

Abstract
Background
Intermittent preventive treatment could help prevent malaria in infants (IPTi) living in areas of moderate to high malaria transmission in sub‐Saharan Africa. The World Health Organization (WHO) policy recommended IPTi in 2010, but its adoption in countries has been limited.
Objectives
To evaluate the effects of intermittent preventive treatment (IPT) with antimalarial drugs to prevent malaria in infants living in malaria‐endemic areas.
Search methods
We searched the following sources up to 3 December 2018: the Cochrane Infectious Diseases Group Specialized Register, CENTRAL (the Cochrane Library), MEDLINE (PubMed), Embase (OVID), LILACS (Bireme), and reference lists of articles. We also searched the metaRegister of Controlled Trials (mRCT) and the WHO International Clinical Trials Registry Platform (ICTRP) portal for ongoing trials up to 3 December 2018.
Selection criteria
We included randomized controlled trials (RCTs) that compared IPT to placebo or no intervention in infants (defined as young children aged between 1 to 12 months) in malaria‐endemic areas.
Data collection and analysis
The primary outcome was clinical malaria (fever plus asexual parasitaemia). Two review authors independently assessed trials for inclusion, evaluated the risk of bias, and extracted data. We summarized dichotomous outcomes and count data using risk ratios (RR) and rate ratios respectively, and presented all measures with 95% confidence intervals (CIs). We extracted protective efficacy values and their 95% CIs; when an included trial did not report this data, we calculated these values from the RR or rate ratio with its 95% CI. Where appropriate, we combined data in meta‐analyses and assessed the certainty of the evidence using the GRADE approach.
Main results
We included 12 trials that enrolled 19,098 infants; all were conducted in sub‐Saharan Africa. Three trials were cluster‐RCTs. IPTi with sulfadoxine‐pyrimethamine (SP) was evaluated in 10 trials from 1999 to 2013 (n = 15,256). Trials evaluating ACTs included dihydroartemisinin‐piperaquine (1 trial, 147 participants; year 2013), amodiaquine‐artesunate (1 study, 684 participants; year 2008), and SP‐artesunate (1 trial, 676 participants; year 2008). The earlier studies evaluated IPTi with SP, and were conducted in Tanzania (in 1999 and 2006), Mozambique (2004), Ghana (2004 to 2005), Gabon (2005), Kenya (2008), and Mali (2009). One trial evaluated IPTi with amodiaquine in Tanzania (2000). Later studies included three conducted in Kenya (2008), Tanzania (2008), and Uganda (2013), evaluating IPTi in multiple trial arms that included artemisinin‐based combination therapy (ACT).
Although the effect size varied over time and between drugs, overall IPTi impacts on the incidence of clinical malaria overall, with a 30% reduction (rate ratio 0.70, 0.62 to 0.80; 10 studies, 10,602 participants). The effect of SP appeared to attenuate over time, with trials conducted after 2009 showing little or no effect of the intervention. IPTi with SP probably resulted in fewer episodes of clinical malaria (rate ratio 0.78, 0.69 to 0.88; 8 trials, 8774 participants, moderate‐certainty evidence), anaemia (rate ratio 0.82, 0.68 to 0.98; 6 trials, 7438 participants, moderate‐certainty evidence), parasitaemia (rate ratio 0.66, 0.56 to 0.79; 1 trial, 1200 participants, moderate‐certainty evidence), and fewer hospital admissions (rate ratio 0.85, 0.78 to 0.93; 7 trials, 7486 participants, moderate‐certainty evidence). IPTi with SP probably made little or no difference to all‐cause mortality (risk ratio 0.93, 0.74 to 1.15; 9 trials, 14,588 participants, moderate‐certainty evidence).
Since 2009, IPTi trials have evaluated ACTs and indicate impact on clinical malaria and parasitaemia. A small trial of DHAP in 2013 shows substantive effects on clinical malaria (RR 0.42, 0.33 to 0.54; 1 trial, 147 participants, moderate‐certainty evidence) and parasitaemia (moderate‐certainty evidence).
Authors' conclusions
In areas of sub‐Saharan Africa, giving antimalarial drugs known to be effective against the malaria parasite at the time to infants as IPT probably reduces the risk of clinical malaria, anaemia, and hospital admission. Evidence from SP studies over a 19‐year period shows declining efficacy, which may be due to increasing drug resistance. Combinations with ACTs appear promising as suitable alternatives for IPTi.
Plain language summary
Administering antimalarial drugs to prevent malaria in infants
What is the aim of the review?
This Cochrane Review aimed to find out if administering repeated doses of antimalarial treatment to infants living in sub‐Saharan Africa can prevent malaria. We found and analysed results from 12 relevant studies conducted between 1999 and 2013 that addressed this question in infants (defined as young children aged between 1 to 12 months).
Key messages
Intermittent preventive treatment with sulfadoxine‐pyrimethamine (SP)
Giving SP as preventive antimalarial treatment to infants probably reduced the risk of clinical malaria, anaemia, and hospital admissions in the African countries it was evaluated. However, this effect was attenuated in more recent studies.
Intermittent preventive treatment with artemisinin‐based combination therapy (ACT)
Giving ACT as preventive antimalarial treatment to infants may reduce the risk of clinical malaria. It may also reduce the proportion of infants with malaria parasites in their blood.
What was studied in the review?
In areas where malaria is common, infants often suffer repeated episodes of malarial illness. In areas where malaria transmission occurs all‐year, some authorities recommend intermittent preventive treatment, which requires giving drugs at regular intervals (at child vaccination visits) regardless of whether the child has malaria symptoms or not to prevent malarial illness.
We studied the effects of IPTi with SP and other medicines (including ACTs) on malaria‐related outcomes. Review outcomes included clinical malaria, severe malaria, death, hospital admission, parasitaemia, anaemia, change in haemoglobin level, and side effects.
What are the main results of the review?
We included 12 studies that enrolled 19,098 infants. All studies were done in sub‐Saharan Africa (Gabon, Ghana, Kenya, Mali, Mozambique, Tanzania, and Uganda). These studies compared infants who received IPTi to those who received placebo pills or nothing. The infants in the IPTi group were given different medicines, in different doses, and for different lengths of time.
Ten studies evaluated IPTi with SP from 1999 to 2013. The effect of SP appear to wane over time, with trials conducted after 2009 showing little or no effect of the intervention. The studies show that IPTi with SP probably resulted in fewer episodes of clinical malaria, anaemia, hospital admission, and blood parasites without symptoms (moderate‐certainty evidence). IPTi with SP probably made little or no difference to the risk of death (moderate‐certainty evidence).
Since 2009, IPTi some small studies have evaluated artemisinin‐based combination medicines and indicate impact on clinical malaria and blood parasites. A small study of IPTi with dihydroartemisinin‐piperaquine in 2013 showed up to 58% reduction in episodes of clinical malaria (moderate‐certainty evidence) and reductions in proportion of infants with blood parasites (moderate‐certainty evidence).
How up‐to‐date is this review?
The review authors searched for studies published up to 3 December 2018.
Summary of findings
Background
Description of the condition
Malaria is caused by infection with the Plasmodium parasite, which is transmitted to humans through the bite of infected female Anopheles mosquitoes. In the human body, the parasites multiply in the liver and then infect red blood cells. Malaria can also be transmitted from a mother to her unborn baby (congenitally) and through blood transfusions. Five Plasmodium species are known to cause this disease in humans. Plasmodium falciparum is the most common worldwide, and is responsible for almost all severe disease cases and deaths (WHO 2018). People visiting or living in areas where malaria transmission is prevalent are at risk of malaria infection; children and pregnant women living in malaria‐endemic areas are particularly at risk. People who are infected with Plasmodium parasites may show no sign of illness (asymptomatic malaria), or may develop symptoms such as fever, chills, weakness, and headache (symptomatic malaria). The severity of malaria infection varies from mild (uncomplicated) to life‐threatening (severe). People with severe malaria may experience severe anaemia, convulsions, unconsciousness, and in some cases can die. Severe malaria is more likely to occur in people with low or no immunity to malaria (Gilles 2000). Children living in malaria‐endemic areas have relatively less acquired immunity to malaria. In 2017, 61% of global cases of malaria were in children under five years of age, most of whom were residing in sub‐Saharan Africa (WHO 2018).
Description of the intervention
Malaria control efforts have been aimed towards reduction of illness and death from Plasmodium infection. The World Health Organization (WHO) global malaria control strategy combines preventive interventions (for example, use of long‐lasting insecticide‐treated nets (LLINs), and indoor residual spraying) with early diagnosis and appropriate treatment of symptomatic people with artemisinin‐based combination therapy (ACT) (WHO 2018). Intermittent preventive treatment (IPT) is one of the interventions recommended for malaria prevention in vulnerable and at‐risk groups (infants, children, and pregnant women) (WHO 2004; WHO 2010; WHO 2012).
IPT is defined as "the administration of a full therapeutic course of an antimalarial or antimalarial combination to a selected target population at specified times without determining whether or not the subject is infected" (Greenwood 2010). IPT in infants (IPTi) is a full therapeutic course of antimalarial medicine delivered to infants through routine immunization services, regardless of whether the child is infected with malaria or not. The WHO recommends IPTi with sulfadoxine‐pyrimethamine (SP) in areas with moderate‐to‐high malaria transmission in sub‐Saharan Africa where the prevalence of the pfdhps‐540E allele of the P falciparum parasite is less than 50% (WHO 2010; WHO 2011). Administration of IPTi is aimed at reducing the risk of clinical malaria, anaemia, and severe malaria in the first year of life. Treatment is given three times during the first year of life at approximately 10 weeks, 14 weeks, and nine months of age, which corresponds to the routine vaccination schedule of the Expanded Programme on Immunization (EPI) (WHO 2011).
IPTi was proposed as an alternative to prophylaxis because of concerns that the latter may impair the acquisition of natural immunity to malaria in infants, making them more vulnerable to severe malaria after prophylaxis is discontinued when they are older (Greenwood 2004; Otoo 1988; WHO 1993). There are also concerns that the widespread use of antimalarial drugs for prophylaxis in infants could increase the resistance of the Plasmodium parasites to these drugs (Alexander 2007; WHO 1990; WHO 1993). Further concerns about chemoprophylaxis include the feasibility and sustainability of the intervention. While the mechanism of IPTi may not be clear, available data does suggest the post‐treatment prophylaxis (longer‐acting drugs) is an important component in areas of high transmission where reinfection is likely. Studies that have tried IPTi with shorter‐acting drugs have not achieved as good a preventive effect. It is unclear whether it is by the intermittent clearance of existing Plasmodium infections or the post‐treatment prophylactic effect of long‐acting drugs (White 2005). There is also the ‘leaky vaccine' hypothesis that a partially effective drug combined with high LLIN coverage may lead to attenuated blood‐stage infections, enabling immunity to develop without leading to clinical disease. This may increase subclinical infection and promote protection in infants, as has been demonstrated in one study (Pombo 2002). The duration of protection from IPTi is limited to periods when the drug has not been eliminated from the body, typically about 1 to 2 months after drug administration (Cairns 2010).
Since 2009, when the policy recommendations were made, only Chad has adopted IPTi as national policy (WHO 2015). However, as of 2015 no countries have reported implementation of an IPTi policy (WHO 2018).This may be due to concerns about dosage and administration to young infants, a limited understanding of the baseline prevalence of molecular markers of anti‐folate resistance. The research capacity to obtain and monitor relevant resistance data is often inadequate in endemic countries of sub‐Saharan Africa. The complexity of the IPTi policy may have also affected the uptake. Moreover, an increase in P falciparum resistance to SP in sub‐Saharan Africa may have also confounded the cost‐effectiveness assessments upon which the policy recommendations for IPTi were based. This has raised concerns for policy makers at country level on the effectiveness of implementing IPTi on a public health scale. However, alternative drugs are being investigated for IPTi. Some of the alternatives studied include single‐drug regimens (such as amodiaquine, mefloquine) and artemisinin‐based combination drug regimens (such as amodiaquine‐artesunate, SP‐artesunate, SP‐amodiaquine).
How the intervention might work
The effects of IPTi may be mediated through chemoprophylaxis (White 2005). The terminal elimination half‐lives of sulfadoxine and pyrimethamine in infants has been shown to be about nine days and 16 days respectively (Salman 2011). The effects wane over time, hence the need for intermittent repeat doses. SP may be useful for IPTi because this drug combination is readily available, relatively affordable, and well‐tolerated in both adults and children. Moreover, it is already recommended for IPT in pregnancy (WHO 2004). The long half‐life of SP and alternative drugs used for IPTi produces a prolonged prophylactic effect. In addition, SP can be administered as a single dose, which is easier to directly observe at health facilities. Also, IPTi is associated with more limited drug exposure than in chemoprophylaxis. Thus the effect of IPTi on the spread of resistance and impairment of immunity development might also be lower. Furthermore, logistical challenges of intervention delivery are almost eliminated by administering IPTi at time points that fit the schedule of routine vaccinations through the WHO EPI.
Why it is important to do this review
Earlier versions of this systematic review addressed the broader question of the effectiveness of chemoprevention (including prophylaxis and IPT) against malaria in preschool children living in malaria‐endemic communities (Meremikwu 2002; Meremikwu 2005; Meremikwu 2008). A previous Cochrane Review documented the evidence for IPT in children (IPTc) (Meremikwu 2012). Although there is a meta‐analysis on IPTi (Aponte 2009), there have been additional studies since its publication. Moreover, these additional studies have evaluated the protective efficacy of alternative drugs for use as IPTi. This Cochrane Review summarizes the updated evidence to inform public health practice and policy.
Objectives
To evaluate the effects of intermittent preventive treatment (IPT) with antimalarial drugs to prevent malaria in infants living in malaria‐endemic areas.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs). The randomization unit could be the individual participant or a cluster, such as a household.
Types of participants
Children aged below 12 months living in an area where malaria was endemic with moderate‐to‐high perennial transmission. Children with unknown infection status (that is, it is unknown whether each child was infected or uninfected) or known infection status were eligible. We excluded trials that, at enrolment, included children aged ≥ 12 months and only anaemic participants.
Types of interventions
Intervention
IPTi
Control
Placebo or no treatment
We included trials that allocated an additional intervention (such as insecticide‐treated nets or iron supplementation) to both the intervention and control group provided the additional intervention was the same for each group. We included trials that compared one drug with another under the IPTi platform.
Types of outcome measures
Primary outcomes
Clinical malaria (fever plus asexual parasitaemia)
Secondary outcomes
Severe malaria (as defined by WHO 2000)
All‐cause mortality
Hospital admission for any reason
Parasitaemia
Anaemia (< 8 g/dL)
Change in haemoglobin (or haematocrit)
Adverse events
Serious adverse effects
Other adverse events, that occur within the follow‐up time of the trial
Search methods for identification of studies
Electronic searches
We attempted to identify all relevant trials regardless of language or publication status (published, in press, and in progress).
Databases
We searched the following databases using the search terms and strategy described in Appendix 1: the Cochrane Infectious Diseases Group Specialized Register; the Cochrane Central Register of Controlled Trials (CENTRAL) on the Cochrane Library, Issue 12, 2018; MEDLINE (PubMed; 1966 to 3 December 2018); Embase (OVID; 1980 to 3 December 2018); and LILACS (Bireme; 1982 to 3 December 2018). We also searched the metaRegister of Controlled Trials (mRCT; www.isrctn.com/) and the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) portal (www.who.int/ictrp/en/) using ‘malaria', ‘infant*', ‘intermittent', ‘prevent*' and ‘IPT' as search terms.
