

Summary
Bacterial CRISPR systems provide acquired immunity against invading nucleic acids by activating RNA-programmable RNases and DNases. Cas13a and Cas12a enzymes bound to CRISPR RNA (crRNA) recognize specific nucleic acid targets, initiating cleavage of the targets as well as non-target (trans) nucleic acids. Here, we examine the kinetics of single-turnover target and multi-turnover trans-nuclease activities of both enzymes. High-turnover, non-specific Cas13a trans-RNase activity is coupled to rapid binding of target RNA. By contrast, low-turnover Cas12a trans-nuclease activity is coupled to relatively slow cleavage of target DNA, selective for DNA over RNA, indifferent to base identity, and preferential for single-stranded substrates. Combining multiple crRNA increases detection sensitivity of targets, an approach we use to quantify pathogen DNA in samples from patients suspected of Buruli ulcer disease. Results reveal that these enzymes are kinetically adapted to play distinct roles in bacterial adaptive immunity and show how kinetic analysis can be applied to CRISPR-based diagnostics.
Subject areas: Chemical reaction kinetics, Biochemistry, Molecular biology
Graphical abstract
Highlights
-
•
Cas13a HEPN trans-RNase activation is directly coupled to rapid target RNA binding
-
•
Cas12a RuvC trans-nuclease activity is coupled to slow target DNA cleavage
-
•
Individual crRNA generate widely varying levels of targeted trans-cleavage
-
•
Pooling multiple crRNA allows pathogen quantification without target amplification
Chemical reaction kinetics; Biochemistry; Molecular biology
Introduction
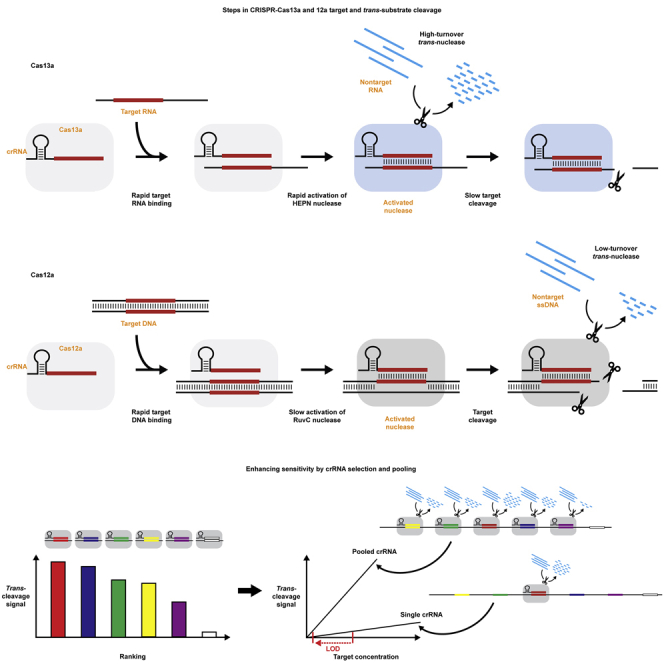
CRISPR-Cas systems provide bacteria and archaea acquired immunity using programmable crRNA to guide nuclease activity against viruses and plasmids. CRISPR-Cas systems are divided into six types by identity and architecture of protein domains, types of nucleic acid polymers they cleave, and mechanisms by which cleavage is catalyzed (Koonin et al., 2017). Cas12a, of the large, diverse type V family (Yan et al., 2019), binds crRNA to form ribonucleoproteins (RNP) that cleave single-stranded (ss) as well as double-stranded (ds) DNA targets containing sequences complementary to the crRNA (Zetsche et al., 2015). This engagement also activates non-specific cleavage of ssDNA, ssRNA, and dsDNA (Chen et al., 2017; Fu et al., 2019; Fuchs et al., 2019; Li et al., 2018; Murugan et al., 2020). Cas13a, a type VI enzyme (Shmakov et al., 2015), binds crRNA to form RNP that cleave specific RNA targets; this interaction activates robust non-specific cleavage of RNA (Abudayyeh et al., 2015; East-Seletsky et al., 2016). Though mechanisms have been proposed, the roles of Cas12a-mediated and Cas13a-mediated trans-activity in viral defense remain incompletely understood (Varble and Marraffini, 2019).
