

Abstract
Background & Aims:
Chronic hepatitis B virus (HBV) infection is characterized clinically by progression through disease phases. The first, labeled immune tolerant (IT), is perceived to lack disease activity. Here, we examined HBV-DNA integration, clonal hepatocyte expansion, HBV antigen expression and HBV-specific immunity in patients considered IT to assess whether this designation is appropriate, or if pathological changes may be present.
Methods:
HBV-DNA integration, clonal hepatocyte expansion, HBsAg and HBcAg expression were studied in liver tissue from CHB patients, (age 14–39 years; median=24.5). These included 9 HBeAg(+) IT patients. Ten HBeAg(+) and 7 HBeAg(−) age-matched patients with active disease served as controls. HBV-specific T cells were quantified in paired peripheral blood lymphocytes.
Results:
HBV antigen expression differed between the patient groups. However, unexpectedly high numbers of HBV-DNA integrations, randomly distributed across most chromosomes, were detectable in all patient groups. Patients considered IT also displayed significant clonal hepatocyte expansion, potentially in response to active HBV-specific T cell immunity. HBV-specific T cell responses were also confirmed in the periphery of these patients.
Conclusions:
A high level of HBV DNA integration and clonal hepatocyte expansion in patients considered IT suggests that events in hepatocarcinogenesis are underway even in the early stages of CHB. The concept that the IT phase is devoid of markers of disease progression and is immunologically inert is unsupported; instead, we propose that high replicative low inflammatory (HRLI) CHB more accurately reflects this early disease phase. The timing of therapeutic intervention to minimize further genetic damage to the hepatocyte population should be reconsidered.
Keywords: HBV, immune tolerance, hepatocyte proliferation
INTRODUCTION
Chronic hepatitis B (CHB) virus infection acquired at birth or in early childhood typically progresses through an early disease phase characterized by normal serum alanine aminotransferase (ALT) and high titer viremia (EASL & AASLD guidelines).1 Patients can remain in this phase of CHB for several decades. Historically perceived as disease-free, these patients are considered ‘immune tolerant’ (IT) and thus excluded from therapy based on international treatment recommendations (EASL & AASLD). Classically, the IT phase is followed by a period of immune active (IA) liver disease, characterized by hepatic flares of increased inflammatory activity with elevated ALT levels, where patients are deemed to meet treatment criteria.
A question in the management of chronic HBV infection is whether antiviral treatment should be withheld until the development of persistently elevated serum ALT. Arguments against treatment in the IT phase have centered on drug cost, potential selection for drug resistant virus, and toxicity associated with long-term therapy.2 Historically, a stronger argument against treatment has been the perceived lack of disease activity and suppression of antiviral immunity, but the validity of these arguments, which in a clinical setting normally rely on serological assays without liver histology, is unclear. For instance, the mechanism of hepatocyte destruction (e.g., apoptosis versus necroptosis) might change during the course of CHB, influencing ALT levels in a manner not reflecting the amount of cell destruction.3, 4
The notion that events potentially leading to cumulative liver damage, including HCC initiation and promotion, are absent in IT patients has been contested by recent immunological data, which do not support clear differences between phases of CHB.1, 4–6 We have previously shown that HBV exposure in utero does not induce a generic state of immunological tolerance,7 and also, that HBV-specific T cell responses in young patients labeled IT are not inferior to those seen in their peers with IA disease differentiated only by ALT elevation.5 Recent data from CHB adults confirms that HBV-specific immunological parameters are no different between these two disease phases.8 Further evidence against an inert immunological response in IT patients has come from a study demonstrating an increased innate immune gene signature in IT patients6 and from virological data showing sequence evolution of HBV with increasing age in a cohort of IT patients.9
The presence of immunological activity and high levels of HBV replication in what is considered the IT phase may promote cumulative liver damage, since hepatocytes appear to constitute a closed, self-renewing cell population, as reported in animal studies investigating both syngeneic hepatocytes and transplanted human hepatocytes.10, 11 First, normal hepatocytes as well as hepatocytes with markers of senescence were able to proliferate to maintain liver mass during injury.12 Second, recent evidence suggests that so-called liver progenitor/stem cells (e.g., oval cells) either do not have a significant role in liver regeneration13–15 or conversely, if they do have a role in regeneration, are first formed via de-differentiation of mature hepatocytes.10 Though some of these issues are still contested,16–18 the overall conclusion that hepatocytes are primarily self-renewing seems valid. Consequently, epigenetic and genetic dysregulation, including damage via HBV-DNA integration, might increase over time.
In the present study, we performed a comprehensive analysis of clinical and virological parameters in patients considered IT, and in age-matched IA non-cirrhotic HBeAg positive(+) and negative(−) CHB patients. We also assessed the frequency of HBV DNA integration and clonal hepatocyte expansion across all patient groups. Integration of HBV DNA into chromosomal DNA during chronic infection is one of the factors believed to contribute to or reflect mutagenesis leading to hepatocarcinogenesis. Importantly, using duck hepatitis B virus, integration was found to occur at double strand breaks, probably due to non-homologous end joining, and the frequency of mutagenesis during repair of double stranded breaks was 10 times as frequent as HBV DNA integration at the site.19 Thus, HBV DNA integration frequency may significantly underestimate the mutation frequency in hepatocytes. Errors during repair of double stranded DNA breaks are considered important in human oncogenesis.20
A recent study showed that virus integration and hepatocyte expansion may be present in the IT phase, but this phenomenon was not studied in detail and age-matched controls were not available.21 In the present study, we compared HBV integration frequency and clonal hepatocyte expansion in young patients considered IT, and aged-matched IA HBeAg(+) and HBeAg(−) controls. Since HBV DNA integration occurs at random sites in host DNA, virus/host DNA junctions serve as markers of hepatocyte lineages, and the multiplicity of virus/cell DNA junctions from liver tissue can be used to calculate clonal hepatocyte expansion. Finally, differential HBV antigen expression in hepatocytes, as well as HBV-specific immune responses were determined across the disease phases to test the validity of what is labeled IT CHB.
