

Abstract
Motivation
To complement experimental efforts, machine learning-based computational methods are playing an increasingly important role to predict human–virus protein–protein interactions (PPIs). Furthermore, transfer learning can effectively apply prior knowledge obtained from a large source dataset/task to a small target dataset/task, improving prediction performance.
Results
To predict interactions between human and viral proteins, we combine evolutionary sequence profile features with a Siamese convolutional neural network (CNN) architecture and a multi-layer perceptron. Our architecture outperforms various feature encodings-based machine learning and state-of-the-art prediction methods. As our main contribution, we introduce two transfer learning methods (i.e. ‘frozen’ type and ‘fine-tuning’ type) that reliably predict interactions in a target human–virus domain based on training in a source human–virus domain, by retraining CNN layers. Finally, we utilize the ‘frozen’ type transfer learning approach to predict human–SARS-CoV-2 PPIs, indicating that our predictions are topologically and functionally similar to experimentally known interactions.
Availability and implementation: The source codes and datasets are available at https://github.com/XiaodiYangCAU/TransPPI/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The detection of human–virus protein–protein interactions (PPIs) is essential for our understanding of the mechanisms that allow viruses to control cellular functions of the human host. Considerable experimental efforts allow the determination of binary interactions between viral and human proteins through yeast two-hybrid (Y2H) assays and mass spectroscopy (MS) techniques (Gordon et al., 2020; Shah et al., 2018). However, maps of interactions between the human host and various viruses remain incomplete, as a consequence of experimental cost, noise and a multitude of potential protein interactions. Although tens of thousands of interactions have been experimentally determined, an immense need still exists for the development of reliable computational methods to predict human–virus PPIs.
The primary amino acid sequence remains the most accessible and complete type of protein information. As a consequence, many sequence-based feature extraction methods have been developed, such as Local Descriptors (LD) (Davies et al., 2008; Yang et al., 2010), Conjoint Triads (CT) (Shen et al., 2007; Sun et al., 2017) and Auto Covariance (AC) (Guo et al., 2008; You et al., 2013). Specifically, such features generally represent physicochemical properties or positional information of amino acids that appear in the protein sequences. In addition, other heterogeneous encoding schemes have been used as well to supplement traditional sequence encodings, including biological functions, protein interaction network properties, domain/motif information, expression profiles, evolutionary information and natural language processing-based sequence embedding techniques (Lian et al., 2021). Based on these features, several traditional machine learning algorithms (Alguwaizani et al., 2018; Cui et al., 2012; Dyer et al., 2011; Eid et al., 2016; Emamjomeh et al., 2014; Lian et al., 2020; Yang et al., 2020) were previously applied to predict human–virus PPIs. Dyer et al. proposed a linear Support Vector Machine (SVM) model to predict human–HIV PPIs based on k-mers composition, properties of human proteins in human PPI networks and domain profile features. Cui et al. utilized CT to encode protein sequences that were fed to an SVM model with a radial basis function kernel to predict human–HPV/HCV interactions. Emamjomeh et al. developed ensemble models to predict human–HCV PPIs including four popular machine learning methods and six different encoding schemes (i.e. amino acid composition, pseudo amino acid composition, evolutionary information, network centrality, expression information and post-translational modification information). Eid et al. introduced a domain/linear motif-based SVM approach called DeNovo to predict human–virus PPIs (Eid et al., 2016). In (Alguwaizani et al., 2018), an SVM model was developed to predict human–virus PPIs based on sequence features representing single amino acid repeats and local amino acid composition. Recently, we proposed a sequence embedding-based Random Forest (RF) method to predict human–virus PPIs with promising performance (Yang et al., 2020). In particular, we applied an unsupervised sequence embedding technique (i.e. doc2vec) to represent interacting protein sequences as low-dimensional vectors. While effectively capturing amino acid-specific information to predict novel human–virus PPIs, such machine learning methods still suffer from several limitations, such as publicly unavailable source codes/web servers, limited sets of virus species and unsatisfactory performance in real applications. Therefore, further method development of human–virus PPI predictions is still in high demand.
In the past decade, deep learning methods have demonstrated improved performance and potential in many fields. In particular, convolutional neural networks (CNNs) (Hashemifar et al., 2018) and recurrent neural networks (RNNs) (Zhang et al., 2016) are comparatively well-established approaches, where CNNs automatically capture local features while RNNs preserve contextualized/long-term ordering information. While deep learning methods (Ahmed et al., 2018; Chen et al., 2019; Du et al., 2017; Hashemifar et al., 2018; Sun et al., 2017) that allow the prediction of PPIs yield excellent performance, such models usually focus on intraspecies interactions. Very recently, Liu-Wei et al. (2021) reported a predictive method called DeepViral that utilized the information of sequences, disease phenotypes and functions as input to train a CNN model for human–virus PPI prediction.