Searching other resources
Reference lists
We also checked the reference lists of all studies identified by the above methods.
Data collection and analysis
Selection of studies
Review author EE and researcher Obiamaka Okafo (OO) independently screened the results of the literature search for potentially relevant trials by title and abstract. We coded articles as either ‘retrieve' if articles potentially fulfilled the inclusion criteria or if it was unclear whether the article fulfilled the inclusion criteria or not; or ‘do not retrieve' for articles that did not fulfil the inclusion criteria. We obtained the full‐text reports of potentially relevant trials. We independently applied the inclusion criteria to the full reports using an eligibility form and scrutinized publications to ensure we included each trial in the review only once. Any disagreements were resolved through discussion with either MM or CO, and when necessary by consulting a member of the Cochrane Infectious Diseases Group (CIDG) editorial team. We listed the excluded studies and the reasons for their exclusion in the ‘Characteristics of excluded studies' table. We illustrated the study selection process in a PRISMA study flow diagram.
Data extraction and management
Two review authors (CO and EE) independently extracted data using a specifically developed piloted data extraction form. We resolved disagreements through discussion among all review authors. We contacted the corresponding publication author in the case of unclear information or missing data.
For each outcome, we aimed to extract the number of participants randomized and the number analysed in each treatment group. For dichotomous outcomes, we recorded the number of participants experiencing the event and the number assessed in each treatment group. For continuous outcomes, we extracted arithmetic means and standard deviations for each treatment group, together with the numbers assessed in each group. For outcomes reported as count data, we extracted the total number of episodes as well as the total time at risk.
For trials that randomized clusters, we recorded the number of clusters in the trial, the average size of clusters, and the randomization unit (for example, household or institution). We attempted to document details about adjustment for clustering or other covariates. When reported, we recorded the estimates of the intracluster correlation (ICC) coefficient for each outcome. If the trials' analyses adjusted for clustering, we extracted the treatment effect and a corresponding measure of variability.
Assessment of risk of bias in included studies
Two review authors (EE and CO) independently assessed the risk of bias in each included trial using a ‘Risk of bias' form. We resolved any disagreements by discussion between the review authors.
For trials that randomized individuals, we assessed six components: generation of the randomization sequence, allocation concealment, blinding, incomplete outcome data, selective outcome reporting, and other biases (such as early termination of the trial). For trials that randomized clusters, we assessed additional components, namely, recruitment bias, baseline imbalances, loss of clusters, incorrect analysis, and comparability with trials that randomized individuals.
We made judgements of either ‘yes', ‘no', or ‘unclear' to indicate a low, high, or unclear risk of bias. We presented the results of the assessment in a ‘Risk of bias' graph, ‘Risk of bias' tables, and a ‘Risk of bias' summary.
Measures of treatment effect
We used the risk ratio (RR) to summarize dichotomous outcomes, reported the mean difference for continuous outcomes, and used the rate ratio for count outcomes. We presented all measures of effect with 95% confidence intervals (CIs). For time‐to‐event data presented as Kaplan‐Meier curves in trial reports, we calculated Peto hazard ratios. We extracted protective efficacy values and their 95% CIs and when an included trial did not report this data, we calculated these values from the RR or rate ratio with its 95% CI.
Unit of analysis issues
If the original trial analyses did not adjust for clustering, we adjusted the results for clustering by multiplying the standard errors of the treatment effect by the square root of the design effect. We calculated the design effect as 1+(m‐1)*ICC where ‘m' is the average cluster size and ICC is the ICC coefficient.
Dealing with missing data
We aimed to perform the analysis according to the intention‐to‐treat (ITT) principle (all randomized participants analysed in the groups to which they were originally assigned). However, when there was loss to follow‐up, we employed a complete‐case analysis, such that, we excluded from the analysis participants for whom no outcome was reported. This analysis assumed that the participants for whom an outcome was available were representative of the originally randomized participants.
Assessment of heterogeneity
We assessed statistical heterogeneity between subgroups by visually inspecting the forest plots for overlapping CIs, applying the Chi² test (where a P value < 0.10 is considered statistically significant), and by using the I² statistic (with values > 40% representing moderate heterogeneity, > 60% substantial heterogeneity, and > 80% considerable heterogeneity).
Assessment of reporting biases
We planned to construct funnel plots to look for evidence of publication bias. However, the number of trials in each meta‐analysis were insufficient to make this informative.
Data synthesis
We analysed the data using Review Manager 5 (RevMan 5) (Review Manager 2014). In the first instance, we applied a fixed‐effect meta‐analysis. However, if we detected moderate heterogeneity but still considered it appropriate to combine the trials, we used a random‐effects approach. Where heterogeneity was very high such that meta‐analysis was inappropriate, we displayed the results in forest plots or tables but did not combine the results.
We stratified the analyses by when the outcome was measured (during intervention and post‐intervention follow‐up). We placed cluster‐RCTs that adjusted effects for clustering in the same forest plots as trials that randomized individual participants. Also, we included footnotes in forest plots to identify cluster‐RCTs. We tabulated the results from non‐adjusted cluster‐RCTs. We used generic inverse variance meta‐analysis.
Certainty of the evidence
We assessed the certainty of the evidence using the GRADE approach (Guyatt 2008). We presented the main results of the review alongside the certainty of the evidence in the ‘Summary of findings' tables. We appraised the certainty of evidence for each outcome against five criteria: risk of bias (an appraisal of the overall risk of bias for trials contributing to the outcome), consistency (an evaluation of explained and unexplained heterogeneity), directness (an appraisal of how directly the included trials address the review question), precision (an assessment of the statistical precision of the result), and publication bias (an assessment of the risk of publication bias). Where we identified deficiencies that were sufficient to decrease our confidence in the estimates of effect, we downgraded the certainty of evidence for RCTs from ‘high' to either ‘moderate', ‘low', or ‘very low' and explained our reasons for doing so. We used the GRADEpro GDT software, GRADEpro 2014, to import data from RevMan 5 (Review Manager 2014). We have presented ‘Summary of findings' tables only for SP and the three drug combinations that are feasible for use as IPTi, given WHO recommendations regarding the use of monotherapy.
Subgroup analysis and investigation of heterogeneity
It was not feasible to undertake subgroup analyses by the length of follow‐up as data were insufficient. There was still insufficient information available on the levels of parasite resistance to SP in the included trials.
Sensitivity analysis
We conducted a sensitivity analysis to investigate the robustness of the results to the risk of bias components by including only trials that concealed the allocation adequately and had low incomplete outcome data (less than 10%). We also excluded cluster‐randomized trials that were at high or unclear risk of bias for one of the additional cluster‐specific risk of bias components.
Results
Description of studies
Results of the search
We conducted the literature search up to 3 December 2018. Searches of various databases yielded 153 records to be screened, after we deleted duplicates. Of these, we found that 114 were irrelevant to the review after screening by title/abstract. We obtained full texts of the remaining 39 studies. Of these, 12 studies (three cluster‐RCTs and nine RCTs) described in 19 articles met our inclusion criteria (Figure 1). We reported reasons for excluding studies in the ‘Characteristics of excluded studies' table.
1.
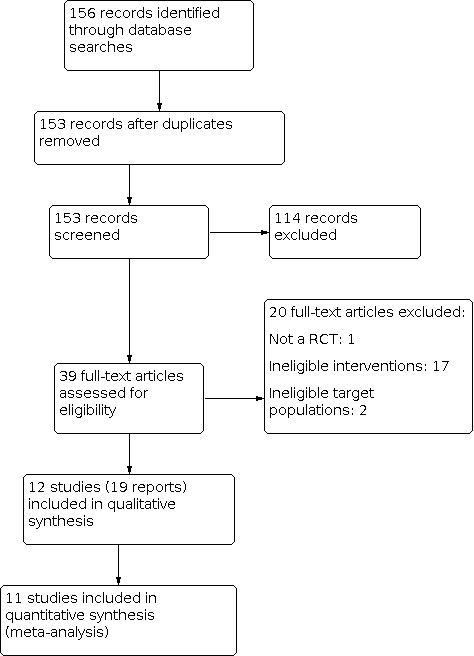
Study flow diagram
Included studies
See the ‘Characteristics of included studies' section for details of the included trials. We included 12 RCTs that enrolled 17,530 infants. Three of the included RCTs had a cluster‐randomized trial design (Armstrong Schellenberg 2010 TZA; Chandramohan 2005 GHA; Dicko 2012 MLI), and the remaining nine RCTs randomized individuals.
Location
The included trials were all conducted in Africa where P falciparum is predominant: four in Tanzania, three in Ghana, and one trial each in Gabon, Kenya, Mali, Mozambique, and Uganda. We have attached a three letter country code to each trial ID to aid forest plot interpretation.
Trial design
Nine trials randomized individuals, while three trials randomized clusters (household units of families living in a compound or villages in subdistricts). All three cluster‐RCTs adjusted for design effect and reported the average cluster size. Armstrong Schellenberg 2010 TZA adjusted for clustering in the analysis but did not provide the intra‐cluster correlation coefficient (ICC) value. Chandramohan 2005 GHA adjusted for design effect using a random‐effects model (REM) to allow for intra‐cluster correlation and other covariates (sex and urban‐rural residence). We obtained ICC values for Chandramohan 2005 GHA as follows: clinical malaria (ICC = 0.075), all‐cause hospital admissions (ICC = 0.000), haematocrit less than 24% (that is, severe anaemia; ICC = 0.006), and all‐cause death (ICC = 0.000).
Interventions
All included trials were conducted between 1999 and 2013. Nine trials compared IPT to placebo, while the remaining three trials had no IPT as the control arm (Armstrong Schellenberg 2010 TZA; Bigira 2014 UGA; Dicko 2012 MLI). Ten trials co‐administered IPT with routine EPI vaccinations (Armstrong Schellenberg 2010 TZA; Chandramohan 2005 GHA; Dicko 2012 MLI; Gosling 2009 TZA; Kobbe 2007 GHA; Macete 2006 MOZ; Massaga 2003 TZA; Mockenhaupt 2007 GHA; Odhiambo 2010 KEN; Schellenberg 2001 TZA). Two trials administered iron to all enrolled infants (Chandramohan 2005 GHA; Schellenberg 2001 TZA). Nine trials administered IPT with sulfadoxine‐pyrimethamine (SP) (Armstrong Schellenberg 2010 TZA; Chandramohan 2005 GHA; Dicko 2012 MLI; Gosling 2009 TZA; Grobusch 2007 GAB; Kobbe 2007 GHA; Macete 2006 MOZ; Mockenhaupt 2007 GHA; Schellenberg 2001 TZA). Alternative drug combinations to SP evaluated in the included trials were amodiaquine (AQ) (Massaga 2003 TZA), chlorproguanil‐dapsone (CD) (Gosling 2009 TZA; Odhiambo 2010 KEN), dihydroartemisinin‐piperaquine (DHAP) (Bigira 2014 UGA), and mefloquine (MQ) (Gosling 2009 TZA). One trial evaluated drug combinations that included SP; SP+ artesunate (AS) (Odhiambo 2010 KEN). Another drug combination evaluated was AQ+AS (Odhiambo 2010 KEN).
The length of follow‐up was until 24 months of age in eight trials (Bigira 2014 UGA; Chandramohan 2005 GHA; Gosling 2009 TZA; Grobusch 2007 GAB; Kobbe 2007 GHA; Mockenhaupt 2007 GHA; Odhiambo 2010 KEN; Schellenberg 2001 TZA). In the remaining four trials infants were followed‐up after the discontinuation of the intervention up to a maximum of 18 months of age (Armstrong Schellenberg 2010 TZA; Dicko 2012 MLI; Macete 2006 MOZ; Massaga 2003 TZA).
Outcome measures
Eleven trials reported on the outcome all‐cause mortality death (Bigira 2014 UGA; Chandramohan 2005 GHA; Dicko 2012 MLI; Gosling 2009 TZA; Grobusch 2007 GAB; Kobbe 2007 GHA; Macete 2006 MOZ; Massaga 2003 TZA; Mockenhaupt 2007 GHA; Odhiambo 2010 KEN; Schellenberg 2001 TZA). Dicko 2012 MLI and Armstrong Schellenberg 2010 TZA were the only trials that did not report anaemia and clinical malaria respectively. Only two trials reported severe malaria (Bigira 2014 UGA; Macete 2006 MOZ). Ten trials reported hospital admissions during the intervention period (Armstrong Schellenberg 2010 TZA; Bigira 2014 UGA; Chandramohan 2005 GHA; Gosling 2009 TZA; Kobbe 2007 GHA; Macete 2006 MOZ; Massaga 2003 TZA; Mockenhaupt 2007 GHA; Odhiambo 2010 KEN; Schellenberg 2001 TZA). Three trials reported changes in haemoglobin (Armstrong Schellenberg 2010 TZA; Chandramohan 2005 GHA; Grobusch 2007 GAB). Four trials reported asymptomatic parasitaemia (Armstrong Schellenberg 2010 TZA; Bigira 2014 UGA; Macete 2006 MOZ; Mockenhaupt 2007 GHA). Nine trials reported on adverse events (Armstrong Schellenberg 2010 TZA; Bigira 2014 UGA; Chandramohan 2005 GHA; Grobusch 2007 GAB; Kobbe 2007 GHA; Macete 2006 MOZ; Massaga 2003 TZA; Odhiambo 2010 KEN; Schellenberg 2001 TZA).
We have listed the outcome definitions that the included trials used in Table 5. Other outcomes reported by trials that we did not include in this Cochrane Review were all‐cause hospital attendance (Armstrong Schellenberg 2010 TZA; Schellenberg 2001 TZA); serological responses to EPI vaccines (Macete 2006 MOZ; Schellenberg 2001 TZA); and aspartate transaminase (AST), creatinine, and white blood cell counts (Grobusch 2007 GAB).
1. Definitions of outcome measures used in the included trials.
| Trial | Clinical malaria | Anaemia |
| Armstrong Schellenberg 2010 TZA | Not reported | Severe anaemia defined as haemoglobin level of < 8 g/dL. Mild anaemia defined as haemoglobin level of < 11 g/dL. |
| Bigira 2014 UGA | Documented fever (tympanic temperature ≥ 38.0°C) or history of fever in the previous 24 hours plus parasitaemia (thick blood smear). | Moderate–severe anaemia was defined as haemoglobin level of < 8.0 g/dL. |
| Chandramohan 2005 GHA | Not reported | Anaemia was defined as packed‐cell volume of < 24%. |
| Dicko 2012 MLI | Not reported | Not reported |
| Gosling 2009 TZA | Either a history of fever during the previous 2 days or an axillary temperature greater than 37.5°C plus parasitaemia of any density | Moderate anaemia was defined as haemoglobin level of < 8.0 g/dL |
| Grobusch 2007 GAB | The presence of any asexual P falciparum parasitaemia and either a rectal temperature of at least 38.5°C or a history of fever during the last 48 hours reported by the mother. | Anemia was defined as a haemoglobin level of < 8.0 g/dL. |
| Kobbe 2007 GHA | A malaria episode was defined as fever (temperature 38.0°C or fever during the preceding 48 hours reported by mothers without being asked), accompanied by asexual P falciparum parasitaemia of 1500 parasites/mL. | Anemia was defined as haemoglobin level of < 7.5 g/dL. |
| Macete 2006 MOZ | An episode of clinical malaria was defined as an axillary temperature of ≥ 37.5°C together with asexual P falciparum parasitaemia of any density. | Severe anaemia was defined as a packed‐cell volume of < 25%. |
| Massaga 2003 TZA | A febrile malarial episode was diagnosed in infants with a reported history of fever within the last 24 to 72 hours or a measured temperature of 37.5°C or greater (or both), who had a positive blood slide with asexual forms of P falciparum at any level of parasite density at time of contact with Maternal and Child Health clinic. | Anaemia was defined as packed‐cell volume of < 24%. |
| Mockenhaupt 2007 GHA | Malaria was defined as parasitaemia of any density plus fever (axillary temperature, ≥ 37.5°C) or a voluntarily reported history of fever within 48 hours of presentation to the clinic. | Severe anaemia was defined as haemoglobin level of < 7.0 g/dL. |
| Odhiambo 2010 KEN | An episode of clinical malaria was defined as an axillary temperature of at least 37.5°C or history of fever in the preceding 48 hours together with asexual P falciparum parasitaemia of any density. | Moderate‐to‐severe anaemia defined as haemoglobin level of < 8 g/dL. |
| Schellenberg 2001 TZA | A clinical malaria episode was defined as an axillary temperature of at least 37.5°C together with asexual P falciparum parasitaemia of any density. | Severe anaemia was defined as a packed‐cell volume of < 25%. |
Excluded studies
The ‘Characteristics of excluded studies' summarizes the reasons why we excluded studies. We excluded the 20 studies (20 reported papers) for the following reasons:
the intervention was intermittent preventive treatment in children (IPTc) (13 studies: Bojang 2010; Cissé 2006; Dicko 2008; Dicko 2011a; Dicko 2011b; Glinz 2015; Konaté 2011a; Konaté 2011b; Kweku 2008; Liljander 2010; Sesay 2011; Tagbor 2011; Tine 2011);
the intervention studied was chemoprophylaxis and not IPTi (4 studies: Greenwood 1988; Lemnge 1997; Menendez 1997; Wolde 1994);
the study was conducted outside sub‐Saharan Africa where IPTi is recommended (Senn 2012);
IPT was given to participants post‐discharge following recovery from malarial anaemia (Phiri 2012);
the study was a meta‐analysis (Aponte 2009).
Risk of bias in included studies
See Figure 2 and Figure 3 for a summary of the ‘Risk of bias' assessments. We have presented further details in the ‘Characteristics of included studies' tables.
2.