MATERIALS AND METHODS
Patient samples & Study design
Twenty-six patients were recruited and categorized into CHB phases using established clinical characteristics: measurements of serum transaminases (ALT), serological parameters, including HBsAg, HBeAg, anti-HBeAg and virus titers (EASL & AASLD): Immune tolerant (IT) (n=9); HBeAg(+) immune active (IA) (n=10); HBeAg(−) immune active (IA) (n=7) (Table 1). The patients were further assessed by liver biopsy. HBV DNA levels (virus titers) in serum samples were quantified by real-time PCR (Roche COBAS AmpliPrep/COBAS Taqman HBV test v2.0-dynamic range 20 to 1.7×108 IU/ml-Roche molecular diagnostics, Pleasanton, CA) and HBsAg by Abbott Architect (Abbott Diagnostics, Abbot Park, IL). Serum was tested for HBeAg and anti-HBe with a chemiluminescent microparticle immunoassay (Abbott Architect). HBV genotype was also recorded. Ishak fibrosis stage (FS) and necro-inflammatory (NI) scores from liver biopsies were also determined. Whole blood was taken at the time of liver biopsy. PBMC were isolated by Ficoll-Hypaque density gradient centrifugation and cryopreserved for immunological analysis. Liver biopsy specimens, surplus to diagnostic requirements, were stored at −80°C for subsequent DNA extraction. Tissue samples taken for diagnostic histological examination were formalin-fixed, paraffin-embedded and used for immune-histochemical staining. Written informed consent was obtained from all patients. The study was approved by the local ethics committee (Barts and The London NHS Trust Ethics Review Board) and the Institutional Review Board of the Fox Chase Cancer Center.
Table 1:
Patient Characteristics
| Group 1: Immune Tolerant | Sex | Age | ALT IU/L | HBV Geno-type | HBeAg/anti-HBe | HBV DNA log IU/ ml | HBsAg titer log IU/ml | Fibrosis Stage (/6) | HAI (/18) | Peripheral T cell analysis* | IHC & Image analysis* |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt. 1 | F | 15 | 36 | E | +/− | 8.69 | 5.22 | 2 | 3 | Yes | Yes |
| Pt. 2 | M | 17 | 29 | C | +/− | 9.17 | 4.59 | 1 | 2 | Yes | Yes |
| Pt. 3 | F | 18 | 18 | B | +/− | 8.42 | 4.68 | 0 | 2 | Yes | Yes |
| Pt. 4 | M | 18 | 38 | D | +/− | 9.71 | 5.16 | 2 | 2 | Yes | Yes |
| Pt. 5 | M | 22 | 40 | E | +/− | 8.66 | 4.36 | 3 | 3 | Yes | Yes |
| Pt. 6 | F | 24 | 38 | C | +/− | 8.58 | 4.52 | 1 | 2 | No | No |
| Pt. 7 | F | 28 | 30 | E | +/− | 7.60 | 4.57 | 1 | 4 | No | Yes |
| Pt. 8 | F | 30 | 32 | C | +/− | 8.51 | 4.89 | 1 | 3 | Yes | Yes |
| Pt. 9 | F | 39 | 31 | B | +/− | 8.52 | 4.55 | 1 | 2 | No | Yes |
| Group 2: HBeAg(+) IA | |||||||||||
| Pt. 10 | M | 14 | 70 | D | +/− | 8.80 | 4.17 | 2 | 3 | Yes | Yes |
| Pt. 11 | M | 14 | 99 | A | +/− | 8.19 | 4.11 | 3 | 4 | No | No |
| Pt. 12 | F | 16 | 63 | D | +/− | 7.06 | 2.67 | 1 | 3 | No | No |
| Pt. 13 | F | 17 | 127 | D | +/− | 7.98 | 3.02 | 3 | 5 | Yes | Yes |
| Pt. 14 | M | 19 | 89 | C | +/− | 8.49 8.32 |
4.82 | 3 | 3 | Yes | Yes |
| Pt. 15 | F | 23 | 172 | A | +/− | 8.32 | 4.19 | 2 | 7 | No | No |
| Pt. 16 | M | 25 | 77 | B | +/− | 8.36 | 4.76 | 1 | 2 | Yes | Yes |
| Pt. 17 | M | 25 | 59 | D | +/− | 8.19 | 5.13 | 1 | 3 | No | No |
| Pt. 18 | F | 28 | 161 | C | +/− | 7.09 | 2.29 | 1 | 6 | Yes | Yes |
| Pt. 19 | F | 29 | 68 | B | +/− | 8.59 | 5.09 | 1 | 4 | No | No |
|
Group 3: HBeAg(−)
IA |
|||||||||||
| Pt. 20 | M | 23 | 113 | D | −/+ | 3.64 | 4.09 | 2 | 2 | Yes | Yes |
| Pt. 21 | F | 25 | 29 | E | −/+ | 4.19 | 3.73 | 1 | 2 | Yes | Yes |
| Pt. 22 | M | 26 | 55 | D | −/+ | 2.62 | 4.15 | 1 | 2 | No | No |
| Pt. 23 | M | 26 | 118 | D | −/+ | 3.94 | 5.09 | 0 | 1 | Yes | Yes |
| Pt. 24 | M | 26 | 110 | C | −/+ | 6.31 | 3.82 | 4 | 5 | No | Yes |
| Pt. 25 | F | 27 | 23 | D | −/+ | 6.70 | 4.03 | 1 | 2 | Yes | Yes |
| Pt. 26 | M | 29 | 81 | C | −/+ | 8.22 | 4.49 | 2 | 5 | No | Yes |
Group 1: HBeAg positive/HBeAb negative; ALT ≤40 (median 32 IU/L); HBV DNA ≥ 7.50 (median 8.58 log IU/ml)
Group 2: HBeAg positive/HBeAb negative; ALT >40 (median 83 IU/L); HBV DNA >7.00 (median 8.26 log IU/ml)
Group 3: HBeAg negative/HBeAb positive; ALT ≥ 40 with HBV DNA at any level, or if ALT ≤40 with HBV DNA >3.3 (median ALT 81 IU/L; median HBV DNA 4.19 log IU/ml)
Columns indicating whether or not peripheral T cell analyses and IHC were carried out. T cell results are presented in Figure 2 and IHC in Figure 3. All samples were analyzed for HBV DNA integration and clonal hepatocyte expansion.
HAI, histological activity index
In vitro expansion of HBV-specific T cells
Frozen PBMCs isolated from fresh heparinized blood by Ficoll-Hypaque density gradient centrifugation were thawed and resuspended in AIM-V medium with 2% pooled human AB serum (serum AIM-V). For HBV-specific T cell expansion, panels of synthetic peptides (15-mers, with 10 amino acids overlap, 313 in total) were pooled in 4 mixtures covering the whole HBV proteome. After 10 days of in vitro expansion, the presence of T cells responding to HBV peptide stimulation were determined by measuring the frequency of T cells producing IFN-γ with intracellular cytokine staining (ICS) or ELISPOT assays as previously described22 (Supplementary Materials & Methods).
