

Abstract
The interphase nuclear envelope (NE) is extensively remodeled during nuclear pore complex (NPC) insertion. How this remodeling occurs and why it requires Torsin ATPases, which also regulate lipid metabolism, remains poorly understood. Here, we show that Drosophila Torsin (dTorsin) affects lipid metabolism via the NEP1R1‐CTDNEP1 phosphatase and the Lipin phosphatidic acid (PA) phosphatase. This includes that Torsins remove NEP1R1‐CTDNEP1 from the NE in fly and mouse cells, leading to subsequent Lipin exclusion from the nucleus. NEP1R1‐CTDNEP1 downregulation also restores nuclear pore membrane fusion in post‐mitotic dTorsinKO fat body cells. However, dTorsin‐associated nuclear pore defects do not correlate with lipidomic abnormalities and are not resolved by silencing of Lipin. Further testing confirmed that membrane fusion continues in cells with hyperactivated Lipin. It also led to the surprising finding that excessive PA metabolism inhibits recruitment of the inner ring complex Nup35 subunit, resulting in elongated channel‐like structures in place of mature nuclear pores. We conclude that the NEP1R1‐CTDNEP1 phosphatase affects interphase NPC biogenesis by lipid‐dependent and lipid‐independent mechanisms, explaining some of the pleiotropic effects of Torsins.
Keywords: Drosophila fat body, Lipin, nuclear membrane, nuclear pore, Torsin
Subject Categories: Membrane & Intracellular Transport, Metabolism
The nuclear envelope phosphatase complex NEP1R1‐CTDNEP1 connects Torsin ATPases to lipid metabolism and membrane remodeling for nuclear pore complex insertion during interphase in Drosophila and mammalian cells.
Introduction
Most cellular lipids are synthesized by enzymes localized on or in the endoplasmic reticulum (ER) membranes. The ER is also continuous with the inner and outer nuclear membranes (INM and ONM), which together with embedded nuclear pore complexes (NPC) comprise the nuclear envelope (NE). For unknown reasons, several lipid synthetic enzymes localize in the nucleus or INM (Jacquemyn et al, 2017; Haider et al, 2018; Romanauska & Köhler, 2018; Sołtysik et al, 2021). This includes the Lipin phosphatidic acid (PA) phosphatase enzyme that catalyzes PA conversion to diacylglycerol (DAG). This is a key step in the glycerol‐ and glycerophospholipid (GL and GPL synthetic network). The DAG produced by Lipin is the main substrate for triacylglycerol (TAG) synthesis (Adeyo et al, 2011) (Fig 1A and Appendix Fig S1A). In parallel, Lipin activity reduces PA levels, which affect the cell by several mechanisms including suppressing membrane lipid synthesis (Santos‐Rosa et al, 2005; Han et al, 2007; Craddock et al, 2015; Grillet et al, 2016; Yang et al, 2019) (Fig 1A and Appendix Fig S1A).
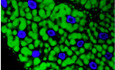
Figure 1. dTor and dLap1 differentially affect the fat body lipidome.
-
AGL and GPL and enzymes. Full abbreviations in Appendix Fig S1A.
-
BdTor and dLap1 topology.
-
C–ESubcellular localization of dLap1mGFP and dTormGFP‐E177Q in the wild‐type (w‐) or dLap1 +/Δ11 fat body of 3rd‐instar fly larvae, when expressed as UAS transgenes.
-
F–HControl, dTorKO, and dLap1 −/− larvae with pseudo‐colored red fat body.
-
IThe relative abundance of individual GL and GPL classes in the wild‐type fat body, as detected by mass spectrometry (MS). n = 10 individual analyses.
-
JTAG content of 3DO larvae measured by enzymatic assay. Bars show mean ± SEM measured in three sets of thirty larvae. One‐way ANOVA with Tukey’s multiple comparison test. ****P < 0.0001.
-
KThe abundance of individual lipid classes (mean ± SEM) within the set of six membrane GL and GPL extracted from 4DO fat body samples. w‐ n = 7, dTor KO n = 7, dLap1 −/− n = 4. One‐way ANOVA with Bonferroni’s post hoc test. *P < 0.05 and **P < 0.01.
-
L–ONeutral lipid staining of the 5DO larval fat body. White arrows (O) indicate nuclear‐localized neutral lipid staining, and blue signal shows DAPI.
-
P% of adult females that hatch after crossing female dTor +/−;; dLap1 −/TM6C with male y/FM7i;; dLap1 −/TM6C flies. Two‐tailed t‐test. ****P < 0.0001, compared with expected Mendelian ratio (n ≥ 3).
Lipin is regulated by numerous mechanisms (O’Hara et al, 2006; Harris et al, 2007; Liu & Gerace, 2009; Shimizu et al, 2017; Li et al, 2018). This includes that it is dephosphorylated by an evolutionarily conserved transmembrane complex formed between the C‐terminal domain nuclear envelope phosphatase 1 (CTDNEP1) and its regulatory subunit 1 (NEP1R1) (Siniossoglou et al, 1998; Santos‐Rosa et al, 2005; Kim et al, 2007; Han et al, 2012). The NEP1R1‐CTDNEP1 phosphatase complex of yeast appears to exclusively target Lipin (Su et al, 2014) while, in contrast, the metazoan complex has multiple substrates including BMP pathway effectors (Satow et al, 2006; Liu et al, 2011; Sakaguchi et al, 2013; Darrigrand et al, 2020).
Torsin AAA+ ATPases of the ER/NE lumen are newly identified regulators of Lipin (Grillet et al, 2016; Cascalho et al, 2020), as well as disease‐associated proteins (Ozelius et al, 1997; Kariminejad et al, 2017; Fichtman et al, 2019). Torsins regulate Lipin by an undefined post‐translational mechanism that transduces information across the ER/NE membrane (Cascalho et al, 2020). Torsins are atypical AAA+ proteins that become active ATPases upon binding a transmembrane activator protein, LAP1 and LULL1 in mammals (Zhao et al, 2013; Sosa et al, 2014; Chase et al, 2017) or, alternatively, Torsins assemble into large stable homo‐oligomers that are not active ATPases (Demircioglu et al, 2019). It is unclear whether Torsin regulates Lipin via its ATPase activity although, intriguingly, it appears that Torsin‐LAP1 ATPase activity is conveyed across the NE membrane (Luithle et al, 2020), and mammalian LAP1 affects TAG, LD, and lipid trafficking in a manner consistent with Lipin regulation (Ugrankar et al, 2011; Schmitt et al, 2015; Grillet et al, 2016; Shin et al, 2019).
Torsins also affect cellular events that have no clear relationship to lipid metabolism. This includes that Torsins have a poorly understood importance for NPC insertion into the NE of post‐mitotic/interphase cells (Goodchild et al, 2005; Liang et al, 2014; VanGompel et al, 2015; Laudermilch et al, 2016; Pappas et al, 2018; Rampello et al, 2020). Interphase NPC biogenesis occurs by less well‐understood mechanisms than its mitotic counterpart. It appears that it is initiated by membrane deforming nucleoporins (Nups) that curve the INM toward the ONM, followed by membrane fusion and the recruitment of the full complement of NPC subunits (Otsuka et al, 2016; Otsuka & Ellenberg, 2018). Torsin loss from worm, fly, and mammalian cells induces characteristic nuclear membrane herniations that are believed to result from failed INM/ONM fusion (Naismith et al, 2004; Goodchild & Dauer, 2005; Kim et al, 2010; Jokhi et al, 2013; Liang et al, 2014; Tanabe et al, 2016; Pappas et al, 2018; Rampello et al, 2020) and these cells have correspondingly fewer mature nuclear pores (VanGompel et al, 2015; Laudermilch et al, 2016; Pappas et al, 2018; Rampello et al, 2020). As a result, it has been proposed that Torsins are part of the membrane scission or fusion machinery (Jokhi et al, 2013). Alternatively, given their role in lipid metabolism, Torsins might maintain NE membrane lipid composition within a range permissive for fusion, a hypothesis supported by the fact that deregulated lipid metabolism impairs INM/ONM remodeling during NPC insertion in yeast (Scarcelli et al, 2007; Hodge et al, 2010).
Here, by analyzing the fat body of flies lacking dTorsin (referred to as dTor), we uncovered that Torsin regulates Lipin and the cellular lipidome via the NEP1R1‐CTDNEP1 phosphatase complex. The fat body function of dTor appears to occur via homo‐oligomerization rather than Torsin ATPase activity. Furthermore, NEP1R1‐CTDNEP1 dysfunction explained why dTor loss impaired INM/ONM fusion during NPC insertion. However, unexpectedly, this was not explained by lipidomic defects, even though excess PAP activity was sufficient to block late‐stage pore membrane remodeling. We therefore conclude that Torsins influence the lipidome and NPC biogenesis through the NEP1R1‐CTDNEP1 phosphatase complex and provide evidence that this complex negatively influences NPC biogenesis by multiple mechanisms.
Results
dTor and the dTor activator (dLap1) differentially affect the fat body lipidome
There is one Torsin activator in Drosophila, dLap1 (Sosa et al, 2014) (also known as Torip), that resides in the NE‐like mammalian LAP1 (Fig 1B and C, Grillet et al, 2016). We introduced an 11 basepair deletions into dLap1 that produced a premature stop codon and triggered dLap1 mRNA removal (Appendix Fig S1B–D). To test whether dLap1 is an NE partner of dTor, we crossed dLap1Δ11 flies and flies expressing dTormGFP carrying the Walker B E→Q mutation that stabilizes the Torsin–activator complex. dTormGFP‐E177Q concentrated in the NE of wild‐type cells as previously shown (Fig 1D; Jokhi et al, 2013). In contrast, there was noticeably less NE‐localized dTormGFP‐E177Q in dLap1Δ11/+ cells (Fig 1E). We therefore conclude that dLap1Δ11 is a null allele and fly dLap1 is the ortholog of mammalian LAP1/TOR1AIP1. Hereafter, we refer to dLap1 Δ11/Δ11 as dLap1 −/−.
dTor is on the X chromosome, and we refer to dTor −/y hemizygous males as dTorKO . Late stage (5 days old; 5DO) third‐instar dTorKO larvae have an abnormally small fat body (a liver and adipose tissue equivalent), concomitant with impaired cell growth, elevated TAG levels, and larger LD (Grillet et al, 2016). We analyzed whether dTor regulates these phenotypes via the ATPase activator by examining whether dLap1 −/− animals have similar defects. dLap1 −/− was viable and fertile as adults, while dTorKO is not (Wakabayashi‐Ito et al, 2011). The dLap1 −/− fat body was also larger than that of dTorKO (Fig 1F–H) and had normally sized cells (Appendix Fig S1E–H).
The relatively normal appearance of dLap1 −/− animals is surprising since LAP1 loss is lethal in mammals (Kim et al, 2010; Fichtman et al, 2019), and elevates hepatic TAG levels (Shin et al, 2019). TAG is also produced at high levels in the fly fat body (Fig 1I). We therefore compared TAG levels between the fat body of control, dLap1 −/−, and dTorKO larvae. This identified that dLap1 animals had abnormally high levels of TAG that even exceeded those associated with dTor loss (Fig 1J). We also examined membrane lipids (Fig 1K). We confirmed the defects we previously reported for the dTorKO fat body (Grillet et al, 2016); compared to controls, dTorKO had lower relative levels of PI, PA, and PE, and increased DAG and PS (Fig 1K; note that Lipin metabolizes PA into DAG). dLap1 loss also reduced PI and PE, but did not significantly affect other lipids. Thus, although both dLap1 and dTor control TAG levels, they differentially affect membrane lipid classes.