‘Risk of bias' summary: review authors' judgements about each ‘Risk of bias' item for each included trial
3.
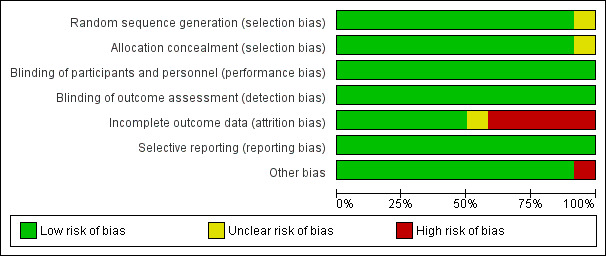
‘Risk of bias' graph: review authors' judgements about each ‘Risk of bias' item presented as percentages across all included trials
Allocation
Eleven trials were at low risk of bias regarding the generation of allocation sequence. One trial, Dicko 2012 MLI, was at unclear risk of bias because the trial authors did not provide enough information to permit us to make a judgement. Eleven trials were at low risk of bias regarding allocation concealment and the remaining trial, Dicko 2012 MLI, was at unclear risk of bias as the trial authors provided insufficient information to make a judgement.
Blinding
In all included trials, investigators and participants were unaware of treatment allocation. This was achieved by the use of clusters, the use of personnel not involved in patient care to perform treatment allocation, and the use of centrally coded drugs and placebos.
Incomplete outcome data
Six trials reported outcome data for at least 90% of randomized participants and were at low risk of bias regarding incomplete outcome data (Bigira 2014 UGA; Dicko 2012 MLI; Macete 2006 MOZ; Mockenhaupt 2007 GHA; Odhiambo 2010 KEN; Schellenberg 2001 TZA). Another five trials reported over 10% attrition in either one or both trial arms (Chandramohan 2005 GHA; Gosling 2009 TZA; Grobusch 2007 GAB; Kobbe 2007 GHA; Massaga 2003 TZA). One trial, Armstrong Schellenberg 2010 TZA, was at unclear risk of bias because different participants were surveyed at baseline and follow‐up.
Selective reporting
We did not detect any evidence of selective outcome reporting in any of the included trials.
Other potential sources of bias
We did not identify any other sources of bias for the individually RCTs. However, for the cluster‐RCTs, we considered recruitment bias, baseline imbalances, incorrect analyses, their comparability with individually RCTs, and the loss of clusters. We considered Armstrong Schellenberg 2010 TZA and Chandramohan 2005 GHA to be at low risk of bias for all of these additional sources of bias. However, we rated Dicko 2012 MLI as at high risk because of the high risk of recruitment bias, baseline imbalances, and incorrect analyses. Also, the trial authors did not provide any information on the loss of clusters.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Summary of findings 1. ‘Summary of findings' table 1.
| Intermittent preventive treatment in infants (IPTi) with sulfadoxine‐pyrimethamine (SP) versus placebo or no IPTi | ||||||
|
Participant or population: children under 12 months of age
Settings: areas with moderate to high malaria transmission (August 1999 to September 2013; Gabon, Ghana, Mali, Mozambique,Tanzania, and Uganda)
Intervention: intermittent preventive treatment (IPT) with SP Comparison: placebo or no IPTi | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (trials) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no IPTi | Risk with IPTi‐SP | |||||
| Clinical malaria | 74 episodes per 100 infants per yeara | 58 episodes per 100 infants per year (51 to 65) |
Rate ratio 0.78 (0.69 to 0.88) | 8774 (8 trials) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐SP probably reduced the risk of clinical malaria compared to placebo or no IPTi |
| Severe malaria | 20 episodes per 1000 infants per yearc | 19 episodes per 1000 infants per year (11 to 31) |
Rate ratio 0.92 (0.47 to 1.81) |
1347 (2 trials) | ⊕⊕⊝⊝
LOWd,e due to inconsistency and imprecision |
IPTi‐SP may have made little or no difference to the risk of severe malaria compared to placebo or no IPTi |
| All‐cause mortality | 23 per 1000 per year | 21 per 1000 per year (17 to 26) | Risk ratio 0.93 (0.74 to 1.15) | 14,588 (9 trials) | ⊕⊕⊕⊝
MODERATEf due to inconsistency |
IPTi‐SP may have made little or no difference to the risk of death compared to placebo or no IPTi |
| Hospital admission for any reason | 37 episodes per 100 infants per yearg | 32 episodes per 100 infants per year (29 to 36) |
Rate ratio 0.85 (0.78 to 0.93) |
7486 (7 trials) | ⊕⊕⊕⊝
MODERATEh due to imprecision |
IPTi‐SP probably slightly reduced hospital admission compared to placebo or no IPTi |
| Parasitaemia | 60 episodes per 100 infants per yeari | 40 episodes per 100 infants per year (34 to 47) |
Rate ratio 0.66 (0.56 to 0.79) |
1200 (1 trial) | ⊕⊕⊕⊝
MODERATEj due to imprecision |
IPTi‐SP probably reduced the risk of parasitaemia compared to placebo or no IPTi |
| Anaemia | 32 episodes per 100 infants per yeark | 26 episodes per 100 infants per year (22 to 31) |
Rate ratio 0.82 (0.68 to 0.98) |
7438 (6 trials) | ⊕⊕⊕⊝
MODERATEl due to inconsistency |
IPTi‐SP probably reduced the risk of anaemia compared to placebo or no IPTi |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; IPT: intermittent preventive treatment: IPTi: intermittent preventive treatment in infants; sulfadoxine‐pyrimethamine: SP. | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe incidence of malaria in the control groups was between 0.16 and 6.41 episodes per child per year. bDowngraded by 1 due to imprecision: these trials and the overall meta‐analysis are underpowered to detect a difference or to prove equivalence. cThe incidence of severe malaria in the control groups was between 0.02 and 0.03 episodes per child per year. dDowngraded by 1 due to inconsistency: there was considerable variation in the size of effect. eDowngraded by 1 for serious imprecision: these trials and the overall meta‐analysis are underpowered to detect a difference or to prove equivalence. Also the 95% CI overlaps and had no effect. fDowngraded by 1 due to inconsistency: wide variance of point estimates observed among the 9 trials in this meta‐analysis. gThe incidence of hospital admissions for any cause in the control groups was between 0.06 and 0.63 episodes per child per year. hDowngraded by 1 due to imprecision: these trials and the overall meta‐analysis are underpowered to detect a difference or to prove equivalence. iThe incidence of parasitaemia in the control group of one trial from Ghana was 0.6 episodes per child per year. jDowngraded by 1 due to imprecision: very small sample included in this analysis and is unlikely to detect differences or prove equivalence. kThe incidence of anaemia in the control groups was between 0.07 and 0.67 episodes per child per year. lDowngraded by 1 due to inconsistency: significant statistical heterogeneity observed in this meta‐analysis (I² statistic = 67%, P = 0.01).
Summary of findings 2. ‘Summary of findings' table 2.
| Intermittent preventive treatment in infants (IPTi) with AQ‐AS compared to placebo or no IPTi for malaria in infants | ||||||
| Patient or population: malaria in infants Setting: areas with moderate to high malaria transmission (March 2004 to March 2008; Kenya) Intervention: IPTi‐AQ‐AS Comparison: placebo or no IPTi | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no IPTi | Risk with IPTi‐AQ‐AS | |||||
| Clinical malaria | 133 episodes per 100 infants per yeara | 100 episodes per 100 infants per year (81 to 125) | Rate ratio 0.75 (0.61 to 0.94) | 547 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐AQ‐AS probably reduces the risk of clinical malaria compared to placebo or no IPTi |
| Severe malaria | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| All‐cause mortality | 36 per 1000 | 43 per 1000 (21 to 91) | Risk ratio 1.21 (0.58 to 2.55) | 684 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐AQ‐AS probably makes little or no difference to the risk of death compared to placebo or no IPTi |
| Hospital admission for any reason | 65 episodes per 100 infants per yearc | 64 episodes per 100 infants per year (49 to 83) | Rate ratio 0.98 (0.76 to 1.27) | 684 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐AQ‐AS probably makes little or no difference to the risk of hospital admission compared to placebo or no IPTi |
| Parasitaemia | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Anaemia | 30 infants per 1000 infantsd | 23 per 100 infants (159 to 336) | Rate ratio 0.77 (0.53 to 1.12) | 684 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐AQ‐AS probably makes little or no difference to the risk of anaemia compared to placebo or no IPTi |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; IPT: intermittent preventive treatment: IPTi: intermittent preventive treatment in infants; AQ‐AS: amodiaquine‐artesunate | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe incidence of malaria in the control group was 1.33 episodes per child per year (Odhiambo 2010 KEN). bDowngraded by 1 due to imprecision: CIs include potential for important harm and benefit. cThe incidence of hospital admissions for any cause in the control group was 0.65 episodes per child per year (Odhiambo 2010 KEN). dThe incidence of anaemia in the control group 0.3 episodes per child per year (Odhiambo 2010 KEN).
Summary of findings 3. ‘Summary of findings' table 3.
| Intermittent preventive treatment in infants (IPTi) with DHAP compared to placebo or no IPTi for malaria in infants | ||||||
| Patient or population: malaria in infants Setting: areas with moderate to high malaria transmission (June 2010 to September 2013; Uganda) Intervention: IPTi‐DHAP Comparison: placebo or no IPTi | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no IPTi | Risk with IPTi‐DHAP | |||||
| Clinical malaria | 641 episodes per 100 infants per yeara | 269 episodes per 100 infants per year (211 to 346) | Rate ratio 0.42 (0.33 to 0.54) | 147 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐DHAP probably reduces the risk of clinical malaria compared to placebo or no IPTi |
| Severe malaria | 29 episodes per 1000 infants per yearc | 37 episodes per 1000 infants per year (8 to 173) | Rate ratio 1.29 (0.28 to 5.98) | 147 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐DHAP probably makes little or no difference to the risk of severe malaria compared to placebo or no IPTi |
| All‐cause mortality | 20 per 1000 | 3 per 1000 (0 to 83) | Risk ratio 0.17 (0.01 to 4.06) | 147 (1 trial) | ⊕⊕⊝⊝
LOWb,d due to imprecision |
IPTi‐DHAP may make little or no difference to the risk of death compared to placebo or no IPTi |
| Hospital admission for any reason | 58 episodes per 1000 infants per yeare | 92 episodes per 1000 infants per year (27 to 314) | Rate ratio 1.58 (0.46 to 5.42) | 147 (1 trial) | ⊕⊕⊝⊝
LOWb,d due to imprecision |
IPTi‐DHAP may make little or no difference to the risk of hospital admission compared to placebo or no IPTi |
| Parasitaemia | The prevalence in the IPTi‐DHAP group was 3% compared to 11% in the control group (P < 0.001) | 147 (1 trial) |
⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐DHAP probably reduces the risk of parasitaemia compared to placebo or no IPTi | ||
| Anaemia | The prevalence in the IPTi‐DHAP group was half the prevalence in the control group (3% versus 6%; P = 0.04) | 147 (1 trial) |
⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐DHAP probably reduces the risk of anaemia compared to placebo or no IPTi | ||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; IPT: intermittent preventive treatment: IPTi: intermittent preventive treatment in infants; DHAP: dihydroartemisinin‐piperaquine | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe incidence of malaria in the control group was 6.41 episodes per child per year (Bigira 2014 UGA). bDowngraded by 1 due to imprecision: very few infants contributed to this analysis. cThe incidence of severe malaria in the control group was 0.029 episodes per child per year (Bigira 2014 UGA). dDowngraded by 1 due to imprecision: CIs include potential for important harm and benefit. eThe incidence of hospital admission in the control group was 0.058 episodes per child per year (Bigira 2014 UGA).
Summary of findings 4. ‘Summary of findings' table 4.
| Intermittent preventive treatment in infants (IPTi) with SP‐AS compared to placebo or no IPTi for malaria in infants | ||||||
| Patient or population: malaria in infants Setting: areas with moderate to high malaria transmission (March 2004 to March 2008; Kenya) Intervention: IPTi‐SP‐AS Comparison: placebo or no IPTi | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo or no IPTi | Risk with IPTi‐SP‐AS | |||||
| Clinical malaria | 133 episodes per 100 infants per yeara | 104 episodes per 100 infants per year (82 to 129) | Rate ratio 0.78 (0.62 to 0.97) | 676 (1 trial) | ⊕⊕⊕⊕ HIGH | IPTi‐SP‐AS reduces the risk of clinical malaria compared to placebo or no IPTi |
| Severe malaria | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| All‐cause mortality | 36 per 1000 | 30 per 1000 (13 to 67) | Risk ratio 0.83 (0.36 to 1.89) | 676 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐SP‐AS probably makes little or no difference to the risk of death compared to placebo or no IPTi |
| Hospital admission for any reason | 65 episodes per 100 infants per yearc | 60 episodes per 100 infants per year (462 to 780) | Rate ratio 0.92 (0.71 to 1.20) | 676 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐SP‐AS probably makes little or no difference to the risk of hospital admission compared to placebo or no IPTi |
| Parasitaemia | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Anaemia | 30 infants per 100 infantsd | 22 per 100 infants per year (15 to 32) | Rate ratio 0.72 (0.49 to 1.07) | 676 (1 trial) | ⊕⊕⊕⊝
MODERATEb due to imprecision |
IPTi‐SP‐AS probably makes little or no difference to the risk of anaemia compared to placebo or no IPTi |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; IPT: intermittent preventive treatment: IPTi: intermittent preventive treatment in infants; SP‐AS: sulfadoxine‐pyrimethamine‐artesunate | ||||||
| GRADE Working Group grades of evidence High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe incidence of malaria in the control group was 1.33 episodes per child per year (Odhiambo 2010 KEN). bDowngraded by 1 for imprecision: CIs include potential for important harm and benefit. cThe incidence of hospital admissions for any cause in the control group was 0.65 episodes per child per year (Odhiambo 2010 KEN). dThe incidence of anaemia in the control group 0.3 episodes per child per year (Odhiambo 2010 KEN).
IPTi versus placebo or no IPTi
Clinical malaria
Sulfadoxine‐pyrimethamine
IPTi with SP at that time probably reduced the risk of clinical malaria (rate ratio 0.78, 95% CI 0.69 to 0.88; 8 trials, 8774 participants; Analysis 1.1). There was substantial statistical heterogeneity as indicated by an I² statistic value of 64%. Sensitivity analysis, which excluded cluster‐randomized trials and studies at high risk of selection bias, did not considerably change the summary effect estimate (rate ratio 0.71, 95% CI 0.55 to 0.92; 4 trials, 3551 participants; Analysis 2.1).
1.1. Analysis.

