

Abstract
The enzyme nitrogenase uses a suite of complex metallocofactors to reduce N2 to ammonia. Mechanistic details of this reaction remain sparse. We report a 1.83-Å crystal structure of the nitrogenase molybdenum-iron protein captured under physiological N2-turnover conditions. This structure reveals asymmetric displacements of the cofactor belt-sulfurs (S2B or S3A/S5A) with distinct dinitrogen species in the two αβ-dimers of the protein. The sulfur-displaced sites are distinct in the ability of protein ligands to donate protons to the bound dinitrogen species, as well as the elongation of either the Mo-O5 (carboxyl) or Mo-O7 (hydroxyl) distance that switches the Mo-homocitrate ligation from bidentate to monodentate. These results highlight the dynamic nature of the cofactor during catalysis, and provide evidence for participation of all belt-sulfur sites in this process.
One Sentence Summary:
Asymmetric belt sulfur displacements of nitrogenase cofactor suggest involvement of all belt sulfur sites in N2 reduction.
Nitrogenase catalyzes a key step in the global nitrogen cycle: the ambient reduction of atmospheric dinitrogen (N2) to the bioavailable ammonia (NH3). The molybdenum (Mo) nitrogenase consists of two component proteins (1–3). One, designated the iron (Fe) protein, is a homodimer that contains a [Fe4S4] cluster at the subunit interface and an adenosine triphosphate (ATP)-binding site within each subunit. The other, designated the molybdenum iron (MoFe) protein, is an α2β2 heterotetramer that contains two complex metalloclusters per αβ dimer: a P-cluster ([Fe8S7]) at each α/β interface and an M-cluster (or FeMoco; [(R-homocitrate)MoFe7S9C]) within each α subunit (4, 5). Catalysis by the Mo-nitrogenase involves repeated association and dissociation between the two component proteins, which permits ATP-dependent electron transfer from the [Fe4S4] cluster of the Fe protein, via the P-cluster, to the M-cluster of the MoFe protein, where substrate reduction occurs (1–3). The ability of nitrogenase to shuttle multiple electrons to its cofactor renders the enzyme highly versatile in substrate reduction. Other than N2 and protons (H+), nitrogenase can reduce a variety of alternative substrates, such as acetylene (C2H2), cyanide (CN−), and carbon monoxide (CO) (1, 6, 7).
Considerable efforts have gone into the mechanistic investigation of nitrogenase since its discovery, with a number of studies focused on the substrate and inhibitor interactions of this enzyme (1, 3, 8–10). However, characterization of the substrate- or intermediate-bound states of nitrogenase has proven to be challenging due to the transient nature of these bound species (11). To circumvent this problem, freeze quench spectroscopic techniques, combined with strategic genetic modifications of the active site, have been applied to accumulate and characterize nitrogenase-bound intermediates, leading to the proposal of a reductive elimination/oxidative addition mechanism (3) and a concurrent refinement of the Thorneley-Lowe model (8, 9) for N2 reduction by Mo-nitrogenase. Moreover, high-resolution crystal structures of Mo- and V-nitrogenases have been obtained with ligands (e.g., CO or N/O species) bound to the enzyme (12, 13), pointing to an intriguing mechanism in which a reactive Fe species is generated upon displacement of a specific belt S (S2B) of the cofactor by a μ2-bridging ligand. An elegant crystallographic pulse-chase study using a Se-reporter further demonstrated a catalysis-dependent migration of belt S (14), highlighting the dynamic nature of the entire belt region of the cofactor during turnover. Together, these studies offered invaluable insights into the reaction mechanism of nitrogenase while providing added incentive for exploration of strategies to specifically capture N2 or related intermediate(s) for mechanistic studies.