In general, traditional machine learning/deep learning only perform well, if training and test set were cut from the same statistical distribution in the feature space (Shao et al., 2015). While the rigid application of a trained model on testing datasets with different distributions usually perform poorly, transfer learning methods utilize prior knowledge from a ‘source’ to train in a ‘target’ task domain (Chang et al., 2018; Shao et al., 2015) to improve performance. Effective transfer learning can improve the generalization of models, reduce the size of labeled datasets and save training time on the target dataset/task. With the development of deep learning networks, a regular phenomenon appears in various training objectives (Lee et al., 2009) in that the first layers of deep neutral networks (DNNs) usually capture standard features of training data, providing a foundation for transfer learning. Specifically, a DNN can be trained on a source task, establishing the parameters of the first layers. Subsequently, parameters of late layers are trained on the target task, striking a balance between the distributions of the different training domains. Depending on the size of the target dataset and number of parameters of the DNN, first layers of the target DNN can either remain unchanged during training on the new dataset or fine-tuned toward the new task, leveling specificity and generality of derived prior knowledge (Taroni et al., 2019).
Here, we focus on the development and application of transfer deep learning approaches to predict human–virus PPIs, an important issue amidst the world-wide COVID-19 pandemic. In particular, we design a deep learning framework through representing interacting protein sequences with a pre-acquired protein sequence profile module followed by a Siamese CNN and a multi-layer perceptron (MLP) module. Based on our deep learning framework, we propose two types of transfer learning methods through freezing/fine-tuning the parameters of the CNN layers trained with a source and retrained with a target human–virus system, showing improved prediction performance and better model generalization. Finally, we use the transfer learning models to predict human–SARS-CoV-2 PPIs and conduct in-depth topological and functional analysis of the obtained interaction network.
2 Materials and methods
2.1 Deep learning network framework
Our end-to-end DNN framework consists of a pre-acquired protein sequence profile module, a Siamese CNN module and a prediction module (Fig. 1). Evolutionary profile features have been used for intraspecies PPI predictions with favorable performance (Hamp and Rost, 2015; Hashemifar et al., 2018). In particular, we represent interacting proteins by protein sequence profile [i.e. position-specific scoring matrix (PSSM)], as input to the Siamese CNN module to generate respective high-dimensional sequence embeddings that captures local features of human and viral proteins such as protein linear binding motif patterns. Finally, output embeddings of two proteins form a sequence pair vector as the input to an MLP with an appropriate loss function to predict the presence/absence of an interaction between a viral and a human protein.
Fig. 1.
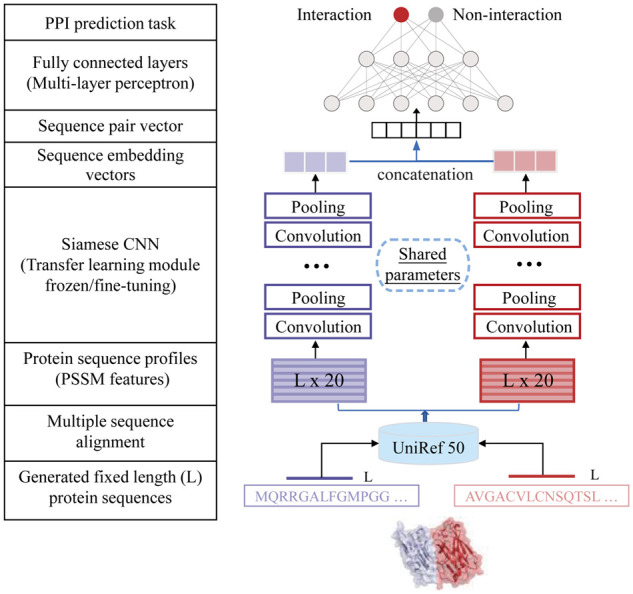
Our proposed deep learning architecture to predict human–virus PPIs combines evolutionary sequence profile features of interacting human and viral proteins with a Siamese CNN architecture and an MLP
2.1.1. Pre-acquired protein sequence profile module
By applying a threshold of E-value < 0.001, we performed PSI-BLAST searches with default parameters in the UniRef50 protein sequence database (Suzek et al., 2015) to discover protein sequences that are evolutionarily linked to the search sequence (Hamp and Rost, 2015; Hashemifar et al., 2018). Sequence profiles (i.e. PSSMs) thus obtained for each search sequence were processed by truncating profiles of long sequences to a fixed length n and zero-padding short sequences, a method widely used for data pre-processing and effective training (Min et al., 2017). As a result, we obtained a dimensional array S for each protein,
where denotes the probability of the jth out of the alphabet of 20 amino acids in the ith position of the sequence.
2.1.2. Siamese CNN module
To capture complex relationship between two proteins, we use a Siamese CNN architecture (Chen et al., 2019; Hashemifar et al., 2018) with two identical CNN sub-networks that share the same parameters for a given pair of protein profiles . Each sub-network produces a sequence embedding of a single protein profile that are subsequently concatenated. While each single CNN module consists of a convolution layer and a pooling layer, we leverage four connected convolutional modules to capture the patterns in an input sequence profile.
