

Abstract
In presymptomatic Alzheimer’s disease (AD), beta-amyloid plaques (Aβ) and tau tangles accumulate in distinct spatiotemporal patterns within the brain, tracking closely with episodic memory decline. Here, we tested whether age-related changes in the segregation of the brain’s intrinsic functional episodic memory networks—anterior-temporal (AT) and posterior-medial (PM) networks—are associated with the accumulation of Aβ, tau, and memory decline using fMRI and PET. We found that AT and PM networks were less segregated in older than that in younger adults and this reduced specialization was associated with more tau and Aβ in the same regions. The effect of network dedifferentiation on memory depended on the amount of Aβ and tau, with low segregation and pathology associated with better performance at baseline and low segregation and high pathology related to worse performance over time. This pattern suggests a compensation phase followed by a degenerative phase in the early, preclinical phase of AD.
Keywords: aging, Alzheimer’s disease, brain networks, dedifferentiation, memory
Introduction
The accumulation of beta-amyloid (Aβ) plaques and neurofibrillary tau tangles is associated with episodic memory loss in both normal and pathological aging (Nelson et al. 2012; Jagust 2018), but the mechanisms underlying this association are not understood. Molecular and animal studies suggest that these pathologies spread through structurally and functionally connected brain regions (de Calignon et al. 2012; Ahmed et al. 2014; Boluda et al. 2015; Wu et al. 2016), and human neuroimaging studies indicate that patterns of tau deposition conform to large-scale brain networks in older adults (OA) (Franzmeier et al. 2019, 2020; Vogel et al. 2020). Given that AD pathology starts to deposit in episodic memory networks, we investigated whether age-related changes in functional connectivity in these resting state networks were associated with the accumulation of Aβ and tau.
Functional connectivity (FC)—the coactivation of brain regions within brain networks—reflects the brain’s large-scale network architecture. Brain networks specialized for different cognitive functions become dedifferentiated, or less segregated from each other, with older age (Koen and Rugg 2019). In task-free functional magnetic resonance imaging (fMRI) studies, this progression is typically characterized by decreased within- and increased between-network FC at rest (Chan et al. 2014; Geerligs et al. 2015; Damoiseaux 2017; Cassady et al. 2019; Cassady, Gagnon, et al. 2020). This decrease in network segregation leads to reduced specialization of neural networks and has been linked to OA’s worse performance relative to younger adults (YA) in several behavioral domains (Chan et al. 2014; King et al. 2017; Iordan et al. 2018; Cassady et al. 2019). In contrast, other studies have found that less segregated networks are associated with better performance, suggesting that dedifferentiation may reflect greater plasticity or compensatory processes that occur during normal aging and neurodegeneration (Cabeza et al. 2002, 2018; Gallen et al. 2016; Grady et al. 2016; Monge et al., 2017, 2018; Wig 2017). One potential reason for this discrepancy may be uncertainty about the molecular and pathological processes that drive network reconfiguration.
During the early stages of neurodegeneration, normal cognitive performance is often maintained despite neuronal loss, changes in network function, or the accumulation of neurodegenerative pathologies (Barulli and Stern 2013; Scheller et al. 2014; Gregory et al. 2017). Such compensation is typically only evident in preclinical or mild cases of neurodegeneration, diminishing once the neurodegenerative pathology becomes too severe. There is also evidence that greater FC can be associated with either better or worse memory performance depending on disease severity. For instance, Van Hooren and colleagues found that greater FC between the default mode network and the dorsal attention network was associated with better memory in a cognitively normal group, but with worse memory in an MCI group (Van Hooren et al. 2018). One potential interpretation of these results is that although increased connectivity between different networks is beneficial early, it may fail to support compensation as pathology increases. In addition, it could provide a means for that pathology to spread.
Events encoded as episodic memories usually combine information about objects/items and scenes/context. Processing of these two types of information depends on distinct cortical pathways in the neocortex and medial temporal lobe (MTL) that converge in the hippocampus (Ranganath and Ritchey 2012; Inhoff and Ranganath 2017; Kim et al. 2018). Object processing involves an anterior-temporal (AT) system that includes fusiform gyrus (FuG)/perirhinal cortex, inferior temporal gyrus (ITG), and amygdala. In contrast, scene processing relies on a posterior-medial system (PM) that includes retrosplenial cortex (RSC), precuneus, and parahippocampal cortex (PHC).
In vivo positron emission tomography (PET) studies have demonstrated that Aβ and tau accumulate in distinct regions within these two subnetworks in the aging brain (Maass et al. 2019). Specifically, tau initially deposits in the transentorhinal region (Braak and Braak 1992, 1995) and appears to spread throughout the AT system in both healthy aging and AD, although it eventually affects the PM system as well. In contrast, Aβ deposition preferentially affects the PM system (Maass et al. 2019). Previous work has shown that the AT and PM functional networks have distinct patterns of resting state FC with entorhinal cortex subregions in YA and that such patterns predict the spatial topography and level of cortical tau deposition in cognitively normal OA (Adams et al. 2019; Berron et al. 2020). However, it remains largely unknown whether these networks change with age and whether the accumulation of Aβ and tau is associated with changes in their modular organization and, consequently, memory decline.
There were two main goals of this study: first, to investigate the effects of age, Aβ, and tau on the resting state functional architecture of the AT and PM memory networks and second, to examine how relationships between pathology and network segregation affect episodic memory performance. To that end, we use resting state fMRI (rsfMRI) to measure the segregation of the AT and PM networks in cognitively healthy YA and OA. After examining the effect of age on network segregation, we then use PET measures of Aβ and tau deposition in OA to explore the relationship between these pathologies and segregation in the AT and PM networks. Finally, we assess the relationship between Aβ and tau, segregation, and episodic memory performance at baseline as well as change in performance over an average of 6 years in OA. We test three hypotheses: 1) AT and PM networks will be less segregated in OA compared to YA; 2) Given their distinct spatial topographies, higher levels of tau in OA will be associated with less segregated AT networks, whereas higher levels of Aβ will be associated with less segregated PM networks; and 3) Network segregation in OA will interact with AD pathology to predict episodic memory performance at baseline as well as change in performance over time.
Materials and Methods
Participants
Fifty-five YA (age 18–35) and 97 cognitively normal OA (age 60+) enrolled in the Berkeley Aging Cohort Study (BACS) were included in this study. All YA and OA participants underwent structural and resting state functional MRI. All OA additionally underwent tau-PET imaging with 18F-Flortaucipir (FTP), Aβ-PET with C-Pittsburgh 11Compound-B (PiB), and a standard neuropsychological assessment. Eligibility requirements included that all participants had a baseline MMSE score of ≥25. We also excluded any participants with a history of significant neurological disease (e.g., stroke, seizure, and loss of consciousness ≥10 min), or any medical illness that could affect cognition, history of substance abuse, depression, or contraindications to MRI or PET. All study procedures were reviewed and approved by the Institutional Review Boards of the University of California, Berkeley, and Lawrence Berkeley National Laboratory (LBNL). All participants provided written informed consent for their involvement in this study. Demographic information for each age group is presented in Table 1.
Table 1.
Cohort demographics
| YA (n = 55) | OA (n = 97) | |
|---|---|---|
| Age | 24.9 ± 4.4 (18–35) | 76.4 ± 6.1 (60–93) |
| Sex (M/F) | 28/27 | 36/61 |
| Education (Years) | 16.3 ± 2.0 | 16.8 ± 1.9 |
| APOE ε4 (C/NC) | N/A | 28/66 (3 N/A) |
| Global PiB DVR | N/A | 1.17 ± 0.24 (0.92–1.89) |
| AT FTP SUVR | N/A | 1.28 ± 0.20 (0.98–2.3) |
| PM FTP SUVR | N/A | 1.18 ± 0.12 (0.94–1.63) |
| Aβ +/− | N/A | 42/54 (1 N/A) |
| Tau +/− | N/A | 30/66 (1 N/A) |
Neuropsychological Assessment
All OA participants in the BACS undergo neuropsychological testing to measure cognitive performance related to verbal and visual memory, working memory, processing speed, executive function, language, and attention. In this study, composite scores were calculated to measure two specific memory domains: episodic memory and working memory. The tests for episodic memory included the California Verbal Learning Test (CVLT) immediate and long delay free recall totals as well as the Visual Reproduction (VR) immediate and delay recall totals. Working memory was assessed with the Digit Span total score. For episodic memory, the composite scores were produced by calculating the average z-score of the tests included in each domain. Please refer to Harrison et al. (2019) for more details of the procedure (Harrison et al. 2019).
