

SUMMARY
Emerging single-cell epigenomic assays are used to investigate the heterogeneity of chromatin activity and its function. However, identifying cells with distinct regulatory elements and clearly visualizing their relationships remains challenging. To this end, we introduce TooManyPeaks to address the need for the simultaneous study of chromatin state heterogeneity in both rare and abundant subpopulations. Our analyses of existing data from three widely used single-cell assays for transposase-accessible chromatin using sequencing (scATAC-seq) show the superior performance of TooManyPeaks in delineating and visualizing pure clusters of rare and abundant subpopulations. Furthermore, the application of TooManyPeaks to new scATAC-seq data from drug-naive and drug-resistant leukemic T cells clearly visualizes relationships among these cells and stratifies a rare “resistant-like” drug-naive sub-clone with distinct cis-regulatory elements.
Graphical Abstract
In brief
Schwartz et al. present TooManyPeaks, a suite of algorithms to explore and visualize heterogeneity of regulatory elements by using single-cell ATAC-seq data. Using TooManyPeaks’s functionalities, they find evidence that heterogeneity of the chromatin accessibility state contributes to the propensity of Notch-mutated T leukemic cells to develop resistance to Notch inhibitors.
INTRODUCTION
Cell-type-specific transcriptional diversity is largely set by the interactions between transcription factors and their cognate cis-regulatory elements within accessible chromatin regions. The emergence of single-cell/single-nucleus assays for transposase-accessible chromatin using sequencing (here, collectively called scATAC-seq) has enabled profiling of accessible cis-regulatory elements (here, interchangeably referred to as the epigenome) for thousands of individual cells. Unique characteristics of scATAC-seq readouts coupled with the increase in data volume have created a need for efficient computational tools for identifying and visualizing cells with similar chromatin accessibility, including rare populations. Although some scATAC-seq data analysis methods have been proposed, it still remains challenging to simultaneously identify and visualize rare and abundant subpopulations with distinct chromatin structures. To address this need, we introduce TooManyPeaks, which is equipped with several functionalities and provides a standalone end-to-end solution for scATAC-seq analysis. We assessed the accuracy and efficiency of TooManyPeaks in identifying and visualizing both rare and abundant populations by using several benchmarks. Given the key role of Notch signals in T cell acute lymphoblastic leukemia (T-ALL), we also used TooManyPeaks to investigate how heterogeneity of cis-regulatory elements influences divergent responses to Notch antagonist gamma-secretase inhibitor (GSI) in T-ALL. TooManyPeaks is open source and available through https://github.com/faryabib/too-many-cells#too-many-peaks.
RESULTS
TooManyPeaks relates cells with distinct chromatin states
To identify and visualize cell subpopulations with distinct cis-regulatory elements from scATAC-seq data, we introduce TooManyPeaks (Figure 1A). TooManyPeaks provides an end-to-end solution for scATAC-seq data analysis from chromatin accessibility readouts to multi-scalar renderings of cell group relationships and is integrated into the TooManyCells suite (Schwartz et al., 2020), a platform originally built for single-cell RNA-seq (scRNA-seq) data analysis. To this end, TooManyPeaks implements a number of graph-based algorithms to extract distinct cis-regulatory elements of both rare and abundant subpopulations and creates cell clade relationships from scATAC-seq data (Figure 1A; see STAR Methods). These cell clades are represented by a nested cluster structure in which relationships among the groups are maintained. In contrast to single-resolution clustering algorithms commonly used for scATAC-seq analysis (Li et al., 2020; Pliner et al., 2018; Bravo González-Blas et al., 2019; Cusanovich et al., 2018; Danese et al., 2019; Stuart et al., 2020; Fang et al., 2021), each inner node of the TooManyPeaks output is a cluster at a given resolution and a leaf node is a finer-grain cluster for which any additional partitioning would be as informative as randomly separating the cells (see STAR Methods).
Figure 1. TooManyPeaks overview and performance comparison.
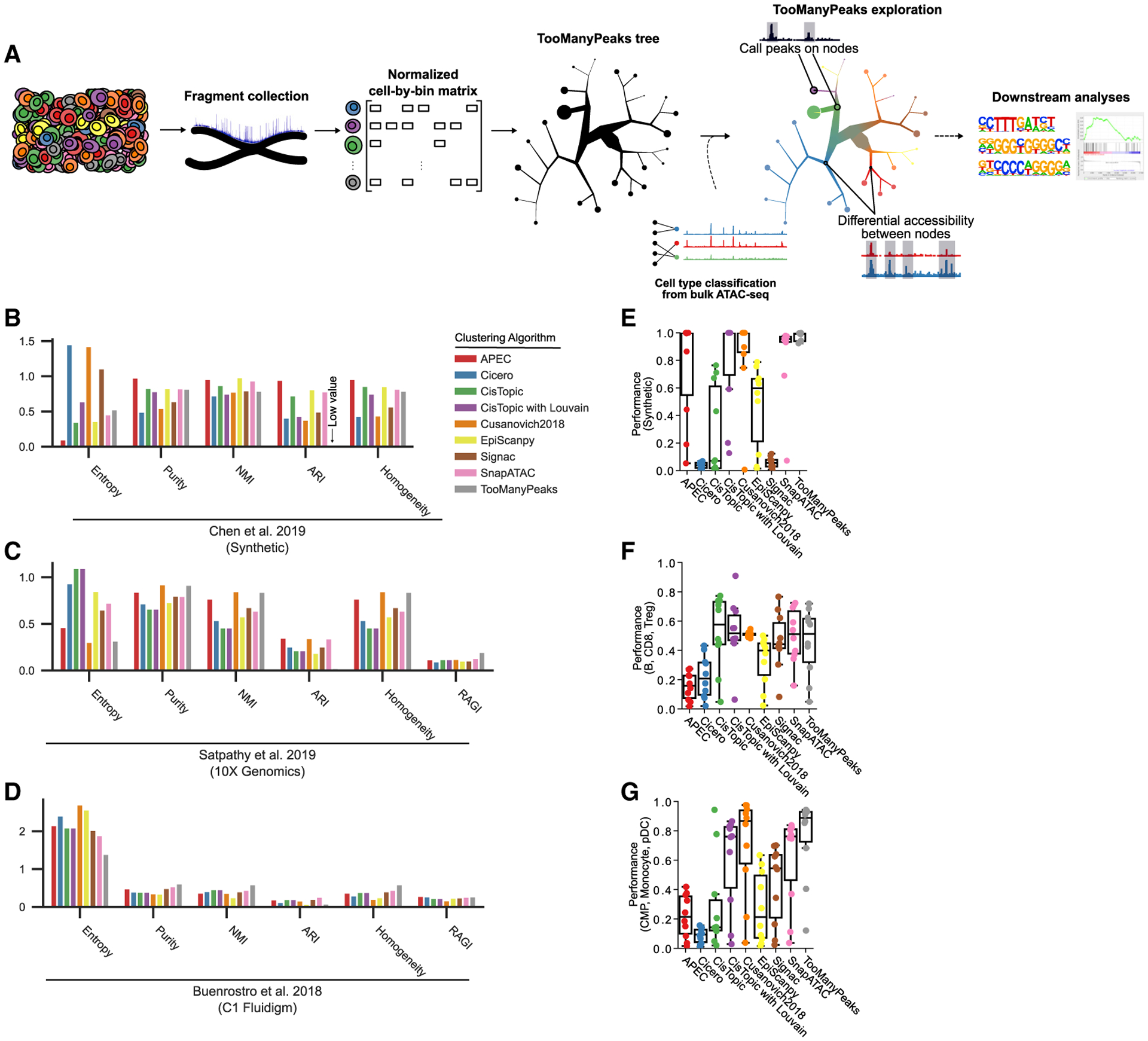
(A) Graphical representation of the TooManyPeaks algorithm. Following the arrows from left to right, TooManyPeaks converts scATAC-seq data to a cell-by-bin matrix, binarizes each value (accessible or inaccessible), and identifies and visualizes cell clade relationships by using matrix-free divisive hierarchical spectral clustering (see STAR Methods). TooManyPeaks trees are interpreted by following the cell groups from the root (the largest inner node) to the leaves. A leaf node here is shown as a pie chart of its cell composition. The sizes of a leaf and branches are proportional to the number of cells in the node. TooManyPeaks may then perform several downstream analyses.
(B–D) Clustering benchmarks with, from left to right, lower entropy, higher purity, higher normalized mutual information (NMI), higher adjusted Rand index (ARI), higher homogeneity, and higher residual average Gini index (RAGI; not applicable to synthetic data) representing more accurate clustering of simulated bone marrow cells with a moderate noise level of 0.2 (Chen et al., 2019) (B), CD34+ hematopoietic progenitor cells profiled using 10x Genomics (n = 7,771 cells) (Satpathy et al., 2019) (C), or Fluidigm C1 (n = 2,954 cells) (Buenrostro et al., 2018) (D).
(E–G) Detection of cells from two “rare” populations mixed with a “common” population was benchmarked. Box-and-whisker plots quantifying the accuracy of rare population detection in controlled admixtures from various datasets (m = 10 admixtures), as follows: n = 1,000 synthetic cells generated by simATAC (Navidi et al., 2021) (E); n = 1,000 B (common), CD8+ T (“rare1”) and Treg cells (“rare2”) (Satpathy et al., 2019) (F); and n = 500 common myeloid progenitors (CMPs) (common), monocytes (rare1), and plasmacytoid dendritic cells (pDC) (rare2) (Buenrostro et al., 2018) (G). Each point represents the average performance of 10 experiments from an admixture (100 admixtures overall). Performance indicates (true rare pairs (cells from the same rare population in the same cluster)/total rare pairs (true rare pairs and cells from different rare populations)). Box-and-whisker plots represent the following: center line, median; box limits, upper (75th) and lower (25th) percentiles; whiskers, 1.5 × interquartile range; points, outliers. See also Figure S1.
