

Abstract
The commensal microbiota has been implicated in the regulation of a diverse array of physiological processes, both within the gastrointestinal tract and at distant tissue sites. Cancer is no exception, and distinct aspects of the microbiota have been reported to have either pro- or anti-tumor effects. The functional role of the microbiota in regulating not only mucosal but also systemic immune responses has led to investigations into the impact on cancer immunotherapies, particularly with agents targeting the immunologic checkpoints PD-1 and CTLA-4. Microbial sequencing and reconstitution of germ-free mice have indicated both positive and negative regulatory bacteria likely exist, which either promote or interfere with immunotherapy efficacy. These collective findings have led to the development of clinical trials pursuing microbiome-based therapeutic interventions, with the hope of expanding immunotherapy efficacy. This review summarizes recent knowledge about the relationship between the host microbiota and cancer and anti-tumor immune response, with implications for cancer therapy.
Keywords: Cancer Immunotherapy, Checkpoint Blockade Therapy, Anti–PD-1, Gut Microbiome, Tumor Microbiome
Graphical Abstract
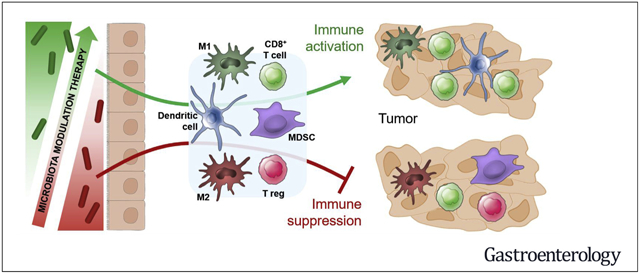
The surface barriers of the human body are inhabited by complex communities of bacteria, yeast, fungi, protozoa, archaea, and viruses. Collectively, these microorganisms make up the human microbiota, and the collection of genomes of these microorganisms make up the human microbiome. The functions encoded in the human microbiome play an important role in various aspects of human physiology. For instance, the gut microbiota plays a central role in regulating host metabolism, and also guides the proper development and function of the immune system.1–4
The bacterial component of the microbiome is the most abundant, and its deeper study in recent years has been enabled by advancements in next-generation DNA and RNA sequencing methods, improvements in bacterial culture techniques, and specialized animal models. The majority of gut bacteria are anaerobic, and improvements in the methods for anaerobic culture have made it feasible to isolate pure cultures of many species that were previously considered unculturable in laboratory conditions.5–9 This has allowed the wide use of gnotobiotic animals (usually mice) as models in microbiome research.10 These are animals harboring a defined microbiota and housed in isolation from environmental microbes. The term gnotobiotic also encompasses germ-free (GF, also called axenic) animals that are completely devoid of microorganisms. GF mice have severely underdeveloped immune system, but these defects are largely corrected by colonization with microbiota from conventional specific pathogen–free (SPF) mice, exemplifying the profound role of the microbiota in guiding immune system maturation.11 The use of gnotobiotic mice has enabled deeper studies of the mechanisms of interaction between specific microbes and both a developing cancer and the host immune system, which, in turn, can influence cancer response to therapy.10 The composition of the gut microbiome in healthy individuals gradually shifts throughout life, which implies that adults vary widely in the microbial communities with which they are colonized. This phenomenon brings into consideration a widely varying commensal microbiota composition as a potential source of phenotypic variation in disease development and/or therapeutic efficacy among individuals. In the cancer context, this variation has implications for carcinogenesis, response and toxicity to therapy, characteristics of anti-tumor immunity, and clinical response to immunotherapy.
Evidence Towards a Functional Role of the Gut Microbiota in Tumor Development, Anti-Tumor Immunity, and Response to Therapy
Local Effects of the Gut Microbiota on Tumor Development in Tissues of the Gastrointestinal Tract
Certain bacterial components of the gut microbiota can drive tumorigenesis in tissues of the gastrointestinal tract (Figure 1). For instance, experimental evidence supports a strong link between Helicobacter pylori and development of atrophic gastritis, metaplasia, dysplasia, and progression to gastric cancer.12,13 Its tumorigenic role is further supported by data showing that eradication of H pylori is an important method to reduce the risk of developing H pylori–driven gastric cancer, including a risk reduction of developing metachronous cancer in patients with endoscopically resected early gastric cancer.14,15 H pylori infection can contribute to development of gastric cancer by various proposed mechanisms, many of which involve release of virulence factors (eg, CagA and VacA) that cause endoplasmic reticulum stress, autophagy, and oxidative stress in gastric epithelium.16 Despite the well-accepted link between H pylori and gastric cancer, other studies have indicated an inverse correlation between H pylori and risk for development of esophageal adenocarcinoma, hypothesizing a protective role of this bacterium in certain contexts.17 Emerging data from high-throughput sequencing techniques have uncovered non–H pylori gastric microbial communities associated with gastric cancer.18–20
Figure 1.
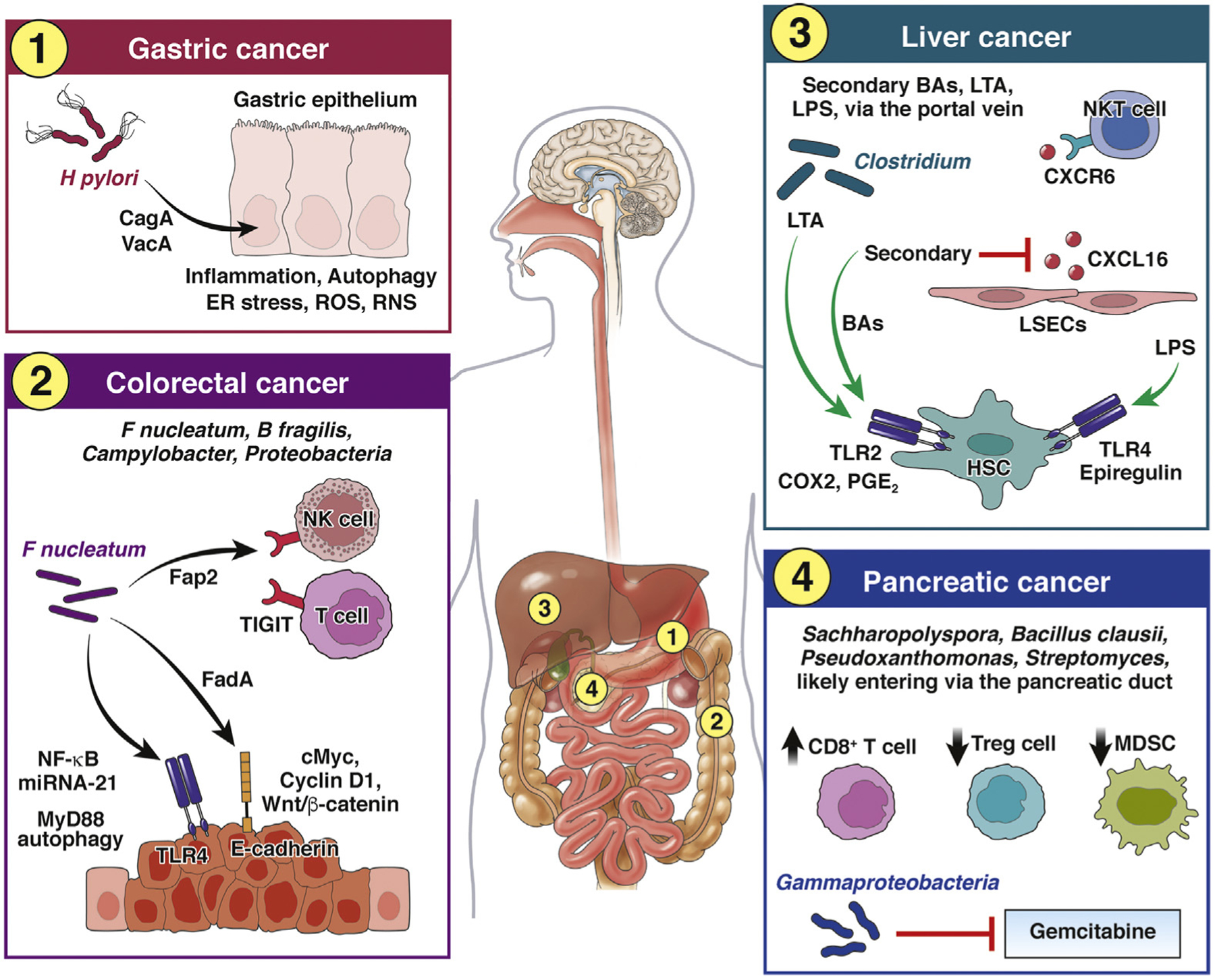
Local effects of the gut microbiota on tumor development. Gastric cancer: H pylori release virulence factors (eg, CagA, VacA) causing endoplasmic reticulum (ER) stress, autophagy, and oxidative stress in gastric epithelial cells, which collectively contribute to cancer development. RNS, reactive nitrogen species; ROS, reactive oxygen species. Colorectal cancer: F nucleatum contributes to tumor development and progression via several mechanisms. The virulence factor FadA can signal through E-cadherin and lead to an increase in annexin A1 expression, activation Wnt/β-catenin signaling, and up-regulation oncogenes c-Myc and cyclin D1. Signaling through TLR4 activates of NF-κB and up-regulation of microRNA-21 (miRNA-21), which also has oncogene functions. Signaling through TLR4 also induces MyD88-driven autophagy in cancer cells, thus aiding in chemoresistance. The outer membrane protein Fap2 binds to inhibitory TIGIT receptor on tumor-infiltrating NK cells and T cells, thus aiding in cancer immune evasion. Other gut bacteria, such as B fragilis, Campylobacter, and Proteobacteria, have also been associated with the development of CRC. Liver cancer: Gut bacterial metabolites (eg, secondary BAs) and microbe-associated molecular patterns, e.g., LTA and LPS) enter the liver via the hepatic portal vein and exert diverse effects on various cells in the liver that collectively contribute to cancer development and immune evasion. LPS-driven TLR4 signaling up-regulates the hepatomitogen epiregulin in hepatic stellate cells (HSCs), which can contribute to cancer development. Enrichment of Clostridium can result in accumulation of LTA and of the secondary BA deoxycholine, which collectively signal via TLR2 and up-regulate COX2, causing increase in prostaglandin E2 (PGE2), which in turn inhibits tumor cell killing by infiltrating CD8+ T cells, thus contributing to cancer immune evasion. Secondary BAs can also inhibit secretion of CXCL16 by liver sinusoidal endothelial cells (LSECs) required for recruitment of NKT cells, thus further contributing to cancer immune evasion. Pancreatic cancer: Gut bacteria found in pancreatic tumors, such as Bacillus clausii, Sachharopolyspora, Streptomyces, and Pseudoxanthomonas, are associated with improved antitumor immunity in pancreatic cancer, and Gammaproteobacteria contribute to resistance to gemcitabine chemotherapy by metabolizing the active form of the drug. The route of translocation from the gut to the pancreas is likely via the pancreatic duct, which opens into the duodenum. MDSC, myeloid-derived suppressor cells.
