

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal non‐cell‐autonomous neurodegenerative disease characterized by the loss of motor neurons (MNs). Mutations in CRMP4 are associated with ALS in patients, and elevated levels of CRMP4 are suggested to affect MN health in the SOD1G93A‐ALS mouse model. However, the mechanism by which CRMP4 mediates toxicity in ALS MNs is poorly understood. Here, by using tissue from human patients with sporadic ALS, MNs derived from C9orf72‐mutant patients, and the SOD1G93A‐ALS mouse model, we demonstrate that subcellular changes in CRMP4 levels promote MN loss in ALS. First, we show that while expression of CRMP4 protein is increased in cell bodies of ALS‐affected MN, CRMP4 levels are decreased in the distal axons. Cellular mislocalization of CRMP4 is caused by increased interaction with the retrograde motor protein, dynein, which mediates CRMP4 transport from distal axons to the soma and thereby promotes MN loss. Blocking the CRMP4‐dynein interaction reduces MN loss in human‐derived MNs (C9orf72) and in ALS model mice. Thus, we demonstrate a novel CRMP4‐dependent retrograde death signal that underlies MN loss in ALS.
Keywords: ALS, axonal transport, CRMP4, dynein, retrograde signaling
Subject Categories: Molecular Biology of Disease, Neuroscience
Dynein‐mediated CRMP4 redistribution from axons into the cell bodies of ALS‐affected motor neurons promotes selective neuronal toxicity in diverse ALS model‐ and patient‐derived cells.
Introduction
Amyotrophic lateral sclerosis (ALS) is a lethal neurodegenerative disease that is characterized by degeneration of upper and lower motor neurons (MN). This process leads to spasticity, muscle atrophy, and paralysis, which develop into respiratory failure and patient death (Frey et al, 2000; Fischer et al, 2004; Boillée et al, 2006; Moloney et al, 2014; Peters et al, 2015). The most common mutations responsible for familial ALS (fALS) include expansions of a repeated DNA element (GGGGCC) in the C9orf72 gene and point mutations in the superoxide dismutase 1 (SOD1) gene (Rosen et al, 1993; DeJesus‐Hernandez et al, 2011; Renton et al, 2011).
A hallmark finding in ALS patients, as well as in ALS mouse models, is alterations in axonal transport (Bilsland et al, 2010; Perlson et al, 2010; Gershoni‐Emek et al, 2015; De Vos & Hafezparast, 2017). In order to survive and function, MNs depend upon the propagation of signaling events along the axons between the synapse and soma (cell body) (Harrington & Ginty, 2013; Millecamps & Julien, 2013; Terenzio et al, 2017; Zahavi et al, 2017). Retrograde and anterograde axonal transport are mediated by the dynein/dynactin and kinesin motor protein families, respectively (Paschal & Vallee, 1987; Howard et al, 1989; Guedes‐Dias & Holzbaur, 2019). Notably, mutations in kinesin and dynein/dynactin are also associated with ALS in humans (LaMonte et al, 2002; Münch et al, 2004; Steinberg et al, 2015; Nicolas et al, 2018). Several studies have suggested that alterations in cross‐talk and long‐distance signaling pathways between neurons and their diverse extracellular cues, which are mediated by axonal transport, contribute to ALS pathology (Boillée et al, 2006; Perlson et al, 2009; Gibbs et al, 2018).
Collapsin response mediator proteins (CRMPs) constitute a family of developmentally regulated phosphoproteins known for their intracellular mediation of class 3 semaphorin signaling (Goshima et al, 1995; Ziak et al, 2020). There are 5 known CRMPs in vertebrates, all of which share high sequence similarities (Schmidt & Strittmatter, 2007). The semaphorin/CRMP signaling pathway involves phosphorylation of CRMPs via various kinases including Rho‐kinase, CDK5, or GSK3β, which leads to microtubule destabilization and axon retraction (Sasaki et al, 2002; Yamashita & Goshima, 2012; Balastik et al, 2015). In addition to their role in mediating semaphorin intrinsic cell responses, CRMPs have been reported to bind dynein and kinesin and modulate their function (Arimura et al, 2009; Rahajeng et al, 2010). Several studies have demonstrated the involvement of CRMPs in neurodegenerative diseases and neuronal injury (Charrier et al, 2003; Jang et al, 2010; Yamashita & Goshima, 2012; Nagai et al, 2017). Specifically, CRMP4 expression levels were found to be elevated in the SOD1G93A mouse spinal cord and were suggested to promote MN death (Duplan et al, 2010; Valdez et al, 2012; Nagai et al, 2015). Interestingly, mutations in CRMP4 are associated with ALS (Blasco et al, 2013). However, the mechanism of CRMP4‐mediated MN cell death in ALS is unknown.
Here, we identify a novel mechanism by which CRMP4 mediates MN toxicity in ALS. We discover a CRMP4‐dependent retrograde signal in ALS that facilitates MN loss. This process is mediated by alterations of CRMP4 expression and the formation of a CRMP4‐dynein complex via a specific motif in the CRMP4 protein in a subtype of ALS‐diseased MN axons.
Results
Alterations in CRMP4 protein levels along ALS‐diseased motor unit
Increased CRMP4 levels in MN cell bodies of the SOD1G93A ALS mouse model were previously reported to be toxic to MNs and lead to cell death (Duplan et al, 2010). Specific mutations in the N‐terminus of CRMP4 are associated with ALS in patients (Duplan et al, 2010; Valdez et al, 2012; Nagai et al, 2015). However, how CRMP4 mediates its toxicity in ALS MNs remains unknown. To address this, we first measured CRMP4 protein levels in the spinal cord of human ALS patients (Fig 1A and B). Consistent with the published results from the SOD1G93A ALS mice model (Duplan et al, 2010), we observed a significant 2.5‐fold increase in the relative CRMP4 expression in sALS patient compared to non‐diseased controls (mean: healthy 1.285 ± 0.184; sALS patient 2.571 ± 0.137) (Fig 1A and B). This increase was also prevalent in SOD1G93A mice spinal cord, as the total number of cells expressing CRMP4 in the SOD1G93A postnatal day 90 (P90) spinal cord compared to their littermate control was elevated (Fig 1C and D) (mean: WT 4.54 ± 4.4%; SOD1G93A 26.43 ± 3.04%). Next, we determined the expression levels of CRMP4 in the distal parts of the motor neuron: (i) intra‐muscular axons and (ii) neuromuscular junctions (NMJs). Unexpectedly, our analysis in human intra‐muscular nerves from sALS patients, revealed a significant 28% decrease in CRMP4 levels within neurofilament heavy chain (NFH)‐positive axons compared with healthy controls and a similar non‐significant trend in MBP‐positive Schwann cells (Fig 2 A,B) (Appendix Fig S1A and B) (mean intensity: non‐ALS 1.00 ± 0.097; ALS 0.719 ± 0.108). Furthermore, analysis of P90 gastrocnemius muscles (GC) revealed a decrease in the number of NMJs expressing CRMP4 in SOD1G93A mice compared to the WT control (Fig 2C and D) (the mean percentage of P90 NMJs expressing CRMP4: WT 90.6595 ± 4.78%; SOD1G93A 60.29 ± 9.00%). Interestingly, we detected early partial NMJ degeneration in CRMP4‐negative NMJs, suggesting an active role for CRMP4 in preservation of distal axons (Fig 2C and E) (the mean percentage of P90 partially innervated SOD1G93A NMJs: CRMP4‐positive 33.33 ± 16.67%; CRMP4‐negative 91.67 ± 8.33%). Lastly, we monitored CRMP4 expression levels in WT and SOD1G93A P90 sciatic nerves by immunostaining. We detected a 3‐fold increase in CRMP4 signal in sciatic nerve axons (NFH‐positive) and a moderate 70% increase in CRMP4 signal also in GFAP‐positive cells in SOD1G93A mice compared to the controls (Fig 2F and G; Appendix Fig S1C and D) (mean area: WT 1.00 ± 0.146; SOD1G93A 2.817 ± 0.32) (mean area: WT 0.763 ± 0.127; SOD1G93A 1.415 ± 0.25). Western blot analysis of SOD1G93A and WT P90 sciatic axoplasm confirmed this overall increase in CRMP4 levels in ALS sciatic nerves (Fig 2H and I) (mean: WT 0.56 ± 0.11; SOD1G93A 1.22 ± 0.14). Thus, our data thus far reveal that, while the expression levels of CRMP4 increase in cell bodies and proximal nerves, the opposite trend occurs in the distal/terminal parts of ALS‐diseased MNs. This suggests that in ALS, distal‐CRMP4 is mislocalized into proximal axons and cell bodies.
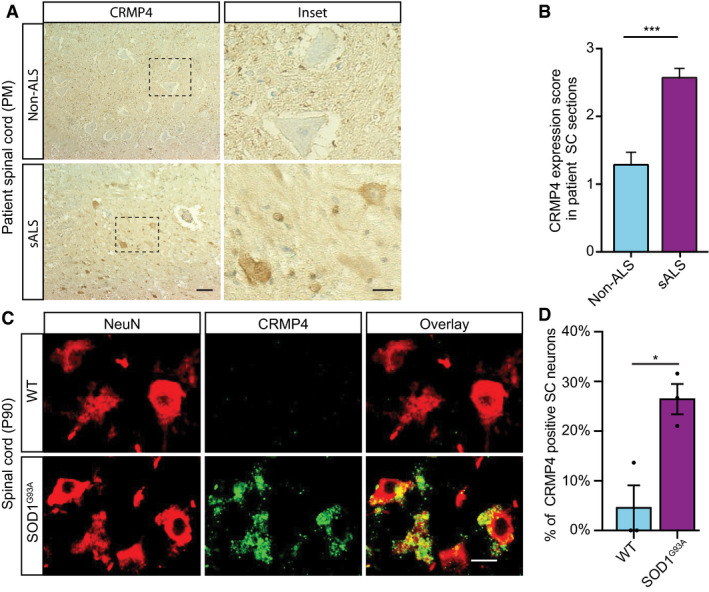
Figure 1. CRMP4 is elevated in ALS‐diseased spinal cord neurons.
-
A, B(A) Representative IHC images and (B) semi quantification of CRMP4 protein in human spinal cords (SC) cross sections from 2 controls and 3 ALS patients. We analyzed total of 7 SC sections of controls and 14 SC sections of ALS patients, data presented as mean ± SE. DAB: labeled CRMP4. Scale bar: left images 20 µm, right insets 10 µm. Mann‐Whitney test ***P = 0.0003.
-
CRepresentative images of P90 SC cross sections of SOD1G93A and WT mice. Red: denotes NeuN, Green: denotes CRMP4. Scale bar: 10 μm.
-
DQuantification of the percentage of CRMP4‐positive SC neurons in 3 WT VS. 3 SOD1G93A mice. We monitored CRMP4 expression in total of 108 cells in WT condition and 123 cells in SOD1G93A, an average of 36 or 41 cells in each repeat respectively. Student's t‐test, n = 3, data presented as mean ± SE, *P = 0.0161.
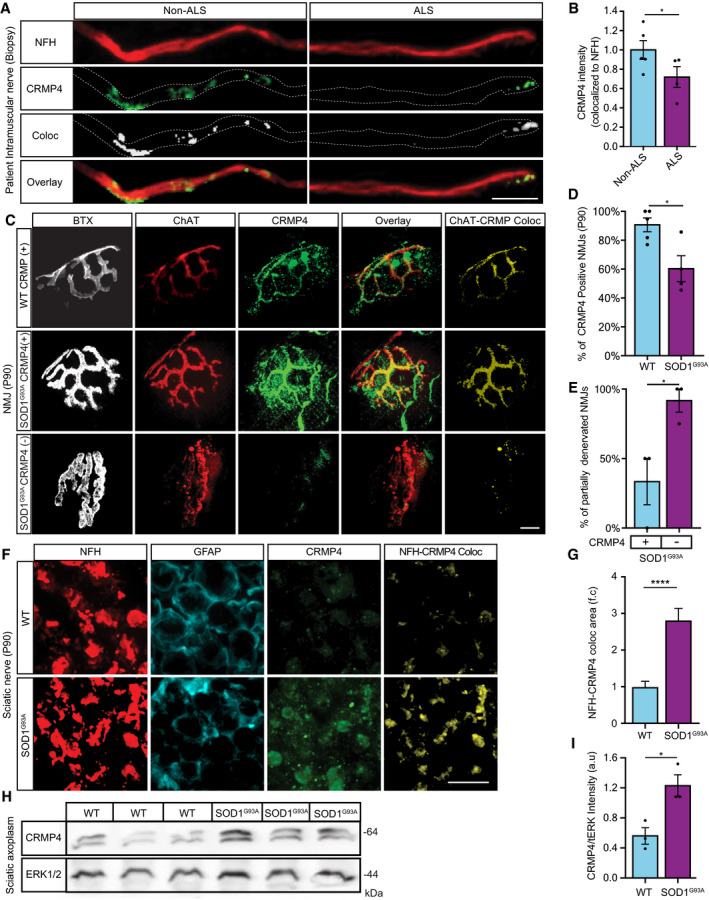
Figure 2. CRMP4 is mislocalized in ALS motor units.