The TooManyPeaks tree-based visualization offers several advantages over “flat” two-dimensional portrayals of data provided by projection-based methods such as t-distributed stochastic neighbor embedding (t-SNE) and uniform manifold approximation and projection (UMAP) (van der Maaten and Hinton, 2008; McInnes et al., 2018). Although frequently used, projection-based methods generally do not report quantitative inter-cluster relationships and lack interpretable visualizations across clustering resolutions (Kobak and Linderman, 2021). To complement these existing single-resolution visualization methods and enable multi-resolution scATAC-seq data exploration, TooManyPeaks provides a fully customizable dendrogram for the visualization of inter-cluster relationships. To facilitate data exploration, we included many features in the TooManyPeaks visualization output including, but not limited to, branch scaling, weighted-average color blending, and statistically driven tree pruning. TooManyPeaks can also display outputs of other scATAC-seq clustering algorithms to quantify the relationships among their identified cell populations.
To enable an end-to-end built-in scATAC-seq analysis solution, TooManyPeaks provides several specialized and commonly used functionalities for scATAC-seq data analysis. For example, TooManyPeaks provides an algorithm for cell-type annotation based on input reference cis-regulatory elements of fluorescence-activated cell sorting (FACS)-purified cells. TooManyPeaks can also perform cluster- and cell-label-specific peak calling, as well as differential accessibility analyses across various clustering resolutions. Furthermore, TooManyPeaks enables several downstream analyses by generating normalized genome browser tracks and incorporating motif analysis methods (Heinz et al., 2010; Bailey et al., 2009) for each population. All TooManyPeaks functionalities can be readily set through its command-line interface (see STAR Methods).
TooManyPeaks accurately segregates and clearly visualizes rare cells
To assess the accuracy of cell clustering, we compared the outputs of TooManyPeaks and seven commonly used scATAC-seq clustering methods, as follows: APEC (Li et al., 2020), Cicero (Pliner et al., 2018), CisTopic (Bravo González-Blas et al., 2019), Cusanovich2018 (Cusanovich et al., 2018), EpiScanpy (Danese et al., 2019), Signac (Stuart et al., 2020), and SnapATAC (Fang et al., 2021). Importantly, these methods use different combinations of features for cell clustering. TooManyPeaks and Cusanovich2018 use latent semantic analysis (LSA) (Deerwester et al., 1990) for producing features in lower dimensional space, whereas CisTopic uses latent Dirichlet allocation (LDA) to identify “topics” as a form of feature definition (Falush et al., 2003). In contrast, Cicero and APEC summarize scATAC-seq signals into gene activity scores and “accessons,” respectively. As CisTopic recommends density peak clustering but other selected algorithms use Louvain clustering, we also included CisTopic topics as features for Louvain clustering (referred to as CisTopic with Louvain) in our comparative analysis (see STAR Methods).
To assess the performance of each method in identifying homogeneous cell label clusters, we used purity (Manning et al., 2008), entropy (Tan et al., 2019), mutual information (Kvålseth, 2017), adjusted rand index (ARI), homogeneity (Rosenberg and Hirschberg, 2007), and residual average Gini index (RAGI) (Chen et al., 2019). More homogeneous clusters result in higher purity, normalized mutual information (NMI), homogeneity, and RAGI, as well as lower entropy (see STAR Methods). We compared the ability of each algorithm to identify pure cell clusters of synthetic data (Chen et al., 2019; Figure 1B) and phenotypically defined cells within bone marrow and blood samples profiled using 10x Genomics (Satpathy et al., 2019; Figure 1C) or Fluidigm C1 (Buenrostro et al., 2018; Figure 1D) scATAC-seq platforms. We chose a 5-kb genomic bin size and 50 LSA dimension due to the low variability of performance across parameter choices (Figures S1A and S1B). As expected, TooManyPeaks resulted in low ARI, a measure that is sensitive to true label uncertainty and is biased against multi-resolution clustering methods (Chen et al., 2019). Nevertheless, TooManyPeaks, Cusanovich2018, and SnapATAC generated the purest clusters in both synthetic and complex real datasets included in this analysis (Figures 1B–1D). Together, these comprehensive analyses indicated the advantage of using TooManyPeaks for clustering individual cells based on their chromatin state, while maintaining their multi-scalar relationships in highly diverse hematopoietic cells.
Although some clustering methods provide resolution parameters to focus on small or large populations, concurrent identification and visualization of rare and abundant cells from scATAC-seq data remain challenging (Lancichinetti and Fortunato, 2011; Fang et al., 2021). Previous clustering benchmarks (Figures 1C and 1D) measured the diversity of cell labels within clusters, yet they did not directly quantify an algorithm’s ability to detect rare subpopulations with distinct regulatory elements. To rigorously assess the ability of various scATAC-seq clustering algorithms to simultaneously identify rare and abundant cells, we adapted our previous scRNA-seq rare population benchmark (Schwartz et al., 2020) to scATAC-seq. We used synthetic data (Navidi et al., 2021), 10x Genomics (Satpathy et al., 2019), and Fluidigm C1 (Buenrostro et al., 2018) scATAC-seq datasets and generated several controlled cell admixtures with various ratios of one “common” and two equally abundant “rare” populations. We then assessed how each algorithm separated the two rare populations from each other and from the common population in 10 controlled cell admixtures with various levels of rare populations. We found that TooManyPeaks outperformed all other tested algorithms or tied with SnapATAC in recovering rare populations (Figures 1E–1G).
Feature choice can significantly affect scATAC-seq analysis outputs. Given that several genomic elements could be involved in the regulation of a gene, scATAC-seq data have orders of magnitude more features than scRNA-seq and cannot necessarily be collapsed to the resolution of genes. As such, genomic bins or peaks, defined as equal-sized genomic windows or loci with enriched accessibility in pseudo-bulk ATAC-seq, respectively, are commonly used as scATAC-seq analysis features (Chen et al., 2019). Alternatively, some algorithms use topic and gene activity features. TooManyPeaks can readily compute on all four types of features (Figures S1C–S1H). More importantly, our data revealed that TooManyPeaks, Cusanovich2018, and SnapATAC, which are algorithms that use genomic bin features, show superior performance in detecting rare cells compared to algorithms using peaks or other features (Figures 1F and 1G). Timing benchmark on a set of 2,954 cells (Figure S1I) further showed that TooManyPeaks operates at a comparable rate or faster than other algorithms, even with its multi-resolution output.
TooManyPeaks classifies and relates cells from mouse bone marrow and spleen
Cell-type classification is one of the major applications of scATAC-seq analysis. To this end, we equipped TooManyPeaks with functionality to annotate individual cells based on input reference cis-regulatory element sets, including those from FACS-sorted bulk ATAC-seq data. Briefly, TooManyPeaks implements a fast bipartite-graph algorithm using cosine similarity to assign each cell to one of the reference cell types with a known cis-regulatory element repertoire (see STAR Methods and Figure 1A). To assess the efficacy of the TooManyPeaks cell-type classification, we annotated murine bone marrow and spleen cells (Cusanovich et al., 2018) based on reference cis-regulatory elements defined by bulk ATAC-seq analysis of 92 FACS-purified progenitor and differentiated hematopoietic cells (Yoshida et al., 2019). Visual inspection of the TooManyPeaks tree showed general separation of major phenotypically defined hematopoietic cell types (e.g., B cells segregate into a single branch) without or with modularity-guided pruning (Schwartz et al., 2020; Figures S2 and 2A).
Figure 2. Stratification and annotation of murine bone marrow and spleen cells.
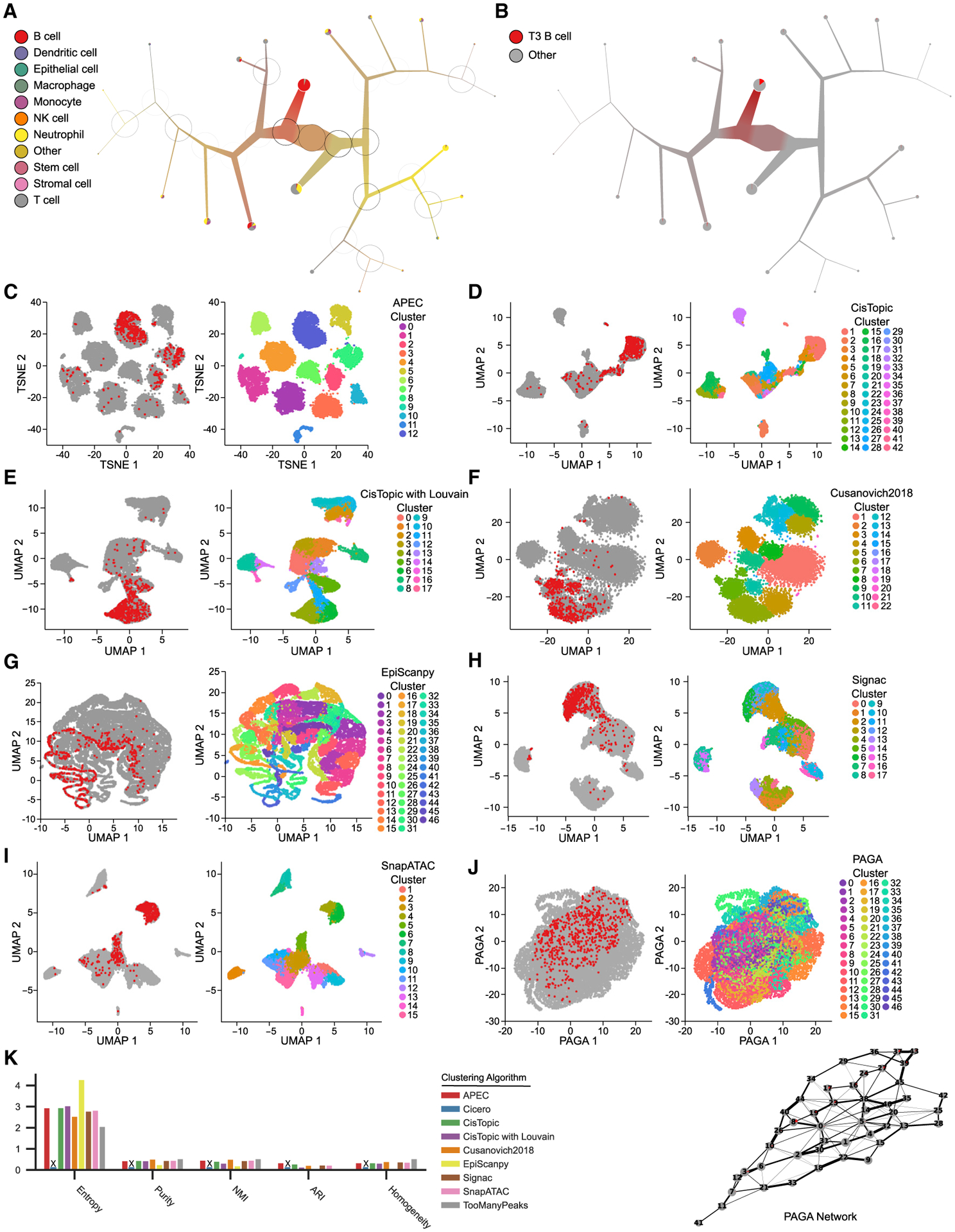
(A) The TooManyPeaks algorithm for cell-type annotation based on input reference cis-regulatory elements is used to predict the cell types in mouse bone marrow and spleen (n = 16,749 cells) (Cusanovich et al., 2018). Reference cis-regulatory elements of 92 phenotypically defined progenitor and differentiated hematopoietic cell types are generated from the analyses of bulk ATAC-seq in FACS-sorted cells (Yoshida et al., 2019). A TooManyPeaks tree pruned at median(modularity) + 15 × MAD (modularity) threshold shows major hematopoietic lineages. At each bipartitioning, a darker circle circumference represents higher modularity.
