

ABSTRACT
Microbial communities are frequently numerically dominated by just a few species. Often, the long “tail” of the rank-abundance plots of microbial communities constitutes the so-called “rare biosphere,” microorganisms that are highly diverse but are typically found in low abundance in these communities. Their presence in microbial communities has only recently become apparent with advances in high-throughput sequencing technologies. Despite their low numbers, they are thought to play important roles in their communities and may function as potential members to keep the communities intact and resilient. Their phylogenetic diversity also means that they are important subjects for better understanding the interplay between microbial diversity and evolution. I propose that more efforts should be put into characterizing these poorly understood and mostly unknown microbial lineages that hold vast potentials for our understanding of microbial diversity, ecology, and evolution of life on this planet.
KEYWORDS: rare biosphere, microbial, diversity, ecology, evolution, microbial ecology, microbial evolution
COMMENTARY
The rare microbial biosphere refers to the microorganisms that are genetically diverse but are typically found in low abundance in various microbial communities (1). Despite their low abundance, they frequently constitute a phylogenetically diverse pool of microbes from all three domains of life. The rare biosphere that persists in the environment may act as seed banks of microbial diversity, allowing them to thrive when conditions are right (1, 2). Conditionally rare taxa may remain in low numbers until optimal conditions for them arise and they increase in numbers (2). Critical reviews of the rare microbial biosphere and their importance have been published previously (3, 4), and this commentary is meant to further highlight the importance of these microbes to advance the fields of microbial diversity, ecology, and evolution.
