

Abstract
Fertilization calcium waves are a conserved trigger for animal development; however, genetic analysis of these waves has been limited due to the difficulty of imaging in vivo fertilization. Here we describe a protocol to image calcium dynamics during in vivo fertilization in the genetic animal model Caenorhabditis elegans. This protocol consists of germline microinjection of a chemical calcium indicator, worm immobilization, live imaging, and image processing that quantifies the calcium fluorescence in the oocyte region moving in the field-of-view during ovulation. This imaging protocol can also be used to image other cellular processes during in vivo fertilization in C. elegans, such as membrane fusion and cytoskeletal dynamics.
Keywords: Fertilization, Fertilization calcium waves, Calcium waves, Caenorhabditis elegans, Live imaging, Calcium imaging, Image processing
Background
Fertilization calcium waves play a pivotal role in egg activation and have been analyzed in various organisms by using in vitro fertilization systems. The nematode Caenorhabditis elegans is amenable to imaging in vivo fertilization because of its translucent body. The fertilization calcium imaging in C. elegans by using a chemical calcium indicator is reported in Samuel et al. (2001) . We describe here a protocol modified from the imaging method by applying high-speed spinning disk confocal microscopy and image processing methods that segment the fertilized oocyte region moving in the field-of-view during ovulation. This protocol enables a precise quantitative description of the temporal dynamics of the calcium waves and genetic analysis of the wave pattern.
Materials and Reagents
Glass capillary with filament (NARISHIGE, catalog number: GDC-1)
24 x 55 mm cover glass (Matsunami Glass, catalog number: C024551)
76 x 26 mm glass slide (Matsunami Glass, catalog number: S011110)
18 x 18 mm cover glass (thickness 0.16-0.19 mm; No. 1S/No. 1.5) (Matsunami Glass, special order product)
Micro spatula (e.g., AS ONE, catalog number: 6-524-01)
1.5-ml brown tubes (e.g., FUKAEKASEI, WATSON, catalog number: 131-915BR)
Aluminum foil (e.g., UACJ Foil Corporation)
Syringe-driven membrane filter (EMD Millipore, catalog number: SLGS033SS)
19 (W) x 0.18 (T) mm vinyl tape (3M, catalog number: 35-WHITE-3/4)
Lint-free wipe (KCWW, Kimwipes, catalog number: S-200)
-
Dark chamber (e.g., Ina-optika, model: FB-35BL)
Note: We make a rectangular hole of approximately 7 x 10.5 cm in the inner lid.
N2 wild-type C. elegans
-
Agarose gel pad for microinjection
Agarose-LE, Classic Type (Nacalai Tesque, catalog number: 01157-66)
Or agarose (Thermo Fisher Scientific, Invitrogen, catalog number: 15510-027)
Note: This product has been discontinued.
Liquid paraffin (Wako Pure Chemical Industries, catalog number: 164-00476)
-
0.10-µm polystyrene microsphere suspension (Polysciences, catalog number: 00876-15)
Notes:
Prepare approximately 500-µl aliquots in 1.5-ml tubes
Store at 4 °C
Serotonin hydrochloride (Sigma-Aldrich, catalog number: H9523)
Calcium Green-1 dextran, potassium salt, 10,000 MW, anionic (Thermo Fisher Scientific, Molecular ProbesTM, catalog number: C3713)
Nuclease-free water (Promega, catalog number: P1193)
Potassium dihydrogen phosphate (KH2PO4) (Wako Pure Chemical Industries, catalog number: 169-04245)
Disodium hydrogenphosphate (Na2HPO4) (Wako Pure Chemical Industries, catalog number: 197-02865)
Sodium chloride (NaCl) (Wako Pure Chemical Industries, catalog number: 191-01665)
Magnesium sulfate heptahydrate (MgSO4.7H2O) (Wako Pure Chemical Industries, catalog number: 131-00405)
NuSieve GTG agarose (Lonza, catalog number: 50080)
Vaseline (white petrolatum) (Kozakai Pharmaceutical)
Lanolin (Sigma-Aldrich, catalog number: L7387)
Paraffin pellets (e.g., Seiwa)
-
D(+)-glucose (Wako Pure Chemical Industries, catalog number: 041-00595)
Note: This product has been discontinued.