(B–J) TooManyPeaks tree (B) and UMAP outputs (C–J) colored by T3 B cells (red, left) or cluster label (right) generated by the noted algorithms.
(K) Clustering benchmarks with, from left to right, lower entropy, higher purity, higher NMI, higher ARI, and higher homogeneity showing more accurate clustering of phenotypically defined progenitor and differentiated hematopoietic cell types in mouse bone marrow and spleen by TooManyPeaks. An “X” marks algorithms that failed to complete. See also Figures S2, S3, and S4.
To further inspect the localization of more refined cellular sub-types, we next overlayed the positions of late transitional T3 B cells on the TooManyPeaks tree and projection outputs of all other algorithms included in this analysis including PAGA, which attempts to conserve and display global topology by using a network (Wolf et al., 2019; Figures 2B–2J). Interestingly, Cicero failed to complete the analysis of 16,749 cells. T3 B cells were mostly compartmentalized within a single TooManyPeaks tree branch (Figure 2B), whereas they were spread across the projection plots (Figures 2C–2J, left panels) and separated into multiple clusters (Figures 2C–2J, right panels) with the other algorithms. Notably, T3 B cells were spread out over 13 nodes of the PAGA network (Figure 2J). Furthermore, quantitative assessment of cell-type classification based on reference cis-regulatory elements of hematopoietic cells showed the improved performance of TooManyPeaks compared to all the other algorithms in accurately detecting (Figure 2K) and clearly visualizing (Figure S3) 92 distinct cell types in murine bone marrow and spleen.
Similar to the T3 B cell analysis of single-cell combinatorial indexing ATAC-seq (sciATAC-seq) data (Cusanovich et al., 2018; Figure 2), human hematopoietic stem cells (HSCs) profiled with the Fluidigm C1 platform (Buenrostro et al., 2018) were clearly distinguishable within the TooManyPeaks tree, but not in the projection plots of other algorithms, and the PAGA network (Figure S4).
TooManyPeaks determines the unique chromatin state of GSI-“resistant-like” drug-naive T-ALL cells
Notch mutations are observed in nearly 60% of patients with T-ALL and correlate with poor prognosis (Marks et al., 2009). These observations provide a compelling rationale for focusing on Notch signaling antagonists, such as GSI, as targeted therapies for Notch-mutated T-ALL. Nevertheless, progress toward targeted treatment of Notch-mutated T-ALL has been stymied partly due to a limited understanding of GSI-resistance acquisition. To investigate the underlying mechanisms of GSI resistance, we selected for GSI-resistant T-ALL cells by prolonged treatment of parental NOTCH1-mutated DND-41 cells with a high GSI dose (Schwartz et al., 2020). Given the genetic homogeneity of DND-41 cells and results of earlier studies showing the reversibility of the GSI resistance phenotype (Knoechel et al., 2014), we hypothesized that epigenetic differences contribute to the divergence of parental cells with resistant-like regulatory programs from non-resistant-like parental cells.
To test this hypothesis, we measured the accessibility of chromatin in 7,989 parental and GSI-resistant DND-41 cells. TooManyPeaks revealed that although parental cells are largely segregated from resistant cells, a rare resistant-like subpopulation of 144 parental cells had a chromatin state similar to that of the GSI-resistant cells (Figures 3A and S5A). Analyses with other selected tools (Figures S5B–S5E and S6A–S6E) showed resistant-like from non-resistant-like parental cells were separated partially. Nevertheless, the flat outputs of these algorithms generally obscured full separation of resistant-like cells from non-resistant-like parental cells. In contrast, the TooManyPeaks tree immediately rendered the relationship between resistant-like parental and resistant cells and clearly placed them within the resistant-cell-dominant subtree (Figure 3A).
Figure 3. TooManyPeaks identifies genomic elements specific to resistant-like parental T-ALL cells.
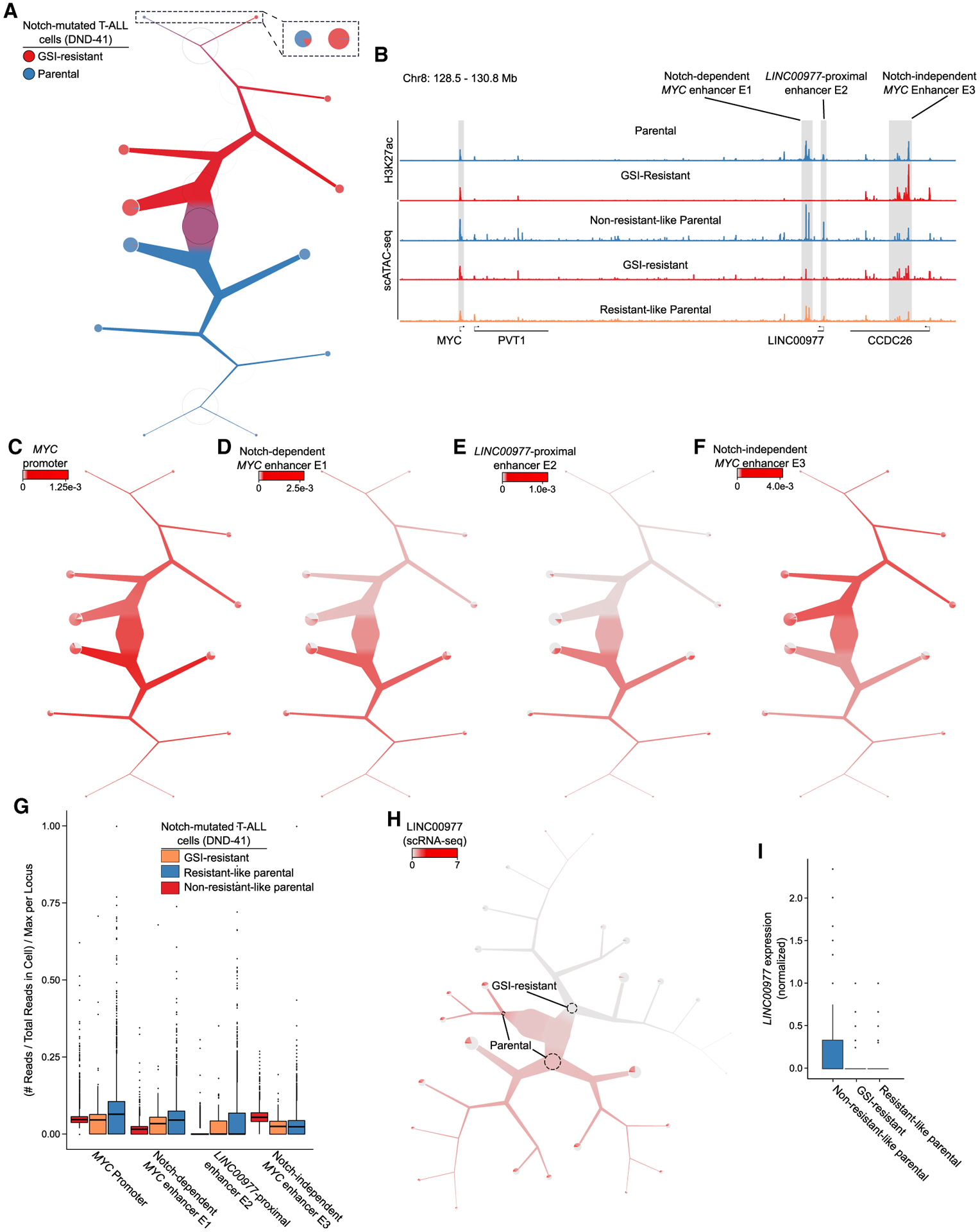
(A) TooManyPeaks tree of parental (n = 3,831 cells) and GSI-resistant (n = 4,158 cells) DND-41 T-ALL cells showing a resistant-like parental subpopulation of n = 144 cells.