Immunohistochemistry & Image Analysis
Adequate specimens of Formalin-fixed and paraffin-embedded tissue from 19/26 patients (Table 1) were available for immunohistochemistry (IHC) (Supplementary Materials & Methods).
Slides were imaged using a Leica DM6000 B microscope (Leica Biosystems, Newcastle, UK) equipped with a Leica DFC300 FX camera (Leica Biosystems, Newcastle, UK). A variable number of serial micrographs were taken from each Sirius red stained slide to cover the entire tissue. Tissue and collagen areas were measured on each micrograph using the ImageJ software (Bethesda, Maryland, USA) (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsbweb.nih.gov/ij/, 1997–2015) and a protocol described on the ImageJ web page (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA,http://rsbweb.nih.gov/ij/docs/examples/stained-sections/index.html, 2015) following previous calibration. Total tissue and collagen areas were then calculated for each biopsy (Supplementary Materials & Methods).
Results were assessed and plotted using GraphPad Prism 6 Trial Version (GraphPad Software, SanDiego, USA). The following tests were performed: Shapiro-Wilk normality test, Mann-Whitney, Kolmogorov-Smirnov, and Spearman correlation.
Extraction and inverse PCR analysis of liver DNA
Two to three ~1 mm pieces of each liver biopsy were cut, and nucleic acids extracted. Inverse PCR was designed to detect the right hand junction of integrations occurring between host DNA and HBV double stranded linear DNA (HBV dslDNA) (Figure 1A), the primary substrate for viral DNA integration.23, 24 To design PCR primers, and determine endonuclease cleavage sites for detection of the right hand virus/cell junction fragments, the predominant HBV sequence in the liver of each patient was determined by PCR amplification and sequencing of fragments covering the region from nts ~1193 to ~1860 on the HBV genome.25 HBV sequences were numbered according to Galibert et al.26 (accession number V01460).
Figure 1: Inverse PCR detection of integrated HBV DNA.

A) Strategy for detection of integrated HBV DNA and clonal hepatocyte expansion. Inverse PCR, as used by Summers et al.,25, 28 was designed to detect the right hand junction of integrations of HBV dslDNA, the predominant precursor for integration, into host DNA.23, 24 Following cleavage and ligation (Figure 1A), the DNA samples were serially diluted and subjected to nested PCR using the indicated forward and reverse primers (Figure 1B). Primers are indicated in Supplementary Table 1 and Materials and Methods. (Figure 1A modified from reference by Mason et al.25). B) Gel electrophoresis of inverse PCR products. Samples from nested PCR, carried out in a 96 well tray, were subjected to gel electrophoresis in a 1.3% agarose gel. PhiX phage DNA digested with HaeIII was used as a size marker (M). The fraction of the initial DNA sample distributed across each row of 12 wells is indicated. Bands were picked from the last 5 rows, not including the negative control, and subjected to DNA sequencing to identify virus/cell DNA junctions. For instance, the circled bands arise from a single hepatocyte clone; other clones were also identified by DNA sequencing (not highlighted).
Prior to inversion, high MW DNA (≥10–20 kbp) was purified by low-melt agarose gel electrophoresis, to reduce cccDNA contamination. The DNA was then digested by addition of NcoI-HF (NEB) and incubation for 30 min at 37°C. NcoI-HF was heat inactivated for 20 min at 80°C, and the DNA recovered using the QIAquick PCR purification kit. The DNA fragments were then circularized by incubation with T4 DNA ligase (Figure 1A).27 Prior to use for PCR, the circularized DNA was suspended in 40μl NEB buffer 4 supplemented with BSA (NEB) and linearized by digestion at 65°C with BsiHKAI (NEB). Molecules potentially derived from intra-molecular ligation of residual cccDNA (e.g., between the authentic NcoI site and a distal NcoI “star” site in cccDNA) or from cccDNA deletion mutants (PCR conditions were not adequate to amplify full-length cccDNA) were cleaved with SphI (NEB) to reduce their amplification during inverse PCR. (For several samples, it was necessary to use different restriction enzymes, because of differences in HBV DNA sequence (Supplementary Table 1 and Figure 1A). See Supplementary Materials & Methods for additional details.
Following inversion, endpoint dilution, and nested PCR, the products were subjected to electrophoresis in 1.3% agarose gels containing E-buffer and 0.5μg/ml of ethidium bromide (Figure 1B). Bands were excised from the gel and sequenced with the F2 or R2 primer, as previously described.28 The junction of viral with cellular DNA was located using the GCG program FASTA. Junctions repeated in different wells were identified by comparing cell sequences immediately adjacent to virus/cell junctions, using Sequencher version 5.0.1 (Gene Codes Corporation)(Supplementary Materials & Methods).
Quantifying host DNA in liver biopsy extracts
Host DNA was quantified by real time qPCR of epsilon globin DNA (accession number M81361), as previously described.25 A PCR amplified epsilon globin DNA was used as a control. The cell equivalents of DNA extracted from each biopsy are summarized in Supplementary Table 2.
Statistical analyses:
Quantifying virus/cell junctions by end-point dilution
As illustrated in Figure 1, inverted DNA samples were serially diluted into 96 well PCR plates. Typically, 5–10μl of inverted DNA, representing a small fraction of the original DNA sample (~5–10%), was added to 170–175μl of PCR reaction mix in well A1. After mixing, 60μl was serially diluted into 120μl of reaction mix in wells B1 through G1. Well H1 contained 120μl of reaction mix, but no DNA sample, and served as a negative control. 10μl aliquots of the reactants in column 1 were then distributed to columns 2 to 12 and subjected to nested PCR. 95% confidence intervals for clone sizes determined using end-point dilution were calculated using the fortran program Sim19 (Supplementary Materials & Methods).
Modeling the Clonal Expansion of hepatocytes
The program Csize8 was devised to predict the size of hepatocyte clones created after birth, as a consequence of liver growth and random hepatocyte turnover. Liver growth was assumed to be linear during the growth phase. Hepatocyte turnover during growth and in the full size liver were assumed to occur as a result of random death of hepatocytes with a rate constant, k. In the adult liver, death and regeneration were assumed to occur at the same rate, to maintain liver size. In the simulations presented here, k was assumed to be the same for the growing and adult liver (Supplementary Materials & Methods).