-
ARepresentative images of ALS patient or non‐ALS human control intra‐muscular nerves. Red: denotes NFH, Green: denotes CRMP4, White: denotes co‐localization area using Imaris software. Scale bar: 20 μm.
-
BQuantification of CRMP4 intensity levels in NFH‐positive intra‐muscular distal nerves from 5 non‐ALS controls and 4 sALS patients. We analyzed 40 terminal axons from the healthy samples (~ 8 axons per sample) and 36 terminal axons from sALS samples (~ 8 axons per sample). Data presented as mean ± SE. Student's t‐test, *P = 0.0475.
-
CRepresentative images of SOD1G93A/ChAT::tdTomato or WTChAT::tdTomato neuromuscular junctions (NMJ's) at P90. White: denotes BTX, Red: denotes direct ChAT, Green: denotes CRMP4, Yellow: denotes Z projection of 3D Imaris co‐localization of CRMP4 and ChAT. Scale bar: 10 µm.
-
DQuantification of CRMP4‐positive NMJ's in gastrocnemius muscles from 3 WT or 3 SOD1G93A P90 mice. Total of 44 NMJ’s in WT condition and 60 NMJ’s in SOD1G93A condition. Student's t‐test, n = 3, data presented as mean ± SE, *P = 0.0157.
-
EQuantification of the percent of partially denervated NMJ’s in the presence or absence of CRMP4 immunostaining in 3 different SOD1G93A mice. We counted 24 NMJ’s in SOD1G93A CRMP4 negatives and 67 NMJ’s in SOD1G93A CRMP4 positives. Student's t‐test, n = 3, data presented as mean ± SE, *P = 0.0352.
-
FRepresentative images of P90 SOD1G93A and WT sciatic nerves. Red: denotes NFH, Cyan: denotes GFAP and green denotes CRMP4, Yellow: denotes the Z projection of 3D Imaris co‐localization of CRMP4 and NFH. Scale bar: 5 μm.
-
GQuantification of the co‐localization area of CRMP4 with NFH in the sciatic nerve in 3 SOD1G93A mice compared to 3 WT mice using Imaris analysis. 14 WT sciatic nerve sections and 11 SOD1G93A sections were monitored. Data presented as mean ± SE. Student's t‐test, n = 3, ****P < 0.0001.
-
H, IWestern blot analysis and quantification of 3 independent repeats of P90 SN tissues for CRMP4 expression levels (size of ~ 64 kDa) in SOD1G93A compared to WT. tERK was used as a loading control (size of ~ 44 kDa). Student's t‐test, n = 3, data presented as mean ± SE, *P = 0.0215.
Source data are available online for this figure.
CRMP4 protein mislocalization via dynein‐dependent activity
To investigate the potentially disease‐relevant function of this intracellular mislocalization of CRMP4, we utilized human iPSC‐derived MNs (iPS‐MN) from healthy controls and C9orf72‐ALS patients (Tank et al, 2018) and plated them in microfluidic chambers (MFCs). First, we immunostained for the neuronal/axonal markers NFH and Tau, along with the MN‐specific marker HB9, to validate the MN cell identity (Appendix Fig S2A–F). Then, we performed immunostaining for CRMP4 to assess changes in its levels in distal axons (Fig 3A–D). Our analysis revealed no difference in CRMP4 intensity levels between healthy and C9orf72 iPS‐MN axons in this system (Fig 3A and B). In our previous study, we demonstrated that muscle‐secreted Sema3A leads to NMJ's disruption and axonal degeneration in ALS (Maimon et al, 2018). The canonical pathway for CRMP activation involves Sema3A‐NRP‐PlexinA interactions. Therefore, we considered whether CRMP4 requires stress activation for differential, disease‐relevant mislocalization in MNs cultures and asked whether Sema3A treatment might differentially affect CRMP4 levels in ALS iPS‐MNs versus Healthy iPS‐MNs. We found that exposing distal axons to Sema3A for 8 h led to significant increase in CRMP4 intensity specifically in axons of C9orf72 MNs (mean: healthy 0.7 ± 0.05; Healthy + Sema3A 0.75 ± 0.047; C9orf72 0.61 ± 0.03; C9orf72 + Sema3A 1.27 ± 0.16) (Fig 3A and B). Given that CRMP4 levels were specifically high in somata and proximal axons of ALS patients and SOD1G93A mice, we tested the CRMP4 levels in somata and proximal axons of C9orf72 and healthy iPSC‐MNs following distal Sema3A treatment. We used cholera toxin B‐647 (CTX) retrograde tracing to specifically examine CRMP4 levels in proximal axons and somata parts of neurons that send their axons into distal compartment of MFC (Fig 3C). Looking specifically at this neuronal population, we detected an increase in CRMP4 intensity post Sema3A treatment in C9orf72 iPS‐MN soma and proximal axons but not in healthy iPS‐MN controls (Fig 3D–G) (CRMP4 mean intensity in somata area, measured by GAPDH outline: healthy 1.00 ± 0.128; Healthy + Sema3A 1.222 ± 0.148; C9orf72 1.00 ± 0.078; C9orf72 + Sema3A 1.258 ± 0.068) (CRMP4 mean intensity proximal axons: Healthy 1.00 ± 0.066; Healthy + Sema3A 1.048 ± 0.059; C9orf72 1.00 ± 0.093; C9orf72 + Sema3A 1.302 ± 0.11).
Figure 3. CRMP4 protein levels are altered via a dynein‐dependent activity.
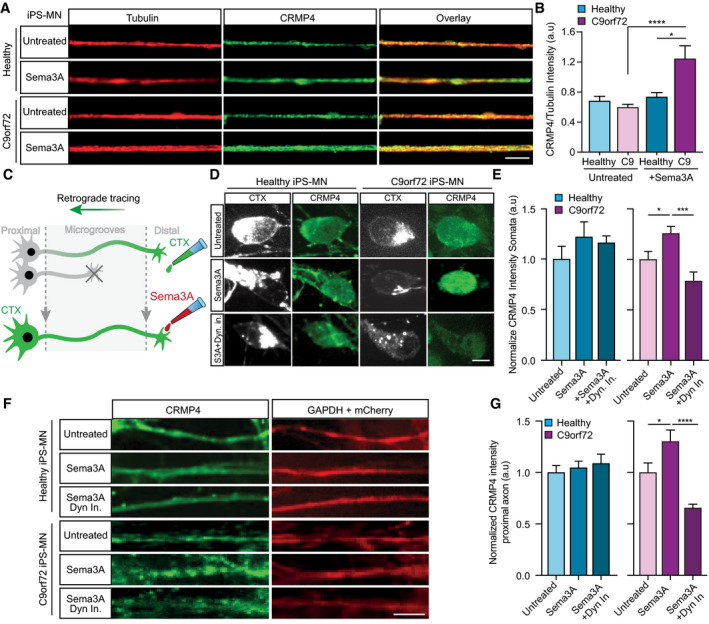
- Representative images of healthy or C9orf72 iPSC‐derived MNs treated with Sema3A or untreated in the distal compartment, 8 h post‐treatment. Red: denotes Tubulin, Green: denotes CRMP4. Scale bar: 5 μm.
- Quantification of CRMP4 intensity levels in healthy or C9orf72 iPSC‐derived MNs with Sema3A treatment or untreated. 14 untreated healthy axons, 12 healthy axons with Sema3A treatment, 49 untreated C9orf72 axons, and 31 C9orf72 axons with Sema3A treatment were monitored from 3 different chambers. One‐way ANOVA, Tukey's multiple comparisons test, n = 3, data presented as mean ± SE, *P = 0.0338; ****P < 0.0001.
- Illustration of the experimental procedure for MNs in an MFC treated with the fluorescently tagged retrograde tracer CTX in the distal compartment. Neuronal cell bodies in the primary neuron whose axons have traversed into the distal compartment were also labeled by the retrograde tracer.
- Representative images of healthy or C9orf72 human‐derived MN cell somata with Sema3A treatment, Sema3A + dynein inhibitor treatment, or untreated. Gray: denotes CTX, Green: denotes CRMP4. Scale bar: 5 μm.
- Quantification of CRMP4 intensity (normalized to GAPDH + mCherry/area) levels at the somata of healthy or C9orf72 human‐derived MN after Sema3A treatment, Sema3A + dynein inhibitor treatment, or untreated. Analysis performed in 3 independent chambers per condition. 19 healthy untreated cell somata, 26 healthy cell somata with Sema3A treatment, 20 healthy cell somata with Sema3A + dynein inhibitor treatment and 14 C9orf72 cell somata from each condition were monitored. One‐way ANOVA, Newman–Keuls multiple comparisons test, n = 3, data presented as mean ± SE, *P = 0.0207; ***P = 0.0004.
- Representative images of healthy or C9orf72 human‐derived MN proximal axons with Sema3A treatment, Sema3A + dynein inhibitor treatment, or untreated control. Green: denotes CRMP4, Red: denotes GAPDH. Scale bar: 5 μm.
- Quantification of CRMP4 intensity levels (normalized to GAPDH + mCherry/area) at the proximal axons in healthy or C9orf72 human‐derived MN after Sema3A treatment, Sema3A + dynein inhibitor treatment, or untreated control. Analysis performed from 3 independent chambers in each condition. 21 healthy untreated proximal axons, 24 healthy proximal axons with Sema3A treatment, 16 healthy proximal axons with Sema3A + dynein inhibitor treatment, 12 C9orf72 untreated proximal axons, 8 C9orf72 proximal axons with Sema3A treatment and 13 C9orf72 proximal axons with Sema3A + dynein inhibitor treatment were monitored. One‐way ANOVA, Newman‐Keuls multiple comparisons test, n = 3, data presented as mean ± SE, *P = 0.0334, ****P < 0.0001.
CRMPs have been reported to bind to the dynein motor protein and modulate its function (Arimura et al, 2009; Rahajeng et al, 2010). Since distal exposure to Sema3A led to CRMP4 elevation in the ALS‐diseased MN soma, we speculated that CRMP4 undergoes retrograde transport mediated by dynein. Therefore, we examined CRMP4 levels in the cell bodies and proximal axons of healthy and C9orf72 iPS‐MNs that were distally exposed to Sema3A with or without the presence of the dynein inhibitor, Ciliobrevin D (herein Dyn‐In) (Firestone et al, 2012) (Fig 3D–G). Inhibiting dynein completely blocked Sema3A‐induced increase in CRMP4 levels in C9orf72 iPS‐MNs axons and somata (Fig 3D–G; Appendix Fig S3A and B) (CRMP4 mean intensity somata—measured by GAPDH outline: Healthy + Sema3A 1.222 ± 0.148; Healthy + Sema3A + Dyn in 1.161 ± 0.067; C9orf72 + Sema3A 1.258 ± 0.068; C9orf72 + Sema3A + Dyn in 0.787 ± 0.088) (CRMP4 mean intensity proximal axons: Healthy + Sema3A 1.048 ± 0.059; Healthy + Sema3A + Dyn in 1.087 ± 0.089; C9orf72 + Sema3A 1.302 ± 0.11; C9orf72 + Sema3A + Dyn in 0.655 ± 0.036). We did not observe this effect in the presence of Dyn‐In alone (Appendix Fig S3A and B). Furthermore, to validate that distal Sema3A treatment has effect on MN soma, we quantified the cell body area (by measuring CTX signal outline) and found that C9orf72 Sema3A‐treated MNs exhibit smaller soma areas compared to untreated or healthy MNs cultures (Appendix Fig S3C). Here again, when dynein activity was inhibited prior to Sema3A application, this effect abolished (mean area: Healthy untreated 302 ± 29; Healthy + Sema3A 285 ± 15; Healthy + Sema3A + Dyn‐In 296 ± 18; C9orf72 untreated 314 ± 10; C9orf72 + Sema3A 263 ± 10; C9orf72 + Sema3A + Dyn‐In 318 ± 12) (Appendix Fig S3C). Importantly, no differences were monitored post‐application of Dyn‐In alone in both healthy and disease conditions. These data indicate that the elevation in CRMP4 in cell bodies and in proximal axons of ALS‐diseased neurons is mediated by dynein, likely by binding to axonal CRMP4 and subsequent retrograde transport to the cell body.