To examine change in behavioral performance over time, our longitudinal analyses included participants (from the 97 OA sample) that had 1) at least one resting state fMRI scan, 2) at least one Aβ and one tau PET scan (both near the time of the resting state scan), and 3) at least two neuropsychological testing sessions (one of which was near the time of the resting state scan). Critically, all participants had resting state fMRI, PET, and neuropsychological data at the same timepoint, which is the timepoint we used to assess all cross-sectional relationships (i.e., “baseline” timepoint). The additional neuropsychological session(s) could be either before or after the baseline timepoint (or both), depending on the participant. Eighty-six of 97 OA participants had longitudinal cognitive data (≥2 testing sessions). These participants had between 2 and 13 testing sessions (mean, 6.1 ± 3.1) over a period of 1–13 years (mean, 6.1 ± 3.5) with an average delay of 1.3 ± 0.6 years between sessions. Sixteen participants had only retrospective data.
MRI Data Acquisition
All YA and OA participants underwent structural and functional MRI acquired on a 3 T TIM/Trio scanner (Siemens Medical System, software version B17A) using a 32-channel head coil. First, a whole-brain high-resolution T1-weighted volumetric magnetization prepared rapid gradient echo image (MPRAGE) structural MRI scan was acquired with the following parameters: voxel size = 1 mm isotropic, TR = 2300 ms, TE = 2.98 ms, matrix = 256 × 240 × 160, FOV = 256 × 240 × 160 mm3, sagittal plane, 160 slices, 5-min acquisition time. This was followed by an rsfMRI scan that was acquired using T2*-weighted echo planar imaging (EPI) with the following parameters: voxel size = 2.6 mm isotropic, TR = 1067 ms, TE = 31.2 ms, FA = 45, matrix = 80 × 80, FOV = 210 mm, sagittal plane, 300 volumes, anterior to posterior phase encoding, ascending acquisition, 5-min acquisition time. A multiband acceleration factor of four was used to acquire whole-brain coverage at high spatial resolution by acquiring four slices at the same time (Feinberg and Setsompop 2013; Todd et al. 2016). During the rsfMRI scan, participants were instructed to remain awake with their eyes open and focused on the screen, which displayed a white asterisk on a black background.
As part of the standard PET processing pipeline, a whole-brain high-resolution T1-weighted volumetric MPRAGE scan was acquired for each participant on a Siemens Magnetom Avanto scanner at LBNL with the following parameters: voxel size = 1 mm isotropic, TR = 2110 ms, TE = 3.58 ms, flip angle = 15°, sagittal slice orientation. These data were used for PET coregistration and to parcellate the brain for PET data analysis.
PET Data Acquisition
All OA participants underwent PET scanning at LBNL using a Biograph PET/CT Truepoint 6 scanner (Siemens, Inc.) with CT scans performed for attenuation correction prior to each emission acquisition and radiotracers synthesized at the LBNL Biomedical Isotope Facility. Tau deposition was measured using 18F-Flortacipir (FTP) with data binned into 4 × 5 min frames from 80 to 100 min postinjection (Adams et al. 2019.; Harrison et al. 2019; Maass et al. 2017). Aβ was measured using 11C-Pittsburgh Compound B (PiB), with data acquired across 35 dynamic frames for 90 min postinjection (4 × 15, 8 × 30, 9 × 60, 2 × 180, 10 × 300, and 2 × 600 s). All PET images were reconstructed using an ordered subset expectation maximization algorithm, with attenuation correction, scatter correction, and smoothing using a Gaussian kernel of 4 mm.
MRI Processing
Structural scans (3 T) were processed with FreeSurfer to derive regions of interest (ROIs) in each subject’s native space using the Desikan–Killiany atlas. The structural images were also segmented into gray matter (GM), white matter (WM), and cerebrospinal fluid (CSF) using Statistical Parametric Mapping software (SPM12; Wellcome Trust Centre for Neuroimaging, London, UK) (default parameters). RsfMRI data were preprocessed using SPM12 and FreeSurfer (v5.3.0). Preprocessing included slice time correction, realignment, coregistration to the T1 image, and outlier volume detection. All functional images were first corrected for differences in slice time acquisition using SPM12. Functional images were then realigned to the first volume, and coregistered to the T1 image. Outliers in average intensity and/or scan-to-scan motion were identified using the artifact detection toolbox (ART; http://www.nitrc.org/projects/artifact_detect) using a conservative movement threshold of >0.5 mm/TR and a global intensity z-score of 3. Outlier volumes were flagged and included as spike regressors during the denoising procedure (Lemieux et al. 2007; Power et al. 2014). Additional denoising on the rsfMRI data was performed using the CONN toolbox (v18a: www.nitrc.org/projects/conn). Temporal and confounding factors were regressed from each voxel BOLD time series and the resulting residual time series were filtered using a temporal band-pass filter of 0.008–0.09 Hz to examine the frequency band of interest and to exclude higher frequency sources of noise such as heart rate and respiration. For noise reduction, we used the anatomical component–based noise correction method aCompCor (using the first five components of the times series signal from white matter and CSF), which models the influence of noise as a voxel-specific linear combination of multiple empirically estimated noise sources by deriving principal components from noise regions of interest (ROIs) and including them as nuisance regressors in the first-level general linear model (GLM) (Behzadi et al. 2007). Residual head movement parameters (three rotations, three translations, and six parameters representing their first-order temporal derivatives) and signals from WM and CSF, and spike regressors from motion detection were regressed out during the computation of functional connectivity maps. We did not exclude any participants from analyses due to excess motion, as all participants had <19% of outlier volumes, with an average of 4.4% ± 3.25% outlier volumes. Although there were significant age differences in both percentage of outlier volumes (t = 3.4, P = 0.001) and mean framewise displacement (t = 5, P < 0.001) between OA and YA, these parameters did not change the significance of any of the results when they were included as covariates in the statistical models (see Supplemental Tables 1–4). Supplemental Table 5 details these movement parameters for each age group.
First-level ROI-to-ROI functional connectivity analysis was performed using the CONN toolbox. For this analysis, we used 12 FreeSurfer ROIs that included unilateral amygdala, fusiform gyrus/perirhinal cortex, and inferior temporal gyrus as part of the AT network and retrosplenial cortex, parahippocampal cortex, and precuneus as part of the PM network (Fig. 1). Semipartial correlations were used for these first-level analyses to determine the unique variance of each (unilateral) seed, controlling for the variance of all other seed regions entered into the same model. For each participant, the rsfMRI time series within each of the ROIs was extracted and the mean time series was computed. Then, the cross-correlation of each ROI’s time course with every other ROI’s time course was computed, creating a 12 × 12 correlation matrix for each subject. Correlation coefficients (i.e., graph edges) were converted to z-values using Fisher’s r-to-z transformation (Zar 1996). As in previous studies (Chan et al. 2014; Cassady et al. 2019; Cassady, Gagnon,et al. 2020), the diagonal of the matrix was removed and negative correlations were set to zero as we were mainly interested in positive connections (Zhan et al. 2017). We also performed the same analyses with inclusion of both positive and negative correlations and observed similar results (Supplemental Fig. 1). Network segregation values were calculated as the difference in mean within-network FC and mean between-network FC divided by mean within-network FC
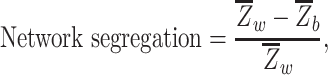 |
where 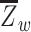 is the mean Fisher z-transformed correlation between ROIs within the same network and
is the mean Fisher z-transformed correlation between ROIs within the same network and 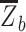 is the mean Fisher z-transformed correlation between ROIs of one network with all ROIs in the other network (Chan et al. 2014). Thus, larger positive values for network segregation indicate that regions within a network (e.g., AT) have higher connectivity with each other compared to their connectivity with regions outside of the network (e.g., PM).
is the mean Fisher z-transformed correlation between ROIs of one network with all ROIs in the other network (Chan et al. 2014). Thus, larger positive values for network segregation indicate that regions within a network (e.g., AT) have higher connectivity with each other compared to their connectivity with regions outside of the network (e.g., PM).