Specifically, we use one-dimensional (1-D) convolution in each convolution layer. For the first convolution layer, we input a array for each protein where the array can be regarded as a vector of length 2000 with 20 channels (i.e. 20 features in each position). Therefore, for each convolution layer, we consider a input array where is the length of the input vector, and is the number of channels. The convolution layer applies a sliding window of length (i.e. the size of filters/kernels) to convert into aarray where represents the number of filters/kernels. denotes the score of filter/kernel , , that corresponds to position of array . Moreover, the convolution layer applies a parameter-sharing kernel, a array where is the coefficient of pattern at position and feature . As a consequence, we define as
Furthermore, the pooling layer immediately follows the convolution layer and further transforms to a ( array where is the size of pooling window, and t is the stride of the sliding window. Array is calculated as the maximum of all positions over each feature where ,
2.1.3. Prediction module
The prediction module concatenates a pair of protein sequence embedding vectors into a sequence pair vector as the input of fully connected layers in an MLP and computes the probability that two proteins interact. The MLP contains three dense layers with leakyReLU where cross-entropy loss is optimized for the binary classification objective defined as
where is numerical class label of the protein pair. The output of the MLP for the protein pair is a probability vector , whose dimensionality is the number of classes . s is normalized by a softmax function, where the normalized probability value for the class is defined as .
2.1.4. Implementation details
As for pre-acquired sequence profile construction, we consider a fixed sequence length of 2000. As for the construction of our learning approach, we use four 1-D convolutional modules, where the input sizes (i.e. the number of channels) of these four convolution layers for each protein sequence are 20, 64, 128 and 256, respectively. As for the size of the CNN models, the numbers of filters in the four layers are 64, 128, 256 and 512, respectively. The convolution kernel size (i.e. the length of the convolution sliding window) is set to 3. Both the length and the stride of the pooling window are set to 2 for three max-pooling layers while the final pooling layer adopts global max-pooling. Each convolution layer is followed by a pooling layer. The detailed network architecture of the deep learning model is provided in Supplementary Figure S1. To optimize cross-entropy loss we use AMSGrad (Reddi et al., 2018), and set the learning rate to 0.0001. The batch size is set to 64, while the number of epochs is 100. The fully connected layers contain three dense layers with input sizes 1024, 512 and 256, respectively, and output a two-dimensional vector with the last softmax layer. We implemented the proposed architecture with Keras (https://keras.io/) using the GPU configuration. The parameter selection and optimization are detailed in Supplementary Table S1.
2.2 Dataset construction and partition
We collected experimentally verified human–virus PPI data from five public databases, including HPIDB (Ammari et al., 2016), VirHostNet (Guirimand et al., 2015), VirusMentha (Calderone et al., 2015), PHISTO (Durmuş Tekir et al., 2013) and PDB (Altunkaya et al., 2017). To obtain high-quality PPIs, we removed interactions from large-scale MS experiments that were detected only once, redundant interactions, non-physical interactions and interactions between proteins without available PSSM features. By performing the above filtering steps, we obtained 31 381 interactions in all viruses, capturing 9880 interactions in HIV, 5966 in Herpes, 5099 in Papilloma, 3044 in Influenza, 1300 in Hepatitis, 927 in Dengue and 709 in Zika (Supplementary Table S2). We took these pre-processed experimentally verified interactions as positive sample sets. As for human–SARS-CoV-2 PPIs, we collected experimental interactions from two high-throughput MS experiments (Gordon et al., 2020; Li et al., 2021), amounting to 568 human–SARS-CoV-2 PPIs as positive samples.
To compile negative samples, we first randomly selected human–virus protein pairs from human proteins in Swiss-Prot (The UniProt Consortium, 2017) and viral proteins in positive samples except those already reported to interact. Utilizing the ‘Dissimilarity-Based Negative Sampling’ method (Eid et al., 2016; Yang et al., 2020, 2021) we further sampled negative samples that were 10 times larger than the positive counterparts in each human–virus system (Supplementary Table S2). As the key strategy of ‘Dissimilarity-Based Negative Sampling’ we stipulate that if the sampled sequence of viral protein B is similar to another viral protein A (sequence identity > 0.3), that is found to interact with human protein C (i.e. A-C is a positive sample), then the pair of the viral protein B and the human protein C is not selected as a negative sample. As for the size of training sets, we surmise that positive interaction examples are far less abundant than negative examples, prompting us to use an unbalanced ratio of positives/negatives (i.e. 1:10) to capture this disparity. Furthermore, we mainly relied on 5-fold cross-validation for evaluating the predictive models in all experimental settings. To this end, all the benchmark datasets were equally divided into five non-overlapping subgroups and each subgroup owns one chance to train/test the model which can provide an unbiased evaluation. Note that the dataset partition was fixed for all experiment settings, providing a reliable basis for an unbiased comparison of different models.