The divergence of the dTor and dLap1 loss‐of‐function phenotypes suggests that, while some functions may be mediated by a Torsin–LAP1 complex, this does not fully explain how Torsin affects the lipidome. We further tested this by intercrossing null alleles and using neutral lipid staining as a read‐out of cellular TAG (Fig 1L–O; Appendix Fig S1I). We failed to find dTorKO ;; dLap1 −/− larvae, but did identify L3 stage dTorKO ;; dLap1 +/−. These had small fat body cells such as the dTorKO that gained the additional defect of nuclear‐localized neutral lipid staining (Fig 1O; white arrows). Moreover, quantification of adult hatching defined a synthetic lethal genetic interaction between the dTor +/− and dLap1 −/− genotypes (Fig 1P). Thus, dTor and dLap1 have an additive effect on TAG metabolism at the NE, further supporting the hypothesis that they (i) independently affect cellular lipid metabolism, and (ii) ATPase activity is not critical for Torsin to regulate Lipin PAP activity.
dTor function in the fat body requires membrane association and a motif responsible for homo‐oligomerization
We further explored the role of ATPase activity by comparing the ability of wild‐type and mutant Torsin cDNAs to revert the (i) reduced cell size and (ii) LD overproduction of dTorKO fat body cells (Fig 2A). We use these phenomena as read‐outs since Lipin PAP activity promotes TAG and LD at the expense of membrane growth in yeast (Santos‐Rosa et al, 2005; Han et al, 2007), plants (Craddock et al, 2015), Drosophila (Grillet et al, 2016), and mammals (Yang et al, 2019). dTormGFP‐WT expression (Fig 2B) restored dTorKO cells to wild‐type size and suppressed LD defects (Fig 2C and D, Appendix Fig S2A and B). We examined dTor cDNAs carrying mutations in the highly conserved motifs of ATP binding (Walker A K114A) or hydrolysis (Walker B E177Q) (Fig 2A and B). These expressed at similar levels to dTormGFP‐WT (Appendix Fig S2C and D). dTormGFP‐K114A and dTormGFP‐E177Q expression significantly increased the size of dTorKO cells compared with the negative control (mGFPSec61b; an unrelated transmembrane ER protein; Fig 2C; Appendix Fig S2A) and normalized excess LD density as effectively as dTormGFP‐WT re‐expression (Fig 2D; Appendix Fig S2B). This suggests that dTor continues to function even when its ATPase activity is severely impaired. We therefore considered Torsin assembly into ATPase inactive homo‐oligomers. This requires a highly conserved glycine residue (G256 of dTor) in the “back‐interface” domain which is dispensable for activator‐binding and ATPase activity (Chase et al, 2017; Demircioglu et al, 2019). dTormGFP‐G256D expressed at similar levels to dTormGFP‐WT (Appendix Fig S2C and D) and similarly localized in the ER/NE (Fig 2B). However, dTormGFP‐G256D expression did not rescue the size of dTorKO cells (Fig 2C; Appendix Fig S2A) or normalize LD density (Fig 2D; Appendix Fig S2B). This suggests that Torsin homo‐oligomerization is required for its effects on TAG production and cell size.
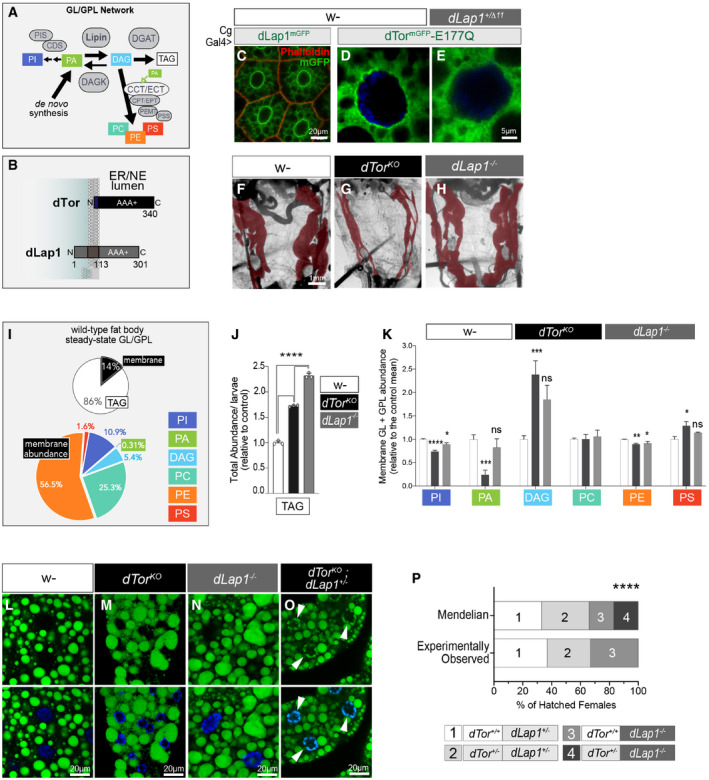
Figure 2. The dTor membrane domain and homo‐oligomerization motif are important for dTor function in the fat body.
-
AdTor mutants and chimeras. Sig Seq, ER targeting signal sequence.
-
BWild‐type and mutant dTormGFP proteins expressed in dTorKO fat body cells.
-
C, DSize of individual fat body cells (C) and LD density of individual fat body cells (D). Bars show the group mean, light gray highlights 2× SD of the control mean (animals expressing the Sec61b control ER protein), and dotted red lines show dTorKO mean. These are results of n ≥ 3 larvae (of 3 independent crosses). One‐way ANOVA, with Bonferroni’s post hoc test. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
-
E–MGFP, Phalloidin, and LipidTOX detect the expression of dTormGFP chimeras (green), cell boundaries (red), and LD (red), respectively, in dTorKO fat body cells.
Source data are available online for this figure.
Torsins are evolutionarily related to HSP100‐type AAA+ proteins that detect substrate proteins through their N‐terminus and use the energy of ATPase activity to drive conformational changes in the substrate (Rosenzweig et al, 2015). However, Torsins diverge because the N‐terminus is a monotopic membrane domain (MMD) (Fig 2A). We examined the functional importance of the MMD. dTormGFP‐ΔMMD expressed poorly (Fig 2E; Appendix Fig S2C and D) and failed to rescue dTorKO cell size or LD defects (Fig 2C, D, F and G). We therefore generated a chimeric Torsin with the native MMD replaced by the unrelated transmembrane domain (TMD) of mouse Sec61b (Fig 2A). dTormGFP‐TMDSec61 expressed and localized in the ER/NE similarly to dTormGFP‐WT (Fig 2H; Appendix Fig S2C and D), but failed to rescue the size or LD content of dTorKO cells (Fig 2C, D, I and J). This suggests that the dTor MMD is (a) required and (b) may encode specific information that couples Torsin to downstream partners. We wondered whether this was conserved in dLap1 given overlapping effects on the fat body lipidome. We replaced the MMD of dTor with the TMD of dLap1 (Fig 2A), which produced a stable chimera that localized in the ER similar to dTormGFP‐WT (Fig 2K). Remarkably, dTormGFP‐TMDdLap1 rescued cell size and LD abnormalities as effectively as dTormGFP‐WT (Fig 2C, D, L and M). This suggests that specific information is encoded in the membrane domains of dTorsin and dLap1 and this is key to how both proteins affect cellular lipid metabolism.
dTor affects the fat body lipidome via the NEP1R1‐CTDNEP1 phosphatase complex and the Lipin PA phosphatase
We searched for the mechanism that couples dTor to the fat body lipidome using an RNAi survey of transmembrane lipid enzymes and regulators of lipid metabolism. We expressed candidates in the dTorKO fat body using the r4‐Gal4 driver that expresses at high levels throughout development (Lee & Park, 2004). While most transgenes had no effect on dTorKO morphology (Fig 3A), RNAi against Ctdnep1 (Dullard; CG1696) or Nep1r1 (CG41106) improved dTorKO fat body size to a similar extent as dTor‐WT re‐expression (Fig 3A; final panels).
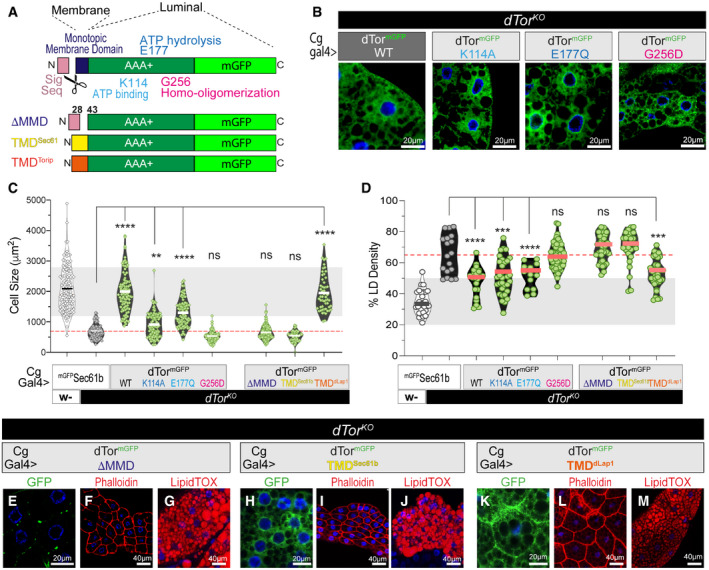
Figure 3. dTor affects the fat body lipidome via Nep1r1, Ctdnep1, and Lipin .
-
AdTorKO larvae expressing UAS RNAi transgenes or expressing a wild‐type dTor cDNA (positive control). The fat body is pseudo‐colored red.
-
BTopology of NEP1R1, CTDNEP1, and Lipin.
-
CFat body cells labeled by BODIPY to detect neutral lipids.
-
DThe relative abundance of each lipid class (mean ± SEM) within the set of six membrane lipids. n = 3 MS analyses of 4DO fat body samples for each genotype. ‡ symbols indicate a significant difference compared with w‐ expressing Luciferase RNAi, * symbols indicate a significant difference compared with dTorKO expressing Luciferase RNAi. Note we only tested the effect of Lipin, Ctdnep1, and Nep1r1 RNAi when a lipid class significantly differed between w‐ and dTorKO . One‐way ANOVA, Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ‡‡‡‡ P < 0.0001.
-
EAs above except that TAG abundance was calculated relative to protein. One‐way ANOVA, Tukey’s multiple comparisons test.
-
F, GBars show the fraction (mean ± SEM) of membrane GL and GPL lipids with each fatty acyl chain characteristic. n = 3 MS analyses of 4DO fat body samples for each genotype. MUFA, mono‐unsaturated; PUFA, poly‐unsaturated. ‡ indicate a significant difference between w‐ and dTorKO expressing Luciferase RNAi, * indicate a significant difference compared with dTorKO expressing Luciferase RNAi. Two‐way ANOVA, Dunnett’s multiple comparison test. ‡ P < 0.05 and *P < 0.05.
NEP1R1 regulates CTDNEP1 that, in turn, dephosphorylates Lipin to activate Lipin PAP activity (Fig 3B) (Kim et al, 2007; Han et al, 2012; Su et al, 2014). We now genetically tested whether the NEP1R1‐CTDNEP1/Lipin pathway accounts for why Torsin loss affects the fat body lipidome. Lipin, Ctdnep1, and Nep1r1 RNAi similarly reduced their respective mRNAs (Appendix Fig S3A), and, consistent with their effect on fat body mass, they increased the size of dTorKO cells toward wild type (Appendix Fig S3B and C). In contrast, none of the three RNAi transgenes increased cell size when expressed in control animals (Appendix Fig S3D). All three RNAi transgenes appeared to normalize neutral lipid staining defects of dTorKO cells (Fig 3C). Quantification detected that Lipin and Ctdnep1 RNAi returned dTorKO fat body LD density and size to the wild‐type state (Appendix Fig S3E and F) and, while Nep1r1 RNAi did not, this was induced by RNAi targeting the second Nep1r gene of Drosophila (CG8009; Nep1r2; Appendix Fig S3F).
We used mass spectrometry (MS) to quantify the lipidome of fat body samples prepared from wild‐type control (w‐) and dTorKO animals expressing RNAi against a control gene (Luciferase), or Lipin, Ctdnep1, and/or Nep1r1 (Table EV1). The dTorKO fat body expressing the control RNAi again displayed the signature of Lipin hyperactivity (low PA and high DAG compared with wild type; Fig 3D). Consistent with earlier analyses, we again identified lower relative levels of PE and PI than the wild‐type fat body (Figs 3D and 1K, Grillet et al, 2016) and a trend to elevated PS (Fig 3D) and TAG (Fig 3E). In comparison, dTorKO animals expressing RNAi against Lipin, Ctdnep1, or Nep1r1 had at least wild‐type levels of PA. Further, the ∼3‐fold elevated DAG levels of the dTorKO expressing Luciferase RNAi were reduced below wild type by Lipin, Ctdnep1, or Nep1r1 RNAi (Fig 3D). The three RNAi transgenes also normalized the PI and PE deficits of the dTorKO lipidome (Fig 3D). As a further control, we examined the impact of RNAi transgenes on the fat body lipidome of w‐ animals. There was no significant difference in the levels of PA, DAG, or PE between w‐ animals expressing Luciferase RNAi versus Lipin or Ctdnep1 RNAi (Fig 3D). Finally, we focused on the fatty acyl chains of membrane GL and GPL that were significantly longer and more saturated in the fat body of the dTorKO compared with w‐ animals (Fig 3F and G, compare first two bars). Lipin, Ctdnep1, and Nep1r1 RNAi transgenes significantly reduced the severity of both acyl chain defects (Fig 3F and G) including that Lipin RNAi normalized 5 out of the 6 measures to wild type levels. We therefore conclude that dTor loss affects the lipidome by a mechanism involving Nep1r1/Ctdnep1/Lipin pathway gain of function (GOF).