A specific CRMP4 motif mediates the CRMP4‐dynein‐dynactin interaction
CRMP2 members were previously found to bind the dynein motor protein (Arimura et al, 2009). Arimura et al (2009) also characterized two specific domains in the CRMP2 protein that are responsible for dynein binding (Arimura et al, 2009). Since CRMP2 and CRMP4 share substantial sequence similarity, we hypothesized that the dynein‐binding domains (100aa–150aa) of CRMP2 would play a similar role in CRMP4. Following this, and on the basis of the CRMP4 protein 3D structure (PDB code 4CNT) (Ponnusamy et al, 2014) (Fig 4A), we overexpressed full‐length GFP‐CRMP4 or CRMP4 lacking amino acids 100–150 (GFP‐CRMP4Δ100–150) in COS7 cells and immunoprecipitated the endogenous dynein intermediate chain (DIC). Western blot analysis of these fractions revealed a clear interaction of DIC with full‐length GFP‐CRMP4 but not with GFP‐CRMP4Δ100–150 (Fig 4B) (mean: GFP‐CRMP4 1.473 ± 0.373; GFP‐CRMP4Δ100–150 0.009 ± 0.001). Since a large deletion in the CRMP4 protein sequence may result in its misfolding and dysfunction, we pursued an alternative strategy by generating small peptides to cover the potential dynein‐binding motif and test whether this could block dynein binding to CRMP4. We designed four short peptides within the 50 amino acid domain, which exhibit the potential to block CRMP4‐dynein interaction, based on the protein structure. Importantly, the peptide sequences were designed to avoid an overlap with CRMP4 homo‐tetramer interfaces, likely preventing a disturbance to the protein’s homomeric assembly (Fig 4A). To test the peptide activity, we pre‐incubated a mixture of peptides 1–4 with lysate from GFP‐CRMP4 overexpressing COS7 cells. This process significantly reduced the CRMP4‐DIC interaction (Fig 4B and C; mean: GFP‐CRMP4 1.473 ± 0.373; GFP‐CRMP4 + peptides 0.3 ± 0.05). We also examined CRMP4 interaction with dynactin (p150), a dynein activator, in COS7 cells overexpressing Flag‐tagged CRMP4. After an overnight incubation of cell lysate with peptides 1–4, we pulled down Flag‐CRMP4 and blotted for dynactin (p150). Application of the peptide mixture resulted in a dramatic decrease in CRMP4 binding to dynactin (Fig 4D and E) (mean: control 0.77 ± 0.1; All peptides 0.17 ± 0.06). We also determined whether introducing the individual peptides might be sufficient to also block the CRMP4‐dynactin interaction. Using the same assay, we incubated each peptide separately and found that only peptide‐4 had a mild but significant ability to block CRMP4 interaction with dynactin (Fig 4F and G) (mean: control 1.92 ± 0.2; peptide‐1 1.44 ± 0.3; peptide‐2 1.34 ± 0.3; peptide‐3 1.8 ± 0.8; peptide‐4 1.13 ± 0.14). Hence, although peptide‐4 was sufficient to block CRMP4‐dynactin binding, blocking CRMP4‐dynactin complex formation requires a combination of all four peptides. Lastly, we generated a genetic tool to block the CRMP4‐dynein interaction using a plasmid with CRMP4‐dynein‐binding motif sequence (corresponding to a.a 100–150 of CRMP4) and determined whether it could act in a dominant‐negative manner. We transfected COS7 cells with GFP or GFP‐expressing 50aa sequence (GFP‐50aa) and then extracted the cells and assayed for CRMP4 that co‐purified with the dynein intermediate chain (DIC) via immunoprecipitation. Our Western blot analysis revealed weaker interaction of CRMP4 with dynein in the presence of GFP‐50aa overexpressing cells compare to the control (mean: GFP‐CRMP4 1.00 ± 0.10; GFP‐50aa 0.54 ± 0.11) (Fig 4H and I). Thus, amino acids 100–150 in the CRMP4 protein are essential for CRMP4/dynein/dynactin binding.
Figure 4. CRMP4 binds dynein via a specific 50 amino acid motif.
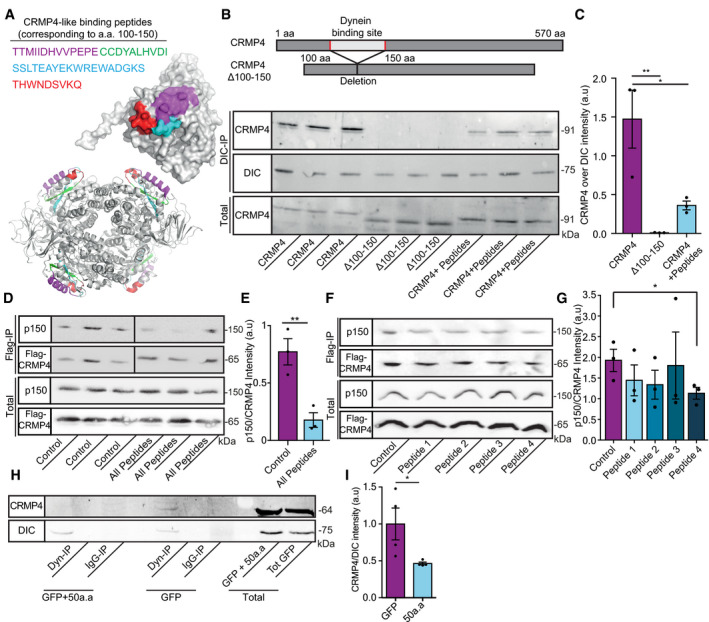
- Crystal structures (PDB code 4CNT) of a CRMP4 monomer (upper panel) and biological tetramer assembly (lower panel). The peptides that were selected to inhibit binding are highlighted and color coded as indicated.
- Upper panel represents the binding site domain of dynein in CRMP4 and its deletion. Middle panel—Immunoprecipitation of DIC followed by western blot analysis of CRMP4 in COS7 cells overexpressing either GFP‐CRMP4, or GFP‐CRMP4 with deletion of amino acid 100–150, or GFP‐CRMP4 overexpressing cells that were pre‐incubated with a 10 µm mixture of peptides 1–4 (size of ~ 91 kDa). Lower panel ‐ Western blot analysis of total protein levels before the pull‐down assay (DIC size: ~ 75 kDa).
- Quantification of the Western blot in B from 3 independent repeats. One‐way ANOVA, Tukey's multiple comparisons test, n = 3, data presented as mean ± SE, **P = 0.007, *P = 0.0261.
- Upper panel ‐ immunoprecipitation assay with anti‐Flag antibody followed by Western blot analysis of dynactin (p150) (size of ~ 150 kDa) in COS7 cells overexpressing Flag‐CRMP4 (size of ~ 65 kDa). Lower panel ‐ total protein input.
- Quantification of the blot in D from 3 independent repeats. The dynactin intensity band was normalized to the Flag‐CRMP4 intensity band in each repeat. Student’s t‐test, n = 3, data presented as mean ± SE, **P = 0.01.
- Upper panel ‐ Immunoprecipitation assay with anti‐Flag antibody followed by Western blot analysis of dynactin (p150) (size of ~ 150 kDa) in COS7 cells overexpressing Flag‐CRMP4 (size of ~ 65 kDa). Lower panel ‐ Total input.
- Quantification of the blot in F. The dynactin intensity band was normalized to the Flag‐CRMP4 intensity band in each repeat. Student’s t‐test, n = 3, data presented as mean ± SE, *P = 0.0299.
- Immunoprecipitation of DIC (size of ~ 75 kDa) followed by Western blot analysis of CRMP4 (size of ~ 64 kDa) in COS7 cells that were transfected with CRMP4 and AAV9‐50aa or its control. IgG antibody was used as a control.
- Quantification of the blot in H from 3 independent repeats. The CRMP4 intensity band was normalized to the DIC intensity band in each repeat. Student’s t‐test, n = 3, data presented as mean ± SE, *P = 0.0479.
Source data are available online for this figure.
Enhanced CRMP4‐dynein complex formation in ALS‐diseased MNs
Our data thus far suggest that CRMP4 levels are (i) increased in the cell soma and (ii) decreased in the NMJs and distal axons of ALS‐diseased MNs (Figs 1 and 2). We further demonstrate that CRMP4 mislocalization in ALS‐diseased MNs can be facilitated by Sema3A and is mediated by dynein (Figs 3 and 4). Since our previous report suggests elevations in Sema3A secretion from ALS muscles (Maimon et al, 2018), we predicted an increase in CRMP4‐dynein complex formation along the axons of ALS models. To test this, we first extracted sciatic nerves axoplasm from WT and SOD1G93A P90 mice and measured the levels of CRMP4 that co‐purified with the dynein intermediate chain (DIC) in vivo using immunoprecipitation. We found a stronger interaction of DIC with CRMP4 in the SOD1G93A mice compared with the control (Fig 5A and B) (mean: WT 0.28 ± 0.11; SOD1G93A 1.48 ± 0.62). Notably, transfecting cells with CRMP4 that carries an ALS‐associated mutation, I141V, also enhanced the formation of the CRMP4‐dynein complex (Fig 5C and D) (mean: WT CRMP4 1.00 ± 0.207; mutated CRMP4 1.625 ± 0.116). Importantly, I141V is located in the same dynein‐binding motif in the CRMP4 protein, suggesting a CRMP4 gain of toxic function in ALS.
Figure 5. CRMP4‐dynein interaction is enhanced in ALS motor neuron axons.
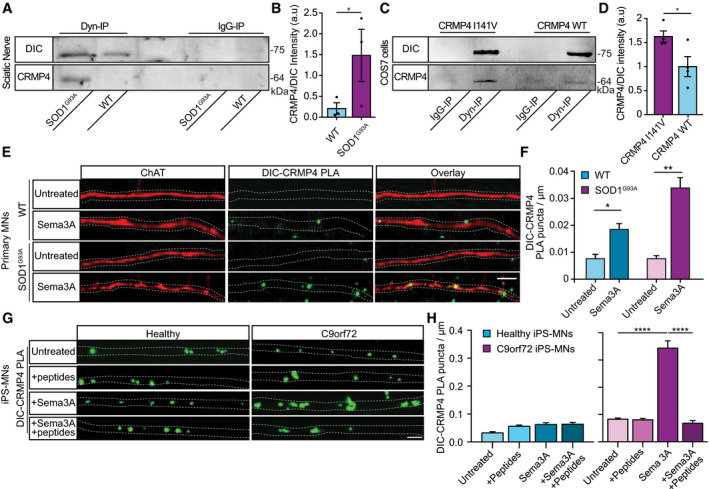
- Immunoprecipitation of DIC followed by Western blot analysis of CRMP4 in SOD1G93A compared to WT P90 sciatic nerves under physiological conditions. IgG antibody was used as a control.
- Quantification of the blot in A. 3 repeats, 12 sciatic nerves per condition were used in each repeat. The CRMP4 intensity band was normalized to the DIC intensity band in each repeat. Data presented as mean ± SE (Ratio Paired t‐test, *P = 0.0416).
- Immunoprecipitation of DIC followed by Western blot analysis of CRMP4 in COS7 cells that were transfected with mutant CRMP4 I141V compared to control. IgG antibody was used as a control.
- Quantification of four repeated pull down in C. The CRMP4 intensity band was normalized to the DIC intensity band for each repeat. (Student’s t‐test, n = 4, data presented as mean ± SE, *P = 0.0393).
- Representative images from the proximity ligation assay (For explanation of PLA technique; please refer to method section) for CRMP4 and dynein in SOD1G93A and WT primary MNs axons that were exposed to either control or Sema3A 8h post‐treatment. Scale bar: 5µm.
- Quantification of the CRMP4‐DIC puncta number per primary motor neuron axon in each condition. We analyzed ~ 20 axons per condition from 3 independent chambers per group (One‐way ANOVA, Tukey's multiple comparisons test, n = 3, data presented as mean ± SE, **P = 0.01, *P = 0.04).
- Representative images of proximity ligation assay for CRMP4 and dynein in healthy and C9orf72 human‐derived proximal axons post peptides treatment, Sema3A treatment, Sema3A + peptides treatment, or untreated controls. Scale bar: 5 µm.
- Quantification of the CRMP4‐DIC puncta number per axon in healthy or C9orf72 human‐derived MN proximal axons after Sema3A treatment, Sema3A + peptides treatment, or untreated controls. Data collected from 3 independent chambers in each condition. Total of 37 healthy untreated proximal axons, 61 healthy proximal axons with peptides treatment, 59 healthy proximal axons with Sema3A treatment and 52 healthy proximal axons with Sema3A + peptides treatment. 67 C9orf72 untreated proximal axons, 63 C9orf72 proximal axons with peptides treatment, 41 C9orf72 proximal axons with Sema3A treatment, and 50 C9orf72 proximal axons with Sema3A + peptides treatment monitored. Data presented as mean ± SE. One‐way ANOVA, Tukey's multiple comparisons test, n = 3, ****P < 0.0001.