2.3 Two types of transfer learning methods
To further improve the performance of our DNN especially when dealing with smaller datasets, we propose two transfer learning methods that keep the parameters of the CNN layers constant (i.e. ‘frozen’) or allow their fine-tuning in the early layers (i.e. ‘fine-tuning’). In more detail, we used the proposed DNN architecture to train the models based on a given source set of human–virus interactions to obtain pre-trained parameters in the CNN layers that learn the representation of the protein sequences. In subsequent transfer learning steps, we kept the parameters of these CNN layers constant (i.e. ‘frozen’) and only trained parameters of the fully connected layers of the MLP to predict interactions in a target human–virus interaction set. As an alternative, our ‘fine-tuning’ approach trained parameters of the fully connected layers of the MLP and retrains the parameters of CNN layers that we obtained from the initial training step and changed such parameters by learning interactions in a target set of human–virus interactions.
3 Results and discussion
3.1 Performance of the proposed deep learning method
Based on our deep learning architecture, we assessed the predicted interactions between proteins of various viruses and the human host through 5-fold cross-validation. While Table 1 indicates generally high prediction performance of our deep learning approach, we observed that small sizes of training datasets such as Dengue, Zika and SARS-CoV-2 decreased prediction performance. As RF outperforms other machine learning methods when applied to binary classification problems (Chen et al., 2019; Wu et al., 2009; Yang et al., 2020), we compared the performance of our deep learning approaches (i.e. PSSM+CNN+MLP) to this representative state-of-the-art classifier. Moreover, we considered three widely used encoding schemes (i.e. LD, CT and AC) for feature representations as input to the RF classifier (see Supplementary Methods and Supplementary Table S1 for method details). By comparing AUPRC (area under the precision–recall curve) values, we observed that our deep learning method generally outperformed other encoding schemes-based RF classifiers especially when applied to comparatively large datasets (Table 2).
Table 1.
Performance of our deep learning architecture (PSSM+CNN+MLP) using 5-fold cross-validationa
| Human–virus PPI dataset | Accuracy (%) | Precision (%) | Sensitivity (%) | Specificity (%) | F1-score (%) | AUPRC |
|---|---|---|---|---|---|---|
| Human–HIV | 98.65 | 95.16 | 89.72 | 99.54 | 92.36 | 0.974 |
| Human–Herpes | 95.26 | 77.11 | 68.10 | 97.98 | 72.33 | 0.768 |
| Human–Papilloma | 95.98 | 82.70 | 70.48 | 98.53 | 76.10 | 0.818 |
| Human–Influenza | 96.10 | 84.22 | 70.30 | 98.68 | 76.63 | 0.834 |
| Human–Hepatitis | 93.43 | 69.27 | 49.77 | 97.79 | 57.92 | 0.636 |
| Human–Dengue | 93.29 | 70.02 | 45.85 | 98.04 | 55.41 | 0.605 |
| Human–Zika | 95.41 | 85.17 | 59.94 | 98.96 | 70.36 | 0.746 |
| Human–SARS-CoV-2 | 90.64 | 45.81 | 16.37 | 98.06 | 24.12 | 0.329 |
The definitions of performance assessment metrics are available in Supplementary Materials.
Table 2.
Performance comparison of our deep learning architecture and three sequence encoding schemes-based RF methods using 5-fold cross-validation
| AUPRC |
||||
|---|---|---|---|---|
| Human–virus PPI dataset | Our method | LD+RF | CT+RF | AC+RF |
| Human–HIV | 0.974 | 0.972 | 0.970 | 0.972 |
| Human–Herpes | 0.768 | 0.741 | 0.737 | 0.699 |
| Human–Papilloma | 0.818 | 0.740 | 0.724 | 0.656 |
| Human–Influenza | 0.834 | 0.813 | 0.795 | 0.713 |
| Human–Hepatitis | 0.636 | 0.571 | 0.580 | 0.537 |
| Human–Dengue | 0.605 | 0.526 | 0.505 | 0.456 |
| Human–Zika | 0.746 | 0.720 | 0.718 | 0.698 |
| Human–SARS-CoV-2 | 0.329 | 0.371 | 0.350 | 0.314 |
To further assess the proposed sequence profile-based encoding scheme, we compared the performance of our deep learning architecture based on PSSM to a different word embedding technique called word2vec+CT one hot. Briefly, word2vec+CT one-hot is the concatenation of two pre-trained amino acid embeddings [i.e. the word2vec encoding method (Chen et al., 2019; Le and Mikolov, 2014) and the CT one-hot encoding scheme of the corresponding sequence], where each protein was represented by a dimensional array. Training our CNN+MLP approach with word2vec+CT one hot encodings of the corresponding protein sequences, we observed that the representation of sequences through PSSM in our approach provided better prediction performance especially in relatively small datasets such as Dengue, Zika and SARS-CoV-2 (Supplementary Table S3).