One strategy worthy of consideration is to limit excess electron supplies that inadvertently drive the reaction of N2-reduction forward, which either reverts the substrate- or intermediate-bound state to the resting state or renders the enzyme in an indiscernible mixed state. This is particularly relevant since nitrogenase proteins are routinely isolated in the presence of excess dithionite, an externally supplied reductant (1). Removal of this artificial electron source, coupled with quick isolation of the protein in the absence of oxygen, could facilitate capture of N2 or intermediate(s) on the protein upon exhaustion of available electrons. To obtain a proof-of-concept for this strategy, we first prepared the crude extract of an Azotobacter vinelandii strain under anaerobic conditions with or without addition of dithionite upon cell disruption. In both cases, the A. vinelandii strain was actively expressing a Mo-nitrogenase comprising a histidine-tagged MoFe protein and a non-tagged Fe protein prior to cell disruption (15, 16). Activity analysis of these samples revealed that, contrary to the dithionite-treated crude extract, the dithionite-free crude extract was nearly inactive in substrate reduction, but its activity could be fully restored upon addition of dithionite and ATP (Fig. S1A). This observation suggests a depletion of electrons (e.g., from ferredoxins) and other metabolites (e.g., ATP) in the crude extract when the metabolic processes that (re)generate these components are interrupted upon cell disruption. Thus, when a nitrogenase-expressing culture that is actively performing N2 fixation is subjected to cell lysis without additional electron supplies, the nitrogenase remains functional but is potentially arrested in a ‘dormant’ N2- or intermediate-bound state due to a withdrawal of electron flow to the M-cluster. Indeed, following a quick one-step purification under N2 from the dithionite-free crude extract, the histidine-tagged MoFe protein (designated Av1*) was not only active in N2 reduction, but also fully functional upon substitution of the gas atmosphere with C2H2 or Ar in an in vitro activity assay where it was combined with the Fe protein (designated Av2), ATP and dithionite (Fig. S1B). More excitingly, GC-MS analysis demonstrated that contrary to Av1 purified with dithionite from a 15N2-grown culture (Fig. S1C, black), Av1* purified without dithionite from the same 15N2-grown culture released 15N2 upon acid quenching (Fig. S1C, red), suggesting capture of N2 or related species on Av1*. Release of 15N2 was not detected from the reductant-free, resting-state Av1 (designated Av1′) upon incubation with 15N2 (Fig. S1C, brown), pointing to the turnover conditions as the prerequisite for capturing N2 on Av1 while providing further support for the physiological relevance of Av1* to catalysis.
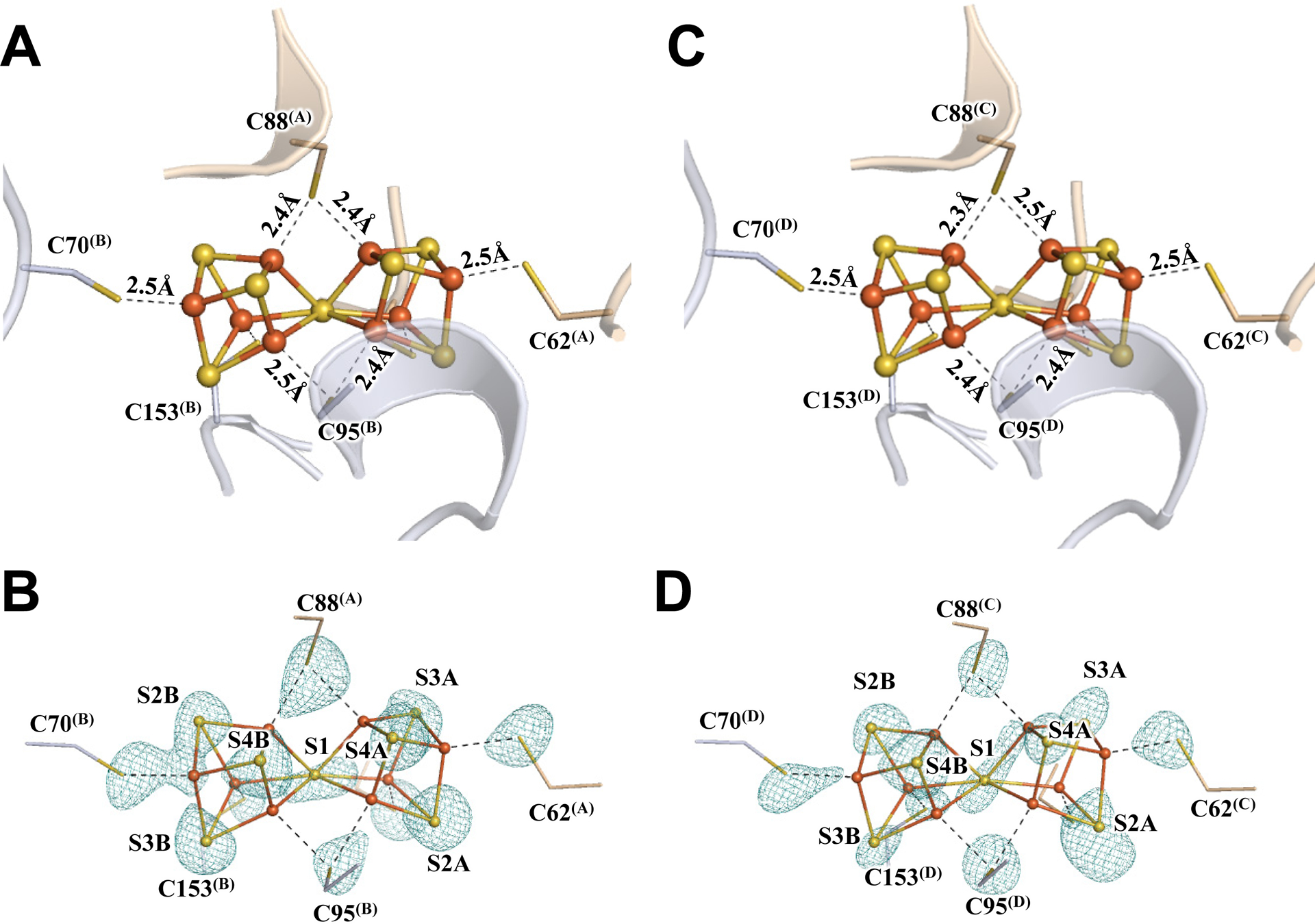