CTDNEP1 and Lipin abnormally concentrate at the NE of fly and mouse cells lacking Torsins
We sought the molecular basis of the Nep1r1/Ctdnep1/Lipin GOF. Nep1r1, Ctdnep1, and Lipin mRNA levels were similar in control and dTorKO animals (Appendix Fig S4A–C), ruling out the possibility of abnormal gene expression. We therefore examined protein localization by imaging NEP1R1 and CTDNEP1 fused to mGFP. In the control fat body, NEP1R1mGFP and CTDNEP1mGFP primarily colocalized with Calnexin in the main ER with minimal enrichment at the NE (Fig 4A and B). In contrast, the NE of dTorKO cells was enriched in NEP1R1mGFP (Fig 4C; Appendix Fig S4D and E) and especially CTDNEP1mGFP (Fig 4D; Appendix Fig S4F and G) relative to the appearance of Calnexin (Fig 4A–D, compare red and green signal). CTDNEP1mGFP was often observed in patches around the dTorKO nucleus, while it uniformly localized around the nucleus of control cells (Fig 4B versus D). We examined the same question in mammalian cells where NEP1R1‐CTDNEP1 and Lipin proteins are better characterized. While CTDNEP1Scarlet signal overlapped with the Sun1 NE marker in control and Tor1aKO /Tor1bKO (TorA/BKO) mouse embryonic fibroblasts (MEFs) (Fig 4E and F; Appendix Fig S4H), there was significantly greater colocalization between Sun1 and CTDNEP1Scarlet at the TorA/BKO NE compared with the control NE (Fig 4E′ versus F′; Appendix Fig S4I). Thus, it appears that Torsin loss from fly or mammalian cells leads to CTDNEP1 and/ or NEP1R1 enrichment in the NE.
Figure 4. CTDNEP1 and Lipin concentrate at the NE of fly and mouse cells lacking Torsins.
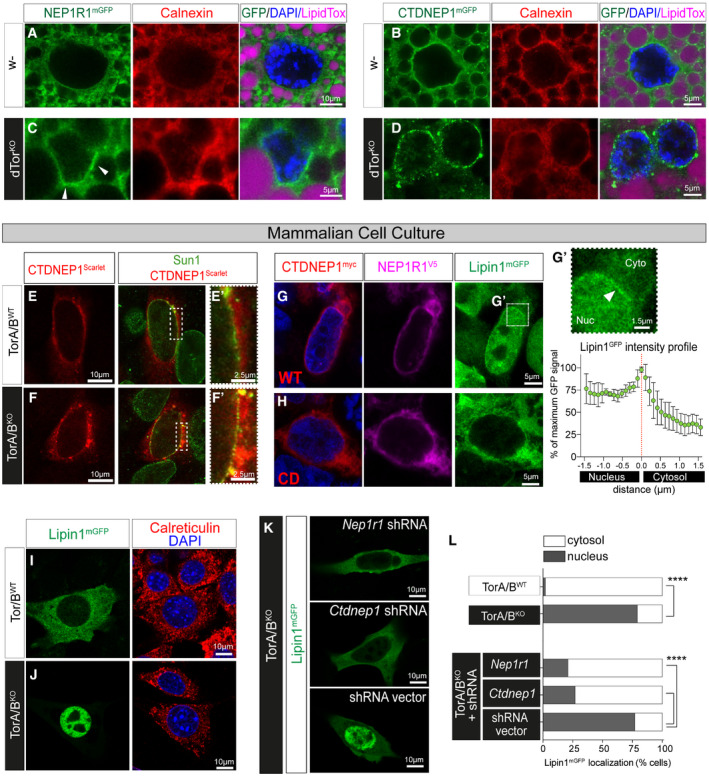
-
A–DControl and dTorKO 5DO fat body cells expressing UAS GFP fusion proteins with the Cg‐Gal4 driver, and labeled with anti‐Calnexin (red) and a neutral lipid dye (LipidTOX, magenta). Note that dTorKO cells are smaller. White arrowhead indicates NE enrichment of LipinmGFP.
-
E, FCTDNEP1Scarlet and anti‐Sun1 localization in MEFs.
-
G, HLipin1mGFP localization in HEK293T cells transfected with a plasmid that co‐expresses CTDNEP1myc‐WT or CTDNEP1myc‐CD (red) and NEP1R1V5 (magenta). (G′) Upper: enlargement of panel G, white arrow highlights NE enrichment. Lower: plot showing the intensity of Lipin1mGFP signal (mean ± SD, n = 5 cells) along a 3 µm profile that transects the NE at 0 µm.
-
I–KMouse Lipin1mGFP expressed in MEFs, red signal shows Calreticulin.
-
LPercentage of MEFs with primarily cytosolic or nuclear Lipin1mGFP. Chi‐square test. ****P < 0.0001.
NEP1R1‐CTDNEP1 dephosphorylation of mammalian Lipin1 activates PAP activity concomitant with Lipin relocalizing from the cytosol to the nucleus (Han et al, 2007). We confirmed previous studies showing that Lipin relocalization could be induced by co‐overexpression of NEP1R1 and CTDNEP1 (Han et al, 2012) (Fig 4G; Appendix Fig S4J and K). This included that Lipin1mGFP appeared at the nuclear periphery of cells co‐overexpressing NEP1R1V5 and CTDNEP1myc, suggestive of INM‐localized PAP activity (Fig 4G′), while Lipin1mGFP remained in the cytosol in cells co‐overexpressing catalytically dead CTDNEP1 (CTDNEP1myc‐CD; Han et al, 2012) with NEP1R1V5 (Fig 4H; Appendix Fig S4J and K). Consistent with Torsin regulation of Lipin through NEP1R1‐CTDNEP1, Lipin1mGFP was cytosolic in control MEFs but localized in the nucleus of TorA/BKO MEFs (Fig 4I and J), similar to previous observations in fly (Grillet et al, 2016). We then co‐transfected MEFs with a control plasmid or plasmids expressing Nep1r1 or Ctdnep1 shRNA. Lipin1mGFP continued to concentrate in the nucleus of TorA/BKO MEFs transfected with empty vector (Fig 4K and L). In contrast, the majority of TorA/BKO cells transfected with Nep1r1 or Ctdnep1 shRNA had cytosolic Lipin1mGFP (Fig 4K and L), showing that NEP1R1‐CTDNEP1 can explain the Lipin dysfunction of cells lacking Torsins.
Nep1r1 and Ctdnep1 downregulation, but not Lipin downregulation, restores NPCs and removes INM/ONM fusion defects from dTorKO cells
Interphase cells lacking Torsins have lower levels of NPC alongside abnormal omega‐shaped membrane profiles that are characteristic of failed INM/ONM membrane fusion (Fig 5A and B) (Scarcelli et al, 2007; Jokhi et al, 2013; Laudermilch et al, 2016; Otsuka et al, 2016; Pappas et al, 2018; Allegretti et al, 2020; Rampello et al, 2020). This has not been examined in the fly fat body but, since fat body cells exit the cell cycle after embryogenesis (Zheng et al, 2016), they are ideal to dissect this role of Torsins.
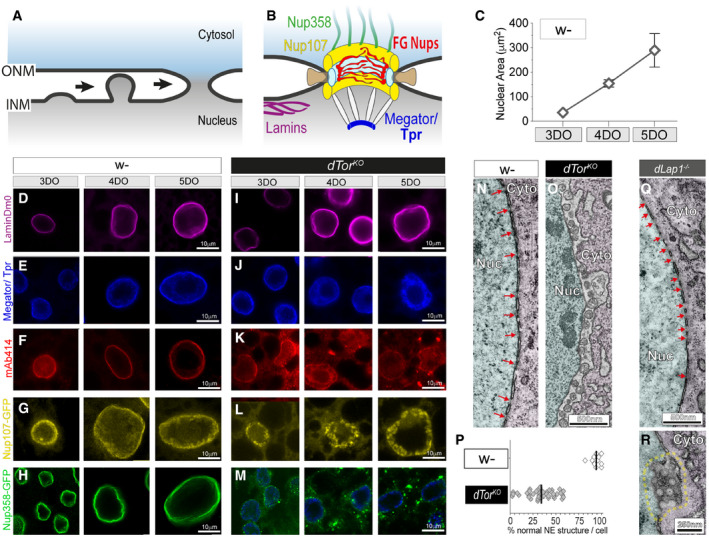
Figure 5. dTor loss impairs NPC insertion during nuclear growth in developing fat body cells.
-
AInterphase NPC insertion occurs via inside–out extrusion of the INM, followed by INM/ONM fission and fusion (Otsuka & Ellenberg, 2018). Nucleoporins are not depicted.
-
BThe mature NPC, highlighting proteins examined in fat body cells. Green, cytosolic filaments; yellow, outer ring subcomplex; pale blue, inner ring subcomplex; brown, transmembrane nucleoporins; red, phenylalanine‐glycine repeats (FG‐Nups) that form the permeability barrier; white/blue, nuclear basket Nups.
-
CFat body nuclei increase in size during development. Graph shows mean ± SD for each day during development of larvae. DO, day‐old.
-
D–MControl (w‐) and dTorKO fat body nuclei labeled with anti‐LaminDm0, Megator/Tpr, mAb414, or expressing UAS Nup107GFP or UAS Nup358GFP with the Cg‐Gal4 driver.
-
N, ORepresentative TEM of the NE of control and dTorKO 5DO fat body cells. Red arrows show nuclear pores. Nuc, nucleus; Cyto, cytosol.
-
PThe percentage of NE in individual 5DO fat body cells that has normal INM, ONM, and NPC morphology. Black lines show the mean of each group.
-
Q, RRepresentative TEM showing the NE of 5DO dLap1 −/− fat body cells. Red arrows in (Q) show nuclear pores. Dashed yellow line highlights unusual circular membrane profiles.
Fat body nuclei expanded over L3 larval development (Fig 5C and D): assuming that nuclei are spherical, surface area increased ∼10‐fold between 3DO and 5DO. Concomitantly, the NE maintained consistent levels of the Megator/Tpr NPC basket component (Fig 5E), FG‐containing Nups detected by the mAb414 antibody (Hampoelz et al, 2019) (Fig 5F), the structural outer ring Nup107 (Fig 5G), and cytoplasmic Nup358 (Otsuka et al, 2016) (Fig 5H), showing that NPCs are inserted as nuclei grow in size.
dTorKO nuclei also expanded over development, and the nuclear lamina was similar to that of controls (Fig 5I). Megator/Tpr was also normally localized at the NE of 3DO dTorKO fat body cells, although was in the nuclear interior at 4DO and 5DO (Fig 5J). Other NPC markers were abnormal at all ages: mAb414 barely labeled the dTorKO NE (Fig 5K), Nup107GFP appeared to aggregate within the nucleus (Fig 5L), and there was less NE‐localized Nup358GFP signal in dTorKO cells compared with controls (Fig 5H versus 5M). We assessed NE ultrastructure at 5DO. The INM and ONM of wild‐type cells were parallel, closely spaced, and frequently interrupted by highly curved pores (Fig 5N, red arrows). In contrast, the dTorKO NE was coated in INM herniations alongside very few nuclear pores (Fig 5O and P). We also examined the NE of dLap1 −/− animals. Although there were occasional unusual circular membrane profiles, dLap1 −/− cells had many nuclear pores and no INM herniations (Fig 5Q and R). The absence of INM herniations in the dLap1 −/− cells continues to support a model where the fat body function of dTor does not depend on its ATPase activator.
We examined cause‐and‐effect between NPC defects and NEP1R1/CTDNEP1/Lipin dysfunction. First we considered this in the TorA/BKO MEFs that have excess Lipin enzyme activity (Cascalho et al, 2020) and mislocalized Lipin (Fig 4I and J), but insert most NPC during mitosis using a process that does not require Torsins (Otsuka & Ellenberg, 2018; Rampello et al, 2020). Consistent with previous studies, there was no striking difference in nucleoporin labeling between control and TorA/BKO MEFs (Appendix Fig S5A–D), suggesting that NEP1R1/CTDNEP1/Lipin dysfunction occurs independently of NPC defects. We then examined if NEP1R1/CTDNEP1/Lipin pathway dysfunction explains the impaired NPC biogenesis in interphase cells lacking Torsin by comparing nucleoporin mAb414 labeling and NE ultrastructure in dTorKO fat body cells expressing control transgenes or Nep1r1 RNAi, Ctdnep1 RNAi, or Lipin RNAi. As expected, dTormGFP‐WT cDNA re‐expression increased the number of dTorKO cells with NE‐localized mAb414 compared with the negative control of Luciferase RNAi expression (Fig 6A and B). dTormGFP‐WT re‐expression also increased the density of nuclear pores detected by TEM (Fig 6C and D), removed INM herniations (Fig 6C and E), and increased the percentage of NE with normal morphology (Fig 6F).
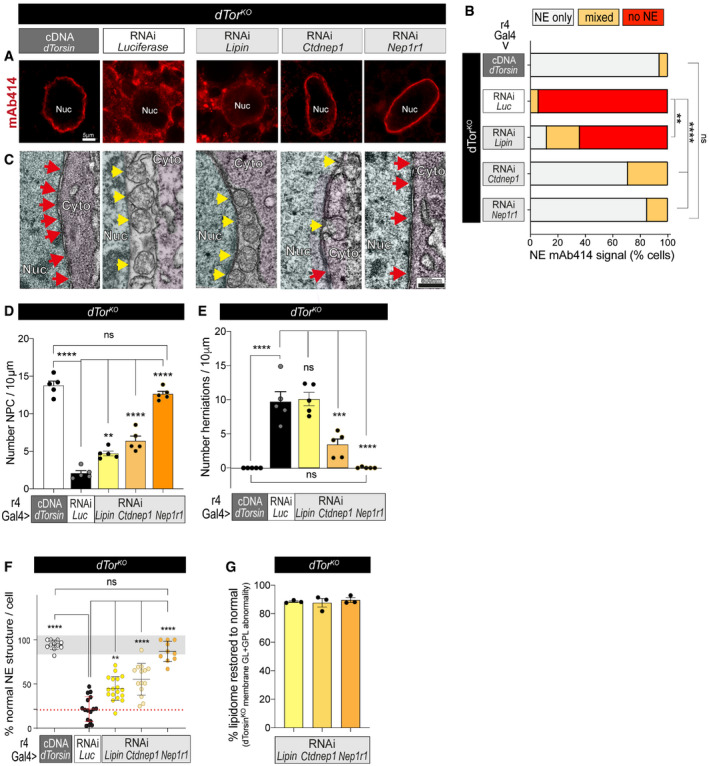
Figure 6. Nep1r1 or Ctdnep1 downregulation restore NPC levels and suppress INM herniations in dTorKO cells.