Comparison 1: IPTi versus placebo or no IPTi (by specific drug combination), Outcome 1: Clinical malaria
2.1. Analysis.

Comparison 2: Sensitivity analysis: IPTi with SP versus placebo or no IPTi, Outcome 1: Clinical malaria
Artemisinin‐combination therapy
IPTi with AQ‐AS probably reduces the risk of clinical malaria (rate ratio 0.75, 95% CI 0.61 to 0.94; 1 trial, 547 participants; Analysis 1.1). IPTi with DHAP probably reduces the risk of clinical malaria (rate ratio 0.42, 95% CI 0.33 to 0.54; 1 trial, 147 participants; Analysis 1.1). We downgraded the certainty of the evidence by one level due to imprecision (very few infants contributed to the analysis).
IPTi with SP‐AS reduces the risk of clinical malaria (rate ratio 0.78, 95% CI 0.62 to 0.97; 1 trial, 676 participants; Analysis 1.1).
Monotherapy
IPTi with amodiaquine may have reduced the risk of clinical malaria episodes at the time (rate ratio 0.35, 95% CI 0.22 to 0.56; 1 trial, 146 participants; Analysis 1.1).
IPTi with mefloquine resulted in a large reduction in the risk of clinical malaria (rate ratio 0.62, 95% CI 0.44 to 0.88; 1 trial, 480 participants; Analysis 1.1).
Severe malaria
Sulfadoxine‐pyrimethamine
IPTi with SP may have made little or no difference on the risk of severe malaria (rate ratio 0.92, 95% CI 0.47 to 1.81; 2 trials, 1347 participants; Analysis 1.2). Another trial also found no difference in the risk of severe malaria (Macete 2006 MOZ; see Table 6). However, overall the sample size was too small to detect or exclude clinically important differences.
1.2. Analysis.

Comparison 1: IPTi versus placebo or no IPTi (by specific drug combination), Outcome 2: Severe malaria
2. Additional data: IPTi versus placebo or no IPTi.
| Prespecified outcome | Trial‐reported outcome | Trial | Number of participants | IPTi | Placebo or no IPTi | Comparative results reported in article |
| Anaemia | Mild anaemia (< 11 g/dL) | Armstrong Schellenberg 2010 TZA | 620 | 277/346 (80%) | 241/274 (88%) | P = 0.02 |
| Severe anaemia (< 8 g/dL) | 620 | 40/346 (12%) | 44/274 (16%) |
P = 0.19 | ||
| Moderate‐to‐severe anaemia (< 8 g/dL) |
Bigira 2014 UGA IPTi SP |
196 | 145/1113 (13%) |
66/1112 (6%) |
P = 0.04 | |
|
Bigira 2014 UGA IPTi DHAP |
196 | 25/899 (3%) | 66/1112 (6%) |
P = 0.04 | ||
| Moderate anaemia (at least one episode) |
Grobusch 2007 GAB IPTI SP |
1011 | 65/504 (13%) | 88/507 (17%) |
P = 0.05 | |
| Severe malaria | Severe malaria (WHO definition) | Macete 2006 MOZ | 1503 | 26/748 (4%) | 29/755 (4%) |
P = 0.66 |
| Parasitaemia | Asymptomatic parasitaemia |
Bigira 2014 UGA IPTi SP |
196 | 59/500 (12%) |
60/528 (11%) |
P = 0.89 |
|
Bigira 2014 UGA IPTi DHAP |
196 | 24/849 (3%) |
60/528 (11%) |
P < 0.001 |
Abbreviations: IPTi: intermittent preventive treatment in infants.
Artemisinin‐combination therapy
IPTi with DHAP probably has little or no effect on the risk of severe malaria (rate ratio 1.29, 95% CI 0.28 to 5.98; 1 trial, 147 participants; Analysis 1.2). We downgraded the certainty of evidence by one level due to imprecision (very few infants contributed to the analysis). No studies that evaluated IPTI with SP‐AS or AQ‐AS reported on this outcome.
Monotherapy
No studies that evaluated IPTI with AQ or MQ reported on this outcome.
All‐cause mortality
Sulfadoxine‐pyrimethamine
IPTi with SP probably made little or no difference to the risk of all‐cause mortality (risk ratio (RR) 0.93, 0.74 to 1.15; 9 trials, 14,588 participants; Analysis 1.3). Sensitivity analysis did not considerably change the summary effect estimate (RR 0.91, 95% CI 0.60 to 1.37; 4 trials, 3551 participants; Analysis 2.3).
1.3. Analysis.

Comparison 1: IPTi versus placebo or no IPTi (by specific drug combination), Outcome 3: All‐cause mortality
2.3. Analysis.

Comparison 2: Sensitivity analysis: IPTi with SP versus placebo or no IPTi, Outcome 3: All‐cause mortality
Artemisinin‐combination therapy
IPTi with AQ‐AS probably does not reduce the risk of all‐cause mortality (RR 1.21, 95% CI 0.58 to 2.55; 1 trial, 684 participants; Analysis 1.3). We downgraded the certainty of the evidence by one level due to imprecision (the CI included potential for important harm and benefit).
IPTi with DHAP may not reduce the risk of all‐cause mortality (RR 0.17, 95% CI 0.01 to 4.06; 1 trial, 147 participants; Analysis 1.3). We downgraded the certainty of evidence by two levels due to imprecision (very few infants contributed to the analysis and the CI included potential for important harm and benefit).
IPTi with SP‐AS probably has little or no effect on all‐cause mortality (risk ratio 0.83, 95% CI 0.36 to 1.89; 1 trial, 676 participants; Analysis 1.3). We downgraded the certainty of the evidence by one level due to imprecision (the CI included potential for important harm and benefit).
Monotherapy
The evidence suggests IPTi with mefloquine may have resulted in little to no difference in all‐cause mortality (risk ratio 0.33, 95% CI 0.06 to 1.97; 1 trial, 480 participants; Analysis 1.3).
However, IPTi with amodiaquine may not have reduced the risk of all‐cause mortality (risk ratio 1.30, 95% CI 0.30 to 5.59; 1 trial, 146 participants; Analysis 1.3).
Hospital admission for any reason
Sulfadoxine‐pyrimethamine
IPTi probably reduced the risk of hospital admission for any reason (rate ratio 0.85, 95% CI 0.78 to 0.93; 7 trials, 7486 participants; Analysis 1.4). Moderate levels of statistical heterogeneity were observed (I² statistic = 53%). Sensitivity analysis did not significantly change the summary effect estimate (rate ratio 0.78, 95% CI 0.68 to 0.88; 4 trials, 3551 participants; Analysis 2.4).
1.4. Analysis.

Comparison 1: IPTi versus placebo or no IPTi (by specific drug combination), Outcome 4: Hospital admission for any reason
2.4. Analysis.

Comparison 2: Sensitivity analysis: IPTi with SP versus placebo or no IPTi, Outcome 4: Hospital admission for any reason
Artemisinin‐combination therapy
IPTi with AQ‐AS probably does not reduce the risk of hospital admission for any reason (rate ratio 0.98, 95% CI 0.76 to 1.27; 1 trial, 684 participants; Analysis 1.4) .We downgraded the certainty of the evidence by one level due to imprecision (the CI included potential for important harm and benefit).
IPTi with DHAP may not reduce the risk of hospital admission for any reason (rate ratio 1.58, 95% CI 0.46 to 5.42; 1 trial, 147 participants; Analysis 1.4). We downgraded the certainty of evidence by two levels due to imprecision (very few infants contributed to the analysis and the CI included potential for important harm and benefit).
IPTi with SP‐AS probably has little or no effect on hospital admission for any reason (rate ratio 0.92, 95% CI 0.71 to 1.20; 1 trial, 676 participants; Analysis 1.4). We downgraded the certainty of the evidence by one level due to imprecision (the CI included potential for important harm and benefit).
Monotherapy
IPTi with amodiaquine may have reduced the risk of hospital admission for any reason (rate ratio 0.40, 95% CI 0.21 to 0.77; 1 trial, 146 participants; Analysis 1.4).
IPTi with mefloquine may not have reduced the risk of hospital admission for any reason (rate ratio 0.98, 95% CI 0.73 to 1.31; 1 trial, 480 participants; Analysis 1.4).
Parasitaemia
Sulfadoxine‐pyrimethamine
IPTi with SP probably reduced the risk of asymptomatic parasitaemia among infants (rate ratio 0.66, 95% CI 0.56 to 0.79; 1 trial, 1200 participants; Analysis 1.5).
1.5. Analysis.

Comparison 1: IPTi versus placebo or no IPTi (by specific drug combination), Outcome 5: Parasitaemia
Artemisinin‐combination therapy
One study evaluated IPTi with DHAP but did not contribute data to the meta‐analysis. This study showed that IPTi with DHAP probably reduces the risk of parasitaemia (prevalence of 3% compared to 11% in the control group P < 0.001; Table 6). We downgraded the certainty of evidence by one level due to imprecision (very few infants contributed to the analysis).
No studies that evaluated IPTi with AQ‐AS or SP‐AS reported on this outcome.
Monotherapy
No studies that evaluated IPTi with AQ or MQ reported this outcome.
Anaemia
Sulfadoxine‐pyrimethamine
IPTi with SP probably reduced the risk of anaemia in infants (rate ratio 0.82, 95% CI 0.68 to 0.98; 6 trials, 7438 participants; Analysis 1.6). Sensitivity analysis did not considerably change the summary effect estimate (rate ratio 0.77, 95% CI 0.62 to 0.95; 3 trials, 3404 participants; Analysis 2.2). One trial, Armstrong Schellenberg 2010 TZA, reported mild (Hb < 11 g/dL) and severe (Hb < 8 g/dL) anaemia. The trial authors reported a significantly lower risk of mild anaemia in the IPTi group (277/346, 80%) compared to controls (241/274, 88%). The risk of severe anaemia was also lower in the IPTi group compared to controls (12% versus 16%) as shown in Table 6. There was no overall difference in mean haemoglobin levels between infants in the IPTi and control groups (mean difference −0.03, 95% CI −0.43 to 0.36; 3 trials, 4295 participants; Analysis 1.7).
1.6. Analysis.

Comparison 1: IPTi versus placebo or no IPTi (by specific drug combination), Outcome 6: Anaemia
2.2. Analysis.

Comparison 2: Sensitivity analysis: IPTi with SP versus placebo or no IPTi, Outcome 2: Anaemia
1.7. Analysis.

Comparison 1: IPTi versus placebo or no IPTi (by specific drug combination), Outcome 7: Change in haemoglobin (or haematocrit)
Artemisinin‐combination therapy
IPTi with AQ‐AS probably does not reduce the risk of anaemia (rate ratio 0.77, 95% CI 0.53 to 1.12; 1 trial, 684 participants; Analysis 1.6). We downgraded the certainty of the evidence by one level due to imprecision (the CI included potential for important harm and benefit). Similarly, the risk of moderate to severe anaemia was lower in the IPTi with DHAP group compared to controls (3% versus 6%), as shown in Table 6. We downgraded the certainty of evidence by two levels due to imprecision (very few infants contributed to the analysis and the CI included potential for important harm and benefit).
We found that IPTi with SP‐AS probably has little or no effect on anaemia (rate ratio 0.72, 95% CI 0.49 to 1.07; 1 trial, 676 participants; Analysis 1.6). We downgraded the certainty of the evidence by one level due to imprecision (the CI included potential for important harm and benefit).
Monotherapy
IPTi with amodiaquine may have reduced the risk of anaemia (rate ratio 0.29, 95% CI 0.13 to 0.63; 1 trial, 146 participants; Analysis 1.6).
IPTi with mefloquine may not have reduced the risk of anaemia (rate ratio 1.06, 95% CI 0.78 to 1.44; 1 trial, 480 participants; Analysis 1.6).
Change in haemoglobin (or haematocrit)
Sulfadoxine‐pyrimethamine
There was no overall difference in mean haemoglobin levels between infants in the IPTi and control groups (mean difference −0.03, 95% CI −0.43 to 0.36; 3 trials, 4295 participants; Analysis 1.7). No other studies were found that reported this outcome.
Post‐intervention follow‐up effects
We evaluated post‐intervention follow‐up effects of IPTi to determine if the effects were sustained beyond the intervention period. We found no evidence of an effect of IPTi on the risk of clinical malaria (Analysis 3.1), risk of death from any cause (Analysis 3.2), in the period after the discontinuation of the intervention. Similarly, IPTi had no effect on the risk of hospital admission (Analysis 3.3) and the risk of anaemia (Analysis 3.4) in the period after the discontinuation of the intervention. This lack of a sustained effect of IPTi in the period after the discontinuation of the intervention was consistent across all medicines.
3.1. Analysis.

Comparison 3: IPTi versus placebo or no IPTi (post‐intervention follow‐up), Outcome 1: Clinical malaria
3.2. Analysis.

Comparison 3: IPTi versus placebo or no IPTi (post‐intervention follow‐up), Outcome 2: All‐cause mortality
3.3. Analysis.

Comparison 3: IPTi versus placebo or no IPTi (post‐intervention follow‐up), Outcome 3: Hospital admission for any reason
3.4. Analysis.

Comparison 3: IPTi versus placebo or no IPTi (post‐intervention follow‐up), Outcome 4: Anaemia
Adverse events
Adverse events reported by trial authors were Stevens‐Johnson syndrome, fever, loss of appetite, weakness, skin reactions, gastrointestinal, and respiratory events. One trial, Bigira 2014 UGA, reported elevated enzyme levels and raised levels of platelets and white blood cells. These adverse events were associated with SP and DHAP. The adverse events reported are shown in Analysis 4.1, Analysis 4.2, and Table 7.
4.1. Analysis.

Comparison 4: IPTi versus placebo or no IPTi (adverse events), Outcome 1: SP
4.2. Analysis.