Subsequent crystallization of Av1* yielded brown crystals that diffracted to a resolution of 1.83 Å (Figs. S2–S4; Tables S1–S3). Strikingly, the two P-clusters in the two αβ-dimers of Av1* (designated P-cluster(A/B) and P-cluster(C.D), respectively) are no longer present in the all-ferrous, resting state (PN); instead, both adopt the conformation of the previously reported two-electron-oxidized state of this cluster (POX) (17, 18), with the central S (designated S1) losing two Fe–S bonds and becoming μ4-coordinated to three Fe atoms of the α-subunit cubane and one Fe atom of the β-subunit partial cubane (Fig. 1; Figs. S5, S6). The structural rearrangement of the P-clusters in Av1*, confirmed by anomalous density data calculated at 7100 eV and 7141 eV, is accompanied by formation of two Fe–X bonds with an O atom of α-Ser188 and the backbone N atom of α-Cys88 (Fig. 1; Figs. S5, S6). EPR analysis provided further support for this assignment, showing a POX-specific, g = 11.8 signal in the spectrum of Av1* (Fig. S1D) (19). The POX signal of Av1* is indistinguishable in intensity to that of an equimolar amount of Av1 treated with excess oxidant, suggesting that all P-clusters of Av1* exist in the POX state (Fig. S1D). The presence of the P-clusters of Av1* in the POX state not only provides the long-sought-after answer to the physiological relevance of this state, but also points to a limited electron flow from the P-cluster to the M-cluster due to the absence of dithionite, leaving the former in an electron-depleted state while permitting the latter to capture N2 and/or intermediate(s) under limited turnover conditions.
Fig. 1. The oxidized P-clusters in Av1*.

Structures of P-clusters at (A–C) Chain-A/B interface (P-cluster(A/B)) and (D–F) Chain-C/D interface (P-cluster(C/D)) of Av1*. Chain-A and Chain-C are the α-subunits, and Chain-B and Chain-D are the β-subunits of the two αβ-dimers of Av1*. (A, D) The P-clusters are shown in ball-and-stick presentation, and the key residues interacting with the P-clusters are indicated as sticks. Chain-A and Chain-C are shown as wheat ribbons, and Chain-B and Chain-D are shown as light-blue ribbons. (B, C) P-cluster(A/B) and (E, F) P-cluster(C/D) superimposed with (B, E) the anomalous density maps calculated at 7100 eV at a resolution of 2.18 Å and contoured at 4.0 σ, showing the position of sulfur atoms (mint-blue mesh); and with (C, F) the anomalous density maps calculated at 7141 eV at a resolution of 2.1 Å and contoured at 15.0 σ, showing the position of iron atoms (red mesh). Color code of atoms: Fe, orange; S, yellow; O, red; N, blue.
Consistent with this suggestion, in the α-subunit (designated Chain-A) of one αβ-dimer of Av1*, the absence of a belt S (S2B) from the M-cluster (designated M-cluster(A)) can be clearly and reproducibly visualized in the anomalous difference Fourier map calculated with diffraction data at 7100 eV (Fig. 2A, B; also see Fig. S7A, B); yet, there is electron density at this location based on analysis of the 2Fo-Fc and Fo-Fc maps (Fig. S8A), which can be modeled as a dinitrogen species that binds in a pseudo μ1,2 bridging mode between Fe2 and Fe6 (Fig. 2C; see Figs. S9, S10 for ligand modeling). The proximal (Nprox) and distal (Ndist) nitrogen atoms of this species are located at 1.8 Å and 2.3 Å, respectively, to Fe2 and Fe6, rendering it roughly parallel to the M-cluster along the Fe1–C–Mo axis (Fig. 2C; Table S3). Additionally, Nprox interacts with the Nε group of α-H195(A), a residue implicated in N2 reduction (20), via hydrogen bonding at 2.9 Å (Fig. 2C).
