correlation of IFN-β responsiveness with IL-27 levels in MS patients support an underlying role of IL-27 in the IFN pathway and the disease (54, 55). APCs are the primary producers of IL-27 and express the IL-27R. In our study, we observe that cGAMP MPs induce STING-dependent IL-10 and IL-27 expression that is enhanced by IFN-I signaling, which is not required. The link between the cGAMP and IL-27 has not been previously appreciated. STING-associated IL-10 production is only a recent finding; although the role of cGAS was not demonstrated in one study, the impact of STING on IL-10 was paradoxically cGAS independent (56–58). In contrast, our study clearly showed that STING was required for the induction of both IL-10 and IL-27 by cGAMP MP and that IL-27 signaling was required for IL-10 induction. Importantly, we further demonstrated that IL-10 and IL-27 can be produced during cGAMP MP stimulation without IFN-I signaling. This observation has not been reported previously. Given the interplay between IFN-I and these two anti-inflammatory cytokines and their roles in several autoimmune disorders, cGAMP MP may provide a unique therapeutic benefit by targeting several immunoregulatory pathways that are IFN dependent and independent, but both involve IL-27 and IL-10.
Several transcription factors have been associated with downstream IL-10 production in innate cells. cGAMP MPs activated STING and greatly promoted STING phosphorylation within an hour of exposure in BMDCs. IFN induces STAT1, so it was not surprising that this signaling followed STING activation by cGAMP MPs. IL-10 signaling via STAT1 in innate immune cells has been conflicting, with both induction and inhibition reported (59, 60). Direct addition of IFN-β on BMDCs has been previously shown to increase CREB phosphorylation and IL-10 production (22). We focused on the CREB pathway given that cGAMP MPs induced high amounts of IFN-I in innate immune cells. ERK1/2 is involved in numerous cellular responses involving innate cell survival (61) and has been linked to IL-10 production in these cells (62, 63). Similarly, it has been reported that certain pathogen-associated molecular patterns, such as zymosan, can induce IL-10 in DCs via CREB by upstream ERK activation (26). ERK has also been reported to bolster IL-10 response (64). In our study, we observed ERK phosphorylation early upon cGAMP MP addition, whereas CREB signaling proceeds soon after. Both ERK and CREB are regulating increased IL-10 production by cGAMP MPs given that ERK or CREB specific inhibitors reduced IL-10 to a basal level.
Our in vivo data indicate that cGAMP MP can increase IFN-β and IL-27 production at the injection site and induce IL-10+ T cells and DCs during disease progression. This implies that cGAMP MP can support immunoregulatory phenotypes, as we observed in vitro. It is noteworthy that throughout the monitoring period in the C57BL/6J model, we did not observe any relapses or EAE worsening after the final dose of cGAMP MPs was given. In the SJL relapsing–remitting model, mice given cGAMP MP also had little disease activity at least 5 d (day 38) after the last dose (day 33). Although our work focused on exogenous cGAMP, dying cells can release self-DNA and induce endogenous 2′,3-cGAMP to trigger STING activation in resident innate immune cells (65). Interrogating the muscle tissue during active EAE revealed substantial baseline IL-10. It is possible that EAE may cause STING activation, but the levels are insufficient to regulate the disease. Supplementing endogenous response with cGAMP MPs showed protection against EAE. We also found that cGAMP MPs were significantly protective in Ifnar−/− mice and are more effective than rIFN-β in the relapsing–remitting model, supporting our hypothesis of both IFN-dependent and -independent mechanisms. However, both Il27ra−/− and Il10−/− mice showed minor response to cGAMP MP treatment, suggesting that these two cytokines were more critical than IFN-I for protection. This would support the notion that the role of IFN-β, IL-27, and IL-10 signaling is complex, relying on independence and interdependence to suppress EAE (24). Moreover, a noteworthy report has shown STING in microglia to be protective in the context of neuroinflammation (66). Thus cGAMP MP therapy likely induces multiple factors that restrain MS and EAE, which may be superior to rIFN-β alone. Thus, our data indicate that the suppression of EAE by cGAMP MP is multifactorial but is primarily dependent on IL-27 and IL-10.