RESULTS
Evidence of HBV-specific T cell responses in patients in the immune tolerant phase of CHB
HBV-specific T cells were detected in all 3 patient groups, IT, HBeAg(+), and HBeAg(−) IA disease (Table 1). Using HBV-specific peptides spanning the entire HBV proteome, T cells were expanded in vitro and assayed for both intracellular cytokine staining and ELISPOT (Figure 2A). The quantity of HBV-specific T cells in terms of magnitude (number of cells recognizing a single HBV peptide mixture, Figure 2B) or the ability to recognize different mixtures of HBV peptides (Figure 2C) were comparable among the three patient cohorts. Consistent with our previous data,5 patients classified as IT did not show any significant difference in circulating HBV-specific T cells in comparison with CHB patients classified as IA in relation to their virological and clinical features (Table 1; Groups 2 & 3). Serum ALT levels were significantly lower in the IT group compared to the other groups. Despite this, differences in immune response of patients across the disease phases were not detected. ALT is often considered a surrogate of immune activity; however, as noted earlier, we and others have previously demonstrated that ALT does not ‘benchmark’ the HBV immune response.5, 8 The comparable levels of peripheral HBV-specific T cell responses in IT patients with those in the other two groups suggested that infected hepatocytes might be targeted for T cell mediated destruction in all patients including those diagnosed as IT. For this reason, we analyzed whether clinical phases could be distinguished by differences in the intrahepatic compartment. Immunohistochemistry analyses, measurements of HBV DNA integration and hepatocyte turnover were performed to determine if IT patients were different from the other patients studied.
Figure 2: Profile of HBV-specific T cell responses in all patient groups.
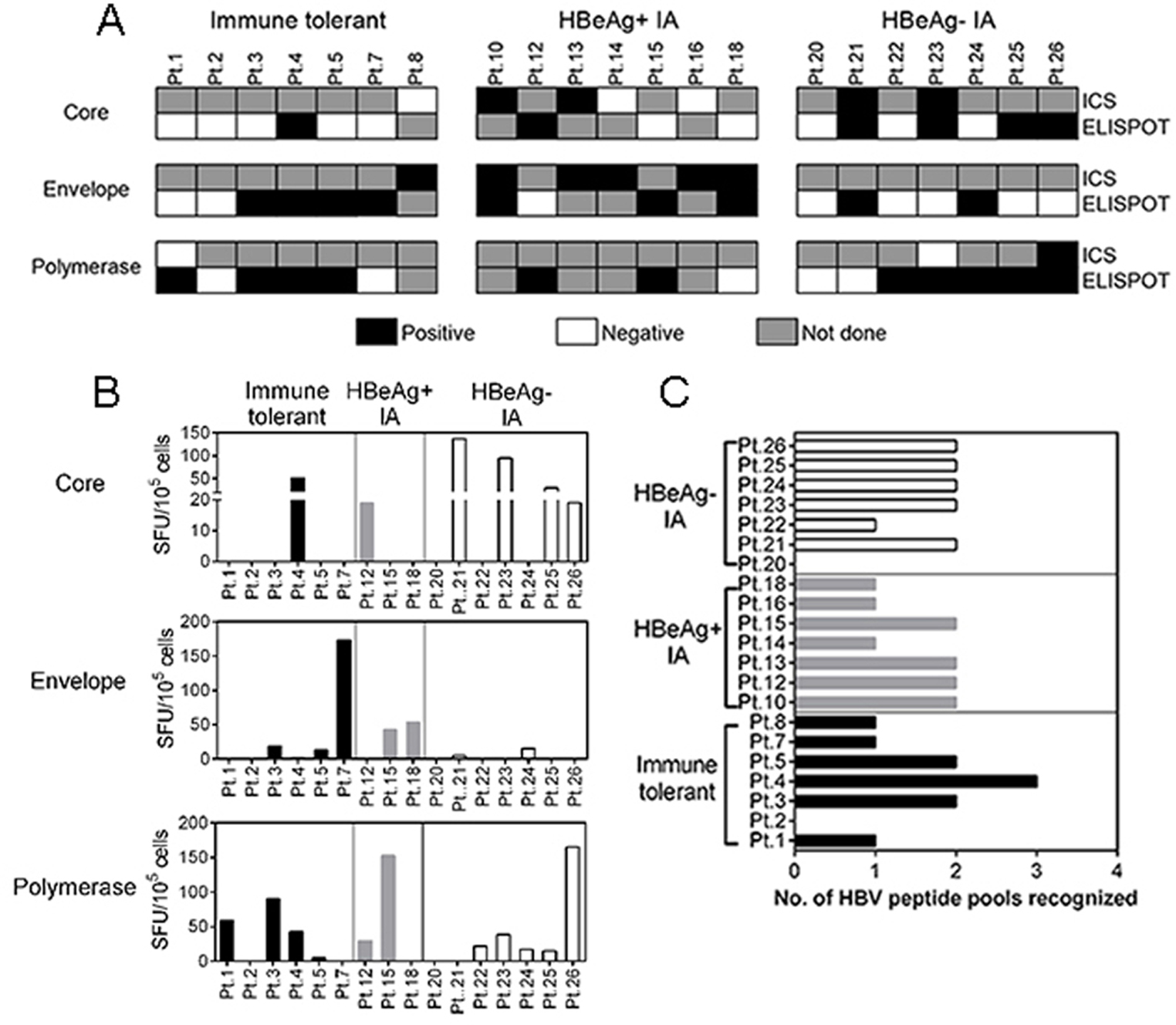
Patient PBMC were analyzed by ELISPOT and intracellular cytokine staining (ICS) for IFN-γ. (A) Evidence of HBV-specific T cell responses by ELISPOT and ICS against the Core, Envelope and Polymerase proteins, for each patient in the groups studied; Shaded black – positive HBV-specific T cell response; unshaded squares – negative HBV-specific T cell response, shaded grey – sample not done. (B) Comparison of spot forming units (SFU) by ELISPOT, in each patient, in the different groups; immune tolerant (IT) (shaded black), HBeAg(+) IA (shaded grey) and HBeAg(−) IA (unshaded). Bars represent the number of SFU cells in response to HBV core, envelope, and polymerase peptide pools. (C) Number of HBV peptide pools recognized by HBV-specific T cells obtained in the indicated patients.
