

Abstract
Using the updated module boundary of polyketide assembly lines, modules from the pikromycin synthase were recombined into engineered synthases that furnish an enantiomeric pair of 2-stereocenter triketide lactones at >99% ee with yields up to 0.39 g per liter of E. coli K207–3 in shake flasks.
Graphical Abstract
Modular polyketide synthases (PKSs) are widely regarded as the enzymatic champions of synthetic organic chemistry because they excel at forging carbon-carbon bonds and setting stereocenters.1, 2 Since their discovery, scientists have endeavored to engineer them to produce designer polyketides.3 However, with only a partial understanding of their biosynthetic logic and architecture, most efforts have resulted in nonfunctional synthases. When the desired molecule was produced, it was usually at a low titer (<5 mg per liter of culture).4, 5
Recent developments in our understanding of assembly line logic and architecture prompted our group to engineer model synthases. A study of related aminopolyol synthases suggested that the set of domains that evolutionarily co-migrate differs from the traditional definition of the module.6–8 The carbon-carbon bond-forming ketosynthase (KS) genetically travels with the processing enzymes [e.g., ketoreductase (KR), dehydratase (DH), enoylreductase (ER)] that are upstream of it, rather than downstream. This was surprising to many since these processing enzymes and the KS downstream of them commonly reside on different polypeptides and solved structures show rigid connections between KS and downstream domains.9–13 However, at the most downstream position of the module, the KS can gatekeep to ensure that reactions, such as dehydration or epimerization, occur before the polyketide chain is passed to the next module. Other studies have also indicated that KSs are most active when paired with the acyl carrier proteins (ACPs) naturally upstream of them and that these domains are genetically connected.14, 15 We experimented with hybrid synthases constructed from the venemycin and pikromycin PKSs and determined that synthases designed with the updated module boundary outperform those with the traditional module boundary.16
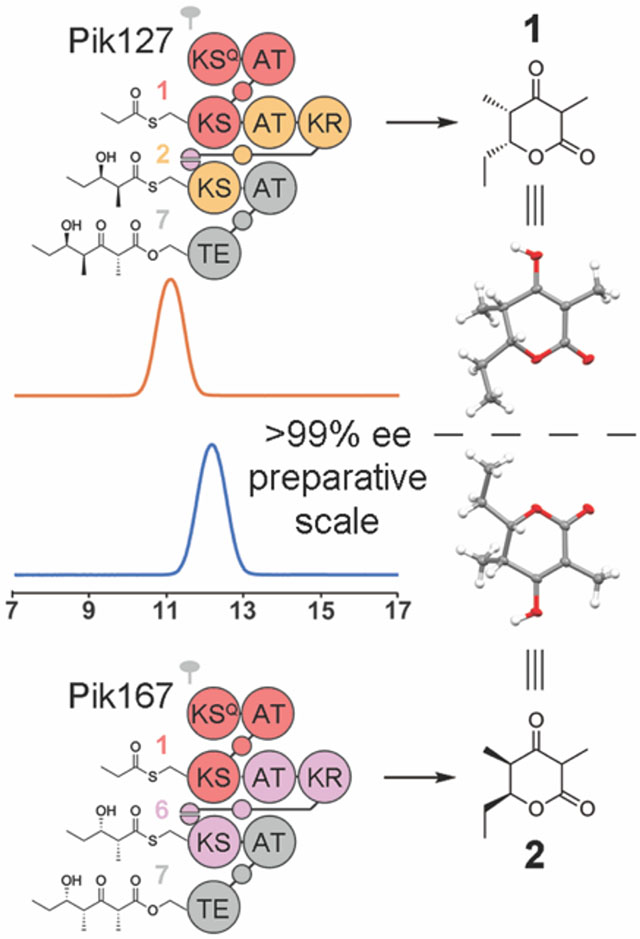
While these previously described hybrid synthases are informative, the aromatic products they produce are not representative of the stereochemically rich compounds biosynthesized by most polyketide assembly lines. Moreover, they do not provide an opportunity to study the enzymes that generate stereochemistry.17 We therefore sought to recombine modules from the pikromycin PKS to engineer KR-containing synthases that produce an enantiomeric pair of chiral triketide lactones.18 Based on how they forge stereocenters within the pikromycin synthase, the KRs from the second and sixth modules would be expected to generate substituents with l- and d-orientations, respectively, in an engineered assembly line.17, 19 Thus, if modules 1, 2, and 7 were combined into one synthase (Pik127) and modules 1, 6, and 7 were combined into another synthase (Pik167), they would theoretically produce the enantiomeric triketide lactones, 1 and 2, respectively (Figure 1).