-
AmAb414 labeling of 5DO dTorKO fat body cells expressing the dTor cDNA, Luciferase RNAi, or RNAi against Nep1r1, Ctdnep1, or Lipin. Nuc, nucleus.
-
BThe percentage of fat body cells with NE‐localized mAb414. ** and **** indicate that Lipin, Ctdnep1, and Nep1r1 RNAi significantly increased the number of dTorKO cells with NE‐specific mAb414 labeling compared with the Luciferase RNAi (Luc). Chi‐square test followed by individual post hoc chi‐square tests. **P < 0.01 and ****P < 0.0001.
-
CNE ultrastructure in 5DO dTorKO cells. Red arrows, nuclear pores; yellow arrows, abnormal INM herniations. Nuc, nucleus; Cyto, cytosol.
-
DThe density of NPC detected by TEM in the 5DO dTorKO NE. Each point shows the mean of multiple measurements from an individual nucleus, and bars show mean ± SD for the group (n = 5). One‐way ANOVA, with Bonferroni’s post hoc test. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
-
ESame as above except that the density of INM herniations is presented.
-
FThe percentage of NE in 5DO fat body cells that has normally appearing INM, ONM, and NPC morphology. Points show values from individual nuclei (n = 5). Bars show mean ± SD. Gray zone shows +/− 2× SD of the positive control mean; dotted red line shows mean of dTorKO expressing Luciferase RNAi. One‐way ANOVA, with Bonferroni’s post hoc test. **P < 0.01 and ****P < 0.0001.
-
GThe percentage that Lipin, Ctdnep1, and Nep1r1 RNAi normalize the abundance of membrane GL and GPL classes in the dTorKO fat body. 0% “normal” reflects lipid abundance in dTorKO expressing Luciferase RNAi, while 100% “normal” reflects lipid abundance in w‐ expressing Luciferase RNAi. Percentage normalization was separately calculated for PI, PA, DAG, PC, PE, and PS (for dTorKO expressing Lipin, Ctdnep1, and Nep1r1 RNAi). Points show the mean of all lipid class normalization per sample, and bars show mean ± SEM for the group.
dTorKO animals expressing Lipin RNAi, Ctdnep1 RNAi, and Nep1r1 RNAi also had more cells with NE‐localized mAb414 than those expressing the control RNAi (Fig 6A and B). However, there was a striking difference between the impact of Lipin RNAi compared with Ctdnep1 and Nep1r1 RNAi. Lipin RNAi only mildly increased the percentage of dTorKO fat body cells with NE‐localized mAb414 labeling (∼0 to ∼10%), while most dTorKO cells expressing the Ctdnep1 and Nep1r1 RNAi transgenes had NE‐localized mAb414 signal (Fig 6A and B). When considering NE ultrastructure, Lipin, Ctdnep1, and Nep1r1 RNAi all increased the density of nuclear pore profiles in the dTorKO , but Nep1r1 RNAi was as effective as dTor re‐expression whereas a substantial defect remained with Lipin RNAi (Fig 6C, red arrows; Fig 6D). Moreover, Lipin RNAi did not suppress the abnormal omega structures of dTorKO fat body cells, while these were reduced and abolished by Ctdnep1 and Nep1r1 RNAi, respectively (Fig 6C, yellow arrows; Fig 6E and F). We therefore revisited the question of how well each RNAi corrected the lipidome of the dTorKO fat body. We compared the cumulative percentage of lipid class defects that remained uncorrected in dTorKO animals expressing each RNAi transgene. Lipin RNAi, Ctdnep1 RNAi, and Nep1r1 RNAi indistinguishably normalized the dTorKO lipidome by this measure (Fig 6G). These data suggest that NEP1R1‐CTDNEP1 dysfunction links Torsin loss to nuclear pore membrane defects, but that this does not involve Lipin‐mediated PA metabolism.
Excess Lipin activity inhibits pore membrane curving
We further explored whether NPC insertion is resistant to abnormal Lipin‐mediated PA metabolism by overexpressing CTDNEP1mGFP in the wild‐type fat body using the r4‐Gal4 driver. Overexpressed CTDNEP1mGFP‐WT and catalytically dead CTDNEP1mGFP‐CD were localized in the NE and ER similar to mGFPSec61, and neither appeared to severely disturb fat body cell morphology (Fig 7A–C) especially considered against the defects they contribute to in dTor KO animals. Nevertheless, TAG was higher in the fat body overexpressing CTDNEP1mGFP‐WT compared with CTDNEP1mGFP‐CD (Fig 7D), and CTDNEP1mGFP‐WT overexpression was associated with more LD (Appendix Fig S6A), to confirm that CTDNEP1mGFP overexpression affects lipid metabolism.
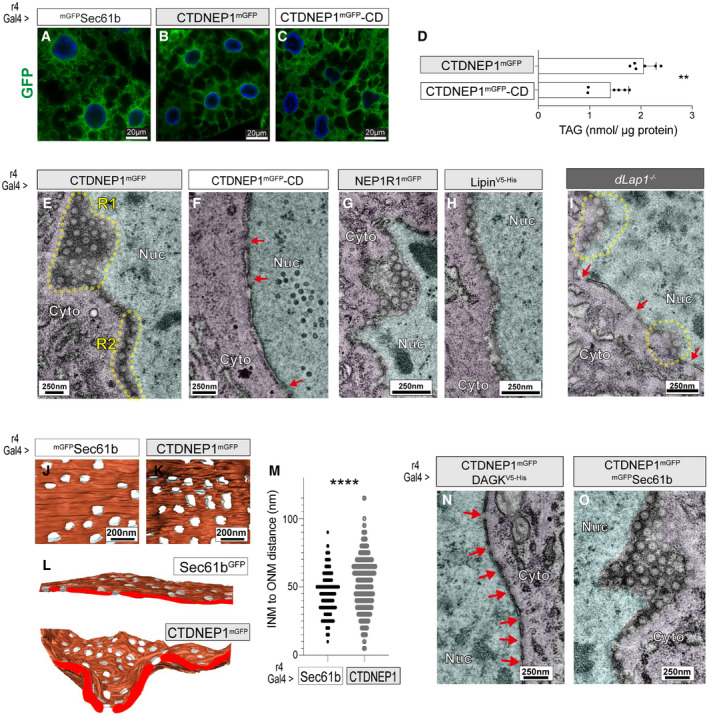
Figure 7. Abnormal nucleocytoplasmic membrane channels occur in fat body cells overexpressing NEP1R1, CTDNEP1, or Lipin, and dLap1 −/− cells.
-
A–CFat body cells expressing GFP tagged UAS transgenes with r4‐Gal4.
-
DTAG (nmol/ µg protein) in the 4 DO fat body expressing UAS transgenes with the r4‐Gal4 driver. Bars show group mean ± SEM (n = 6), and points show values from individual MS runs. Unpaired t‐test. **P < 0.01.
-
E–INE ultrastructure of 5DO fat body cells overexpressing UAS transgenes, or from dLap1 −/− animals. Yellow dotted lines highlight NE regions with subtly abnormal membrane architecture. Red arrows indicate nuclear pores. R1, region 1; R2, region 2; Nuc, nucleus; Cyto, cytosol.
-
J–MData from FIB‐SEM imaging of fat body nuclei expressing Sec61b (control) or CTDNEP1mGFP. (J–L) 3D reconstructions, (M) distance between the INM and ONM at NPC in wild‐type cells versus CTDNEP1mGFP‐induced structures. Kolmogorov–Smirnov non‐parametric t‐test. ****P < 0.0001.
-
N, OTEM analysis of the NE in 5DO fat body cells expressing the CTDNEP1 cDNA with DAGK or Sec61 cDNAs (control). Red arrows indicate nuclear pores.
We examined whether this relatively mild CTDNEP1 GOF affected NPC biogenesis. TEM did not detect the omega‐shaped structures. Instead, surprisingly, cells overexpressing CTDNEP1mGFP‐WT displayed circular membrane profiles within the NE (Fig 7E, R1) or, if the NE was obtusely bisected by the TEM thin section, hemispheric membrane profiles were continuous with the cytosol or nucleus (Fig 7E, R2). These did not occur in cells overexpressing CTDNEP1mGFP‐CD (Fig 7F), but were present in cells overexpressing NEP1R1mGFP (Fig 7G) or LipinV5‐His (Fig 7H). They also resembled the abnormalities we observed in some dLap1 −/− cells, which we confirmed in additional dLap1 −/− nuclei (Fig 7I). We explored the relationship between CTDNEP1mGFP localization and the abnormal structures using correlative light and electron microscopy (CLEM). This identified that abnormal membrane profiles coincided with sites of NE‐localized CTDNEP1mGFP signal, suggesting they are a relatively direct consequence of CTDNEP1 protein phosphatase activity (Appendix Fig S6B).
We used FIB‐SEM to better define the structures, which revealed that the NE of CTDNEP1mGFP overexpressing cells was perforated by nucleocytoplasmic channels. They occurred at a similar density to that of nuclear pores in wild‐type cells (Fig 7J–L). However, CTDNEP1‐induced channels lacked the highly curved membrane that characterizes the mature nuclear pore, and thus linked the nucleus and cytosol over a longer distance: At times, the INM and ONM were separated by 100 nm in CTDNEP1 overexpressing cells and had a tunnel‐like appearance (Fig 7M). The length of CTDNEP1‐induced structures can explain why they were frequently bisected as circular profiles in TEM, while thin sections rarely bisect the compressed mature nuclear pore.
We further tested whether Lipin‐mediated PA metabolism was responsible for the appearance of membrane channels using DAG kinase (DAGK) as a tool: DAGK re‐converts DAG to PA, and some overexpressed DAGK was present in the nucleus of fat body cells (Appendix Fig S6C). The circular membrane profiles were absent from cells co‐overexpressing CTDNEP1mGFP‐WT with DAGKV5‐His, which instead had normal nuclear pores with highly curved membranes (Fig 7N; red arrows). As controls, we confirmed abnormal circular profiles still occurred in cells co‐overexpressing CTDNEP1mGFP‐WT with mGFPSec61 (Fig 7O), and single overexpression of DAGKV5‐His or mGFPSec61 had no impact on NE structure (Appendix Fig S6D and E). Thus, unlike the omega profiles of impaired INM/ONM fusion, the elongated channels do appear to result from excess Lipin PA metabolism.
These nucleocytoplasmic channels occur at a similar density to nuclear pores and appear to replace nuclear pores, suggesting they derive from defective NPC insertion. We explored this by examining Nup localization and levels in cells overexpressing CTDNEP1mGFP‐WT (where channels predominate over mature NPC) using the array of NPC subcomplex antibodies (Fig 8A). Megator/Tpr and Nup107RFP indistinguishably concentrated at the NE of control and CTDNEP1mGFP‐WT overexpressing cells, and Nup214 signal was also NE‐localized in both (Fig 8B–D). In contrast, Nup35 and mAb414 were exclusively at the NE in control cells, but partially or entirely relocalized to the cytosol and/or nucleus in CTDNEP1mGFP‐WT overexpressing cells (Fig 8E and F; Appendix Fig S7A). We again examined the connection to Lipin PA metabolism. This confirmed that mAb414 was abnormally localized in many cells overexpressing NEP1R1mGFP and LipinV5 (Fig 8G and H) and that DAGK co‐overexpression prevented the mAb414 defects induced by CTDNEP1mGFP‐WT (Appendix Fig S7B–D). Moreover, mAb414 defects occurred in ∼50% of dLap1 −/− cells (Fig 8G and H), further linking the loss of FG‐Nups to the appearance of abnormal membrane channels.
Figure 8. Impaired Nup35 recruitment can explain the abnormal membrane channels caused by excess PA to DAG conversion.