A quick inspection of the taxonomic classification of the small subunit rRNA (SSU rRNA) also known as the 16S rRNA of nonredundant Archaea, Bacteria, and Eukarya from the Silva database (5) reveals a disparity in taxonomic representation. Most of the bacterial 16S sequences are from just a few phyla such as Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes (Fig. 1) (Note that there are some differences between Silva and NCBI taxonomies.) Similarly, archaeal 16S rRNA sequences are mostly from Crenarchaeota and Euryarchaeota (Fig. 1). Mitochondrial and plastid sequences from eukaryotes are also unevenly distributed. As seen in Fig. 1, the long “tails” of these rank-abundance plots reveal a vast diversity of low-abundant organisms from various habitats. The Silva database typically contains longer, near-full-length 16S rRNA sequences obtained by Sanger sequencing. More taxonomically diverse lineages are likely hidden in microbial community surveys using short-read high-throughput Illumina sequencing, but they are likely discarded as noise (4).
FIG 1.
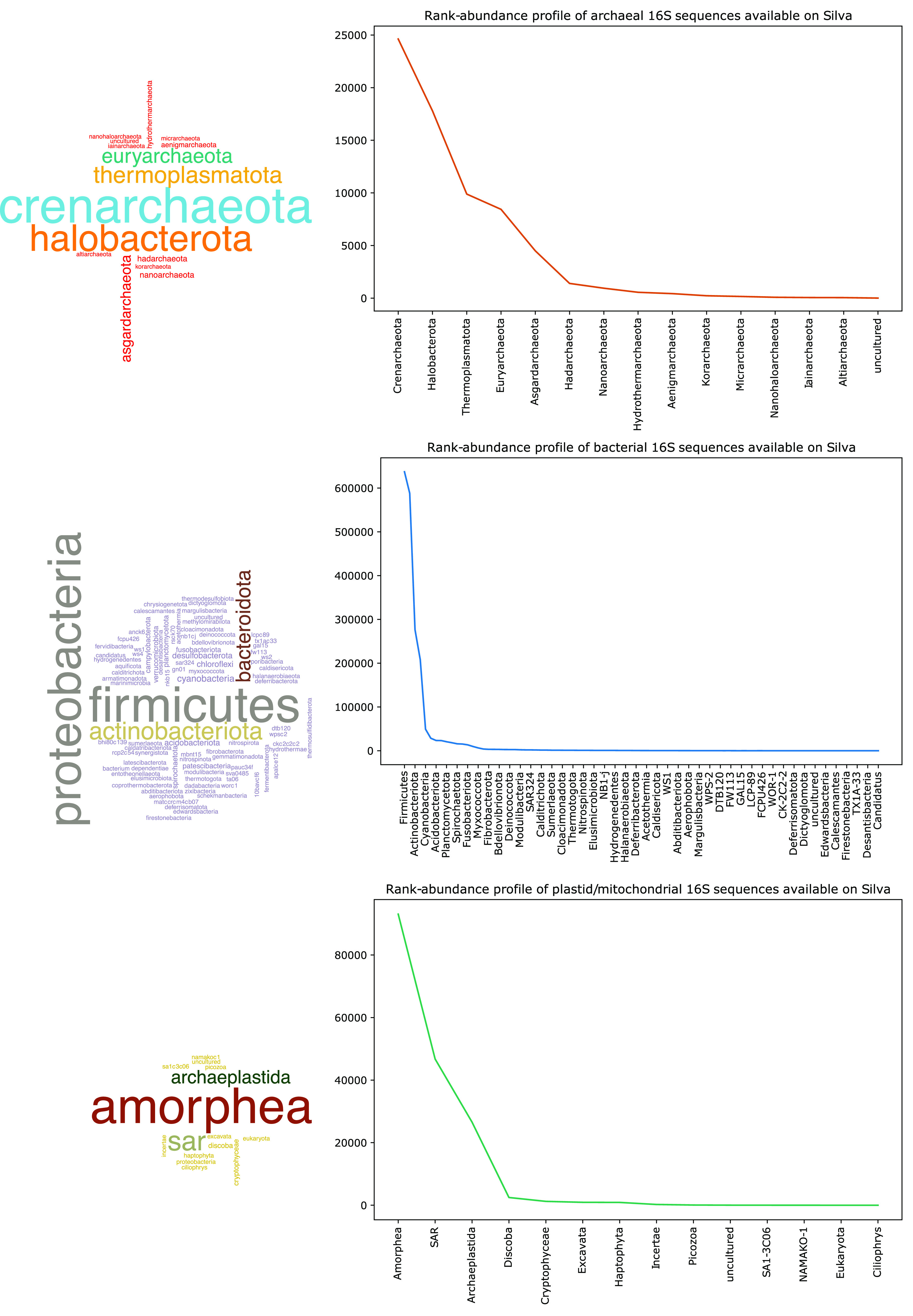
Word cloud and rank-abundance plots of archaeal, bacterial, and eukaryal phyla identified in the Silva database release 138.1. Word clouds were created from taxonomic classification of the nonredundant 16S rRNA sequences obtained from the Silva database. Note that only one out of every two bacterial phyla is shown in the figure.
Figure 2 illustrates rank-abundance and word cloud plots of taxonomic classification of a set of curated archaeal and bacterial genomes from the Genome Taxonomy Database (GTDB) (6). As seen in this figure, a vast majority of the genomes are from just a small number of archaeal and bacterial phyla: Halobacteriota, Thermoproteota, Thermoplasmatota, Methanobacteriota, Proteobacteria, etc.
FIG 2.
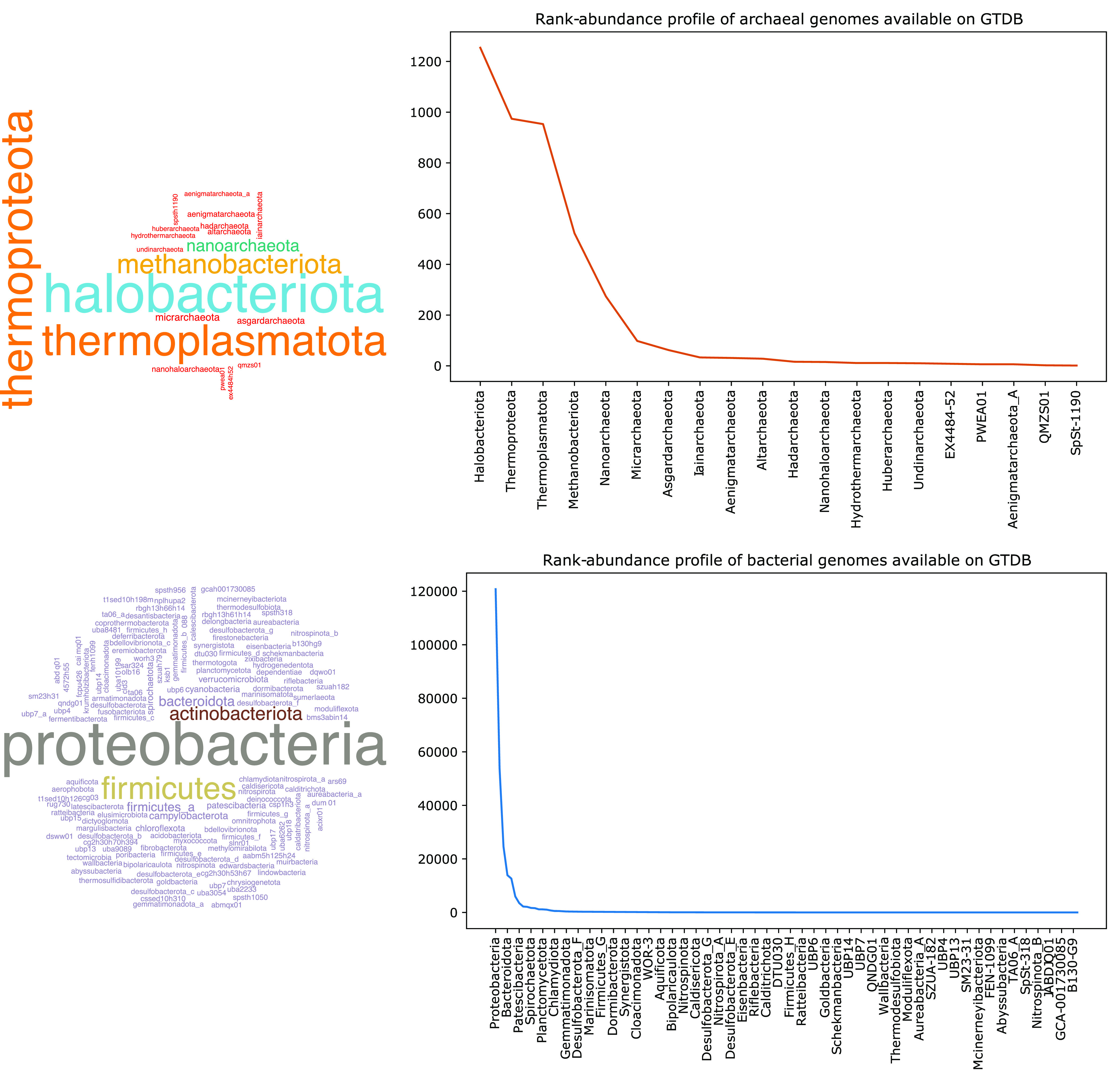
Word cloud and rank-abundance plots of archaeal and bacterial phyla identified in the Genome Taxonomy Database (GTDB). Word clouds were created from GTDB taxonomic classification of curated genomes used in the database. Note that only one out of every three bacterial phyla is shown in the figure.
A FEW EXAMPLES OF RARE TAXA AND THEIR IMPORTANCE
Here, I will highlight some of the microbial lineages that I consider to belong to the rare biosphere and are important for a number of reasons.