Comparison 4: IPTi versus placebo or no IPTi (adverse events), Outcome 2: DHAP
3. Adverse event information not appropriate for meta‐analysis.
| Type of antimalarial drug | Trial | Adverse event | Comments |
| Sulfadoxine‐pyrimethamine (SP) | Macete 2006 MOZ | Chest indrawing | RR 0.57, 95% CI 0.34 to 0.94, P = 0.025 |
| Splenomegaly | RR 0.06, 95% CI 0.01 to 0.47, P < 0.001 | ||
| Diarrhoea | RR 0.09, 95% CI 0.01 to 0.69, P = 0.002 | ||
| Skin | No severe cutaneous reactions | ||
| Schellenberg 2001 TZA | Fever | PE 13%, 95% CI 0.1 to 24.3, P = 0.048 | |
| Vomiting | "The frequency of vomiting after each dose was low (1%) and similar in each group." | ||
| Skin | "No severe skin reactions were reported in any child at any stage." | ||
| Armstrong Schellenberg 2010 TZA | Skin | "No children aged 2–11 months were admitted because of a rash associated with SP in either IPTi or comparison divisions." | |
| Fever | "Fever in the 2 weeks before the survey was similar in the two groups, being reported for 38% children in the intervention areas and 41% children in comparison areas ( P = 0.24)." | ||
| Chandramohan 2005 GHA | Vomiting | "The proportions of children who vomited after administration of drugs was similar between the two groups (0.4% in the placebo group versus 0.3% in the sulfadoxine‐pyrimethamine group)" | |
| Amodiaquine + artesunate | Odhiambo 2010 KEN | Skin and haematological | "No serious cutaneous adverse events were noted, and no cases of severe haemolysis were recorded." |
| SP in combination | Odhiambo 2010 KEN | Skin and haematological | |
| Amodiaquine | Massaga 2003 TZA | Haematological | “No clinical adverse effects such as sore throat or agranulocytosis were reported or observed during the study.” “No significant difference in mean leucocyte counts between the groups.” |
Abbreviations: CI: confidence interval; PE: protective efficacy; SP: sulfadoxine‐pyrimethamine. RR: risk ratio; IPTi: intermittent preventive treatment in infants.
Discussion
See ‘Summary of findings' tables 1 to 4 (Table 1; Table 2, Table 3 and Table 4). We have presented results for the review outcomes under three headings: sulfadoxine‐pyrimethamine (SP), artemisinin‐combination therapy (ACT), and monotherapy.
Summary of main results
We included 12 trials (19,098 participants) that were conducted in Africa.
IPTi with sulfadoxine‐pyrimethamine (SP) versus placebo or no IPTi
These trials suggest that at the time, IPTi with SP probably reduced the risk of clinical malaria episodes, hospital admissions, anaemia, and the risk of asymptomatic parasitaemia (moderate‐certainty evidence). IPTi with SP probably made little or no difference to the risk of all‐cause mortality (moderate‐certainty evidence). Also IPTi with SP may have made little or no difference to the risk of severe malaria (low‐certainty evidence).
IPTi with artemisinin combination treatments (ACTs) versus placebo or no IPTi
IPTi with amodiaquine plus artesunate probably reduces the risk of clinical malaria (moderate‐certainty evidence). However, IPTi with amodiaquine plus artesunate probably does not reduce the risk of all‐cause mortality, hospital admission for any reason, and anaemia (moderate‐certainty evidence).
IPTi with dihydroartemisinin‐piperaquine (DHAP) probably reduces the risk of clinical malaria, anaemia, and parasitaemia (moderate‐certainty evidence). However, IPTi with DHAP probably makes little or no difference to the risk of severe malaria (moderate‐certainty evidence) and may not reduce the risk of all‐cause mortality and hospital admission for any reason (low‐certainty evidence).
IPTi with SP plus artesunate reduces the risk of clinical malaria (high‐certainty evidence). However, IPTi with SP plus artesunate probably does not reduce the risk of all‐cause mortality, hospital admission for any reason, and anaemia (moderate‐certainty evidence). Severe malaria and parasitaemia were not reported for IPTi with SP plus artesunate.
Post‐intervention follow‐up effects
IPTi did not have sustained effects in the post‐intervention follow‐up period. There was no apparent effect of IPTi on the risk of clinical malaria, all‐cause mortality, hospital admission for any reason, and anaemia.
Overall completeness and applicability of evidence
This Cochrane Review included trials from several countries in East and West Africa where P falciparum malaria is predominant. IPTi is a policy recommendation for sub‐Saharan Africa, and thus it would be reasonable to generalize these findings to all sub‐Saharan countries of Africa with moderate‐to‐high malaria transmission. We included all published studies that evaluated IPTi in sub‐Saharan Africa with the currently recommended drug SP and alternative medicines. We found no ongoing studies. However, most included trials were not adequately powered to detect clinical differences for several outcomes.
Levels of parasite drug resistance to SP across Africa have increased and have led most countries to abandon SP as a monotherapy in first‐line treatment. This has raised questions regarding the efficacy of SP in the prevention of malaria given this increasing parasite resistance levels. However, SP has a proven safety profile, is low‐cost. Moreover, studies in pregnant women have demonstrated that SP could still be effective even in the presence of high levels of SP resistance (Desai 2015; Likwela 2012).
IPTi with SP probably reduced the risk of clinical malaria episodes, anaemia, and hospital admissions for any reason in infants. The artemisinin‐based combination medicines evaluated for use as IPTi appear to have demonstrated a better protective effect against clinical malaria. Albeit from a few trials that enrolled a small number of infants. However, although the review shows that IPTi with SP probably had a protective effect against clinical malaria, hospital admission, and anaemia; the finding is based on trials conducted over a 14‐year period. A close look at the meta‐analysis shows an attenuation of the effect of IPTi‐SP over time with the most recent trials showing no effect.
Current levels of SP resistance in Africa, suggest that the period over which SP remains useful as the drug of choice for IPTi may be very limited. The current World Health Organization (WHO) recommendations on IPTi with SP recommend a ≥ 50% cut‐off of dhps 540E gene mutation in the population as a benchmark for discouraging IPTi‐SP use. From a programmatic perspective, this portends additional challenges and a constant need to monitor SP molecular markers of resistance. Some of the antimalarial drug combination options evaluated for use as IPTi include some artemisinin‐combination therapy formulations currently included in national malaria treatment policies as first‐line treatment for uncomplicated falciparum malaria.
The WHO recommendation advises against treating a patient who has malaria using the same drug they were using for prophylaxis. This is to minimize the risk of overdosing and also to prolong the usefulness of the drugs reserved for treatment of uncomplicated malaria. Now, most countries are on artemether‐lumefantrine as first‐line treatment of uncomplicated malaria. There are also many trials that have used DHAP for mass drug administration. Seasonal malaria chemoprevention (SMC) now uses artesunate‐amodiaquine for children aged 3 to 59 months in the Sahel subregion. Thus, artemether‐lumefantrine and DHAP may not be appropriate for use as IPTi in countries where their components are part of the first‐line treatment of uncomplicated malaria. Also, in areas where malaria transmission is intense, it may be judicious to restrict ACTs for the treatment of cases, and not overexpose the drug for prophylactic purposes given the limited number of ACTs currently available. Also, Bigira 2014 UGA reported a low adherence to DHAP which may be related to the three‐day course of treatment. There have also been reports of the emergence of piperaquine‐resistant P falciparum infections in Southeast Asia (Amaratunga 2016). This calls to question the suitability of DHAP as a potential candidate for use as IPTi.
Certainty of the evidence
The included trials were generally well‐conducted with adequate methods for random sequence generation, allocation concealment, and blinding. There was also no evidence of selective reporting in the included studies.
For IPTi with SP, we have moderate certainty that the intervention probably reduced the risk of clinical malaria, anaemia, and hospital admission for any reason. As described above, for anaemia we downgraded the certainty of the evidence for inconsistency due to statistically significant heterogeneity observed. For clinical malaria, asymptomatic parasitaemia, and hospital admission for any reason, we downgraded the certainty to moderate for ‘imprecision', as the trials were underpowered to exclude the possibility of small but clinically important effects. For the finding of no effect on death from any cause, we downgraded the certainty to moderate as a result of inconsistency (wide variation in the size of the effect). For severe malaria, the finding of no effect was downgraded to low certainty for reasons also related to inconsistency and ‘imprecision'.
Although it was not feasible to undertake a priori specified subgroup analyses, in post‐hoc analyses we found that for clinical malaria (Analysis 1.1), excluding the earliest conducted trial (Schellenberg 2001 TZA) from the meta‐analysis reduced the I2 from 64% to 0%. This may be related to the time at which this trial was performed (August 1999 to April 2000). At this time in Tanazia, SP was not associated with any late treatment failures and was still first‐line treatment for uncomplicated malaria. This can be contrasted with the other trials which were conducted afterwards when SP resistance was becoming more widespread across sub‐Saharan Africa. Similarly, in post‐hoc analyses excluding the most recently conducted trials (Bigira 2014 UGA; Gosling 2009 TZA) from the meta‐analysis for hospital admission for any reason (Analysis 1.4), the I2 reduced from 53% to 0%. These two studies are the only multi‐arm randomized controlled trials in the meta‐analysis.
Potential biases in the review process
We only included peer‐reviewed and published clinical trials in this review. We also searched clinical trial registers and found no ongoing studies. It is very unlikely that we missed papers that were unpublished. We did not identify any potential biases in the review process. We included three cluster‐RCTs. However, only two reported that they took account of the cluster randomization. Intraclass correlation coefficients (ICCs) were available for one trial (Chandramohan 2005 GHA), and the other trial reported adjusting for clustering in the sample size determination (Dicko 2012 MLI). However, we did not include the third cluster‐RCT, which did not provide details, in the meta‐analyses (Armstrong Schellenberg 2010 TZA).
Agreements and disagreements with other studies or reviews
The conclusions of this Cochrane Review are consistent with a previously published meta‐analysis of trials that evaluated IPT in African infants (Aponte 2009). This meta‐analysis, like our review, found that IPTi had a substantial protective effect against clinical malaria, anaemia, and hospital admissions. Both reviews also did not find significant effects of IPTi on all‐cause mortality.
The main difference between the previous meta‐analysis (Aponte 2009), and this Cochrane Review is that we included clinical trials that evaluated other antimalarial drug combination options used as IPTi in this Cochrane Review. We found ACT options had substantial protective effect against clinical malaria.
Authors' conclusions
Implications for practice.
On the basis of the more recently conducted trials that showed no effect of IPTi with SP, the prospects for the continued use of SP as IPTi are limited. This is likely due to widespread resistance to SP. Several antimalarial drug combination options have been evaluated and show high levels of effectiveness. IPTi with other antimalarial drug combination options may reduce the risk of clinical malaria and asymptomatic parasitaemia. However, as long as SP remains the drug of choice for IPTi, resistance monitoring should be integrated into relevant epidemiological studies and surveillance programmes within national malaria control programmes in sub‐Saharan Africa.
Implications for research.
The evidence for the benefit of IPTi with SP is mainly from trials conducted up to 10 years ago. Questions remain regarding the efficacy of SP in the prevention of malaria in the face of widespread parasite resistance especially with the emergence of mutant P falciparum isolates carrying sulfadoxine resistance associated A437G and K540E mutations in the Pfdhps gene across West Africa. Concerns also remain about the potential for IPTi to increase the carriage and spread of drug‐resistant P falciparum parasites.
There are a few trials that evaluated other drug combination options for use as IPTi with some evidence of effectiveness (Bigira 2014 UGA; Gosling 2009 TZA; Massaga 2003 TZA; Odhiambo 2010 KEN). However, larger adequately powered trials are needed to provide more robust evidence for or against IPTi. Additional trials would most likely improve our confidence in the effect estimates for the effectiveness of IPTi. Also, as more trials evaluate alternative drug options for IPTi, subgroup analyses based on the type of antimalarial drug would become more robust and informative.
Future studies should investigate the efficacy, safety, operational feasibility, and cost‐effectiveness of IPTi with multi‐day antimalarial drugs in a programmatic setting.
What's new
| Date | Event | Description |
|---|---|---|
| 7 July 2021 | New citation required but conclusions have not changed | Author team addressed minor comments submitted via Cochrane Comments system |
| 7 July 2021 | Amended | Feedback incorporated into the review to clarify methods used to assess heterogeneity, and correct minor inconsistencies between sections. |
History
Protocol first published: Issue 2, 2015 Review first published: Issue 12, 2019
Acknowledgements
The Academic Editor of this review is Dr Joseph Okebe.
The editorial base of the Cochrane Infectious Diseases Group is funded by UK aid from the UK government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed do not necessarily reflect the UK government’s official policies.
The review was supported by the Research, Evidence, and Development Initiative (READ‐It) (project number 300342‐104). The review author team was supported by the grant previous to READ‐It (Grant: 5242).
This Cochrane Review is dedicated to the memory of our wonderful colleague, Ms Obiamaka Okafo, who passed away in 2019.
Appendices
Appendix 1. Search strategies
| Search set | CIDG SRa | CENTRAL | MEDLINEb | Embaseb | LILACSb |
| 1 | malaria | Malaria [Mesh, ti, ab] | Malaria [Mesh, ti, ab] | Malaria (Emtree, ti,ab) | malaria |
| 2 | prophylaxis | Prophylaxis ti,ab | Prophylaxis ti,ab | Prophylaxis ti,ab | prophylaxis |
| 3 | intermittent treatment | intermittent treatment ti, ab | Chemoprophylaxis ti,ab | Chemoprophylaxis ti,ab | intermittent treatment |
| 4 | IPT* | Prevention ti, ab | Prevention ti, ab | Prevention ti, ab | IPT$ |
| 5 | Infant* OR newborn* OR neonatal | presumptive treatment ti, ab | intermittent treatment ti,ab | intermittent treatment ti,ab | Infant$ OR newborn$ OR neonatal |
| 6 | 2 or 3 or 4 | IPT* ti, ab | presumptive treatment ti, ab | presumptive treatment ti, ab | 2 or 3 or 4 |
| 7 | 1 and 5 and 6 | Infant* OR newborn* OR neonatal ti,ab | IPT* ti, ab | IPT* ti, ab | 1 and 5 and 6 |
| 8 | — | 2 or 3 or 4 or 5 or 6 | 2 or 3 or 4 or 5 or 6 or 7 | 2 or 3 or 4 or 5 or 6 or 7 | — |
| 9 | — | 1 and 7 and 8 | Infant* OR newborn* OR neonatal ti,ab | Infant* OR newborn* OR neonatal ti,ab | — |
| 10 | — | — | 1 and 8 and 9 | 1 and 8 and 9 | — |
aCochrane Infectious Diseases Group Specialized Register. bSearch terms used in combination with the search strategy for retrieving trials developed by the Cochrane Collaboration (Lefebvre 2011).
Data and analyses
Comparison 1. IPTi versus placebo or no IPTi (by specific drug combination).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Clinical malaria | 10 | 10602 | Rate Ratio (IV, Random, 95% CI) | 0.70 [0.62, 0.80] |
| 1.1.1 IPTi AQ | 1 | 146 | Rate Ratio (IV, Random, 95% CI) | 0.35 [0.22, 0.56] |
| 1.1.2 IPTi MQ | 1 | 480 | Rate Ratio (IV, Random, 95% CI) | 0.62 [0.44, 0.88] |
| 1.1.3 IPTi SP | 8 | 8774 | Rate Ratio (IV, Random, 95% CI) | 0.78 [0.69, 0.88] |
| 1.1.4 IPTi AQ‐AS | 1 | 547 | Rate Ratio (IV, Random, 95% CI) | 0.75 [0.61, 0.94] |
| 1.1.5 IPTi DHAP | 1 | 147 | Rate Ratio (IV, Random, 95% CI) | 0.42 [0.33, 0.54] |
| 1.1.6 IPTi SP‐AS | 1 | 508 | Rate Ratio (IV, Random, 95% CI) | 0.78 [0.62, 0.97] |
| 1.2 Severe malaria | 2 | Rate Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 IPTi SP | 2 | 1347 | Rate Ratio (IV, Fixed, 95% CI) | 0.92 [0.47, 1.81] |
| 1.2.2 IPTi DHAP | 1 | 147 | Rate Ratio (IV, Fixed, 95% CI) | 1.29 [0.28, 5.98] |
| 1.3 All‐cause mortality | 11 | 16930 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.77, 1.14] |
| 1.3.1 IPTi AQ | 1 | 146 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.30, 5.59] |
| 1.3.2 IPTi MQ | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.11, 3.96] |
| 1.3.3 IPTi SP | 9 | 14588 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.74, 1.15] |
| 1.3.4 IPTi AQ‐AS | 1 | 684 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.58, 2.55] |
| 1.3.5 IPTi DHAP | 1 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.08] |
| 1.3.6 IPTi SP‐AS | 1 | 676 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.36, 1.89] |
| 1.4 Hospital admission for any reason | 9 | Rate Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 1.4.1 IPTi AQ | 1 | 146 | Rate Ratio (IV, Fixed, 95% CI) | 0.40 [0.21, 0.77] |