Another example of a bacterial driver of tumorigenesis in the gut is Fusobacterium nucleatum, which has been studied extensively in the setting of colorectal cancer (CRC). An investigation of 3 European cohorts found an increased level of this species in diseased tissue compared with matched normal tissue. In addition, enrichment of F nucleatum in CRC tumors was linked to the malignant transformation of adenomas to carcinomas.21 In the same study, patients with high levels of intratumoral F nucleatum showed decreased survival compared with those whose tumors had low levels (2 years vs 3 years, respectively). This is consistent with other studies correlating levels of F nucleatum DNA found in CRC tumors and survival.22,23 F nucleatum, and also certain strains of Bacteroides fragilis, have been mechanistically linked to the development of CRC by activating β-catenin signaling and by driving inflammatory responses.24–27 Intratumoral F nucleatum can also promote immune evasion of colon cancer due to its ability to bind inhibitory receptor TIGIT on human natural killer (NK) cells and T cells.28 F nucleatum has also been implicated in resistance to chemotherapy in the setting of CRC and its higher abundance was predictive of disease recurrence.29 In vitro co-culture experiments, xenograft CRC models in immunodeficient mice, and real-time polymerase chain reaction analyses of CRC biopsies from patients pointed toward a Toll-like receptor (TLR) 4/MyD88–driven activation of autophagy in tumor cells as a mechanism of Fusobacterium-mediated chemoresistance.29 Fusobacterium has also been found in tumors of the larynx, esophagus, pancreas, breast, and bladder, suggesting a possible role in modulating tumor progression and response to therapy in these cancers.30–34 However, the route of Fusobacterium colonization of tumors outside of the gastrointestinal tract and the possible contribution of the gut microbiota in this process are not well understood.
In some instances, enrichment of Fusobacterium in the gut microbiome or within tumor tissues has also been observed to be accompanied by increased abundance of Campylobacter.34–37 Some invasive Campylobacter species have been suggested to contribute to tumor progression by inducing a pro-inflammatory response driven by IL-18.36,38 Another bacteria-driven process promoting CRC progression is proposed to be the activation of senescence-associated secretory phenotype in malignant or premalignant epithelial cells by the genotoxic metabolite colibactin produced by some Proteobacteria, such as Escherichia coli.39 Senescence-associated secretory phenotype is associated with secretion of various growth factors, cytokines, chemokines, and enzymes. Some of these factors could be protective by contributing to immune-mediated tumor control, but prolonged senescence-associated secretory phenotype could cause immunosuppression.40 In addition, enzymes secreted by senescent tumor cells, such as matrix metalloproteinases, could promote tumor invasion and metastasis.40
Distant Effects of the Gut Microbiota on Tumor Development in Tissues Outside of the Gastrointestinal Tract
Translocation of gut bacteria to pancreatic tumors.
Bacteria from the gut can translocate to other closely associated tissues and affect tumor progression (Figure 1). For instance, bacterial translocation from the gut luminal compartment to the pancreas has been demonstrated by gavaging mice with fluorescently labeled commensal bacteria.41 Gammaproteobacteria have been suggested to translocate from the gut to pancreatic tumors, where they metabolized the active form of the drug gemcitabine, thus decreasing its efficacy.42 The most likely route for this translocation has been suggested to be via the pancreatic duct, which communicates with the duodenum.42
In a genetically engineered mouse model of pancreatic ductal adenocarcinoma (PDAC), the growth of Kras-driven tumors was accelerated by the presence of commensal microbiota, as evidenced by comparing SPF with GF animals, as well as bacterial ablation with oral antibiotics.41 Antibiotic treatment resulted in improved spontaneous anti-tumor immunity and also enabled the efficacy of anti–PD-1 therapy. Interestingly, in this genetic model, Kras expression in the pancreas seemed to result over time not only in the development of pancreatic tumors, but also in a divergence of gut microbiome composition compared with wild-type littermates. This microbiome alteration likely involved enrichment in tumor-promoting bacteria; recolonization of antibiotic-treated or GF mice before tumor onset with fecal microbiota from genetically engineered mice with late-stage tumors, but not from their tumor-free wild-type littermates, resulted in accelerated tumor progression in the recipient mice.41
Although these data demonstrated that a cross-talk with the gut microbiome modulated progression of pancreatic cancer in a preclinical model, another study by Riquelme et al43 found that the composition and diversity of the tumor microbiome were also associated with improved CD8+ T cell response and prolonged survival in patients with PDAC. Sequencing of 16S rDNA revealed that the tumor microbiome composition was distinct from that of adjacent healthy pancreatic tissue and only a fraction of the bacteria detected in the tumors were also detectable in the stool. Specifically, enrichment of Sachharopolyspora, Pseudoxanthomonas, Streptomyces, and Bacillus clausii within the tumor was strongly predictive of a prolonged survival. Building on their findings, the authors explored whether the gut microbiome could modify the intratumoral microbiome and tumor growth using fecal microbial transplantation (FMT) from long-term PDAC survivors (ie, tumors resected at least 5 years prior with no apparent disease at present), short-term PDAC survivors, and healthy controls into mice challenged with syngeneic tumors. Indeed, gut microbiota modification via FMT modulated the composition of the tumor microbiome, anti-tumor immune responses, and tumor growth kinetics in the recipient mice. Intratumoral bacterial composition in the recipient mice mimicked gut signatures from the 3 different patient groups. Mice that received FMT from long-term survivors showed increased tumor infiltration and activation of CD8+ T cells, higher levels of serum interferon (IFN) gamma and IL-2, and improved tumor control, and mice that received FMT from short-term survivors had increased tumor infiltration of regulatory T (Treg) cells and myeloid-derived suppressor cells and faster tumor growth.43
Together, these studies suggest a cross-talk among the gut microbiota, tumor microbiota, and tumor progression in the setting of pancreatic cancer. On the one hand, tumor progression might influence tumor and gut microbiota composition through mechanisms that remain to be determined. On the other hand, the gut microbiome could also shape the tumor microbiome and the tumor microenvironment, thus modulating the quality of anti-tumor immune responses. These findings open avenues for using the gut and PDAC microbiota composition as a biomarker predictive of survival or for selection of therapeutic approaches. In addition, modification of gut microbiota can be envisioned as a potential therapeutic approach to improve chemotherapy or immunotherapy efficacy in PDAC patients.