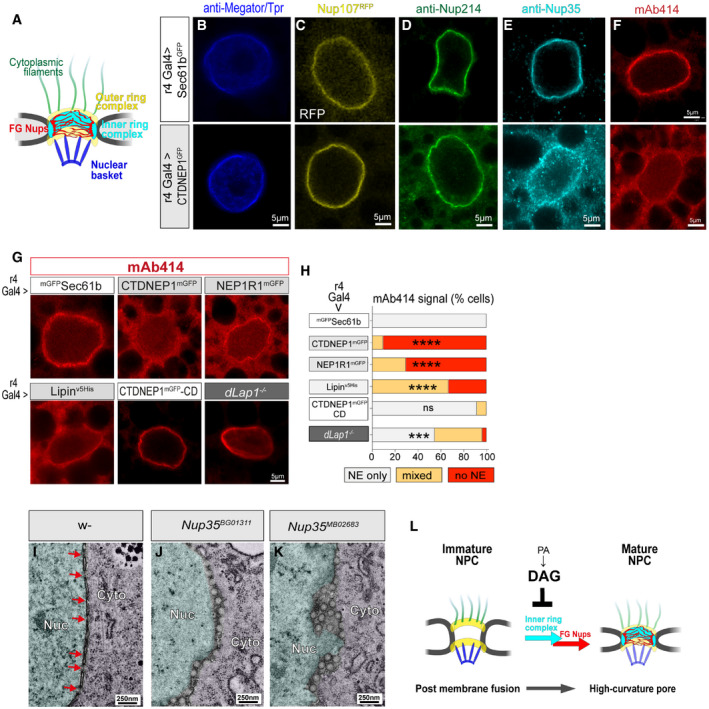
-
AThe NPC is comprised of several subcomplexes.
-
B–GConfocal imaging of 5DO fat body nuclei expressing UAS transgenes and labeled by anti‐Megator/Tpr, Nup214, Nup35, or mAb414, or expressing Nup107RFP.
-
HmAb414 localization in 5DO fat body cells expressing the indicated UAS transgenes or from a dLap1 −/− animal. *** and **** indicate a significant difference compared with control mGFPSec61b‐expressing cells. Chi‐square test followed by individual post hoc chi‐square tests. ***P < 0.001 and ****P < 0.0001.
-
I–KNE ultrastructure of 5DO fat body cells from a (I) control animal, (J) homozygous Nup35BG01311, and (K) homozygous Nup35MB02683 animals. Nuc, nucleus; Cyto, cytosol.
-
LModel of the relationship between nucleocytoplasmic channels, PA metabolism, and NPC maturation.
The labeling patterns seen with different Nups suggest that NE channels contain basket, cytosolic, and outer ring subcomplexes of the NPC, while the inner ring complex and FG‐Nups are absent. Nup35 is known as Nup53 in other species. It has been shown to directly bind membrane, induce membrane curvature, and anchor the inner ring complex and, indirectly, FG‐Nups to the nuclear pore (Vollmer et al, 2012; Otsuka & Ellenberg, 2018). We therefore hypothesized that failed Nup35 recruitment during NPC biogenesis was responsible for the channel‐like structures. We examined NE morphology in animals carrying Nup35 mutations (Appendix Fig S7E) by TEM, which identified the same abnormal membrane profiles associated with CTDNEP1/NEP1R1/Lipin pathway hyperactivity (Fig 8I–K). From this, we conclude that abnormal nucleocytoplasmic channels develop because excess PA to DAG conversion prevents Nup35 recruitment during late‐stage NPC maturation (Fig 8L).
Discussion
Here, we show evidence that the NEP1R1‐CTDNEP1 phosphatase complex connects Torsins to the Lipin PA phosphatase and the cellular lipidome. The data also define that Torsin/Nep1r1/Ctdnep1, but not Lipin, are a genetic network that interferes with INM/ONM fusion during NPC insertion. Finally, although unconnected with membrane fusion, excess Lipin‐mediated PA metabolism impairs NPC membrane curving and maturation. Considered as a whole, these data provide new insight into the mechanisms by which the Torsin protein family affects the cell, including their relationship with interphase NPC insertion.
CTDNEP1 is a transmembrane ER/NE protein with a catalytic phosphatase domain facing the cytosol or nucleus. NEP1R1 is its transmembrane‐binding partner that stabilizes CTDNEP1 against degradation (Han et al, 2012). The NEP1R1‐CTDNEP1 complex is known to dephosphorylate Lipin to, in turn, target Lipin to the nucleus and activate Lipin PA phosphatase activity (Santos‐Rosa et al, 2005; Kim et al, 2007; Han et al, 2012; Bahmanyar et al, 2014). Our data now extend this knowledge by indicating that NEP1R1‐CTDNEP1 connects Torsins of the NE lumen to Lipin. The evidence includes that Torsin loss led to elevated NE‐localized levels of CTDNEP1 and/or NEP1R1 in mammalian and fly fat body cells, that NEP1R1‐CTDNEP1 overexpression in mammalian cells mimicked how Torsin loss affects Lipin localization, and that NEP1R1‐CTDNEP1 expression was required for mammalian Torsins to affect Lipin localization. Further, Nep1r1 or Ctdnep1 RNAi in fly almost completely reversed the lipidomic defects associated with dTor loss, thus indicating that Torsin only affects Lipin PA phosphatase activity and the cellular lipidome when NEP1R1 and CTDNEP1 are also present.
The genetic analyses showed that NEP1R1‐CTDNEP1 is key to why Torsin loss blocks INM/ONM fusion during NPC insertion. However, this does not appear to involve Lipin or lipid metabolism, given that Lipin, Nep1r1, and Ctdnep1 RNAi similarly rescued dTorKO ‐associated lipidomic defects, but only Nep1r1 and Ctdnep1 RNAi robustly prevented NPC‐associated membrane herniations and restored normal NPC levels. Additional work is needed to define why NEP1R1‐CTDNEP1 removal allows INM/ONM fusion to proceed in the absence of Torsins. There are many outstanding questions, including whether Nep1r1 and Ctdnep1 RNAi act by reducing NEP1R1‐CTDNEP1 phosphatase activity or by releasing proteins that interact with NEP1R1 and/or CTDNEP1. Further, while our data define that dTor acts upstream of Nep1r1 and Ctdnep1 within a genetic network that interferes with INM/ONM fusion during NPC insertion, the mechanism could be indirect whereby none of these three proteins directly affect the NPC.
The analyses of NPC biogenesis led to the unexpected discovery that independent molecular events drive the membrane fusion versus the membrane curving of interphase NPC insertion. This includes that, while we found no evidence that Lipin affected fusion, NPC membrane curving was impaired in fat body cells overexpressing Lipin. Moreover, PA metabolism appears responsible given that co‐overexpression of DAG kinase restored the normal highly curved morphology of mature pores. We also present evidence that Nup35/53 recruitment is the key driver of membrane curving, which is consistent with its known roles in membrane deformation and anchoring the inner ring complex/FG‐Nups to the NPC (Vollmer et al, 2012; Otsuka et al, 2016). Further, the presence of elongated channels in the dLap1 −/− suggests that the cell may actively regulate lipid metabolism to facilitate pore membrane curving. Additional work is needed to uncover the relationship between dLap1 and NEP1R1‐CTDNEP1/Lipin and to dissect why excess PA to DAG conversion impairs Nup35 recruitment; potentially insufficient PA and/or NE‐localized DAG or TAG accumulation is responsible. The post‐mitotic cells of the larval fat body will be ideal to address this, as well as to further define the molecular processes and machinery of interphase NPC insertion.
The AAA+ ATPase superfamily has numerous members that assemble into oligomeric structures and use the energy of ATP hydrolysis to dissociate otherwise stable protein complexes (Hanson & Whiteheart, 2005). However, Torsins are atypical members that form at least two different structures, a Torsin–activator complex that has ATPase activity or Torsin homo‐oligomers that lack ATPase activity but have membrane deforming properties (Chase et al, 2017; Demircioglu et al, 2019). Here, we find that dTorKO and dLap1 −/− differentially affect fat body lipid homeostasis and NPC insertion, which suggests that dTor retains at least some function when its ATPase activity is lost. This is further supported by the fact a dTor residue involved in homo‐oligomerization is essential for fat body development (Chase et al, 2017; Demircioglu et al, 2019). We therefore hypothesize that homo‐oligomer formation is key to dTor function in the developing fat body.
Additional work is required to understand the mechanistic and structural basis by which Torsin homo‐oligomers suppress NEP1R1‐CTDNEP1 activity. One possibility is that they dissociate NEP1R1 binding to CTDNEP1 since this would, in turn, trigger CTDNEP1 turnover. This mechanism is suggested by the data showing that NEP1R1‐CTDNEP1 overexpression mimics the effect of Torsin loss on Lipin localization and that cells lacking Torsins have elevated levels of NE‐localized CTDNEP1. However, altered CTDNEP1 protein levels may not be the only Nep1r1/Ctdnep1 gain‐of‐function mechanism in cells lacking Torsins. It is also unclear whether Torsins directly bind NEP1R1 or CTDNEP1 and, if so, which domains are responsible. It is alternatively feasible that the interaction is indirect and, given the importance of dTor membrane association, involves structural changes within or via the nuclear membrane. Finally, it is unlikely that all Torsin functions are mediated by its homo‐oligomeric structures. There is substantial evidence that Torsin–activator ATPase activity modulates NE‐localized LINC complexes (Nery et al, 2008; Van der Heyden et al, 2009; Jungwirth et al, 2011; Saunders et al, 2017; Dominiguez Gonzalez et al, 2018; Chalfant et al, 2019) and modifies LAP1 chromatin binding (Luithle et al, 2020). It therefore appears that Torsin function varies by cellular context, and it will be interesting to discover how the cell controls whether Torsin homo‐oligomer or Torsin–activator complexes predominate.
The finding that NEP1R1‐CTDNEP1 downregulation reverses the cellular defects of Torsin loss also has clinical relevance. TorsinA is associated with at least two diseases; a glutamic acid residue deletion causes autosomal dominant childhood‐onset dystonia (Ozelius et al, 1997), while homozygous mutations in TOR1A/TorsinA or TOR1AIP1/LAP1 cause autosomal recessive congenital syndromes (Kariminejad et al, 2017; Fichtman et al, 2019). While the connection between TorsinA loss and disease remains poorly defined, abnormal Lipin PA phosphatase activity and NPC abnormalities occur in mouse models and/or patient cells (Goodchild et al, 2005; Liang et al, 2014; Tanabe et al, 2016; Shin et al, 2019; Cascalho et al, 2020). Thus, the data we present here that NEP1R1‐CTDNEP1 downregulation normalizes the lipidomic and NPC defects of Torsin loss define NEP1R1‐CTDNEP1 as a therapeutic target for a series of incurable human diseases.
Materials and Methods
Fly stocks, maintenance, and tissue collection
Drosophila stocks (Appendix Table S1) were maintained using standard protocols and fed a standard diet consisting of cornmeal, agar, yeast, sucrose, and dextrose. Experimental crosses were kept at 25°C. Developmentally staged larvae were collected (randomized) 3, 4, or 5 days after placing male and female flies in fresh vials for a 12‐h period. The sample size that examined whether Nep1r1/Ctdnep1/Lipin RNAi reverted abnormalities in the dTorKO was based on that which detected statistically significant differences between control and dTorKO flies in this study or a previous study (Grillet et al, 2016). The fat body was exposed in individual larvae using standard methods and was either imaged under bright field with a Zeiss Discovery V12 stereomicroscope, snap‐frozen on dry ice, or fixed as described below.
New Drosophila lines
Microinjections were performed by BestGene Inc. (CA, USA). We used CRISPR to disrupt genomic sequences by injecting vas‐cas9 (X) flies (Norrander et al, 1983) with pCFD3‐dU6:3gRNA (Addgene #49410 (Port et al, 2014)) carrying guide RNA sequences. Transgenic UAS fly lines were produced by injecting pUAST plasmids into attP2+ or attP40+ stocks for PhiC31 integrase‐mediated site‐specific transgenesis.
dLap1 −/− (Torip; CG14103)
The flyCRISPR Target Finder Tool (https://flycrispr.org/) predicted a guide RNA 5′‐gCCGATCAGTCCAGAAGACG‐3′ that would introduce a double‐stranded break into exon 1 of dLap1, 191bp after the start codon. G0 flies were crossed with w/y; Tub‐Gal4/TM6C, Tb flies. 158 offspring with white eyes and short bristles were expanded into lines. The dLap1 locus of each line was PCR amplified and sequenced to identify lines where non‐homologous end joining introduced frameshift mutations. We identified two lines with an 11‐nucleotide deletion (Δ11) for further characterization.
UAS lines
One or more cDNA sequences were cloned between the EcoRI and XhoI sites of pUAST‐attB (Bischof et al, 2007) using the Gibson NEBuilder® Hifi DNA assembly kit. We also generated a modified pUAST_attB‐V5‐His backbone by assembling a 135bp PCR‐amplified fragment containing the V5 and His tag from pBudCE4.1 (Invitrogen™, V53220) with pUAST‐attB linearized with XbaI. The coding region of all pUAST plasmids was fully verified by Sanger sequencing before further use.
pUAST‐mGFP‐Sec61b was produced by assembling pUAST with (i) a 5′ PCR fragment encoding mGFP (amplified from pCR8‐dTormGFP) and (ii) a 3′ PCR fragment encoding mouse Sec61b amplified from pcDNA3 SEC61B‐V5‐APEX (Addgene #83411 (Lee et al, 2016)).
pUAST‐dTor plasmids with mutagenized dTor were produced by assembling two PCR‐amplified DNA fragments into pUAST. Both reactions used pUAST‐dTormGFP as the template (Grillet et al, 2016). PCR 1 generated the 5′ sequence from the ATG of dTor until the mutation and PCR 2 generated a 3′ fragment extending from immediately after the mutation to the stop codon of mGFP. 3′ and 5′ primers of PCR 1 and PCR 2, respectively, carried the mutation. dTormGFP‐ΔMMD was produced by Quikchange site‐directed mutagenesis to delete codons 2–42. For dTor membrane domain chimeras, these again used Gibson Assembly to integrate 2 PCR‐derived fragments into pUAST: PCR 1 generated sequence encoding the TMD of Sec61b or dLap1 using pcDNA3 Sec61b‐V5‐APEX (Addgene #83411 (Lee et al, 2016)) and dLap1 (Grillet et al, 2016) as templates, respectively. PCR 2 generated dTormGFP sequence from codon 43 of dTor and using a dTormGFP‐WT (Grillet et al, 2016) as template.