(B) Genome tracks highlight key genomic elements at the MYC locus from 5′ to 3′, as follows: MYC promoter, Notch-dependent MYC enhancer E1, LINC00977-proximal enhancer E2, and Notch-independent MYC enhancer E3. The top two and bottom two tracks show H3K27ac and aggregated scATAC-seq of DND-41 populations in (A), respectively.
(C–F) TooManyPeaks tree as in (A) showing the accessibility of the MYC promotor (C) and enhancers E1 (D), E2 (E), and E3 (F).
(G) Box-and-whisker plot showing normalized accessibility at each locus in (B) for each population from (A).
(H) TooManyCells tree of gene expression showing elevated LINC00977 levels in the parental population (n = 7,371 cells).
(I) Box-and-whisker plot quantifying upper-quartile-normalized LINC00977 expression in each population from (H). See also Figures S5, S6, and S7 and Tables S1, S2, S3, S4, and S5.
To gain insights into transcriptional regulatory programs conferring resistance to GSI, we used TooManyPeaks to directly compare the chromatin accessibility of resistant-like and non-resistant-like parental cells. We identified 28,593 genomic elements with significantly higher accessibility in resistant-like cells (q < 0.05; see STAR Methods), which were collectively enriched with motifs associated with transcription factors with known functions in T cell development, transformation, and malignancies, such as GATA3, RUNX1, and MYC (Figure S7A; Table S1). Integration of scATAC-seq and scRNA-seq data (Table S1) further revealed that MYC had both significantly elevated expression (Figure S7B; Table S1) and higher accessible consensus binding sequences in the resistant-like parental cells (Figure S7C).
Guided by the differential activity of MYC in resistant-like cells, we used TooManyPeaks to map putative MYC regulatory elements in GSI-resistant and non-resistant-like parental cells. Concordant with transcriptional levels, the MYC promoter was active in both non-resistant-like parental and GSI-resistant cells (Figures 3B and 3C; Table S2). Our scATAC-seq data of non-resistant-like parental cells delineated clusters of accessible elements within ~2-Mb region 3′ of the MYC promoter (Figure 3B). Importantly, we observed marked differences in accessibility of three chromatin regions flanking the MYC promoter when comparing non-resistant-like parental and GSI-resistant cells (Figure 3B). Accessibility of genomic element E1 (~1.42-Mb 3′ of the MYC promoter), and E2 (~1.5-Mb 3′ of the MYC promoter and proximal to the long non-protein coding gene LINC00977) were significantly (p < 0.05 and q < 0.05) reduced in the GSI-resistant cells (Figures 3B, 3D, 3E, 3G, and S7D; Table S2; E1: log2FC = − 1.74 and E2: log2FC = − 2.78). In contrast, genomic element cluster E3 (~1.85-Mb 3′ of the MYC promoter) significantly gained accessibility in the GSI-resistant cells (Figures 3B, 3F, 3G, and S7D; Table S2; log2FC = 0.924). Together, this scATAC-seq analysis revealed significant chromatin restructuring of the MYC locus during GSI resistance development.
To further elucidate the function of differentially accessible elements at the MYC locus, we complemented our single-cell measurements with chromatin immunoprecipitation sequencing (ChIP-seq) analysis of enhancer histone mark H3K27ac. In concordance with the scATAC-seq data (Figures 3B and 3G), we recapitulated the loss of activity at E1 and E2 and gain of activity in E3 in GSI-resistant cells (Figure 3B). Interestingly, earlier studies showed that although genomic element E1 binds the Notch transcription complex and functions as a Notch-dependent MYC enhancer, E2 does not bind Notch and functions as a Notch-independent MYC enhancer (Yashiro-Ohtani et al., 2014; Herranz et al., 2014; Shi et al., 2013). Together, our bulk ChIP-seq analysis confirmed our scATAC-seq results and further showed differential activity of Notch-dependent and Notch-independent MYC distal enhancers E1 and E3, as well as uncharacterized LINC00977-proximal putative MYC enhancer E2, in GSI-sensitive and GSI-resistant DND-41 cells.
To more directly test whether chromatin accessibility differences in drug-naive cells contribute to the GSI-resistant phenotype, we next benefited from our scATAC-seq data to identify potential chromatin changes underpinning differential MYC expression in the resistant-like compared to non-resistant-like parental cells (Figure S7B). Notch-independent MYC enhancer E3 was similarly accessible in the resistant-like and non-resistant-like parental cells (Figures 3B and 3G; Table S3). Similarly, the accessibility of Notch-dependent MYC enhancer E1 was comparable in these two subpopulations of parental cells (Figures 3B and 3G; Table S3; log2FC = − 0.316, q = 2.65×10−3). In contrast, enhancer E2 accessibility was markedly different between these two parental subpopulations (Figures 3B and 3G). Similar to GSI-resistant cells, enhancer E2 was significantly less accessible in resistant-like than in non-resistant-like subpopulation of parental cells (Figures 3B and 3G; Table S3; log2FC = − 0.802, q = 0.0286). To assess if the loss of enhancer E2 accessibility may further affect LINC00977 expression, we used TooManyCells (Schwartz et al., 2020) to quantify LINC00977 transcript levels in 7,371 parental and resistant cells (Figures 3H and 3I). This scRNA-seq analysis revealed that LINC00977 expression was markedly lower in the GSI-resistant cells than in non-resistant-like parental cells (Figures 3H and 3I; Table S4; log2FC = − 1.84, q < 2.22×10−16). Notably, in concordance with enhancer E2 accessibility loss (Figures 3B and 3G), we also observed reduced LINC00977 expression in resistant-like compared to non-resistant-like parental cells (Figures 3H and 3I, and Table S1; log2FC = − 0.568, p = 0.117).
To further elucidate the underlying mechanisms of differential LINC00977-proximal enhancer E2 activity in the two parental sub-populations, we used motif search to explore transcription factors that potentially bound enhancer E2 in non-resistant-like but not resistant-like parental cells (Figure S7E; Table S5). These data revealed the presence of consensus binding motifs of TCF high-mobility group (HMG) family of proteins in the sequences of enhancer E2. Notably, scRNA-seq data showed significant downregulation of TCF-7, the gene encoding for T cell-lineage determinant factor TCF-1 (Johnson et al., 2018), in both GSI-resistant and resistant-like parental compared to non-resistant-like parental cells (Figures S7E and S7F; log2FC = − 2.63, q = 9.30×10−5). Together, these data suggest that in addition to MYC, differential activity of TCF-1 and its cognate regulatory elements such as LINC00977-proximal enhancer E2 may play a role in setting disparate epigenetic transcriptional regulatory programs in resistant-like and non-resistant-like parental sub-populations.
DISCUSSION
We developed TooManyPeaks, which provides complementary algorithms for clustering and visualizing scATAC-seq data. TooManyPeaks visualization and clustering are fundamentally different from projection-based visualization and single-resolution clustering. In addition to various visualization features, TooManyPeaks provides other capabilities including, but not limited to, flexible genomic feature options and cell type classification based on reference cis-regulatory elements. To enhance usability, TooManyPeaks is extensively documented (https://github.com/faryabib/too-many-cells#too-many-peaks) and is available as an easy-to-install standalone program through Nix or Docker.