Odinarchaeota are one of the few thermophilic members of the Asgard archaea, and they have thus far been recovered only from hot springs in two very remote locations: Yellowstone National Park in the United States and Taupo Volcanic Zone in New Zealand (7). Only two metagenome-assembled genomes have been constructed so far in published peer-reviewed studies. Despite being geographically separated by thousands of miles, the first-described metagenome-assembled genomes (MAGs) of Odinarchaeota from the United States and New Zealand encode bona fide copies of tubulin and have the smallest genome sizes among the Asgard archaea (7). They are present in only a small fraction of their respective communities based on 16S rRNA gene abundance estimates and metagenomic read recovery (7). More genomes of the Odinarchaeota are needed to better understand the Asgard archaeal evolution and to answer important questions on the origins of eukaryotes.
Aigarchaeota represent the letter A of the TACK superphylum (8), and “Candidatus Caldiarchaeum subterraneum” at one time was the sole member of this enigmatic archaeal phylum. It was first identified in a deep subsurface gold mine in South Africa but subsequently found in several geothermal habitats through metagenomics (9). These archaea are present in very low abundance in hot spring sediments and geothermal habitats. Similar to the Aigarchaeota, Korarchaeota represent the letter K of the TACK superphylum, and “Candidatus Korarchaeum cryptofilum” used to represent the sole member of the phylum for more than a decade (10). Again, thanks to metagenomics, additional members of the Korarchaeota are being discovered (11). Due to the position it occupies within the archaeal tree of life, thermophilic lifestyle, and lack of representation, the use of the sole member of this archaeal group tends to introduce phylogenetic artifacts that may result in wrong topologies of phylogenomic trees constructed (12). Most of the problems stem from codon biases present in the thermophilic members of these archaeal groups (13). Therefore, it is very important to obtain additional members of these enigmatic archaeal groups to improve taxonomic representation and to facilitate construction of more accurate phylogenetic trees.
