

Abstract
Objective
To examine outcomes in people with multiple sclerosis (PwMS) treated with autologous hematopoietic stem cell transplantation (AHSCT) in a real-world setting.
Methods
This was a retrospective cohort study of PwMS treated with AHSCT at 2 centers in London, UK, consecutively between 2012 and 2019 who had ≥6 months of follow-up or died at any time. Primary outcomes were survival free of multiple sclerosis (MS) relapses, MRI new lesions, and worsening of Expanded Disability Status Scale (EDSS) score. Adverse events rates were also examined.
Results
The cohort includes 120 PwMS; 52% had progressive MS (primary or secondary) and 48% had relapsing-remitting MS. At baseline, the median EDSS score was 6.0; 90% of the evaluable cases showed MRI activity in the 12 months preceding AHSCT. Median follow-up after AHSCT was 21 months (range 6–85 months). MS relapse-free survival was 93% at 2 years and 87% at 4 years after AHSCT. No new MRI lesions were detected in 90% of participants at 2 years and in 85% at 4 years. EDSS score progression–free survival (PFS) was 75% at 2 years and 65% at 4 years. Epstein-Barr virus reactivation and monoclonal paraproteinemia were associated with worse PFS. There were 3 transplantation-related deaths within 100 days (2.5%), all after fluid overload and cardiac or respiratory failure.
Conclusions
Efficacy outcomes of AHSCT in this real-world cohort are similar to those reported in more stringently selected clinical trial populations, although the risks may be higher.
Classification of Evidence
This study is rated Class IV because of the uncontrolled, open-label design.
Autologous hematopoietic stem cell transplantation (AHSCT) has increasingly been used in recent years as treatment for multiple sclerosis (MS) and other severe autoimmune diseases, on the basis of the hypothesis that the procedure may reset the immune system and stop the inflammatory attack.1 Clinical studies have demonstrated profound suppression of MS activity2-4 and long-term clinical stabilisation.5 Refinement of patient selection and treatment protocols has reduced treatment-related toxicity and mortality.6 Clinically based criteria have helped to define the optimal patient profile for whom AHSCT could be considered an appropriate treatment option.1 A recent position statement from the American Society for Blood and Marrow Transplantation and treatment guidelines from the European Bone Marrow Transplantation Society (EBMT) recommend considering AHSCT in selected people with MS (PwMS).7,8 Two small randomized controlled trials (RCTs) in relapsing MS provide proof of principle of higher efficacy of AHSCT compared to standard treatment on MRI9 and clinical outcomes.10
The objective of this observational study is to examine clinical efficacy outcomes and adverse events in a cohort of PwMS treated with AHSCT according to standard clinical practice at the 2 lead centers in London, UK. We also investigated the variables associated with the outcomes. Because symptomatic Epstein-Barr virus (EBV) reactivation increases the risk of neurologic sequelae and the monitoring of EBV-associated monoclonal paraprotein after AHSCT has recently been recommended,11,12 we also examined the relationship between EBV reactivation and monoclonal paraprotein formation in the AHSCT-treated MS cohort. Furthermore, we explored risk factors, including treatment-related morbidity, on efficacy outcomes.
Methods
Patient Selection
Data were collected retrospectively on consecutive PwMS who underwent AHSCT for treatment of MS between February 15, 2012, and January 2019 at Kings College Hospital (KCH) and from April 12, 2016, to January 2019 at Hammersmith Hospital (HH) (London) and had at least 6 months of follow-up or died at any time. All eligible patients were included in the retrospective analysis. Initially (2012–2015), the indication for AHSCT was based on the agreement of at least 2 neurologists and 1 hematologist with expertise in AHSCT that the treatment was in the patient's best interest in the absence of appropriate treatment alternatives. Eligibility criteria for treatment were formally defined in September 2015 to select patients with a profile consistent with inflammatory active MS and, for relapsing-remitting MS (RRMS), treatment-refractory disease. The treatment inclusion and exclusion criteria for AHSCT that were implemented since September 2015 are summarized in table 1. Eligibility had to be approved by a multidisciplinary team (MDT) comprising neurologists and transplantation hematologists from a number of independent centers in London and vicinity with established or developing expertise in AHSCT for MS. The MDT reviewed the clinical information collected in a referral form and discussed each case to assess eligibility by consensus. For some cases who did not strictly meet all the inclusion criteria but convincingly fulfilled the overall profile of eligibility (table 1), the MDT made the clinical decision to offer AHSCT, documenting the specific basis for the approval. For this study, MRI reports were audited, and in some cases, scans were reviewed to examine any inconsistency. The patient flow, including the reasons for exclusion from treatment and from study analysis, is shown in the Consolidated Standards of Reporting Trials diagram (figure 1). The database was locked for analysis in July 2019, and statistical analysis was completed in December 2019.
Table 1.