Source data are available online for this figure.
To further characterize CRMP4‐dynein interactions in ALS, we attempted to track CRMP4‐GFP retrograde movement in healthy and ALS‐diseased MN axons. However, overexpressing CRMP4‐GFP resulted in a uniformly diffuse distribution (Appendix Fig S4). Thus, in order to demonstrate retrograde transport of endogenous CRMP4, we immunostained distinct cellular compartments (dynein and CRMP4) using proximity ligation assay (PLA), as previously performed (Olenick et al, 2019). First, by using PLA, we observed that indeed there is an increase in the CRMP4‐dynein co‐localization along cultured WT MNs axons, post‐Sema3A treatment compared to untreated WT cultures (Fig 5E). Importantly, this co‐localization was twice as high in the Sema3A‐treated SOD1G93A MNs axons (Fig 5E and F). Furthermore, when comparing co‐localization patterns of the CRMP4‐dynein puncta in human iPS‐MNs in the presence or absence of Sema3A, we obtained similar results: In naive, untreated axons, the number of CRMP4‐dynein puncta was similar between healthy and C9orf72 iPS‐MNs. However, following Sema3A treatment, the number of CRMP4‐dynein puncta in C9orf72 axons was significantly higher compared to treated healthy control (Fig 5G and H) (mean puncta per axon: healthy untreated 0.032 ± 0.004; Healthy + Sema3A 0.062 ± 0.006; C9orf72 untreated 0.082 ± 0.004; C9orf72 + Sema3A 0.344 ± 0.026). Next, in order to determine whether the formation of CRMP4‐dynein complexes in MN axons is reversible, we aimed to block the CRMP4‐dynein interaction in iPS‐MN distal axons. To this end, we plated healthy and C9orf72 iPS‐MNs in MFCs and exclusively introduced a mix of all peptides (1–4) into axons in the distal compartment. Using TAMRA peptides as a positive control for uptake, we observed that peptides 1–4 peptides were successfully taken up by distal axons (Appendix Fig S5A and B). Strikingly, application of peptides 1–4 significantly interfered with the interaction of CRMP4 and dynein in distal C9orf72 iPS‐MN axons as determined by PLA. CRMP4‐dynein interaction in healthy iPS‐MN remained unaffected by either Sema3A, peptides 1–4, or by both (Fig 5G and H) (mean puncta per axon: Healthy + Sema3A 0.062 ± 0.006; Healthy + Sema3A + peptide 0.063 ± 0.006; C9orf72 + Sema3A 0.344 ± 0.026; C9orf72 + Sema3A + peptide 0.067 ± 0.009). No differences were monitored when peptides were inserted without Sema3A activation in both conditions (Fig 5G and H) (mean puncta per axon: healthy untreated 0.032 ± 0.004; Healthy + peptides 0.056 ± 0.004; C9orf72 untreated 0.082 ± 0.004; C9orf72 + peptides 0.080 ± 0.005). Together, we have demonstrated both in vivo and in vitro that the CRMP4‐dynein interaction is elevated in ALS‐mutated MN axons. Importantly, this strong interaction can be blocked in ALS MN axons by interfering with the CRMP4‐dynein‐binding domain using both genetic and pharmacological tools.
The CRMP4‐dynein complex facilitates selective neuronal loss in ALS
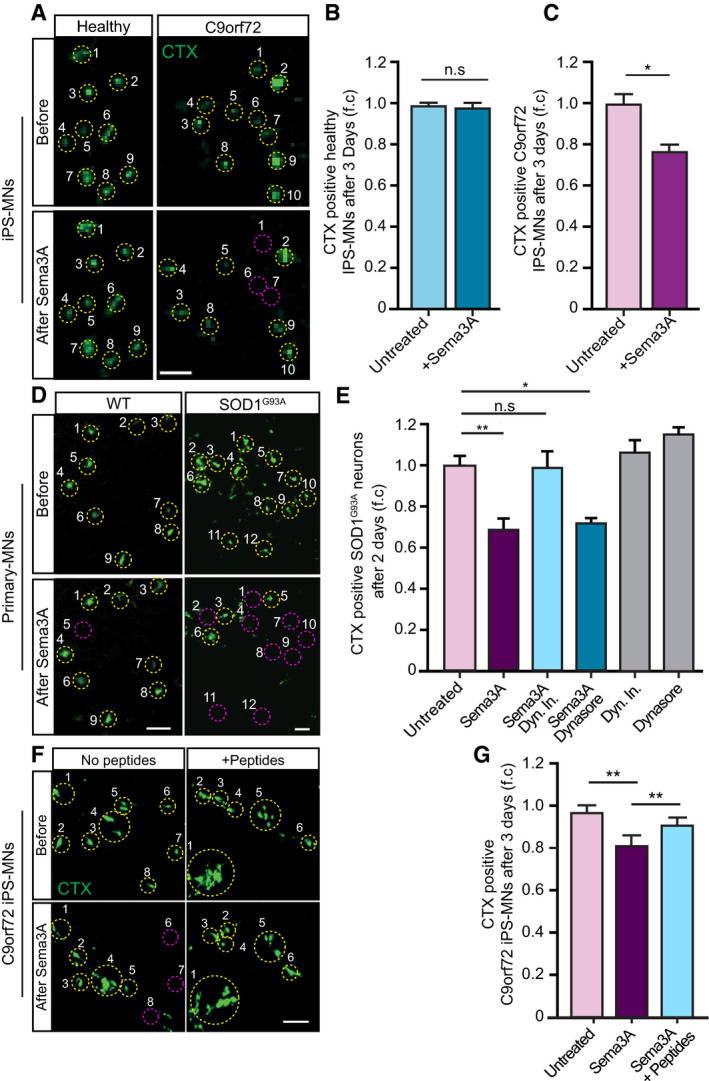
MNs undergo apoptosis and degenerate in ALS (Reyes et al, 2010). Downregulation of CRMP4 was previously suggested to inhibit neurodegeneration in vitro and in vivo in ALS models (Charrier et al, 2003; Duplan et al, 2010). Thus, we examined whether enhancement of the CRMP4‐dynein interaction by Sema3A would lead to neuronal cell death in ALS. Similar to the experimental design in Fig 3C, we applied CTX to the distal compartment of the MFCs in order to label only the cell bodies of neurons whose axons traversed into the distal compartment. The number of CTX+ iPS‐MNs was quantified before and 2 days after Sema3A was applied to the distal compartment. Our analysis did not detect any significant loss of CTX+ cells in healthy iPS‐MNs in response to Sema3A, whereas in C9orf72 iPS‐MNs we detected a ~ 25 percent decrease in CTX+ MNs upon treatment with Sema3A (Fig 6A–C). Similarly, applying Sema3A to spinal cord cultures from SOD1G93A embryos resulted in a ~ 30% reduction in CTX+ MNs 3 days after treatment, compared with ~ 5% in the control and WT explants (Fig 6D and E; Appendix Fig S6) (mean fold change over control: Sema3A 0.68 ± 0.06; control 1 ± 0.04). In order to determine whether distal stress such as Sema3A application triggers MN loss in ALS‐diseased MNs via retrograde signaling, we used dynein inhibitor to block all retrograde transport events (Firestone et al, 2012). Inhibiting retrograde transport in the distal axon prevented MN loss in the SOD1G93A primary cultures (Fig 6D and E; Appendix Fig S6A–E) (mean fold change over control: Sema3A + Dyn‐In 0.92 ± 0.09; control 1 ± 0.04). Sema3A was previously shown to internalize together with its receptor, Plexin A1 (PLXNA1) in a dynamin‐dependent manner (Fournier et al, 2000; Castellani et al, 2004). To further determine whether endocytosis of Sema3A is important for the apparent retrograde death signal, as shown before in different neurons (Wehner et al, 2016), we applied Dynasore, a dynamin‐dependent endocytosis inhibitor (Macia et al, 2006), to the distal axons prior to application of Sema3A. Inhibiting Sema3A + PLAXNA1 (Appendix Fig S7) internalization did not inhibit the loss of SOD1G93A CTX+ MNs (Fig 6E; Appendix Fig S6) (mean fold change over control: Sema3A + Dynasore 0.71 ± 0.01; control 1 ± 0.04). Thus, Sema3A internalization at the distal axons is not required for the observed toxicity. Importantly, interfering with the CRMP4‐dynein interaction by introducing peptides 1–4 into diseased iPS‐MN axons prior to applying Sema3A completely abolished the CTX+ signal loss (mean: control 0.97 ± 0.009; Sema3A 0.813 ± 0.036; Sema3A + Pep 0.94 ± 0.019) (Fig 6F and G). Our findings demonstrate that death of ALS MNs following Sema3A application can be prevented by inhibiting the formation of CRMP4‐dynein complexes.
Figure 6. Retrograde CRMP4‐dynein complex formation mediates MN loss in ALS.
-
ARepresentative images of CTX signal in healthy and C9orf72 human IPSC‐derived MNs before and after Sema3A application. Green: denotes CTX‐positive cells. Yellow circles are numbered CTX‐positive cells. Purple circles are cells that are missing post Sema3A treatment. Scale bar: 40 μm.
-
B, CQuantification of CTX signal in healthy and C9orf72 IPSC‐derived MNs before and 3 days after applying Sema3A to distal compartment, compared with untreated control. 3 independent chambers in each condition were analyzed. Average of ~ 150 neurons per condition monitored. Student’s t‐test, n = 3, data presented as mean ± SE, *P < 0.05.
-
DRepresentative images of CTX signal in WT or SOD1G93A primary MNs before and 2 days after Sema3A application to the distal compartment in the presence of either Dynein inhibitor + Sema3A, Dynasore + Sema3A, or untreated. Green: denotes CTX‐positive cells. Yellow circles are numbered CTX‐positive cells. Purple circles are cells that are missing post Sema3A treatment. Scale bar: 30 μm.
-
EQuantification of CTX signal in a SOD1G93A explant before and 2 days after Sema3A application to the distal compartment in the presence of either Dynein inhibitor + Sema3A, Dynasore + Sema3A. 3 independent chambers in each condition were analyzed. ~ 200 neurons were monitored per each condition. One‐way ANOVA, Tukey's multiple comparisons test, n = 3, data presented as mean ± SE, *P < 0.05, **P < 0.01. Dynein inhibitor and Dynasore treatments were used as a negative control.
-
F, GRepresentative images and quantification of C9orf72 iPSC‐derived MNs in the proximal compartment of an MFC before and after Sema3A treatment with and without 10 μg peptides 1–4. Green: denotes CTX‐positive cells. Yellow circles are numbered CTX‐positive cells. Purple circles are cells that are missing post Sema3A treatment. 3 independent chambers in each condition were analyzed. ~ 200 neurons per condition monitored. Scale bar: 40µm. One‐way ANOVA Tukey's multiple comparisons test, n = 3, data presented as mean ± SE, **P = 0.004.
We further examined whether impairing the CRMP4‐dynein interaction in vivo reduces MN death, which is a hallmark in the SOD1G93A mouse model. To this end, we chose to prevent CRMP4‐dynein interaction using intrathecal injections of AAV9 viruses to insert CRMP4‐dominant‐negative construct (Fig 4H and I) into spinal cord MNs. First, to demonstrate injection efficacy, we immunostained spinal cord and sciatic nerve tissues that were infected with a control AAV9‐GFP, by intrathecal injections (Appendix Fig S8A) and monitored the percentage of infected neurons by analyzing co‐localization of the neuronal markers NeuN or NFH with GFP in spinal cords and axons along the sciatic nerves (Appendix Fig S8B and D). We found that 65% of spinal cord neurons as well as 50% sciatic nerve axons expressed GFP signal in AAV9‐GFP injected mice compared to non‐injected control (Appendix Fig S8C and D) (infected SC mean: non‐injected 0.25 ± 0.19%; AAV9‐GFP 70.31 ± 0.97%; infected SN mean: non‐injected 0.25 ± 0.163%; AAV9‐GFP 53.98 ± 4.233%). We then delivered AAV9‐GFP‐50aa/AAV‐GFP as a dominant‐negative approach, into pre‐symptomatic ~ P60 SOD1G93A CSF by lumbar intrathecal injection and monitored for the activation of the apoptotic marker caspase 3 (Pasinelli et al, 1998; Porter, 1999; Reyes et al, 2010) 4 weeks post‐injection (Fig 7A–E). Importantly, GFP signal was detected in similar number of spinal cord neurons and in similar intensity in both AAV treatments, meaning no difference in infection effectiveness between the GFP and GFP‐50aa constructs (Appendix Fig S8E and F) (infected cells mean: AAV9‐GFP 70.31 ± 4.024%; 50aa‐AAV9‐GFP 76.38 ± 2.965%) (GFP intensity mean: AAV9‐GFP 3,955 ± 318; 50aa‐AAV9‐GFP 3,625 ± 383). We measured the degree of activated caspase 3 in P90 SOD1G93A and compared it to the age‐matched control. As expected, ~ 90% of NeuN‐positive cells with MN morphology in the ventral horn were positive for activated caspase 3 in the spinal cord of SOD1G93A mice, while only few caspase 3‐positive neurons were detected in the control mice (Fig 7A and B) (mean: WT 12.18 ± 5.666; SOD1G93A 92.67 ± 3.167). Injection with AAV9‐GFP‐50aa resulted in a ~ 25% decrease in the percentage of activated caspase 3‐positive cells in the spinal cord of SOD1G93A mice, compared to injection with AAV9‐GFP (Fig 7C and D)(mean: AAV9‐GFP 84.44 ± 1.47%; AAV9‐GFP‐50aa 73.24 ± 0.39%). Additionally, the number of NeuN‐positive cells in the ventral horn of the spinal cord was significantly higher in the 50 aa‐injected group compared with GFP control (Fig 7E) (mean: AAV9‐GFP 1 ± 0.12 AAV9‐GFP‐50aa 1.37 ± 0.12).