A larger fraction of nuclear HBcAg positive hepatocytes are found in immune tolerant CHB
Liver tissue from 19/26 patients [Group 1, IT n=8; Group 2, HBeAg(+) IA n=5; HBeAg(−) IA n=6] was double stained for detection of HBcAg and HBsAg. Significant differences were found in the level of nuclear HBcAg positive hepatocytes in IT patients (Group 1; mean 30.1%) compared to the other groups (mean 0.92% and 0%; Groups 2 and 3 respectively) (IT vs. IA, P<.005) (Figure 3A, B). Interestingly 7/8 IT patients had >18% nuclear-HBcAg positive hepatocytes (Figure 3A); conversely, no patient exceeded ~3% positivity in the other groups irrespective of virus titer. HBsAg staining alone, the classical ground glass appearance reported on HBV tissue, was significantly higher in HBeAg(−) IA disease (Group 3) compared with IT (Group 1) (P=.004), but was not significantly different between Groups 1 and 2 (Figure 3A, B). These findings are consistent with previous work, which reported that nuclear HBcAg positive hepatocytes predominated in the IT phase in children.29 The reason for this finding remains unclear.
Figure 3: Differential nuclear core antigen staining but similar fibrosis and inflammatory indices between CHB phases.

Formalin-fixed and paraffin-embedded tissue was analyzed with immunohistochemistry for HBcAg and HBsAg positive hepatocytes, along with quantification of fibrosis and histological activity indices for each patient. (A) Percentage of HBcAg positive hepatocytes (left panel) and HBsAg positive hepatocytes (right panel) in each group; IT (open circles), HBeAg(+) IA (open squares) and HBeAg(−) IA (open triangles). Each point represents 1 patient, data shown as mean with SEM, as error bars. (B) Immunostaining identifying HBcAg positive hepatocytes (brown) and HBsAg positive hepatocytes (pink) from representative patients from each patient group (Table 1) (100x); IT (left panel), HBeAg(+) IA (middle panel) and HBeAg(−) IA (right panel). Inset shows magnified image (400x). (C) Ishak Fibrosis stage (left panel) and collagen proportionate area (right panel) of patients studied in each phase of CHB, data shown as mean with SEM, as error bars. (D) Sirius red staining of liver tissue from representative patients in each phase; IT (left panel), HBeAg(+) IA (middle panel) and HBeAg(−) IA (right panel). (E) Histological activity index scores; (from left to right – Interface hepatitis, Confluent necrosis score, Focal lytic necrosis, apoptosis & focal inflammation score and Portal inflammation score) of patients studied in each phase of CHB, data shown as mean with SEM, as error bars, (F) Identification of the inflammatory infiltrate as shown in (E) from representative patients in each phase of CHB; IT (left panel), HBeAg(+) IA (middle panel) and HBeAg(−) IA (right panel). Significant changes marked with asterisks, *P<.05; **P<.01; ***P<.001; ns=not significant
Despite the significant difference in nuclear HBcAg positive hepatocytes between patient groups, there was no overall difference in Ishak fibrosis stage (Table 1), collagen proportionate area (CPA) (Figure 3C, D) or histological activity index (HAI) (Table 1; Figure 3E, F) underscoring the limitations of standard histological assessment and clinical parameters used alone or even in combination to define phases of CHB.
Integrated HBV DNA was identified in chromosomes of all patients
Five hundred and ninety two different virus/cell junctions were detected overall, using inverse PCR. 500 could be mapped to unique sites on human chromosomal DNA (208 for group 1, 195 for group 2, 97 for group 3) (Supplementary Table 3; Supplementary Results). Of these 500 integration sites, 246 were located within potentially transcribed regions, including 217 mRNA encoding regions and 29 non-coding RNAs. 231 of the integration sites mapped to introns, 13 to exons, one at an intron/exon boundary, and one mapped within a gene (uncertainty in the exact junction site precluded the exon/intron distinction). Of the protein coding genes with integrated HBV DNA, ~70% appeared to be transcribed in the liver. Protein expression in the liver has been reported for ~45% of these (www.genecards.org).30 4/29 regions with integrated HBV DNA that specified non-coding RNAs also appeared to be expressed in liver. For most, it was unclear if expression occurred in hepatocytes or other liver cells. The remaining 92/592 integrations were located in repeated DNA sequences and/or could not be mapped. Our results are likely to underestimate the true number of unique HBV integration sites in the DNA samples; that is, single or low copy clones might be obscured by competing amplification of high copy number clones (Figure 1B). Notably, multiple integrations were found on every chromosome except Y (Table 2; Supplementary Table 3). Integration sites are illustrated in Figure 4A. Using the Chi-Squared Test, we were unable to reject the null hypothesis that the integration frequency on chromosomes (Figure 4A; Supplementary Table 3), including Y (13 of 26 patients were male), was proportional to their length (P=0.195). No significant differences were seen between patient groups (Figure 4B; IT patients).
Table 2:
Observed and Expected Integration Sites per Chromosome
| Chromosome | Chromosome length | Integration Sites Observed Expected |
|
|---|---|---|---|
| 1 | 2.49×108 | 44 | 40.9 |
| 2 | 2.43×108 | 47 | 39.9 |
| 3 | 1.98×108 | 34 | 32.6 |
| 4 | 1.90×108 | 39 | 31.2 |
| 5 | 1.82×108 | 26 | 29.8 |
| 6 | 1.71×108 | 28 | 28.1 |
| 7 | 1.59×108 | 25 | 26.2 |
| 8 | 1.45×108 | 19 | 23.8 |
| 9 | 1.38×108 | 21 | 22.7 |
| 10 | 1.34×108 | 21 | 22.0 |
| 11 | 1.35×108 | 19 | 22.2 |
| 12 | 1.33×108 | 18 | 21.9 |
| 13 | 1.14×108 | 20 | 18.8 |
| 14 | 1.07×108 | 16 | 17.6 |
| 15 | 1.02×108 | 13 | 16.8 |
| 16 | 9.03×107 | 9 | 14.8 |
| 17 | 8.33×107 | 12 | 13.7 |
| 18 | 8.04×107 | 10 | 13.2 |
| 19 | 5.86×107 | 19 | 9.6 |
| 20 | 6.44×107 | 13 | 10.6 |
| 21 | 4.67×107 | 7 | 7.7 |
| 22 | 5.08×107 | 8 | 8.3 |
| X | 1.56×108 | 25 | 19.2 |
| Y | 5.72×107 | 1 | 2.3 |
Expected integration sites per chromosome were calculated assuming that the incidence of integration was proportional to chromosome length. Integration incidence for the X and Y chromosome were adjusted to account for the equal numbers of males and females in the patient population.
Figure 4: Sites of HBV DNA integration on human chromosomes.