Ctdnep1 (Dd, NM_134605) and Nep1r1 (CG41106, NM_001042844) cDNAs lacking the stop codon were PCR amplified from third‐instar larvae cDNA. These were assembled with a PCR fragment encoding a linker and mGFP. We generated phosphatase‐dead CTDNEP1 by mutating the catalytic LDLD motif to LELE (Kim et al, 2007), via assembling two PCR‐amplified DNA fragments into pUAST. Both reactions used pUAST‐CTDNEP1mGFP as the template. PCR 1 generated the 5′ sequence from the ATG to the site of the mutation, and PCR 2 generated the 3′ sequence extending from immediately after the mutation to the stop codon of mGFP. 3′ and 5′ primers of PCR 1 and PCR 2, respectively, carried the mutation.
Dgk (DgK, DAGK, NM_078930.4) and Lipin (Lpin, NM_136515) cDNA sequences lacking the stop codon were amplified from EST clones (BT003323 and GH19076 Source BioScience) and assembled as individual fragments with pUAST_attB‐V5‐His.
Cell lines
Cell culturing, electroporation, and transfection were performed according to the manufacturer’s instructions, unless otherwise stated.
Plasmid cloning
Cloning was performed using the Gibson NEBuilder® Hifi DNA assembly kit. Primer sequences, including overhangs for Gibson cloning, are provided in Appendix Table S4. Mouse Lipin1 and GFP were amplified and inserted into pcDNA5⁄FRT⁄TO (Invitrogen) linearized by HindIII and XhoI, as a three‐fragment Gibson cloning reaction. Lipin1 was PCR amplified from “pRK5 FLAG wild type, catalytic active Lipin1” or “pRK5 FLAG 17xS/T‐>A, catalytic active Lipin1” that were gifts from David Sabatini (Addgene plasmids # 32005, 32007 (Peterson et al, 2011)). mGFP with a short linker was amplified from a modified pEGFP‐N1 (Clontech).
We produced dual‐promoter pBudCE4.1‐CTDNEP1myc‐His/ NEP1R1V5‐His as a four‐fragment Gibson assembly. Human CTDNEP1 was amplified from cDNA clone MGC:16648 IMAGE:4123279, human NEP1R1 was amplified from cDNA clone MGC:41938 IMAGE:5266084, and pBudCE4.1 (Invitrogen) was the template for PCR amplification of the two backbone fragments. CTDNEP1‐CD was produced by mutating the catalytic LDLD motif to LELE (Kim et al, 2007). The pBudCE4.1‐CTDNEP1myc‐His‐CD/ NEP1R1V5‐His plasmid was generated by excising wild‐type CTDNEP1 from pBudCE4.1‐CTDNEP1myc‐His/NEP1R1V5‐His as a HindIII and XbaI fragment. The backbone was then re‐assembled with CTDNEP1‐CD produced as two PCR fragments. CTDNEP1 PCR 1 generated the 5′ DNA fragment from the ATG to the site of the mutation, and PCR 2 generated the 3′ CTDNEP1 fragment from immediately after the mutation to the penultimate codon. 3′ and 5′ primers of PCR 1 and PCR 2, respectively, carried the “LELE” mutation. pBudCE4.1‐CTDNEP1Scarlet and pBudCE4.1‐NEP1R1Scarlet plasmids were produced in two steps. Scarlet sequence was amplified from pmScarlet_H2A_C1 (a gift from Dorus Gadella (Addgene plasmid #85051, Bindels et al, 2017) and inserted into pBudCE4.1‐CTDNEP1myc‐His/ NEP1R1V5‐His linearized with BamH1 (to produce the CTDNEP1 fusion) or SfiI (to produce the NEP1R1 fusion). Then, CTDNEP1Scarlet and NEP1R1Scarlet sequences were amplified and re‐cloned by Gibson assembly in front of the pEF1α promoter of empty pBudCE4.1 linearized by Xho1 and BglII.
Flp‐In HEK293T
HEK293T lines stably expressing mouse Lipin1mGFP were produced using the Invitrogen Flp‐In system (Thermo Fisher Scientific). pBudCE4.1 plasmids were introduced into the new cell line using Lipofectamine 2000 (Thermo Fisher Scientific), after plating cells onto coverslips precoated with 0.1 mg/ml poly‐L‐lysine (Sigma). Lipin1mGFP expression was induced by incubation with 1 µg/ml tetracycline for 24 h.
MEF lines
Tor1a+/+ and Tor1a Δgag/Δgag lines have been previously described (Cascalho et al, 2020). We targeted Tor1b in the Tor1a Δgag/Δgag line (Tor1aKO ) using predesigned Alt‐R® CRISPR‐Cas9 guide RNA (Integrated DNA Technologies) against exon1 (5′‐g GGAACGGCCCCTCAACACGTCGG‐3′), cloned into pX459 V2.0;pSpCas9(BB)‐2A‐Hygro (Addgene #62988, puromycin cassette replaced by hygromycin) according to the Zhang lab protocol (Cong et al, 2013). Cells were transfected and placed under selection for 2 weeks, and then DNA was extracted and amplified to identify clonal lines carrying frameshift mutations. Additional details are provided in the Appendix. Single plasmids were introduced into TorA/BWT and TorA/BKO MEF lines by electroporation using the T20 program of a Nucleofector™ II/2b device (Lonza), then immediately plated onto coverslips and fixed 24 h later. For shRNA experiments, plasmids containing Ctdnep1 and Nep1r1 shRNA (TRCN0000247433 and TRCN0000253537 (Sigma)) or a scrambled shRNA (gift from David Sabatini (Addgene #1864) were electroporated as above and plated onto coverslips. 48 h later, each coverslip was transfected with 400 ng pcDNA5⁄FRT⁄TO‐Lipin1mGFP using Lipofectamine 2000 and fixed after another 24 h of culturing.
qRT–PCR
RNA was extracted using the RNeasy Mini Kit (Qiagen), followed by DNAse1 treatment, and cDNA synthesis with the SuperScript II system (Life Technologies) and a mix of oligo(dt) and random hexamer primers. 100 ng of cDNA (or equivalent volume of negative control sample) was added with primers to the LightCycler 480 SYBR Green I Master Mix (Roche) and quantitative PCR performed and analyzed with a LightCycler 480 (Roche). All qPCR runs were performed in triplicate. The ribosomal house‐keeping gene, RpL32, was used to normalize the data for loading differences. The fold change in mRNA expression was determined for each gene using the 2–∆∆Ct method, where the expression of the genes was compared with the control sample. Primers are listed in Appendix Table S4.
Fluorescent labeling
Procedures were performed at room temperature unless otherwise stated.
Larval fat body
Larval preparations were fixed for 20 min in 4% formaldehyde (Sigma #47608) / phosphate‐buffered saline (PBS; pH 7.4) and then washed with PBS containing 0.4% Triton X‐100 (PBS‐X). Immunolabeling was performed as previously described (Soldano et al, 2013). Cell borders and lipid droplets were labeled by a 45‐min incubation with PBS‐X containing 0.03 mg/ml Alexa Fluor® 488 or 594 Phalloidin (Thermo Fisher Scientific #A12379 and #A12381), followed by 3 × 10‐min washes in PBS‐X, and then a 30‐min incubation with either (i) 1 μg/ml BODIPY493/503 (Invitrogen #D‐3922) or (ii) HCS LipidTox Neutral Red 577/609 or Deep red 650/670 (1:300 dilution). Samples were then further washed in PBS‐X and prepared for microscopy by placing the larval preparation (3DO) or isolated fat body (4DO and 5DO) into VECTASHIELD containing DAPI (Vector Laboratories #H‐1200).
Cultured cells
Cells cultured on coverslips were fixed with 4% formaldehyde (Sigma #47608) in PBS for 20 min or with pre‐chilled 100% methanol for 5 min at −20°C, followed by three 10‐min washes in PBS. Cells were incubated with blocking buffer (PBS, 0.25% Triton X‐100, 10% Normal Donkey Serum (Jackson ImmunoResearch)) for 1 h in a humidified chamber, which was then replaced by chilled blocking buffer containing diluted antibody. Samples were incubated for 16 h at 4°C, then washed three to five times for 10 min with PBS, followed by 2‐h incubation with blocking buffer containing diluted secondary antibodies. After a final series of three to five 10‐min washes with PBS, coverslips were mounted with VECTASHIELD with DAPI (Vector Laboratories #H‐1200).
Lipid measurements
TAG was measured by enzymatic assay as described in Grillet et al (2016), unless otherwise stated. MS was performed on samples of twenty 4DO or 5DO fat bodies homogenized in 100 µl D‐PBS (Dulbecco’s phosphate‐buffered saline without Mg2+ and Ca2+) by Lipotype using previously described methods (Grillet et al, 2016). Pmol values of individual lipid species (determined by MS) were transformed into a fraction of the total PI, PA, DAG, PC, PE, and PS lipids in the sample. The comparison of relative lipid class levels in control, dTorKO, and dLap1 −/− fat bodies included a reanalysis of previously reported data (Grillet et al, 2016). Protein was measured by BCA assay.
Western Blotting
Cell pellets and tissue were lysed in T‐PER (Thermo Fisher Scientific) containing protease inhibitors (Sigma). 30 μg of protein was subjected to standard SDS–PAGE and Western blotting with HRP‐conjugated secondary antibodies. Signal was visualized by West Pico Plus chemiluminescent reagent (Thermo Fisher Scientific) and an ImageQuant LAS 4000 device.
Electron microscopy
TEM
Isolated fat bodies from several animals were fixed in 2.5% glutaraldehyde (Electron Microscopy Services (EMS) #16220), 4% paraformaldehyde (PFA) (EMS #15714), 0.2% picric acid (EMS #19554), 1% sucrose in 0.1 M PB (phosphate buffer, pH 7.4) at 4°C for at least 24 h, and up to 3 weeks. The tissue was then condensed by embedding in 4% low gelling temperature agarose (Sigma, #A9414) and replaced in fixative at 4°C overnight. Samples were washed three times in 0.1 M SCB (sodium cacodylate trihydrate buffer (pH 7.6); EMS #12300), post‐stained with 1% OsO4 (2% Aqueous solution, EMS #19152) and 1.5% potassium ferrocyanide (Sigma #455989) diluted in 0.1 M SCB (pH 7.6) for 1hr. Then, samples were incubated with 0.2% tannic acid (EMS #2170) and stained with 0.5% uranyl acetate (EMS #22400) in 25% methanol overnight, followed by washing and staining with lead aspartate (EMS #17900) en bloc for 30 min at 60°C. Finally, samples were dehydrated through a graded series of ethanol solutions, infiltrated, and embedded in Epon (Agar100) and cured at 60°C for 48 h. Sections (70 nm) were cut with a DuPont Diamond Knife on a Leica UCT ultra‐microtome and collected on copper grids.
3D‐EM
Larval filets were fixed in 4% PFA, 0.5% glutaraldehyde in 0.1 M PB for 1 h at room temperature. After samples were rinsed in 0.1 M PB, near‐infrared branding (NIRB) was performed using a Zeiss LSM 780 inverted confocal microscope equipped with a Mai Tai DeepSee two‐photon laser (Spectra‐Physics). Branding marks were introduced as a square around nuclei of interest using the two‐photon laser at 800 nm and 40% maximal power. Two Z‐stacks with a 63× objective and a 25× objective 5× zoom were taken of the region of interest after NIRB together with an overview with the 25× objective.
Samples were then trimmed, post‐fixed overnight at 4°C in freshly prepared 4% PFA, 2.5% glutaraldehyde, 0.2% picric acid in 0.1 M PB, and stored in this solution until further processing. Next, samples were incubated in 1% OsO4, 1.5% potassium ferrocyanide in ultrapure water for 1 h at room temperature, followed by 30 min in 0.2% tannic acid. Then, samples were incubated in 0.5% uranyl acetate in 25% methanol overnight at 4°C. After rinsing with ultrapure water, samples were stained en bloc with lead aspartate and dehydrated using ice‐cold solutions of increasing ethanol concentration followed by flat embedding in Epon resin. Embedded samples were mounted on aluminum pin stubs (Gatan) with conductive epoxy glue (Circuit Works) and inserted in a Zeiss Sigma Variable pressure SBF‐SEM with 3 View technology (Gatan) to approach the region of interest. Once nuclei of interest were located, based on the branding marks and fat body morphology, the pins were inserted in FIB‐SEM. Samples were coated with ˜20 nm of platinum in a sputtercoater (Quorum Q150T ES).