Profile of Eligibility for Treatment With AHSCT
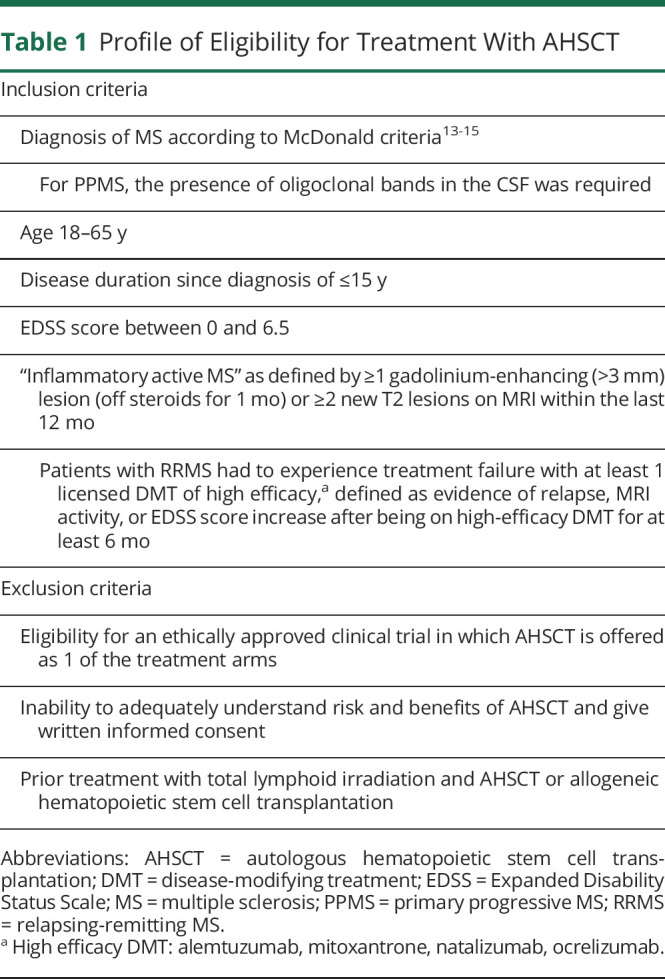
Figure 1. Consolidated Standards of Reporting Trials Diagram of Enrollment in the Study.

Patient disposition is shown in the flowchart with information available about the reasons for exclusion from treatment with autologous hematopoietic stem cell transplantation (AHSCT) or from the cohort analysis. DMT = disease-modifying treatment.
Standard Protocol Approvals, Registrations, and Patient Consents
All patients signed informed consent forms before initiating the treatment procedure and to give agreement to data collection and analysis. In line with standard practice, all data were also reported to the EBMT registry database.
Treatment Procedure
Patients underwent AHSCT according to the approved protocols at the 2 centers. At KCH, peripheral blood stem cells (PBSCs) were mobilized after administration of cyclophosphamide 4 g/m2 over 2 days (62 patients) or cyclophosphamide 2 g/m2 over 1 day (3 patients) after a modification in the protocol in November 2018, with granulocyte colony-stimulating factor (G-CSF; 5 μg/kg SC) for 7 days until leukapheresis. Conditioning was performed with a standard protocol of cyclophosphamide (50 mg/kg for 4 days) and rabbit anti-thymocyte globulin (rATG; 2.5 mg/kg/d for 3 days, total dose 7.5 mg/kg) for in vivo lymphodepletion followed by stem cell infusion. One patient, at the start of the KCH program, was conditioned with a carmustine/etoposide/cytarabine/melphalan regimen plus an equivalent dose of rATG (BEAM [BiCNU (carmustine), etoposide, Ara-C (cytarabine), melphalan]-ATG) before stem cell infusion. The median CD34 stem cell dose returned was 7.17 × 106/kg (range 4.0–17.1 × 106/kg). At HH, PBSCs were mobilized (52 patients) with cyclophosphamide 2 g/m2 and daily G-CSF (5 μg/kg SC) starting from day +3 from cyclophosphamide until leukapheresis. Transplantation conditioning used cyclophosphamide (50 mg/kg for 4 days) and rATG (2 mg/kg/d, total dose 6 mg/kg in 48 cases or 2.5 mg/kg/d, total dose 7.5 mg/kg in 4 cases after a protocol change in August 2018). One patient was mobilized with cyclophosphamide 1 g/m2 followed by G-CSF and plerixafor (2 doses) and conditioned with BEAM-ATG due to intolerance of cyclophosphamide. At both centers, the collected product was not CD34 selected or otherwise manipulated ex vivo. The median CD34+ cell dose in the cryopreserved PBSC product was 7.75 × 106/kg (range 2.2–24.3 × 106/kg). After conditioning and reinfusion of the autologous PBSC product, G-CSF was administered starting from day 7 after AHCST until engraftment to half of the patients from KCH and to all the patients from HH.
Supportive medical treatments (including platelet and packed red cell transfusions, antimicrobial prophylaxis, dietetics, and physiotherapy support) were provided during the inpatient period as per standard institutional protocols. Psychological support was available for all inpatients.
Clinical and MRI Assessments
Clinical assessments were performed according to standard clinical practice at the centers. To qualify as events, relapses had to be confirmed by a clinician and recorded in the case notes. Changes in the Expanded Disability Status Scale (EDSS) scores in the short term before/after AHSCT were evaluated over the 24-month period comprising 12 months before AHSCT to 12 months after treatment. EDSS score progression was defined as an increase in EDSS score by 0.5 point if baseline EDSS score was ≥6 and by 1 point if baseline EDSS score was <6. MRI scans were performed as per local protocols. MRI analysis, including new lesion counts, was based on the neuroradiology clinical reports. Patient underwent the first posttransplantation MRI scan on average 6 months (range 1–13 months) after AHSCT. For analyses requiring rebaselining of MRI, the first posttransplantation MRI as described above was considered the new baseline scan, and post-AHSCT MRI activity was evaluated by comparing the subsequent scans obtained during follow-up with this scan.