A) Integration sites are summarized from all three patient groups (Table 1) by vertical lines. Results include the 208 from IT disease patients (Group 1), 195 from HBeAg(+) IA disease (Group 2), and 97 from HBeAg(−) IA disease (Group 3). Groups 1 (IT) and 2 [HBeAg(+)]; integrations were found on all chromosomes except Y. The single Y chromosome integration was from a patient from group 3. No group 3 patient integration sites were mapped to chromosomes 15 and 16. B) Integration sites in Group 1 patients - IT phase. Integration site details are shown in Supplementary Table 3. Clone sizes: *>5,000 and #>20,000.
The average frequency of total integrations in groups 1 through 3, respectively, including those in hepatocyte clones, ranged from 1.5×109 to 5×109 per liver of 5×1011 hepatocytes (see Supplementary Table 2 for individual patients). Importantly, integration is prevalent in patients considered IT. Because just a small fraction of each DNA sample was assayed, we could only make a minimum estimate of the unique integration sites among the total. The data suggested at least ~5×106 distinct integration sites are present in a liver of 5×1011 hepatocytes in each patient group. This high number of possible sites means that a liver of 5×1011 hepatocytes would contain at least one hepatocyte in which a particular gene would be mutated in each patient group including those characterized as IT, not just age matched controls with more advanced liver disease. (We could not demonstrate any correlation with HBsAg or HBV DNA levels and total integrations in the whole study cohort; thus, the extent to which integration might contribute to HBsAg production in the 3 patient groups remains unclear).
Clonal hepatocyte expansion
Because the hepatocyte population appears self-renewing, death and regeneration will lead to loss of some cell lineages and clonal expansion of others to maintain liver mass. To determine if IT patients have elevated hepatocyte turnover, possibly due to anti-HBV immune killing, we investigated if these patients had evidence of hepatocyte clones that were similar in size to those found in late phases of CHB with HCC.28 Simultaneously, we asked if similar levels of clonal hepatocyte expansion were present in our three age-matched patient groups. Insertional mutagenesis and expression of HBV genes from integrated DNA are potential initiation events in hepatocarcinogenesis, as is repair of double stranded DNA breaks by non-homologous end joining in the absence of HBV integration.19 Enhanced hepatocyte turnover could be promotional,31 by facilitating clonal expansion of subsets of hepatocytes, including but not limited those with preneoplastic mutations. Large hepatocyte clones were seen in all three patient groups (Figure 5). The difference in maximum clone sizes between groups 1 and 3 was statistically significant (P=.0015), as was the difference between groups 2 and 3 (P=.014) (Supplementary Table 4; Figure 5); the difference between Groups 1 and 2 did not reach statistical significance (P=0.36) (Wilcoxon 2-sided Rank Sum Test).
Figure 5: Hepatocyte clones detected in all patient groups.

Hepatocyte clones in (A) IT disease (Group 1), (B) HBeAg(+) IA disease (Group 2) and (C) HBeAg(−) IA disease (Group 3). Clone sizes were estimated as described. (Figure 1, Materials and Methods and Supplementary Materials & Methods). The point estimates for clone size were calculated using the program Sim19 (Supplementary Materials & Methods). Clones are grouped by increasing size for each patient, and patients within a group are arranged by increasing age from left to right. D) Mean of the maximum clone size for each patient within a group. Geometric means were calculated using the point estimates in Supplementary Table 4. HCC data are from a published analysis of clone sizes in non-tumorous liver from a group of 5 non-cirrhotic HCC patients.25 (E) Predicted maximum clone sizes vs. age. These were calculated using the Csize8 program (Materials and Methods and Supplementary Materials & Methods), for 3 different daily rate constants for hepatocyte turnover; k=0.0015/day (0.15%) - (black dashed line); k=0.004/day (0.40%) – (grey dashed line) and k=0.01/day (1.00%) - (solid black line). The adjacent corresponding bars indicate the geometric mean hepatocyte clone size, for each patient group in (D), for comparison against the predicted maximum clone size.
Hepatocyte clone sizes were larger in immune tolerant patients than predicted by a model of random hepatocyte turnover
As discussed, hepatocyte turnover in the liver should lead to increasing clonality, with loss of some hepatocyte lineages and expansion of others. To determine if the large clones (Figure 5A-C) could be explained by random death and compensatory division of hepatocytes, to maintain liver mass, a computer simulation, Csize8 (Supplementary Materials & Methods), was used. We assumed that hepatocytes proliferate (and die) in the adult liver with a rate constant k=0.0015/day (0.15%/day), 3 times the fraction of hepatocytes in the S phase (0.0005) in healthy adult liver at any given time.32 We also assumed that infection occurred at birth and that the liver size increases 10-fold during maturation. The maximum expected clone sizes in the patients studied (age range 14–39 years) increased, with age, from ~400 to ~600 hepatocytes (Figure 5E). This range would increase from ~800 to ~1200 if the rate constant for hepatocyte death increased to k=0.004/day (0.4%/day), and ~1600 to 2800 with a rate constant of k=0.01/day (~1% of hepatocytes killed/day) (Figure 5E).
Maximum observed clone sizes exceeded clone sizes predicted for random liver turnover (0.015%/day)32 for 6/9 IT patients (Group 1), 6/10 in HBeAg(+) IA patients (Group 2), and 7/7 in HBeAg(−) IA patients (Group 3) (Figure 5E; Supplementary Table 4). For a turnover of 0.04% per day, excess turnover was observed in 2/9 patients in Group 1, 5/10 in Group 2, and 7/7 in Group 3. 3/10 patient samples in Group 2 and 5/7 in Group 3 exceeded predictions even for a daily turnover of 1%. While differences in maximum predicted clone sizes and observed sizes may appear small, it is important to note that the amount of hepatocyte destruction and replacement in the model that is necessary, for example, to give a maximum clone size of 600 (k=0.0015) vs. 2800 (k=0.01) hepatocytes, after 39 years, is 21 vs. 142 livers worth of hepatocyte death and replacement. In summary, a model of random death and regeneration of hepatocytes at a level estimated for healthy liver did not provide a consistent explanation for maximum clone sizes observed in 6/9 IT patients, which was also true of 6/10 patients in Group 2 and all patients in Group 3. The differences might be more extreme, because the modeling assumes all clones are detected, not just those with integrations. These analyses suggest a selective process for hepatocyte turnover can occur in all groups. This might result, for example, from emergence of hepatocyte clones that are resistant to T cell killing, or because some hepatocyte lineages are more responsive to growth signals to divide, to maintain liver mass.