Figure 1 .
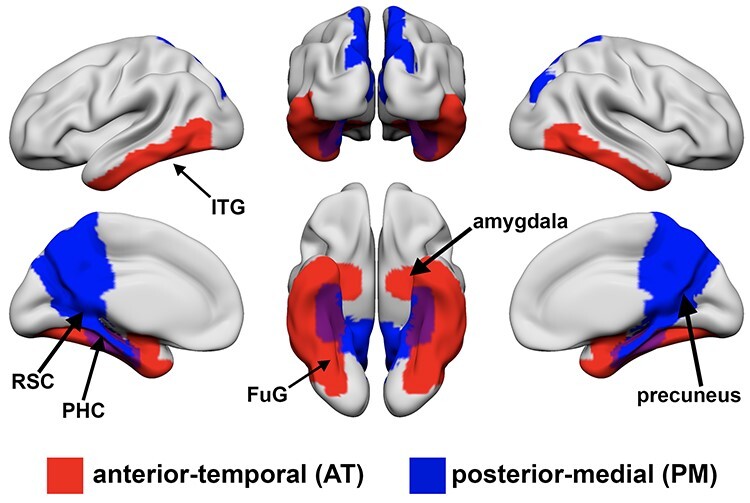
A priori defined regions of interest in anterior-temporal (AT; red) and posterior-medial (PM; blue) networks. AT regions include bilateral amygdala, fusiform gyrus (FuG)/perirhinal cortex, and inferior temporal gyrus (ITG). PM regions include bilateral retrosplenial cortex (RSC), parahippocampal cortex (PHC) and precuneus.
PET Data Processing
As part of our standard PET preprocessing procedure, 1.5 T structural MRI data were preprocessed with FreeSurfer to derive ROIs in subject’s native space. These ROIs were then used for the calculation of PiB-PET global distribution volume ratio (DVR) and region-specific, partial volume corrected (PVC) (Baker, Maass, et al. 2017) FTP standardized uptake value ratio (SUVR) measures. FTP images were processed with SPM12. Images were realigned, averaged, and coregistered to each participant’s 1.5 T structural MRI scan. SUVR images were calculated by averaging the mean tracer uptake over the 80–100 min data and normalized by an inferior cerebellar gray reference region (Baker, Lockhart, et al. 2017). The mean SUVR of each (FreeSurfer segmented) ROI was extracted from the native space images. These data were then partial volume corrected using a modified Geometric Transfer Matrix approach (Rousset et al. 1998) as previously described (Baker, Maass, et al. 2017). The weighted mean (by region size), partial volume corrected FTP SUVR of all AT (amygdala, FuG/perirhinal cortex, ITG) and PM (RSC, PHC, and precuneus) ROIs were used in subsequent analyses. Tau positivity was defined as the mean SUVR in a BraakIII-IV composite ROI (cutoff 1.26) that included regions from both AT (amygdala, FuG, and ITG) and PM (PHC, RSC) systems. APOE was not related to segregation (Ps > 0.63); therefore, we did not control for APOE in these analyses.
PiB images were also processed with SPM12. Images were realigned, averaged across frames from the first 20 min of acquisition, and coregistered to each participant’s 1.5 T structural MRI image. DVR values for PiB-PET images were calculated with Logan graphical analysis over 35–90 min data and normalized by a cerebellar gray matter reference region (Logan et al. 1996; Price et al. 2005). Global PiB was calculated across cortical FreeSurfer ROIs as previously described (Mormino et al. 2012), and a threshold of 1.065 was used to classify participants into Aβ− and Aβ+ groups (Villeneuve et al. 2015). One participant was missing PiB DVR data and therefore was excluded from all analyses involving measures of Aβ. There were 209 (cognitive) time points for Aβ+ and 249 time points for Aβ− older adults. There were 60 time points for Aβ + OA participants versus 62 timepoints for Aβ− OA participants during or after the “baseline” scan. There were 148 time points for Aβ+ OA participants versus 182 timepoints for Aβ− OA participants before the “baseline” scan. Supplemental Table 2 contains demographic information for the older adult group split by Aβ− and tau status.
Statistical Analyses
Statistical analyses were conducted using R (http://www.R-project.org/) and SPSS (SPSS Inc.) software. Independent sample t-tests were used to test for age group differences in within- and between-network FC and segregation (corrected for multiple comparisons using FDR). Multiple regression models were used to assess the relationship between segregation, Aβ and tau, and baseline cognitive performance.
Because our episodic memory composite measure included both object- and spatial-related memory domains, we computed a single segregation measure by averaging the AT and PM segregation values for associations with behavior (follow-up analyses showed essentially the same results for AT and PM networks; see Supplemental Fig. 2). As a control analysis, we also examined the association between segregation and working memory performance using the same analysis procedure as above.
Longitudinal cognitive measures were modeled using linear mixed-effects regression with a random intercept (to account for variability in baseline measurements) and slope (to account for variability in slopes) using the lme4 package in R v3.6.3 (www.r-project.org). In order to examine the relationship between baseline segregation, Aβ and tau, and change in cognitive performance, the models included two-way interactions between baseline segregation and time, baseline Aβ and time as well as tau and time. We were specifically interested in the segregation x time interaction to determine whether baseline segregation was associated with longitudinal episodic memory decline. As a control analysis, we also examined change in working memory performance over time using the same model. All predictor variables were standardized before entered into the model.
All statistical models were adjusted for age and sex, and years of education (for models including cognitive measures). Table 2 provides a description of the time (in years) between all PET, fMRI, and cognitive sessions. All statistical analyses used a two-tailed level of 0.05 for determining statistical significance. Reported P-values were corrected for multiple comparisons (using FDR) where relevant, including the associations between age and segregation.
Table 2.
Mean, standard deviation, and range of time (in years) between PET, fMRI, and (MRI baseline) cognitive sessions
| Sessions | Time between (Years) |
|---|---|
| PiB- and Tau-PET | 0.04 ± 0.13 (0–0.76) |
| PiB and rsfMRI | 0.13 ± 0.12 (0–0.52) |
| Tau and rsfMRI | 0.12 ± 0.11 (0–0.4) |
| Tau and (Baseline) Cog | 0.28 ± 0.44 (0.02–4.1) |
| PiB and (Baseline) Cog | 0.28 ± 0.45 (0.02–4.1) |
| rsfMRI and (Baseline) Cog | 0.28 ± 0.47 (0–4.4) |
Results
AT and PM Networks Are Less Segregated with Older Age
As hypothesized, OA exhibited lower within-network (AT: Fig. 2A, t(150) = 6.7, P = 0.002; PM: Fig. 2B, t(150) = 3.1, P = 0.002) and greater between-network (AT: Fig. 2C, t(150) = −2.5, P = 0.01; PM: Fig. 2D, t(150) = −3, P = 0.006) functional connectivity and decreased segregation (AT: Fig. 2E, t(150) = 4.9, P < 0.001; PM: Fig. 2F, t(150) = 4.3, P < 0.001) in the AT and PM networks compared to YA. Of particular importance, the relationship between age group and segregation was assessed across multiple analysis approaches related to matrix thresholding (i.e., inclusion of positive only vs. negative correlations), bivariate versus semi-partial correlations, various network metrics of intersystem relationships (i.e., segregation, participation coefficient, and modularity), and network labeling (i.e., the regions included to define AT and PM networks). The age group differences in segregation were found to be robust in all instances (see Supplemental Fig. 1).
Figure 2 .