-
0.2% BSA-fluorescein solution (dissolved in nuclease-free water)
Note: Prepare immediately before use.
2.0% gelatin solution
BSA-fluorescein (Thermo Fisher Scientific, Molecular ProbesTM, catalog-number: A23015)
100 µM Calcium Green-1 dextran solution (see Recipes)
Sterilized 1 M MgSO4 solution (see Recipes)
M9 buffer (see Recipes)
10% NuSieve GTG agarose/M9 pad (see Recipes)
VALAP (see Recipes)
Injection recovery solution (see Recipes)
20 mg/ml serotonin hydrochloride/M9 buffer (see Recipes)
Fluorescence reference slide (see Recipes)
Equipment
Needle puller (NARISHIGE, model: PC-10)
Microinjector (Eppendorf, model: FemtoJet® Microinjector)
Inverted microscope with manipulator (e.g., Leica Microsystems, model: DMIRB; NARISHIGE models: MMO-220A, MO-202U, MN-4)
Hot plate (e.g., IKA, catalog number: C-MAG HP7)
-
Microwave oven (e.g., Panasonic, model: NE-EH212)
Note: This product has been discontinued.
Microscope (e.g., Leica Microsystems, model: DMRE; Nikon Instruments, model: Ti-E)
50-ml screw-cap glass bottle (e.g., AS ONE, catalog number: 1-4568-01)
100-ml beaker (e.g., ASAHI Glass, AGC, catalog number: 1000BK100)
50-ml beaker (e.g., ASAHI Glass, AGC, catalog number: 1000BK50)
Diamond pen (e.g., Ogura Jewel Industry, model: D-point pen)
High-magnification water-immersion objective lens (e.g., Leica Microsystems, model: HCX PL Apo 63x/1.20 W corr.; Nikon Instruments, model: Plan Apo VC 60XA/1.20 W)
Spinning disk confocal unit (Yokogawa Electric, model: CSU-X1)
EM-CCD camera (Andor Technology, model: iXon DU-897)
Ar/Kr laser (IDEX, Melles Griot, model: 643-YB-A01)
Filter/shutter controller (Ludl Electronic Products, model: Mac 6000, catalog numbers: 73006081, 73006042, 73006001)
488-nm laser line filter (IDEX, Semrock, catalog number: LL01-488)
Dichroic mirror (IDEX, Semrock, catalog number: Di01-T488)
Long-pass barrier filter (IDEX, Semrock, catalog number: BLP01-488R)
Windows 7 computer for image acquisition
Mac computer for image processing (e.g., ver. 10.6.8 or later)
Interactive pen display (Wacom, model: Cintiq 21UX)
Software
ImageJ (ver. 1.49v or later) (https://imagej.nih.gov/ij/download.html)
Image-acquisition software (e.g., Andor Technology, iQ [ver. 1.10.3 or later] or iQ2 [ver. 2.9.1 or later])
Procedure
Note: This protocol is modified from the original method described in Samuel et al. (2001).
-
Microinjection of chemical calcium indicator dye
-
Inject 100 µM Calcium Green-1 dextran solution into the distal arm of the anterior gonad of a gravid adult hermaphrodite by using a standard method for C. elegans DNA microinjection (see Evans, 2006). We use a GDC-1 glass capillary with filament pulled by the PC-10 puller in the double-pulling mode. In addition, we use a 2% or 3.5% agarose gel pad on a 24 x 55 mm cover glass for worm immobilization and liquid paraffin to protect the specimen from drying.