CLEM
Larval filets were fixed in 4% PFA, 0.5% glutaraldehyde in 0.1 M PB for 30 min at room temperature. After samples were rinsed in 0.1 M PB, a 1.35‐mm disk was cut out of larval FB and submerged in 20% BSA in DMEM medium with 10% fetal calf serum (FCS). Next tissue disks were loaded in membrane carriers (0.1 mm thick, Leica‐microsystems), frozen with a high‐pressure freezer (Leica EMPACT2), and vitrified at 2,050 bar. The quick freeze substitution protocol was started by transferring the membrane carriers to cryotubes (72.694.005, Sarstedt, Germany) with freeze substitution medium with 0.2% uranyl acetate (#02624‐AB, SPI) in acetone (#1002990500, Merck) and 5% Milli‐Q water at −180°C. Once the cryotubes reached −80°C, the cryotube‐holder was placed on its side and agitated. At −50°C, the carriers were transferred to the pre‐cooled −50°C Leica AFS2 apparatus, washed in 100% acetone, and after a final rinsing in ethanol, infiltrated with lowicryl HM20 (#02628‐AB, SPI), and finally polymerized at −50°C with UV. 200‐nm sections were cut on a Leica Ultracut S ultramicrotome and collected on microscopic slides and immediately followed by 70‐nm sections collected on slot grids (#01805‐F, Ted Pella, USA). The fluorescence image of the last 200‐nm section was overlaid with EM images of the first 70‐nm section using GNU Image Manipulation Program (GIMP).
Microscopy and image analysis
Fat body cells were imaged in 5DO animals, unless otherwise stated, by a researcher blind to genotype. Fly and cell line image analysis was performed using FIJI unless otherwise stated, by a researcher blind to animal genotype and/or plasmid transfection. At least three animals and/or independently generated cell preparations were examined in each analysis.
Size and density
Images were collected using a Nikon A1R Eclipse Ti microscope and 40x or 60x objectives. We measured the size of fat body cells when the optical section bisected the nucleus, by outlining the cell boundary (as defined by phalloidin) with the freehand selection tool. We measured LD density within each cell by determining the percentage of pixels that had LD‐associated fluorescence. LD size was measured using an optimized macro that detected single LD particles (analyze particles size = 5‐infinity) after smoothening and applying a threshold that was manually verified to specifically detect LD. Nuclear size was measured from images of anti‐LaminDm0‐labeled tissue imaged on a Zeiss Airyscan microscope at 63× magnification, with the Z‐plane bisecting the widest part of a nucleus. The area of each nucleus in an image (> 30 nuclei in total for each age) was measured by manual outlining.
Protein localization
HEK293T‐Lipin1mGFP cells transfected with plasmids co‐expressing human CTDNEP1myc‐WT or CTDNEP1myc‐CD with NEP1R1V5, and labeled with anti‐myc and anti‐V5, were imaged at 60× on a C2 Nikon confocal microscope. MEF lines expressing Lipin1mGFP were imaged at 60× with oil on a A1R1 Nikon confocal microscope. GFP was scored as primarily nuclear or primarily cytosolic by an observer blind to condition, plasmid, and cell genotype. In the case of HEK cell analyses, only myc/V5‐positive cells were considered. More than 50 transfected cells were examined per condition. Cells with obvious abnormalities, including multi‐nucleated cells, were not considered.
The intensity of fluorescent signal at the nuclear periphery was assessed by quantifying pixel intensities along a 3 µm profile that spanned the NE (defined by DAPI labeling). These were transformed to a percentage of the maximum intensity detected along the line, or to the maximum value from control cells for Nup35 labeling. Three lines were assessed for each cell, and a total of five cells were examined.
MEF lines expressing CTDNEP1mScarlet and labeled with anti‐Sun1 were imaged at 60x with oil on a A1R1 Nikon confocal microscope. NE colocalization was examined by manually outlining the NE (defined by the Sun1 marker) and using the Coloc 2 plug‐in of Fiji to calculate Pearson’s linear correlation coefficient.
Fat body tissue labeled with mAb414 and DAPI was examined with a Zeiss Airyscan and 63× magnification. Individual nuclei were scored for whether mAb414 specifically labeled the NE (strong NE staining), labeled the NE and the cytosol/nucleus, or there was no clear NE labeling. At least 60 cells per genotype were examined.
Nuclear membrane ultrastructure
Tissue was prepared for TEM using variations of standard methods that are described in detail in the Appendix Supplementary Methods. Thin sections were imaged with JEOL JEM1400 transmission electron microscope operated at 80 kV and equipped with an Olympus SIS Quemesa (11 Mpxl) camera. Quantification of NE ultrastructure was performed with images of 2,500× and 10,000× magnification. NE structure was considered normal when the INM and ONM were adjacent and ran approximately parallel, and no additional membrane was present within the NE lumen. An observer determined the percentage of NE that fulfilled these criteria for at least 10 nuclei. The density of mature nuclear pores and attached herniations was calculated by counting their number and measuring the length of the NE.
FIB‐SEM imaging was performed using a Zeiss Crossbeam 540 system with Atlas5 software. The Focused Ion Beam (FIB) was set to remove 5‐nm sections by propelling gallium ions at the surface. Atlas software was set to image a 10 × 5 µm area at 5‐nm pixels (5 µs dwell time & line averaging 4) using an ESB (back‐scattered electron) detector with the electron beam at 1.5 kV and 1 nA. FIB‐SEM data were first denoised using Fiji and the Tikhonov algorithm using following parameters; lambda = 2, iterations = 50, and sigma=1.058. Next, data were segmented using Microscopy Image Browser (MIB) software and 3D modeling was performed using Amira software (FEI/Thermo Fisher Scientific, France). The distance between INM and ONM adjacent to pores/ channels was calculated from the 3D reconstruction of FIB‐SEM images.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 8.2.1 software. Data are presented as mean and SD, unless otherwise stated. Differences between two groups were assessed using a two‐tailed t‐test, differences between three or more groups were assessed by one‐way ANOVA, with Bonferroni’s post hoc test, unless otherwise stated. The criteria for significance are as follows: ns (not significant), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Author contributions
JJ and REG developed the project concept, designed experiments, collected and analyzed data, wrote and edited the text, and assembled the figures. JJ, PV, and REG supervised and/or analyzed experiments. REG wrote the manuscript revision and revised figures with input from JJ and PV, while JF, SFG, JS, KV, and NVG developed concepts for specific data sets, developed methods, collected, and analyzed data. PV and REG supervised Drosophila genetics. PV, NVG, and REG supervised and/or interpreted EM experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Table EV1
Source Data for Appendix
Source Data for Figure 2
{kind=link}
Acknowledgements
This work was only possible by the broad support provided by the Foundation for Dystonia Research. R.G. does not agree with the manner that VIB and the KUL used Foundation for Dystonia Research funding during 2020. J.J. and J.F. held FWO Sb fellowships (1S07317N and 1S54121N). We thank the Dystonia Medical Research Foundation for an award. Data storage was supported by an ISPAMM project grant (AKUL/13/39). The Nikon A1R Eclipse Ti was acquired through a Hercules type I grant (AKUL/09/037) to Wim Annaert. We thank Jens Rummens for many discussions with many authors, as well as work with cell lines, to Sergio Hernandez Diaz for supporting fly husbandry, to Nils Schoovaerts for plasmids, and to visiting students Noémie De Broeck, Sara Garcia Garrote, and Laura Ordonez Cuerva. We thank Professor Samakovlis for the kind gift of the anti‐Nup214 antibody. We also thank the Gent VIB Bio imaging Core, especially Anneke Kremer, and the Leuven VIB Bio imaging Core, especially Benjamin Pavie. We also wish to acknowledge the advice of Ragna Sannerud and the Annaert Lab, as well as the Schmucker Lab, for cell lines. The KU Leuven did not provide financial or logistical support to RG to write and revise the manuscript or for her supervision of KU Leuven students after Feb 2020. This work is supported by the NIH grant U2C‐DK119886.
The EMBO Journal (2021) 40: e106914.
Data availability
Detailed information is provided in tables for Drosophila melanogaster lines (Appendix Table S1), plasmids (Appendix Table S2), antibodies (Appendix Table S3), and primers (Appendix Table S4) used in this study. Individual lipid species data (as a fraction of total membrane lipids) are provided in Table EV1. The lipidomic data generated by Lipotype is available at the NIH Common Fund's National Metabolomics Data Repository (NMDR) Web site, the Metabolomics Workbench, https://www.metabolomicsworkbench.org where it has been assigned Project ID PR001172. The data can be accessed directly via its Project DOI: http://dx.doi.org/10.21228/M86T2S (Sud et al, 2016).
References
- Adeyo O, Horn PJ, Lee S, Binns DD, Chandrahas A, Chapman KD, Goodman JM (2011) The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J Cell Biol 192: 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M, Zimmerli C, Rantos V, Wilfling F, Ronchi P, Fung H, Lee C‐W, Hagen W, Turonova B, Karius Ket al (2020) In‐cell architecture of the nuclear pore complex and snapshots of its turnover. Nature 586: 796–800 [DOI] [PubMed] [Google Scholar]
- Bahmanyar S, Biggs R, Schuh AL, Desai A, Müller‐Reichert T, Audhya A, Dixon JE, Oegema K (2014) Spatial control of phospholipid flux restricts endoplasmic reticulum sheet formation to allow nuclear envelope breakdown. Genes Dev 28: 121–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MAet al (2017) MScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods 14: 53–56 [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ‐line‐specific phiC31 integrases. Proc Natl Acad Sci USA 104: 3312–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascalho A, Foroozandeh J, Hennebel L, Swerts J, Klein C, Rous S, Dominguez Gonzalez B, Pisani A, Meringolo M, Gallego SFet al (2020) Excess Lipin enzyme activity contributes to TOR1A recessive disease and DYT‐TOR1A dystonia. Brain 143: 1746–1765 [DOI] [PubMed] [Google Scholar]
- Chalfant M, Barber KW, Borah S, Thaller D, Lusk CP (2019) Expression of TorsinA in a heterologous yeast system reveals interactions with lumenal domains of LINC and nuclear pore complex components. Mol Biol Cell 30: 530–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase AR, Laudermilch E, Wang J, Shigematsu H, Yokoyama T, Schlieker C (2017) Dynamic functional assembly of the Torsin AAA+ ATPase and its modulation by LAP1. Mol Biol Cell 28: 2765–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran Fa, Cox D, Lin S, Barretto R, Habib N, Hsu Pd, Wu X, Jiang W, Marraffini Laet al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock CP, Adams N, Bryant FM, Kurup S, Eastmond PJ (2015) Phosphatidic acid phosphohydrolase regulates phosphatidylcholine biosynthesis in Arabidopsis by phosphatidic acid‐mediated activation of CTP: phosphocholine cytidylyltransferase activity. Plant Cell 27: 1251–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrigrand J‐F, Valente M, Comai G, Martinez P, Petit M, Nishinakamura R, Osorio DS, Renault G, Marchiol C, Ribes Vet al (2020) Dullard‐mediated smad1/5/8 inhibition controls mouse cardiac neural crest cells condensation and outflow tract septation. Elife 9: 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demircioglu FE, Zheng W, Mcquown AJ, Maier N, Watson N, Cheeseman M, Denic V, Egelman EH, Schwartz TU (2019) The AAA+ ATPase TorsinA polymerizes into hollow helical tubes with 8.5 subunits per turn. Nat Commun 10: 3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominiguez Gonzalez B, Billion K, Rous S, Pavie B, Lange C, Goodchild R (2018) Excess LINC complexes impair brain morphogenesis in a mouse model of recessive TOR1A disease. Hum Mol Genet 27: 2154–2170 [DOI] [PubMed] [Google Scholar]
- Fichtman B, Zagairy F, Biran N, Barsheshet Y, Chervinsky E, Ben Neriah Z, Shaag A, Assa M, Elpeleg O, Harel Aet al (2019) Combined loss of LAP1B and LAP1C results in an early onset multisystemic nuclear envelopathy. Nat Commun 10: 605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT (2005) The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol 168: 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Kim CE, Dauer WT (2005) Loss of the dystonia‐associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 48: 923–932 [DOI] [PubMed] [Google Scholar]
- Grillet M, Dominguez Gonzalez B, Sicart A, Pöttler M, Cascalho A, Billion K, Hernandez Diaz S, Swerts J, Naismith TV, Gounko NVet al (2016) Torsins are essential regulators of cellular lipid metabolism. Dev Cell 38: 235–247 [DOI] [PubMed] [Google Scholar]