3.2 Comparison with other existing human–virus PPI prediction methods
We further compared the performance of our method to four existing human–virus PPI prediction approaches [i.e. our previous RF-based method (Yang et al., 2020), DeepViral (Liu-Wei et al., 2021), the method of (Alguwaizani et al., 2018) and the DeNovo method (Eid et al., 2016)]. Allowing a fair comparison, we first constructed the PSSMs of the protein sequences in DeNovo’s PPI dataset and used their training set to retrain our Siamese-based CNN model. Finally, we assessed the performance of our reconstructed deep learning model on the test set provided in Eid et al. (2016) including 425 positive and 425 negative samples. Furthermore, we used DeNovo’s PPI interaction dataset to assess the prediction performance of our RF-based method, DeepViral and Alguwaizani et al.’s method utilizing their corresponding performance metrics. As shown in Supplementary Table S4, our deep learning and previously published RF-based method clearly outperformed or were comparable with other approaches, emphasizing that our deep learning method is fully competitive compared to the newly developed method DeepViral using sequence alone or together with phenotype and functional features (Supplementary Table S4).
3.3 Cross-viral tests and transfer learning
To explore potential factors that affect prediction performance in a cross-viral setting, we trained our deep learning model on four subgroups of one human–virus PPI dataset, predicted protein interactions in one subgroup of a different human–virus system and repeated these steps five times, implementing a 5-fold cross-validation of a naïve cross-viral test. As expected, we observed that the prediction performance dropped considerably compared to training and testing in the same human–virus system (Fig. 2a). To allow reliable cross-viral predictions of PPIs, we introduced two transfer learning methods (i.e. ‘frozen’ and ‘fine-tuning’). To comprehensively test our transfer learning approaches, we considered each combination of human–virus PPI sets as source and target domains. Similar to the previous naïve 5-fold cross-validation setting, we first trained the parameters of CNN layers on four randomly sampled subgroups of a source domain. Subsequently, we transferred all parameters of CNN layers to initialize a new model (‘frozen’ or ‘fine tuning’) with randomly initialized MLP layers to train on the corresponding four subgroups of a target domain and test the predictive model on the remaining subgroup in the target domain. Figure 2b indicates that a relatively rigid transfer learning methodology by keeping the parameters of the CNN module untouched (i.e. ‘frozen’) and training the MLP layers strongly outperformed the naïve baseline performance as shown in Figure 2a. In turn, fine-tuning parameters in the CNN module and training the MLP layers as well with a given target human–virus domain allowed for another increase in performance (Fig. 2c). As for individual pairs of human–virus domains, we also observed that independently from the training domain the ‘frozen’ transfer methodology worked better compared to the ‘fine-tuning’ approach when the target domain dataset was extremely small (i.e. human–SARS-CoV-2). In turn, performance of the ‘frozen’ transfer learning approach dropped compared to ‘fine-tuning’ when the target human–virus domain datasets of PPIs were larger such as human–Hepatitis, human–Dengue and human–Zika.
Fig. 2.

(a) Investigating prediction performance we trained our deep learning model on one human–virus PPI dataset (rows) and predicted protein interactions in a different human–virus system (columns). In (b) and (c) we show the corresponding performance of the ‘frozen’ and the ‘fine-tuning’ transfer learning methods
3.4 Prediction and analysis of human–SARS-CoV-2 PPIs based on transfer learning models
To predict a genome-wide map of potential PPIs between the human host and SARS-CoV-2, we first trained parameters of the CNN layers of our deep learning model utilizing all human–virus protein interactions. Subsequently, we used our two transfer learning approaches to train our set of interactions between proteins of human and SARS-CoV-2. Applying 5-fold cross-validations, we observed that the AUPRC of 0.483 with the ‘frozen’ transfer learning approach outperformed the corresponding value of 0.435 when we used the ‘fine-tuning’ method. In addition, training on all source human–virus PPI datasets showed best performance compared to separately training with virus-specific source PPI datasets (data not shown). Therefore, we used five ‘frozen’ models in a 5-fold cross-validation setting based on human–all virus source dataset to predict human–SARS-CoV-2 PPIs and averaged the scores of the five models as the prediction result. At a false positive rate control of 0.01, we identified 946 high-confidence interactions between 21 SARS-CoV-2 proteins and 551 human proteins (Supplementary Table S5).