Effects of the gut microbiota on liver cancer.
Metabolites, pathogen-associated molecular patterns, and antigens derived from gut bacteria can be transported to the liver via the hepatic portal vein. Approximately 70% of the blood in the liver is contributed by the intestinal circulation. This organ is heavily populated by immune cells, which can make it an important site of interaction between gut microbiota– derived signals and the immune system. Among the gut bacteria–derived molecules that have been shown to either drive carcinogenesis or to suppress anti-tumor immunity in the liver are secondary bile acids (BAs); lipopolysaccharide (LPS), a component of the cell wall of gram-negative bacteria, which can signal via TLR4; and lipoteichoic acid (LTA), a component of gram-positive bacteria, which can signal via TLR2 (Figure 1).44–48
Primary BAs are synthesized from cholesterol in the liver, where they undergo conjugation to amino acids, such as taurine and glycine, and are secreted from the gall bladder into the duodenum to aid in lipid absorption. Gut bacteria in the distal small intestine and colon can metabolize BA conjugates to various secondary BAs and the composition of gut bacteria shapes the secondary BA profile in the host. Secondary BAs can be passively absorbed in the colon and returned to the liver.47 This recirculation of BAs from the intestines back to the liver allows for their reuse, but additionally, the secondary BAs can exert immunoregulatory functions. It has been suggested that primary and secondary BAs could, in some instances, carry out opposing functions within the host.44 The majority of secondary BAs are derived via a 7α-dehydroxylation reaction carried out by gram-positive Clostridium cluster XVI.47,49 Indeed, the production of secondary BAs, such as lithocholic acid or the murinespecific ω-muricholic acid, by Clostridium species has been shown to contribute to tumor progression in different mouse models of primary or metastatic liver cancer.44 The pro-tumor effect was carried out by secondary BA-driven down-regulation of CXCL16 in liver sinusoidal endothelial cells, which resulted in reduced recruitment of NK T cells to the liver.44 Depletion of Clostridium by oral vancomycin or dietary administration of primary BAs increased CXCL16 expression in liver sinusoidal endothelial cells and led to increased accumulation and IFN-γ production by NK T cells, increased numbers of CXCR6+CD62LlowCD44hi effector memory CD4+ and CD8+ T cells, and improved tumor control.44
In another study, certain Clostridium species became enriched in the murine gut in response to a high-fat diet, leading to overproduction of the secondary BA deoxycholic acid. Accumulation of deoxycholic acid and LTA in the liver accelerated the progression of carcinogen-induced liver cancer.45 The mechanism included a cooperative stimulation of the TLR2 pathway by LTA and deoxycholic acid, causing induction of senescence in hepatic stellate cells and overexpression of COX2.45 COX2 is the rate-limiting enzyme in the prostaglandin cascade and its accumulation resulted in overproduction of prostaglandin E2, which in turn augmented carcinogen-induced tumor development by inhibition of antitumor immunity. This effect was mediated by engagement of the prostaglandin E receptor 4 within the tumor microenvironment, which was expressed primarily by CD8+ T cells. Blockade of prostaglandin E receptor 4 resulted in increased infiltration of CD103+ dendritic cells and a decrease in regulatory CD4+FoxP3+ T cells. These effects were accompanied by an increase in the number of CD8+ T cells expressing the activation marker CD69, reduction of those expressing the inhibitory receptor PD-1, and ultimately improved tumor control.45
Another mechanism of microbiota-driven tumorigenesis in the liver has been shown to be mediated by LPS. TLR4 stimulation up-regulated the expression of the hepatomitogen epiregulin in stellate cells and had a pro-tumorigenic effect in a model of chronic injury-induced liver cancer.48
Systemic effects of the gut microbiota on cancer biology.
The gut microbiota can affect functions and processes in virtually all organs and systems in the host via a multitude of signaling mechanisms that are just beginning to be understood. For instance, the gut microbiota can regulate estrogen levels in the circulation, thereby affecting the risk for development of sex hormone–driven cancers, such as endometrial, ovarian, prostate, and breast cancer.50–52 Other products of bacterial metabolism that can access the host circulation and affect cancer progression include short-chain fatty acids (SCFAs), secondary BAs, cadaverine, and enterolignans.53,54
The gut microbiota is intimately linked with the central nervous system through a reciprocal circuitry called the gut–brain axis.4,55 Bacteria can signal to the central nervous system directly by producing a variety of hormones, but also indirectly through the activation of immune-mediated signals, such as inflammasome activation and induction of type I interferons. These signals constitute an important aspect of the gut–brain axis and have been implicated in the pathogenesis of various central nervous system disorders.4,55 A clear mechanistic link between gut microbiota and brain tumor development has not been established; however, it could be envisioned that neuroinflammation driven by the gut microbiota could be a contributing factor. Interestingly, it has been suggested that the gut–brain axis could also constitute a route through which the gut microbiota can regulate cancer progression systemically.56 This provocative idea is supported by a study demonstrating that activation of the reward system of the brain inhibited the growth of lung tumors in mice by alleviating the immunosuppressive phenotype of myeloid-derived suppressor cells.57 Although this study did not include investigating the effects of the gut microbiota on the elements of the reward system, other investigations have pointed toward such a link.58,59
Impact of the gut microbiota on anti-tumor immunity and immunotherapy efficacy.
Tumor development is intricately linked with the immune system60 and it follows that notable systemic effects of the gut microbiota on tumor progression and therapy would be mediated via modulation of the immune system. Indeed, preclinical and clinical evidence supports a role for gut bacteria in modulating the efficacy of chemotherapy and immunotherapy in various cancers.41,42,61–75 Initial studies in mouse models established a role of the gut microbiota in supporting the efficacy of CpG-oligonucleotide immunotherapy64 and of immune-stimulatory cyclophosphamide chemotherapy.62 An immunostimulatory role was further demonstrated for specific bacteria, such as Bifidobacterium and B fragilis, which were shown to augment the efficacy of checkpoint blockade immunotherapy (anti–PD-L1 and anti–CTLA-4, respectively) in mouse models.67,68 Specifically, oral administration of Bifidobacterium increased dendritic cell activation and improved the tumor-specific CD8+ T cell response,68 whereas B fragilis activated anti-tumor TH1 cells with suspected cross-reactivity to bacterial antigens and tumor neoantigens.67 Another mouse study using adoptive T cell therapy linked an increase in CD8α+ dendritic cells, up-regulation of IL-12, and improved anti-tumor immunity to a higher abundance of the Bacteroidales S24–7 family.76
These mouse studies motivated analogous pursuits for identifying mechanistic links between microbiota composition and anti–PD-1 immunotherapy efficacy in cancer patients.69–73 Analyses of fecal DNA by 16S and shotgun sequencing revealed differences in microbiota composition between responder and nonresponder patients, and reconstitution of GF mice with patient microbiota confirmed a mechanistic link between microbiota composition and anti-tumor immunity.71–73 Clinical studies have also confirmed an impact of the gut microbiota in patients after hematopoietic stem cell transplantation.77,78 A higher diversity in gut bacterial populations were associated with lower risk of transplantation-related death. Further analysis revealed that an abundance of Proteobacteria was associated with higher mortality, and abundance of Enterococcus was associated with higher risk of graft-vs-host disease.
A common concept in the majority of the studies mentioned is that the gut microbiota can exert systemic effects on the immune system that would shape therapeutic outcomes. Delineating the mechanisms of the gut microbiota–mediated immunomodulation is an active area of research and requires understanding of the pathways of communication between the gut microbiota and systemic regulation of the immune system (Figure 2).
Figure 2.