Figure 7. Blocking the formation of the CRMP4‐dynein complexes reduces motor neuron toxicity in vivo .
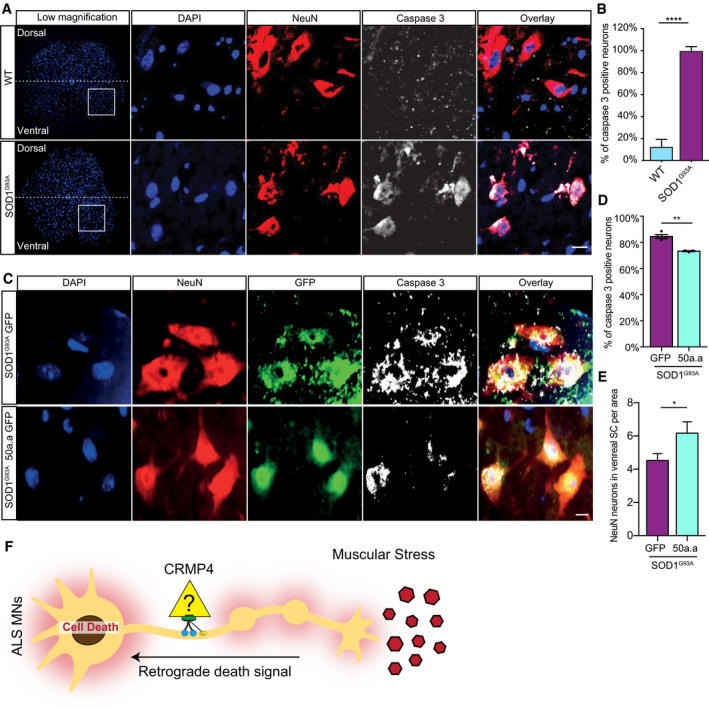
- Representative images and insets of WT and SOD1G93A SC cross sections at P90. Blue: denotes DAPI, Red: denoted NeuN, and White: denotes activated caspase 3. Scale bar: 20 µm.
- Quantification of active caspase 3‐positive cells in P90 WT and SOD1G93A SC. 3 different mice in each condition analyzed. We monitored active caspase 3 expression in total of 108 cells in WT SC and 123 cells in SOD1G93A. Student’s t‐test, n = 3, data presented as mean ± SE, ****P < 0.0001.
- Representative images of P90 SOD1G93A mice SC cross sections that were injected with AAV9‐GFP/AAV9‐50aa‐GFP. Blue: denotes DAPI, Red: denoted NeuN, and White: denotes activated caspase 3. Scale bar: 10 µm.
- Quantification of caspase 3‐positive cells in P90 SOD1G93A mice SC cross sections that were injected with either AAV9‐GFP or AAV9‐50aa‐GFP. Data collected from 3 different mice in each condition. We monitored active caspase 3 expression in total of 228 cells in P90 SOD1G93A mice SC cross sections that were injected with AAV9‐GFP and 179 cells P90 SOD1G93A mice SC cross sections that were injected with AAV9‐50aa‐GFP. Student’s t‐test, n = 3, data presented as mean ± SE, **P = 0.0019.
- Quantification of the number of NeuN‐positive cells in P90 SOD1G93A mice SC cross sections that were injected with either AAV9‐GFP or AAV9‐50aa‐GFP. Data collected from 3 different mice in each condition. We monitored the number of NeuN‐positive cells from total of 228 cells in P90 SOD1G93A mice SC cross sections that were injected with AAV9‐GFP and 179 cells P90 SOD1G93A mice SC cross sections that were injected with AAV9‐50aa‐GFP. Unpaired t‐test with Welch's correction, n = 3, data presented as mean ± SE, *P = 0.0484.
- Working model—CRMP4‐dynein complex formation is enhanced in ALS disease and leads to subtype‐specific neuronal loss.
Taken together, our in vivo and in vitro data demonstrate that a CRMP4‐dynein complex contribute to motor neuron loss in ALS disease. Importantly, this process is reversible and can be prevented by blocking the CRMP4 and dynein interaction.
Discussion
Duplan et al (2010) previously reported that CRMP4 is elevate in SOD1G93A MNs both in vivo and in vitro and lead to their loss. By reducing CRMP4 levels, the group demonstrated protective effect on MN health (Duplan et al, 2010). However, the mechanism by which CRMP4 mediates MN toxicity, its involvement in other ALS models, and its relevance to human ALS disease were unknown. In this work, we discover subcellular alterations in CRMP4 levels in sALS human patients, in C9orf72 human‐derived MNs, and in the SOD1G93A mice CRMP4 alterations dependent on dynein activity. Specifically, CRMP4‐dynein interactions are mediated by amino acids 100–150 in the CRMP4 protein, a region in which mutation was indeed correlated with ALS (Blasco et al, 2013). Notably, CRMP4‐dynein complexes are enriched in ALS‐diseased MNs and lead to ~ 25% cell death observed in ALS‐diseased spinal cord. Finally, we show that blocking the CRMP4‐dynein interaction rescued this MN population, both in vitro and in vivo (Fig 7F). These results pose many important open questions:
What is the cause for CRMP4 alterations in ALS‐diseased MN axons?
Here, we reported that CRMP4 is elevated in the soma of several ALS model MNs but is decreased near distal axons. We further demonstrated that Sema3A facilitates an increase in CRMP4 protein levels specifically in ALS‐diseased MN somata and proximal axons. Since in our previous report we demonstrated that ALS disease muscles secrete Sema3A, we assume that CRMP4 elevations in somata and proximal axons are due to the nearby presence of Sema3A. However, the mechanism responsible for the permanent elevation of CRMP4 specifically in ALS‐diseased MNs is unknown. miRNA downregulation and defects in local protein synthesis are common features in several ALS models (Haramati et al, 2010; Costa & Willis, 2018). Along with that, Sema3A was shown to induce axonal local synthesis in several neuronal systems (Campbell & Holt, 2001; Wu et al, 2005; Manns et al, 2012; Cagnetta et al, 2018, 2019). Thus, we hypothesize that the permanent elevation in CRMP4 that we observed in diseased MN somata and proximal axons are possibly due to increase in axonal protein synthesis. Specifically, our recent published work suggests that miR126‐5p is downregulated in both muscles and MN axons in several ALS models (Rotem et al, 2017; Maimon et al, 2018). Thus, it is tempting to speculate further that miR126‐5p downregulation mediates CRMP4 increases in ALS‐diseased MNs via local protein synthesis and consolidation of retrograde death signals in ALS models. Another possibility for CRMP4 alterations in ALS disease is a proteolytic degradation of CRMP4 at the injured site of the neuron, as has been shown before (Jang et al, 2010). Further experiments are needed to test the probability of those ideas.
How does CRMP4‐dynein activate caspase 3 in ALS‐diseased MNs?
Our data further suggest that CRMP4 forms complexes with dynein along ALS‐diseased MNs and leads to their loss via a caspase 3‐dependent cascade. However, it is still not clear whether CRMP4 itself activates the apoptotic program or whether it plays a regulatory role in recruiting the death complex. Since CRMP members have not yet been reported to act as transcription factors, we assume that CRMP4 is a critical part of a retrograde signaling complex that might contain additional proteins. For example, DLK regulation of JNK and c‐Jun might also be a part of this death signal mediated by Sema3A in ALS MNs, since it was previously shown that both DLK and JNK signaling are elevated in ALS models and that they are part of a retrograde death signal (Perlson et al, 2010; Sengupta Ghosh et al, 2011; Siu et al, 2018; Escudero et al, 2019). Another possibility is that the neurotrophic receptor p75NTR is also involved in this process. p75NTR regulates a diverse range of cellular functions including axon pruning (Singh et al, 2008) and neuronal death (Bamji et al, 1998; Kenchappa et al, 2010; Pathak et al, 2018). p75NTR is retrogradely transported along the axon (Deinhardt et al, 2006; Harrington & Ginty, 2013; Cosker & Segal, 2014) and plays a role in generating a retrograde apoptotic signal that activates JNK (Kenchappa et al, 2010). It is noteworthy that the activity of Sema3A and its receptor was previously linked to p75NTR (Ben‐Zvi et al, 2007). Another possible candidate that was shown to be coupled with Sema3A is PTEN (Chadborn et al, 2006). Furthermore, it was established that the p75NTR‐dependent apoptosis signal is promoted by PTEN activation (Song et al, 2010). Thus, future experiments should examine whether PTEN, p75NTR, and JNK indeed participate in Sema3A‐dependent retrograde death signals in ALS.
CRMP4 gain of toxicity in ALS disease
CRMP4 overexpression has been suggested to cause MN death in ALS models (Charrier et al, 2003; Duplan et al, 2010). Furthermore, a CRMP4 mutation was associated with ALS in patients (Blasco et al, 2013), However, the mechanism of CRMP4 toxicity in ALS MNs is unknown. Here, we demonstrate a specific mechanism by which CRMP4 toxicity to MNs is dependent on dynein activity. Moreover, we demonstrate that amino acids 100–150 of CRMP4 are responsible for the CRMP4 and dynein interaction. Importantly, the ALS‐associated mutation in CRMP4 is located in this motif. Our data show a stronger dynein interaction for the mutant CRMP4, which is in accordance with CRMP4 gain of toxicity. Alternatively, CRMP4 has been documented before, in a number of experimental models to elevate post‐neuronal injury (near injury site). Thus, it is possible that the results in this manuscript reflect a response of motor neurons to stress rather than an ALS‐specific mechanism (Jang et al, 2010). Additional studies will be needed to dissect in detail the mutant CRMP4 activity in ALS‐diseased MNs.
Materials and Methods
Animals
SOD1G93A (Stock No. 002726) mice were originally obtained from Jackson Laboratories and maintained by breeding with C57BL/6J mice.