By analyzing the 551 targeted human proteins we found several network patterns that are in line with previous observations (Supplementary Fig. S2a–d). In particular, the power-law distribution of the number of viral proteins that interact with a given human protein suggests that a majority of human proteins are targeted by one viral protein, while a minority interacts with many viral proteins (Wuchty et al., 2010). Collecting 365 284 human PPIs from the HIPPIE database (Alanis-Lobato et al., 2017) we observed that targeted human proteins are enriched in bins of increasing degree, a result that is consistent with previous findings as well (Dyer et al., 2008; Wuchty et al., 2010). Considering 2916 human protein complexes from the CORUM database (Giurgiu et al., 2019) we found that viral targets are enriched in sets of proteins that participate in an increasing number of protein complexes (Wuchty et al., 2010). To illustrate viral similarities, we compared the experimentally known human–SARS-CoV-2 interactome and our predicted interactome with their counterparts of seven other viruses. While our predictions show similar overlaps of viral targets with the experimentally obtained interactomes, we further found that Dengue and Influenza had the most similar interacting partners in both predictions and experimentally known interactions (P-value 0.05, hypergeometric test). Notably, the association with Influenza is of particular interest as this virus also induces respiratory disease (i.e. pneumonia).
3.5 Comparative analysis of known and our predicted human–SARS-CoV-2 PPIs
Comparing our predicted and experimentally obtained human–SARS-CoV-2 PPIs, we found considerable overlaps. In particular, 298 out of 946 predicted PPIs were identified through previous experimental efforts that amount to 52.5% of known interactions in SARS-CoV-2, while 648 were specifically identified through our deep learning approach (Fig. 3a, Supplementary Table S5), indicating the reliability and specificity of our model for the identification of novel interactions. Moreover, we performed functional and pathway enrichment for experimentally known and predicted viral targets, respectively. Considering hypergeometric tests (Bonferroni corrected P-value 0.01), we observed a relatively large number of shared GO enrichment terms/KEGG enrichment pathways of experimentally confirmed targets and predicted targets, further indicating the reliability of our predictions (Fig. 3b, Supplementary Tables S6 and S7). In more detail, we found that GO BP enrichment of experimental and predicted viral targets both point to the involvement of viral targets in protein transport, protein import and mRNA export from the nucleus (Fig. 3c). Notably, our predictions augment such functions, indicating that the virus may also interfere with nuclear pore organization and assembly as well as protein export from the nucleus.
Fig. 3.
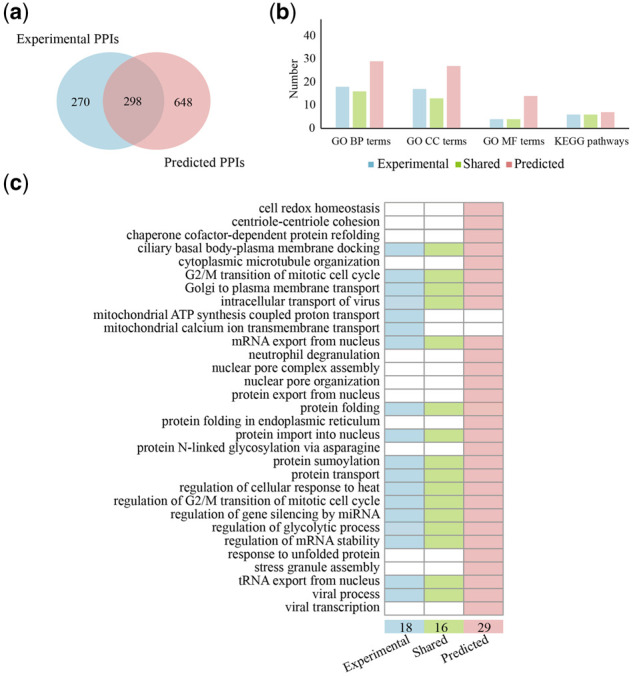
(a) Overlap of experimentally observed and predicted interactions between proteins of the human host and SARS-CoV-2. (b) In a quantitative functional analysis of targeted human host proteins, we considered the enrichment of GO terms and KEGG pathways through a hypergeometric test (Bonferroni corrected P-value 0.01). We found that a relatively large share of functional terms in groups of host proteins appearing in the experimentally known PPIs and predictions. (c) In more detail, we observed that enriched GO BP terms in host proteins appearing in the experimental and predicted PPIs were functionally similar
3.6 Modular analysis of human–SARS-CoV-2 PPI network
To further explore potential functional modules that can reveal SARS-CoV-2 biology, we combined our predicted 946 human–SARS-CoV-2 PPIs with known human-specific PPIs as of the HIPPIE database (Alanis-Lobato et al., 2017) (Fig. 4a). Specifically, we identified nine topological modules based on connectivity between human proteins (Fig. 4a and b; Supplementary Methods), utilizing the MCODE algorithm (Bader and Hogue, 2003). Investigating the enrichment of GO BP terms and KEGG pathways through hypergeometric tests (Bonferroni adjusted P-value 0.05; Supplementary Methods), we observed that these modules largely revolved around ribosome biogenesis, retrograde protein transport, elastic fiber assembly, mitochondrial translation, protein processing in endoplasmic reticulum, stress granule regulation, protein folding in endoplasmic reticulum, centrosome and gene splicing (Fig. 4b, Supplementary Table S8).
Fig. 4.