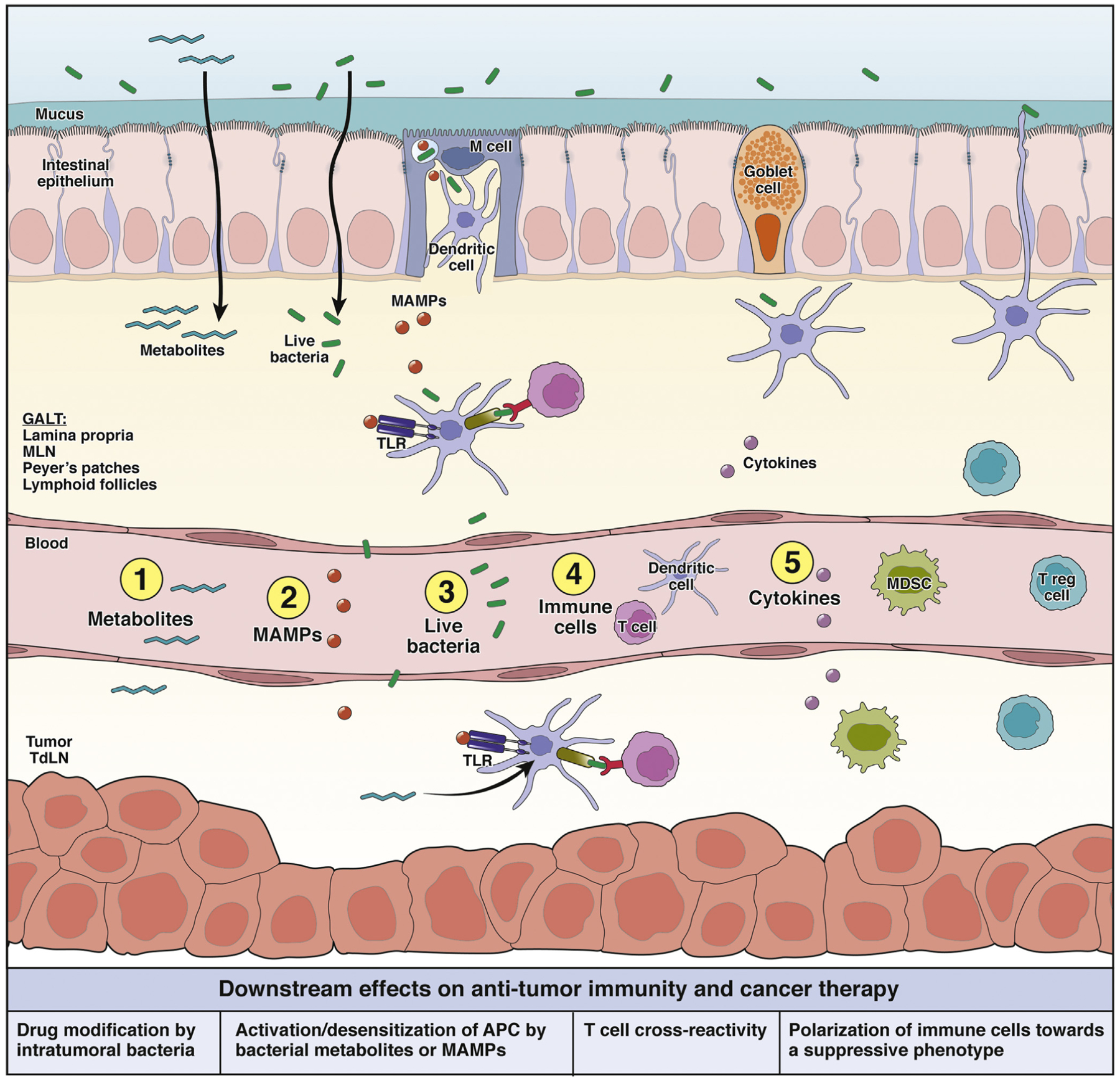
Systemic effects of the gut microbiota on antitumor immunity and immunotherapy. The gut microbiota could modulate immunotherapy outcomes by stimulating or inhibiting antitumor immunity. The messengers that could carry signals from the gut and/or gut-associated lymphoid tissues (GALT) to a distant tumor site include (1) bacterial metabolites that enter the circulation and regulate gene expression in various cells; (2) microbe-associated molecular patterns (MAMPs), which can modulate innate immunity by signaling through pattern recognition receptors, such as TLRs; (3) whole viable bacteria could potentially translocate and affect immune responses or drug activity in distant tumor tissues; (4) immune cells conditioned by sensing microbiota signals in the GALT could migrate and carry out immune-stimulatory or -inhibitory functions in distant tumors; and (5) cytokines could be released in the GALT in response to microbial stimuli and potentially enter the circulation and modulate downstream immune functions systemically. APC, antigen presenting cell; MDSC, myeloid-derived suppressor cell; MLN, mesenteric lymph node.
The gut microbiota and the host immune system constantly interact and influence each other in order to prevent infection and maintain immune homeostasis. Gut commensals can signal to immune cells within the gut-associated lymphoid tissue and mesenteric lymph nodes by engaging pattern recognition receptors, which can sense a wide variety of microbial components (microbe-associated molecular patterns), and also host-derived molecules from damaged cells (damage-associated molecular patterns).79 Pattern recognition receptors are a large group of receptors that are expressed on the cell surface or intracellularly, including TLRs, RIG-I–like receptors, NOD-like receptors, C-type lectin receptors, AIM2-like receptors.79–81 They can recognize microbe-associated molecular pattern signatures characteristic of bacteria, fungi, viruses, and protozoa and initiate inflammatory cytokine signaling aimed at recruitment of additional immune cell subsets and elimination of infection. However, prolonged pattern recognition receptor signaling can result in chronic inflammation and tissue damage that could promote tumor development.
Gut commensals are also a source of antigens that can be sampled by M cells and transported into Peyer’s patches, where they initiate antigen-specific B cell responses leading to the expansion of IgA-secreting plasma cells.82 Goblet cells, the primary function of which is mucus secretion, have also been shown to channel bacterial antigens from the lumen to antigen-presenting cells, such as dendritic cells and macrophages in the lamina propria.83 Dendritic cells can also directly sample bacterial antigens via transepithelial dendrites projecting into the intestinal lumen.84 Antigen-loaded antigen-presenting cells can then transport the antigens to lymphoid follicles within the lamina propria or to the mesenteric lymph nodes to activate antigen-specific T cell responses. Antigen-presenting cells under homeostatic conditions are capable of exiting the gut-associated lymphoid tissue and entering the circulation85 and, therefore, in theory, are capable of transporting bacterial antigens systemically. The significance of microbial antigens on tumor progression and therapy is exemplified in the context of cycloheximide chemotherapy, which can induce translocation of Enterococcus hirae from the gut to the mesenteric lymph nodes and spleen, which improved anti-tumor immune responses in mice.61,62 Recently, a follow-up study demonstrated that this antitumor effect could be mediated by a bacteriophage antigen carried by E hirae, which was cross-reactive to a tumor-expressed antigen.63 The concept of antigen cross-reactivity as a factor augmenting anti-tumor immunity could have wider implications beyond the context of cycloheximide therapy. Indeed, expression of tumor neoantigens homologous to infectious disease–derived peptides was one of the factors predictive of long-term survival of pancreatic cancer patients.86
The gut microbiota can also influence the immune system by releasing various metabolites that can enter the circulation. A prominent example is the effect of SCFAs, which are produced by fermentation of dietary fiber and can signal via G-protein–coupled receptors or act as histone deacetylase inhibitors and activate gene expression. SCFAs have versatile local effects on the differentiation and activation of anti-inflammatory Treg cells or pro-inflammatory TH1 and TH17 cells, differentiation and IgA secretion by plasma cells, and polarization of proinflammatory M1 vs anti-inflammatory M2 macrophages.87 Other examples of microbial metabolites with immunomodulatory roles include some bacterial amino acid derivatives and secondary BAs.
An active area of interest is the identification of bacterial factors that might be involved in modulating the clinical outcome of immune checkpoint blockade therapy. Recent work by McCoy and colleagues75 identified a role for the metabolite inosine in altering the effectiveness of immunotherapy in a mouse model of CRC by focusing on the bacterium Bifidobacterium pseudolongum. Inosine alone induced expression of TH1 regulating genes in CD4+ T cells, and the addition of immune checkpoint inhibitors caused a concomitant increase in IFN-γ production. Interestingly, in vivo anti-tumor activity of checkpoint blockade therapy and inosine required co-stimulation, delivered in this form by CpG administration, as well as IL-12 from dendritic cells. Another bacterium identified in their initial screen, Akkermansia muciniphila, produced the same metabolite, which had the same anti-tumor effect, possibly providing an explanation for the number of differing bacteria identified as having a positive effect on immunotherapy, that is, common immune-modulating metabolites.