Fig. 2. The nitrogen ligand-bound M-clusters in Av1*.
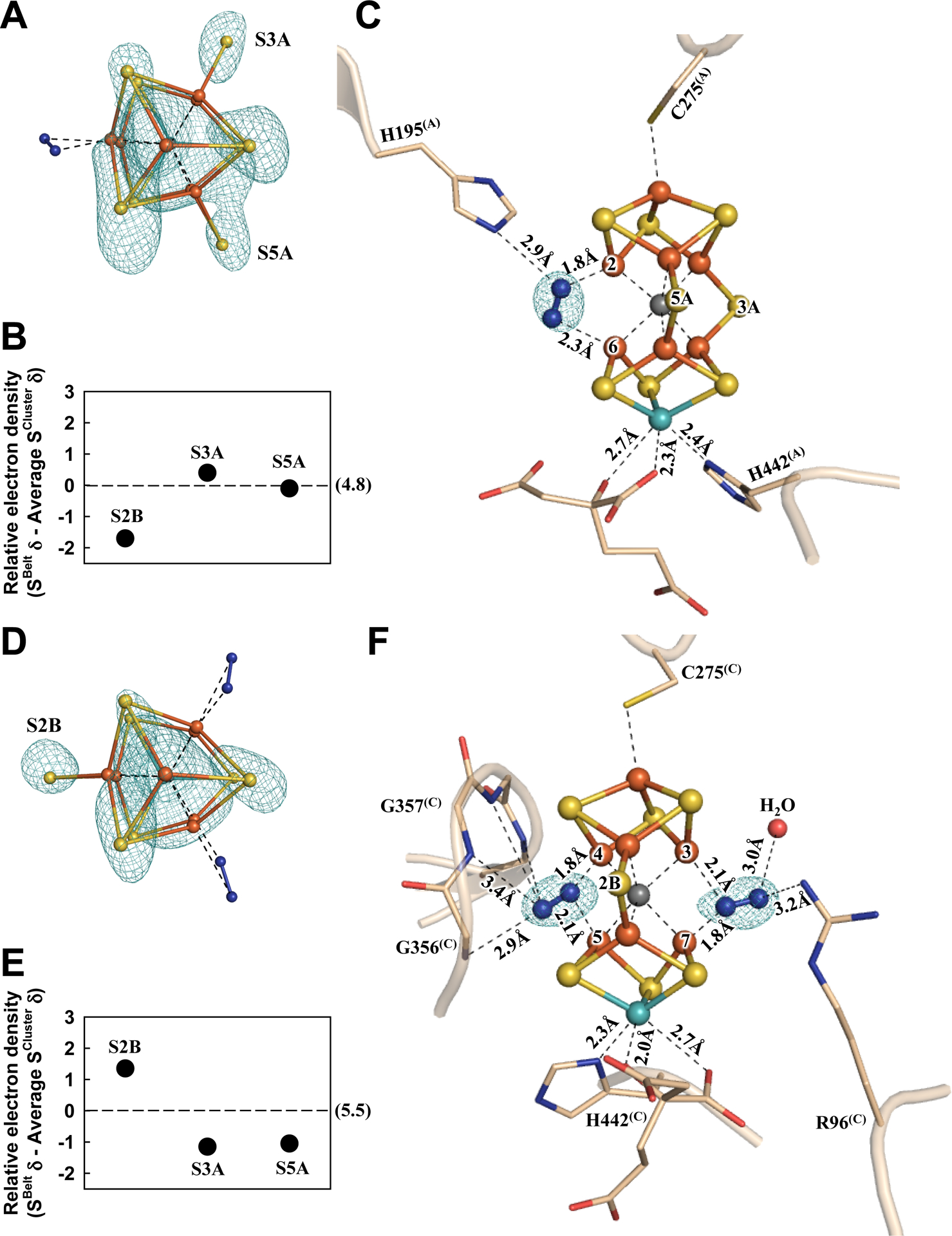
Structures of M-clusters in (A–C) Chain-A (M-cluster(A)) and (D–F) Chain-C (M-cluster(C)) refined at a resolution of 1.83 Å. View along the Fe1-C-Mo direction of (A) M-cluster(A) and (D) M-cluster(C) superimposed with the anomalous density maps (mint-blue mesh) calculated at 7100 eV at a resolution of 2.18 Å and contoured at 4.0 σ (A) and 5.3 σ (D), respectively, showing the displacement of sulfur at the S2B site in M-cluster(A) (A) and at the S3A and S5A sites in M-cluster(C) (D). Electron densities of the belt sulfurs (SBelt) relative to the average density of cluster sulfurs (SCluster) in (B) Chain-A and (E) Chain-C, respectively, expressed in sigma values. The average sigma values of the cluster sulfurs in Chain-A and Chain-C are 4.8 and 5.5, respectively (B, E). Side view of (C) M-cluster(A) and (F) M-cluster(C) with key residues interacting with the clusters and the bound nitrogen ligands indicated as sticks. M-cluster(A) and M-cluster(C) are superimposed with the Fo-Fc omit map of the nitrogen ligands contoured at 10 σ (mint-blue mesh). The peptides and atoms are colored as those in Fig. 1.