In conclusion, this study demonstrates that cGAMP incorporated into MPs induces anti-inflammatory cytokines that mitigate EAE at the peak of disease. Multiple injections of cGAMP MPs appear safe, and significantly improve disease outcome over soluble cGAMP in a STING-dependent and both IFN-I–dependent and –independent manner. cGAMP MP induces the immunosuppressive cytokine, IL-10, in both APCs and CD4 T cells in a process that is ERK- and CREB-dependent. IL-10 induction by cGAMP MPs is primarily IL-27-dependent, and cGAMP MP–induced IL-10 and IL-27 are crucial for protection against EAE. Importantly, cGAMP MPs promote monocytes and cocultures of monocytes and T cells from RRMS patients to produce IFN-I, IL-27 and IL-10, associated with immune regulation and disease control. This study links the cGAS-cGAMP-STING pathway to the mitigation of EAE through the induction of immune-regulatory, anti-inflammatory cytokines and indicates a potential for such use in RRMS therapy.
Supplementary Material
Acknowledgments
The authors thank The University of North Carolina at Chapel Hill School of Medicine Histology Research Core Facility and the Lineberger Cancer Center National Cancer Institute Core Grant for providing technical expertise in spinal sectioning and staining.
This work was supported by National Multiple Sclerosis Society (NMSS) Grant CA10068 and NMSS fellowship (to W.-C.C.) and Foundation for the National Institutes of Health Grants R01-AI141333, AI029564, CA232109, U54CA198999, T32NHLBI7106-39 (to B.M.J.), T32CA009156 (to S.A.G. and J.W.T.), 1R01AI131238-01A1 (to S.M.-P.), R01AI137525-01A1 (to E.M.B., K.M.A., S.M.-P.), and SAP4100083100 (to S.M.-P.).
Abbreviations used in this article:
- Ace-DEX
acetalated dextran
- BMDC
bone marrow dendritic cell
- BMDM
bone marrow–derived macrophage
- cGAMP
cyclic GMP–AMP
- cGAMP MP
cGAMP encapsulated in MP
- cGAS
cGAMP synthase
- DC
dendritic cell
- EAE
experimental autoimmune encephalomyelitis
- IFN-I
type I IFN
- Il27ra−/−
IL-27 receptor α
- LN
lymph node
- MP
microparticle
- MS
multiple sclerosis
- RRMS
relapsing–remitting MS
- STING
stimulator of IFN genes
- WT
wild-type
Footnotes
Disclosures
K.M.A., E.M.B., and J.P.-Y.T. are cofounders of IMMvention Therapeutix, Inc. After contributing to the current work, M.D.G. was hired at IMMvention Therapeutix, Inc., as a research scientist. Although the positive findings of this paper could serve towards a financial conflict of interest, the research in this study is no longer related to the interests of IMMvention Therapeutix, Inc. The University of North Carolina at Chapel Hill has reviewed this arrangement and has deemed it in accord with its policy on objectivity in research. The other authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Sun L, Wu J, Du F, Chen X, and Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies BW, Bogard RW, Young TS, and Mekalanos JJ. 2012. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149: 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn J, Gutman D, Saijo S, and Barber GN. 2012. STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. USA 109: 19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Škrnjug I, Guzmán CA, and Rueckert C. 2014. Cyclic GMP-AMP displays mucosal adjuvant activity in mice. [Published erratum appears in 2015 PLoS One 10: e0123605.] PLoS One 9: e110150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaauboer SM, Mansouri S, Tucker HR, Wang HL, Gabrielle VD, and Jin L. 2015. The mucosal adjuvant cyclic di-GMP enhances antigen uptake and selectively activates pinocytosis-efficient cells in vivo. eLife 4: e06670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, et al. 2015. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 112: 15408–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, Xu P, Tan J, Rui Y, Li P, and Tan X. 2016. Antitumor Activity of cGAMP via stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci. Rep. 6: 19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.González-Navajas JM, Lee J, David M, and Raz E. 2012. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 12: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrie RA, and Rudick RA. 2006. Drug insight: interferon treatment in multiple sclerosis. Nat. Clin. Pract. Neurol. 2: 34–44. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Hu F, Zhang Y, and Li K. 2020. Comparative efficacy and acceptability of disease-modifying therapies in patients with relapsing-remitting multiple sclerosis: a systematic review and network meta-analysis. J. Neurol. 267: 3489–3498. [DOI] [PubMed] [Google Scholar]