It has been known that bacterial metabolites can alter the inflammatory landscape of the host in the absence of immunotherapy. SCFAs such as butyrate and propionate can induce peripheral Treg differentiation through epigenetic modification of the FoxP3 locus,88 and others have shown butyrate’s effect on increasing IFN-γ and granzyme B expression in CD8+ T cells,89 as well as inducing the transition of an effector to memory phenotype for CD8+ T cells after butyrate exposure.90 The differential effects of butyrate could be concentration-dependent, that is, lower levels induce differentiation of FoxP3 Treg cells, and higher levels increase CD8+ T cell effector functions and phenotypes. Indeed, a recent study of PD-1–responsive vs nonresponsive patients with solid tumors showed higher SCFA, including butyrate, in responsive patients.91
Therapeutic Strategies for Microbiome Modulation in the Treatment of Cancer
Largely based on the identified modulatory role of the gut microbiota on anti-tumor immunity and immunotherapy efficacy, clinical trials have been initiated that are attempting to manipulate the microbiota with therapeutic intent. The overall goal is to augment the effect of immune-potentiating bacteria, while decreasing the effects of immune-regulatory bacteria. Multiple studies have been initiated using either fecal microbial transplantation or defined bacterial isolates, and a major focus is on improving the efficacy of checkpoint blockade immunotherapy.
Fecal Microbiota Transplantation
One strategy for manipulating the gut microbiome involves FMT. This process, whereby fecal material from a given donor is transferred to a recipient, has been used to treat several clinical indications, including Clostridium difficile infection, ulcerative colitis, and other gastrointestinal conditions.92–98 FMT has been well studied in C difficile and is indicated for recurrent C difficile infections, having been shown to improve patient status and resolve clinical symptoms.99–102
More recently, FMT has begun to be investigated in combination with checkpoint blockade therapy. The rationale is based on preclinical studies in which FMT from human cancer patients into GF mice was able to recapitulate the responder/nonresponder phenotype. In the majority of cases, when GF mice were reconstituted using fecal material from patients who responded to anti–PD-1 therapy, the corresponding mice also were responders to PD-1 blockade. In contrast, GF mice reconstituted with nonresponder fecal material failed to respond to PD-1 blockade.71–73 This nonresponder phenotype in mice could be reversed by additional FMT from responder patients. Another clinical correlation is that anti–PD-1 efficacy appeared to be reduced in patients who received broad-spectrum antibiotics.71 With these observations as a foundation, clinical interventions to restore favorable microbiota are underway.
Several clinical studies, mostly in patients with metastatic melanoma, are underway evaluating the potential for FMT to enhance immune checkpoint blockade therapy (Table 1). Trials are also underway in prostate and gastrointestinal cancer and in mesothelioma. A general strategy in these clinical protocols is to identify candidate fecal donors among patients who had experienced a complete response to anti–PD-1, and who have stool samples and a microbial composition that pass defined quality control testing. Preliminary readouts from these trials have been described, and although the studies are early and with small sample sizes, the preliminary data are provocative. Early data from a trial at the University of Pittsburgh evaluating FMT in combination with pembrolizumab in melanoma patients refractory to anti–PD-1 treatment reported stable disease or tumor regression in 2 of 3 patients (ClinicalTrials.gov, number NCT 03341143).103 Another melanoma trial out of Sheba Medical Center in Israel, evaluating FMT from responder patients followed by anti–PD-1 treatment in patients already resistant to PD-1 inhibitor therapy, reported disease regression in 1 patient at the time of first scan, but progression on follow-up monitoring. A second patient had a marked decrease in disease burden (45%), with continued survival after 8 months. Tumor biopsies from these patients revealed an increase in immune cell infiltrate, commensurate with a change in their gut microbiome reflected by repeat bacterial sequencing (ClinicalTrials.gov, number NCT03353402).104
Table 1.
Ongoing Cancer Clinical Trials Investigating Gut Microbiome Interventions and Immunotherapy
| ClinicalTrials.gov no. | Phase | Cancer type | Intervention | Investigator | Status |
|---|---|---|---|---|---|
| FMT interventions | |||||
| NCT03772899 | I | Melanoma | FMT + immune checkpoint inhibitors | Lawson Health Research Institute | Recruiting |
| NCT03353402 | I | Melanoma | FMT (via colonoscopy and stool capsules) + immune checkpoint inhibitors | Sheba Medical Center | Recruiting |
| NCT03341143 | II | Melanoma | FMT (via colonoscopy) + immune checkpoint inhibitors | UPMC Hillman Cancer Center | Suspended due to COVID-19 |
| NCT04577729 | – | Melanoma | Autologous or allogeneic FMT + immune checkpoint inhibitors | Medical University of Graz | Not yet recruiting |
| NCT04521075 | I/II | Melanoma NSCLC | FMT (stool capsules) + immune checkpoint inhibitors | Sheba Medical Center | Not yet recruiting |
| NCT04056026 | I | Mesothelioma | FMT (via colonoscopy) + immune checkpoint inhibitors | ProgenaBiome | Completed |
| NCT04116775 | II | Prostate | FMT (via endoscopy) + immune checkpoint inhibitors + enzalutamide | VA Portland Health Care System | Recruiting |
| NCT04130763 | I | Gastrointestinal | FMT (stool capsules) + immune checkpoint inhibitors | Peking University | Recruiting |
| Non-FMT microbiome interventions | |||||
| NCT03686202 | I | Solid tumors | MET-4 (defined bacterial consortia) + immune checkpoint inhibitors | University Health Network, Toronto | Recruiting |
| NCT03817125 | I | Melanoma | SER-401 (defined bacterial consortia) + immune checkpoint inhibitors | Parker Institute for Cancer Immunotherapy | Recruiting |
| NCT03637803 | I/II | Solid tumors | MRx0518 (E. gallinarum capsule) + immune checkpoint inhibitors | 4D pharma plc | Recruiting |
| NCT03775850 | I/II | Solid tumors | EDP1503 (Bifidobacterium capsule) + immune checkpoint inhibitors | Evelo Biosciences | Recruiting |
| NCT03595683 | II | Melanoma | EDP1503 (Bifidobacterium capsule) + immune checkpoint inhibitors | Evelo Biosciences | Active, not recruiting |
| NCT03829111 | I | Renal cell carcinoma | CBM 588 (Clostridium butyricum probiotic) + immune checkpoint inhibitors | City of Hope Medical Center | Recruiting |
COVID-19, coronavirus 2019; NSCLC, non–small cell lung cancer; UPMC, University of Pittsburgh Medical Center; VA, Veterans Affair.
Although early results from FMT-treated patients receiving immunotherapy have shown promise, investigators need to be cautious when using FMT. Pathogenic bacteria, parasites, bacteriophage, and multidrug-resistant bacteria can be unwittingly transferred within the fecal material. Two patients receiving FMT outside of these studies, one for hepatic encephalopathy and the other for myelodysplastic syndrome, were reported to develop severe bacterial infections and died as a consequence of their treatment.105 This observation led to a safety bulletin issued by the US Food and Drug Administration warning of the risk of infections from FMT therapy.106 Future protocols using FMT would ideally disqualify donors harboring these pathogenic organisms. A recent study evaluating the stool of candidate FMT donors to determine their fitness for clinical use by several criteria, including detection of pathogens and antibiotic-resistant bacteria, suggest that only 3% of donors would pass such quality control assessments.107 An additional concern with FMT involves identification of which patients fail anti–PD-1 therapy specifically because of microbiota defects. Primary resistance to anti–PD-1 can be impacted not only by the composition of the gut microbiota, but also by the involvement of tumor cell–intrinsic oncogenic events that mediate immune cell exclusion and also by germline polymorphisms in immune regulatory genes that impact the magnitude of the endogenous immune response.108–115 In addition, a given FMT donor might have the necessary microbial composition to correct a microbiota defect in one patient but not another patient. As the biochemical and cellular mechanisms by which the gut microbiota regulate host anti-tumor immunity become better defined, and as the identity of functionally relevant bacteria (both positive and negative) becomes better understood, the ideal selection of FMT donor for a given patient should become facilitated.