Sulfur anomalous density (designated S(A/B)) is observed at the same sulfur-binding-site (SBS) at the α/β-subunit interface as that identified earlier in the S2B-displaced, CO-bound structure of the M-cluster (12). Located ~22 Å away from the S2B-site in Chain-A of Av1* (Fig. S11A, B), the relevance of this sulfur species to the displaced S2B-sulfur is yet to be established. Displacement of S2B with the dinitrogen species, like that of S2B with CO, does not have much impact on the structure of the M-cluster, as both ligand-bound clusters show an overall conservation of the Fe–S, Fe---Fe, and Mo–S distances as those in the resting-state cofactor (Fig. S12; Table S3) (5). However, contrary to its CO-bound and resting-state counterparts (12), there is a significant change in the ligation of Mo by homocitrate in Chain-A of Av1*. While the Mo-O5 (carboxyl) distance remains largely unchanged, there is a notable elongation of the Mo-O7 (hydroxyl) distance, thereby switching the ligation of Mo by homocitrate from bidentate in the resting-state and CO-bound structures (Mo–O5, 2.2 Å; Mo–O7, 2.2 Å) to monodentate in Chain-A of Av1* (Mo–O5, 2.3 Å; Mo–O7, 2.7 Å) (Fig. 2C).
Surprisingly, in the M-cluster (designated M-cluster(C)) of the α-subunit (designated Chain-C) of the second αβ-dimer, sulfur is clearly and reproducibly visualized at the S2B position but not at the S3A and S5A positions in the anomalous difference Fourier map calculated from diffraction data at 7100 eV (Fig. 2D, E; also see Fig. S7C, D). Like the S2B position in Chain-A, the electron densities observed at the S3A and S5A positions based on analysis of the 2Fo-Fc and Fo-Fc maps of Chain-C (Fig. S8B) can each be modeled with a dinitrogen species with a proximal (Nprox) and a distal (Ndist) nitrogen relative to the cluster Fe atoms (Fig. 2F; also see Figs. S9, S10). At the S3A site, Nprox is located at 1.8 Å and 2.1 Å, respectively, to Fe4 and Fe5; whereas at the S5A site, Nprox is located at 2.1 Å and 1.8 Å, respectively, to Fe3 and Fe7 (Fig. 2F; Table S3). As such, the dinitrogen species at S3A or S5A appears to form an asymmetric μ1,1 bridge between two Fe centers, with a stronger (1.8 Å) and a weaker (2.1 Å) interaction of Nprox with a primary (Fe4 or Fe7) and a secondary (Fe5 or Fe3) Fe center. In both cases, the distal nitrogen atoms are directed away from the M-cluster, rendering the two nitrogen atoms in a somewhat linear alignment with the primary Fe center. Other than interacting with the cluster, the dinitrogen species at the S3A site has potential hydrogen bonding interactions with the amide groups of the protein backbone (α-G356(C) and α-G357(C); Ndist−Namide=2.9–3.4 Å); whereas the dinitrogen species at the S5A site has potential hydrogen bonding interactions with the side chain of α-R96(C) (Ndis−NR96=3.2 Å), a residue implicated in N2 reduction (21), and a water molecule (Fig. 2F). As observed in Chain-A/B, sulfur anomalous density (designated S(C/D)) appears at the same SBS at the α/β-subunit interface of Chain-C/D, which is ~19 Å and 25 Å, respectively, away from the S5A- and S3A-sites (Fig. S11C, D). Additionally, the M-cluster in Chain-C adopts a conformation that closely resembles the resting-state structure (Fig. S13) (5). Finally, there is also a clear switch of the ligation of Mo by homocitrate from bidentate to monodentate in Chain-C. However, the two Mo–O distances in Chain-C undergo changes opposite to those in Chain-A, with the Mo–O5 (carboxyl) distance lengthened to 2.7 Å and the Mo–O7 (hydroxyl) distance shortened to 2.0 Å (Fig. 2F).