Using the unique capabilities of TooManyPeaks, we identified a rare resistant-like population of Notch-mutated T-ALL DND-41 cells with chromatin accessibility more similar to GSI-resistant cells than non-resistant-like parental cells. Our new scATAC-seq data also suggested regulatory element markers of cells with a propensity for developing GSI resistance and signify potential transcription factor drivers of the resistance phenotype.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Robert B. Faryabi (faryabi@pennmedicine.upenn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
DND-41 T-ALL scATAC-seq and ChIP-seq data have been deposited at the Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3 acetyl-K27 | Active Motif | Cat# 39133; RRID:AB_2561016 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Protein G Agarose | Invitrogen | Cat# 15920-010 |
| Proteinase K | Invitrogen | Cat# 25530-049 |
| RNase A | Roche | Cat# 10109169001 |
| γ-Secretase Inhibitor XXI (compound E) | Calbiochem | Cat# 565790 |
| RPMI 1640 | Corning | Cat# 10-040-CM |
| HyClone Fetal bovine serum | Thermo Fisher Scientific | Cat# SH30070.03 |
| L-glutamine | Corning | Cat# 25-005-CI |
| Penicillin-Streptomycin | Corning | Cat# 30-002-CI |
| MEM Non-Essential Amino Acids | GIBCO | Cat# 11140-050 |
| Sodium Pyruvate | GIBCO | Cat# 11360-070 |
| Glycine | Invitrogen | Cat# 15527-013 |
| Pierce 16% Formaldehyde | Thermo Fisher Scientific | Cat# 28908 |
| Trizma Hydrochloride Solution, pH 7.4 | Sigma-Aldrich | Cat# T2194-100ml |
| Sodium Chloride Solution, 5M | Sigma-Aldrich | Cat# 59222C-500ml |
| Magnesium Chloride Solution, 1M | Sigma-Aldrich | Cat# M1028-100ml |
| Nonidet P40 Substitute | Sigma-Aldrich | Cat# 74385-5l |
| MACS BSA Stock Solution | Miltenyi Biotec | Cat# 130-091-376 |
| Flowmi Cell Strainer, 40 mm | Bel-Art | Cat# H13680-0040 |
| Digitonin | Thermo Fisher Scientific | Cat# BN2006 |
| Dulbecco’s Phosphate-Buffered Salt Solution 1X | Corning | Cat# 21031CV |
| Critical commercial assays | ||
| KAPA Library Quant Kit | Roche | Cat# KK4824 |
| D1000 ScreenTape | Agilent | Cat# 5067-5582 |
| D1000 Reagents | Agilent | Cat# 5067-5583 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067-5584 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067-5585 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| NEBNext Ultra II DNA Library Prep Kit | NEB | Cat# E7645S |
| Chromium Single Cell ATAC Library & Gel Bead Kit, 4 rxns | 10X GENOMICS | Cat# PN-1000111 |
| Chromium i7 Multiplex Kit N, Set A | 10X GENOMICS | Cat# PN-1000084 |
| Chromium Chip E Single Cell ATAC Kit, 48 rxns | 10X GENOMICS | Cat# PN-1000082 |
| NextSeq® 500/550 High Output Kit v2 (75 cycles) | Illumina | Cat# FC-404-2005 |
| NextSeq® 500/550 High Output Kit v2 (150 cycles) | Illumina | Cat# FC-404-2002 |
| Deposited data | ||
| Raw and analyzed scATAC-seq data | This paper | GEO: GSE155916 |
| Raw and analyzed ChIP-seq data | This paper | GEO: GSE171098 |
| Bulk ATAC-seq of purified progenitor and differentiated hematopoietic cells | Yoshida et al., 2019; https://doi.org/10.1016/j.cell.2018.12.036 | GEO: GSE100738 |
| 10x Genomics scATAC-seq of CD34\textsuperscript{+} hematopoietic progenitor cells | Satpathy et al., 2019; https://doi.org/10.1038/s41587-019-0206-z | GEO: GSE129785 |
| Fluidigm C1 scATAC-seq of CD34\textsuperscript{+} hematopoietic progenitor cells | Buenrostro et al., 2018; https://doi.org/10.1016/j.cell.2018.03.074 | GEO: GSE96769 |
| sciATAC-seq of murine marrow and spleen cells | Cusanovich et al., 2018; https://doi.org/10.1016/j.cell.2018.06.052 | GEO: GSE111586 |
| scRNA-seq of GSI-resistant DND-41 cells | Schwartz et al., 2020; https://doi.org/10.1038/s41592-020-0748-5 | GEO: GSE138892 |
| Experimental models: Cell lines | ||
| DND-41 | DSMZ | ACC 525 |
| Software and algorithms | ||
| APEC v1.2.2 | Li et al., 2020; https://doi.org/10.1186/s13059-020-02034-y | https://github.com/QuKunLab/APEC |
| Cicero v1.9.1 | Pliner et al., 2018; https://doi.org/10.1016/j.molcel.2018.06.044 | https://github.com/cole-trapnell-lab/cicero-release |
| CisTopic v0.3.0 | Bravo González-Blas et al., 2019; https://doi.org/10.1038/s41592-019-0367-1 | https://github.com/aertslab/cisTopic |
| Cusanovich2018 | Cusanovich et al., 2018; https://doi.org/10.1016/j.cell.2018.06.052 | This paper https://github.com/faryabib/CellReports_TooManyPeaks_analysis |
| EpiScanpy v0.3.0 | Danese et al., 2019; https://doi.org/10.1101/648097 | https://github.com/colomemaria/epiScanpy |
| Seurat v3.2.3 | Butler et al., 2018; https://doi.org/10.1038/nbt.4096 | https://github.com/satijalab/seurat |
| Signac v1.1.0 | Stuart et al., 2020; https://doi.org/10.1101/2020.11.09.373613 | https://github.com/timoast/signac |
| SnapATAC v1.0.0 | Fang et al., 2021; https://doi.org/10.1038/s41467-021-21583-9 | https://github.com/r3fang/SnapATAC |
| tsne v0.1.3 | van der Maaten and Hinton, 2008 | https://github.com/jdonaldson/rtsne/ |
| TooManyPeaks v2.2.0.0 | This paper https://doi.org/10.5281/zenodo.5130671 | https://github.com/faryabib/too-many-cells#too-many-peaks |
| TooManyPeaks analysis code | This paper https://doi.org/10.5281/zenodo.5130655 | https://github.com/faryabib/CellReports_TooManyPeaks_analysis |
| R wrapper for TooManyCells v0.1.1.0 | Schwartz et al., 2020; https://doi.org/10.1038/s41592-020-0748-5 | https://github.com/GregorySchwartz/tooManyCellsR |
| umap-learn v0.4.6 | McInnes et al., 2018; https://doi.org/10.21105/joss.00861 | https://github.com/lmcinnes/ |
| HOMER v4.9 | Heinz et al., 2010; https://doi.org/10.1016/j.molcel.2010.05.004 | http://homer.ucsd.edu/homer |
| bedtools v2.30.0 | Quinlan and Hall, 2010; https://doi.org/10.1093/bioinformatics/btq033 | http://bedtools.readthedocs.io/en/stable |
| BWAv0.7.13 | Li and Durbin, 2009; https://doi.org/10.1093/bioinformatics/btp324 | http://bio-bwa.sourceforge.net |
| Cell Ranger ATAC v1.2.0 | Satpathy et al., 2019; https://doi.org/10.1038/s41587-019-0206-z | https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/what-is-cell-ranger-atac |
| Picard v2.1.0 | Broad Institute | https://github.com/broadinstitute/picard |
| Trim Galore v0.4.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| UCSC tools v404 | Kent et al., 2010; https://doi.org/10.1093/bioinformatics/btq351 | https://github.com/ucscGenomeBrowser/kent |
In addition, Bulk ATAC-seq of purified progenitor and differentiated hematopoietic cells, 10x Genomics scATAC-seq, Fluidigm C1 scATAC-seq, sciATAC-seq, and GSI-resistant scRNA-seq data are existing, publicly available data. The accession numbers for these datasets are listed in the Key resources table.
All original software code has been deposited at https://github.com/faryabib/too-many-cells#too-many-peaks (source), https://hub.docker.com/repository/docker/gregoryschwartz/too-many-cells/ (Docker), https://cran.r-project.org/web/packages/TooManyCellsR (R wrapper), and https://github.com/faryabib/CellReports_TooManyPeaks_analysis (analysis code) and is publicly available as of the date of publication. DOIs are listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
GSI-resistant T-ALL cell culture
DND-41 cells (DSMZ, cat# ACC525) were purchased from the Leibniz-Institute DSMZ-German Collection of Microorganisms and Cell Lines. These male cells were cultured in RPMI 1,640 (Corning, cat# 10-040-CM) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, cat# SH30070.03), 2 mM L-glutamine (Corning, cat# 25-005-CI), 100 Ug/mL and 100 μg mL−1 penicillin/streptomycin (Corning, cat# 30-002-CI), 100 mM nonessential amino acids (GIBCO, cat# 11140-050), 1 mM sodium pyruvate (GIBCO, cat# 11360-070) and 0.1 mM of 2-mercaptoethanol (Sigma, cat# M6250). All cells were grown at 37°C and 5% CO2 with media refreshed every 3–4 days. Cells were regularly tested for mycoplasma contamination.
IC50 values for gamma-secretase inhibitor (GSI) compound E (Calbiochem, cat# 565790) were calculated from dose-response curves using CellTiter Glo Luminescent Cell Viability Assay (Promega, cat# G7571). Briefly, 1,000 treatment-naive DND-41 cells in 5 replicates/condition were plated in 96-well plates with vehicle or increasing concentrations of GSI (0.016, 0.031, 0.062, 0.125, 0.25, 0.5, 1, 2 μM). Luminescence was measured on day 7 with CellTiter Glo Luminescent Cell Viability Assay according to the manufacturer’s instructions. DND-41 IC50 of GSI was determined to be 5 nM.
To generate GSI-resistant cells, DND-41 treatment-naive cells were cultured in the presence of 125 nM GSI for at least six weeks. The establishment of GSI-resistance was determined with IC50 assay as described above. GSI-resistant DND-41 cells can tolerate 10 mM GSI with less than 20% cell death. Short-term DMSO treatment was performed on treatment-naive DND-41 cells with 125 nM DMSO for 24 hours.
METHOD DETAILS
GSI-resistant T-ALL single-cell ATAC-sequencing
We performed single-cell ATACseq for parental and GSI-resistant DND-41 cells following manufacture’s instructions for Chromium Single Cell ATAC Library & Gel Bead Kit and Chromium Chip E Single Cell ATAC Kit (10x Genomics). Briefly, we loaded cells onto independent channels of a Chromium Controller for targeted recovery of 4,000 cells per condition. We assessed libraries with Agilent TapeStation using High sensitivity D1000 chip and quantified using KAPA Library Quantification Kits for Illumina platform (KAPA Bio-systems, Roche, cat# KK4824). We performed paired-end sequencing on NextSeq 550 using 150 cycles High Output kit.
We performed FASTQ file generation and alignment to hg19 using Cell Ranger ATAC v1.2.0 (Satpathy et al., 2019) default arguments. We aggregated these cells using Cell Ranger. We sequenced parental and GSI-resistant cells at 253,594,800 and 252,343,254 read pair depth, respectively. In total, 8,041 cells passed the Cell Ranger QC and showed the typical “knee” plots indicating high quality from DMSO-treated parental (3,887) and GSI-resistant (4,154). We used sequence fragments of parental and resistant cells, with median of 13,238 and 33,523 per cell respectively, either directly as TooManyPeaks and SnapATAC inputs, or indirectly as Cicero, CisTopic, EpiScanpy, Cusanovich2018, APEC, Signac, and PAGA via pseudo-bulk ATAC-seq peak calling.
H3K27ac ChIP-seq
We performed H3K27ac ChIP-seq as previously described (Petrovic et al., 2019). Briefly, we sonicated and cleared chromatin samples prepared from 107 fixed cells with recombinant protein G–conjugated Agarose beads (Invitrogen, cat# 15920-010) and subsequently immunoprecipitated these cells with antibodies recognizing H3K27ac (Active Motif, cat# 39133). We captured Antibody-chromatin complexes with recombinant protein G–conjugated Agarose beads, washed them with Low Salt Wash Buffer, High Salt Wash Buffer, LiCl Wash Buffer and TE buffer with 50mM NaCl and eluted them. After reversal of cross-linking, we performed RNase and Proteinase K (Invitrogen, cat# 25530-049) treatment and purified DNA with QIAquick PCR Purification Kit (QIAGEN, cat# 28106). We then prepared libraries using the NEBNext Ultra II DNA library Prep Kit for Illumina (NEB, cat# E7645S). We validated indexed libraries for quality and size distribution using a TapeStation 2200 (Agilent). We performed paired-end sequencing (38 bp+38 bp) on a NextSeq 550.