- 11.Koshy ST, Cheung AS, Gu L, Graveline AR, and Mooney DJ. 2017. Liposomal delivery enhances immune activation by STING agonists for cancer immunotherapy. Adv. Biosyst. 1: 1600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N, David CN, McGee CE, Sempowski GD, Shterev I, et al. 2018. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J. Control. Release 270: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee E, Jang HE, Kang YY, Kim J, Ahn JH, and Mok H. 2016. Submicron-sized hydrogels incorporating cyclic dinucleotides for selective delivery and elevated cytokine release in macrophages. Acta Biomater. 29: 271–281. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura T, Miyabe H, Hyodo M, Sato Y, Hayakawa Y, and Harashima H. 2015. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J. Control. Release 216: 149–157. [DOI] [PubMed] [Google Scholar]
- 15.Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N, and Schuler G. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223: 77–92. [DOI] [PubMed] [Google Scholar]
- 16.Weischenfeldt J, and Porse B. 2008. Bone marrow-derived macrophages (BMM): isolation and applications. CSH protocols 2008: pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 17.Aroh C, Wang Z, Dobbs N, Luo M, Chen Z, Gao J, and Yan N. 2017. Innate immune activation by cGMP-AMP nanoparticles leads to potent and long-acting antiretroviral response against HIV-1. J. Immunol. 199: 3840–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harari D, Kuhn N, Abramovich R, Sasson K, Zozulya AL, Smith P, Schlapschy M, Aharoni R, Köster M, Eilam R, et al. 2014. Enhanced in vivo efficacy of a type I interferon superagonist with extended plasma half-life in a mouse model of multiple sclerosis. J. Biol. Chem. 289: 29014–29029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, Pacholczyk G, Barber GN, Hayakawa Y, Munn DH, and Mellor AL. 2014. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J. Immunol. 192: 5571–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zimmermann M, Arruda-Silva F, Bianchetto-Aguilera F, Finotti G, Calzetti F, Scapini P, Lunardi C, Cassatella MA, and Tamassia N. 2016. IFNα enhances the production of IL-6 by human neutrophils activated via TLR8. Sci. Rep. 6: 19674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levesque N, Mitchinson K, Lawrie D, Fedorak L, Macdonald D, Normand C, and Pouliot JF. 2008. Health management program: factors influencing completion of therapy with high-dose interferon alfa-2b for high-risk melanoma. Curr. Oncol. 15: 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Brown J, Garcia CA, Tang Y, Benakanakere MR, Greenway T, Alard P, Kinane DF, and Martin M. 2011. The role of glycogen synthase kinase 3 in regulating IFN-β-mediated IL-10 production. J. Immunol. 186: 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Yuan S, Cheng G, and Guo B. 2011. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One 6: e28432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald DC, Fonseca-Kelly Z, Cullimore ML, Safabakhsh P, Saris CJ, Zhang GX, and Rostami A. 2013. Independent and interdependent immunoregulatory effects of IL-27, IFN-β, and IL-10 in the suppression of human Th17 cells and murine experimental autoimmune encephalomyelitis. J. Immunol. 190: 3225–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saraiva M, and O’Garra A. 2010. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10: 170–181. [DOI] [PubMed] [Google Scholar]
- 26.Kelly EK, Wang L, and Ivashkiv LB. 2010. Calcium-activated pathways and oxidative burst mediate zymosan-induced signaling and IL-10 production in human macrophages. J. Immunol. 184: 5545–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gee K, Angel JB, Ma W, Mishra S, Gajanayaka N, Parato K, and Kumar A. 2006. Intracellular HIV-Tat expression induces IL-10 synthesis by the CREB-1 transcription factor through Ser133 phosphorylation and its regulation by the ERK1/2 MAPK in human monocytic cells. J. Biol. Chem. 281: 31647–31658. [DOI] [PubMed] [Google Scholar]