| 1.4.2 IPTi MQ | 1 | 480 | Rate Ratio (IV, Fixed, 95% CI) | 0.98 [0.73, 1.31] |
| 1.4.3 IPTi SP | 7 | 7486 | Rate Ratio (IV, Fixed, 95% CI) | 0.85 [0.78, 0.93] |
| 1.4.4 IPTi AQ‐AS | 1 | 684 | Rate Ratio (IV, Fixed, 95% CI) | 0.98 [0.76, 1.27] |
| 1.4.5 IPTi DHAP | 1 | 147 | Rate Ratio (IV, Fixed, 95% CI) | 1.58 [0.46, 5.42] |
| 1.4.6 IPTi SP‐AS | 1 | 676 | Rate Ratio (IV, Fixed, 95% CI) | 0.92 [0.71, 1.20] |
| 1.5 Parasitaemia | 1 | Rate Ratio (IV, Random, 95% CI) | Subtotals only | |
| 1.5.1 IPTi SP | 1 | 1200 | Rate Ratio (IV, Random, 95% CI) | 0.66 [0.56, 0.79] |
| 1.6 Anaemia | 8 | Rate Ratio (IV, Random, 95% CI) | Subtotals only | |
| 1.6.1 IPTi AQ | 1 | 146 | Rate Ratio (IV, Random, 95% CI) | 0.29 [0.13, 0.63] |
| 1.6.2 IPTi MQ | 1 | 480 | Rate Ratio (IV, Random, 95% CI) | 1.06 [0.78, 1.44] |
| 1.6.3 IPTi SP | 6 | 7438 | Rate Ratio (IV, Random, 95% CI) | 0.82 [0.68, 0.98] |
| 1.6.4 IPTi AQ‐AS | 1 | 684 | Rate Ratio (IV, Random, 95% CI) | 0.77 [0.53, 1.12] |
| 1.6.5 IPTi SP‐AS | 1 | 676 | Rate Ratio (IV, Random, 95% CI) | 0.72 [0.49, 1.07] |
| 1.7 Change in haemoglobin (or haematocrit) | 3 | 4295 | Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.43, 0.36] |
Comparison 2. Sensitivity analysis: IPTi with SP versus placebo or no IPTi.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Clinical malaria | 4 | 3551 | Rate Ratio (IV, Random, 95% CI) | 0.71 [0.55, 0.92] |
| 2.2 Anaemia | 3 | 3404 | Rate Ratio (IV, Random, 95% CI) | 0.77 [0.62, 0.95] |
| 2.3 All‐cause mortality | 4 | 3551 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.60, 1.37] |
| 2.4 Hospital admission for any reason | 4 | 3551 | Rate Ratio (IV, Fixed, 95% CI) | 0.78 [0.68, 0.88] |
Comparison 3. IPTi versus placebo or no IPTi (post‐intervention follow‐up).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Clinical malaria | 6 | Rate Ratio (IV, Random, 95% CI) | Subtotals only | |
| 3.1.1 IPTi MQ | 1 | 451 | Rate Ratio (IV, Random, 95% CI) | 1.00 [0.80, 1.26] |
| 3.1.2 IPTi SP | 5 | 5359 | Rate Ratio (IV, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 3.1.3 IPTi AQ‐AS | 1 | 520 | Rate Ratio (IV, Random, 95% CI) | 0.99 [0.82, 1.20] |
| 3.1.4 IPTi SP‐AS | 1 | 520 | Rate Ratio (IV, Random, 95% CI) | 0.99 [0.81, 1.20] |
| 3.2 All‐cause mortality | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.2.1 IPTi MQ | 1 | 449 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.12, 2.39] |
| 3.2.2 IPTi SP | 3 | 2106 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.24, 1.13] |
| 3.3 Hospital admission for any reason | 2 | Rate Ratio (IV, Random, 95% CI) | Subtotals only | |
| 3.3.1 IPTi MQ | 1 | 450 | Rate Ratio (IV, Random, 95% CI) | 1.37 [1.01, 1.87] |
| 3.3.2 IPTi SP | 2 | 1337 | Rate Ratio (IV, Random, 95% CI) | 1.09 [0.84, 1.42] |
| 3.4 Anaemia | 4 | Rate Ratio (IV, Random, 95% CI) | Subtotals only | |
| 3.4.1 IPTi MQ | 1 | 395 | Rate Ratio (IV, Random, 95% CI) | 0.97 [0.68, 1.36] |
| 3.4.2 IPTi SP | 3 | 3479 | Rate Ratio (IV, Random, 95% CI) | 0.89 [0.73, 1.08] |
| 3.4.3 IPTi AQ‐AS | 1 | 684 | Rate Ratio (IV, Random, 95% CI) | 0.89 [0.63, 1.26] |
| 3.4.4 IPTi SP‐AS | 1 | 676 | Rate Ratio (IV, Random, 95% CI) | 0.78 [0.54, 1.12] |
Comparison 4. IPTi versus placebo or no IPTi (adverse events).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 SP | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.1 Stevens‐Johnson syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.2 Fever | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.3 Loss of appetite | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.4 Weakness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.5 Skin | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.6 Gastrointestinal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.7 Respiratory | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.8 Laboratory abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.9 Thrombocytopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.10 Elevated aspartate aminotransferase | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.11 Elevated alanine aminotransferase | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.12 Neutropenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2 DHAP | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2.1 Fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2.2 Thrombocytopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2.3 Elevated aspartate aminotransferase | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2.4 Elevated alanine aminotransferase | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2.5 Neutropenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Armstrong Schellenberg 2010 TZA.
| Study characteristics | ||
| Methods |
Trial design: cluster‐RCT Unit of randomization: administrative divisions Trial dates: April 2005 to August 2006 Length of follow‐up: 11 months of age Average cluster size = 30; intracluster correlation coefficient (ICC) not given |
|
| Participants |
Number of participants: 600 infants from 24 health divisions Inclusion criteria: infants aged 2 to 11 months in the study area Exclusion criteria: none stated |
|
| Interventions |
|
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review
|
|
| Notes |
Location: Lindi and Mtwara regions of southern Tanzania (192 clusters, 5760 households) Malaria transmission: perennial transmission Funding: Bill and Melinda Gates Foundation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial authors used restricted randomization |
| Allocation concealment (selection bias) | Low risk | The trial authors performed randomization centrally with computer programmes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The trial used clusters, which minimized the risk of performance bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The trial used clusters, which minimized the risk of performance bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The trial surveyed different participants at follow‐up |
| Selective reporting (reporting bias) | Low risk | The published trial report included all expected outcomes |
| Other bias | Low risk | The study appears to be free of other sources of bias. Recruitment bias: "We allocated the 24 divisions to the two study arms using restricted randomization to assure adequate balance in terms of baseline mortality, overall population size, and geographic area (and hence district health management team)." Baseline imbalances: "We used a program in Stata version 8.0 (Stata Corp., College Station, TX) to test whether each of these possibilities satisfied balance criteria, including the following.
Incorrect analysis: "Statistical testing of household survey data was based on the t test, using a summary measure of the data from each of the 12 intervention and 12 comparison divisions. This adjusts both for the survey design and for the study design, which was randomized by division" Loss of clusters: all clusters included in final analysis |
Bigira 2014 UGA.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: June 2010 to September 2013 Length of follow‐up: 36 months of age |
|
| Participants |
Number of participants: 393 infants at 6 months of age Inclusion criteria: (1) born to HIV uninfected mothers, (2) residency within 30 km of the study clinic with no intention of moving outside the study area, (3) agreement to come to the study clinic for any illness and to avoid medications outside the study protocol, (4) provision of informed consent by parent/guardian Exclusion criteria: (1) no history of allergy or sensitivity to any study drugs, (2) absence of active medical problem requiring inpatient evaluation or chronic medical conditions requiring frequent attention, and (3) absence of clinically significant electrocardiogram (ECG) abnormalities, family history of long QT syndrome, and current use of drugs that prolong the QTc interval. |
|
| Interventions |
At the time of treatment allocation and during each visit to the study clinic, parents/guardians were given a 2‐month supply of drugs and a diary with dates for dosing and check‐offs to indicate administration. |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review: none |
|
| Notes |
Location: Tororo District, Uganda Malaria transmission: perennial transmission; entomological inoculation rate (EIR) = 562 infectious bites/person/year (2002) Funding: National Institutes of Health (HD059454). Holley‐Cotec provided the DHAP |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used permuted block randomization with computer to generate the randomization list |
| Allocation concealment (selection bias) | Low risk | "Study participants were randomised to their assigned treatment group at 6 mo of age using pre made, consecutively numbered, sealed envelopes" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Treatment allocation was performed by nurses not involved with patient care" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Treatment allocation was performed by nurses not involved with patient care" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The trial authors included 90% of infants in the analyses postintervention |
| Selective reporting (reporting bias) | Low risk | The published trial report included all expected outcomes |
| Other bias | Low risk | The trial appears to be free of other sources of bias |
Chandramohan 2005 GHA.
| Study characteristics | ||
| Methods |
Trial design: cluster‐RCT Unit of randomization: households Average cluster size = 26, ICCs and additional data provided by trial authors Trial dates: September 2000 to June 2004 Length of follow‐up: 24 months of age |
|
| Participants |
Number of participants: 96 clusters comprising a total of 2485 infants Inclusion criteria: infants living in selected clusters attending routine immunization clinics for second (DPT‐2) and third doses of diphtheria‐pertussis‐tetanus (DPT) vaccine (DPT‐3), measles vaccine (usually at age 9 months) and at age 12 months Exclusion criteria: allergy to SP |
|
| Interventions |
In addition, all infants received 1 month's supply of iron supplement (2.5 mL, 15 mg elemental iron, twice weekly for 4 weeks) when they received each vaccine. |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review: none |
|
| Notes |
Location: Kassena‐Nankana District, Upper East Region, Ghana Malaria transmission: high/seasonal; EIR = 418 infective bites/person/year (almost all between June and November) Funding: Department for International Development (DFID) UK (grant No R7602). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial computer‐generated random numbers |
| Allocation concealment (selection bias) | Low risk | The trial used identical and centrally coded drugs and placebo |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The study team and caretakers of study children were blinded to the drug codes." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The study team and caretakers of study children were blinded to the drug codes." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Loss to follow up in the per protocol population was 11.8% |
| Selective reporting (reporting bias) | Low risk | Published report includes most expected outcomes |
| Other bias | Low risk | The trial appears to be free of other sources of bias Recruitment bias: "To increase blinding, we assigned clusters allocated to sulfadoxine‐pyrimethamine or placebo to eight different drug codes (four sulfadoxine‐pyrimethamine and four placebo)." Baseline imbalances: there was baseline comparability of clusters from the data presented Incorrect analysis: results adjusted for clustering Loss of clusters: no loss of clusters identified |
Dicko 2012 MLI.
| Study characteristics | ||
| Methods |
Trial design: cluster‐RCT Unit of randomization: sub districts Average cluster size = 13 villages; cluster effect = 1.5 Trial dates: December 2006 to March 2009 Length of follow‐up: 18 months of age |
|
| Participants |
Number of participants: 22 health sub districts comprising a total of 5882 infants Inclusion criteria: infants living in health sub district attending routine immunization clinics for second (DPT‐2) and third doses of diphtheria‐pertussis‐tetanus (DPT) vaccine (DPT‐3), at age 3 and 4 months respectively and measles vaccine at age 9 months Exclusion criteria: none stated |
|
| Interventions |
All participants concurrently received routine immunization with DPT, measles, and yellow fever vaccines |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review: none |
|
| Notes |
Location: Kolokani District, Mali Malaria transmission: hyperendemic; malaria prevalence in children under 5 years of age= 45 and > 70% (dry and rainy seasons respectively) Funding: Institut de Recherche Biomédicale des Armées IRBA ‐ ex‐ IMTSSA & UMR6236‐URMITE, Marseille, France. We only extracted and included data from Cohort 2 for this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The 22 health sub‐districts were randomised in a 1:1 ratio with the intervention in 11 health areas and the other 11 serving as controls". The trial authors did not provide any further details |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not provide any details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Individuals who were not involved in the implementation of IPTi and who were not aware if a locality was in the intervention or non‐intervention zone collected the data. Use of clusters minimizes the risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Individuals who were not involved in the implementation of IPTi and who were not aware if a locality was in the intervention or non‐intervention zone collected the data. Use of clusters also minimizes the risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Losses in intervention and control sites were less than 10% |
| Selective reporting (reporting bias) | Low risk | The trial authors reported the prespecified outcomes that were in the protocol |
| Other bias | High risk | The trial appears to have several other sources of bias Recruitment bias: individuals recruited after clusters were randomized Baseline imbalances: no report on baseline comparability of clusters Incorrect analysis: no adjustment for clustering in the analysis was reported Loss of clusters: no information was provided |
Gosling 2009 TZA.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: December 2004 to May 2008 Length of follow‐up: 24 months of age |
|
| Participants |
Number of participants: 2419 infants Inclusion criteria: all infants aged 8 to 16 weeks who attended clinics for WHO’s Extended Program on Immunization (EPI) at the ten study health facilities (five in each site) for DPT2 and polio vaccination were eligible for inclusion. Exclusion criteria: infants who had any of the following conditions: history of allergy to study drugs; history of convulsions; clinical features of severe malnutrition or chronic illness, including infants with signs of HIV/AIDS; plans to leave the study area before 12 months of age; weight less than 4.5 kg at enrolment; and no witnessed, written consent from the caretaker. |
|
| Interventions |
Interventions: IPTi with one of the following.
The 1st and 2nd doses of IPTi were either:
The 3rd dose of IPTi at 9 months of age were either:
Placebo: identical placebos given at the same time points with iPTi All treatments at the health facility were observed and administered with routine immunizations. Field workers visited participants on days 2 and 3 to ensure doses were taken. |
|
| Outcomes |
Outcomes included in review
Outcomes not included in the review: none |
|
| Notes |
Location: Korogwe and Same Districts, Tanzania Malaria transmission: moderate transmission site (Korogwe District, Tanga region) and a neighbouring low‐transmission site (Same District, Kilimanjaro region). High SP resistance reported. EIR in neighbouring district (Muheza) was 148 infective bites per year (2000). Funding: IPTi Consortium and Gates Malaria Partnership (both supported by Bill & Melinda Gates Foundation) Additional notes: enrolment was prematurely suspended in the low‐transmission site after interim analysis (low malaria incidence resulting in lower power) thus only data from moderate‐transmission site is reported. Witnessed bed net coverage: 87% at enrolment Reported insecticide‐treated net (ITN) coverage: 53% at enrolment |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used computer‐generated numbers for sequence generation |
| Allocation concealment (selection bias) | Low risk | The trial administered drugs to participants in a secluded cubicle |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Both research team and child were masked to treatment allocation." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Both research team and child were masked to treatment allocation." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Percentage loss 15.3% (per protocol) |
| Selective reporting (reporting bias) | Low risk | The trial authors reported relevant outcomes |
| Other bias | Low risk | The trial appears to be free of other sources of bias |
Grobusch 2007 GAB.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: December 2002 to February 2005 Length of follow‐up: 30 months of age |
|
| Participants |
Number of participants: 1189 infants Inclusion criteria: provision of parental written informed consent or witnessed oral consent in the case of illiteracy and permanent residentship in the study area. Exclusion criteria: known or suspected allergy to sulphonamides or pyrimethamine or signs and symptoms thereof and history of severe hepatic or renal dysfunction |
|
| Interventions |
|
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review
|
|
| Notes |