Reads from H3K27ac ChIP-seq experiments were trimmed with Trim Galore (version 0.4.1, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with parameters -q 15–phred33–gzip–stringency 5 -e 0.1–length 20. Trimmed reads were aligned to the Ensembl GRCh37.75 primary assembly including chromosome 1–22, chrX, chrY, chrM and contigs using BWA (version 0.7.13) (Li and Durbin, 2009) with parameters bwa aln -q 5 -l 32 -k 2 -t 6 and paired-end reads were group with bwa sampe -P -o 1000000. Reads mapped to contigs, ENCODE blacklist and marked as duplicates by Picard (version 2.1.0, https://broadinstitute.github.io/picard/) were discarded and the remaining reads were used in downstream analyses and visualization. Bedgraph of reads normalized to reads per million (RPM) from ChIP-seq were generated with bedtools genomecov (Quinlan and Hall, 2010). Genome-wide uploadable bigWig files were generated with UCSC tools (version 329) (Kent et al., 2010) bedGraphToBigWig.
QUANTIFICATION AND STATISTICAL ANALYSIS
TooManyPeaks analysis of scATAC-seq data
TooManyPeaks is a collection of specialized functionalities and entry points for the parent suite TooManyCells, and extends TooManyCells to analyze chromatin accessibility of individual cells. TooManyPeaks provides many additional functionalities such as processing genomic region features (e.g., parsing regions for merging features) for sequence fragment file and / or peak matrix input, binning of regions, filtering out “black list” regions, and dimensionality reduction with LSA. In addition TooManyPeaks provides TooManyCells with several new entry points: peaks for peak finding with MACS2 (customizable with any other program), motifs for de novo or known motif search with MEME or HOMER (customizable with any other program), and classify for cell-type assignment from bulk ATAC-seq. While both MACS2 and MEME are included in the Nix derivation, any command line program can be integrated into the TooManyPeaks framework for use with these entry points.
Accessibility matrix
Given a sequence fragment file where each line consists of an initial three BED columns followed by cell barcode and duplicate count columns, TooManyPeaks initially generates an m×n matrix M of m observations (cells) and n genomic loci (equal-size genomic bins or pseudo-bulk ATAC-seq peaks) features, where M(i,j) is the number of counts for cell i at feature j. When starting from a fragments file, “black list” regions of the genome are known to have high signal (Amemiya et al., 2019). All analyses presented here that originate from a sequence fragment file filter out known “black list” regions with erroneously high signal (Amemiya et al., 2019), unless otherwise specified using–blacklist-regions-file. The width of genomic bin features across all the cells specified using–binwidth, which is set here to 5000 bp unless stated otherwise. By default, TooManyPeaks converts the matrix into a binary matrix to represent accessible or inaccessible sites.
Tree of single-cell clades
Potentially due to the “curse of dimensionality,” the large number of features in scATAC-seq data may result in every cell being an outlier, which in turn leads to low modularity in the initial bi-partitioning and stops the tree generation prematurely. To avoid this situation, we use latent semantic analysis for dimensionality reduction (here using 50 dimensions with–lsa) (Deerwester et al., 1990). TooManyPeaks passes this reduced feature space matrix to TooManyCells, which generates a tree of single-cell relationships based on the accessibility of their chromatins. Briefly, we generate a tree of cell clade relationships by recursively bi-partitioning the cells using an efficient matrix-free divisive hierarchical spectral clustering (Schwartz et al., 2020). To simultaneously detect large and small populations and avoid creating arbitrary small clusters, we use Newman-Girvan modularity (Newman and Girvan, 2004) as a stopping criterion for recursive cell bi-partitioning. The TooManyPeaks divisive hierarchical spectral clustering algorithm produces a nested cluster structure where relationships among the groups are maintained.
Peak calling and downstream analyses
Each node in the tree contains a collection of cells. The TooManyPeaks peaks entry point can be used to directly call peaks using MACS2 that is integrated into TooManyPeaks. TooManyPeaks calls peaks at each node specified (or all nodes) for downstream differential peak calculations. TooManyPeaks can be instructed with the–bedgraph option to generate bedGraph and bigWig files to visualize chromatin accessibility of each node on a genome browser. Given labels such as cell type or disease state, TooManyPeaks can also make tracks filtered for a label for a set of nodes. Transcription factor binding sequence motif search programs MEME and HOMER are integrated into TooManyPeaks and can be used to identify de novo motifs and search for known motifs for each node. TooManyPeaks motif analysis options can be controlled from the motifs entry point.
Classification based on reference elements
To assign cell types to individual cells in the TooManyPeaks tree, TooManyPeaks can use peaks from pseudo-bulk scATAC-seq or bulk ATAC-seq data from FACS-purified cells as reference cis-regulatory elements. Here, we annotated each murine bone marrow and spleen cells (Cusanovich et al., 2018) based on reference cis-regulatory elements. We generated reference cis-regulatory elements of 92 phenotypically defined FACS-sorted progenitor and differentiated hematopoietic cell types by analyzing their bulk ATAC-seq. To this end, fastq files for 186 samples were obtained from ImmGenn GSE100738 (Yoshida et al., 2019), and aligned to mm9 genome with BWA (version 0.7.13) (Li and Durbin, 2009) with parameters bwa aln -q 5 -l 32 -k 2 -t 6, after trimming with Trim Galore (version 0.4.1) with parameters -q 15–phred33–gzip–stringency 5 -e 0.1–length 20. Reads mapped to contigs, ENCODE blacklist, and marked as duplicates by Picard (version 2.1.0) were discarded and the remaining reads were used for peak calling and creating genome tracks.
Reproducible peaks in ATAC-seq replicates were identified following an implementation of ENCODE Irreproducible Discovery Rate (IDR) pipeline. Peaks in true replicates, pseudoreplicates, and pooled samples were identified using MACS (version 2.0.9) (Zhang et al., 2008) with parameters -p 1E-5 -g mm9–nomodel–format = BAM–bw = 300–keep-dup = 1. IDR cutoffs for true replicates, pseudoreplicates, and pooled samples were 0.05, 0.05 and 0.005 respectively. Replicates with Np/Nt, 2 and N1/N2, 2 were considered reproducible. The resulting ATAC-seq peaks were used as reference cis-regulatory elements of 92 phenotypically defined progenitor and differentiated hematopoietic cell types.
Given a set of observations (cells) O = {1…m}, a set of features (regions or genes) F = {1…n}, a set of reference regulatory elements (pseudo-bulk scATAC-seq or bulk ATAC-seq FACS-purified populations) R = 1…r, an m×n observation feature matrix M and a new r×n reference matrix R, TooManyPeaks first normalizes each row in M and R such that for some m×n matrix X,
| (Equation 1) |
where is the Euclidean norm of X row i. Then we can generate a new matrix S representing a bipartite graph of relationships between the observations in M and bulk populations in R with
| (Equation 2) |
where −1≤S(i,j)≤1 is the score (cosine similarity) of relatedness between observation 1≤i≤m to reference 1≤j≤r, with higher score indicating higher relatedness. Then the set of cell-type assignments A of length m by maximum score is defined by
| (Equation 3) |
Clustering benchmarks
We adapted the clustering benchmark for scRNA-seq as previously described (Schwartz et al., 2020) to scATAC-seq . Briefly, using TooManyPeaks, APEC (Li et al., 2020), Cicero (Pliner et al., 2018), CisTopic (Bravo González-Blas et al., 2019), CisTopic with Louvain, Cusanovich2018 (Cusanovich et al., 2018), EpiScanpy (Danese et al., 2019), Signac (Stuart et al., 2020), and SnapATAC (Fang et al., 2021), we clustered separately two datasets of phenotypically defined cells within bone marrow and blood samples profiled using 10x Genomics (Satpathy et al., 2019) (starting from a cell-by-peak file generated by TooManyPeaks to keep cells consistent) or Fluidigm C1 (Buenrostro et al., 2018) (starting from peaks as given in the dataset) scATAC-seq platforms. To increase the robustness of our benchmark, we additionally clustered a simulated bone marrow dataset with a moderate noise level of 0.2 (Chen et al., 2019). As Cicero and CisTopic focused on generating features, we also ensured a version of CisTopic using Louvain clustering from Signac instead of densityClust. We based this clustering benchmark on the assumption that similar cell types should cluster together. As such, we used purity (Manning et al., 2008), entropy (Tan et al., 2019), mutual information (Kvålseth, 2017), adjusted rand index (ARI), homogeneity (Rosenberg and Hirschberg, 2007), and residual average Gini index (RAGI) (Chen et al., 2019) to compare clustering performances between algorithms. In summary, entropy and homogeneity assess the extent of cell-type label diversity within clusters. Purity evaluates the extent of the dominant cell-type labels within the clusters. RAGI evaluates cluster-specificity of enrichment for marker accessible elements. Finally, NMI measures dependency of information of the cell-type labels given the cluster labels. RAGI requires gene activities as well as a list of known marker genes and housekeeping genes, so we used Cicero to generate the gene activity matrix and the gene lists originally given with RAGI’s introduction (Chen et al., 2019).
Purity is based on the frequency of the most abundant class (e.g., cell type) in a cluster. Let Ω = {ω1, ω2, …, ωK} be the set of clusters and be the set of classes. Then purity is defined as
where N is the total number of cells, ωk is the set of cells in cluster k, and cj is the set of cells in class j (Manning et al., 2008). This measure ranges from 0, poor clustering, to 1, perfect clustering.
Entropy as a measure of cluster accuracy uses Shannon entropy (Shannon, 1948) to measure the expected amount of information from the clusters. The entropy of each cluster k is defined by
where ωkj is the set of cells from ωk∩cj. Then the entropy for the entire clustering is (Tan et al., 2019)
Here, lower entropy of a clustering indicates higher accuracy.