Statistics
The database of transplantations and outcomes was built in Microsoft Excel 2016 (Microsoft Corp, Redmond, WA), and statistical analyses were performed with IBM SPSS Statistics version 24.0 (IBM Corp, Armonk, NY). Patient characteristics are presented as medians (with interquartile ranges) for data with nonnormal distribution. Comparisons of baseline characteristics were performed with the Mann-Whitney U test, Fisher exact test, or χ2 test for trend as appropriate. The changes in EDSS scores between the 12 months preceding treatment and baseline (premobilization) and between baseline and 12 months after AHSCT were calculated. Pretransplantation and posttransplantation relapse rates were compared by a Wilcoxon test for paired data. Time to first relapse and time to first MRI activity were studied by Kaplan-Meier (KM) survival curves and compared between patients with RRMS and progressive MS by the log-rank test. Confirmed disability worsening was defined when the EDSS score changes defined above were confirmed at 6 months and was assessed by KM curves for univariate analysis and by a Cox model for multivariate analysis. The variables included in the analysis were age, sex, progressive vs relapsing MS subtype, total number of previous disease-modifying treatments (DMTs), number high-efficacy DMT, EBV reactivation, time of EBV reactivation, highest EBV copy number, and detection of serum paraprotein (paraproteinemia). The effect of baseline variables on the risk of developing paraproteinemia was studied by a multivariate Cox model.
Data Availability
Any data not published within the article will be shared in anonymized form on request from a qualified investigator.
Results
Pre-AHSCT MS Disease Characteristics
One hundred twenty PwMS were included in the study. Their demographic and clinical features at baseline are presented in table 2. Sixty-two (52%) cases had a progressive MS phenotype (primary progressive [PPMS] or secondary progressive [SPMS]). At acceptance for AHSCT, 90% of the patients for whom data were available had evidence of MRI activity in the preceding 12 months, demonstrated by new T2 or gadolinium-enhancing lesions (table 2); 85% of the evaluable patients had developed ≥1 T2 lesions, and 59% had a gadolinium-enhancing lesion on MRI.
Table 2.
Demographics and Disease Characteristics at Baseline

The cohort had a mean age of 42.3 years and median EDSS score of 6.0. There was no difference in the mean age or EDSS score in the PwMS treated at the 2 centers. Patients with RRMS had received an average of 2.3 previous DMTs (SD 1.3); those with SPMS, 1.6 treatments (SD 1.1). Seventy (58%) patients had been treated before AHSCT with ≥1 DMT regarded as high efficacy (alemtuzumab, mitoxantrone, natalizumab, ocrelizumab), among whom 19 patients had alemtuzumab and 58 had natalizumab (7 had both). The median duration of follow-up after AHSCT, with the autologous graft infusion being day 0, was 21 months (range 6–85 months).
AHSCT Admission and Engraftment
The median duration of hospital inpatient admission was 22 days (mean ± SD 25.3 ± 9.7 days). Median time to neutrophil engraftment was 12 days (mean 12.3 ± 9.2.55 days). There were center differences: the duration of inpatient admission was longer at KCH (median 26 days, mean 27.5 ± 9.7 days) than at HH (median 20 days, p = 0.00097, Wilcoxon test [w = 2786.5]; mean 21.7 ± 8.0 days, p = 0.00052, t test). The mean time to neutrophil engraftment was also longer at KCH (median 13 days, mean 13.2 ± 2.5 days; 1 missing) than at HH (median 11 days, p = 0.00072, Wilcoxon test [w = 2695.5]; mean 11.1 ± 2.1 days, p = 0.0064, t test; 1 missing).
Neurologic Outcomes After AHSCT
Relapses
The relapse rate in the study population was compared before and after AHSCT. The overall annualized relapse rate dropped from 0.46 ± 0.57 in the 2 years before AHSCT to 0.08 ± 0.38 in the post-AHSCT follow-up at 4 years (p < 0.001, Wilcoxon test; figure 2A). Ninety-three percent of all cases were free from relapse at 2 years after AHSCT; 87% were free from relapse at 4 years after AHSCT. Relapses after AHSCT occurred only in patients with RRMS, and in that subgroup, relapse-free survival was 87% at 2 years and 77% at 4 years.
Figure 2. MS Disease Outcomes: Relapse Rate, MRI New Lesions, and Change in EDSS Score.

(A) Annualized relapse rates (ARRs) over 2 years before autologous hematopoietic stem cell transplantation (AHSCT) and over up to 4 years after AHSCT demonstrate a significant reduction (mean ± 95% confidence interval, p < 0.001). (B) New MRI T2 lesion development over 1 year before and over up to 4 years was categorized, and the comparison demonstrated a significant reduction (χ2, p < 0.001). (C) Change in Expanded Disability Status Scale (EDSS) score before and after AHSCT over 12 months before transplantation/12 months after transplantation compared to treatment baseline (day 0 being transplantation day) examined in the total population. (D) Subgroup analysis of change in EDSS score shows a difference in the relapsing vs progressive multiple sclerosis (MS) subgroups (p < 0.05, Mann-Whitney test).
MRI Lesions
Annualized MRI new T2 lesion numbers without rebaselining were compared between the 12 months before AHSCT and the available follow-up of up to 4 years after AHSCT; the last MRI was performed after a mean of 22 months (SD 17.6 months). There was a significant reduction in new T2 lesions after AHSCT in the whole evaluable population (p < 0.0001, χ2 test; figure 2B). At survival analysis, 90% of participants were free of new lesions at 2 years and 85% at 4 years. In contrast to relapses, there was no difference in the development of new T2 lesions between the RRMS and progressive MS subgroups (data not shown).