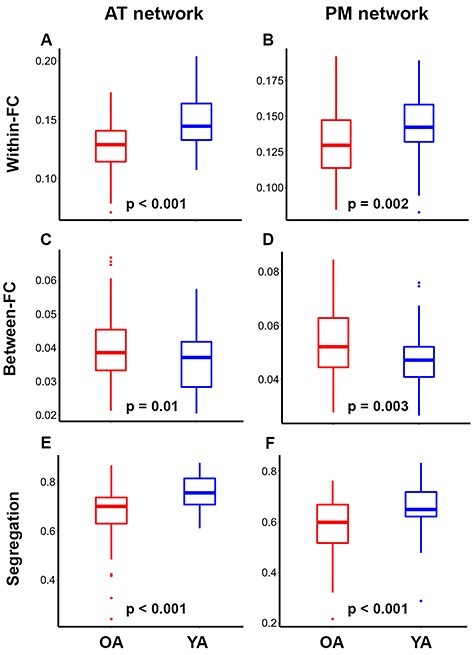
Anterior-temporal (AT; left) and posterior-medial (PM; right) networks are less segregated in older (red) relative to younger (blue) adults. Independent samples t-test indicated that OA have lower within-network (A and B) and greater between-network (C and D) functional connectivity (FC), and lower network segregation (E and F) compared to YA. T-tests were corrected for multiple comparisons using FDR. On each box, the central mark indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers.
Tau Relates to at Segregation and Aβ Relates to PM Segregation
To assess the relationship between Aβ, tau, and segregation, we performed two multiple regressions including age, sex, Aβ, tau (AT-tau for predicting AT-segregation and PM-tau for predicting PM segregation), and the interaction between Aβ and tau in the models. The fit for the overall model predicting AT segregation was not significant (F(5, 89) = 1.96, P = 0.092) with an R2 of 0.099 and an adjusted R2 of 0.049. However, we did observe a main effect of AT-tau (B = −0.25, t = −2.1, P = 0.04), but not for Aβ, and no significant interaction of Aβ and AT-tau (Table 3). This indicates that greater levels of tau in AT regions were associated with less segregated AT networks. However, because the relationship between AT tau and AT segregation appeared to be influenced by a few high-tau individuals, we performed a follow-up robust regression, which is less affected by more extreme data points (Rousseeuw and Leroy 2005). This analysis was performed using the “fitlm” function with the “RobustOpts” name-value pair in Matlab to create a model that limits the influence of outliers and heteroscedasticity. The relationship between tau and AT segregation was no longer significant across the whole group (t = 0.95, P = 0.34), but remained significant in the Aβ+ group (t = 3.1, P = 0.004). These results should thus be interpreted in light of these disparate results. The fit for the overall model predicting PM-segregation was significant (F(4, 90) = 2.6, P = 0.042) with an R2 of 0.1 and an adjusted R2 of 0.063. We also observed a main effect of Aβ (B = −0.32, t = −2.8, P = 0.006), but not for tau, and no significant interaction of Aβ and PM-tau (Table 3). This indicates that greater levels of global Aβ were associated with less segregated PM networks. Figure 3 displays these results as partial correlations for visual purposes.
Table 3.
Multiple regression results for AT-Tau predicting AT-segregation and Global Aβ predicting PM-segregation
| AT-seg | PM-seg | |||
|---|---|---|---|---|
| Predictor | t | P | t | P |
| Age | −0.34 | 0.73 | −1.3 | 0.19 |
| Global Aβ | 0.13 | 0.9 | −2.8 | 0.006 |
| Sex | −0.54 | 0.59 | 0.068 | 0.95 |
| Tau* | −2.1 | 0.04 | 1.6 | 0.12 |
| Aβ × Tau* | −1.4 | 0.18 | 0.44 | 0.66 |
Note: *AT-tau was used for the regression predicting AT-segregation, whereas PM-tau was used for predicting PM-segregation.
Figure 3 .
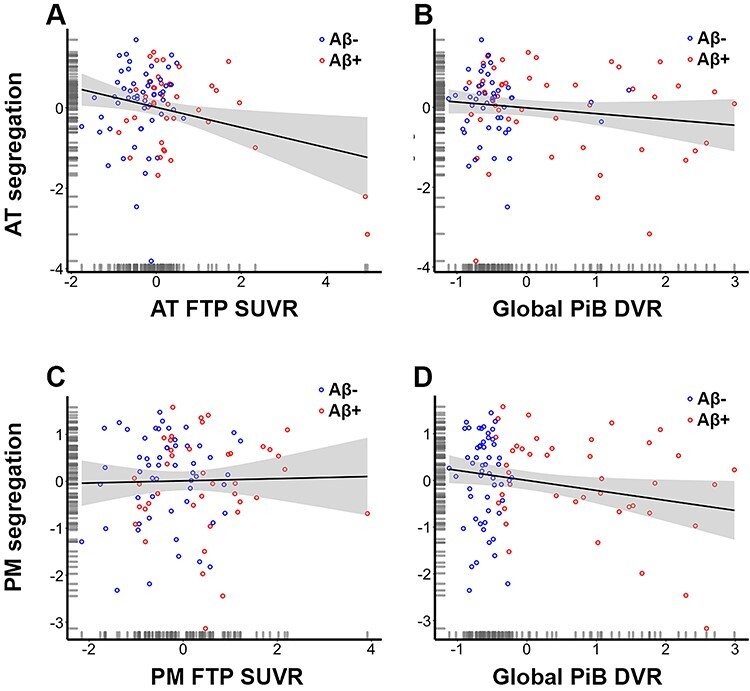
Tau and beta amyloid (Aβ) deposition are related to the segregation of anterior-temporal (AT; first row) and posterior-medial (PM; second row) networks, respectively. Less segregated AT networks are associated with higher levels of tau in AT regions* (A) but are not associated with global Aβ (B). Less segregated PM networks are not associated with tau in PM regions (C) but are associated with higher levels of global Aβ (D). Plots are illustrated as partial correlations, controlling for the effects of age and sex. The x- and y-axes reflect the residuals from the model. *The relationship depicted in Figure 3A was no longer significant after using robust regression; this relationship should therefore be interpreted cautiously.
AD Pathology Moderates the Association between Segregation and Episodic Memory
We performed a multiple regression to assess the effects of segregation and Aβ-status on episodic memory performance in OA. The fit for the overall model was significant (F(6, 89) = 2.65, P = 0.021) with an R2 of 0.15 and an adjusted R2 of 0.094. We observed main effects of segregation (B = −0.4, t = −2.9, P = 0.005), Aβ-status (B = −1.7, t = −2.5, P = 0.013), and age (B = −0.27, t = −2.7, P = 0.008) on episodic memory. This analysis also revealed a significant interaction between Aβ-status and segregation on episodic memory performance (B = 1.7, t = 2.5, P = 0.014). Specifically, less segregated networks were associated with better performance among all OA (Fig. 4A) and in Aβ- OA (Fig. 4B), but segregation was not associated with performance in Aβ + OA (Fig. 4C). Table 4 reports the results of this regression.
Figure 4 .
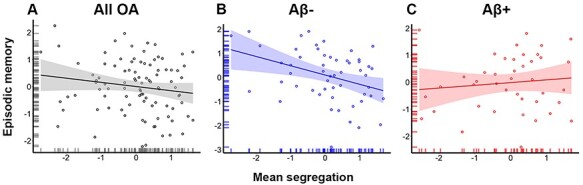
Alzheimer’s disease pathology moderates the association between mean network segregation and episodic memory performance. (A) Multiple regression results showed that less segregated networks were associated with better performance in OA. They also demonstrated an interaction between segregation and memory performance such that (B) less segregated networks were associated with better performance in Aβ- OA, whereas (C) segregation was not associated with performance in Aβ + OA (C). Plots are illustrated as partial correlations, controlling for the effects of age, sex, and education; the x- and y-axes reflect the residuals from the model.
Table 4.
Multiple regression results for mean segregation and its interaction with Aβ-status predicting episodic memory at baseline
| Predictor | t | P |
|---|---|---|
| Age | −2.7 | 0.008 |
| Aβ-status | −2.5 | 0.013 |
| Sex | 0.86 | 0.39 |
| Education | 1.3 | 0.2 |
| Segregation | −2.9 | 0.005 |
| Aβ-status × Seg | 2.5 | 0.014 |
As a follow-up analysis, we also explored the effects of segregation and tau-status on episodic memory performance. The fit for the overall model was not significant (F(6, 89) = 1.86, P = 0.097) with an R2 of 0.11 and an adjusted R2 of 0.05. We again found main effects of segregation (B = −0.26, t = −2.1 P = 0.043) and age (B = −0.26, t = −2.5, P = 0.014), but not tau-status (B = −0.92, t = −1.4, P = 0.16) on episodic memory. There was not a significant interaction between tau-status and mean segregation on performance, (B = 0.96, t = 1.5, P = 0.14). Table 5 reports the results of this follow-up regression.