Normalized mutual information (NMI) measures the normalized dependency of the class labels on the cluster labels, or the amount of information about the class labels gained when the cluster labels are given. Mutual information is defined by
To compare mutual information across clusterings, is normalized to the interval [0, 1]. As is bounded by where
is the entropy of Ω along with the analogous , total normalization NMI can be defined by
where higher values indicate more accurate clustering based on (Kvålseth, 2017).
Homogeneity makes the assumption that clusterings assign all members within a single cluster a single label. Therefore, the label distribution within a single cluster should result in zero entropy. Thus, the perfect case of homogeneity would be the Shannon entropy of . Then, instead of the raw entropy, homogeneity produces the normalized entropy by the maximum reduction in entropy from the clustering, namely . As 1 would be desirable as a maximum rather than 0, homogeneity is thus defined as (Rosenberg and Hirschberg, 2007)
where
Adjusted Rand Index (ARI) is calculated based on the number of pairings between two data clusterings, then adjusted for chance (Hubert and Arabie, 1985). Specifically, we first compute the Rand index
where TP and FP is the number of true or false positives respectively, while TN and FN is the number of true or false negatives respectively, based on cells in the clustering pairs. Then, we can define the adjustment for chance as (Hubert and Arabie, 1985)
For single-cell clustering accuracy, this measure requires a “ground truth” which was based on the given labels from each published dataset which defines coarse labels which some algorithms, such as TooManyPeaks, attempt to further delineate. As such, TooManyPeaks tends to perform poorly when using this type of measure with ambiguous “ground truth” clusterings.
Residual Average Gini Index (RAGI) is a recently proposed measure to define accuracy based on accessibility between known housekeeping genes and marker genes (Chen et al., 2019). This method first requires a gene activity matrix generated from the accessibility data, which we created using Cicero. Next, we calculate the mean accessibility values for all cells in each cluster. We use the Gini index on this vector of values based on either housekeeping genes or marker genes, both of which we used as previously reported (Chen et al., 2019). The Gini index (Gini, 1997) measures dispersion based on inequality among values in a distribution. Briefly, if xi is the mean accessibility of i of all cells in a cluster, then the Gini index of n accessibility sites would be
Then, we define the RAGI value as the difference in Gini index means between the housekeeping and marker genes for a clustering (Chen et al., 2019).
We used all algorithms with either default parameters as outlined in their function definitions or associated vignettes throughout the entire study which includes preprocessing with Seurat (Butler et al., 2018), with the exception of: knn = False in epi.pp.neighbors followed by episcanpy.tl.diffmap and another neighbor identification round for EpiScanpy and PAGA for visualizations to avoid low nearest neighbor errors as suggested by the Scanpy vignettes, and RunSVD with 3 dimensions to have fewer dimensions than topics in CisTopic with Louvain in the rare population benchmarks to also avoid an error. Furthermore, while the recommended latent semantic indexing (LSI, analogous to LSA) transformation through Signac was done to maintain a standard between Cicero and CisTopic with Louvain clusterings, topics were directly inputted into Signac UMAP for visualization of CisTopic with Louvain to avoid UMAP artifacts. All UMAP projections of the same data from different tools used the same seed.
Rare population benchmarks
We adapted the rare population benchmark for scRNA-seq as previously described to scATAC-seq (Schwartz et al., 2020). To this end, we generated ten random datasets each from two immune cell datasets using subsampling. The first set of ten samples included 1000 cells each with one common B cell population (ranging from 900 to 990 cells), one rare CD8+ T population (5 to 50 cells), and one rare T regulatory cell (Treg) population (5 to 50 cells) (starting from sequencing fragments or peaks, depending on which algorithm accepts which format as all cells were included) (Satpathy et al., 2019). The second set of ten samples included 500 cells each with one frequent common myeloid progenitor population (400 to 450 cells), one rare monocyte population (5 to 25 cells), and one rare plasmacytoid dendritic cell population (5 to 25 cells), with fewer cells due to a smaller dataset (starting from peaks as given in the dataset) (Buenrostro et al., 2018). Additionally, we benchmarked on synthetic data generated using simATAC (Navidi et al., 2021), where each of the common and two rare populations were generated from different seeds.
To quantify these benchmarks, we calculated a contingency table of the fraction of pairwise labels. For all rare cell pairs, we called a true pair if the two cells were of the same cell type (e.g., a Treg with another Treg or a CD8+ T cell with another CD8+ T), while we assigned a false pair if the two cells were of different cell types (e.g., a Treg with a CD8+ T cell). Then, the measure for accuracy in this benchmark was the fraction of true pairs in all pairs.
Timing benchmark
We ran each algorithm three times on a dataset of 2,954 cells (Buenrostro et al., 2018) using a machine with Ubuntu 20.04, 512GiB Memory, Intel® Xeon® CPU E5–2670 v3 @ 2.30GHz, 2 physical processors 24 cores, and 48 threads.
T-ALL scATAC-seq statistical analyses
We used the Kruskal-Wallis test with the Benjamini–Hochberg method for multiple-hypothesis correction (Benjamini and Hochberg, 1995) for differential accessibility between populations normalized by total sequence fragment. For the differential expression analysis, we used edgeR (Robinson et al., 2010) for normalization and the Benjamini–Hochberg multiple-hypothesis correction with quasi-likelihood (QL) F-test p value.
T-ALL scATAC-seq motif analyses
We used HOMER findMotifsGenome.pl (Heinz et al., 2010) on the differential accessibility list of resistant-like / other parental cells, keeping peaks that were considered significant at q < 0.05. This process generated a list HOMER identified as known motifs differential between these subpopulations. To understand the ontology of these motifs, we then performed a Metascape (Zhou et al., 2019) analysis on the motifs significant at q < 0.05. To identify putative regulatory elements that correlated with differential expression, we intersected these found elements with the differential gene expression between resistant-like and non-resistant-like parental cells of this system (Schwartz et al., 2020).
In order to identify motifs for putative regulatory elements at the LINC00977 locus, we used FIMO (Bailey et al., 2009) on the LINC00977 peak using the JASPAR reference database (Sandelin et al., 2004). To identify regulatory elements correlating with differential gene expression as with the global analysis above, we intersected this candidate list with the differential gene expression between resistant-like and non-resistant-like parental cells (Schwartz et al., 2020).
Statistical parameter definitions
Definitions of statistical parameters such as n and box-plot notations are defined in their respective figure legends.
Supplementary Material
Highlights.
TooManyPeaks identifies genomic element heterogeneity from single-cell ATAC-seq
TooManyPeaks tree shows relationships among cells based on genomic elements
Genomic element heterogeneity contributes to leukemia drug resistance
Drug-naive leukemic cells exist with accessibility similar to that of resistant cells
ACKNOWLEDGMENTS
We thank Golnaz Vahedi and her laboratory for their support. This work was supported by R01-CA230800 and Susan G. Komen CCR185472448 (to R.B.F.), R01-CA215518 (to W.S.P.), and T32-CA009140 (to G.W.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109575.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol 57, 289–300. [Google Scholar]
- Bravo González-Blas C, Minnoye L, Papasokrati D, Aibar S, Hulselmans G, Christiaens V, Davie K, Wouters J, and Aerts S (2019). cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods 16, 397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 173, 1535–1548.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lareau C, Andreani T, Vinyard ME, Garcia SP, Clement K, Andrade-Navarro MA, Buenrostro JD, and Pinello L (2019). Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 20, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, et al. (2018). A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Richter ML, Fischer DS, Theis FJ, and Colomé-Tatché M (2019). EpiScanpy: Integrated single-cell epigenomic analysis. bioRxiv. 10.1101/648097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerwester S, Dumais ST, Furnas GW, Landauer TK, and Harshman R (1990). Indexing by latent semantic analysis. J. Am. Soc. Inf. Sci 41, 391–407. [Google Scholar]
- Falush D, Stephens M, and Pritchard JK (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, et al. (2021). Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun 12, 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gini C (1997). Concentration and dependency ratios. Riv. Polit. Econ 87, 769–792. [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navás D, Cordon-Cardo C, et al. (2014). A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med 20, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert L, and Arabie P (1985). Comparing partitions. J. Classif 2, 193–218. [Google Scholar]
- Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, Pear WS, Bhandoola A, Wherry EJ, and Vahedi G (2018). Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cells. Immunity 48, 243–257.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H, et al. (2014). An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet 46, 364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobak D, and Linderman GC (2021). Initialization is critical for preserving global data structure in both t-SNE and UMAP. Nat. Biotechnol 39, 156–157. [DOI] [PubMed] [Google Scholar]