Notes:
Fill about half the gonad as described in Samuel et al. (2001). Injecting too much dye often results in an abnormal gonad shape after recovery.
We always place only one worm per agarose pad for injection to minimize the time that the worm is immobilized and hence minimize the damage.
We typically inject four to six worms in a single set of experiments (e.g., one genotype) and perform two sets of experiments in a day with a 2-h interval. Approximately half of the injected worms can be imaged for each set of experiments.
To recover the injected worm, apply approximately 1 µl of injection recovery solution onto the worm by using a P20 micropipette.
-
By using a P20 micropipette, suck up the injected worm in the drop of injection recovery solution and transfer it from the agarose pad to a 3.5-cm OP50/NGM plate.
Note: A single injection procedure from picking to transfer (steps A1-A3) should be completed within approximately 5 min.
Allow the injected worm to recover on the plate in a dark chamber for 4-6 h at room temperature.
-
Check whether the injected dye is incorporated into the oocytes of the worm by using a stereo fluorescence microscope.
Note: Sloppy injection can result in leakage and incorporation of the dye into the coelomocytes (Samuel et al., 2001).
-
-
Worm preparation and imaging
Note: The worm-immobilization method is modified from the original method described in Kim et al. (2013).
Melt VALAP at 130 °C on a hot plate.
-
Prepare a 10% agarose gel pad.
Prepare two double-taped glass slides (Figure 1A). The thickness of the tape stack in this image was 0.36 mm.
Melt the 10% NuSieve GTG agarose/M9 buffer in a microwave oven.
Apply a drop of the melted 10% NuSieve GTG agarose/M9 buffer to a glass slide between the two taped glass slides by using a micro spatula (Figure 1A; Video 1).
Cover the melted agarose drop with another glass slide (Figure 1B; Video 1).
Incubate for at least 30 min at room temperature to solidify the agarose pad. Keep the agarose pad covered by the glass slide until use.
Allow the polystyrene microsphere suspension and the serotonin hydrochloride/M9 buffer to warm at room temperature for at least 30 min before use.
Check the shape of the leading oocyte of the injected worm to determine the timing of ovulation by using the stereo fluorescence microscope. The shape of the leading oocyte should appear rectangular, and its nucleus should be in the cortical region distal to the spermatheca (Figure 2).
Remove the glass slide covering the agarose pad and apply 0.5 µl of the 0.10-µm polystyrene microsphere suspension to the center of the agarose pad (Video 2).
-
Apply 0.5 µl of 20 mg/ml serotonin hydrochloride/M9 buffer to the polystyrene microsphere suspension on the agarose pad (Figure 1C; Video 2). Serotonin is believed to promote fertilization ( Samuel et al., 2001 ).
Notes:
Do not mix the polystyrene microspheres and the serotonin in advance because mixing causes the microspheres to aggregate.
Although we usually use serotonin, it is not necessary, and we have observed normal ovulation and fertilization in worms immobilized without serotonin (Takayama and Onami, 2016).
Transfer the worm that is due to ovulate within a few minutes from the 3.5-cm OP50/NGM plate to the drop on the agarose pad by using a worm pick (Video 2).
Arrange the orientation of the worm by using a worm pick such that the oocytes with the injected dye will be situated just beneath the cover glass. Because of the asymmetrical configuration of the anterior and posterior gonads, only one proximal gonad can usually be situated just beneath the cover glass.
Gently place an 18 x 18 mm cover glass on the agarose pad (Video 2).
Stabilize the cover glass by applying four drops of melted VALAP on the four corners of the cover glass by using a P20 micropipette (Figure 1D; Video 2).
Position the sample slide glass on the spinning disk confocal microscope.
Find the worm by using low-magnification objective lenses (e.g., 10x or 20x).
Use a high-magnification water-immersion objective lens to set the field-of-view and the focal plane that best capture the leading sperm in the spermatheca that is expected to fertilize the oocyte.