- 28.Severa M, Rizzo F, Giacomini E, Salvetti M, and Coccia EM. 2015. IFN-β and multiple sclerosis: cross-talking of immune cells and integration of immunoregulatory networks. Cytokine Growth Factor Rev. 26: 229–239. [DOI] [PubMed] [Google Scholar]
- 29.Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, and Weiner HL. 2007. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat. Immunol. 8: 1380–1389. [DOI] [PubMed] [Google Scholar]
- 30.Hall AOH, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, et al. 2012. The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cua DJ, Hutchins B, LaFace DM, Stohlman SA, and Coffman RL. 2001. Central nervous system expression of IL-10 inhibits autoimmune encephalomyelitis. J. Immunol. 166: 602–608. [DOI] [PubMed] [Google Scholar]
- 32.Isaacs A, and Lindenmann J. 1957. Virus interference. I. the interferon. Proc. R. Soc. Lond. B Biol. Sci. 147: 258–267. [PubMed] [Google Scholar]
- 33.Wenner CA, Guler ML, Macatonia SE, O’Garra A, and Murphy KM. 1996. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J. Immunol. 156: 1442–1447. [PubMed] [Google Scholar]
- 34.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, et al. 2014. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41: 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei J, Waithman J, Lata R, Mifsud NA, Cebon J, Kay T, Smyth MJ, Sadler AJ, and Chen W. 2010. Influenza A infection enhances cross-priming of CD8+ T cells to cell-associated antigens in a TLR7- and type I IFN-dependent fashion. J. Immunol. 185: 6013–6022. [DOI] [PubMed] [Google Scholar]
- 36.Aoyagi S, Hata H, Homma E, and Shimizu H. 2012. Sequential local injection of low-dose interferon-beta for maintenance therapy in stage II and III melanoma: a single-institution matched case-control study. Oncology 82: 139–146. [DOI] [PubMed] [Google Scholar]
- 37.Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, de la Torre JC, and Brooks D. 2014. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. USA 111: 7409–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478: 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishikawa H, and Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. [Published erratum appears in 2008 Nature 456: 274.] Nature 455: 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.López C, Comabella M, Al-zayat H, Tintoré M, and Montalban X. 2006. Altered maturation of circulating dendritic cells in primary progressive MS patients. J. Neuroimmunol. 175: 183–191. [DOI] [PubMed] [Google Scholar]
- 41.Tsai VW, Mohammad MG, Tolhurst O, Breit SN, Sawchenko PE, and Brown DA. 2011. CCAAT/enhancer binding protein-δ expression by dendritic cells regulates CNS autoimmune inflammatory disease. J. Neurosci. 31: 17612–17621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohane DS 2007. Microparticles and nanoparticles for drug delivery. Biotechnol. Bioeng. 96: 203–209. [DOI] [PubMed] [Google Scholar]
- 43.Hauschild A, Gogas H, Tarhini A, Middleton MR, Testori A, Dréno B, and Kirkwood JM. 2008. Practical guidelines for the management of interferon-alpha-2b side effects in patients receiving adjuvant treatment for melanoma: expert opinion. Cancer 112: 982–994. [DOI] [PubMed] [Google Scholar]
- 44.Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, and Poltorak A. 2017. Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunol. 199: 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braun DA, Fribourg M, and Sealfon SC. 2013. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J. Biol. Chem. 288: 2986–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Forero I, Garcia-Munoz R, Martinez-Pasamar S, Inoges S, Lopez-Diaz de Cerio A, Palacios R, Sepulcre J, Moreno B, Gonzalez Z, Fernandez-Diez B, et al. 2008. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur. J. Immunol. 38: 576–586. [DOI] [PubMed] [Google Scholar]
- 47.Kole A, He J, Rivollier A, Silveira DD, Kitamura K, Maloy KJ, and Kelsall BL. 2013. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J. Immunol. 191: 2771–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O’Shea JJ, and Hunter CA. 2007. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat. Immunol. 8: 1363–1371. [DOI] [PubMed] [Google Scholar]