Figure 1.

The pikromycin PKS and triketide synthases engineered from it. Each polyketide assembly line is colored using the updated module boundary (the traditional boundary is upstream of the KS domain). The pikromycin PKS generates narbonolide, the carbon scaffold of pikromycin, while the engineered synthases, Pik127 and Pik167, are anticipated to produce the triketide enantiomers, 1 and 2, respectively. Unlabeled circles and half-circles represent acyl carrier protein (ACP) domains and docking domain (DD) motifs, respectively. The twofold symbol indicates that modular PKSs are homodimeric.
Expression plasmids encoding these synthases were constructed using amplicons from S. venezuelae ATCC 15439 genomic DNA (Tables S1–2).20 To facilitate protein purification, histidine tags were fused to the upstream and downstream termini of Pik127.21 Since the sixth module is split between 2 polypeptides that noncovalently assemble through docking domain (DD) motifs, Pik167 is encoded on 2 genes.22 These were placed in vectors such that the histidine tags used in the purification are at the assembly line termini, distant from the DD interface.
Proteins were expressed in E. coli K207–3 cells, which harbor the PKS-activating Bacillus subtilis phosphopantetheinyl-transferase Sfp, and purified by nickel chromatography (Figure S1).23, 24 For in vitro functional assessment, methylmalonyl-CoA extender units and NADPH were generated in situ using Streptomyces coelicolor MatB and B. subtilis glucose dehydrogenase (GDH), respectively.25, 26 After incubating the synthases with methylmalonate, CoA, ATP, MgCl2, NADP+, d-glucose, MatB, and GDH, the expected masses were detected by high-resolution mass spectrometry (HRMS): found [M+H]+, m/z 171.10236 for the Pik127 product and 171.10166 for the Pik167 product; expected [M+H]+, m/z 171.1021 (Figure 2). As confirmation that these masses were from triketide lactones, a +9 shift was observed for both products when 13C4-methylmalonate was used.
Figure 2.

High-resolution mass spectra of triketide lactones from in vitro reactions. Pik127 and Pik167 incubated with unlabeled methylmalonate (left) and 13C4-labeled methylmalonate (right). The [M−H2O+H]+, [M+H]+, and [M+Na]+ adducts were observed for each product.
E. coli K207–3 was also employed to obtain preparative quantities of polyketide products, as this engineered strain converts propionate supplied to the media to (2S)-methylmalonyl-CoA. Similar conditions to those used to produce 0.2 g of 6-dEB per liter of culture in shake flasks were used for Pik167.24, 27 HPLC analysis of ethyl acetate extracts showed that the area of the major peak (λmax=247 nm) exponentially increased over 6 d (Figures 3 and S2a). After the corresponding compound was purified from the culture broth, 1H-NMR analysis confirmed the isolation of a molecule possessing the splitting patterns expected for 2.28 As only tens of milligrams were produced per liter of culture, the composition of the media and culture conditions were optimized. The buffering agent HEPES was replaced with cost-effective potassium phosphate without loss in productivity (Figure S2b). Covering the flasks with milk filter disks instead of aluminum foil increased production 4-fold (Figure S2c). A culture volume of 300 mL was found to be optimal for non-baffled 2.8 L Fernbach flasks (Figure S2d). Lowering the temperature from 22 °C to 19 °C also boosted production 3-fold (Figure S2e). The optimized culture conditions enabled the production of 0.39 ± 0.02 g L−1 (Figures 3 and S3), superior to the highest triketide production reported for actinomycete hosts [0.10 g L−1 from the first polypeptide of the erythromycin PKS fused to its thioesterase (TE)].29–33 In contrast, only 0.011 ± 0.001 g L−1 of a molecule consistent with 1 was produced from cells expressing Pik127. As the large size of the polypeptide (412 kDa) could adversely affect its expression or folding, the DD motifs from the sixth module were inserted into the second module. The resulting 2-polypeptide Pik127 generated 0.22 ± 0.02 g L−1 of a molecule consistent with 1. The products from the 2-polypeptide Pik127 and Pik167 were purified by silica gel flash chromatography and characterized by NMR (Figures S4–7).
Figure 3.