Bacterial Isolates and Consortia
Additional therapeutic efforts are centered on the administration of defined bacterial species that have been selected based on experimental evidence for augmenting anti-tumor immunity and immunotherapy efficacy preclinically. Focusing on defined bacteria has the potential to mitigate the concerns of transferring pathogens using whole FMT, and also can serve to deliver a consistent therapeutic product with less variability compared with different donor gut microbiota. Based on preclinical evidence as well as clinical correlative data indicating that Bifidobacteria species have the potential to facilitate anti-tumor T cell responses and anti–PD-1 efficacy, a strain of Bifidobacterium animalis lactis has been developed for clinical testing (called EDP1503).116 When administered to mice, this strain acts at the level of the small intestine, does not have to colonize to have immune-potentiating activity, and induces a broad immune response that includes induction of inflammatory cytokines, T cell responses, and NK cell activation. Early data from a phase 1/2 study were recently reported in 29 patients with solid tumors, using a 2-week run-in of EDP1503 followed by combination treatment with pembrolizumab.116 A cohort of patients with triple negative breast cancer showed interesting preliminary results, with 2 responders seen among 6 evaluable patients, which is encouraging, given that response with single-agent anti–PD-1 therapy in this patient group ranges between 5% and 10%. Preliminary biomarker analysis after the 2-week course of EDP1503 revealed increases in CD8+ T cells, consistent with the expected immunologic mechanism of action. This and other clinical trials are continuing with this agent (ClinicalTrials.gov, numbers NCT03775850 and NCT03775850).
In a separate clinical development, a combination of 7 Bacteroidales and 4 non-Bacteroidales strains from healthy individuals are being explored, which have been shown preclinically to increase the number of cytotoxic CD8+ T cells and to enhance the anti-PD1 response in mouse tumor models.117 Further analysis of these 11 strains showed a true consortium effect; the 7 Bacteroidales strains had no effect on enhancement of CD8+ T cell response and the 4 non-Bacteroidales did, however, the effect was much greater when the 11 strains were combined. These encouraging preclinical data have led to a clinical study with this specific bacterial consortium.118
Probiotics
Altering the microbiota using over-the-counter probiotics or empirically determined clinical probiotic candidates is another strategy that is being investigated in the context of tumor therapy using a combination of retrospective studies and prospective clinical trials. Preliminary data from retrospective analysis of melanoma patient’s lifestyle surveys show over-the-counter probiotics negatively impact the response to immune checkpoint inhibitor therapy.119 A clinical trial evaluating an over-the-counter probiotic is underway in breast cancer—it remains to be seen whether the therapeutic effect of over-the-counter probiotics is dependent on tumor indication, as well as the makeup of the specific probiotic (ClinicalTrials.gov, number NCT03358511). In colorectal cancer, probiotics have been shown to modulate tumor microbiota, as well as alter the inflammatory cytokine profile found in tumors, and there are other trials underway evaluating specific probiotic mixtures in the setting of immunotherapy for melanoma and other solid indications.120,121
Intratumoral Bacteria: The Microbiome of Other Tissues Could Modulate Local Tumor Progression Independent of the Gut Microbiome
The microbiome of other mucosal sites can also play a role in local tumor development independently of the gut microbiota. In a genetically engineered mouse model of inducible lung adenocarcinoma, conventional SPF mice developed higher lung tumor burden than GF mice, and experimental evidence suggested that this effect was specifically driven by the local effect of the lung microbiota.122 The lung microbiome of tumor-bearing mice was characterized by a higher bacterial load, distinct taxonomic composition, and a lower diversity compared with non–tumor-bearing mice. Intratracheal administration of a bacterial consortium isolated from late-stage lung tumors accelerated tumor progression.122 Mechanistically, this effect was attributed to the expansion of tissue-resident γδ T cells, which produced IL-17 and drove neutrophil recruitment. These lung γδ T cells might also be able to directly increase tumor cell proliferation by secretion of IL-22 and amphiregulin, a ligand for the epidermal growth factor receptor.122
Breast tissue, originally thought to be sterile, also harbors its own microbiota.53,54,123–125 A number of clinical studies have found that breast cancer biopsies have a distinct bacterial composition compared with healthy breast tissue, although there is no clear agreement on the bacterial taxa associated with breast cancer.54 Some bacteria within the breast microbiota, such as E coli and Staphylococcus epidermidis, have been hypothesized to contribute to breast cancer initiation in susceptible individuals by causing double-stranded DNA breaks in host cells, whereas others, such as Fusobacterium genus could contribute to tumor development by creating a chronic pro-inflammatory environment.33,126
Additional studies have also suggested that bacteria are present in more types of tumor tissue than appreciated previously, where they can play a role in tumor progression and immunity. Analysis of whole-genome sequencing data from The Cancer Genome Atlas127 revealed that microbial gene signatures in cancer tissues and in the blood of cancer patients were associated with various cancer types, including cancer tissues in body sites previously considered to be sterile.124 By using multiple methods for detection of bacteria, Nejman et al125 demonstrated the presence of bacteria within tumor cells and tumor-infiltrating immune cells in various cancer settings, including breast, bone, lung, ovary, brain, pancreas, and melanoma. Although the taxonomic composition of the tumor microbiome largely reflected that of adjacent normal tissues, there were instances of enrichment of total bacterial load and of certain taxa and bacterial metabolic pathways within the tumor tissues, suggesting that the tumor microenvironment could constitute a unique niche for their growth. Breast tumors had the most diverse microbiome and at least a fraction of the bacteria detected within breast tumors were viable and culturable.125 Interestingly, electron microscopy revealed that these intracellular bacteria were largely devoid of cell walls,125 which is indicative of an L-form–like state.125,128 Various bacterial species are known to be able to switch to cell wall–free L-forms in certain conditions. This switch can render them resistant to antibiotics targeting cell wall synthesis and it has been suggested that inefficient clearance of pathogens in their L-form state could be the cause of recurring infections.128 What the original source of these intratumoral bacteria is, and whether or which of them are involved in tumorigenesis, tumor progression, or response to therapy remains to be determined.
Conclusions
The commensal microbiota has a profound impact on the host immune system and is being recognized as one of the factors that can influence anti-tumor immunity and therapeutic outcomes. Clinical efforts to augment the efficacy of cancer immunotherapy are based primarily on administration of microbial preparations with immunostimulatory properties (such as FMT, more defined bacterial consortia, or probiotic bacterial strains) and are still in their infancy. Deeper understanding of the mechanisms of microbiota-mediated immunomodulation and identification of precise immunostimulatory and immune-inhibiting bacterial strains or pathways could lead to increased precision in therapeutic approaches that could help avoid undesirable outcomes, such as the infection risk associated with FMT. In addition to administration of immunostimulatory microbial preparations or metabolites, it is conceivable that targeted depletion of immune-inhibiting bacteria could also augment immunotherapeutic outcomes. In a more personalized approach, better characterization of the recipient patient microbiota by sequencing, quantitative polymerase chain reaction, or functional screens could help select the optimal therapy. The commensal microbiota also impacts tumor development, which suggests that microbiota composition screens could identify individuals with high risk for developing cancer, and that microbiota modulation in these individuals could also have implications for cancer prevention. Toward this objective, recent studies have shown that commensal bacteria can be found in tissues that were previously considered to be sterile, and much remains to be understood about the sources of microbial colonization and the microbiota composition and function at these sites.
Funding
This work was supported by NIH grant R35 CA210098, a Team Science Award from the Melanoma Research Alliance, the American Cancer Society -Jules L. Plangere Jr. Family Foundation Professorship in Cancer Immunotherapy (T.F.G), University of Chicago Basic Medical Research Training in Oncology T32CA009566 (C.S.F.), and NIH Postdoctoral Fellowship Award F32CA236296 (V.M.).
Conflicts of interest
T.F.G. is an advisory board member for Roche-Genentech, Merck, Abbvie, Bayer, Aduro, and Fog Pharma. T.F.G. receives research support from Roche-Genentech, BMS, Merck, Incyte, Seattle Genetics, and Ono. T.F.G. is a shareholder/cofounder of Jounce Therapeutics. The University of Chicago holds a licensing arrangement with Evelo. T.F.G. and V.M. are inventors on U.S. patent US20160354416 A1 submitted by the University of Chicago that covers the use the microbiota to improve cancer immunotherapy.