Cyanobacteria of the genus Gloeobacter are important and may represent one of the conditionally rare taxa. First of all, they are depauperate, and only two species have been identified: one from a limestone rock in Switzerland (14) and another from a volcanic cave in Hawaii (15). Interestingly, they are numerically dominant in biofilm communities found near steam vents of Hawaii (unpublished data) and may be considered conditionally rare microbes. It has been suggested that they are common rock-dwelling cyanobacteria (16), but very few genomes of Gloeobacter and related deeply branching cyanobacteria currently exist in databases. There are only two cultivated species of Gloeobacter and a cultivated species of a sister group of Gloeobacter known as Anthocerotibacter isolated from Panama (17). A few metagenome-assembled genomes of related species were identified from Lake Vanda in Antarctica (18). These cyanobacteria occupy the deepest nodes within the cyanobacterial tree of life and are thought to be descendants of the cyanobacteria that first innovated oxygenic photosynthesis and are key to understanding how oxygenic photosynthesis evolved.
Another important lineage is a group of cyanobacteria known as Vampirovibrionia (formerly Melainabacteria) that lack the essential genes needed to perform photosynthesis (19, 20). Only a few representative genomes have been obtained from a few locations, and none of them have been isolated or cultivated yet. These rare taxa also are important to understanding the origin and evolution of oxygenic photosynthesis. Besides these examples, additional phylum-level novel lineages exist, such as WPS-2/Eremiobacterota, which may be important to understand the evolution of anoxygenic phototrophy (21), and GAL15 and Fervidibacteria, which are poorly known due to lack of representative genomes in databases but shown to be metabolically active in hot springs (22).
Here, I note an example of why increased recovery of the genomes of the rare biosphere is important. Previously known as the SAGMEG (South Africa Gold Mine Euryarchaeotic Group) (23) and later classified as a novel class, Hadesarchaea, based on metagenomic information (24), these archaea have been reclassified into a new phylum of their own, Hadarchaeota, due to increased recovery of their genomes from environmental samples (25). Phylogenomic trees constructed can only be as good as the taxa included in the tree inference, and lack of representation can mean a world of difference to the correct outcome of these studies. Therefore, an expanded genomic information of these rare taxa is very important to accurately construct phylogenetic or phylogenomic trees to understand microbial evolution.
HOW DO WE INCREASE THE TAXONOMIC DIVERSITY OF THE RARE BIOSPHERE?
As I have highlighted in Fig. 1 and 2, the vast majority of 16S rRNA sequences and draft or complete genomes of Archaea and Bacteria belong to just a few dominant phyla. These phyla are also typically overrepresented in most microbial communities, and it is not surprising that they make up the major part of these sequence repositories.
Therefore, it is imperative that we obtain more genomic information on the vastly underrepresented but phylogenetically diverse lineages from various habitats. I propose a few approaches to increase the recovery of the genomes of these rare taxa:
Targeted genomic and metagenomic sequencing and exploration of the rare biosphere.
Targeted enrichment and cultivation of the rare biosphere based on genomic information.
Fluorescent-activated cell sorting (FACS) or similar approaches to enrich and sequence the rare biosphere.
In situ or mesocosm experiments to understand their physiological roles in the environment.
Targeted metagenomic sequencing of environmental samples will be crucial to recover these rare taxa and to obtain more representative genomes from various habitats. From high-throughput Illumina amplicon sequences deposited to the Sequence Read Archive (SRA) database, one can identify rare microbial lineages and identify the samples or locations they came from. Most of these studies are 16S rRNA-based surveys and do not have accompanying metagenomic data. Contacting the original authors of these studies to obtain DNA or samples to perform targeted deep sequencing of these samples would be a good start to explore and recover genomic information of these rare taxa without indiscriminately sequencing everything in sight.
Enrichment and cultivation of microbes of interest are currently having a resurgence in microbial ecology and physiology studies. More concerted cultivation efforts combined with physiological experiments will be needed to characterize these poorly represented and understood microbes and their roles in various habitats. However, cultivation of previously uncultivated microbes is still a tremendous challenge, and more efforts will need to be put in to obtain pure cultures. Various approaches to cultivate previously uncultivated microbes have been reported, and they are promising methods to target isolation of the rare biosphere (26, 27). Single-cell genomics is one of the solutions to the problem with cultivating the rare biosphere, but it can be quite expensive and single-cell amplified genomes (SAGs) tend to be highly incomplete. Because we are targeting the rare biosphere that tends to be found in low abundance, chances of sorting them out of the pool of abundant but less interesting microbes are very limited.