(a) Combining predictions from the transfer learning approach and known human PPIs we determined connectivity-based modules that were subjected to functional interpretation. (b) Human–SARS-CoV-2 PPI network with enriched GO BP terms and KEGG pathways for each topological module. (c) SARS-CoV-2 targets a module that involves the centrosome, cell cycle and interferon pathway. Through multiple sequence alignment, we observed a potential conserved binding motif shared by nsp13 of SARS-CoV-2 and proteins of other viral pathogens, suggesting that SARS-CoV-2 nsp13 protein may interfere with the regulation processes of IFN to support antiviral innate immune response
Considering a module that was enriched with centrosome functions through interactions with nsp13 and cell cycle functions through interactions with orf6, we also found that this module harbors human genes that allow SARS-CoV-2 to interact with innate immune pathways which is consistent with previous findings (Gordon et al., 2020). As shown in the module, the interferon (IFN) pathway is targeted through TBK1 by nsp8, nsp13 and orf6, a serine/threonine-protein kinase that plays an important role in the induction of the antiviral IFN response to foreign agents such as viruses. A number of viral proteins bind to TBK1 and regulate their kinase activity to reduce TBK1-mediated secretion of IFN and induction of an antiviral state, such as Borna disease virus (BDV) P protein (Unterstab et al., 2005), Human herpesvirus 1 (HSV-1) ICTP34.5 protein (Manivanh et al., 2017) and Ebola virus (EBOV) VP35 protein (Prins et al., 2009). BDV P protein itself is phosphorylated by TBK1, suggesting that P functions as a viral decoy substrate that prevents activation of cellular target proteins of TBK1. Furthermore, residues from 87 to 106 in HSV-1 ICTP34.5 protein interact with TBK1 to modulate type I IFN signaling (Manivanh et al., 2017; Verpooten et al., 2009). Considering the multiple sequence alignment of these viral proteins and nsp13 of SARS-CoV and SARS-CoV-2 we found a potential conserved binding motif (Fig. 4c), corroborating our assumption that SARS-CoV-2 nsp13 protein may also interfere with the regulation processes of IFN that support antiviral innate immune response.
4 Conclusion
We designed a Siamese-based multi-scale CNN architecture by using PSSMs to represent sequences of interacting proteins, allowing us to predict human–virus PPIs with an MLP approach. We observed that our model outperformed previous state-of-the-art prediction methods as well as combinations of other machine learning and pre-trained feature embeddings. Moreover, we introduced two transfer learning methods (i.e. ‘frozen’ type and ‘fine-tuning’ type), which allowed us to train on a source human–virus domain and retrain the layers of CNN with data of a target domain. Notably, our methods increased the cross-viral prediction performance dramatically, compared to the naïve baseline model. Finally, we used our ‘frozen’ transfer learning method to predict human–SARS-CoV-2 PPIs and performed in-depth network analysis based on the identified interactions. Our transfer learning model resembled closely the functions and characteristics of experimentally obtained interactions and indicated novel functions that the virus potentially targets. Taken together, our transfer learning method can be effectively applied to predict human–virus PPIs in a cross-viral setting and the study of viral infection mechanism.
Supplementary Material
Acknowledgement
The authors thank the support of the high-performance computing platform of the State Key Laboratory of Agrobiotechnology.
Funding
This work was supported by the National Key Research and Development Program of China [2017YFC1200205 and 2017YFD0500404].
Conflict of Interest: none declared.
References
- Ahmed I.et al. (2018) Prediction of human-Bacillus anthracis protein–protein interactions using multi-layer neural network. Bioinformatics, 34, 4159–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanis-Lobato G.et al. (2017) HIPPIE v2.0: enhancing meaningfulness and reliability of protein–protein interaction networks. Nucleic Acids Res., 45, D408–D414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alguwaizani S.et al. (2018) Predicting interactions between virus and host proteins using repeat patterns and composition of amino acids. J. Healthc. Eng., 2018, 1391265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altunkaya A.et al. (2017) The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res., 45, D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammari M.G.et al. (2016) HPIDB 2.0: a curated database for host-pathogen interactions. Database, 2016, baw103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G.D., Hogue C.W.V. (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A.et al. (2015) VirusMentha: a new resource for virus-host protein interactions. Nucleic Acids Res., 43, D588–D592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.et al. (2018) Unsupervised transfer learning via multi-scale convolutional sparse coding for biomedical applications. IEEE Trans. Pattern Anal. Mach. Intell., 40, 1182–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.et al. (2019) Multifaceted protein–protein interaction prediction based on Siamese residual RCNN. Bioinformatics, 35, i305–i314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G.et al. (2012) Prediction of protein–protein interactions between viruses and human by an SVM model. BMC Bioinformatics, 13, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M.N.et al. (2008) Optimizing amino acid groupings for GPCR classification. Bioinformatics, 24, 1980–1986. [DOI] [PubMed] [Google Scholar]
- Du X.et al. (2017) DeepPPI: boosting prediction of protein–protein interactions with deep neural networks. J. Chem. Inf. Model., 57, 1499–1510. [DOI] [PubMed] [Google Scholar]
- Durmuş Tekir S.et al. (2013) PHISTO: pathogen–host interaction search tool. Bioinformatics, 29, 1357–1358. [DOI] [PubMed] [Google Scholar]