Table 5.
Multiple regression results for mean segregation and its interaction with Tau-status predicting episodic memory at baseline
| Predictor | t | P |
|---|---|---|
| Age | −2.5 | 0.014 |
| Tau-status | −1.4 | 0.16 |
| Sex | 0.75 | 0.45 |
| Education | 1.2 | 0.22 |
| Segregation | −2.9 | 0.043 |
| Tau-status × Seg | 2.5 | 0.14 |
As a control analysis, we also examined the relationship between segregation, Aβ-status, and baseline working memory. There were no significant relationships between segregation and working memory performance, nor was there an interaction between Aβ-status and segregation on performance (Ps > 0.41). Including tau-status in place of Aβ-status, we similarly found no significant relationships between segregation and working memory performance in either group, nor was there an interaction between tau-status and segregation on performance (Ps > 0.67).
Baseline Segregation Predicts Longitudinal Memory Decline
Longitudinal cognitive measures were modeled using linear mixed-effects regression with a random intercept (variance = 0.52) and slope (variance = 0.04). In order to examine the relationship between baseline (at MRI) segregation, baseline Aβ and tau, and change in (both retrospective and prospective) cognitive performance in one model, the model included two-way interactions between baseline segregation and time, global Aβ and time, and tau and time. We report the results using continuous measures of global Aβ and BraakIII-IV tau as they retain more statistical power in the model. The results were very similar whether we used BraakIII-IV tau, AT-tau (Supplemental Table 3), or PM tau (Supplemental Table 4) and whether we used dichotomous (Supplemental Table 5) or continuous Aβ and tau in the model. Segregation was a continuous variable in the model but is displayed graphically using tertiles. Figure 5A displays each participant’s trajectory in longitudinal episodic memory performance over time.
Figure 5 .
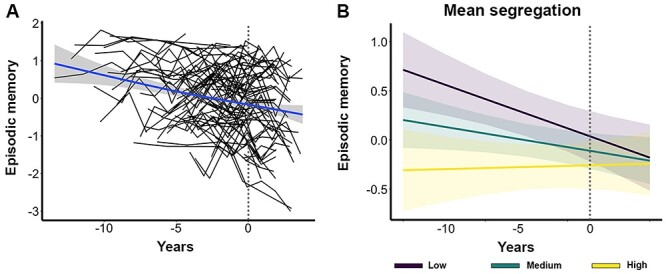
Relationship between network segregation and longitudinal episodic memory change. (A) Individual participant trajectories in longitudinal episodic memory change over time. Each black line represents one participant. The blue trendline reflects the participant average change in memory over time. The dotted gray line (at X = 0) represents the “baseline” timepoint in each plot. (B) Plot of estimated curves for three groups with different baseline network segregation (low, medium, and high) and episodic memory outcomes over time. Note that segregation was modeled as a continuous variable but is shown as a categorical variable for illustration purposes only. Lower baseline segregation was associated with a steeper decline rate in episodic memory over time.
We found that individuals with lower segregation at baseline showed a steeper decline rate in episodic memory over time (β = 0.08, SE = 0.03, P = 0.02; Fig. 5B; Table 6). We also found that more tau at baseline was associated with a steeper decline rate in memory over time (β = −0.15, SE = 0.04, P = 0.002). There was no interaction of Aβ and time predicting memory change (β = −0.003, SE = 0.04, P = 0.94). To examine whether Aβ or tau moderated the effect of segregation on cognitive change, follow-up analyses included the same factors in addition to three-way interactions between baseline segregation, baseline Aβ and tau, and time. These analyses did not show a significant three-way interaction between segregation, tau, and time (β = 0.03, SE = 0.05, P = 0.58) nor between segregation, Aβ, and time (β = −0.04, SE = 0.04, P = 0.34) on memory change. As a control analysis, we also examined change in working memory performance over time using the same model (not including three-way interactions). Baseline segregation was not associated with longitudinal change in working memory (β = −0.02, SE = 0.04, P = 0.57).
Table 6.
Linear mixed-model results for segregation and pathology predicting longitudinal episodic memory change
| Predictor | Estimate | P |
|---|---|---|
| Age | −0.19 | 0.03 |
| Sex | −0.01 | 0.93 |
| Education | 0.13 | 0.13 |
| Segregation | −0.20 | 0.02 |
| Time | −0.07 | 0.06 |
| Tau | −0.13 | 0.17 |
| Aβ | 0.03 | 0.8 |
| Tau × Time | −0.15 | 0.002 |
| Aβ × Time | −0.003 | 0.94 |
| Segregation × Time | 0.08 | 0.02 |
Discussion
The goal of this study was to investigate the effects of Aβ and tau on the intrinsic functional architecture of episodic memory networks and episodic memory ability in cognitively normal OA. OA showed reduced segregation of AT and PM networks compared to YA. This effect was driven by lower within-network FC and greater between-network FC between the two systems. Higher levels of tau in AT regions were associated with less segregated AT networks, whereas higher levels of global Aβ were associated with less segregated PM networks, demonstrating a regional dissociation of these AD pathologies to the large-scale organization of the AT and PM systems. Finally, less segregated networks were associated with better memory ability at baseline (at the time of the MRI) in OA with low levels of AD pathology but with a steeper decline in memory performance over time, independent of baseline pathology. These results suggest different phases in the long-term interaction of network organization and AD pathology on episodic memory.
We interpreted our findings based on a model that includes both age- and AD pathology-related effects. We found that age was associated with changes in the functional segregation of the AT and PM resting state networks. This finding is consistent with studies of age-related neural dedifferentiation demonstrating that older age is associated with less distinct neural activation patterns (Carp et al. 2011; Lalwani et al. 2018; Cassady et al. 2019; Koen et al. 2019; Koen and Rugg 2019; Cassady, Ruitenberg, et al. 2020) and, more recently, with less distinct large-scale resting state networks (Betzel et al. 2014; Cao et al. 2014; Chan et al. 2014; Geerligs et al. 2015; Damoiseaux 2017; Cassady et al. 2019; Cassady, Gagnon, et al. 2020). While a majority of these prior studies explored the organization of the brain’s canonical resting state networks (e.g., the default mode, frontoparietal and cingulo-opercular networks), we demonstrated a robust age effect in two neural networks that are associated with episodic memory and AD pathology. Recent work from our laboratory also showed that less differentiated activation in AT and PM regions during an object/scene discrimination task was associated with more tau deposition (Maass et al. 2019). These findings, in conjunction with the present results, suggest a neuropathological correlate of dedifferentiation in the episodic memory system.
We found that the modular organization of the AT and PM brain networks was selectively vulnerable to tau and Aβ deposition. Specifically, greater tau in AT regions was associated with less segregated AT networks but was not associated with PM network segregation. In contrast, greater cortical Aβ was associated with less segregated PM networks but was not associated with AT network segregation. Since between-network FC was the same in the AT and PM networks, these results indicate that this relationship was driven by within-network FC. This is consistent with previous investigations that have demonstrated relationships between within-network FC and AD pathology (Schultz et al. 2017; Adams et al. 2019; Franzmeier et al. 2020). The findings of a double dissociation between AD pathology and network segregation are in accordance with previous work from our lab demonstrating differential selective vulnerability to these two networks participating in episodic memory function. Specifically, Maass et al. (2019) showed that tau deposits mainly in AT regions, resulting in object discrimination deficits, whereas Aβ deposits preferentially in PM regions, resulting in impaired scene discrimination (Maass et al. 2019).
There is no agreement on a precise region where Aβ deposition begins, and existing data suggest that this pathology appears multifocally and quickly accumulates throughout most of association cortex (Braak and Braak 1991; Palmqvist et al. 2017; Thal et al. 2002; Whittington et al. 2018). For example, while we used a global measure of cortical Aβ to define positivity, Aβ in the PM network is highly correlated with this measure as are most regions throughout the brain (Lockhart et al. 2017). In contrast, tau initially deposits in the entorhinal cortex and then progresses in a distinct spatiotemporal pattern first to anterior temporal and limbic regions and then throughout association cortex (Braak and Braak 1985, 1991; Kaufman et al. 2018). Cellular and molecular data reveal that tau can spread trans-synaptically and in relation to neural activity (de Calignon et al. 2012; Pooler et al. 2013; Yamada et al. 2014; Wu et al. 2016), suggesting that this specific AD pathology accumulates through the brain along neural connections.