Start the time-lapse recording when the worm starts ovulating. Typical time-lapse settings for 2D time-lapse imaging are as follows: exposure, 178.6 msec; camera binning, 2; time interval, 0.2 sec (fastest); and duration, 6 min. For 3D time-lapse imaging, the typical settings are as follows: exposure, 50 msec; camera binning, 2; time interval, 1.0 sec; duration, 6 min; z-plane interval, 1 µm; and number of z-planes, 12. The EM-gain is set so that the intensities of the image will be in the predefined range (e.g., 0-4,095 for 12 bit).
Save images as a 16-bit multi-tiff image file.
Capture fluorescence reference images for uneven illumination correction by using a fluorescence reference slide. We typically capture at least 40 different images in the protein film region for the foreground and 5 different images for the background in the 14-bit range (0-16,383).
Figure 1. Sample preparation.

A. A drop of 10% agarose is applied to a glass slide between the two double-taped spacers by using a micro spatula. B. A slide glass is placed on the 10% agarose drop. C. A drop of microsphere suspension and a drop of serotonin/M9 solution are applied to the agarose pad on the stage of a stereo microscope. D. A sample ready for imaging, in which the cover glass is stabilized by VALAP on the agarose pad (a non-injected worm is immobilized in this image).
Video 1. Preparation of a 10% agarose gel pad.

Figure 2. Inference of the timing of ovulation on the basis of the oocyte shape.

A. An oocyte that appears ready to be ovulated within a few minutes. The oocyte shape is rectangular, the boundary facing the next oocyte looks like a straight line (indicated by the black line), and the nucleus is close to the cortical region distal to the spermatheca. B. An oocyte not ready for imaging. The boundary has an arc-like appearance (indicated by the black line), and the nucleus is not close to the distal cortex. Differential interference contrast images of non-injected wild-type worms are shown. Images are flipped to be in the same direction as the confocal microscopy images in Figure 3. Arrowheads indicate the nucleus. The black lines were drawn to emphasize the boundaries. Scale bar = 20 µm.
Video 2. Worm-immobilization.

Data analysis
-
Installation of plugins for image processing
Download and install ImageJ from https://imagej.nih.gov/ij/download.html.
Download and install mbf-plugins.zip from https://imagej.nih.gov/ij/plugins/mbf/, which contains the BG_Subtraction_from_ROI plugin.
-
Download, (compile), and install the following plugins from http://so.qbic.riken.jp/cawave/index.html:
Calculator_Plus3.java (Calculator_Plus3.class)
ContourTrack3Stack_.java (ContourTrack3Stack_.class)
Layer_Test2M.java (Layer_Test2M.class, Layer_Test2M$LayeredImageM.class)
ContourTrack4Stack_.java (ContourTrack4Stack_.class)
TrackOocyte_.java (TrackOocyte_.class, LabelHolder.class)
RoughQuantify_.java (RoughQuantify_.class)
-
Image processing to correct for uneven illumination
Notes:
The protocol for uneven illumination correction is modified from the original method described by Wolf et al. (2007).
The procedure described below is also presented as a video (Video 3).
Convert the 16-bit tiff images linearly from 0-16,383 (14-bit range) to 8-bit by using the /Image/Adjust/Brightness/Contrast function and Image/Type/8-bit function.
Average the foreground image (the 40 images of the protein film region in the fluorescence reference slide) by using the Average Intensity projection operation of the Image/Stack/ZProject... function. The resulting file is the averaged foreground image.
Average the background image (the 5 images of the regions outside of the protein film in the fluorescence reference slide) by using the Average Intensity projection operation of the Image/Stack/ZProject... function. The resulting file is the averaged background image.
Subtract the averaged background image from the averaged foreground image by using the Subtract operation of the Calculator_Plus plugin. Open the averaged foreground image and the averaged background image, start the Calculator_Plus plugin, select the averaged foreground image as the i1 and the averaged background image as the i2, select the Subtract operation, and click the OK button. The resulting image file is the reference image.