To further validate the sulfur-displaced conformation captured in Av1* under a limited electron flux, Av1* was incubated with Av2, ATP and excess dithionite, and re-isolated from the incubation mixture. Crystallization of this re-isolated Av1 protein (designated Av1*(TOD); TOD, TurnOver with Dithionite) yielded brown crystals that diffracted to a resolution of 1.73 Å (Figs. S14, S15; Tables S1, S3, S4). Both P-clusters of Av1*(TOD) adopt the conformation of the reduced PN state (Fig. 3; Figs. S16, S17); whereas both M-clusters of Av1*(TOD) adopt the resting-state conformation, with all three belt sulfurs in place and homocitrate assuming bidentate ligation to Mo via O5 and O7 (Fig. 4; Figs. S18, S19). These observations are consistent with the bound dinitrogen species being turned over and subsequently released from the M-clusters, as well as the inability of the M-clusters to (re)capture N2 or N2-derived intermediates in the presence of excess reductant, which inadvertently reverts both P- and M-clusters in Av1*(TOD) to the well-established resting-state conformations. The ability of Av1* to resume turnover further illustrates the physiological relevance of this conformation to catalysis. Moreover, the ‘return’ of belt sulfurs to both M-clusters in Av1*(TOD) provides strong validation for the asymmetric displacement of the belt sulfurs in the two M-clusters of Av1*.
Fig. 3. The reduced P-clusters in Av1*(TOD).
Structures of P-clusters at (A, B) Chain-A/B interface (P-cluster(A/B)) and (C, D) Chain-C/D interface (P-cluster(C/D)) of Av1*(TOD). Chain-A and Chain-C are the α-subunits, and Chain-B and Chain-D are the β-subunits of the two αβ-dimers of Av1*(TOD). (B, D) P-clusters superimposed with the anomalous density maps calculated at 7100 eV at a resolution of 2.17 Å and contoured at 4.5 σ, showing the position of sulfur atoms (mint-blue mesh). The peptides and atoms are colored as those in Fig. 1.
Fig. 4. The M-clusters in Av1*(TOD).
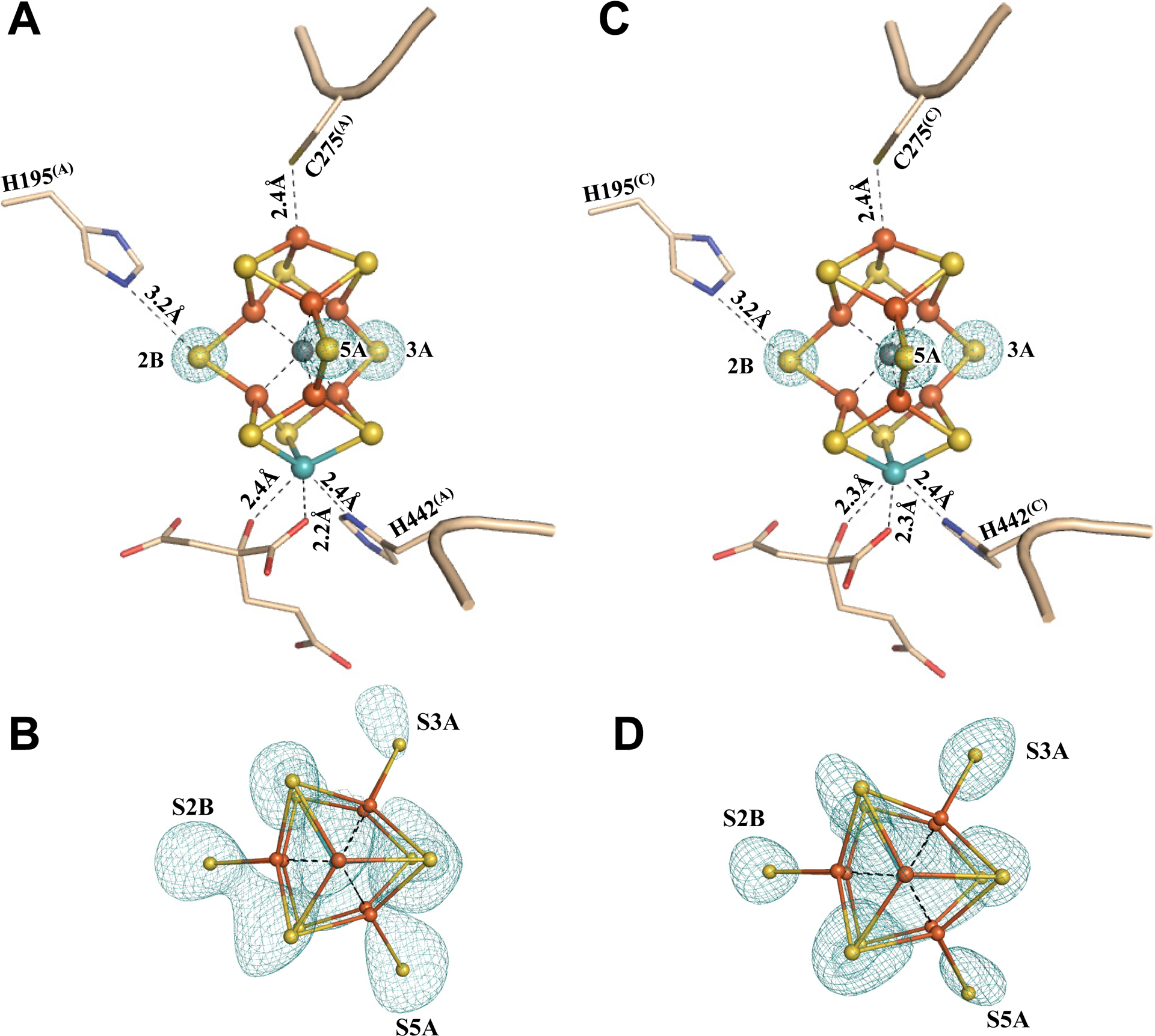
Structures of M-clusters in (A, B) Chain-A (M-cluster(A)) and (C, D) Chain-C (M-cluster(C)) refined at a resolution of 1.73 Å. Side view of (A) M-cluster(A) and (C) M-cluster(C) with key residues interacting with the clusters and the bound nitrogen ligands indicated as sticks. M-cluster(A) and M-cluster(C) are superimposed with the Fo-Fc omit maps of the belt sulfurs contoured at 13 σ (mint-blue mesh). View along the Fe1-C-Mo direction of (B) M-cluster(A) and (D) M-cluster(C) superimposed with the anomalous density maps calculated at 7100 eV at a resolution of 2.17 Å and contoured at 4.0 σ, showing the presence of the anomalous sulfur density (mint-blue mesh) at all belt sulfur positions (S2B, S3A and S5A) in (B) M-cluster(A) and (D) M-cluster(C). The peptides and atoms are colored as those in Fig. 1.