Abbreviations used in this paper:
- BA
bile acid
- CRC
colorectal cancer
- FMT
fecal microbial transplantation
- GF
germ-free
- LPS
lipopolysaccharide
- LTA
lipoteichoic acid
- NK
natural killer
- NSCLC
non–small-cell lung carcinoma
- PDAC
pancreatic ductal adenocarcinoma
- SCFAs
short-chain fatty acids
- SPF
specific pathogen–free
- TLR
Toll-like receptor
- Treg
regulatory T cell
Biographies
Vyara Matson

Carolina Soto Chervin

Thomas F. Gajewski

References
- 1.Martin AM, Sun EW, Rogers GB, et al. The influence of the gut microbiome on host metabolism through the regulation of gut hormone release. Front Physiol 2019; 10:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell 2014;157:121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res 2020;30:492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Q, Xing C, Long W, et al. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflammation 2019;16:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browne HP, Forster SC, Anonye BO, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 2016;533:543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodman AL, Kallstrom G, Faith JJ, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 2011;108:6252–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parida S, Sharma D. The power of small changes: comprehensive analyses of microbial dysbiosis in breast cancer. Biochim Biophys Acta Rev Cancer 2019; 1871:392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagier J-C, Hugon P, Khelaifia S, et al. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin Microbiol Rev 2015;28:237–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lagier J-C, Khelaifia S, Alou MT, et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol 2016;1:16203. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Rogala AR, Oka A, Sartor RB. Strategies to dissect host-microbial immune interactions that determine mucosal homeostasis vs intestinal inflammation in gnotobiotic mice. Front Immunol 2020;11:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 2019;7:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amieva M, Peek RM. Pathobiology of Helicobacter pylori–induced gastric cancer. Gastroenterology 2016; 150:64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moss SF. The clinical evidence linking Helicobacter pylori to gastric cancer. Cell Mol Gastroenterol Hepatol 2017;3:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi IJ, Kook M-C, Kim Y-I, et al. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N Engl J Med 2018;378:1085–1095. [DOI] [PubMed] [Google Scholar]
- 15.Lee Y-C, Chiang T-H, Chou C-K, et al. Association between Helicobacter pylori eradication and gastric cancer incidence: a systematic review and meta-analysis. Gastroenterology 2016;150:1113–1124.e5. [DOI] [PubMed] [Google Scholar]
- 16.Díaz P, Valenzuela Valderrama M, Bravo J, et al. Helicobacter pylori and gastric cancer: adaptive cellular mechanisms involved in disease progression. Front Microbiol 2018;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polyzos SA. Helicobacter pylori infection and esophageal adenocarcinoma: a review and a personal view. Ann Gastroenterol 2018;31:8–13; Available at: http://www.annalsgastro.gr/files/journals/1/earlyview/2017/ev-11-2017-06-AG3213-0213.pdf.AccessedNovember 4, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 2018;67:226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coker OO, Dai Z, Nie Y, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018;67:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Shao L, Liu X, et al. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine 2019;40:336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanagan L, Schmid J, Ebert M, et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur J Clin Microbiol Infect Dis 2014;33:1381–1390. [DOI] [PubMed] [Google Scholar]
- 22.Mima K, Sukawa Y, Nishihara R, et al. Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncol 2015;1:653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mima K, Nishihara R, Qian ZR, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016;65:1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubinstein MR, Wang X, Liu W, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013;14:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kostic AD, Chun E, Robertson L, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013;14:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu S, Morin PJ, Maouyo D, et al. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 2003;124:392–400. [DOI] [PubMed] [Google Scholar]
- 27.Wu S, Rhee K-J, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009; 15:1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gur C, Ibrahim Y, Isaacson B, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015;42:344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu T, Guo F, Yu Y, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017;170:548–563.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong H, Shi Y, Xiao X, et al. Alterations of microbiota structure in the larynx relevant to laryngeal carcinoma. Sci Rep 2017;7:5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamura K, Baba Y, Nakagawa S, et al. Human microbiome Fusobacterium nucleatum in esophageal cancer tissue is associated with prognosis. Clin Cancer Res 2016;22:5574–5581. [DOI] [PubMed] [Google Scholar]
- 32.Mitsuhashi K, Nosho K, Sukawa Y, et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 2015; 6:7209–7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hieken TJ, Chen J, Hoskin TL, et al. The microbiome of aseptically collected human breast tissue in benign and malignant disease. Sci Rep 2016;6:30751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bučević Popović V, Šitum M, Chow C-ET, et al. The urinary microbiome associated with bladder cancer. Sci Rep 2018;8:12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu N, Yang X, Zhang R, et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol 2013;66:462–470. [DOI] [PubMed] [Google Scholar]
- 36.Kaakoush NO, Castaño-Rodríguez N, Man SM, et al. Is Campylobacter to esophageal adenocarcinoma as Helicobacter is to gastric adenocarcinoma? Trends Microbiol 2015;23:455–462. [DOI] [PubMed] [Google Scholar]
- 37.Warren RL, Freeman DJ, Pleasance S, et al. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 2013;1:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaakoush NO, Deshpande NP, Man SM, et al. Transcriptomic and proteomic analyses reveal key innate immune signatures in the host response to the gastrointestinal pathogen Campylobacter concisus. Infect Immun 2015;83:832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalmasso G, Cougnoux A, Delmas J, et al. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014;5:675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lasry A, Ben-Neriah Y. Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol 2015;36:217–228. [DOI] [PubMed] [Google Scholar]
- 41.Pushalkar S, Hundeyin M, Daley D, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 2018;8:403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geller LT, Barzily-Rokni M, Danino T, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017; 357:1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riquelme E, Zhang Y, Zhang L, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 2019;178:795–806.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma C, Han M, Heinrich B, et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018;360:eaan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loo TM, Kamachi F, Watanabe Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov 2017;7:522–538. [DOI] [PubMed] [Google Scholar]
- 46.Singh V, Yeoh BS, Chassaing B, et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell 2018;175:679–694.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ridlon JM, Kang D-J, Hylemon PB. Bile salt bio-transformations by human intestinal bacteria. J Lipid Res 2006;47:241–259. [DOI] [PubMed] [Google Scholar]
- 48.Dapito DH, Mencin A, Gwak G-Y, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012;21:504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhowmik S, Chiu H-P, Jones DH, et al. Structure and functional characterization of a bile acid 7α dehydratase BaiE in secondary bile acid synthesis. Proteins 2016; 84:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baker JM, Al-Nakkash L, Herbst-Kralovetz MM. Estrogen–gut microbiome axis: physiological and clinical implications. Maturitas 2017;103:45–53. [DOI] [PubMed] [Google Scholar]
- 51.Flores R, Shi J, Fuhrman B, et al. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med 2012; 10:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwa M, Plottel CS, Blaser MJ, et al. The intestinal microbiome and estrogen receptor-positive female breast cancer. J Natl Cancer Inst 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao VP, Poutahidis T, Ge Z, et al. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res 2006;66:7395–7400. [DOI] [PubMed] [Google Scholar]
- 54.Eslami-S Z, Majidzadeh -AK, Halvaei S, et al. Microbiome and breast cancer: new role for an ancient population. Front Oncol 2020;10:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abdul-Aziz MA, Cooper A, Weyrich LS. Exploring relationships between host genome and microbiome: new insights from genome-wide association studies. Front Microbiol 2016;7:1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xavier JB, Young VB, Skufca J, et al. The cancer microbiome: distinguishing direct and indirect effects requires a systemic view. Trends Cancer 2020;6:192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ben-Shaanan TL, Schiller M, Azulay-Debby H, et al. Modulation of anti-tumor immunity by the brain’s reward system. Nat Commun 2018;9:2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alcock J, Maley CC, Aktipis CA. Is eating behavior manipulated by the gastrointestinal microbiota? Evolutionary pressures and potential mechanisms. BioEssays News Rev Mol Cell Dev Biol 2014;36:940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee K, Vuong HE, Nusbaum DJ, et al. The gut microbiota mediates reward and sensory responses associated with regimen-selective morphine dependence. Neuropsychopharmacology 2018;43:2606–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 61.Daillère R, Vétizou M, Waldschmitt N, et al. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 2016;45:931–943. [DOI] [PubMed] [Google Scholar]