B6;129S6‐ChATtm2(cre)Lowl/J (Stock No. 006410) and B6;129S6Gt(ROSA)26 Sortm14(CAG−tdTomato)Hze/J (Stock No. 007908) mice were originally obtained from Jackson Laboratories. Animals were cross‐bred in the Tel‐Aviv SPF animal unit to yield homozygous ChAT::RosatdTomato mice. The ChAT::RosatdTomato colony was maintained by in‐breeding males and females from the colony. The ChAT::RosatdTomato colony was cross‐bred with SOD1G93A to yield SOD1G93A/ChAT::tdTomato mice. C57BL/6J mice were used as a WT mouse strain. Mice were genotyped using the PCR (KAPA Bio Systems—Wilmington, MA, USA). DNA samples were generated from mouse ear or tail. Animal experiments were performed under the supervision and approval of the Tel‐Aviv University Committee for Animal Ethics.
iPSc cultures
Healthy/Control iPSC lines, provided by Dr. Sami Barmada, were created and characterized as before (Tank et al, 2018). Two lines from fALS patients carrying the C9orf72 mutation, and two lines from healthy controls, were used for all experiments.
| Name | Age donated | Age of onset | Gender | |
|---|---|---|---|---|
| ALS883 | 51 | 49 (Lumbar) | M | (> 44 repeats) per Athena 7/26/2012 |
| ALS312 | 54 | 52 (Lumbar) | M | (44 and 2 repeats) by Athena Diagnostics 10/03/2012 |
| Control746 | 58 | M | Healthy | |
| Control1021 | 54 | F | Healthy |
Colonies were groomed daily until each well of the 6‐well plate was between 30 and 40% confluence and no spontaneously differentiated cells were observed. At this point, we used the direct iMN (diMN) differentiation in monolayers from hiPSCs protocol published by Cedar Sinai and approved by Dhruv Sareen (protocol number: CSMNC‐SOP‐C‐005) for our experiments. Briefly, we induced MN differentiation by MN Differentiation Stage 1 media: prepared with—IMDM (LifeTech), F12 (LifeTech), NEAA (Gibco), B27 (LifeTech), N2 (LifeTech), PSA (LifeTech), LDN193189 0.2 µM (Selleck), SB431542 10 µM (Tocris), and CHIR99021 3 µM (Cayman Chemicals). The media was gently added to the wells, and colonies were grown with it for 5 days. MN differentiation Stage 2: At day 6, the colonies were dissociated, using Accutase, and 100K cells were plated in the proximal compartment of our micro fluidic device. Stage 2 media, which contains IMDM, F12, NEAA, B27, N2, PSA, LDN193189 0.2 µM (Selleck), SB431542 10 µM (Tocris), and CHIR99021 3 µM (Cayman Chemicals), All‐trans RA 0.1 µM (Stemgent), and SAG 1 µM (Sonic Hedgehog Agonist—Cayman Chemicals) media was added to both the distal and proximal compartments of the MFC and refreshed every 2 days until day 11. MN differentiation Stage 3: At day 12 the media was changed to stage 3 media prepared with IMDM, F12, NEAA, B27, N2, PSA, Compound E 0.1 µM (Calbiochem), DAPT 2.5 µM (Cayman Chemicals), db‐cAMP 0.1 μM (Millipore), All‐trans RA 0.5 µM (Stemgent), SAG 0.1 µM, ascorbic acid 200 ng/ml (Sigma), BDNF 10 ng/ml (Alomone Labs), and GDNF 10 ng/ml (Alomone Labs) and was refreshed every 2 days until cells exhibited MN neuronal morphology and positive markers. Human iPSC experiments were performed under the supervision and approval of the Tel‐Aviv University Committee for Human Ethics.
Microfluidic chamber preparation
Polydimethylsilxane (PDMS) microfluidic chambers (MFCs) were designed and cast as described previously (Ionescu et al, 2016). Briefly, MFCs were fabricated from our designed templates and made from PDMS mixture at 70°C. After the wells were punched, a small “cave” was made in the explant well near the grooves using a 25G needle, keeping the explant in place. Microfluidic devices were cleaned of surface particles using adhesive tape and were sterilized in 70% ethanol for 15 min. Devices were completely dried under sterile conditions using UV radiation, attached to a sterile 60‐mm plastic dishes (Nunc) with gentle pressure and margins were sealed with PDMS before incubation at 60°C for 30 min to prevent the chamber from detaching. The wells and channels were filled with 150 µl of 1.5 ng/ml polyornithine (P‐8638, Sigma) in PBS overnight and then replaced with 150 µl laminin (l‐2020, Sigma), 1:333 in deionized distilled water (DDW) overnight. One day before plating the spinal cord explant, laminin was replaced with explant medium containing Neurobasal (Life Technologies) supplemented with 2% B27 (Invitrogen), 1% penicillin–streptomycin (Biological Industries), 1% Glutamax (Life Technologies), and 25 ng/ml brain‐derived neurotrophic factor (Alomone Labs), until the day on which co‐culturing began.
Motor neuron cell culture
Primary spinal cord neurons were cultured using E12.5 mouse embryos of either sex as previously described (Zahavi et al, 2015). Briefly, spinal cords were excised, trypsinized, and triturated. Supernatant was collected and centrifuged through a 4% BSA cushion. The pellet was resuspended and centrifuged through an Optiprep gradient (10.4% Optiprep (Sigma‐Aldrich), 10 mM Tricine, 4% glucose) for 20 min at 760 g with the brake turned off. Cells were collected from the interface, washed once in complete medium, and then plated in coated growth chambers. Cells were maintained in complete neurobasal medium (Gibco) containing B27 (Gibco), 10% (v/v) horse serum (Biological Industries), 25 nM beta‐mercaptoethanol, 1% penicillin–streptomycin (PS; Biological Industries), and 1% GlutaMAX (Gibco) supplemented with 1 ng/ml glial‐derived neurotrophic factor (GDNF), 0.5 ng/ml ciliary neurotrophic factor (CNTF), and 1 ng/ml brain‐derived neurotrophic factor (BDNF) (Alomone Labs). Prior to plating, growth plates were coated with 1.5 g/ml poly d‐l‐ornithine (PLO; Sigma‐Aldrich) overnight at 37°C and with 3 µg/ml Laminin (Sigma‐Aldrich) for 2 h at 37°C. For immunofluorescence staining, 10,000 cells were plated on cover slides in 24‐well plates. Cells were grown at 37°C in 5% CO2.
Spinal cord explants
Spinal cords were dissected from E12.5 mouse embryos of both sexes, either using HB9::GFP or SOD1G93A stripped of meninges and dorsal root ganglia. The ventral horn was separated from the dorsal horn by longitudinal cuts along the spinal cord, and transverse sections up to 1 mm were placed in the explant well. Prior to plating, growth chambers were coated with 1.5 g/ml PLO overnight at 37°C and 3 µg/ml Laminin overnight at 37°C. Explants were maintained in spinal cord explant medium containing Neurobasal, 2% B27, 1% PS, and 1% GlutaMAX, supplemented with 25 ng/ml BDNF. Explants were grown at 37°C in 5% CO2.
Fluorescence microscopy and image analysis
All confocal images were captured using a Nikon Ti microscope equipped with a Yokogawa CSU X‐1 spinning disk and an Andor iXon897 EMCCD camera controlled by Andor IQ3 software. All live‐imaging assays were performed in a humidified incubation chamber at 37°C, 5% CO2. Images were analyzed using ImageJ software.
Rertrograde labeling of cell bodies in the MFC
Alexa Fluor 647‐conjugated cholera toxin subunit B (CTX; Thermo Fisher C‐347777) at 500 ng/ml was applied to the distal compartment of an MFC system while maintaining a higher liquid volume in the proximal compartment to prevent unspecific labeling by diffusion. After 8 h, only somata whose axons traversed to the distal compartment were labeled.
Recombinant Sema3A application
Recombinant Sema3A (R&D, 1250‐S3‐025) at 500 ng/ml was used in our experiments. We dilute the Sema3A in poor neurobasal (PNB) containing Neurobasal medium (Gibco) with 1% PS and 1% Glutamax.
Retrograde transport inhibition
In order to inhibit dynein‐dependent retrograde transport, Cilliobrevin‐D (Merck‐Millipore, 250401) at 10 µM was applied to the distal compartment of the MFC while maintaining a proximal‐to‐distal volume gradient.
Inhibition of dynamin‐dependent endocytosis
Dynasore (Sigma‐Aldrich, D7693) at 100nM was added to the distal compartment of the MFC while maintaining a proximal‐to‐distal volume gradient.
Pull‐down assays
For the cell cultures pull‐down experiments, 2 × 106 COS7 cells were plated in 10 cm culture dishes. The following day, cells were transfected using calcium phosphate protocol with Flag‐CRMP4/ GFP‐CRMP4/ GFP‐Delta‐CRMP4/Mutated‐Flag‐CRMP4/AAV9‐GFP/AAV9‐GFP‐50aa vector. The next day, cells were lysed, and proteins were extracted using lysis buffer containing PBS, 1% Triton X‐100, and 1% protease, and phosphatase inhibitors (Roche), followed by centrifugation and collection of the supernatant. At this point, immunoprecipitation preparation of the lysate was precleared with protein A agarose beads (Roche). Following overnight incubation with primary anti‐flag antibody/anti‐DIC antibody, complexes were incubated with protein A agarose beads for 2 h at 4°C and then precipitated and washed with PBS with 0.1% Triton X‐100 (Sigma). Proteins were eluted by boiling in sample buffer and then subjected to western blot precipitation analysis with CRMP4/dynactin p150/Flag antibody (Sigma‐Aldrich F3165)/DIC. We used mouse IgG antibody as a control (SC‐2025). For sciatic nerve pull downs, 12 sciatic nerves were pooled for each experiment. Here as well, the P90 sciatic nerve samples were first excised and homogenized in lysis buffer containing PBS and 1% protease and phosphatase inhibitors (Roche), followed by centrifugation and collection of the supernatant. Then, we performed the pull‐down assay using the technique described above. Under these conditions, pull downs were performed using DIC (Millipore MAB1618) and CRMP4 (Millipore AB5454) antibodies.
Western blotting
Sciatic nerve axoplasm was isolated by excising and cutting sciatic nerves into short segments, followed by detergent‐free buffer homogenized with PBS X1 protease and phosphatase inhibitors (Roche), followed by centrifugation and collection of the supernatant. Complete sciatic nerve extracts were achieved in the same manner with the exception of adding 1% Triton X‐100. The protein concentration was determined using the Bio‐Rad Protein Assay. Protein samples were denatured by boiling in SDS sample buffer and then electrophoresed in 8% polyacrylamide gels (SDS–PAGE). Proteins were transferred to a nitrocellulose membrane and then immunoblotted with appropriate primary antibodies: anti‐CRMP4—1:2,000 (Millipore AB5454), anti‐DIC—1:1,000 (Millipore MAB1618), anti‐p150 1:250 (BD Bioscience 611003), anti‐Flag 1:4,000 (Sigma‐Aldrich F3165), anti‐Tubulin 1:10,000 (ab7291), and anti‐tERK 1:10,000 (M5670), diluted in 5% (w/v) skim milk (BD Difco) in TBS‐T, followed by species‐specific HRP‐conjugated secondary antibodies (Jackson Laboratories) and visualized using a myECL imager (Thermo), according to the manufacturer’s instructions. ImageJ software was used for quantification.
AAV production
We used AAV serotype 9 (AAV9) for overexpression experiments. The AAV9 produced in AAVpro 293T cells (Takara‐Clontech, #632273), with the AAVpro® Purification Kit (All Serotypes) from TaKaRa (#6666). For each construct four 15‐cm plates were transfected with 20 μg of DNA (AAVplasmid containing the construct of interest and two AAV9 helper plasmids) using jetPEITM (Polyplus‐transfection) in DMEM medium without serum or antibiotics. pAdDeltaF6 and pPHP.S helper vectors were kind gift from Prof. Fainzilber. Medium (DMEM, 20% FBS, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin) was added on the following day to a final concentration of 10% FBS, and extraction was done at 3 days post‐transfection. Purification was performed according to the manufacturer’s instructions. For all constructs, we obtained titers in the range of 1013–1014 viral genomes/ml.
Vector injections
The injection procedure was performed on pre‐symptomatic ~ P60 mice. Mice were first anesthetized using a mixture of xylazine and ketamine. Then, a thin incision was performed in the mouse skin in order to expose the area of the L5 and L6 vertebrae. Next, 5 µl of AAV9‐GFP (6.5 × 1014 vg/ml) or AAV‐GFP‐50aa (1.21 × 1013 vg/ml) were injected by intrathecal injection to L5–L6 vertebrae in the spinal cord using a 25 µl Hamilton syringe and a 30G Hamilton needle. All animal experimentations were approved by the Tel‐Aviv University Animal Ethics Committee. This method was conducted with the help of Dr. Michael Tolmasov. All the tissues were taken 4 weeks post‐injection.
Human muscle biopsy for intra‐muscular nerve staining
Intra‐muscular nerve staining was performed on muscle biopsies from ALS patients and non‐ALS patients. All clinical and muscle biopsy materials used in this study were obtained with written informed consent during 2016–2020 for diagnostic purposes followed by research application, approved by the institutional review board. Deltoid, quadriceps, or gastrocnemius skeletal muscle samples were excised via open biopsies, and pathological analysis was performed at the neuromuscular pathology laboratory at Sheba Medical Center, Ramat Gan, Israel. All ALS patients were diagnosed with clinically definite or probable ALS according to Awaji criteria (de Carvalho et al, 2008) Control muscles included a variation of findings, which were consistent with a diagnosis of normal muscle, severe, chronic ongoing denervation and reinnervation due to spinal stenosis, necrotic autoimmune myopathy, type 2 fiber atrophy due to disuse, and overlap myositis syndrome.
Frozen muscle biopsies were cryo‐sectioned to 10µm thick slices, mounted onto slides, and air dried for 30 min in room temperature (RT). Sections were washed in PBS, fixed in 4% PFA for 20 min, and permeabilized with 0.1% Triton, and blocked with 5% goat serum (Jackson Laboratories) and 1 mg/ml BSA (Amresco). Sections were than incubated with appropriate antibodies overnight at 4°C in blocking solution Rabbit anti‐CRMP4 (Millipore AB5454, 1:250), Chicken anti‐NFH (Abcam, 1:1,000). Sections were washed again and incubated for 2 h with secondary antibodies (1:1,000, Jackson Laboratories and Thermo Fisher), washed, and mounted with ProLong Gold (Life Technologies).