Neurologic Disability
To first evaluate the short-term evolution of neurologic disability, the change in EDSS scores between 12 months before AHSCT and baseline (premobilization) was compared with the change between baseline and 12 months after AHSCT. In the whole population analysis, the average EDSS score change was 0.25 during the 12 months before AHSCT and 0.02 in the 12 months after therapy (figure 2C). In the subgroup analysis, a clear difference emerged, with patients with RRMS showing on average a small improvement 12 months after AHSCT compared to baseline; in contrast, the progressive MS subgroup showed further deterioration (RRMS subgroup: EDSS score change 0.39 before AHSCT and −0.17 after transplantation; progressive subgroup: 0.11 before AHSCT and 0.24 after AHSCT; p < 0.05; figure 2D).
We next examined the evolution of disability in the longer term, assessed as EDSS score worsening confirmed at 6 months or death during the entire follow-up. Seventy-five percent of the whole population did not have confirmed EDSS score worsening at 2 years; the proportion decreased to 65% at the 4-year follow-up. There was no significant difference between the RRMS and progressive MS subgroups (p = 0.487, log-rank test; figure 3A). In the RRMS subgroup, 13 participants (including 1 death) had confirmed disability worsening. In the progressive MS subgroup, 15 participants had confirmed disability worsening: 8 with SPMS and 7 (including 2 deaths) with PPMS. Using confirmed EDSS score worsening as an outcome, we explored factors that could predict failure of AHSCT. Demographics, disease phenotype, number and type of previous treatments, and adverse events were included as variables in the analyses. Univariate and multivariate Cox analyses identified high (>5 g/L) paraproteinemia as the only significant variable associated with confirmed EDSS score progression over 4 years (odds ratio 1.07 [95% confidence interval 1.03–1.10], p < 0.001; KM plot in figure 3B). Further modeling indicated that paraprotein levels were not predictive of relapses (odds ratio 0.96 [95% confidence interval 0.76–1.20], p = 0.67) or new T2 lesions on rebaselined MRI for up to 4 years (0.93 [95% confidence interval 0.72–1.21]).
Figure 3. Survival Analyses of MS Outcomes in the Longer Term.

(A) Kaplan-Meier (KM) analysis of time to confirmed Expanded Disability Status Scale (EDSS) score worsening in the relapsing-remitting (RR) multiple sclerosis (MS), secondary progressive MS (SPMS), and primary progressive (PPMS) subtypes. (B) KM of time to confirmed EDSS score worsening according to paraproteinemia (red line, none; green line, <5 g/L; blue line, ≥5 g/L) illustrates the association detected by multivariate analysis (reported in Results). (C) KM of no evidence of disease activity-3 (NEDA-3) and its components: time to relapse, to new T2 MRI lesion, and to confirmed EDSS score progression in the whole population. (D) KM of NEDA-3 in the RRMS, SPMS, and PPMS subgroups.
No Evidence of Disease Activity
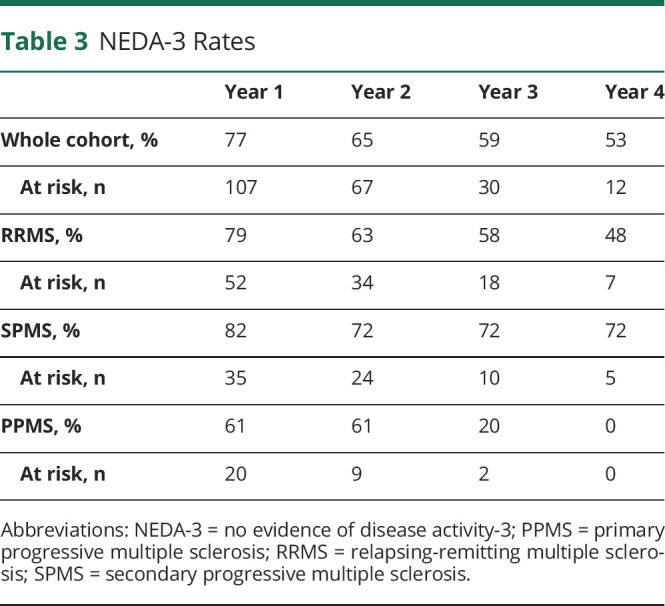
No evidence of disease activity (NEDA) has increasingly been used to demonstrate effects of treatment in MS. NEDA fulfilling the 3 endpoints—no relapses, no new MRI lesions, and no worsening of neurologic disability—is denominated NEDA-3. In this study, data enabling calculation of NEDA-3 (without MRI rebaselining) were available for the majority of cases (107 of 120, including the 3 deaths, considered events in the analysis). The survival analysis of NEDA-3 and its components in the whole cohort is shown in figure 3C. The survival analysis of time to loss of NEDA-3 in the RRMS, SPMS, and PPMS subgroups is presented in figure 3D; the yearly NEDA-3 rates to year 4 after AHSCT are supplied in table 3.
Table 3.