Engineered synthases and their productivities in vivo. Ethyl acetate extracts of culture broths were analyzed by HPLC with a C18-column. Diamonds indicate triketide lactone peaks. a) The 1-polypeptide Pik127, 2-polypeptide Pik127, and Pik167 were designed with the updated and traditional module boundaries (left and right, respectively). Those designed with the updated boundaries were >10-, 3.3-, and 2-fold more active, with the updated Pik167 producing 0.39 g L-1. Cultivations were performed in triplicate (measurements and standard deviations in Source Data 2), and productivity was calculated from peak areas using a standard curve (Figure S3, Source Data 1).
The triketide lactones from Pik127 and Pik167 migrate distinctly from one another on a Chiralcel OD-RH column connected to a TOF LC/MS, and, based on the chromatograms, were generated at >99% ee (Figure 4). No diastereomers were observed by liquid chromatography or NMR spectroscopy. A diketide shunt product is detectable from each of the synthases; however, these β-hydroxy acids are produced in low quantities (Figure S8). More experiments are needed to determine the mechanisms of their formation. The absolute configuration of each triketide lactone was determined by X-ray crystallography through the anomalous scattering of the oxygen atoms (Figures 4 and S9).
Figure 4.

Chiral LC/MS analysis and crystal structures of triketide lactones from 2-polypeptide synthases designed with the updated and traditional module boundary (left and right, respectively). Extracted ion chromatograms (EIC, m/z 171–172) from positive ion mode electrospray ionization show the enantiomeric purity of 1 and 2.
Pik127 and Pik167 serve as platforms to study not only the function of PKSs but also their engineering. We sought to compare synthases engineered with the updated and traditional module boundaries. The 1- and 2-polypeptide Pik127 as well as Pik167 were constructed with the traditional module boundary, and their productivities were compared with their counterparts constructed with the updated module boundary (Figure 3, Tables S1–2, Source Data 1–2). The synthases designed with the traditional module boundary generated the same products as those designed with the updated module boundaries but at a slower rate. The 1-polypeptide Pik127 designed with the traditional module boundary was >10-fold less active. The 2-polypeptide Pik127 and Pik167 designed with the traditional module boundary were also less active (3.3- and 2.0-fold). Synthases designed with the updated boundaries also produced proportionally less diketide shunt product (Figure S8). If the synthetic power of modular PKSs could be reliably harnessed, the production of stereochemically complex polyketide fragments would be revolutionized. The presented engineered synthases achieve >99% ee in the syntheses of an enantiomeric pair of 2-stereocenter triketide lactones. Few synthetic reactions that set a single stereocenter surpass 99% ee.34 In addition to being stereocontrolled, the syntheses are cost-effective, performed under ambient conditions, enviromentally friendly, and scalable.
As our understanding of the collaboration between PKS domains and modules improves, so does our ability to engineer assembly lines. For example, applying the updated module boundary has afforded greater polyketide titers. In comparison with our previous studies on hybrid venemycin/pikromycin synthases that showed up to 48-fold greater activity for synthases designed with the updated module boundary, the 2- to 10-fold difference reported here may seem incongruous. We hypothesize that differences in activity between synthases constructed with the traditional boundary and those constructed with the updated boundary primarily result from KS gatekeeping. In the 2-polypeptide Pik127 designed with the traditional boundary the KS from the sixth pikromycin module accepts a diketide with the α-methyl and β-hydroxyl groups in l-orientations, rather than its native hexaketide containing the same substituents in d-orientations. In Pik167 designed with the traditional boundary the KS from the fifth pikromycin module accepts a propionyl group, rather than its native pentaketide substrate. That these synthases still generate over 60 mg L−1 with >99% ee indicates these KSs are not highly selective for stereochemistry or length. Studies have already shown that the KS from the sixth pikromycin module is tolerant to unnatural intermediates.5 Some KSs (e.g., those of the mycolactone and rapamycin PKSs) may be less selective gatekeepers and desirable within engineered synthases.35–37 Structural studies that reveal which ACP/KS contacts should be preserved in hybrid synthases will also aid PKS engineering.38–41
Titers from Pik127 and Pik167 in E. coli K207–3 can be further increased. The culture conditions reported here are still being optimized. The stoichiometry of synthase polypeptides could be balanced, perhaps through adjusting promoter strengths. Moving from shake flasks to bioreactors will also likely boost polyketide levels, since the 6-dEB production studies showed increased titers in this format (1.1 vs. 0.2 g L−1).27 The conditions reported here could also be used to augment polyketide production from other PKSs heterologously expressed in E. coli.42
We rationally constructed triketide synthases that produce the anticipated enantiomeric products on the gram scale. This work opens up many directions for further studies and applications. While Pik127 and Pik167 can serve as platforms to investigate KRs, other model synthases containing DHs and ERs should be constructed to learn more about these stereochemistry-generating enzymes. Preparative levels of larger polyketides should also be obtainable from engineered synthases since E. coli K207–3 has already been used to generate gram-scale quantities of the heptaketide 6-dEB.27 The incorporation of modules from trans-AT and nonribosomal peptide assembly lines may further increase the functional diversity of the products.2, 43 While the generation of diketide and triketide chiral building blocks as starting materials can already aid total synthesis, it may soon be possible to directly access derivatives of polyketide drug leads to accelerate the process of medicinal chemistry.