IHC of CRMP4 in human spinal cord tissue
The Dako Autostainer Link 48 (Agilent, USA) was used for all human spinal cord immunohistochemistry. The CRMP4 antibody (Millipore AB5454) was used at 1:700 for 30 min at room temperature. Heat‐induced epitope retrieval was used prior to staining with Dako’s EnV Flex Low pH TRS. The Dako Envision Flex Plus Mouse Link Kit (Agilent, USA) to detect the antibody along with the Dako DAB (Agilent, USA). CRMP4 relative expression was semi‐quantified by scoring IHC spinal cord sections between 1 and 3, blindly: 1 = low expression, 2 = middle expression, and 3 = high expression.
Sciatic nerve sectioning and immunostaining
Sciatic nerves of P90 mice were isolated and immediately fixed by using 4% PFA followed by 20% sucrose incubation. Then, the samples were embedded by freezing in Tissue‐Tek® OCT. Next, 10 µm lumbar sciatic nerve sections were prepared using Cryotome™ FSE cryostat (Thermo Fisher Scientific). Sections were rinsed in PBS and then permeabilized with 0.1% Triton X‐100, 5% Goat Serum (GS), 1 mg/ml bovine serum albumin IgG, and protease free (BSA) in PBS. Primary antibodies against NFH 1:500(Abcam ab72996/ Covance smi31p/ Covance smi32p)/CRMP4 1:400 (Millipore AB5454)/GFP 1:400 (Abcam ab13970) were diluted in blocking solution, 5% GS, 1 mg/ml BSA in PBS, and incubated overnight at 4°C. Samples were incubated with species‐specific fluorescent secondary antibodies for 2 h at room temperature. ProLong antifade medium (Molecular Probes) was added, and the samples were covered with a #1.5, 18 × 18 mm cover slide.
Spinal cord sectioning and immunostaining
Spinal cord of P90 mice was isolated and immediately fixed by using 4% PFA followed by 20% sucrose incubation. Then, the samples were embedded by freezing in TissueTek® OCT. Next, 10 µm lumbar spinal cord sections were prepared using Cryotome™ FSE cryostat (Thermo Fisher Scientific). Sections were rinsed in PBS and then permeabilized with 0.1% Triton X‐100, 5% Goat Serum (GS), 1 mg/ml bovine serum albumin IgG, and protease free (BSA) in PBS. Primary antibodies against NeuN 1:500 (Milipore MAB377)/CRMP4 1:400 (Millipore AB5454)/GFP 1:400 (Abcam ab13970)/activated Caspase 3 1:15 (Biovision 3015‐100) were diluted in blocking solution, 5% GS, 1 mg/ml BSA in PBS, and incubated overnight at 4°C. Samples were incubated with species‐specific fluorescent secondary antibodies for 2 h at room temperature. ProLong antifade medium with Dapi (Molecular Probes) was added, and the samples were covered with a #1.5, 18 × 18 mm cover slide.
Immunostaining of cell cultures
Cultures were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X‐100, 5% GS, 1 mg/ml BSA in PBS. Samples were blocked for 1 h with blocking medium containing 5% GS and 1 mg/ml BSA in PBS. Primary antibodies against Tau 1:100 (abcam, ab80579) NFH‐1:500 (Sigma‐Aldrich N4142), PlexinA1‐1:100 (Alomone lab, APR‐081‐F), CRMP4‐1:100 (Millipore, AB5454), GAPDH 1:500 (abcam, ab9484), Tubulin 1:500 (abcam, ab7291), HB9 1:100 (IMGENEX, IMG‐6549A) were diluted in blocking solution and incubated overnight at 4°C. Samples were incubated with species‐specific fluorescent secondary antibodies for 2 h at room temperature. For visualizing nuclei in myotubes, DAPI was used. In the MFC, after the staining protocol was completed, the MFC was peeled from the dish by gently pulling it from the proximal to the distal side.
Whole mount NMJ immunofluorescence staining
Gastrocnemius muscles of P60/P90 SOD1G93A/ChAT::tdTomatoor WTChAT::tdTomatomice were dissected from mice, washed with cold PBS, and cut in longitudinal sections along the fiber before fixed in 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature, while rocking. From fixation until the end of staining protocol (mounting), muscle fibers were washed three times with PBS after each step, except between blocking and primary antibodies staining. After been fixated, muscle fibers were further dissected into smaller section, along fiber orientation. For postsynaptic AChR labeling, the fibers were then stained with αBTX (TMR‐α‐bungarotoxin; T0195 Sigma) 2 μg/ml in PBS for 15min at RT while rocking. Fibers were then permeabilized in −20°C methanol for 5min, then blocked for 1 h at RT with blocking solution (2% BSA, 0.4% Triton X‐100 in PBS), followed by the application of primary antibodies diluted in blocking solution; NFH (1:500; ab72996, Abcam) and CRMP4 (1:250; Millipore, AB5454) and incubation overnight at RT while rocking. On the next day, samples were incubated with species‐specific fluorescent secondary antibodies for 4 h at room temperature while rocking. Muscle fibers were then placed on a cover slide suitable for imaging, mounted with Vectashield (Vector Laboratories) and sealed with clear nail polish. Slides were kept in RT until completely dried, then stored at 4oC until imaged in the microscope.
Proximity ligation assay
The proximity ligation assay (PLA) was used to visualize the co‐localization of selected proteins; it was performed as previously described (Söderberg et al, 2008). Briefly, iPSc‐derived MNs and murine‐MN cultures were grown in the MFC on glass dishes for 18 and 5 DIV, respectively, and were then fixed in 4% PFA, at 4°C for 20 min. Subsequently, the samples were blocked and permeabilized with 5% donkey serum, 1% BSA, and 0.1% Triton X‐100 in PBS for 1h and incubated with anti‐CRMP4 and anti‐DIC antibodies overnight at 4°C. Interactions (range ~40 nm) were detected by the proximity ligation assay Duolink kit (Sigma: PLA probe anti‐mouse minus DUO92004, anti‐rabbit plus DUO92002, and the detection kit Far Red). PLA was performed according to the manufacturer's instructions. Coverslips were washed, mounted, and imaged by confocal microscopy. Half ligation samples were used as a negative control. The axonal PLA signal was quantified with ImageJ software using an axonal mask based on an endogenous mCherry/Rosa signal. The PLA puncta signal was quantified with the analyzed particle function of the software.
CRMP4‐like peptide design and insertion into MNs
The Dynein‐CRMP4 blocking peptide design was based on previous findings by Arimura et al (2009), which pointed to 50 specific amino acid sequences responsible for CRMP2 binding to dynein (Arimura et al, 2009). Peptides were prepared by Alomone labs and GL Biochem. Peptide sequences are as follows:
| Name | Sequence | MW (Da) |
|---|---|---|
| Peptide‐1 | TTMIIDHVVPEPE | 1,480 Da |
| Peptide‐2 | SSLTEAYEKWREWADGKS | 2,143 Da |
| Peptide‐3 | CCDYALHVDI | 1,151 Da |
| Peptide‐4 | THWNDSVKQ | 1,114 Da |
The peptides were inserted into axons by harsh pipetting. Final concentration of 10 μM of each peptide were inserted. Tamra peptide was generously donated by Dr. Mike Fainzilber’s laboratory (10 μM final concentration).
Vectors
CRMP4 and CRMP4Δ100–150 (containing deletion of the coding sequence 301‐450bp) were sub‐cloned in frame into the pLL3.7‐GFP (Addgene) mammalian expression vector. Flag‐CRMP4 and mutated Flag‐CRMP4‐I141V, used in the pull‐down assays, was cloned into pCDNA3 vector (Invitrogen). GFP and GFP‐50aa (containing the coding sequence of CRMP4 301–450 bp) were sub‐cloned in frame into the pAAV‐CBh (Vector Builder) mammalian gene expression vector.
Experimental design and statistical analysis
All statistical analyses were performed using GraphPad Prism v6.0. For two‐group analysis, Student’s t‐test or the Mann‐Whitney test was used, as determined by a normality test. For multiple comparisons, ANOVA was used with the Tukey or Holm–Sidak post hoc tests. All experiments include at least 3 biologically independent repeats; Significance was set at P < 0.05.
Ethics approval and consent to participate
Animal experiments were performed under the supervision and approval of the Tel‐Aviv University Committee for Animal Ethics. Human iPSC experiments were performed under the supervision and approval of the Tel‐Aviv University Committee for Human Ethics.
Author contributions
Project conceptualization by RM, LA, TGP, MB, and EP; Data curation by RM, LA, TGP, TA, AI; Formal analysis by RM, LA, TGP, AI, MO, EP; Investigation by RM, LA, TGP, TA, RW, MO, ET, GA, NS, AD, SB; Methodology by RM, LA, TGP, TA, AI, RW, MO, ET, GA, NS, YA, AD, SB, MB, EP; Resource obtain from ET, GA, NS, AD, SB; Funding acquisition MB, EP; Supervision MB, EP; Writing—original draft, review and editing by RM, LA, TGP, TA, AI, YA, AD, SB, MB, EP and approved by all.
Conflict of interest
The authors declare that they have no conflict of interest point.
Supporting information
Review Process File
Appendix
Source Data for Figures
Acknowledgments
This work was supported by IsrALS Foundation, the Israel Science Foundation (735/19), Ministry of Science and Technology State of Israel, and the European Research Council (grant number 309377) to E.P, Czech Health Research Council grant no. NV18‐04‐00085 to MB, Czech Science Foundation grant no. 21‐24571S to MB and RW, and Grant Agency of the Charles University grants no. 524218 to RW. We thank Prof. Mike Fainzilber for the Tamra peptides, and help with AAV9 design. We thank Prof. Eva Feldman and Prof. Stephen Goutman for obtaining the fibroblasts for the IPSC lines. We thank Dr. Michael Tolmasov for performing the intrathecal injections. We thanks Michigan Brain Bank (5P30 AG053760 University of Michigan Alzheimer’s Disease Core Center) for providing patients spinal cord sections. Immunohistochemistry (IHC) was performed at the Rogel Cancer Center Tissue and Molecular Pathology Shared Resource Laboratory (funding support: NIH P30 CA04659229).
The EMBO Journal (2021) 40: e107586.
Data availability
This study includes no data deposited in external repositories. All data generated or analyzed during this study are included in this published article.