NEDA-3 Rates
Adverse Events After AHSCT
Early Complications
Almost 90% of the treated patients experienced at least 1 early complication after AHSCT. There were differences in the rates of adverse events between the 2 centers, described in table 4. After mobilization, fever/positive culture/neutropenia and readmission rates were higher in the KCH cohort, possibly related to the higher cyclophosphamide dose (4 g/m2 vs 2 g/m2). For conditioning/HSCT, the KCH cohort more frequently experienced fever, diarrhea, and EBV reactivation, whereas severe nausea and vomiting were higher in the HH cohort.
Table 4.
Side Effects of Mobilization and Conditioning Treatments at the 2 Study Centers

Transplantation-Related Mortality
There were 3 deaths (2.5%) within 100 days from transplantation. Two of the participants had PPMS (1 female and 1 male patient, age at death 58 and 42 years, respectively) and 1 had RRMS (female, age 51 years), and all 3 had an EDSS score of 6.5 at baseline. Further clinical and treatment details are available from Dryad (table e1, doi.org/10.5061/dryad.k0p2ngf82). Two deaths occurred the day before the planned autologous stem cell infusion; in both cases, the primary cause was cardiac arrest, and secondary causes were recent pulmonary edema in 1 case and blood electrolyte abnormalities in the other. In the third case, death occurred 32 days after stem cells had been reinfused and was caused by acute respiratory distress syndrome secondary to chest infection and sepsis. To investigate the pathophysiologic basis of these events, a detailed retrospective analysis was undertaken in the whole treated cohort. With potential relevance to the mortality events, fluid overload (defined by >5% weight gain ± peripheral or central edema and need for additional diuretics) was recorded in 78 of 118 patients (66%) and presented at a mean of 3 days (±2.2 days SD) after the first dose of ATG. Further data on fluid overload are available from Dryad (table e2).
Viral Reactivations and Paraprotein Formation in the Patient Cohort
Cytomegalovirus reactivation was detected in 26 cases, and preemptive treatment with valganciclovir or ganciclovir was required in 12 of 26 (46%) cases with no CMV disease observed. EBV serologic status before AHSCT was assessed in a subset of 66 of 120 patients (55%), mainly due to different testing policies in KCH and HH, and was positive in all cases except for 1 indeterminate participant and 1 negative participant. In a subset of 85 participants, EBV DNA copy numbers in blood and paraprotein were measured regularly by standardized laboratory techniques at both sites. EBV reactivation (defined by viremia >10 DNA copies/mL consecutively, as previously described11) was demonstrated in 87 of 109 (80%; 11 missing/not tested) of participants after AHSCT. Of the 87 EBV reactivation cases, 20 (23%) cases were treated with rituximab in a median of 4 courses (range 2–4). Hypogammaglobulinemia was detected in 7 of 20 rituximab-treated cases. In a stepwise multivariate Cox analysis, the following variables were associated with risk of developing paraprotein: lower baseline EDSS score (p = 0.018), symptoms consistent with viral reactivation (p = 0.001), lower EBV DNAemia at the date of first reactivation (<500,000 copies/mL; p = 0.036), and peak EBV DNAemia (>500,000 copies/mL; p = 0.017). The multivariate analysis is available from Dryad (table e3, doi.org/10.5061/dryad.k0p2ngf82).
Late Adverse Events
Seven patients (5.8%) developed secondary autoimmune diseases (6 thyroiditis and 1 case of autoimmune thrombocytopenia) after a median of 17.5 months (range 6–36 months) after transplantation. One of these patients had previously been treated with alemtuzumab. One patient was diagnosed with melanoma 16 months after AHSCT; this patient had previously received 44 weekly doses of natalizumab. Apart from these individual cases, there was no association of prior DMT with adverse events.
Discussion
Increasing evidence supports considering AHSCT as a treatment for patients with aggressive, inflammatory forms of MS.7,8 Studies in RRMS have demonstrated that AHSCT markedly reduces relapse rates and lesion development and improves disability.1 Two RCTs have been reported with encouraging results,9,10 and more definitive RCTs comparing AHSCT with contemporary therapies, including high-efficacy biologicals, are underway. The American Society for Blood and Marrow Transplantation and the EBMT recommend AHSCT as a clinical option for treatment of patients with active relapsing MS, particularly when standard therapy has failed.7,8 The role of AHSCT is less clear in progressive MS, with poor outcomes in participants with advanced disease,16,17 although some evidence led to the question of whether, among patients with earlier progressive disease and ongoing inflammatory MS, the rate of progression of disability might be attenuated after therapy.5 Regarding the safety of AHSCT, current data suggest that the treatment-related risk is higher than with standard DMT but is largely front-loaded as opposed to the poorly understood long-term risk of chronic immune suppression induced by biological therapies, and the risk may be partly offset by higher efficacy against neuroinflammation.18
In this study, we report the results of a retrospective analysis of data from 120 patients treated with AHSCT as part of standard care. Different from recent and ongoing clinical trials, our cohort included a substantial proportion (≈50%) of patients with progressive MS, and any type and number of prior treatments were allowed, with the exclusion of total lymphoid irradiation and AHSCT or allogeneic HSCT. Selection of patients who could be offered AHSCT evolved during the survey period and became more restrictive with the introduction in September 2015 of an eligibility profile that included upper limits to age (65 years), disease duration (15 years), and neurologic disability (EDSS score 6.5). The criteria also required evidence of inflammatory disease activity by MRI demonstrating new T2 lesions or the presence of gadolinium enhancement. Even with these refinements, the patient cohort was less stringently selected than in most trials of treatments in MS.