Location: Lambaréné, Gabon Malaria transmission: perennial, with little seasonal variation and entomological inoculation rate of 50 infective bites/person/year. Funding: Bill & Melinda Gates Foundation (grant 28574), German Ministry of Education and Research (grant 01KA0202), German Academic Exchange Service |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used computer‐generated numbers for sequence generation |
| Allocation concealment (selection bias) | Low risk | The trial used identical centrally coded drug packages |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Two copies of the code were stored separately, accessible only to the principal investigator or a delegate." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This was a placebo controlled trial and drug packages were centrally coded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Percentage loss was 15.5% |
| Selective reporting (reporting bias) | Low risk | The trial reported key outcomes |
| Other bias | Low risk | The trial appeared to be free of other sources of bias |
Kobbe 2007 GHA.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: January 2003 to September 2005 Length of follow‐up: 24 months of age |
|
| Participants |
Number of participants: 1070 infants (535 infants in each arm) Inclusion criteria: age 3 months (4 weeks tolerance accepted); permanent residence in study area Exclusion criteria: severe illness |
|
| Interventions |
All participants concurrently received routine immunization with diphtheria‐pertussis‐tetanus (DPT) and measles vaccines |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review: none |
|
| Notes |
Location: Afigya Sekyere district, Ghana Malaria transmission: holoendemic, intense perennial (with seasonal peaks), EIR = 400 infective bites/person/year Funding: the Bundesministerium für Bildung und Forschung (grant 01KA0202) The German Academic Exchange Service La Roche (Basel, Switzerland) manufactured study drugs free of charge Sanofi‐Aventis donated artesunate tablets for treatment of uncomplicated malaria episodes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used computer‐generated random numbers |
| Allocation concealment (selection bias) | Low risk | The trial used identical and centrally coded drugs and placebo |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "A study team of 2 doctors, a nurse, a technician, and a field worker, all blinded to group assignment, was responsible for recruitment, treatment, and subsequent visits" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A study team of 2 doctors, a nurse, a technician, and a field worker, all blinded to group assignment, was responsible for recruitment, treatment, and subsequent visits" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Percentage loss of 18.5% (per protocol) |
| Selective reporting (reporting bias) | Low risk | The trial reported most of the expected outcomes |
| Other bias | Low risk | The trial appeared to be free of other sources of bias |
Macete 2006 MOZ.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: September 2002 to February 2004 Length of follow‐up: 12 months of age |
|
| Participants |
Number of participants: 1503 infants Inclusion criteria: infants (age 3 months at first dose); permanent residence in study area Exclusion criteria: allergy to sulfa drugs; illness that required admission to hospital |
|
| Interventions |
All participants received routine immunization with diphtheria‐pertussis‐tetanus (DPT) and measles vaccines |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review
|
|
| Notes |
Location: Manhica District (Maputo Province) Mozambique Malaria transmission: perennial transmission with EIR of 38 infective bites/person/year Funding: Hoffman‐La Roche provided SP (Fansidar) and placebo |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used computer‐generated random numbers |
| Allocation concealment (selection bias) | Low risk | The trial used identical and centrally coded drugs and placebo |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The trial used placebo and centrally‐coded drugs limits the chance of performance bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A computer‐generated treatment‐allocation list was used by the health assistant to ensure that subsequent doses were administered from the bottle with the same treatment identification letter as the first dose" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Percentage loss of 8.5% |
| Selective reporting (reporting bias) | Low risk | The published trial report included all expected outcomes |
| Other bias | Low risk | The trial appeared to be free of other sources of bias |
Massaga 2003 TZA.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: June 1999 to May 2000 Length of follow‐up: 9 to 10 months of age |
|
| Participants |
Number of participants: 291 infants Inclusion criteria: infants aged 12 to 16 weeks attending Maternal and Child Health (MCH) clinics for growth monitoring or to receive their third diphtheria‐pertussis‐tetanus (DPT) and oral poliovirus vaccine Exclusion criteria: congenital malformation; severe conditions that needed treatment in hospital; fever within past 2 days; packed‐cell volume < 24%; taking chemoprophylaxis |
|
| Interventions |
Interventions
Infants received 2.5 mL daily supplementation of iron (3 mg of ferric ammonium citrate mixture/mL) or placebo for 6 months. The first dose was given by the team and mothers were instructed how to administer the drug at home. |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review: none |
|
| Notes |
Location: Muheza district, north‐eastern Tanzania Malaria transmission: perennial/Holoendemic Funding: Danish International Development Agency |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used computer‐generated random numbers |
| Allocation concealment (selection bias) | Low risk | The trial used identical and centrally coded drugs and placebo |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "To ensure that treatment allocation was concealed from parents and the research team, and to ensure that infants received the right dose of medication, the trial drugs were coded and pre‐packed." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "To ensure that treatment allocation was concealed from parents and the research team, and to ensure that infants received the right dose of medication, the trial drugs were coded and pre‐packed." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The percentage loss was 21% |
| Selective reporting (reporting bias) | Low risk | The trial authors reported most expected outcomes |
| Other bias | Low risk | The trial appeared to be free of other sources of bias |
Mockenhaupt 2007 GHA.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: March 2003 to July 2005 Length of follow‐up: 24 months of age |
|
| Participants |
Number of participants: 1200 infants Inclusion criteria: parental informed consent and permanent residence in the study area Exclusion criteria: conditions requiring hospital admission, signs of hepatic or renal dysfunction, and reported allergy to sulfa‐containing drugs |
|
| Interventions |
All participants received routine immunization with diphtheria‐pertussis‐tetanus‐Haemophilus influenzae type b‐hepatitis B virus dose 2, measles, and yellow fever vaccinations. |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review: none |
|
| Notes |
Location: Tamale, Ghana Malaria transmission: hyperendemic/perennial transmission and modest seasonal variation Funding: German Ministry of Education and Research (grant 01KA0202), the German Academic Exchange Service (DAAD), and Charite´—University Medicine Berlin (grant 2005‐543) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used block randomization |
| Allocation concealment (selection bias) | Low risk | The trial used identical, centrally coded drug containers |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The study team and caretakers of children were blinded to the treatment regimen. The randomisation and drug code lists were kept by an individual not involved in the analysis of the study" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The study team and caretakers of children were blinded to the treatment regimen. The randomisation and drug code lists were kept by an individual not involved in the analysis of the study" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was no missing outcome data. The percentage loss was 5.5% |
| Selective reporting (reporting bias) | Low risk | The published report included key outcomes |
| Other bias | Low risk | The trial appears free of other sources of bias |
Odhiambo 2010 KEN.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: March 2004 to March 2008 Length of follow‐up: 24 months of age |
|
| Participants |
Number of participants: 1365 infants Inclusion criteria: children aged 5 to 16 weeks resident in the trial area attending clinic prior to first OPV/PENT vaccination. Exclusion criteria: infants with known allergy to any of the trial drugs, receiving cotrimoxazole prophylaxis for opportunistic infections, suffering concomitant illness requiring hospitalization or transfusion, or planning to be away from the study area for more than 6 months. |
|
| Interventions |
Interventions: IPTi with one of the following.
Placebo: 2 placebo tablets co‐administered once daily for 3 days. Treatments at the health facility were observed and administered with routine immunizations. Supplies of iron sulphate (2 mg/kg/day) were given at the first and second IPTi courses, and 1 month later at the fourth scheduled visit to the parent/guardian of study children for home administration during a 4‐month period from 2.5 to 6.5 months of age. |
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review: none |
|
| Notes |
Location: Asembo, Kenya Malaria transmission: Perennial with marked seasonal variation Funding: Bill & Melinda Gates Foundation Global Health Program, Grant ID# 28578. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used permuted block randomization for sequence generation |
| Allocation concealment (selection bias) | Low risk | The trial used centrally labelled and colour coded drug containers |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The colour‐arm assignment of the study identification numbers remained concealed to everyone except the technician. The technician did not have access to names of participants" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The colour‐arm assignment of the study identification numbers remained concealed to everyone except the technician. The technician did not have access to names of participants" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The percentage loss was 10% |
| Selective reporting (reporting bias) | Low risk | The trial protocol was available |
| Other bias | Low risk | The trial appears free of other sources of bias |
Schellenberg 2001 TZA.
| Study characteristics | ||
| Methods |
Trial design: RCT Trial dates: August 1999 to April 2000 Length of follow‐up: 24 months of age |
|
| Participants |
Number of participants: 701 infants (350 versus 351) Inclusion criteria: infants have just received second dose of DPT and oral poliovirus vaccine Exclusion criteria: illness requiring hospital admission |
|
| Interventions |
|
|
| Outcomes |
Outcomes included in the review
Outcomes not included in the review
|
|
| Notes |
Location: Ifakara, Tanzania Malaria transmission: perennial/holoendemic Funding: UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR); Spanish Agency for International Cooperation (AECI); Fondo de Investigaciones Sanitarias (FIS number 00/0803); Swiss Agency for Development and Cooperation; Hoffman‐La Roche provided the SP and placebo, and UNICEF provided the iron syrup. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial computer‐generated the sequence generation |
| Allocation concealment (selection bias) | Low risk | The trial used sealed, opaque envelopes and identical, centrally coded drugs and placebo |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No other project staff had ready access to treatment allocation information besides the health assistant who was not involved in the trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No other project staff had ready access to treatment allocation information besides the health assistant who was not involved in the trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The percentage loss was 3% |
| Selective reporting (reporting bias) | Low risk | The trial authors reported key outcomes |
| Other bias | Low risk | The trial appeared to be free of other sources of bias |
Abbreviations: AIDS: acquired immunodeficiency syndrome; AQ: amodiaquine; AS: artesunate; DHAP: dihydroartemisinin‐piperaquine; DPT:diphtheria‐pertussis‐tetanus; ECG: electrocardiogram; EIR: entomological inoculation rate; EPI: expanded programme on immunization; HIV: human immunodeficiency virus; ICC: intracluster correlation coefficient; IPTi: intermittent preventive treatment in infants; ITN: insecticide‐treated net; OPV: oral poliovirus vaccine; PENT: pentavalent vaccine; RCT: randomized controlled trial; SP: sulfadoxine pyrimethamine.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aponte 2009 | A pooled analysis of 6 trials |
| Bojang 2010 | Intermittent preventive treatment in children (IPTc) was the intervention studied and control arm was not randomized |
| Cissé 2006 | IPTc was the intervention studied |
| Dicko 2008 | IPTc was the intervention studied |
| Dicko 2011a | IPTc was the intervention studied |
| Dicko 2011b | IPTc was the intervention studied |
| Glinz 2015 | Age of participants at enrolment was 12 to 36 months |
| Greenwood 1988 | Chemoprophylaxis, not intermittent preventive treatment (IPT) |
| Konaté 2011a | IPTc was the intervention studied |
| Konaté 2011b | IPTc was the intervention studied |
| Kweku 2008 | IPTc was the intervention studied |
| Lemnge 1997 | Chemoprophylaxis (not IPT) |
| Liljander 2010 | IPTc was the intervention studied |
| Menendez 1997 | Chemoprophylaxis (not IPT) |
| Phiri 2012 | IPT given to participants post discharge following recovery from malarial anaemia |
| Senn 2012 | Study conducted outside sub‐Saharan Africa |
| Sesay 2011 | IPTc was the intervention studied |
| Tagbor 2011 | IPTc was the intervention studied |
| Tine 2011 | IPTc was the intervention studied |
| Wolde 1994 | Chemoprophylaxis (not IPT) |
Abbreviations: IPT: intermittent preventive treatment; IPTc: intermittent preventive treatment in children.
Differences between protocol and review
There are no differences between the protocol and the review.
Contributions of authors
Martin Meremikwu (MM) conceived the idea for the review. Ekpereonne Esu (EE) assessed the eligibility of trials. Chioma Oringanje (CO) and EE extracted data and assessed the methodological quality of eligible trials. EE entered data into Review Manager 5 (Review Manager 2014). EE prepared the ‘Summary of findings tables' and GRADE assessments. All review authors read, provided input, and approved the final version.
Sources of support
Internal sources
University Of Calabar, Nigeria
Liverpool School Of Tropical Medicine, UK
External sources
-
Department for International Development, UK
Project number 300342‐104
Declarations of interest
Ekpereonne Esu has no known conflicts of interest.
Chioma Oringanje has no known conflicts of interest.
Martin Meremikwu has no known conflicts of interest.
Edited (no change to conclusions)
References
References to studies included in this review
Armstrong Schellenberg 2010 TZA {published data only}
- Armstrong Schellenberg JRM, Shirima K, Maokola W, Manzi F, Mrisho M, Mushi A, et al. Community effectiveness of intermittent preventive treatment for infants (IPTi) in rural southern Tanzania. American Journal of Tropical Medicine and Hygiene 2010;82(5):772-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg JRM, Maokola W, Shirima K, Manzi F, Mrisho M, Mushi A, et al. Cluster-randomized study of intermittent preventive treatment for malaria in infants (IPTi) in southern Tanzania: evaluation of impact on survival. Malaria Journal 2011;10:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willey BA, Armstrong Schellenberg JR, Maokola W, Shirima K, Chemba M, Mshinda H, et al. Evaluating the effectiveness of IPTi on malaria using routine health information from sentinel health centres in southern Tanzania. Malaria Journal 2011;10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bigira 2014 UGA {published data only}
- Bigira V, Kapisi J, Clark TD, Kinara S, Mwangwa F, Muhindo MK, et al. Protective efficacy and safety of three antimalarial regimens for the prevention of malaria in young Ugandan children: a randomised controlled trial. PLOS Medicine 2014;11(8):e1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamya MR, Kapisi J, Bigira V, Clark TD, Kinara S, Mwangwa F, et al. Efficacy and safety of three regimens for the prevention of malaria in young HIV-exposed Ugandan children: a randomized controlled trial. AIDS 2014;28(18):2701-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapisi J, Bigira V, Clark T, Kinara S, Mwangwa F, Achan J, et al. Efficacy and safety of artemether-lumefantrine for the treatment of uncomplicated malaria in the setting of three different chemopreventive regimens. Malaria Journal 2015;14:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundell K, Jagannathan P, Huang L, Bigira V, Kapisi J, Kakuru MM, et al. Variable piperaquine exposure significantly impacts protective efficacy of monthly dihydroartemisinin-piperaquine for the prevention of malaria in Ugandan children. Malaria Journal 2015;14:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chandramohan 2005 GHA {published data only}