References
- Arimura N, Hattori A, Kimura T, Nakamuta S, Funahashi Y, Hirotsune S, Furuta K, Urano T, Toyoshima YY, Kaibuchi K (2009) CRMP‐2 directly binds to cytoplasmic dynein and interferes with its activity. J Neurochem 111: 380–390 [DOI] [PubMed] [Google Scholar]
- Balastik M, Zhou X, Alberich‐Jorda M, Weissova R, Žiak J, Pazyra‐Murphy M, Cosker K, Machonova O, Kozmikova I, Chen C‐Het al (2015) Prolyl isomerase Pin1 regulates axon guidance by stabilizing CRMP2A selectively in distal axons. Cell Rep 13: 812–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD (1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Zvi A, Ben‐Gigi L, Klein H, Behar O (2007) Modulation of semaphorin3A activity by p75 neurotrophin receptor influences peripheral axon patterning. J Neurosci 27: 13000–13011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G (2010) Deficits in axonal transport precede ALS symptoms in vivo . Proc Natl Acad Sci USA 107: 20523–20528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco H, Bernard‐Marissal N, Vourc'h P, Guettard YO, Sunyach C, Augereau O, Khederchah J, Mouzat K, Antar C, Gordon PHet al (2013) A rare motor neuron deleterious missense mutation in the DPYSL3 (CRMP4) gene is associated with ALS. Hum Mutat 34: 953–960 [DOI] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52: 39–59 [DOI] [PubMed] [Google Scholar]
- Cagnetta R, Frese CK, Shigeoka T, Krijgsveld J, Holt CE (2018) Rapid cue‐specific remodeling of the nascent axonal proteome. Neuron 99: 29–46.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnetta R, Wong HH‐W, Frese CK, Mallucci GR, Krijgsveld J, Holt CE (2019) Noncanonical modulation of the eIF2 pathway controls an increase in local translation during neural wiring. Mol Cell 73: 474–489.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE (2001) Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32: 1013–1026 [DOI] [PubMed] [Google Scholar]
- de Carvalho M , Dengler R, Eisen A, England JD, Kaji R, Kimura J, Mills K, Mitsumoto H, Nodera H, Shefner Jet al (2008) Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 119: 497–503 [DOI] [PubMed] [Google Scholar]
- Castellani V, Falk J, Rougon G (2004) Semaphorin3A‐induced receptor endocytosis during axon guidance responses is mediated by L1 CAM. Mol Cell Neurosci 26: 89–100 [DOI] [PubMed] [Google Scholar]
- Chadborn NH, Ahmed AI, Holt MR, Prinjha R, Dunn GA, Jones GE, Eickholt BJ (2006) PTEN couples Sema3A signalling to growth cone collapse. J Cell Sci 119: 951–957 [DOI] [PubMed] [Google Scholar]
- Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J (2003) Collapsin Response Mediator Proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Mol Neurobiol 28: 51–64 [DOI] [PubMed] [Google Scholar]
- Cosker KE, Segal RA (2014) Neuronal signaling through endocytosis. Cold Spring Harb Perspect Biol 6: a020669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa CJ, Willis DE (2018) To the end of the line: axonal mRNA transport and local translation in health and neurodegenerative disease. Dev Neurobiol 78: 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Hafezparast M (2017) Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol Dis 105: 283–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G (2006) Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52: 293–305 [DOI] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Mackenzie I, Boeve B, Boxer A, Baker M, Rutherford N, Nicholson A, Finch NA, Flynn H, Adamson Jet al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duplan L, Bernard N, Casseron W, Dudley K, Thouvenot E, Honnorat J, Rogemond V, De Bovis B, Aebischer P, Marin Pet al (2010) Collapsin response mediator protein 4a (CRMP4a) is upregulated in motoneurons of mutant SOD1 mice and can trigger motoneuron axonal degeneration and cell death. J Neurosci 30: 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero CA, Cabeza C, Moya‐Alvarado G, Maloney MT, Flores CM, Wu C, Court FA, Mobley WC, Bronfman FC (2019) c‐Jun N‐terminal kinase (JNK)‐dependent internalization and Rab5‐dependent endocytic sorting mediate long‐distance retrograde neuronal death induced by axonal BDNF‐p75 signaling. Sci Rep 9: 6070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O’Donnell M, Gelfand VI, Kapoor TM, Chen JK (2012) Small‐molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484: 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano‐Sanchez A, Khan J, Polak MA, Glass JD (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185: 232–240 [DOI] [PubMed] [Google Scholar]
- Fournier AE, Nakamura F, Kawamoto S, Goshima Y, Kalb RG, Strittmatter SM (2000) Semaphorin3a enhances endocytosis at sites of receptor–F‐actin colocalization during growth cone collapse. J Cell Biol 149: 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P (2000) Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20: 2534–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni‐Emek N, Chein M, Gluska S, Perlson E (2015) Amyotrophic lateral sclerosis as a spatiotemporal mislocalization disease: location, location, location. Int Rev Cell Mol Biol 315: 23–71 [DOI] [PubMed] [Google Scholar]
- Gibbs KL, Kalmar B, Rhymes ER, Fellows AD, Ahmed M, Whiting P, Davies CH, Greensmith L, Schiavo G (2018) Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis 9: 596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM (1995) Collapsin‐induced growth cone collapse mediated by an intracellular protein related to UNC‐33. Nature 376: 509–514 [DOI] [PubMed] [Google Scholar]
- Guedes‐Dias P, Holzbaur ELF (2019) Axonal transport: driving synaptic function. Science 366: eaaw9997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haramati S, Chapnik E, Sztainberg Y, Eilam R, Zwang R, Gershoni N, McGlinn E, Heiser PW, Wills A‐M, Wirguin Iet al (2010) miRNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci U S A 107: 13111–13116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AW, Ginty DD (2013) Long‐distance retrograde neurotrophic factor signalling in neurons. Nat Rev Neurosci 14: 177–187 [DOI] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ, Vale RD (1989) Movement of microtubules by single kinesin molecules. Nature 342: 154–158 [DOI] [PubMed] [Google Scholar]
- Ionescu A, Zahavi EE, Gradus T, Ben‐Yaakov K, Perlson E (2016) Compartmental microfluidic system for studying muscle–neuron communication and neuromuscular junction maintenance. Eur J Cell Biol 95: 69–88 [DOI] [PubMed] [Google Scholar]
- Jang SY, Shin YK, Jung J, Lee SH, Seo SY, Suh DJ, Park HT (2010) Injury‐induced CRMP4 expression in adult sensory neurons; A possible target gene for ciliary neurotrophic factor. Neurosci Lett 485: 37–42 [DOI] [PubMed] [Google Scholar]
- Kenchappa RS, Tep C, Korade Z, Urra S, Bronfman FC, Yoon SO, Carter BD (2010) p75 neurotrophin receptor‐mediated apoptosis in sympathetic neurons involves a biphasic activation of JNK and up‐regulation of tumor necrosis factor‐alpha‐converting enzyme/ADAM17. J Biol Chem 285: 20358–20368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascaño J, Tokito M, Van Winkle T, Howland DS, Holzbaur ELF (2002) Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late‐onset progressive degeneration. Neuron 34: 715–727 [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T (2006) Dynasore, a cell‐permeable inhibitor of dynamin. Dev Cell 10: 839–850 [DOI] [PubMed] [Google Scholar]
- Maimon R, Ionescu A, Bonnie A, Sweetat S, Wald‐Altman S, Inbar S, Gradus T, Trotti D, Weil M, Behar Oet al (2018) miR126‐5p downregulation facilitates axon degeneration and NMJ disruption via a non‐cell‐autonomous mechanism in ALS. J Neurosci 38: 5478–5494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns RPC, Cook GMW, Holt CE, Keynes RJ (2012) Differing semaphorin 3A concentrations trigger distinct signaling mechanisms in growth cone collapse. J Neurosci 32: 8554–8559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien J‐P (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14: 161–176 [DOI] [PubMed] [Google Scholar]
- Moloney EB, de Winter F , Verhaagen J (2014) ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci 8: 252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann Co, Stumm Get al (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63: 724–726 [DOI] [PubMed] [Google Scholar]
- Nagai J, Baba R, Ohshima T (2017) CRMPs function in neurons and glial cells: potential therapeutic targets for neurodegenerative diseases and CNS injury. Mol Neurobiol 54: 4243–4256 [DOI] [PubMed] [Google Scholar]
- Nagai J, Kitamura Y, Owada K, Yamashita N, Takei K, Goshima Y, Ohshima T (2015) Crmp4 deletion promotes recovery from spinal cord injury by neuroprotection and limited scar formation. Sci Rep 5: 8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle Pet al (2018) Genome‐wide analyses identify KIF5A as a novel ALS gene. Neuron 97: 1268–1283.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenick MA, Dominguez R, Holzbaur ELF (2019) Dynein activator Hook1 is required for trafficking of BDNF‐signaling endosomes in neurons. J Cell Biol 218: 220–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Vallee RB (1987) Retrograde transport by the microtubule‐associated protein MAP 1C. Nature 330: 181–183 [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Borchelt DR, Houseweart MK, Cleveland DW, Brown RH (1998) Caspase‐1 is activated in neural cells and tissue with amyotrophic lateral sclerosis‐associated mutations in copper‐zinc superoxide dismutase. Proc Natl Acad Sci U S A 95: 15763–15768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak A, Stanley EM, Hickman FE, Wallace N, Brewer B, Li D, Gluska S, Perlson E, Fuhrmann S, Akassoglou Ket al (2018) Retrograde degenerative signaling mediated by the p75 neurotrophin receptor requires p150Glued deacetylation by axonal HDAC1. Dev Cell 46: 376–387.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Jeong G‐B, Ross JL, Dixit R, Wallace KE, Kalb RG, Holzbaur ELF (2009) A switch in retrograde signaling from survival to stress in rapid‐onset neurodegeneration. J Neurosci 29: 9903–9917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Maday S, Fu M‐M, Moughamian AJ, Holzbaur ELF (2010) Retrograde axonal transport: pathways to cell death? Trends Neurosci 33: 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters OM, Ghasemi M, Brown RH (2015) Emerging mechanisms of molecular pathology in ALS. J Clin Invest 125: 1767–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy R, Lebedev A, Pahlow S, Lohkamp B (2014) Crystal structure of human CRMP‐4: correction of intensities for lattice‐translocation disorder. Acta Crystallogr, Sect D 70: 1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter AG (1999) Protein translocation in apoptosis. Trends Cell Biol 9: 394–401 [DOI] [PubMed] [Google Scholar]
- Rahajeng J, Giridharan SSP, Naslavsky N, Caplan S (2010) Collapsin Response Mediator Protein‐2 (Crmp2) regulates trafficking by linking endocytic regulatory proteins to dynein motors. J Biol Chem 285: 31918–31922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs J, Schymick J, Laaksovirta H, van Swieten J, Myllykangas Let al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes NA, Fisher JK, Austgen K, VandenBerg S, Huang EJ, Oakes SA (2010) Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis. J Clin Invest 120: 3673–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng H‐Xet al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59–62 [DOI] [PubMed] [Google Scholar]
- Rotem N, Magen I, Ionescu A, Gershoni‐Emek N, Altman T, Costa CJ, Gradus T, Pasmanik‐Chor M, Willis DE, Ben‐Dov IZet al (2017) ALS along the axons – expression of coding and noncoding RNA differs in axons of ALS models. Sci Rep 7: 44500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Cheng C, Uchida Y, Nakajima O, Ohshima T, Yagi T, Taniguchi M, Nakayama T, Kishida R, Kudo Yet al (2002) Fyn and Cdk5 mediate semaphorin‐3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 35: 907–920 [DOI] [PubMed] [Google Scholar]
- Schmidt EF, Strittmatter SM (2007) The CRMP family of proteins and their role in Sema3A signaling. In Semaphorins: receptor and intracellular signaling mechanisms, Pasterkamp RJ (eds), pp 1–11. New York, NY: Springer; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta Ghosh A, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW (2011) DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol 194: 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, Park KJ, Hong EJ, Kramer BM, Greenberg ME, Kaplan DR, Miller FD (2008) Developmental axon pruning mediated by BDNF‐p75NTR‐dependent axon degeneration. Nat Neurosci 11: 649–658 [DOI] [PubMed] [Google Scholar]
- Siu M, Sengupta Ghosh A, Lewcock JW (2018) Dual leucine zipper kinase inhibitors for the treatment of neurodegeneration. J Med Chem 61: 8078–8087 [DOI] [PubMed] [Google Scholar]
- Söderberg O, Leuchowius K‐J, Gullberg M, Jarvius M, Weibrecht I, Larsson L‐G, Landegren U (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45: 227–232 [DOI] [PubMed] [Google Scholar]
- Song W, Volosin M, Cragnolini AB, Hempstead BL, Friedman WJ (2010) ProNGF induces PTEN via p75NTR to suppress Trk‐mediated survival signaling in brain neurons. J Neurosci 30: 15608–15615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg KM, Yu B, Koboldt DC, Mardis ER, Pamphlett R (2015) Exome sequencing of case‐unaffected‐parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep 5: 9124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank EM, Figueroa‐Romero C, Hinder LM, Bedi K, Archbold HC, Li X, Weskamp K, Safren N, Paez‐Colasante X, Pacut Cet al (2018) Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun 9: 2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenzio M, Schiavo G, Fainzilber M (2017) Compartmentalized signaling in neurons: from cell biology to neuroscience. Neuron 96: 667–679 [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR (2012) Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7: e34640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner AB, Abdesselem H, Dickendesher TL, Imai F, Yoshida Y, Giger RJ, Pierchala BA (2016) Semaphorin 3A is a retrograde cell death signal in developing sympathetic neurons. Development 143: 1560–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Hengst U, Cox LJ, Macosko EZ, Jeromin A, Urquhart ER, Jaffrey SR (2005) Local translation of RhoA regulates growth cone collapse. Nature 436: 1020–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N, Goshima Y (2012) Collapsin response mediator proteins regulate neuronal development and plasticity by switching their phosphorylation status. Mol Neurobiol 45: 234–246 [DOI] [PubMed] [Google Scholar]
- Zahavi EE, Ionescu A, Gluska S, Gradus T, Ben‐Yaakov K, Perlson E (2015) A compartmentalized microfluidic neuromuscular co‐culture system reveals spatial aspects of GDNF functions. J Cell Sci 128: 1241–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahavi EE, Maimon R, Perlson E (2017) Spatial‐specific functions in retrograde neuronal signalling. Traffic 18: 415–424 [DOI] [PubMed] [Google Scholar]
- Ziak J, Weissova R, Jeřábková K, Janikova M, Maimon R, Petrasek T, Pukajova B, Kleisnerova M, Wang M, Brill MSet al (2020) CRMP 2 mediates Sema3F‐dependent axon pruning and dendritic spine remodeling. EMBO Rep 21: e48512 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Source Data for Figures
Data Availability Statement
This study includes no data deposited in external repositories. All data generated or analyzed during this study are included in this published article.