As efficacy outcomes, we examined MS relapses and MRI and EDSS score evolution. Relapses were significantly suppressed after AHSCT compared to before transplantation. In this real-world treated cohort, the relapse-free proportion of individuals with RRMS (≈80% for up to 4 years after HSCT) was not substantially different from those achieved in clinical trials.4,10,19 In addition, MRI demonstrated almost complete suppression of new lesion development after AHSCT in both the RRMS and progressive MS subgroups. Because persistence or reactivation of MRI would be expected more frequently in patients with RRMS, the results are consistent with a floor effect in both subgroups, reflecting high efficacy of AHSCT against MRI-detectable inflammation in the CNS, as previously demonstrated.20 In regard to neurologic disability, the clinical relevance of ongoing inflammation in patients with RRMS and its radical suppression after AHSCT are the most plausible reasons for the improvement of neurologic function we observed in the RRMS subgroup after AHSCT, consistent with previous reports.2,10
In the longer term, freedom from EDSS score worsening (75% at 2 years and 65% at 4 years) was encouraging, and rates were similar in the RRMS and progressive MS subgroups. This observation does not demonstrate a benefit of AHSCT in patients with progressive MS, but it does suggest a question. After the licensing of DMTs for progressive MS, including ocrelizumab for PPMS and siponimod for SPMS, an RCT could be designed to compare the efficacy, safety, and cost-effectiveness of AHSCT and approved therapy in patients with inflammatory active, progressive MS forms.
We investigated factors associated with progression of EDSS score, and the analysis revealed that high paraproteinemia (≥5 g/L) was a significant factor for EDSS score progression, together with symptomatic EBV reactivation. Particularly in light of the putative association of EBV in the pathogenesis of MS,21 we speculate that in some patients the reactivation of EBV with high viral loads after AHSCT may predispose and contribute, together with other as-yet unknown susceptibility factors, to continued worsening after treatment.11 Development of monoclonal paraprotein could be of interest as a marker of immune dysregulation after EBV reactivation, as well as its potential impact on neurologic disability after AHSCT, as also observed in our cohort, and monitoring is now recommended.11,12
We examined NEDA, and the rates of 65% at 2 years and 53% at 4 years after AHSCT are slightly below the ranges reported in a pooled analysis of AHSCT trials, in which the proportion of individuals with NEDA was 83.4% (range 70%–92%) at 2 years after AHSCT and 67% (range 59%–70%) at 5 years.22 Even in our cohort, of whom half were patients with RRMS who had failed previous treatments that included high-efficacy biologics, the year 2 NEDA rate of 65% was better than that of any other DMT, among which even the most effective did not exceed 50%.1 Of course, these are indirect comparisons and should be used only to generate hypotheses.
In the study cohort, 3 deaths were recorded, which were treatment related and constitute a higher treatment-related mortality (TRM) than reported in recent case series and in any published report that included only patients with RRMS. Two of the patients who died had PPMS; all 3 were at the upper limit of allowed disability with an EDSS score of 6.5, were middle-aged (42, 51, and 58 years), and had comorbid conditions, although minor. All 3 received the same cyclophosphamide conditioning regimen, with some variation of ATG dosage, as shown in data available from Dryad (table e1, doi.org/10.5061/dryad.k0p2ngf82). Higher baseline EDSS score levels and a lower proportion of cases with RRMS have previously been reported as 2 factors associated with TRM.6 In the same meta-analysis, older age and the conditioning regimen intensity were also considered and were not confirmed as significant in the multivariate analysis.22 In a multicenter cohort study of long-term outcomes, higher baseline EDSS score was found to be independently associated with worse overall survival.23 These associations support the notion that patients with higher EDSS score and progressive MS forms are at higher risk of mortality during and after AHSCT.
In the evaluation of the causes of the 3 deaths in our cohort, cardiac adverse events and fluid overload were identified as factors, even though none of these patients were shown to have any impaired cardiorespiratory function at baseline before AHSCT. Fluid overload is an important side effect of conditioning that has not emerged clearly from earlier studies of AHSCT for MS but has recently been identified in a cancer population as a factor contributing to HSCT outcome.24 In our cohort of PwMS, a high incidence of clinically significant fluid overload was seen, likely related to the conditioning regimen used with ATG. To put this in context, the incidence was significantly higher compared to a cohort of patients with acquired aplastic anemia (n = 40) at KCH, which is predominantly an autoimmune disorder and in which, after treatment with horse ATG 40 mg/kg/d for 4 days and cyclosporin A (5–mg/kg daily dose), only 22% developed significant fluid overload after ATG (p < 0.001) despite the equivalent immunosuppressive and fluid retention properties of treatment with ATG and cyclosporin A and the higher median age (52 years) of the patients with aplastic anemia. The reason for the higher incidence of fluid overload in the MS cohort is unclear. We speculate that previous cardiotoxic DMT (mitoxantrone and cyclophosphamide), use of rATG formulation in the MS AHSCT procedures with added high-dose steroids to reduce risk of ATG reaction, and potentially a subclinical form of neuroautonomic dysfunction in patients with MS might be risk factors. For the last hypothesis, because no significant cardiac comorbid conditions were identified in pretreatment standard organ assessments in this cohort, we suggest that a more detailed cardiac (e.g., cardiac injury biomarker monitoring, stress echocardiogram and/or cardiac MRI for detailed structural and functional assessments) and autonomic evaluations (R-R and tilt test ECG) could help identify PwMS at excess risk. Compared to cancer, any TRM of AHSCT in MS is regarded as less acceptable because, in the majority of patients, untreated or ineffectively treated MS is not immediately life threatening. At the population level, in PwMS, survival is reduced on average by 7 years.25 However, at an individual level, the reduction in life expectancy could be considerably more severe particularly in those with aggressive forms of MS such as those included in this cohort. In addition, standard therapy is not free from risks, and although the risk in the short term is almost certainly lower, safety concerns have emerged from longer-term follow-up, prompting withdrawal and limitations of the use of licensed MS therapies.26,27 Early non-TRM complications such as fever, neutropenia, and diarrhea were common in this AHSCT cohort, as expected. Some of the differences observed in adverse event rates between the 2 centers could be related to the higher ATG dose at KCH (7.5 mg/kg vs 6 mg/kg at HH) where a higher incidence of fever, diarrhea, and EBV reactivation was observed. Among late adverse events, secondary autoimmune disease was observed at a rate similar to the ≈5% reported in a larger multicenter cohort study.23
This study has several limitations, including its retrospective design, some variation in treatment protocols, a relatively short follow-up, and lack of a treatment control arm. The heterogeneity of clinical phenotypes, age, disease duration, and EDSS score level is also a challenge in the analysis of outcome data, although such heterogeneity also provides the opportunity to explore AHSCT outcomes in a broader patient population and to examine factors potentially associated with the outcomes, which could not be revealed in the selected populations usually enrolled in clinical trials.