- Dyer M.D.et al. (2011) Supervised learning and prediction of physical interactions between human and HIV proteins. Infect. Genet. Evol., 11, 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer M.D.et al. (2008) The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog., 4, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid F.et al. (2016) DeNovo: virus-host sequence-based protein–protein interaction prediction. Bioinformatics, 32, 1144–1150. [DOI] [PubMed] [Google Scholar]
- Emamjomeh A.et al. (2014) Predicting protein–protein interactions between human and hepatitis C virus via an ensemble learning method. Mol. Biosyst., 10, 3147–3154. [DOI] [PubMed] [Google Scholar]
- Giurgiu M.et al. (2019) CORUM: the comprehensive resource of mammalian protein complexes – 2019. Nucleic Acids Res., 47, D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E.et al. (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature, 583, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirimand T.et al. (2015) VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res., 43, D583–D587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.et al. (2008) Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res., 36, 3025–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamp T., Rost B. (2015) Evolutionary profiles improve protein–protein interaction prediction from sequence. Bioinformatics, 31, 1945–1950. [DOI] [PubMed] [Google Scholar]
- Hashemifar S.et al. (2018) Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics, 34, i802–i810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Q., Mikolov T. (2014) Distributed representations of sentences and documents. Proc. Int. Conf. Mach. Learn., 14, 1188–1196. [Google Scholar]
- Lee H.et al. (2009) Convolutional deep belief networks for scalable unsupervised learning of hierarchical representations. Proc. 26th Int. Conf. Mach. Learn., 54, 609–616. [Google Scholar]
- Li J.et al. (2021) Virus-host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med, 2, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X.et al. (2021) Current status and future perspectives of computational studies on human-virus protein–protein interactions. Brief. Bioinform., doi:10.1093/bib/bbab029. [DOI] [PubMed] [Google Scholar]
- Lian X.et al. (2020) Prediction and analysis of human-herpes simplex virus type 1 protein–protein interactions by integrating multiple methods. Quant. Biol., 8, 312–324. [Google Scholar]
- Liu-Wei W.et al. (2021) DeepViral: prediction of novel virus–host interactions from protein sequences and infectious disease phenotypes. Bioinformatics, doi: 10.1093/bioinformatics/btab147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manivanh R.et al. (2017) Role of herpes simplex virus 1 γ34.5 in the regulation of IRF3 signaling. J. Virol., 91, e01156-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min X.et al. (2017) Chromatin accessibility prediction via convolutional long short-term memory networks with k-mer embedding. Bioinformatics, 33, i92–i101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins K.C.et al. (2009) Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKε and TBK-1. J. Virol., 83, 3069–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi S.J.et al. (2018) On the convergence of Adam and Beyond. In: Int. Conf. Learn. Represent. OpenReview, Amherst, MA, pp. 1–23.
- Shah P.S.et al. (2018) Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell, 175, 1931–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L.et al. (2015) Transfer learning for visual categorization: a survey. IEEE Trans. Neural Networks Learn. Syst., 26, 1019–1034. [DOI] [PubMed] [Google Scholar]
- Shen J.et al. (2007) Predicting protein–protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA, 104, 4337–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T.et al. (2017) Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinformatics, 18, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek B.E.et al. (2015) UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics, 31, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taroni J.N.et al. (2019) MultiPLIER: a transfer learning framework for transcriptomics reveals systemic features of rare disease. Cell Syst., 8, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res., 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterstab G.et al. (2005) Viral targeting of the interferon-β-inducing Traf family member-associated NF-κB activator (TANK)-binding kinase-1. Proc. Natl. Acad. Sci. USA, 102, 13640–13645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpooten D.et al. (2009) Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J. Biol. Chem., 284, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.et al. (2009) Prediction of DNA-binding residues in proteins from amino acid sequences using a random forest model with a hybrid feature. Bioinformatics, 25, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S.et al. (2010) Viral organization of human proteins. PLoS One, 5, e11796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.et al. (2010) Prediction of protein–protein interactions from protein sequence using local descriptors. Protein Pept. Lett., 17, 1085–1090. [DOI] [PubMed] [Google Scholar]
- Yang X.et al. (2021) HVIDB: a comprehensive database for human-virus protein–protein interactions. Brief. Bioinform., 22, 832–844. [DOI] [PubMed] [Google Scholar]
- Yang X.et al. (2020) Prediction of human-virus protein–protein interactions through a sequence embedding-based machine learning method. Comput. Struct. Biotechnol. J., 18, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z.-H.et al. (2013) Prediction of protein–protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinformatics, 14, S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.et al. (2016) A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res., 44, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.