- 49.Kim G, Shinnakasu R, Saris CJ, Cheroutre H, and Kronenberg M. 2013. A novel role for IL-27 in mediating the survival of activated mouse CD4 T lymphocytes. J. Immunol. 190: 1510–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Z, Liu JQ, Talebian F, Wu LC, Li S, and Bai XF. 2013. IL-27 enhances the survival of tumor antigen-specific CD8+ T cells and programs them into IL-10-producing, memory precursor-like effector cells. Eur. J. Immunol. 43: 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pot C, Jin H, Awasthi A, Liu SM, Lai CY, Madan R, Sharpe AH, Karp CL, Miaw SC, Ho IC, and Kuchroo VK. 2009. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 183: 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura D, Miyakoda M, Kimura K, Honma K, Hara H, Yoshida H, and Yui K. 2016. Interleukin-27-producing CD4(+) T cells regulate protective immunity during malaria parasite infection. Immunity 44: 672–682. [DOI] [PubMed] [Google Scholar]
- 53.Ramgolam VS, Sha Y, Jin J, Zhang X, and Markovic-Plese S. 2009. IFN-beta inhibits human Th17 cell differentiation. J. Immunol. 183: 5418–5427. [DOI] [PubMed] [Google Scholar]
- 54.Tang SC, Fan XH, Pan QM, Sun QS, and Liu Y. 2015. Decreased expression of IL-27 and its correlation with Th1 and Th17 cells in progressive multiple sclerosis. J. Neurol. Sci. 348: 174–180. [DOI] [PubMed] [Google Scholar]
- 55.Sweeney CM, Lonergan R, Basdeo SA, Kinsella K, Dungan LS, Higgins SC, Kelly PJ, Costelloe L, Tubridy N, Mills KH, and Fletcher JM. 2011. IL-27 mediates the response to IFN-β therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav. Immun. 25: 1170–1181. [DOI] [PubMed] [Google Scholar]
- 56.Movert E, Lienard J, Valfridsson C, Nordström T, Johansson-Lindbom B, and Carlsson F. 2018. Streptococcal M protein promotes IL-10 production by cGAS-independent activation of the STING signaling pathway. PLoS Pathog. 14: e1006969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang D, Xiao-Feng H, Guan-Jun D, Er-Ling H, Sheng C, Ting-Ting W, Qin-Gang H, Yan-Hong N, and Ya-Yi H. 2015. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the cjun/CCL22 signal. Biochim. Biophys. Acta 1852: 2494–2503. [DOI] [PubMed] [Google Scholar]
- 58.Ahn J, Son S, Oliveira SC, and Barber GN. 2017. STING-dependent signaling underlies IL-10 controlled inflammatory colitis. Cell Rep. 21: 3873–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iyer SS, Ghaffari AA, and Cheng G. 2010. Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J. Immunol. 185: 6599–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalliolias GD, and Ivashkiv LB. 2008. IL-27 activates human monocytes via STAT1 and suppresses IL-10 production but the inflammatory functions of IL-27 are abrogated by TLRs and p38. J. Immunol. 180: 6325–6333. [DOI] [PubMed] [Google Scholar]
- 61.Sanin DE, Prendergast CT, and Mountford AP. 2015. IL-10 production in macrophages is regulated by a TLR-driven CREB-mediated mechanism that is linked to genes involved in cell metabolism. J. Immunol. 195: 1218–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, Maliszewski C, Akira S, and Pulendran B. 2004. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J. Immunol. 172: 4733–4743. [DOI] [PubMed] [Google Scholar]
- 63.Yi AK, Yoon JG, Yeo SJ, Hong SC, English BK, and Krieg AM. 2002. Role of mitogen-activated protein kinases in CpG DNA-mediated IL-10 and IL-12 production: central role of extracellular signal-regulated kinase in the negative feedback loop of the CpG DNA-mediated Th1 response. J. Immunol. 168: 4711–4720. [DOI] [PubMed] [Google Scholar]
- 64.Kaiser F, Cook D, Papoutsopoulou S, Rajsbaum R, Wu X, Yang H-T, Grant S, Ricciardi-Castagnoli P, Tsichlis PN, Ley SC, and O’Garra A. 2009. TPL-2 negatively regulates interferon-β production in macrophages and myeloid dendritic cells. J. Exp. Med. 206: 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, Mistry KN, Do D, Marsh SE, Abud EM, et al. 2017. Activation of the STING-dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron 96: 1290–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.