- Kvålseth TO (2017). On normalized mutual information: Measure derivations and properties. Entropy (Basel) 19, 1–14. [Google Scholar]
- Lancichinetti A, and Fortunato S (2011). Limits of modularity maximization in community detection. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 84, 066122. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Li Y, Li K, Zhu L, Yu Q, Cai P, Fang J, Zhang W, Du P, Jiang C, et al. (2020). APEC: an accesson-based method for single-cell chromatin accessibility analysis. Genome Biol. 21, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CD, Raghavan P, and Schütze H (2008). Introduction to Information Retrieval (Cambridge University Press; ). [Google Scholar]
- Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, Ferrando A, Fielding AK, Goldstone AH, Ketterling RP, et al. (2009). T-cell acute lymphoblastic leukemia in adults: clinical features, immunopheno-type, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 114, 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). Umap: Uniform manifold approximation and projection for dimension reduction. arXiv, 1802.03426. https://arxiv.org/abs/1802.03426. [Google Scholar]
- Navidi Z, Zhang L, and Wang B (2021). simatac: a single-cell atac-seq simulation framework. bioRxiv. 10.1101/2020.08.14.251488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MEJ, and Girvan M (2004). Finding and evaluating community structure in networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 69, 026113. [DOI] [PubMed] [Google Scholar]
- Petrovic J, Zhou Y, Fasolino M, Goldman N, Schwartz GW, Mumbach MR, Nguyen SC, Rome KS, Sela Y, Zapataro Z, et al. (2019). Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol. Cell 73, 1174–1190.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, et al. (2018). Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71, 858–871.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg A, and Hirschberg J (2007). V-measure: A conditional entropy-based external cluster evaluation measure. In Proceedings of the 2007 Joint Conference on Empirical Methods in Natural Language Processing and Computational Natural Language Learning (EMNLP-CoNLL), Association for Computational Linguistics, pp. 410–420. [Google Scholar]
- Sandelin A, Alkema W, Engström P, Wasserman WW, and Lenhard B (2004). JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32, D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GW, Zhou Y, Petrovic J, Fasolino M, Xu L, Shaffer SM, Pear WS, Vahedi G, and Faryabi RB (2020). TooManyCells identifies and visualizes relationships of single-cell clades. Nat. Methods 17, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE (1948). A Mathematical Theory of Communication. Bell Syst. Tech. J 27, 623–656. [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe J-S, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. (2013). Role of swi/snf in acute leukemia maintenance and enhancer-mediated myc regulation. Genes Dev. 27, 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Srivastava A, Lareau C, and Satija R (2020). Multimodal single-cell chromatin analysis with signac. bioRxiv. 10.1101/2020.11.09.373613. [DOI] [Google Scholar]
- Tan P-N, Steinbach M, Karpatne A, and Kumar V (2019). Introduction to Data Mining, second edition (Pearson; ). [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing data using t-sne. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, and Theis FJ (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, Knoechel B, Lanauze C, Louis L, Forsyth KS, et al. (2014). Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 111, E4946–E4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, et al. ; Immuno-logical Genome Project (2019). The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DND-41 T-ALL scATAC-seq and ChIP-seq data have been deposited at the Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3 acetyl-K27 | Active Motif | Cat# 39133; RRID:AB_2561016 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Protein G Agarose | Invitrogen | Cat# 15920-010 |
| Proteinase K | Invitrogen | Cat# 25530-049 |
| RNase A | Roche | Cat# 10109169001 |
| γ-Secretase Inhibitor XXI (compound E) | Calbiochem | Cat# 565790 |
| RPMI 1640 | Corning | Cat# 10-040-CM |
| HyClone Fetal bovine serum | Thermo Fisher Scientific | Cat# SH30070.03 |
| L-glutamine | Corning | Cat# 25-005-CI |
| Penicillin-Streptomycin | Corning | Cat# 30-002-CI |
| MEM Non-Essential Amino Acids | GIBCO | Cat# 11140-050 |
| Sodium Pyruvate | GIBCO | Cat# 11360-070 |
| Glycine | Invitrogen | Cat# 15527-013 |
| Pierce 16% Formaldehyde | Thermo Fisher Scientific | Cat# 28908 |
| Trizma Hydrochloride Solution, pH 7.4 | Sigma-Aldrich | Cat# T2194-100ml |
| Sodium Chloride Solution, 5M | Sigma-Aldrich | Cat# 59222C-500ml |
| Magnesium Chloride Solution, 1M | Sigma-Aldrich | Cat# M1028-100ml |
| Nonidet P40 Substitute | Sigma-Aldrich | Cat# 74385-5l |
| MACS BSA Stock Solution | Miltenyi Biotec | Cat# 130-091-376 |
| Flowmi Cell Strainer, 40 mm | Bel-Art | Cat# H13680-0040 |
| Digitonin | Thermo Fisher Scientific | Cat# BN2006 |
| Dulbecco’s Phosphate-Buffered Salt Solution 1X | Corning | Cat# 21031CV |
| Critical commercial assays | ||
| KAPA Library Quant Kit | Roche | Cat# KK4824 |
| D1000 ScreenTape | Agilent | Cat# 5067-5582 |
| D1000 Reagents | Agilent | Cat# 5067-5583 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067-5584 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067-5585 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| NEBNext Ultra II DNA Library Prep Kit | NEB | Cat# E7645S |
| Chromium Single Cell ATAC Library & Gel Bead Kit, 4 rxns | 10X GENOMICS | Cat# PN-1000111 |
| Chromium i7 Multiplex Kit N, Set A | 10X GENOMICS | Cat# PN-1000084 |
| Chromium Chip E Single Cell ATAC Kit, 48 rxns | 10X GENOMICS | Cat# PN-1000082 |
| NextSeq® 500/550 High Output Kit v2 (75 cycles) | Illumina | Cat# FC-404-2005 |
| NextSeq® 500/550 High Output Kit v2 (150 cycles) | Illumina | Cat# FC-404-2002 |
| Deposited data | ||
| Raw and analyzed scATAC-seq data | This paper | GEO: GSE155916 |
| Raw and analyzed ChIP-seq data | This paper | GEO: GSE171098 |
| Bulk ATAC-seq of purified progenitor and differentiated hematopoietic cells | Yoshida et al., 2019; https://doi.org/10.1016/j.cell.2018.12.036 | GEO: GSE100738 |
| 10x Genomics scATAC-seq of CD34\textsuperscript{+} hematopoietic progenitor cells | Satpathy et al., 2019; https://doi.org/10.1038/s41587-019-0206-z | GEO: GSE129785 |
| Fluidigm C1 scATAC-seq of CD34\textsuperscript{+} hematopoietic progenitor cells | Buenrostro et al., 2018; https://doi.org/10.1016/j.cell.2018.03.074 | GEO: GSE96769 |
| sciATAC-seq of murine marrow and spleen cells | Cusanovich et al., 2018; https://doi.org/10.1016/j.cell.2018.06.052 | GEO: GSE111586 |
| scRNA-seq of GSI-resistant DND-41 cells | Schwartz et al., 2020; https://doi.org/10.1038/s41592-020-0748-5 | GEO: GSE138892 |
| Experimental models: Cell lines | ||
| DND-41 | DSMZ | ACC 525 |
| Software and algorithms | ||
| APEC v1.2.2 | Li et al., 2020; https://doi.org/10.1186/s13059-020-02034-y | https://github.com/QuKunLab/APEC |
| Cicero v1.9.1 | Pliner et al., 2018; https://doi.org/10.1016/j.molcel.2018.06.044 | https://github.com/cole-trapnell-lab/cicero-release |
| CisTopic v0.3.0 | Bravo González-Blas et al., 2019; https://doi.org/10.1038/s41592-019-0367-1 | https://github.com/aertslab/cisTopic |
| Cusanovich2018 | Cusanovich et al., 2018; https://doi.org/10.1016/j.cell.2018.06.052 | This paper https://github.com/faryabib/CellReports_TooManyPeaks_analysis |
| EpiScanpy v0.3.0 | Danese et al., 2019; https://doi.org/10.1101/648097 | https://github.com/colomemaria/epiScanpy |
| Seurat v3.2.3 | Butler et al., 2018; https://doi.org/10.1038/nbt.4096 | https://github.com/satijalab/seurat |
| Signac v1.1.0 | Stuart et al., 2020; https://doi.org/10.1101/2020.11.09.373613 | https://github.com/timoast/signac |
| SnapATAC v1.0.0 | Fang et al., 2021; https://doi.org/10.1038/s41467-021-21583-9 | https://github.com/r3fang/SnapATAC |
| tsne v0.1.3 | van der Maaten and Hinton, 2008 | https://github.com/jdonaldson/rtsne/ |
| TooManyPeaks v2.2.0.0 | This paper https://doi.org/10.5281/zenodo.5130671 | https://github.com/faryabib/too-many-cells#too-many-peaks |
| TooManyPeaks analysis code | This paper https://doi.org/10.5281/zenodo.5130655 | https://github.com/faryabib/CellReports_TooManyPeaks_analysis |
| R wrapper for TooManyCells v0.1.1.0 | Schwartz et al., 2020; https://doi.org/10.1038/s41592-020-0748-5 | https://github.com/GregorySchwartz/tooManyCellsR |
| umap-learn v0.4.6 | McInnes et al., 2018; https://doi.org/10.21105/joss.00861 | https://github.com/lmcinnes/ |
| HOMER v4.9 | Heinz et al., 2010; https://doi.org/10.1016/j.molcel.2010.05.004 | http://homer.ucsd.edu/homer |
| bedtools v2.30.0 | Quinlan and Hall, 2010; https://doi.org/10.1093/bioinformatics/btq033 | http://bedtools.readthedocs.io/en/stable |
| BWAv0.7.13 | Li and Durbin, 2009; https://doi.org/10.1093/bioinformatics/btp324 | http://bio-bwa.sourceforge.net |
| Cell Ranger ATAC v1.2.0 | Satpathy et al., 2019; https://doi.org/10.1038/s41587-019-0206-z | https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/what-is-cell-ranger-atac |
| Picard v2.1.0 | Broad Institute | https://github.com/broadinstitute/picard |
| Trim Galore v0.4.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| UCSC tools v404 | Kent et al., 2010; https://doi.org/10.1093/bioinformatics/btq351 | https://github.com/ucscGenomeBrowser/kent |
In addition, Bulk ATAC-seq of purified progenitor and differentiated hematopoietic cells, 10x Genomics scATAC-seq, Fluidigm C1 scATAC-seq, sciATAC-seq, and GSI-resistant scRNA-seq data are existing, publicly available data. The accession numbers for these datasets are listed in the Key resources table.
All original software code has been deposited at https://github.com/faryabib/too-many-cells#too-many-peaks (source), https://hub.docker.com/repository/docker/gregoryschwartz/too-many-cells/ (Docker), https://cran.r-project.org/web/packages/TooManyCellsR (R wrapper), and https://github.com/faryabib/CellReports_TooManyPeaks_analysis (analysis code) and is publicly available as of the date of publication. DOIs are listed in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.