One solution might be to use methods such as FACS to size-fractionate microbial cells and sequence minimetagenomes to exclude more abundant lineages in the metagenomic sequences. If one can flow-sort a microbial population by size and other properties, it might be possible to reduce the number of dominant taxa from the sequencing pool. There is still a chance that the dominant and the rare taxa may have similar cell sizes in certain communities, but if we couple it with fluorescent tags bound to cells of interest, the method might be feasible. Lastly, we should not forget that physiological experiments are important to understanding what the rare biosphere does in its natural environment. Polyphasic approaches using a combination of targeted sequencing, isolation of target species, and physiological experiments will help us understand more about the roles of the rare biosphere in microbial communities.
Data availability.
Data and code used to create Fig. 1 and 2 can be accessed at https://github.com/SawLabGW/mSystems_ECSC2021.
ACKNOWLEDGMENTS
I thank Michael Rappé and Jack Gilbert for giving me the opportunity to contribute to this special series.
This work was supported by startup funds provided by The George Washington University to J.H.W.S.
The views expressed in this article do not necessarily reflect the views of the journal or of ASM.
This article is part of a special series sponsored by Floré.
REFERENCES
- 1.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci USA 103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. 2014. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5:e01371-14. doi: 10.1128/mBio.01371-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pedrós-Alió C. 2012. The rare bacterial biosphere. Annu Rev Mar Sci 4:449–466. doi: 10.1146/annurev-marine-120710-100948. [DOI] [PubMed] [Google Scholar]
- 4.Lynch MDJ, Neufeld JD. 2015. Ecology and exploration of the rare biosphere. Nat Rev Microbiol 13:217–229. doi: 10.1038/nrmicro3400. [DOI] [PubMed] [Google Scholar]
- 5.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, Hugenholtz P. 2020. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol 38:1079–1086. doi: 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- 7.Zaremba-Niedzwiedzka K, Caceres EF, Saw JH, Bäckström D, Juzokaite L, Vancaester E, Seitz KW, Anantharaman K, Starnawski P, Kjeldsen KU, Stott MB, Nunoura T, Banfield JF, Schramm A, Baker BJ, Spang A, Ettema TJG. 2017. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541:353–358. doi: 10.1038/nature21031. [DOI] [PubMed] [Google Scholar]
- 8.Guy L, Ettema TJG. 2011. The archaeal “TACK” superphylum and the origin of eukaryotes. Trends Microbiol 19:580–587. doi: 10.1016/j.tim.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Nunoura T, Takaki Y, Kakuta J, Nishi S, Sugahara J, Kazama H, Chee G-J, Hattori M, Kanai A, Atomi H, Takai K, Takami H. 2011. Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res 39:3204–3223. doi: 10.1093/nar/gkq1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elkins JG, Podar M, Graham DE, Makarova KS, Wolf Y, Randau L, Hedlund BP, Brochier-Armanet C, Kunin V, Anderson I, Lapidus A, Goltsman E, Barry K, Koonin EV, Hugenholtz P, Kyrpides N, Wanner G, Richardson P, Keller M, Stetter KO. 2008. A korarchaeal genome reveals insights into the evolution of the Archaea. Proc Natl Acad Sci USA 105:8102–8107. doi: 10.1073/pnas.0801980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKay LJ, Dlakić M, Fields MW, Delmont TO, Eren AM, Jay ZJ, Klingelsmith KB, Rusch DB, Inskeep WP. 2019. Co-occurring genomic capacity for anaerobic methane and dissimilatory sulfur metabolisms discovered in the Korarchaeota. Nat Microbiol 4:614–622. doi: 10.1038/s41564-019-0362-4. [DOI] [PubMed] [Google Scholar]