-
Image processing for quantification of the time course.
Notes:
The original data are presented in Takayama and Onami (2016). The example image file can be obtained from the SSBD database (Tohsato et al., 2016)
The entire procedure described below is also presented as five videos (Videos 4-8).
Convert the 16-bit multi-tiff image stack linearly to an 8-bit multi-tiff image stack by using the /Image/Adjust/Brightness/Contrast function and Image/Type/8-bit function. We typically use ranges of 0-2,047 (11 bit), 0-4,095 (12 bit), 0-8,191 (13 bit), or 0-16,383 (14 bit), depending on the intensity range of the image, and convert them to 0-255 (8 bit).
Subtract the background from the ROI by using the BG_subtraction_from_ROI plugin. The background ROI should be a rectangle region that does not include any parts of the worm throughout the time-lapse image stack. We typically set a 60 (w) x 60 (h) pixel region at one corner of the image stack as the ROI and the stdev value as 0. The resulting image stack is the background-corrected image stack.
-
Normalize the image stack with the reference image by using the Normalize operation of the Calculator_Plus3 plugin. Open the background-corrected image stack and the reference image, start the Calculator_Plus3 plugin, select the background-corrected image stack as the i1 and the reference image as the i2, select Normalize operation, and click the OK button. The resulting image stack is the normalized 8-bit image stack.
Video 4. Conversion to an 8-bit image stack, background subtraction, and uneven illumination correction (steps C1-C3).
Download video file (2.1MB, mp4)
-
Determine the time slice number of the sperm entry T by visually checking the morphological change in the oocyte tip (Figure 3). The time slice of T-1 is defined as the time 0 slice.
Note: A sudden protrusion of the oocyte tip is a signature of sperm entry. When performing 3D time-lapse imaging, determine the z slice of sperm entry also.
(Optional) Cut out (1) the time slices earlier than -50 sec and (2) those later than 300 sec because typically they do not have relevant information about fertilization calcium waves.
-
(Optional) Thin out the time slices to facilitate image processing with an 8-slice interval by using the Deinterleave function. Use the image stack that contains the time 0 slice for subsequent analyses.
Video 5. Determination of the time 0 slice, cutting slices (earlier than -19.6 sec and later than 250.2 sec in this video), and thinning of the image series (steps C4-C6).
Download video file (3.9MB, mp4)
Make a black-white image stack by using the /Image/Adjust/Threshold function. The upper threshold should be set as 255 and the lower threshold to a value that best separates the oocyte region from the background. Use the lower threshold value through the image stack. Check the black background checkbox. The resulting image stack is the black-white image stack.
Remove small debris regions by using the ContourTrack3Stack_ plugin (Figure 4). Open the black-white image stack, start the ContourTrack3Stack_ plugin, and input the upper limit value of the area (pixels) to be removed. A typical input value is 2,000, which means remove a foreground region with an area of less than 2,000 x 0.9 = 1,800. The resulting image stack is the debris-removed black-white image stack.
-
Check the shape of the segmented oocyte region by human eye.
Note: Usually, the oocyte before ovulation cannot be segmented from the other oocytes or the distal gonad regions (Figure 4B).
Video 6. Conversion to a black-white image stack and removal of debris (steps C7-C9).
Download video file (1.8MB, mp4)
-
Manually correct the segmentation by using the Layer_Test2M plugin (Figure 5). Start the Layer_Test2M plugin, click the Open button, select the normalized 8-bit image stack as the background layer, and select the debris-removed black-white image stack as the foreground layer. Draw or erase by clicking the Erase check box, and manually correct the segmentation by using the pen tool and an interactive pen display. The resulting black-white image stack is the manually corrected black-white image stack.
Note: The Save button will save the result as a sequence of single-tiff files, whereas the /File/Save as/Tiff... function will save the image stack as a multi-tiff file.