Our observation that the S2B, S3A and S5A sites can each be displaced by a dinitrogen species is consistent with the previously observed ‘migration’ of a Se-reporter in the entire belt region upon turnover (14) and points to a possible involvement of all three sites in N2 reduction. At the S3A site, the backbone amides of α-G356(C) and α-G357(C) that interact with the dinitrogen species are unlikely to serve as proton donors for N2 reduction. At the S2B site, however, there is a hydrogen bond between α-H195(A) and the dinitrogen species that could provide protons for N2 reduction. Additionally, an elongation of the Mo–O7 (hydroxyl) distance to 2.7 Å would be consistent with a protonation event that breaks this bond in exchange for N2 reduction. At the S5A site, the dinitrogen species seems primed to accept protons from hydrogen bonds with α-R96(C) and a nearby water molecule. Moreover, an elongation of the Mo–O5 (carboxyl) distance to 2.7 Å allows O5 to participate in a hydrogen bond with a nearby water molecule that is positioned at 2.5 Å from O5 and ~4 Å from the S5A site. With ample proton sources available, the dinitrogen species at S2B and S5A are likely more protonated/reduced than N2. Interestingly, the binding conformation of dinitrogen species at S2B is similar to the cis-(μ1,2) binding mode of a diazene adduct to a synthetic compound (22, 23). Moreover, modelling of allowable N–N bond distances suggests the possibility of having diazene-level species bound at both the S2B and S5A sites while disfavoring the presence of a hydrazine-level species at the S5A site (Fig. S9). While many mechanisms can be proposed to explain our observations (Figs. S20–S24; also see discussion in Supplementary Materials), experimental support is yet to be acquired for any of these proposals. The possibility that all belt sulfur sites are involved in catalysis, stemming from our observation asymmetric belt sulfur displacements in the two αβ-dimers of Av1, should provoke a (re)calibration in the mechanistic thinking of nitrogenase, with an ultimate goal to elucidate the intricate mechanism of enzymatic N2 reduction.
Supplementary Material
Acknowledgments
We thank Professor Douglas Rees (Caltech) for insightful discussions and Dr. Jens Keiser (Molecular Observatory at Caltech) for technical assistance.
Funding:
This work was supported by NIH-NIGMS grant GM67626 (to M.W.R. and Y.H.), which funded the crystallographic set-up in the Ribbe/Hu laboratories and the development of reductant-free protein purification strategies for work related to the assembly and catalysis of nitrogenase. The authors were also supported by the Department of Energy grant DOE(BES) DE-SC0016510 (to M.W.R. and Y.H.), which funded work related to the mechanistic investigation of ammonia synthesis by nitrogenase and its homologs. We also wish to acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis Bioengineering Research Program at Caltech for their generous support of the Molecular Observatory at Caltech. Operations at SSRL are supported by DOE and NIH.