- 12.Guy L, Saw JH, Ettema TJG. 2014. The archaeal legacy of eukaryotes: a phylogenomic perspective. Cold Spring Harb Perspect Biol 6:a016022. doi: 10.1101/cshperspect.a016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arella D, Dilucca M, Giansanti A. 2021. Codon usage bias and environmental adaptation in microbial organisms. Mol Genet Genomics 296:751–762. doi: 10.1007/s00438-021-01771-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rippka R, Waterbury J, Cohen-Bazire G. 1974. A cyanobacterium which lacks thylakoids. Arch Microbiol 100:419–436. doi: 10.1007/BF00446333. [DOI] [Google Scholar]
- 15.Saw JHW, Schatz M, Brown MV, Kunkel DD, Foster JS, Shick H, Christensen S, Hou S, Wan X, Donachie SP. 2013. Cultivation and complete genome sequencing of Gloeobacter kilaueensis sp. nov., from a lava cave in Kīlauea Caldera, Hawai’i. PLoS One 8:e76376. doi: 10.1371/journal.pone.0076376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mareš J, Hrouzek P, Kaňa R, Ventura S, Strunecký O, Komárek J. 2013. The primitive thylakoid-less cyanobacterium Gloeobacter is a common rock-dwelling organism. PLoS One 8:e66323. doi: 10.1371/journal.pone.0066323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahmatpour N, Hauser DA, Nelson JM, Chen PY, Villarreal AJ, Ho M-Y, Li F-W. 2021. A novel thylakoid-less isolate fills a billion-year gap in the evolution of Cyanobacteria. Curr Biol 31:2857–2867.e4. doi: 10.1016/j.cub.2021.04.042. [DOI] [PubMed] [Google Scholar]
- 18.Grettenberger CL, Sumner DY, Wall K, Brown CT, Eisen JA, Mackey TJ, Hawes I, Jospin G, Jungblut AD. 2020. A phylogenetically novel cyanobacterium most closely related to Gloeobacter. ISME J 14:2142–2152. doi: 10.1038/s41396-020-0668-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soo RM, Skennerton CT, Sekiguchi Y, Imelfort M, Paech SJ, Dennis PG, Steen JA, Parks DH, Tyson GW, Hugenholtz P. 2014. An expanded genomic representation of the phylum cyanobacteria. Genome Biol Evol 6:1031–1045. doi: 10.1093/gbe/evu073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Rienzi SC, Sharon I, Wrighton KC, Koren O, Hug LA, Thomas BC, Goodrich JK, Bell JT, Spector TD, Banfield JF, Ley RE. 2013. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife 2:e01102. doi: 10.7554/eLife.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward LM, Cardona T, Holland-Moritz H. 2019. Evolutionary implications of anoxygenic phototrophy in the bacterial phylum Candidatus Eremiobacterota (WPS-2). Front Microbiol 10:1658. doi: 10.3389/fmicb.2019.01658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reichart NJ, Jay ZJ, Krukenberg V, Parker AE, Spietz RL, Hatzenpichler R. 2020. Activity-based cell sorting reveals responses of uncultured archaea and bacteria to substrate amendment. ISME J 14:2851–2861. doi: 10.1038/s41396-020-00749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takai K, Moser DP, DeFlaun M, Onstott TC, Fredrickson JK. 2001. Archaeal diversity in waters from deep South African gold mines. Appl Environ Microbiol 67:5750–5760. doi: 10.1128/AEM.67.21.5750-5760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker BJ, Saw JH, Lind AE, Lazar CS, Hinrichs K-U, Teske AP, Ettema TJG. 2016. Genomic inference of the metabolism of cosmopolitan subsurface Archaea, Hadesarchaea. Nat Microbiol 1:16002. doi: 10.1038/nmicrobiol.2016.2. [DOI] [PubMed] [Google Scholar]
- 25.Chuvochina M, Rinke C, Parks DH, Rappé MS, Tyson GW, Yilmaz P, Whitman WB, Hugenholtz P. 2019. The importance of designating type material for uncultured taxa. Syst Appl Microbiol 42:15–21. doi: 10.1016/j.syapm.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Cross KL, Campbell JH, Balachandran M, Campbell AG, Cooper SJ, Griffen A, Heaton M, Joshi S, Klingeman D, Leys E, Yang Z, Parks JM, Podar M. 2019. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat Biotechnol 37:1314–1321. doi: 10.1038/s41587-019-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis WH, Tahon G, Geesink P, Sousa DZ, Ettema TJG. 2021. Innovations to culturing the uncultured microbial majority. Nat Rev Microbiol 19:225–240. doi: 10.1038/s41579-020-00458-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and code used to create Fig. 1 and 2 can be accessed at https://github.com/SawLabGW/mSystems_ECSC2021.