Supplementary Material
Acknowledgments
Funding was provided by the NIH (GM106112 and GM077437) and the Welch Foundation (F-1712). Kaan Kumru helped construct the expression plasmids. Vincent M. Lynch, the director of the X-ray Facility at The University of Texas at Austin, solved the triketide lactone crystal structures.
Footnotes
Conflict of interest
The authors have no conflicts of interest to declare.
Electronic Supplementary Information (ESI) available: Methods and characterization. See DOI: 10.1039/d1cc03073f
Notes and references
- 1.Keatinge-Clay AT, Chem Rev, 2017, 117, 5334–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helfrich EJN and Piel J, Nat Prod Rep, 2016, 33, 231–316. [DOI] [PubMed] [Google Scholar]
- 3.Weissman KJ and Leadlay PF, Nat Rev Microbiol, 2005, 3, 925–936. [DOI] [PubMed] [Google Scholar]
- 4.Klaus M and Grininger M, Nat Prod Rep, 2018, 35, 1070–1081. [DOI] [PubMed] [Google Scholar]
- 5.Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, Hopwood DA and Santi DV, Nat Biotechnol, 2005, 23, 1171–1176. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Hashimoto T, Qin B, Hashimoto J, Kozone I, Kawahara T, Okada M, Awakawa T, Ito T, Asakawa Y, Ueki M, Takahashi S, Osada H, Wakimoto T, Ikeda H, Shin-Ya K and Abe I, Angew Chem Int Ed Engl, 2017, 56, 1740–1745. [DOI] [PubMed] [Google Scholar]
- 7.Keatinge-Clay AT, Angew Chem Int Ed Engl, 2017, 56, 4658–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donadio S, Staver MJ, McAlpine JB, Swanson SJ and Katz L, Science, 1991, 252, 675–679. [DOI] [PubMed] [Google Scholar]
- 9.Tang Y, Kim CY, Mathews II, Cane DE and Khosla C, Proc Natl Acad Sci U S A, 2006, 103, 11124–11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang Y, Chen AY, Kim CY, Cane DE and Khosla C, Chem Biol, 2007, 14, 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whicher JR, Smaga SS, Hansen DA, Brown WC, Gerwick WH, Sherman DH and Smith JL, Chem Biol, 2013, 20, 1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bretschneider T, Heim JB, Heine D, Winkler R, Busch B, Kusebauch B, Stehle T, Zocher G and Hertweck C, Nature, 2013, 502, 124–128. [DOI] [PubMed] [Google Scholar]
- 13.Wang JL, J.; Chen L; Zhang W; Kong L; Peng C; Su C; Tang Y; Deng Z; Wang Z, Nat Commun, 2021, 12, 867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandran SS, Menzella HG, Carney JR and Santi DV, Chem Biol, 2006, 13, 469–474. [DOI] [PubMed] [Google Scholar]
- 15.Vander Wood DA and Keatinge-Clay AT, Proteins, 2018, 86, 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazawa T, Hirsch M, Zhang Z and Keatinge-Clay AT, Nat Commun, 2020, 11, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keatinge-Clay AT, Nat Prod Rep, 2016, 33, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue Y, Zhao L, Liu HW and Sherman DH, Proc Natl Acad Sci U S A, 1998, 95, 12111–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng JT and Keatinge-Clay AT, Medchemcomm, 2013, 4, 34–40. [Google Scholar]
- 20.Motohashi K, Methods Mol Biol, 2017, 1498, 349–357. [DOI] [PubMed] [Google Scholar]