Video 7. Manual correction of the segmented region (step C10).
Download video file (2.9MB, mp4)
Remove the foreground regions except for the segmented oocyte region by using the ContourTrack4Stack_ plugin (Figure 6). Open the manually corrected black-white image stack, start the ContourTrack4Stack_ plugin, and input the approximate value of the area (pixels) of the oocyte region. A typical input value is 6,000, which means remove foreground regions with areas of less than 6,000 x 2/3 = 4,000 and greater than 6,000 x 4/3 = 8,000.
(Optional) If the image stack has foreground regions other than the oocyte region, track the oocyte region through the image stack by using the TrackOocyte_ plugin (Figure 7). Start the TrackOocyte_ plugin, and click the oocyte region of interest at the first time slice of the image stack. The resulting image stack is the tracked image stack.
If the segmented region has holes (small background regions in the segmented region), fill the holes by using the Process/Binary/Fill_Holes function. The resulting image stack is the black-white mask image stack.
Mask the normalized 8-bit image stack with the black-white mask image stack by using Mask operation of the Calculator_Plus3 plugin. Open the normalized 8-bit image stack and the black-white mask image stack, start the Calculator_Plus3 plugin, and select the normalized 8-bit image stack as the i1 and the black-white mask image stack as the i2, select the Mask operation, and click the OK button. The resulting image stack is the masked 8-bit image stack that contains the segmented oocyte.
-
Quantify the time course of the calcium fluorescence by using the RoughQuantify_ plugin (Figure 8). Open the masked 8-bit image stack, start the RoughQuantify_ plugin, and input the time 0 slice number and the interval time (sec). The resulting numerical data will consist of the slice number, the mean intensity of the segmented region (F), the time, the ratio (F/F0), the sum of the intensities, the area of the segmented region, the variance, and the standard deviation of the intensities. Save the data as an .xls file for subsequent analyses.
Video 8. Segmentation, tracking, and quantification (steps C11-C15).
Download video file (4MB, mp4)
Video 3. Image processing of the reference image to correct for uneven illumination.
Figure 3. Morphological changes in the oocyte tip upon sperm entry.
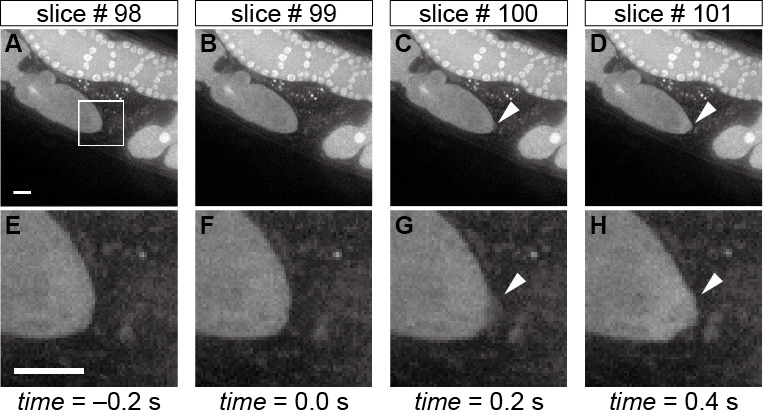
Time-lapse images of the oocyte shape during fertilization in the whole field-of-view (A-D) and in the corresponding magnified region (E-H) indicated by the white box in panel A. The arrowheads in panels C, D, G, and H indicate the sudden protrusion of the oocyte tip upon sperm entry, and hence slice #99 is defined as the time 0 slice. Scale bars in panels A and E = 10 µm.
Figure 4. The ContourTrack3Stack_ plugin.

A. The opened black-white image stack, and the input window of the ContourTrack3Stack_ plugin. B. The resulting image file does not contain debris regions.
Figure 5. The Layer_Test2M plugin.

A. The window of the Layer_Test2M plugin; B. The layered image stack, in which the semitransparent red foreground image is layered on to the background image.