Against the limitations, some important conclusions can be made. The results demonstrate the feasibility of this treatment strategy and provide new information on the potential benefits and risks in a real-world social health care setting. Efficacy outcomes similar to those of clinical trials also can be achieved in real-life patient populations, although risks can be higher, especially in patients with more advanced disease. Furthermore, our study exemplifies a model of service development in the NHS in which innovation can be initiated via MDT collaboration even with minimal funding. Scaling up, long-term sustainability, and optimization of patient pathways, however, require adequate resources.
Acknowledgment
The authors thank Adrian Quintela, Shauncy Fernandez, and Angharad Pryce of the BMT Coordinating Team at HH, Imperial NHS Trust for their help with clinical data retrieval.
Glossary
- AHSCT
autologous hematopoietic stem cell transplantation
- BEAM
BiCNU (carmustine), etoposide, Ara-C (cytarabine), melphalan
- DMT
disease-modifying treatment
- EBMT
European Bone Marrow Transplantation Society
- EDSS
Expanded Disability Status Scale
- G-CSF
granulocyte colony-stimulating factor
- HH
Hammersmith Hospital
- KCH
Kings College Hospital
- KM
Kaplan-Meier
- MDT
multidisciplinary team
- MS
multiple sclerosis
- NEDA
no evidence of disease activity
- PBSC
peripheral blood stem cell
- PPMS
primary progressive MS
- PwMS
people with MS
- rATG
rabbit anti-thymocyte globulin
- RCT
randomized controlled trial
- RRMS
relapsing-remitting MS
- SPMS
secondary progressive MS
- TRM
treatment-related mortality
Appendix 1. Authors
Appendix 2. Coinvestigators
Footnotes
Class of Evidence: NPub.org/coe
Study Funding
The authors are grateful for support from the Multiple Sclerosis Trust to Dr. Silber to audit the HSCT Program.
Disclosure
R.S. Nicholas reports support from advisory boards and travel from Novartis, Roche, and Biogen. He has grant support from the UK MS Society and is a member of a National Institute for Health and Care Excellence Health Technology Assessment Committee. A. Mariottini reports personal fees from Merck-Serono and nonfinancial support from Biogen Idec, Tevan and Novartis. B. Turner has received honoraria and travel grants from and has been a member of advisory boards for Biogen, F. Hoffmann-La Roche Ltd, Merck Serono, Novartis, and Sanofi Genzyme. V. Mehra reports speaker honoraria from Gilead, Pfizer, Jazz Pharma, and Novartis; and research grants from MSD, outside the submitted work. V. Singh-Curry received honoraria or travel bursaries from Biogen, Sanofi, Merck, Celgene, and Roche. A. Scalfari received travel support and compensation for participating to advisory boards from Biogen, Teva, Roche, Sanofi, and Merck. P. Muraro discloses travel support and speaker honoraria from unrestricted educational activities organized by Novartis, Bayer HealthCare, Bayer Pharma, Biogen Idec, Merck-Serono, and Sanofi Aventis; he also discloses consulting to Magenta Therapeutics and Jasper Therapeutics, all activities unrelated to this report. All other authors report no disclosures in connection with this work. Go to Neurology.org/N for full disclosures.