Upon crRNA-targeted binding of Cas12a to protospacers juxtaposed to consensus protospacer adjacent motifs (PAM) that mark targets as foreign, DNA strands within the protospacer are separated—leading to hybridization of the crRNA spacer to the displaced target strand (TS) to form an R-loop structure—followed by cleavage of the two strands. Cas12a RuvC-like nuclease first cleaves the separated non-target strand (NTS) in the R-loop within the protospacer 18-nt from the PAM via a proposed two-metal ion mechanism, which leads to further denaturation of protospacer strands beyond the PAM-proximal ends of the R-loop and cleavage of the separated TS outside the protospacer (Cofsky et al., 2020; Stella et al., 2018). The cleaved PAM-proximal fragments dissociate very slowly, effectively limiting Cas12a to single-turnover of targets (Singh et al., 2018; Sternberg et al., 2014) but allowing multiple rounds of additional cleavage of non-target nucleic acids supplied in trans (Chen et al., 2017; Li et al., 2018). Likewise, in Cas13a, hybridization of crRNA to target RNA allosterically activates conformational changes, bringing together two HEPN domains to form a composite catalytic site that cleaves both target RNA as well non-target ssRNA in a divalent cation-dependent manner (Abudayyeh et al., 2015; East-Seletsky et al., 2016, 2017). Though much is known about target sequence requirements and steps leading to cleavage of targets by Cas12a and Cas13a, steps critical for activation of downstream effector nuclease activities are relatively uncharacterized.
The explosion of reports using Cas12a and Cas13a to detect various nucleic acid sequences indicates their potential as powerful new diagnostic tools (Li et al., 2019). To date, certain biochemical properties have prevented them from reaching their full potential in field-deployable diagnostic assays that could serve to improve global health. For instance, both appear to lack the sensitivity required to detect biologically-relevant concentrations of nucleic acids, a shortcoming typically overcome in Cas-based assays by pre-amplifying target sequences using various nucleic acid amplification techniques (NAAT). However, target amplification poses risks of carryover contamination, especially in clinical settings where assays are often run in a limited number of dedicated spaces (Aslanzadeh, 2004; Borst et al., 2004). Since high rates of false positivity are untenable for most clinical applications, commercial NAAT platforms resort to integrated design features—such as enclosed housings—to limit cross-contamination, adding to assay cost and complexity and limiting uptake, especially in low- and middle-income countries (Aslanzadeh, 2004; Bissonnette and Bergeron, 2006). Furthermore, target pre-amplification convolutes the Cas-generated quantitative response to the original target, constraining analyte detection to a binary output.
Given the central role in bacterial immunity that Cas enzymes play and their growing use in gene editing and diagnostics (Li et al., 2018, 2019), a more complete understanding of the relationship between target recognition and nuclease activities in Cas12a and Cas13a is needed. We examined substrate specificity for trans-nuclease activity, including the influence of length, base composition and sugar backbone, role of crRNA processing on nucleolytic activity, and temporal relationship between target engagement and triggering of nucleolytic activity. We developed a substrate-capture approach to perform endpoint and steady-state kinetic examination of single-turnover target and multi-turnover trans-substrate nuclease activities of crRNA-activated Leptotrichia buccalis Cas13a (LbuCas13a) and Lachnospiraceae bacterium Cas12a (LbCas12a). We used the knowledge gained to tune conditions, enabling direct, amplification-free quantification of pathogen DNA in clinical specimens. Our findings provide insight into mechanisms by which these enzymes perform nucleolytic destruction and their role in adaptive immunity and demonstrate how such analysis can guide the development of CRISPR-based diagnostics.