The idea that large-scale brain network connectivity may underlie the spatiotemporal patterns of AD pathology has support from other laboratories. For instance, Franzmeier et al. (2019) found that canonical network regions with higher FC showed higher covariance of tau deposition. In addition, Jacobs et al. found that Aβ facilitated the spread of tau from the hippocampus to the posterior cingulate via structural connectivity (Jacobs et al. 2018), and Adams et al. reported that FC of the entorhinal cortex was related to Aβ-facilitated neocortical tau deposition (Adams et al. 2019). Previous data suggesting preferential involvement of the AT network by tau (Maass et al. 2019), along with these results showing dedifferentiation, raise the possibility that tau may spread from the AT to the PM network as these networks become less segregated.
Indeed, segregated brain networks are characterized by a fine balance of dense within-system relationships among brain regions that have highly related processing roles, as well as sparser relationships between areas in networks with diverse processing roles. This pattern of brain network organization facilitates communication among brain regions that have related sets of processing operations and also reinforces the functional specialization of networks that perform different sets of processing roles. This distinction in the proportion of functional connections within and between communities is necessary for maintaining a fine balance between functional segregation and global integration across networks. Importantly, alterations to the connections that maintain effective network organization can have negative consequences. For example, too much integration or increased connectivity between systems (as is the case in the present study with the AT and PM networks) could potentially lead to the spread of pathological protein aggregates (Salathé and Jones 2010; Wig 2017).
Our results revealed complex interactions between segregation, Aβ and tau pathology, and memory performance. Our cross-sectional data showed that AD pathology moderated the relationship between segregation and baseline performance. Specifically, less segregated networks were associated with better performance in OA with low levels of Aβ pathology but not in those with high levels of Aβ pathology. Additionally, our longitudinal results revealed that less segregated networks and more tau at baseline independently predicted a steeper decline in memory performance over time. These findings are consistent with previous studies demonstrating that neurodegenerative pathology interacts with FC to influence performance (Van Hooren et al. 2018; Lin et al. 2020). For instance, Lin et al. (2020) showed an interactive effect of Aβ deposition and FC on cognition such that increased FC between left middle frontal gyrus and a memory encoding network was associated with better attention/processing speed and executive function in those with low levels of Aβ but with worse function in those with high levels of Aβ (Lin et al. 2020).
Overall, our findings may suggest different phases in the long-term interaction of network segregation and AD pathology on episodic memory ability. OA with low pathology may compensate, either for normal aging processes or for the start of AD pathology, by increasing communication between AT and PM networks. As functionality in one system declines, recruiting the other system may help performance. Over time, however, this increased between-network FC in the context of increasing pathology could become detrimental, as well as providing a route for AD disease pathology to spread from one network to the other, leading to more decline in memory ability. Based on this model, it is likely that AT and PM networks continue to de-differentiate over time, especially in the transition phase from cognitively normal to cognitively impaired. This would further the spread of AD pathology, eventually resulting in the hallmark episodic memory impairments observed in MCI and AD. Future studies that include patient data as well as longitudinal measures of FC, AD pathology, and memory function are crucial in testing this hypothesis.
The cross-sectional PET and MRI data limit our interpretations of causality as well as long-term changes in this study. Although it is possible that Aβ and tau spread lead to disruptions in large-scale network FC (rather than the reverse), several studies suggest that Aβ and tau propagation is a multifactorial process that depends on both neural connectivity and regional vulnerability (Yamada et al. 2014; Wu et al. 2016; Franzmeier et al. 2020). Hence, the relationship between Aβ and tau and FC is likely bidirectional such that age-related disruptions in network FC guide pathology spread and this, in turn, leads to further changes in the network architecture. Longitudinal designs are critical in determining the order of age-related changes as well as elucidating the sequence of neural events leading to episodic memory decline. Another limitation of this study was that the longitudinal cognitive data included different numbers of time points before and after the “baseline” time point for different participants. This design feature complicates our interpretation of the longitudinal effects of segregation and Aβ and tau on performance because our analyses were both retrospective and prospective. However, this design feature allowed us to examine memory change over a longer period of time (average of ~6 years) compared with many previous studies (O’Brien et al. 2010; Woodard et al. 2010; Amariglio et al. 2018). Furthermore, we were able to include more participants from our sample with longitudinal data using this design. Longitudinal studies are often unable to observe any significant change in cognition in OA given the relatively short time periods of observation (Salthouse 2009; Reisberg et al. 2010). We believe that having a greater number of time points for more participants outweighs the disadvantage of this design feature. Another limitation of this study is that some relationships, particularly those involving AD biomarker measures, were strongly influenced by a few participants. In particular, the relationship between AT-tau and AT network segregation appeared to be influenced by a few individuals with high levels of tau. Although we believe this requires cautious interpretation, these participants also appeared to be more cognitively impaired and closer to the transition of disease symptoms. Excluding them is not biologically justified and would limit the range of biomarker results in our cognitively normal cohort. Finally, the duration of our resting state scan was only 5 min long. This is an important limitation because research indicates that scan length can have a significant impact on the reliability of resting state functional connectivity estimates (Birn et al. 2013). Future studies should include longer resting state scans.
Taken together, our data support a model whereby network dedifferentiation performs a neural compensatory function that fails over time as AD pathology accumulates. The effect of network dedifferentiation on episodic memory ability is helpful to performance when pathology levels are low but is harmful to performance over time as pathology presumably spreads. This research provides an important step in elucidating the neural mechanisms associated with episodic memory decline in healthy and pathological aging. By studying this episodic memory system in healthy OA, we can advance our understanding of healthy aging and its similarities to and differences from pathological aging, which could serve as a crucial building block for the early detection of AD.
Funding
National Institutes of Health (grants AG034570, AG062542, AG057107, and AG062090).
Notes
Avid Radiopharmaceuticals enabled the use of the 18F-Flortaucipir tracer but did not provide direct funding and were not involved in data analysis or interpretation. Conflict of Interest: Dr Jagust has served as a consultant to Genentech, Biogen, Bioclinica, Grifols, and CuraSen.
Supplementary Material
Contributor Information
Kaitlin E Cassady, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA; Helen Wills Neuroscience Institute, University of California Berkeley, Berkeley, CA 94720, USA.
Jenna N Adams, Helen Wills Neuroscience Institute, University of California Berkeley, Berkeley, CA 94720, USA.
Xi Chen, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA; Helen Wills Neuroscience Institute, University of California Berkeley, Berkeley, CA 94720, USA.
Anne Maass, German Center for Neurodegenerative Disease, Magdeburg 39120, Germany.
Theresa M Harrison, Helen Wills Neuroscience Institute, University of California Berkeley, Berkeley, CA 94720, USA.
Susan Landau, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA; Helen Wills Neuroscience Institute, University of California Berkeley, Berkeley, CA 94720, USA.
Suzanne Baker, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
William Jagust, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA; Helen Wills Neuroscience Institute, University of California Berkeley, Berkeley, CA 94720, USA.