References
- 1.Muraro PA, Martin R, Mancardi GL, Nicholas R, Sormani MP, Saccardi R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat Rev Neurol. 2017;13(7):391-405. [DOI] [PubMed] [Google Scholar]
- 2.Burt RK, Balabanov R, Han X, et al. Association of nonmyeloablative hematopoietic stem cell transplantation with neurological disability in patients with relapsing-remitting multiple sclerosis. JAMA. 2015;313(3):275-284. [DOI] [PubMed] [Google Scholar]
- 3.Atkins HL, Bowman M, Allan D, et al. Immunoablation and autologous haemopoietic stem-cell transplantation for aggressive multiple sclerosis: a multicentre single-group phase 2 trial. Lancet. 2016;388(10044):576-585. [DOI] [PubMed] [Google Scholar]
- 4.Nash RA, Hutton GJ, Racke MK, et al. High-dose immunosuppressive therapy and autologous HCT for relapsing-remitting MS. Neurology. 2017;88(9):842-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muraro PA, Pasquini M, Atkins HL, et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. 2017;74(4):459-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sormani M, Muraro PA, Schiavetti I, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a meta-analysis. Neurology. 2017;88(22):2115-2122. [DOI] [PubMed] [Google Scholar]
- 7.Cohen JA, Baldassari LE, Atkins HL, et al. Autologous hematopoietic cell transplantation for treatment-refractory relapsing multiple sclerosis: position statement from the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transpl. 2019;25(5):845-854. [DOI] [PubMed] [Google Scholar]
- 8.Sharrack B, Saccardi R, Alexander T, et al. Autologous haematopoietic stem cell transplantation and other cellular therapy in multiple sclerosis and immune-mediated neurological diseases: updated guidelines and recommendations from the EBMT Autoimmune Diseases Working Party (ADWP) and the Joint Accreditation Committee of EBMT and ISCT (JACIE). Bone Marrow Transpl. 2020;55(2):283-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mancardi GL, Sormani MP, Gualandi F, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology. 2015;84(10):981-988. [DOI] [PubMed] [Google Scholar]
- 10.Burt RK, Balabanov R, Burman J, et al. Effect of nonmyeloablative hematopoietic stem cell transplantation vs continued disease-modifying therapy on disease progression in patients with relapsing-remitting multiple sclerosis: a randomized clinical trial. JAMA. 2019;321(2):165-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehra V, Rhone E, Widya S, et al. Epstein-Barr virus and monoclonal gammopathy of clinical significance in autologous stem cell transplantation for multiple sclerosis. Clin Infect Dis. 2019;69(10):1757-1763. [DOI] [PubMed] [Google Scholar]
- 12.Christopeit M, Schmidt-Hieber M, Sprute R, et al. Prophylaxis, diagnosis and therapy of infections in patients undergoing high-dose chemotherapy and autologous haematopoietic stem cell transplantation: 2020 update of the recommendations of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Medical Oncology (DGHO). Ann Hematol. 2021;100(2):321-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the Diagnosis of Multiple Sclerosis. Ann Neurol. 2001;50(1):121-127. [DOI] [PubMed] [Google Scholar]
- 14.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria.” Ann Neurol. 2005;58(6):840-846. [DOI] [PubMed] [Google Scholar]
- 16.Burt RK, Cohen BA, Russell E, et al. Hematopoietic stem cell transplantation for progressive multiple sclerosis: failure of a total body irradiation-based conditioning regimen to prevent disease progression in patients with high disability scores. Blood. 2003;102(7):2373-2378. [DOI] [PubMed] [Google Scholar]
- 17.Samijn JP, te Boekhorst PA, Mondria T, et al. Intense T cell depletion followed by autologous bone marrow transplantation for severe multiple sclerosis. J Neurol Neurosurg Psychiatry. 2006;77(1):46-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sormani MP, Muraro PA, Saccardi R, Mancardi G. NEDA status in highly active MS can be more easily obtained with autologous hematopoietic stem cell transplantation than other drugs. Mult Scler. 2017;23(2):201-204. [DOI] [PubMed] [Google Scholar]
- 19.Burman J, Iacobaeus E, Svenningsson A, et al. Autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: the Swedish experience. J Neurol Neurosurg Psychiatry. 2014;85(10):1116-1121. [DOI] [PubMed] [Google Scholar]
- 20.Mancardi GL, Saccardi R, Filippi M, et al. Autologous hematopoietic stem cell transplantation suppresses Gd-enhanced MRI activity in MS. Neurology. 2001;57(1):62-68. [DOI] [PubMed] [Google Scholar]
- 21.Ascherio A, Munger KL, Lünemann JD. The initiation and prevention of multiple sclerosis. Nat Rev Neurol. 2012;8(11):602-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sormani MP, Muraro PA, Schiavetti I, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a meta-analysis. Neurology. 2017;88(22):2115-2122. [DOI] [PubMed] [Google Scholar]
- 23.Muraro PA, Pasquini M, Atkins HL, et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. 2017;74(4):459-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimomura Y, Morita M, Hashimoto H, Ishikawa T. Fluid overload early after allogeneic hematopoietic stem cell transplantation was associated with poor survival. Blood. 2017;130(suppl 1):3233. [Google Scholar]
- 25.Lunde HMB, Assmus J, Myhr KM, Bø L, Grytten N. Survival and cause of death in multiple sclerosis: a 60-year longitudinal population study. J Neurol Neurosurg Psychiatry. 2017;88(8):621-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.European Medicines Agency. Use of multiple sclerosis medicine Lemtrada restricted while EMA review is ongoing. Available at: www.ema.europa.eu/en/documents/press-release/use-multiple-sclerosis-medicine-lemtrada-restricted-while-ema-review-ongoing_en.pdf.
- 27.European Medicines Agency. Zinbryta: withdrawal of the marketing authorisation in the European Union. Available at: www.ema.europa.eu/en/documents/public-statement/public-statement-zinbryta-withdrawal-marketing-authorisation-european-union_en.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data not published within the article will be shared in anonymized form on request from a qualified investigator.