- Chandramohan D, Owusu-Agyei S, Carneiro I, Awine T, Amponsa-Achiano K, Mensah N, et al. Cluster randomised trial of intermittent preventive treatment for malaria in infants in area of high, seasonal transmission in Ghana. BMJ 2005;331(7519):727-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dicko 2012 MLI {published data only}
- Dicko A, Konare M, Traore D, Testa J, Salamon R, Doumbo O, et al. The implementation of malaria intermittent preventive trialtreatment with sulphadoxine-pyrimethamine in infants reduced all-cause mortality in the district of Kolokani, Mali: results from a cluster randomized control trial. Malaria Journal 2012;11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gosling 2009 TZA {published data only}
- Gosling RD, Gesase S, Mosha JF, Carneiro I, Hashim R, Lemnge M, et al. Protective efficacy and safety of three antimalarial regimens for intermittent preventive treatment for malaria in infants: a randomised, double-blind, placebo-controlled trial. Lancet 2009;374(9700):1521-32. [DOI] [PubMed] [Google Scholar]
Grobusch 2007 GAB {published data only}
- Grobusch MP, Lell B, Schwarz NG, Gabor J, Dornemann J, Potschke M, et al. Intermittent preventive treatment against malaria in infants in Gabon - a randomized, double-blind, placebo-controlled trial. Journal of Infectious Diseases 2007;196(11):1595-602. [DOI] [PubMed] [Google Scholar]
Kobbe 2007 GHA {published data only}
- Kobbe R, Kreuzberg C, Adjei S, Thompson B, Langefeld I, Thompson PA, et al. A randomized controlled trial of extended intermittent preventive antimalarial treatment in infants. Journal of Infectious Diseases 2007;45(1):16-25. [DOI] [PubMed] [Google Scholar]
- Marks F, Kalckreuth V, Kobbe R, Adjei S, Adjei O, Horstmann RD, et al. Parasitological rebound effect and emergence of pyrimethamine resistance in Plasmodium falciparum after single-dose sulfadoxine-pyrimethamine. Journal of Infectious Diseases 2005;192(11):1962-5. [DOI] [PubMed] [Google Scholar]
Macete 2006 MOZ {published data only}
- Macete E, Aide P, Aponte JJ, Sanz S, Mandomando I, Espasa M, et al. Intermittent preventive treatment for malaria control administered at the time of routine vaccination in Mozambican infants: a randomized, placebo-controlled trial. Journal of Infectious Diseases 2006;194(3):276-85. [DOI] [PubMed] [Google Scholar]
Massaga 2003 TZA {published data only}
- Massaga JJ, Kitua AY, Lemnge MM, Akida JA, Malle LN, Rønn AM, et al. Effect of intermittent treatment with amodiaquine on anaemia and malarial fevers in infants in Tanzania: a randomised placebo-controlled trial. Lancet 2003;361(9372):1853-60. [DOI] [PubMed] [Google Scholar]
Mockenhaupt 2007 GHA {published data only}
- Mockenhaupt FP, Reither K, Zanger P, Roepcke F, Danquah I, Saad E, et al. Intermittent preventive treatment in infants as a means of malaria control: a randomized, double-blind, placebo-controlled trial in northern Ghana. Antimicrobial Agents and Chemotherapy 2007;51(9):3273-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Odhiambo 2010 KEN {published data only}
- Odhiambo FO, Hamel MJ, Williamson J, Lindblade K, ter Kuile FO, Peterson E, et al. Intermittent preventive treatment in infants for the prevention of malaria in rural Western Kenya: a randomized, double-blind placebo-controlled trial. PLOS One 2010;5(4):e10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schellenberg 2001 TZA {published data only}
- Schellenberg D, Menendez C, Aponte JJ, Kahigwa E, Tanner M, Mshinda H, et al. Intermittent preventive antimalarial treatment for Tanzanian infants: follow-up to age 2 years of a randomised, placebo-controlled trial. Lancet 2005;365(9469):1481-3. [DOI] [PubMed] [Google Scholar]
- Schellenberg D, Menendez C, Kahigwa E, Aponte J, Vidal J, Tanner M, et al. Intermittent treatment for malaria and anaemia control at time of routine vaccinations in Tanzanian infants: a randomised, placebo-controlled trial. Lancet 2001;357(9267):1471-7. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aponte 2009 {published data only}
- Aponte JJ, Schellenberg D, Egan A, Breckenridge A, Carneiro I, Critchley J, et al. Efficacy and safety of intermittent preventive treatment with sulfadoxine-pyrimethamine for malaria in African infants: a pooled analysis of six randomised, placebo-controlled trials. Lancet 2009;374(9700):1533-42. [DOI] [PubMed] [Google Scholar]
Bojang 2010 {published data only}
- Bojang K, Akor F, Bittaye O, Conway D, Bottomley C, Milligan P, et al. A randomized trial to compare the safety, tolerability and efficacy of three drug combinations for intermittent preventive treatment in children. PLOS One 2010;5(6):e11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cissé 2006 {published data only}
- Cissé B, Sokhna C, Boulanger D, Milet J, Bâ el H, Richardson K, et al. Seasonal intermittent preventive treatment with artesunate and sulfadoxine-pyrimethamine for prevention of malaria in Senegalese children: a randomised, placebo-controlled, double-blind trial. Lancet 2006;367(9511):659-67. [DOI] [PubMed] [Google Scholar]
Dicko 2008 {published data only}
- Dicko A, Sagara I, Sissoko MS, Guindo O, Diallo AI, Kone M, et al. Impact of intermittent preventive treatment with sulphadoxine-pyrimethamine targeting the transmission season on the incidence of clinical malaria in children in Mali. Malaria Journal 2008;7:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dicko 2011a {published data only}
- Dicko A, Diallo AI, Tembine I, Dicko Y, Dara N, Sidibe Y, et al. Intermittent preventive treatment of malaria provides substantial protection against malaria in children already protected by an insecticide-treated bednet in Mali: a randomised, double-blind, placebo-controlled trial. PLOS Medicine 2011;8(2):e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dicko 2011b {published data only}
- Dicko A, Barry A, Dicko M, Diallo AI, Tembine I, Dicko Y, et al. Malaria morbidity in children in the year after they had received intermittent preventive treatment of malaria in Mali: a randomized control trial. PLOS One 2011;6(8):e23390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Glinz 2015 {published data only}
- Glinz D, Hurrell RF, Ouattara M, Zimmermann MB, Brittenham GM, Adiossan LG, et al. The effect of iron-fortified complementary food and intermittent preventive treatment of malaria on anaemia in 12- to 36-month-old children: a cluster-randomised controlled trial. Malaria Journal 2015;14:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Greenwood 1988 {published data only}
- Greenwood BM, Greenwood AM, Bradley AK, Snow RW, Byass P, Hayes RJ, et al. Comparison of two strategies for control of malaria within a primary health care programme in the Gambia. Lancet 1988;1(8595):1121-7. [DOI] [PubMed] [Google Scholar]
Konaté 2011a {published data only}
- Konaté AT, Yaro JB, Ouédraogo AZ, Diarra A, Gansané A, Soulama I, et al. Intermittent preventive treatment of malaria provides substantial protection against malaria in children already protected by an insecticide-treated bednet in Burkina Faso: a randomised, double-blind, placebo-controlled trial. PLOS Medicine 2011;8(2):e1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Konaté 2011b {published data only}
- Konaté AT, Yaro JB, Ouédraogo AZ, Diarra A, Gansané A, Soulama I, et al. Morbidity from malaria in children in the year after they had received intermittent preventive treatment of malaria: a randomised trial. PLOS One 2011;6(8):e23391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kweku 2008 {published data only}
- Kweku M, Liu D, Adjuik M, Binka F, Seidu M, Greenwood B, et al. Seasonal intermittent preventive treatment for the prevention of anaemia and malaria in Ghanaian children: a randomized, placebo controlled trial. PLOS One 2008;3(12):e4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lemnge 1997 {published data only}
- Lemnge MM, Msangeni HA, Rønn AM, Salum FM, Jakobsen PH, Mhina JI, et al. Maloprim malaria prophylaxis in children living in a holoendemic village in north-eastern Tanzania. Transactions of the Royal Society of Tropical Medicine and Hygiene 1997;91(1):68-73. [DOI] [PubMed] [Google Scholar]
Liljander 2010 {published data only}
- Liljander A, Chandramohan D, Kweku M, Olsson D, Montgomery SM, Greenwood B, et al. Influences of intermittent preventive treatment and persistent multiclonal Plasmodium falciparum infections on clinical malaria risk. PLOS One 2010;5(10):e13649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Menendez 1997 {published data only}
- Menendez C, Kahigwa E, Hirt R, Vounatsou P, Aponte JJ, Font F, et al. Randomised placebo-controlled trial of iron supplementation and malaria chemoprophylaxis for prevention of severe anaemia and malaria in Tanzanian infants. Lancet 1997;350(9081):844-50. [DOI] [PubMed] [Google Scholar]
Phiri 2012 {published data only}
- Phiri K, Esan M, Hensbroek MB, Khairallah C, Faragher B, ter Kuile FO. Intermittent preventive therapy for malaria with monthly artemether-lumefantrine for the post-discharge management of severe anaemia in children aged 4-59 months in southern Malawi: a multicentre, randomised, placebo-controlled trial. Lancet Infectious Diseases 2012;12(3):191-200. [DOI] [PubMed] [Google Scholar]
Senn 2012 {published data only}
- Senn N, Rarau P, Stanisic DI, Robinson L, Barnadas C, Manong D, et al. Intermittent preventive treatment for malaria in Papua New Guinean infants exposed to Plasmodium falciparum and P. vivax: a randomized controlled trial. PLOS Medicine 2012;9(3):e1001195. [DOI: 10.1371/journal.pmed.1001195] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sesay 2011 {published data only}
- Sesay S, Milligan P, Touray E, Sowe M, Webb EL, Greenwood BM, et al. A trial of intermittent preventive treatment and home-based management of malaria in a rural area of The Gambia. Malaria Journal 2011;10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tagbor 2011 {published data only}
- Tagbor H, Cairns M, Nakwa E, Browne E, Sarkodie B, Counihan H, et al. The clinical impact of combining intermittent preventive treatment with home management of malaria in children aged below 5 years: cluster randomised trial. Tropical Medicine & International Health 2011;16(3):280-9. [DOI] [PubMed] [Google Scholar]
Tine 2011 {published data only}
- Tine RC, Faye B, Ndour CT, Ndiaye JL, Ndiaye M, Bassene C, et al. Impact of combining intermittent preventive treatment with home management of malaria in children less than 10 years in a rural area of Senegal: a cluster randomized trial. Malaria Journal 2011;10:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolde 1994 {published data only}
- Wolde B, Pickering J, Wotton K. Chloroquine chemoprophylaxis in children during peak transmission period in Ethopia. Journal of Tropical Medicine and Hygiene 1994;97(4):215-8. [PubMed] [Google Scholar]
Additional references
Alexander 2007
- Alexander N, Sutherland C, Roper C, Cissé B, Schellenberg D. Modelling the impact of intermittent preventive treatment for malaria on selection pressure for drug resistance. Malaria Journal 2007;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Amaratunga 2016
- Amaratunga C, Lim P, Suon S, Sreng S, Mao S, Sopha C, et al. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Diseases 2016;16(3):357-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cairns 2010
- Cairns M, Gosling R, Carneiro I, Gesase S, Mosha JF, Hashim R, et al. Duration of protection against clinical malaria provided by three regimens of intermittent preventive treatment in Tanzanian infants. PLOS One 2010;5(3):e9467. [DOI: 10.1371/journal.pone.0009467] [DOI] [PMC free article] [PubMed] [Google Scholar]
Desai 2015
- Desai M, Gutman J, L'lanziva A, Otieno K, Juma E, Kariuki S, et al. Intermittent screening and treatment or intermittent preventive treatment with dihydroartemisinin–piperaquine versus intermittent preventive treatment with sulfadoxine–pyrimethamine for the control of malaria during pregnancy in western Kenya: an open-label, three-group, randomised controlled superiority trial. Lancet 2015;386(10012):2507-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gilles 2000
- Gilles HM. Management of Severe Malaria: a Practical Handbook. 2nd edition. Geneva: World Health Organization, 2000. [Google Scholar]
GRADEpro 2014 [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Version accessed 1 October 2016. Hamilton (ON): McMaster University (developed by Evidence Prime), 2014. Available at gradepro.org.
Greenwood 2004
- Greenwood B. The use of anti-malarial drugs to prevent malaria in the population of malaria-endemic areas. American Journal of Tropical Medicine and Hygiene 2004;70(1):1-7. [PubMed] [Google Scholar]
Greenwood 2010
- Greenwood B. Anti-malarial drugs and the prevention of malaria in the population of malaria endemic areas. Malaria Journal 2010;9(Suppl 3):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Likwela 2012
- Likwela JL, D’Alessandro U, Lokwa BL, Meuris S, Dramaix MW. Sulfadoxine–pyrimethamine resistance and intermittent preventive treatment during pregnancy: a retrospective analysis of birth weight data in the Democratic Republic of Congo (DRC). Tropical Medicine & International Health 2012;17(3):322-9. [DOI] [PubMed] [Google Scholar]
Otoo 1988
- Otoo LN, Snow RW, Menon A, Byass P, Greenwood BM. Immunity to malaria in young Gambian children after a two-year period of chemoprophylaxis. Transactions of the Royal Society of Tropical Medicine and Hygiene 1988;82(1):59-65. [DOI] [PubMed] [Google Scholar]
Pombo 2002
- Pombo DJ, Lawrence G, Hirunpetcharat C, Rzepczyk C, Bryden M, Cloonan N, et al. Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet 2002;360(9333):610-7. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Salman 2011
- Salman S, Griffin S, Kose K, Pitus N, Winmai J, Moore B, et al. Pharmacokinetic properties of conventional and double-dose sulfadoxine-pyrimethamine given as intermittent preventive treatment in infancy. Antimicrobial Agents and Chemotherapy 2011;55(4):1693-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
White 2005
- White NJ. Intermittent presumptive treatment for malaria. PLOS Medicine 2005;2(1):e3. [DOI: 10.1371/journal.pmed.0020003] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 1990
- World Health Organization Scientific Group on the Chemotherapy of Malaria. Practical chemotherapy of malaria: report of a WHO scientific group [meeting held in Geneva from 5 to 12 June 1989]. World Health Organization Technical Report Series; no. 805. Geneva: World Health Organization, 1990. [Google Scholar]
WHO 1993
- World Health Organization Study Group on the Implementation of the Global Plan of Action for Malaria Control. Implementation of the global malaria control strategy: report of a WHO Study Group on the Implementation of the Global Plan of Action for Malaria Control 1993-2000 [meeting held in Geneva from 8 to 12 February 1993]. WHO Technical Report Series; no. 839. Geneva: World Health Organization, 1993. [PubMed] [Google Scholar]
WHO 2000
- World Health Organization. Severe falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 2000;94(Suppl 1):1-90. [PubMed] [Google Scholar]
WHO 2004
- World Health Organization. A strategic framework for malaria prevention and control during pregnancy in the African region. WHO Regional Office for Africa, Brazzaville, Democratic Republic of Congo. Report AFR/MAL/04/01. 2004. www.who.int/malaria/publications/atoz/afr_mal_04_01/en/ (accessed 3 January 2015).
WHO 2010
- World Health Organization. WHO policy recommendation on Intermittent preventive treatment during infancy with sulfadoxine-pyrimethamine (SP-IPTi) for Plasmodium falciparum malaria control in Africa. March 2010. www.who.int/malaria/news/WHO_policy_recommendation_IPTi_032010.pdf (accessed 27 February 2014).
WHO 2011
- World Health Organization. Intermittent preventive treatment for infants using sulfadoxine-pyrimethamine (SP-IPTi) for malaria control in Africa: Implementation field guide. 2011. http://whqlibdoc.who.int/hq/2011/WHO_IVB_11.07_eng.pdf?ua=1 (accessed 23 February 2014).
WHO 2012
- World Health Organization. WHO Policy Recommendation: Seasonal Malaria Chemoprevention (SMC) for Plasmodium falciparum malaria control in highly seasonal transmission areas of the Sahel sub-region in Africa. www.who.int/malaria/publications/atoz/smc_policy_recommendation_en_032012.pdf?ua=1 (accessed 8 April 2017).
WHO 2015
- World Health Organization. World Malaria Report 2015. www.who.int/malaria/publications/world-malaria-report-2015/en/ (accessed 8 March 2016).
WHO 2018
- World Health Organization. World Malaria Report 2018. www.who.int/malaria/publications/world-malaria-report-2018/en/ (accessed 1 August 2019).
References to other published versions of this review
Meremikwu 2002
- Meremikwu M, Omari AAA. Antimalaria drugs given at regular intervals for preventing clinical malaria and severe anaemia in preschool children. Cochrane Database of Systematic Reviews 2002, Issue 3. Art. No: CD003756. [DOI: 10.1002/14651858.CD003756] [DOI] [Google Scholar]
Meremikwu 2005
- Meremikwu MM, Omari AAA, Garner P. Chemoprophylaxis and intermittent treatment for preventing malaria in children. Cochrane Database of Systematic Reviews 2005, Issue 4. Art. No: CD003756. [DOI: 10.1002/14651858.CD003756.pub2] [DOI] [PubMed] [Google Scholar]
Meremikwu 2008
- Meremikwu MM, Donegan S, Esu E. Chemoprophylaxis and intermittent treatment for preventing malaria in children. Cochrane Database of Systematic Reviews 2008, Issue 2. Art. No: CD003756. [DOI: 10.1002/14651858.CD003756.pub3] [DOI] [PubMed] [Google Scholar]
Meremikwu 2012
- Meremikwu MM, Donegan S, Sinclair D, Esu E, Oringanje C. Intermittent preventive treatment for malaria in children living in areas with seasonal transmission. Cochrane Database of Systematic Reviews 2012, Issue 2. Art. No: CD003756. [DOI: 10.1002/14651858.CD003756.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]