Figure 6. The ContourTrack4Stack_ plugin.

A. The manually corrected black-white image stack and the input window of the ContourTrack4Stack_ plugin. B. A resulting image stack that does not contain the distal gonad region but does contain other embryonic regions.
Figure 7. The TrackOocyte_ plugin.

A. By starting the TrackOocyte_ plugin and then clicking the oocyte region (left), the region is tracked through the image stack. B. The resulting image stack of the TrackOocyte_ plugin. Regions other than the clicked region are removed.
Figure 8. The RoughQuantify_ plugin.

A. The masked 8-bit image stack, and the input window for the time 0 slice number and the time interval. B. The output of the RoughQuantify_ plugin.
Notes
The worm immobilization protocol can be used to image other cellular processes during fertilization such as membrane fusion or cytoskeleton dynamics. For membrane fusion imaging, use the OD58 [oocyte GFP::PH] strain ( Audhya et al., 2005 ) and males of EG4883 [sperm mCherry::histone] (Frøkjaer- Jensen et al., 2008 ) or the ONA18 [sperm TRP-3::tagRFP-T] strain (Takayama and Onami, 2016) instead of the wild-type N2. For filamentous actin imaging, use the BV67 [lifeact::gfp] strain (Pohl and Bao, 2010).
Recipes
-
100 µM Calcium Green-1 dextran solution
Weigh the Calcium Green-1 dextran powder in a 1.5-ml brown tube
-
Add nuclease-free water to the 1.5-ml brown tube to prepare a 1 mM solution
Note: Use the dye mol value to calculate the concentration.
Dilute the 1 mM solution to 100 µM with nuclease-free water
Store as 20-µl aliquots in 1.5-ml brown tubes in the dark at -20 °C
-
Agarose gel pad for microinjection
Prepare 2% or 3.5% agarose/pure water (EMD Millipore, Elix) solution in a 50-ml screw-cap bottle
Make agarose pads on 24 x 55 mm cover glasses
Note: We use Agarose-LE, Classic Type (Nacalai Tesque) or Agarose (Thermo Fisher Scientific, Invitrogen) for the 2% solution or 3.5% solution, respectively.
-
Sterilized 1 M MgSO4 solution
Dissolve 123.24 g MgSO4.7H2O in 500 ml of pure (Elix) water
Autoclave at 121 °C for 20 min
Store at room temperature
-
M9 buffer
-
Dissolve the following in 1 L of pure (Elix) water
3 g KH2PO4
6 g Na2HPO4
5 g NaCl
Autoclave at 121 °C for 20 min, then add 1 ml of sterilized 1 M MgSO4 solution
Store at room temperature
-
-
10% NuSieve GTG agarose pad
Mix well 1.0 g of NuSieve GTG agarose with 10 ml of M9 buffer in a 50-ml screw-cap glass bottle by using a micro spatula
Melt the NuSieve GTG agarose/M9 buffer mixture by using a microwave oven
Cool the 10% NuSieve GTG agarose/M9 buffer mix at room temperature to allow it to solidify
Store at room temperature
-
VALAP
Mix Vaseline, lanolin, and paraffin at a 2:2:1 (weight) ratio in a 100-ml beaker
Cover the beaker with aluminum foil
Melt the contents of the beaker at 130 °C on a hot plate
Store at room temperature
-
Injection recovery solution
Dissolve 2 g of glucose in 50 ml of M9 buffer
Sterilize by membrane filtration
Prepare 500-µl aliquots in 1.5-ml tubes
-
Add 5 µl of 2.0% gelatin solution to the 500-µl aliquot when needed
Note: Gelatin is added to prevent the worm from sticking to the tip.
Store at room temperature
-
20 mg/ml serotonin hydrochloride/M9 buffer
Weigh the serotonin hydrochloride in a 1.5-ml tube
Add M9 buffer to the 1.5-ml tube to prepare a 20 mg/ml solution
Prepare 10-µl aliquots in 1.5-ml tubes
Store in a sealed bag at -20 °C
Note: Storing in a sealed bag prevents the solution from acquiring a brownish color.