- 21.Miyazawa T, Hirsch M, Zhang ZC and Keatinge-Clay AT, Nat Commun, 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchholz TJ, Geders TW, Bartley FE 3rd, Reynolds KA, Smith JL and Sherman DH, Acs Chem Biol, 2009, 4, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P and Walsh CT, Biochemistry, 1998, 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- 24.Murli S, Kennedy J, Dayem LC, Carney JR and Kealey JT, J Ind Microbiol Biotechnol, 2003, 30, 500–509. [DOI] [PubMed] [Google Scholar]
- 25.Hughes AJ and Keatinge-Clay A, Chem Biol, 2011, 18, 165–176. [DOI] [PubMed] [Google Scholar]
- 26.Piasecki SK, Taylor CA, Detelich JF, Liu JN, Zheng JT, Komsoukaniants A, Siegel DR and Keatinge-Clay AT, Chemistry & Biology, 2011, 18, 1331–1340. [DOI] [PubMed] [Google Scholar]
- 27.Lau J, Tran C, Licari P and Galazzo J, J Biotechnol, 2004, 110, 95–103. [DOI] [PubMed] [Google Scholar]
- 28.Harper AD, Bailey CB, Edwards AD, Detelich JF and Keatinge-Clay AT, Chembiochem, 2012, 13, 2200–2203. [DOI] [PubMed] [Google Scholar]
- 29.Massicard JM, Soligot C, Weissman KJ and Jacob C, Chem Commun, 2020, 56, 12749–12752. [DOI] [PubMed] [Google Scholar]
- 30.Cortes J, Wiesmann KE, Roberts GA, Brown MJ, Staunton J and Leadlay PF, Science, 1995, 268, 1487–1489. [DOI] [PubMed] [Google Scholar]
- 31.Kao CM, Luo G, Katz L, Cane DE and Khosla C, Journal of the American Chemical Society, 1995, 117, 9105–9106. [Google Scholar]
- 32.Annaval T, Paris C, Leadlay PF, Jacob C and Weissman KJ, Chembiochem, 2015, 16, 1357–1364. [DOI] [PubMed] [Google Scholar]
- 33.Kellenberger L, Galloway IS, Sauter G, Bohm G, Hanefeld U, Cortes J, Staunton J and Leadlay PF, Chembiochem, 2008, 9, 2740–2749. [DOI] [PubMed] [Google Scholar]
- 34.Breuer M, Ditrich K, Habicher T, Hauer B, Kesseler M, Sturmer R and Zelinski T, Angew Chem Int Ed Engl, 2004, 43, 788–824. [DOI] [PubMed] [Google Scholar]
- 35.Pidot SJ, Hong H, Seemann T, Porter JL, Yip MJ, Men A, Johnson M, Wilson P, Davies JK, Leadlay PF and Stinear TP, BMC Genomics, 2008, 9, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wlodek A, Kendrew SG, Coates NJ, Hold A, Pogwizd J, Rudder S, Sheehan LS, Higginbotham SJ, Stanley-Smith AE, Warneck T, Nur EAM, Radzom M, Martin CJ, Overvoorde L, Samborskyy M, Alt S, Heine D, Carter GT, Graziani EI, Koehn FE, McDonald L, Alanine A, Rodriguez Sarmiento RM, Chao SK, Ratni H, Steward L, Norville IH, Sarkar-Tyson M, Moss SJ, Leadlay PF, Wilkinson B and Gregory MA, Nat Commun, 2017, 8, 1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy AC, Hong H, Vance S, Broadhurst RW and Leadlay PF, Chem Commun, 2016, 52, 8373–8376. [DOI] [PubMed] [Google Scholar]
- 38.Klaus M, Buyachuihan L and Grininger M, Acs Chem Biol, 2020, 15, 2422–2432. [DOI] [PubMed] [Google Scholar]
- 39.Klaus M, Ostrowski MP, Austerjost J, Robbins T, Lowry B, Cane DE and Khosla C, J Biol Chem, 2016, 291, 16404–16415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapur S, Chen AY, Cane DE and Khosla C, P Natl Acad Sci USA, 2010, 107, 22066–22071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapur S, Lowry B, Yuzawa S, Kenthirapalan S, Chen AY, Cane DE and Khosla C, P Natl Acad Sci USA, 2012, 109, 4110–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuet KP, Liu CW, Lynch SR, Kuo J, Michaels W, Lee RB, McShane AE, Zhong BL, Fischer CR and Khosla C, J Am Chem Soc, 2020, 142, 5952–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beck C, Garzon JFG and Weber T, Biotechnol Bioproc E, 2020, 25, 886–894. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.