- 62.Viaud S, Saccheri F, Mignot G, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013;342:971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fluckiger A, Daillère R, Sassi M, et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 2020;369:936–942. [DOI] [PubMed] [Google Scholar]
- 64.Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013;342:967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paulos CM, Wrzesinski C, Kaiser A, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest 2007;117:2197–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peled JU, Devlin SM, Staffas A, et al. Intestinal microbiota and relapse after hematopoietic-cell transplantation. J Clin Oncol 2017;35:1650–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vétizou M, Pitt JM, Daillère R, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015;350:1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015;350:1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chaput N, Lepage P, Coutzac C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 2017;28:1368–1379. [DOI] [PubMed] [Google Scholar]
- 70.Frankel AE, Coughlin LA, Kim J, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 2017;19:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359:91–97. [DOI] [PubMed] [Google Scholar]
- 72.Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018;359:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018;359:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borgerding JN, Shang J, Britton GJ, et al. Human microbial transplant restores T cell cytotoxicity and anti-tumor response to PD-L1 blockade in gnotobiotic mice. Preprint. Published online August 7, 2020.bioRxiv 2020.08.07.242040. doi: 10.1101/2020.08.07.242040. [DOI] [Google Scholar]
- 75.Mager LF, Burkhard R, Pett N, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020;369:1481–1489. [DOI] [PubMed] [Google Scholar]
- 76.Uribe-Herranz M, Bittinger K, Rafail S, et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight 2018;3:e94952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taur Y, Jenq RR, Perales M-A, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 2014;124:1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peled JU, Gomes ALC Devlin SM, et al. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N Engl J Med 2020;382:822–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bermejo-Jambrina M, Eder J, Helgers LC, et al. C-type lectin receptors in antiviral immunity and viral escape. Front Immunol 2018;9:590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 2009;22:240–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014; 14:667–685. [DOI] [PubMed] [Google Scholar]
- 83.McDole JR, Wheeler LW, McDonald KG, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012;483:345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rescigno M, Urbano M, Valzasina B, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 2001;2:361–367. [DOI] [PubMed] [Google Scholar]
- 85.Morton AM, Sefik E, Upadhyay R, et al. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc Natl Acad Sci U S A 2014;111:6696–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Z, Tang H, Chen P, et al. Demystifying the manipulation of host immunity, metabolism, and extra-intestinal tumors by the gut microbiome. Signal Trans-duct Target Ther 2019;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013;504:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luu M, Weigand K, Wedi F, et al. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci Rep 2018;8:14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bachem A, Makhlouf C, Binger KJ, et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T cells. Immunity 2019;51:285–297.e5. [DOI] [PubMed] [Google Scholar]
- 91.Nomura M, Nagatomo R, Doi K, et al. Association of short-chain fatty acids in the gut microbiome with clinical response to treatment with nivolumab or pembrolizumab in patients with solid cancer tumors. JAMA Netw Open 2020;3:e202895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tian Y, Zhou Y, Huang S, et al. Fecal microbiota transplantation for ulcerative colitis: a prospective clinical study. BMC Gastroenterol 2019;19:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Costello SP, Hughes PA, Waters O, et al. Effect of Fecal microbiota transplantation on 8-week remission in patients with ulcerative colitis: a randomized clinical trial. JAMA 2019;321:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cammarota G, Ianiro G. FMT for ulcerative colitis: closer to the turning point. Nat Rev Gastroenterol Hepatol 2019;16:266–268. [DOI] [PubMed] [Google Scholar]
- 95.Xu D, Chen VL, Steiner CA, et al. Efficacy of fecal microbiota transplantation in irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol 2019;114:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y, Wiesnoski DH, Helmink BA, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med 2018; 24:1804–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rohlke F, Stollman N. Fecal microbiota transplantation in relapsing Clostridium difficile infection. Ther Adv Gastroenterol 2012;5:403–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Allegretti JR, Kelly CR, Grinspan A, et al. Outcomes of fecal microbiota transplantation in patients with inflammatory bowel diseases and recurrent Clostridioides difficile infection. Gastroenterology 2020;159:1982–1984. [DOI] [PubMed] [Google Scholar]
- 99.McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis 2018;66:e1–e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nood E van, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 2013;368:407–415. [DOI] [PubMed] [Google Scholar]
- 101.Khoruts A, Staley C, Sadowsky MJ. Faecal microbiota transplantation for Clostridioides difficile: mechanisms and pharmacology. Nat Rev Gastroenterol Hepatol 2020. Available at: http://www.nature.com/articles/s41575-020-0350-4.AccessedNovember 4, 2020. [DOI] [PubMed]
- 102.Cheng Y-W, Phelps E, Ganapini V, et al. Fecal microbiota transplantation for the treatment of recurrent and severe Clostridium difficile infection in solid organ transplant recipients: a multicenter experience. Am J Transplant 2019;19:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Trinchieri G The microbiome in cancer therapy. Available at: https://www.abstractsonline.com/pp8/#!/6812/presentation/7757; 2019. AccessedMarch 30, 2020; 2019.
- 104.Baruch EN, Youngster I, Ortenberg R, et al. Abstract CT042: fecal microbiota transplantation (FMT) and re-induction of anti-PD-1 therapy in refractory metastatic melanoma patients—preliminary results from a phase I clinical trial (NCT03353402). Cancer Res 2019;79. CT042–CT042. [Google Scholar]
- 105.DeFilipp Z, Bloom PP, Torres Soto M, et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med 2019;381:2043–2050. [DOI] [PubMed] [Google Scholar]
- 106.US Food and Drug Administration. Important safety alert regarding use of fecal microbiota for transplantation and risk of serious adverse reactions due to transmission of multi-drug resistant organisms. Available at: http://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/important-safety-alert-regarding-use-fecal-microbiotatransplantation-and-risk-serious-adverse.AccessedSeptember 18, 2019. Published June 13, 2019.
- 107.Kassam Z, Dubois N, Ramakrishna B, et al. Donor screening for fecal microbiota transplantation. N Engl J Med 2019;381:2070–2072. [DOI] [PubMed] [Google Scholar]
- 108.Trujillo JA, Luke JJ, Zha Y, et al. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer 2019;7:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 2018;118:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017;7:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Spranger S, Gajewski TF. Tumor-intrinsic oncogene pathways mediating immune avoidance. OncoImmunology 2016;5:e1086862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 2018;18:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lim YW, Chen-Harris H, Mayba O, et al. Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc Natl Acad Sci U S A 2018;115:E11701–E11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gardner HA, Kashyap S, Ponichtera H, et al. Monoclonal microbial EDP1503 to induce antitumor responses via gut-mediated activation of both innate and adaptive immunity. J Clin Oncol 2019;37. e14241–e14241. [Google Scholar]
- 117.Tanoue T, Morita S, Plichta DR, et al. A defined commensal consortium elicits CD8 T cells and anticancer immunity. Nature 2019;565:600–605. [DOI] [PubMed] [Google Scholar]
- 118.Bristol-Myers Squibb and Vedanta Biosciences Announce a New Clinical Collaboration to Evaluate OPDIVO® (nivolumab) and VE800 in Patients with Advanced or Metastatic Cancers: Vedanta Biosci Inc. Available at: https://www.vedantabio.com/news-media/press-releases/detail/2492.AccessedNovember 9, 2020.
- 119.Spencer CN, Gopalakrishnan V, McQuade J, et al. Abstract 2838: the gut microbiome (GM) and immunotherapy response are influenced by host lifestyle factors. Cancer Res 2019;79(Suppl):2838; Available at: http://cancerres.aacrjournals.org/lookup/doi/10.1158/1538-7445.AM2019-2838. AccessedNovember 5, 2020. [Google Scholar]
- 120.Hibberd AA, Lyra A, Ouwehand AC, et al. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol 2017;4:e000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Consoli MLD, da Silva RS, Nicoli JR, et al. Randomized clinical trial: impact of oral administration of Saccharomyces boulardii on gene expression of intestinal cytokines in patients undergoing colon resection. J Parenter Enter Nutr 2016;40:1114–1121. [DOI] [PubMed] [Google Scholar]
- 122.Jin C, Lagoudas GK, Zhao C, et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019;176:998–1013.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Urbaniak C, Cummins J, Brackstone M, et al. Microbiota of human breast tissue. Appl Environ Microbiol 2014; 80:3007–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Poore GD, Kopylova E, Zhu Q, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020;579:567–574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 125.Nejman D, Livyatan I, Fuks G, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Urbaniak C, Gloor GB, Brackstone M, et al. The microbiota of breast tissue and its association with breast cancer. Appl Environ Microbiol 2016;82:5039–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Errington J, Mickiewicz K, Kawai Y, et al. L-form bacteria, chronic diseases and the origins of life. Philos Trans R Soc B Biol Sci 2016;371:20150494. [DOI] [PMC free article] [PubMed] [Google Scholar]