Footnotes
Data and materials availability:
All data are available in the manuscript or the Supplementary Materials. The structural models have been deposited in the Protein Data Bank with accession codes 6UG0 and 6VXT. The anomalous structure factors for 6UG0 and 6VXT are archived in Zenodo (24).
References and Notes
- 1.Burgess BK, Lowe DJ, Chem. Rev 96, 2983–3012 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Rees DC et al. , Phil. Trans. R. Soc. A 363, 971–984 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC, Chem. Rev 114, 4041–4062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiig JA, Hu Y, Lee CC, Ribbe MW, Science 337, 1672–1675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spatzal T et al. , Science 334, 334 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee CC, Hu Y, Ribbe MW, Science 329, 642–642 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Y, Lee CC, Ribbe MW, Science 333, 753–755 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowe DJ, Thorneley RN, Biochem J. 224, 877–886 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorneley RN, Lowe DJ, Biochem J. 224, 887–894 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cameron LM, Hales BJ, Biochemistry 37, 9449–9456 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Lee SC, Holm RH, Proc. Natl. Acad. Sci. U.S.A 100, 3595–3600 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC, Science 345, 1620–1623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sippel D et al. , Science 359, 1484–1489 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Spatzal T, Perez KA, Howard JB, Rees DC, Elife 4, e11620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. See Supplementary Materials online for more details.
- 16.Hu Y, Fay AW, Schmid B, Makar B, Ribbe MW, J, Biol, Chem. 281, 30534–30541 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Peters JW et al. , Biochemistry 36, 1181–1187 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Owens CP, Katz FE, Carter CH, Oswald VF, Tezcan FA, J. Am. Chem. Soc 138, 10124–10127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierik AJ, Wassink H, Haaker H, Hagen WR, Eur. J. Biochem 212, 51–61 (1993). [DOI] [PubMed] [Google Scholar]
- 20.Kim C-H, Newton WE, Dean DR, Biochemistry 34, 2798–2808 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Benton PM et al. , Biochemistry 40, 13816–13825 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Chen Y et al. , J. Am. Chem. Soc 133, 1147–1149 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Li Y et al. , Nat. Chem 5, 320–326 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Kang W, Lee CC, Jasniewski AJ, Ribbe MW, Hu Y, Version 1.0, Zenodo (2020); 10.5281/zenodo.3756201. doi: 10.5281/zenodo.3756201 [DOI] [Google Scholar]
- 25.Ribbe MW, Hu Y, Guo M, Schmid B, B. K., Burgess, J. Biol. Chem 277, 23469–23476 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Burgess BK, Jacobs DB, Stiefel EI, Biochim. Biophys. Acta 614, 196–209 (1980). [DOI] [PubMed] [Google Scholar]
- 27.Lee CC, Ribbe MW, Hu Y, Methods Mol. Biol 1876, 111–124 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Gavini N, Burgess BK, J. Biol. Chem 267, 21179–21186 (1992). [PubMed] [Google Scholar]
- 29.Fishman MJ, Skougstad MV, Anal. Chem 36, 1643–1647 (1964). [Google Scholar]
- 30.Chaikuad A, Knapp S, von Delft F, Acta Crystallogr. D Biol. Crystallogr 71, 1627–1239 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W, Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Karplus PA, Diederichs K, Science 336, 1030–1033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winn MD et al. , Acta Crystallogr. D Biol. Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams PD et al. , Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Lohkamp B, Scott WG, Cowtan K, Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheldrick GM, Acta Crystallogr. D Biol. Crystallogr 66, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorn A, Sheldrick GM, J. Appl. Crystallogr 44, 1285–1287 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao L, Caldararu O, Ryde U, J. Phys. Chem. B 121, 8242–8262 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Siegbahn PEM, Inorg. Chem 57, 1090–1095 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Harris TV, Szilagyi RK, Inorg. Chem 50, 4811–4024 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Junge W, Nelson N, Annu Rev Biochem. 84, 631–657 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Guo H, Bueler SA, Rubinstein JL, Science 358, 936–940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Y, Ribbe MW, Annu. Rev. Biochem 85, 455–483 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Ribbe MW, Hu Y, Hodgson KO, Hedman B, Chem. Rev 114, 4063–4080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siegbahn PEM, Phys. Chem. Chem. Phys 21, 15747–15759 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the manuscript or the Supplementary Materials. The structural models have been deposited in the Protein Data Bank with accession codes 6UG0 and 6VXT. The anomalous structure factors for 6UG0 and 6VXT are archived in Zenodo (24).