-
Fluorescence reference slide
Draw a circle (approximately 5 mm in diameter) on an 18 x 18 mm cover glass by using a diamond pen to indicate the region of the fluorescent protein film. The side with the circle is the upper side of the cover glass
Apply 10 µl of 0.2% BSA-fluorescein solution to the circle on the upper side of the cover glass
Incubate the cover glass in a dark humidified chamber for 30 min at room temperature
Dip the cover glass into approximately 40 ml of pure (Elix) water in a 50-ml beaker to wash it. Repeat this washing step with another approximately 40 ml of water
-
Carefully remove the water drops with a lint-free wipe (Kimwipes)
Note: Do not touch the protein film region with the wipe.
Leave the cover glass to dry at room temperature
Flip and place the cover glass on a glass slide so that the upper side of the cover glass faces the glass slide
Seal the edges of the cover glass with transparent (Scotch) tape
Store at -20 °C in the dark
Acknowledgments
The protocol is used in Takayama and Onami (2016). The protocol for the microinjection of a chemical calcium indicator dye is based on that described by Samuel et al. (2001). The immobilization method is based on the method of Kim et al. (2013). The uneven illumination correction is derived from the work of Wolf et al. (2007). The Calculator_Plus3 plugin was modified from the Calculator_Plus plugin, which was developed by Wayne Rasband and Gabriel Landini. Portions of the ContourTrack3Stack_, ContourTrack4Stack_, and TrackOocyte_ plugin codes were reused from a program written by Takuya Maeda. We thank Asako Sugimoto for her advice regarding the choice of microscopy filters. We also thank Koji Kyoda and Rie Furushima for their assistance. This work was supported, in part, by the National Bioscience Database Center (NBDC) of the Japan Science and Technology Agency (S.O.), by the Strategic Programs for R&D (President’s Discretionary Fund) of RIKEN (S.O.), and by JSPS KAKENHI Grant Number 15K18547 (J.T.). The authors declare that they have no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1.Audhya A., Hyndman F., McLeod I. X., Maddox A. S., Yates J. R. 3rd Desai A. and Oegema K.(2005). A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans . J Cell Biol 171(2): 267-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans T. C.(2006). Transformation and microinjection. In: WormBook(Ed.). The C. elegans Research Community . WormBook. [Google Scholar]
- 3.Frøkjaer-Jensen C., Davis M. W., Hopkins C. E., Newman B. J., Thummel J. M., Olesen S. P., Grunnet M. and Jorgensen E. M.(2008). Single-copy insertion of transgenes in Caenorhabditis elegans . Nat Genet 40(11): 1375-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim E., Sun L., Gabel C. V. and Fang-Yen C.(2013). Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization . PLoS One 8(1): e53419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pohl C. and Bao Z.(2010). Chiral forces organize left-right patterning in C. elegans by uncoupling midline and anteroposterior axis . Dev Cell 19(3): 402-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuel A. D., Murthy V. N. and Hengartner M. O.(2001). Calcium dynamics during fertilization in C. elegans . BMC Dev Biol 1: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takayama J. and Onami S.(2016). The sperm TRP-3 channel mediates the onset of a Ca2+ wave in the fertilized C. elegans oocyte . Cell Rep 15(3): 625-637. [DOI] [PubMed] [Google Scholar]
- 8.Tohsato Y., Ho K. H., Kyoda K. and Onami S.(2016). SSBD: a database of quantitative data of spatiotemporal dynamics of biological phenomena. Bioinformatics 32(22): 3471-3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolf D. E., Samarasekera C. and Swedlow J. R.(2007). Quantitative analysis of digital microscope images. Methods Cell Biol 81: 365-396. [DOI] [PubMed] [Google Scholar]