References
- Adams JN, Maass A, Harrison TM, Baker SL, Jagust WJ. 2019. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. Elife. 8:1–22. doi: 10.7554/eLife.49132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, et al. 2014. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127(5):667–683. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amariglio RE, Buckley RF, Mormino EC, Marshall GA, Johnson KA, Rentz DM, Sperling RA. 2018. Amyloid-associated increases in longitudinal report of subjective cognitive complaints. Alzheimer Dement. 4:444–449. doi: 10.1016/j.trci.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Lockhart SN, Price JC, He M, Huesman RH, Schonhaut D, Faria J, Rabinovici G, Jagust WJ. 2017. Reference tissue–based kinetic evaluation of 18F-AV-1451 for tau imaging. J Nucl Med. 58(2):332–338. doi: 10.2967/jnumed.116.175273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Maass A, Jagust WJ. 2017. Considerations and code for partial volume correcting [18F]-AV-1451 tau PET data. Data Brief. 15:648–657. doi: 10.1016/j.dib.2017.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barulli D, Stern Y. 2013. Efficiency, capacity, compensation, maintenance, plasticity: emerging concepts in cognitive reserve. Trends Cogn Sci. 17(10):502–509. doi: 10.1016/j.tics.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behzadi Y, Restom K, Liau J, Liu TT. 2007. A component based noise correction method (CompCor) for BOLD and perfusion based fMRI. Neuroimage. 37(1):90–101. doi: 10.1016/j.neuroimage.2007.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berron D, van Westen D, Ossenkoppele R, Strandberg O, Hansson O. 2020. Medial temporal lobe connectivity and its associations with cognition in early Alzheimer’s disease. Brain. 143(4):1233–1248. doi: 10.1093/brain/awaa068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzel RF, Byrge L, He Y, Goñi J, Zuo X-N, Sporns O. 2014. Changes in structural and functional connectivity among resting-state networks across the human lifespan. Neuroimage. 102:345–357. doi: 10.1016/j.neuroimage.2014.07.067. [DOI] [PubMed] [Google Scholar]
- Birn RM, Molloy EK, Patriat R, Parker T, Meier TB, Kirk GR, Nair VA, Meyerand ME, Prabhakaran V. 2013. The effect of scan length on the reliability of resting-state fMRI connectivity estimates. Neuroimage. 83:550–558. doi: 10.1016/j.neuroimage.2013.05.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boluda S, Iba M, Zhang B, Raible KM, Lee VM-Y, Trojanowski JQ. 2015. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 129(2):221–237. doi: 10.1007/s00401-014-1373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. 1985. On areas of transition between entorhinal allocortex and temporal isocortex in the human brain. Normal morphology and lamina-specific pathology in Alzheimer’s disease. Acta Neuropathol. 68(4):325–332. doi: 10.1007/BF00690836. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. 1992. The human entorhinal cortex: normal morphology and lamina-specific pathology in various diseases. Neurosci Res. 15(1):6–31. doi: 10.1016/0168-0102(92)90014-4. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. 1995. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 16(3):271–278. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- Cabeza R, Anderson ND, Locantore JK, McIntosh AR. 2002. Aging gracefully: compensatory brain activity in high-performing older adults. Neuroimage. 17(3):1394–1402. doi: 10.1006/nimg.2002.1280. [DOI] [PubMed] [Google Scholar]
- Cabeza R, Albert M, Belleville S, Craik FIM, Duarte A, Grady CL, Lindenberger U, Nyberg L, Park DC, Reuter-Lorenz PA, et al. 2018. Maintenance, reserve and compensation: the cognitive neuroscience of healthy ageing. Nat Rev Neurosci. 19(11):701. doi: 10.1038/s41583-018-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Wang J-H, Dai Z-J, Cao X-Y, Jiang L-L, Fan F-M, Song X-W, Xia M-R, Shu N, Dong Q, et al. 2014. Topological organization of the human brain functional connectome across the lifespan. Dev Cogn Neurosci. 7:76–93. doi: 10.1016/j.dcn.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carp J, Park J, Hebrank A, Park DC, Polk TA. 2011. Age-related neural dedifferentiation in the motor system. Plos One. 6(12):Article 12. doi: 10.1371/journal.pone.0029411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassady K, Gagnon H, Freiburger E, Lalwani P, Simmonite M, Park DC, Peltier SJ, Taylor SF, Weissman DH, Seidler RD, et al. 2020. Network segregation varies with neural distinctiveness in sensorimotor cortex. Neuroimage. 212:116663. doi: 10.1016/j.neuroimage.2020.116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassady K, Gagnon H, Lalwani P, Simmonite M, Foerster B, Park D, Peltier SJ, Petrou M, Taylor SF, Weissman DH, et al. 2019. Sensorimotor network segregation declines with age and is linked to GABA and to sensorimotor performance. Neuroimage. 186:234–244. doi: 10.1016/j.neuroimage.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassady K, Ruitenberg MFL, Reuter-Lorenz PA, Tommerdahl M, Seidler RD. 2020. Neural dedifferentiation across the lifespan in the motor and somatosensory systems. Cereb Cortex. 30(6):3704–3716. doi: 10.1093/cercor/bhz336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MY, Park DC, Savalia NK, Petersen SE, Wig GS. 2014. Decreased segregation of brain systems across the healthy adult lifespan. Proc Natl Acad Sci U S A. 111(46):E4997–E5006. doi: 10.1073/pnas.1415122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoiseaux JS. 2017. Effects of aging on functional and structural brain connectivity. Neuroimage. 160:32–40 doi: 10.1016/j.neuroimage.2017.01.077. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, et al. 2012. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 73(4):685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg DA, Setsompop K. 2013. Ultra-fast MRI of the human brain with simultaneous multi-slice imaging. J Magn Reson. 229:90–100. doi: 10.1016/j.jmr.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmeier N, Neitzel J, Rubinski A, Smith R, Strandberg O, Ossenkoppele R, Hansson O, Ewers M. 2020. Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat Commun. 11(1):1–17. doi: 10.1038/s41467-019-14159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmeier N, Rubinski A, Neitzel J, Kim Y, Damm A, Na DL, Kim HJ, Lyoo CH, Cho H, Finsterwalder S, et al. 2019. Functional connectivity associated with tau levels in ageing, Alzheimer’s, and small vessel disease. Brain. 142(4):1093–1107. doi: 10.1093/brain/awz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallen CL, Turner GR, Adnan A, D’Esposito M. 2016. Reconfiguration of brain network architecture to support executive control in aging. Neurobiol Aging. 44:42–52. doi: 10.1016/j.neurobiolaging.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerligs L, Renken RJ, Saliasi E, Maurits NM, Lorist MM. 2015. A brain-wide study of age-related changes in functional connectivity. Cereb Cortex. 25(7):1987–1999. doi: 10.1093/cercor/bhu012. [DOI] [PubMed] [Google Scholar]
- Grady C, Sarraf S, Saverino C, Campbell K. 2016. Age differences in the functional interactions among the default, frontoparietal control, and dorsal attention networks. Neurobiol Aging. 41:159–172. doi: 10.1016/j.neurobiolaging.2016.02.020. [DOI] [PubMed] [Google Scholar]
- Gregory S, Long JD, Tabrizi SJ, Rees G. 2017. Measuring compensation in neurodegeneration using MRI. Curr Opin Neurol. 30(4):380–387. doi: 10.1097/WCO.0000000000000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison TM, Maass A, Adams JN, Du R, Baker SL, Jagust WJ. 2019. Tau deposition is associated with functional isolation of the hippocampus in aging. Nat Commun. 10(1):1–12. doi: 10.1038/s41467-019-12921-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inhoff MC, Ranganath C. 2017. Dynamic cortico-hippocampal networks underlying memory and cognition: the PMAT framework. In The Hippocampus from Cells to Systems. Cham: Springer. pp. 559–589. doi: 10.1007/978-3-319-50406-3_18 [DOI] [Google Scholar]