DISCUSSION
We have demonstrated that HBV DNA integration and clonal hepatocyte expansion were similar in patients considered IT to those that have HBeAg(+) IA CHB. These results raise questions about the perception that the IT phase is ‘disease-free’, as well as the premise upon which treatment decisions are made. In line with our previous work and recent publications in the field, we feel that the term ‘high replicative low-inflammatory’ (HRLI) CHB more accurately reflects this early disease phase, and thus should now be adopted into clinical practice.4–6, 8, 33
CHB is the leading cause of primary liver cancer worldwide and despite the lack of robust data to support this notion,1 the current consensus is that HCC risk does not increase in the majority of patients until there is perturbation in serum ALT, interpreted as a sign of immune activity. There are, however, studies supporting the development of HCC in the absence of advanced liver disease. The REVEAL study demonstrated an association between high viral load and HCC development, independent of cirrhosis, thus pertinent to the study population here.34 The data presented here suggest that an approach to management which excludes HRLI patients (formerly considered IT) from treatment may be flawed, as HBV specific T cells as well as extensive clonal hepatocyte expansion are already present in this early phase of CHB. Evidence from both animal and human studies demonstrate that clonal hepatocyte expansion is a major risk factor for HCC,35 moreover, HBV DNA integration, a potential initiating event for HCC, was found to be prevalent not just in later stages of CHB, but also in the HRLI phase. The presence of both HBV DNA integration and clonal hepatocyte expansion in this early phase of CHB are thus at odds with the concept of a ‘disease-free’ state.
These data are consistent with recent studies and our previous findings,5, 6, 8 which dispute the idea that so-called IT patients are immunologically inert and, therefore, fundamentally distinct from HBeAg(+) IA disease. In the current study, we confirmed the presence of HBV-specific responses in the HRLI and later phases of CHB by dual modality (ELISPOT and Intracellular Cytokine Staining) (Figure 2). These data reinforce the fact that there is no quantifiable difference in antiviral immunity between the HRLI phase and HBeAg(+) IA patients. Furthermore, these findings were verified by detailed analysis of the liver compartment of the patients studied. In keeping with the HBV specific response in the periphery, we demonstrated few if any differences in liver histology (Figure 3). Based on serological assessment, patients labeled IT had similar levels of fibrosis, CPA and HAI as those considered to have IA disease.
In addition, we could demonstrate differences in the level of nuclear core expression; being significantly higher in those considered IT compared with HBeAg(+) and HBeAg(−) IA patients; in contrast, HBsAg positive hepatocytes were preferentially found in HBeAg(−) IA patients (Figure 3). This mosaic distribution of HBV antigens in different hepatocytes and phases of HBV infection might reflect different virological or immunological features that need further characterization. A recent study suggested that hepatocytes expressing high HBcAg may have higher level of HBV replication and higher cccDNA content than HBsAg expressing hepatocytes.36 However, the biological significance of the diverse HBV antigen patterns detectable in the different categories of CHB remains unclear.
An important issue is the number of different HBV DNA integration sites in the livers of the three patient groups, which will determine the numbers of host genes potentially mutated by HBV integration, and also may be an indirect indicator of the number of double strand DNA breaks repaired by non-homologous end joining, which is also potentially mutagenic.19, 20 Our primary goal was to estimate hepatocyte clone size using end point dilution assays; thus, we can only make a minimum estimate for the number of unique HBV integration events. The real number may in fact be much larger, but interestingly, the number estimated for all three patient groups (at least ~5×106 per liver in all three groups) would be sufficient, if uniformly distributed across the human genome in a liver of 5×1011 hepatocytes, to place integrated HBV DNA within any 1000 nt region in the genome of at least one hepatocyte. (Note that mutation by incorrect repair of double stranded DNA breaks may be 10-times more frequent.19) Thus, the potential to mutate and alter expression of any host gene in at least one hepatocyte appears very high across the disease phases. Some of these integrations may be procarcinogenic.
To explore the concept that patients considered IT may require earlier treatment we also investigated clonal hepatocyte expansion in these patients and compared it to IA CHB (Figure 5). The rationale was that clonal hepatocyte expansion in mutated hepatocytes would contribute to tumor promotion.31, 35 To the extent that the hepatocyte population is self-renewing, and undergoing random death and regeneration, it is possible to relate cumulative hepatocyte turnover to maximum hepatocyte clone sizes. Compared to our predictions, actual clone sizes in HBeAg(−) IA disease (group 3) appeared excessive, similar to those in HCC patients, even assuming a relatively high hepatocyte death rate of 1.0% per day (Figure 5).32 In contrast, HBeAg(+) IA patients (Group 2) appeared to have much lower hepatocyte turnover and were not significantly different than those considered IT (Figure 5). Nonetheless, average hepatocyte clone sizes in both groups 1 and 2, exceeded predictions for normal liver turnover (k=0.0015). Indeed, in some of these patients, very large clone sizes were detected, which can only be explained by assuming a selective growth or survival advantage for hepatocytes (Supplementary Table 4). This was also noted in a study of non-tumorous liver samples from non-cirrhotic HCC patients.21, 25 In brief, our data suggest that clonal hepatocyte expansion, an HCC risk factor,31, 35 is active across all the phases of CHB studied here.
This study confirms the presence of HBV-specific T cell responses and the significant extent of HBV DNA integration/cell mutagenesis along with clonal hepatocyte expansion in the HRLI phase and across the disease phases. These findings further challenge the notion of an IT phase devoid of disease progression, raising questions about the timing of therapeutic intervention to minimize genetic damage to the hepatocyte population and reduce the promotional role in carcinogenesis of elevated hepatocyte turnover. As the risk of HCC may already be present in the HRLI phase, these data make a compelling case to consider antiviral therapy in these patients. Future studies are required to explore the merits of earlier treatment to prevent disease progression and the development of HCC in CHB.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for helpful discussions and advice from Drs. Richard Katz, Christoph Seeger and Glenn Rall (Fox Chase Cancer Center) and Jesse Summers (University of New Mexico), and to Anita Cywinski (Fox Chase Cancer Center DNA sequencing facility) for the DNA sequencing required to carry out this study.
WSM acknowledges the kind support of Christoph Seeger for providing laboratory space and to Christoph Seeger, John Gricoski, and Robert Beck for encouragement to carry out his portion of the study.
PTFK acknowledges the patients, their families and the staff at The Royal London Hospital who have supported and contributed to this work.