- Haider A, Wei Y‐C, Lim K, Barbosa AD, Liu C‐H, Weber U, Mlodzik M, Oras K, Collier S, Hussain MMet al (2018) PCYT1A regulates phosphatidylcholine homeostasis from the inner nuclear membrane in response to membrane stored curvature elastic stress. Dev Cell 45: 481–495.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B, Schwarz A, Ronchi P, Bragulat‐Teixidor H, Tischer C, Gaspar I, Ephrussi A, Schwab Y, Beck M (2019) Nuclear pores assemble from nucleoporin condensates during oogenesis. Cell 179: 671–686.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G‐S, Siniossoglou S, Carman GM (2007) The cellular functions of the yeast lipin homolog PAH1p are dependent on its phosphatidate phosphatase activity. J Biol Chem 282: 37026–37035 [DOI] [PubMed] [Google Scholar]
- Han S, Bahmanyar S, Zhang P, Grishin N, Oegema K, Crooke R, Graham M, Reue K, Dixon JE, Goodman JM (2012) Nuclear envelope phosphatase 1‐regulatory subunit 1 (formerly TMEM188) is the metazoan Spo7p ortholog and functions in the lipin activation pathway. J Biol Chem 287: 3123–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW (2005) AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6: 519–529 [DOI] [PubMed] [Google Scholar]
- Harris TE, Huffman TA, Chi A, Shabanowitz J, Hunt DF, Kumar A, Lawrence JCJ (2007) Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J Biol Chem 282: 277–286 [DOI] [PubMed] [Google Scholar]
- Hodge CA, Choudhary V, Wolyniak MJ, Scarcelli JJ, Schneiter R, Cole CN (2010) Integral membrane proteins Brr6 and Apq12 link assembly of the nuclear pore complex to lipid homeostasis in the endoplasmic reticulum. J Cell Sci 123: 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemyn J, Cascalho A, Goodchild RE (2017) The ins and outs of endoplasmic reticulum‐controlled lipid biosynthesis. EMBO Rep 18: 1905–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokhi V, Ashley J, Nunnari J, Noma A, Ito N, Wakabayashi‐Ito N, Moore MJ, Budnik V (2013) Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep 3: 988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungwirth MT, Kumar D, Jeong DY, Goodchild RE (2011) The nuclear envelope localization of DYT1 dystonia torsinA‐DeltaE requires the SUN1 LINC complex component. BMC Cell Biol 12: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariminejad A, Dahl‐Halvarsson M, Ravenscroft G, Afroozan F, Keshavarz E, Goullée H, Davis MR, Faraji Zonooz M, Najmabadi H, Laing NGet al (2017) TOR1A variants cause a severe arthrogryposis with developmental delay, strabismus and tremor. Brain 140: 2851–2859 [DOI] [PubMed] [Google Scholar]
- Kim CE, Perez A, Perkins G, Ellisman MH, Dauer WT (2010) A molecular mechanism underlying the neural‐specific defect in torsinA mutant mice. Proc Natl Acad Sci USA 107: 9861–9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Gentry MS, Harris TE, Wiley SE, Lawrence JC, Dixon JE (2007) A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc Natl Acad Sci USA 104: 6596–6601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudermilch E, Tsai PL, Graham M, Turner E, Zhao C, Schlieker C (2016) Dissecting Torsin/cofactor function at the nuclear envelope: a genetic study. MBoC 27: 3964–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Park JH (2004) Hemolymph sugar homeostasis and starvation‐induced hyperactivity affected by genetic manipulations of the adipokinetic hormone‐encoding gene in Drosophila melanogaster . Genetics 167: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Kang MG, Park JS, Lee G, Ting AY, Rhee HW (2016) APEX fingerprinting reveals the subcellular localization of proteins of interest. Cell Rep 15: 1837–1847 [DOI] [PubMed] [Google Scholar]
- Li TY, Song L, Sun Yu, Li J, Yi C, Lam SM, Xu D, Zhou L, Li X, Yang Yet al (2018) Tip60‐mediated lipin 1 acetylation and ER translocation determine triacylglycerol synthesis rate. Nat Commun 9: 1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Tanabe LM, Jou S, Chi F, Dauer WT (2014) TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest 124: 3080–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G‐H, Gerace L (2009) Sumoylation regulates nuclear localization of lipin‐1alpha in neuronal cells. PLoS One 4: e7031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Matsuoka S, Enoki A, Yamamoto T, Furukawa K, Yamasaki Y, Nishida Y, Sugiyama S (2011) Negative modulation of bone morphogenetic protein signaling by Dullard during wing vein formation in Drosophila . Dev Growth Differ 53: 822–841 [DOI] [PubMed] [Google Scholar]
- Luithle N, De Bos JU, Hovius R, Maslennikova D, Lewis RTM, Ungricht R, Fierz B, Kutay U (2020) Torsin ATPases influence chromatin interaction of the torsin regulator LAP1. Elife 9: 1–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naismith TV, Heuser JE, Breakefield XO, Hanson PI (2004) TorsinA in the nuclear envelope. Proc Natl Acad Sci USA 101: 7612–7617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery FC, Zeng J, Niland BP, Hewett J, Farley J, Irimia D, Li Y, Wiche G, Sonnenberg A, Breakefield XO (2008) TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J Cell Sci 121: 3476–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrander J, Kempe T, Messing J (1983) Construction of improved M13 vectors using oligodeoxynucleotide‐directed mutagenesis. Gene 26: 101–106 [DOI] [PubMed] [Google Scholar]
- O’Hara L, Han G‐S, Peak‐Chew S, Grimsey N, Carman GM, Siniossoglou S (2006) Control of phospholipid synthesis by phosphorylation of the yeast lipin Pah1p/Smp2p Mg2+‐dependent phosphatidate phosphatase. J Biol Chem 281: 34537–34548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Bui KH, Schorb M, Julius Hossain M, Politi AZ, Koch B, Eltsov M, Beck M, Ellenberg J (2016) Nuclear pore assembly proceeds by an inside‐out extrusion of the nuclear envelope. Elife 5: e19071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Ellenberg J (2018) Mechanisms of nuclear pore complex assembly – two different ways of building one molecular machine. FEBS Lett 592: 475–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DPet al (1997) The early‐onset torsion dystonia gene (DYT1) encodes an ATP‐binding protein. Nat Genet 17: 40–48 [DOI] [PubMed] [Google Scholar]
- Pappas SS, Liang CC, Kim S, Rivera CAO, Dauer WT (2018) TorsinA dysfunction causes persistent neuronal nuclear pore defects. Hum Mol Genet 27: 407–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson T, Sengupta S, Harris T, Carmack A, Kang S, Balderas E, Guertin D, Madden K, Carpenter A, Finck Bet al (2011) mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146: 408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port F, Chen HM, Lee T, Bullock SL (2014) Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila . Proc Natl Acad Sci USA 111: E2967–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampello AJ, Laudermilch E, Vishnoi N, Prophet SM, Shao L, Zhao C, Lusk CP, Schlieker C (2020) Torsin ATPase deficiency leads to defects in nuclear pore biogenesis and sequestration of MLF2. J Cell Biol 219: e201910185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanauska A, Köhler A (2018) The inner nuclear membrane is a metabolically active territory that generates nuclear lipid droplets. Cell 174: 700–715.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Farber P, Velyvis A, Rennella E, Latham MP, Kay LE (2015) ClpB N‐terminal domain plays a regulatory role in protein disaggregation. Proc Natl Acad Sci USA 112: E6872–E6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Sharmin S, Taguchi A, Ohmori T, Fujimura S, Abe T, Kiyonari H, Komatsu Y, Mishina Y, Asashima Met al (2013) The phosphatase Dullard negatively regulates BMP signalling and is essential for nephron maintenance after birth. Nat Commun 4: 1398 [DOI] [PubMed] [Google Scholar]
- Santos‐Rosa H, Leung J, Grimsey N, Peak‐Chew S, Siniossoglou S (2005) The yeast lipin Smp2 couples phospholipid biosynthesis to nuclear membrane growth. EMBO J 24: 1931–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satow R, Kurisaki A, Chan TC, Hamazaki TS, Asashima M (2006) Dullard promotes degradation and dephosphorylation of BMP receptors and is required for neural induction. Dev Cell 11: 763–774 [DOI] [PubMed] [Google Scholar]
- Saunders CA, Harris NJ, Willey PT, Woolums BM, Wang Y, McQuown AJ, Schoenhofen A, Worman HJ, Dauer WT, Gundersen GGet al (2017) TorsinA controls TAN line assembly and the retrograde flow of dorsal perinuclear actin cables during rearward nuclear movement. J Cell Biol 216: 657–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarcelli JJ, Hodge CA, Cole CN (2007) The yeast integral membrane protein Apq12 potentially links membrane dynamics to assembly of nuclear pore complexes. J Cell Biol 178: 799–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S, Ugrankar R, Greene SE, Prajapati M, Lehmann M (2015) Drosophila Lipin interacts with insulin and TOR signaling pathways in the control of growth and lipid metabolism. J Cell Sci 128: 4395–4406 [DOI] [PubMed] [Google Scholar]
- Shimizu K, Fukushima H, Ogura K, Lien EC, Nihira NT, Zhang J, North BJ, Guo A, Nagashima K, Nakagawa Tet al (2017) The SCFβ‐TRCP E3 ubiquitin ligase complex targets Lipin1 for ubiquitination and degradation to promote hepatic lipogenesis. Sci Signal 10: eaah4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Hernandez‐Ono A, Fedotova T, Östlund C, Lee MJ, Gibeley SB, Liang CC, Dauer WT, Ginsberg HN, Worman HJ (2019) Nuclear envelope–localized torsinA‐LAP1 complex regulates hepatic VLDL secretion and steatosis. J Clin Invest 129: 4885–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniossoglou S, Santos‐Rosa H, Rappsilber J, Mann M, Hurt E (1998) A novel complex of membrane proteins required for formation of a spherical nucleus. EMBO J 17: 6449–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldano A, Okray Z, Tmejova K, Reynaud E, Claeys A, Yan J, Atak ZK, De Strooper B, Dura JM (2013) The Drosophila homologue of the amyloid precursor protein is a conserved modulator of Wnt PCP signaling. PLoS Biol 11: e1001562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sołtysik K, Ohsaki Y, Tatematsu T, Cheng J, Maeda A, Morita SY, Fujimoto T (2021) Nuclear lipid droplets form in the inner nuclear membrane in a seipin‐independent manner. J Cell Biol 220: e202005026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa BA, Esra Demircioglu F, Chen JZ, Ingram J, Ploegh HL, Schwartz TU (2014) How lamina‐associated polypeptide 1 (LAP1) activates Torsin. Elife 3: e03239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W‐M, Han G‐S, Carman GM (2014) Yeast Nem1‐Spo7 protein phosphatase activity on Pah1 phosphatidate phosphatase is specific for the Pho85‐Pho80 protein kinase phosphorylation sites. J Biol Chem 289: 34699–34708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Sreekumaran Nair Ket al (2016) Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res 44: D463–D470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe LM, Liang CC, Dauer WT (2016) Neuronal nuclear membrane budding occurs during a developmental window modulated by torsin paralogs. Cell Rep 16: 3322–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugrankar R, Liu Y, Provaznik J, Schmitt S, Lehmann M (2011) Lipin is a central regulator of adipose tissue development and function in Drosophila melanogaster . Mol Cell Biol 31: 1646–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Heyden AB, Naismith TV, Snapp EL, Hodzic D, Hanson PI (2009) LULL1 retargets TorsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol Biol Cell 20: 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGompel MJW, Nguyen KCQ, Hall DH, Dauer WT, Rose LS (2015) A novel function for the Caenorhabditis elegans torsin OOC‐5 in nucleoporin localization and nuclear import. Mol Biol Cell 26: 1752–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer B, Schooley A, Sachdev R, Eisenhardt N, Schneider AM, Sieverding C, Madlung J, Gerken U, Macek B, Antonin W (2012) Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J 31: 4072–4084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi‐Ito N, Doherty OM, Moriyama H, Breakefield XO, Gusella JF, O’Donnell JM, Ito N (2011) Dtorsin, the Drosophila ortholog of the early‐onset dystonia TOR1A (DYT1), plays a novel role in dopamine metabolism. PLoS One 6: e26183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wang X, Wang J, Wang X, Chen W, Lu N, Siniossoglou S, Yao Z, Liu K (2019) Rewiring neuronal glycerolipid metabolism determines the extent of axon regeneration. Neuron 105: 276–292.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Brown RS, Chase AR, Eisele MR, Schlieker C (2013) Regulation of torsin ATPases by LAP1 and LULL1. Proc Natl Acad Sci USA 110: E1545–E1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Yang X, Xi Y (2016) Fat body remodeling and homeostasis control in Drosophila . Life Sci 167: 22–31 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Table EV1
Source Data for Appendix
Source Data for Figure 2
Data Availability Statement
Detailed information is provided in tables for Drosophila melanogaster lines (Appendix Table S1), plasmids (Appendix Table S2), antibodies (Appendix Table S3), and primers (Appendix Table S4) used in this study. Individual lipid species data (as a fraction of total membrane lipids) are provided in Table EV1. The lipidomic data generated by Lipotype is available at the NIH Common Fund's National Metabolomics Data Repository (NMDR) Web site, the Metabolomics Workbench, https://www.metabolomicsworkbench.org where it has been assigned Project ID PR001172. The data can be accessed directly via its Project DOI: http://dx.doi.org/10.21228/M86T2S (Sud et al, 2016).