- Iordan AD, Cooke KA, Moored KD, Katz B, Buschkuehl M, Jaeggi SM, Jonides J, Peltier SJ, Polk TA, Reuter-Lorenz PA. 2018. Aging and network properties: stability over time and links with learning during working memory training. Front Aging Neurosci. 9:1–18. doi: 10.3389/fnagi.2017.00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs HIL, Hedden T, Schultz AP, Sepulcre J, Perea RD, Amariglio RE, Papp KV, Rentz DM, Sperling RA, Johnson KA. 2018. Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat Neurosci. 21(3):424–431. doi: 10.1038/s41593-018-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. 2018. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci. 19(11):687. doi: 10.1038/s41583-018-0067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI. 2018. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 136(1):57–67. doi: 10.1007/s00401-018-1855-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Nilakantan AS, Hermiller MS, Palumbo RT, VanHaerents S, Voss JL. 2018. Selective and coherent activity increases due to stimulation indicate functional distinctions between episodic memory networks. Sci Adv. 4(8):eaar2768. doi: 10.1126/sciadv.aar2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BR, van Ruitenbeek P, Leunissen I, Cuypers K, Heise K-F, Santos Monteiro T, Hermans L, Levin O, Albouy G, Mantini D, et al. 2018. Age-related declines in motor performance are associated with decreased segregation of large-scale resting state brain networks. Cereb Cortex. 28(12):4390–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koen JD, Hauck N, Rugg MD. 2019. The relationship between age, neural differentiation, and memory performance. J Neurosci. 39(1):149–162. doi: 10.1523/JNEUROSCI.1498-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koen JD, Rugg MD. 2019. Neural dedifferentiation in the aging brain. Trends Cogn Sci. 23(7):547–559. doi: 10.1016/j.tics.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalwani P, Gagnon H, Cassady K, Simmonite M, Peltier S, Seidler RD, Taylor SF, Weissman DH, Polk TA. 2019. Neural distinctiveness declines with age in auditory cortex and is associated with auditory GABA levels. NeuroImage, 201116033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux L, Salek-Haddadi A, Lund TE, Laufs H, Carmichael D. 2007. Modelling large motion events in fMRI studies of patients with epilepsy. Magn Reson Imaging. 25(6):894–901. doi: 10.1016/j.mri.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Lin C, Ly M, Karim HT, Wei W, Snitz BE, Klunk WE, Aizenstein HJ. 2020. The effect of amyloid deposition on longitudinal resting-state functional connectivity in cognitively normal older adults. Alzheimer’s Res Ther. 12(1):7. doi: 10.1186/s13195-019-0573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart SN, Schöll M, Baker SL, Ayakta N, Swinnerton KN, Bell RK, Mellinger TJ, Shah VD, O’Neil JP, Janabi M, et al. 2017. Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage. 150:191–199. doi: 10.1016/j.neuroimage.2017.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. 1996. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 16(5):834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Maass A, Berron D, Harrison TM, Adams JN, La Joie R, Baker S, Mellinger T, Bell RK, Swinnerton K, Inglis B, et al. 2019. Alzheimer’s pathology targets distinct memory networks in the ageing brain. Brain. 142(8):2492–2509. doi: 10.1093/brain/awz154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, Rabinovici GD, Jagust WJ. 2017. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage. 157:448–463. doi: 10.1016/j.neuroimage.2017.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monge ZA, Geib BR, Siciliano RE, Packard LE, Tallman CW, Madden DJ. 2017. Functional modular architecture underlying attentional control in aging. Neuroimage. 155:257–270. doi: 10.1016/j.neuroimage.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monge ZA, Stanley ML, Geib BR, Davis SW, Cabeza R. 2018. Functional networks underlying item and source memory: shared and distinct network components and age-related differences. Neurobiol Aging. 69:140–150. doi: 10.1016/j.neurobiolaging.2018.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Brandel MG, Madison CM, Rabinovici GD, Marks S, Baker SL, Jagust WJ. 2012. Not quite PIB-positive, not quite PIB-negative: slight PIB elevations in elderly normal control subjects are biologically relevant. Neuroimage. 59(2):1152–1160. doi: 10.1016/j.neuroimage.2011.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Tredici KD, et al. 2012. Correlation of Alzheimer disease Neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 71(5):362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JL, O’Keefe KM, LaViolette PS, DeLuca AN, Blacker D, Dickerson BC, Sperling RA. 2010. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline(e–pub ahead of print). Neurology. 74(24):1969–1976. doi: 10.1212/WNL.0b013e3181e3966e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist S, Schöll M, Strandberg O, Mattsson N, Stomrud E, Zetterberg H, Blennow K, Landau S, Jagust W, Hansson O. 2017. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun. 8(1):1214. doi: 10.1038/s41467-017-01150-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DHW, Noble W, Hanger DP. 2013. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14(4):389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Mitra A, Laumann TO, Snyder AZ, Schlaggar BL, Petersen SE. 2014. Methods to detect, characterize, and remove motion artifact in resting state fMRI. Neuroimage. 84:1–45. doi: 10.1016/j.neuroimage.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, DeKosky ST, Mathis CA. 2005. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh compound-B. J Cereb Blood Flow Metab. 25(11):1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- Ranganath C, Ritchey M. 2012. Two cortical systems for memory-guided behaviour. Nat Rev Neurosci. 13(10):713–726. doi: 10.1038/nrn3338. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Shulman MB, Torossian C, Leng L, Zhu W. 2010. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 6(1):11–24. doi: 10.1016/j.jalz.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw PJ, Leroy AM. 2005. Robust regression and outlier detection (vol. 589). John Wiley & Sons. [Google Scholar]
- Rousset OG, Ma Y, Evans AC. 1998. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 39(5):904–911. [PubMed] [Google Scholar]
- Salathé M, Jones JH. 2010. Dynamics and control of diseases in networks with community structure. PLoS Comput Biol. 6(4):e1000736. doi: 10.1371/journal.pcbi.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salthouse TA. 2009. When does age-related cognitive decline begin? Neurobiol Aging. 30(4):507–514. doi: 10.1016/j.neurobiolaging.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller E, Minkova L, Leitner M, Klöppel S. 2014. Attempted and successful compensation in preclinical and early manifest neurodegeneration—a review of task FMRI studies. Front Psych. 5:132. doi: 10.3389/fpsyt.2014.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz AP, Chhatwal JP, Hedden T, Mormino EC, Hanseeuw BJ, Sepulcre J, Huijbers W, LaPoint M, Buckley RF, Johnson KA, et al. 2017. Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J Neurosci. 37(16):4323–4331. doi: 10.1523/JNEUROSCI.3263-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Rüb U, Orantes M, Braak H. 2002. Phases of a beta-deposition in the human brain and its relevance for the development of AD. Neurology. 58(12):1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Todd N, Moeller S, Auerbach EJ, Yacoub E, Flandin G, Weiskopf N. 2016. Evaluation of 2D multiband EPI imaging for high-resolution, whole-brain, task-based fMRI studies at 3T: sensitivity and slice leakage artifacts. Neuroimage. 124(Pt A):32–42. doi: 10.1016/j.neuroimage.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hooren RWE, Riphagen JM, Jacobs HIL, Initiative A’s DN. 2018. Inter-network connectivity and amyloid-beta linked to cognitive decline in preclinical Alzheimer’s disease: a longitudinal cohort study. Alzheimer’s Res Ther. 10(1):88. doi: 10.1186/s13195-018-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, Madison C, Ayakta N, Ghosh PM, La Joie R, Arthur-Bentil SK, Vogel JW, Marks SM, et al. 2015. Existing Pittsburgh compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain. 138(7):2020–2033. doi: 10.1093/brain/awv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JW, Iturria-Medina Y, Strandberg OT, Smith R, Levitis E, Evans AC, Hansson O. 2020. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat Commun. 11(1):2612. doi: 10.1038/s41467-020-15701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington A, Sharp DJ, Gunn RN, Alzheimer’s Disease Neuroimaging Initiative . 2018. Spatiotemporal distribution of β-amyloid in Alzheimer disease is the result of heterogeneous regional carrying capacities. J Nucl Med. 59(5):822–827. doi: 10.2967/jnumed.117.194720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wig GS. 2017. Segregated systems of human brain networks. Trends Cogn Sci. 21(12):981–996. doi: 10.1016/j.tics.2017.09.006. [DOI] [PubMed] [Google Scholar]
- Woodard JL, Seidenberg M, Nielson KA, Smith JC, Antuono P, Durgerian S, Guidotti L, Zhang Q, Butts A, Hantke N, et al. 2010. Prediction of cognitive decline in healthy older adults using fMRI. J Alzheimer’s Dis. 21(3):871–885. doi: 10.3233/JAD-2010-091693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RACM, et al. 2016. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 19(8):1085–1092. doi: 10.1038/nn.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgräfe K, Mandelkow E-M, et al. 2014. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 211(3):387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar JH. 1996. Biostatistical analysis. Upper Saddle River: Prentice Hall, Inc. [Google Scholar]
- Zhan L, Jenkins LM, Wolfson OE, GadElkarim JJ, Nocito K, Thompson PM, Ajilore OA, Chung MK, Leow AD. 2017. The significance of negative correlations in brain connectivity. J Comp Neurol. 525(15):3251–3265. doi: 10.1002/cne.24274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.