GRANT SUPPORT: Institutional funding from Fox Chase Cancer Center to WSM; Wellcome Trust Clinical Research Training Fellowship (107389/Z/15/Z) to USG; Fox Chase Cancer Center Core Grant (CA006927) to SL, YZ and SP; Singapore Translational Research (STaR) Investigator Award (NMRC/STaR/013/2012) to AB; Barts and The London Charity Large Project grant (723/1795) to PTFK.
ABBREVIATIONS:
- CHB
chronic hepatitis B
- CPA
collagen proportionate area
- FS
Ishak fibrosis state
- IA
immune active
- IT
immune tolerant
- HAI
histological activity index
- HBeAg
hepatitis B envelope antigen
- HBcAg
hepatitis B core antigen
- HBsAg
hepatitis B surface antigen
- HCC
hepatocellular carcinoma
- NI
necro-inflammatory stage
- PBMC
peripheral blood mononuclear cells
Footnotes
DISCLOSURES: The authors have no relevant disclosures or conflicts of interest
REFERENCES
Author names in bold designate shared co-first authorship.
- 1.Zoulim F, Mason WS. Reasons to consider earlier treatment of chronic HBV infections. Gut 2012;61:333–336. [DOI] [PubMed] [Google Scholar]
- 2.Gill US, Zissimopoulos A, Al-Shamma S, et al. Assessment of bone mineral density in tenofovir-treated patients with chronic hepatitis B: can the fracture risk assessment tool identify those at greatest risk? J Infect Dis 2015;211:374–382. [DOI] [PubMed] [Google Scholar]
- 3.Maini MK, Boni C, Lee CK, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med 2000;191:1269–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertoletti A, Kennedy PT. The immune tolerant phase of chronic HBV infection: new perspectives on an old concept. Cell Mol Immunol 2015;12:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennedy PT, Sandalova E, Jo J, et al. Preserved T-cell function in children and young adults with immune-tolerant chronic hepatitis B. Gastroenterology 2012;143:637–645. [DOI] [PubMed] [Google Scholar]
- 6.Vanwolleghem T, Hou J, van Oord G, et al. Re-evaluation of hepatitis B virus clinical phases by systems biology identifies unappreciated roles for the innate immune response and B cells. Hepatology 2015;62:87–100. [DOI] [PubMed] [Google Scholar]
- 7.Hong M, Sandalova E, Low D, et al. Trained immunity in newborn infants of HBV-infected mothers. Nat Commun 2015;6:6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park JJ, Wong DK, Wahed AS, et al. Hepatitis B Virus-Specific and Global T-Cell Dysfunction in Chronic Hepatitis B. Gastroenterology 2016;150:684–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang HY, Chien MH, Huang HP, et al. Distinct hepatitis B virus dynamics in the immunotolerant and early immunoclearance phases. J Virol 2010;84:3454–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarlow BD, Pelz C, Naugler WE, et al. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014;15:605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grompe M. Liver stem cells, where art thou? Cell Stem Cell 2014;15:257–258. [DOI] [PubMed] [Google Scholar]
- 12.Wang MJ, Chen F, Li JX, et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology 2014;60:349–361. [DOI] [PubMed] [Google Scholar]
- 13.Schaub JR, Malato Y, Gormond C, et al. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Reports 2014;8:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yanger K, Knigin D, Zong Y, et al. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell 2014;15:340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marongiu F, Serra MP, Sini M, et al. Cell turnover in the repopulated rat liver: distinct lineages for hepatocytes and the biliary epithelium. Cell Tissue Res 2014;356:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Font-Burgada J, Shalapour S, Ramaswamy S, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015;162:766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Zhao LL, Fish M, et al. Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature 2015;524:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyajima A, Tankaka M, Itoh T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprograming. Cell Stem Cell 2014;14:561–574. [DOI] [PubMed] [Google Scholar]
- 19.Bill C, Summers J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc Natl Acad Sci USA 2004;101:11135–11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA 2003;100:12871–12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tu T, Mason WS, Clouston AD, et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J Viral Hep 2015;22:737–753. [DOI] [PubMed] [Google Scholar]
- 22.Tan AT, Loggi E, Boni C, et al. Host ethnicity and virus genotype shape the hepatitis B virus-specific T-cell repertoire. J Virol 2008;82:10986–10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W, Summers J. Integration of hepadnavirus DNA in infected liver: evidence from a linear precursor. J Virol 1999;73:9710–9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong SS, Jensen AD, Chang CJ, et al. Double-stranded linear duck hepatitis B virus (DHBV) stably integrates at a higher frequency than wild-type DHBV in LMH chicken hepatoma cells. J Virol 1999;73:1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason WS, Liu C, Aldrich CE, et al. Clonal expansion of normal-appearing human hepatocytes during chronic hepatitis B virus infection. J Virol 2010;84:8308–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galibert F, Mandart E, Fitoussi F, et al. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E.coli. Nature 1979;281:646–650. [DOI] [PubMed] [Google Scholar]
- 27.Mason WS, Low HC, Xu C, et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J Virol 2009;83:8396–8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Summers J, Jilbert AR, Yang W, et al. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003;100:11652–11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu H-Y, Lin Y-H, Chang M-H, et al. Pathology of chronic hepatitis B infection in children: With special reference to the intrahepatic expression of hepatitis B virus antigens. Hepatology 1988;8:378–382. [DOI] [PubMed] [Google Scholar]
- 30.Stelzer G, Dalah I, Stein TI, et al. In-silico human genomics with GeneCards. Hum Genomics 2011;5:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farber E, Sarma DS. Hepatocarcinogenesis: a dynamic cellular perspective. Lab Invest 1987;56:4–22. [PubMed] [Google Scholar]
- 32.Mancini R, Marucci L, Benedetti A, et al. Immunohistochemical analysis of S-phase cells in normal human and rat liver by PC10 monoclonal antibody. Liver 1994;14:57–64. [DOI] [PubMed] [Google Scholar]
- 33.Gish RG, Given BD, Lai CL, et al. Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antiviral Res 2015;121:47–58. [DOI] [PubMed] [Google Scholar]
- 34.Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65–73. [DOI] [PubMed] [Google Scholar]
- 35.Marongiu F, Doratiotto S, Montisci S, et al. Liver repopulation and carcinogenesis: Two sides of the same coin? Am J Pathol 2008;172:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Lu W, Zheng Y, et al. In situ analysis of intrahepatic virological events in chronic hepatitis B virus infection. J Clin Invest 2016;126:1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.