Results
Cas13a: target cleavage and trans-nuclease activation
Despite the widespread use of Cas13a in diagnostic applications, relatively little is known about the kinetic properties of its HEPN RNase. We created substrate-capture assays to measure crRNA-guided nucleolytic activities of Cas enzymes against targets in cis and non-specific substrates in trans (Figure 1), whereupon cleaved products are quantified using standard curves of uncleaved substrate (Figure S1; Table S2). Cas13a target cleavage was measured using an RNA target modified by extending its 5′ end with U10, since uridine is the preferred substrate for this ortholog (East-Seletsky et al., 2017), internally labeling one base with a biotin capture tag and 5′ end-labeling with Alexa 488 (Figure 2A). Cas13a trans-RNase activity was measured using polyuridine substrates labeled at opposite ends with biotin and Alexa 488 (Figure 2B). As expected, both target and trans cleavage required complementarity between target and crRNA and the presence of divalent cations. Cas13a also cleaved RNaseAlert (Figure S2A), widely used to record Cas13a activation, but not DNA (Figure S2B).
Figure 1.

Substrate-capture to measure Cas nucleolytic activity
Figure 2.

Cas13a cleavage of target RNA and activation of trans-RNase
(A) Cas13a cleavage (yellow triangle) of dual-labeled target. Components tested on Fluorescent Target RNA-1 containing sequence targeted by crRNA-1 but not −2. (B) Target-activated Cas13a cleavage of dual-labeled trans-RNA U10. (C–E) Single-turnover target cleavage using 1.0 nM Fluorescent Target RNA-1 reacted with varied Cas13a bound to mature (C) or precursor (D) crRNA-1 or varied Fluorescent Target RNA-1 reacted with 5.0 nM Cas13a bound to precursor crRNA-1 (E). Lines show single-exponential fit to yield kobs. (F–G) Simultaneously recorded target and trans cleavage: RNP reacted with a mixture of Alexa 488 (F1)-tagged target RNA and Alexa 594 (F2)-tagged trans-substrate. Time courses were recorded on 5.0 nM Cas13a bound to mature or precursor crRNA-1, then reacted with a mixture of 1.0 nM Alexa 488-labeled Target RNA-1 and varied Alexa 594-labeled trans-RNA U10. Lines show single-exponential fit (where applicable) for target (top) or trans cleavage (bottom) to yield kobs. In panels (A–C and E) data represent mean ± standard deviation (SD). See also Figures S2 and S3.
Target cleavage was monitored under single-turnover conditions using Cas13a RNP in excess of target: cleavage at 37°C proceeded with a first-order rate constant kobs of 0.005 s−1 and slowed to 0.002 s−1 at 25°C (Figure S3A), the temperature chosen for further studies. Target cleavage reached a maximal rate of 0.0018 s−1 as RNP concentration was raised from 2.5 to 10 nM (Figure 2C), indicating target binding to RNP achieves a rapid, non-rate-limiting equilibrium with Kd considerably <2.5 nM and cleavage just 5′ to the protospacer is much slower than binding. This observed rate can be attributed to slow, post-target binding steps leading up to and including chemical rates of target cleavage. Replacing the salt-EDTA quench solution with denaturant gave similar results (Figure S3B), confirming that the assay monitors target cleavage rather than capture tag sequestration. RNP formed from precursor crRNA containing additional 6-nt 5′ to the processing site cleaved target more rapidly, raising kobs to 0.0028 s−1 (Figure 2D), also approaching a maximal rate as target concentration was raised from 0.25 to 2.0 nM (Figure 2E), confirming slow target cleavage follows rapid binding equilibrium and precursor crRNA activates a faster target RNase.
To investigate the relationship between target binding and cleavage in cis and activation of trans-RNase activity, both cleavage reactions were measured simultaneously by employing distinct fluorophores on target and trans-substrate (Figure 2F). Cas13a bound with mature, processed crRNA cleaved target at rates unaffected by the presence of trans-substrate, itself cleaved at least five-fold faster than target (kobs 0.011–0.012 s−1), and replacing mature with precursor crRNA again enhanced both target and trans-cleavage rates (Figure 2G). Extending the 5′ end of dual-labeled target from U10 to U20 only modestly increased the target cleavage rate (kobs 0.0024–0.0025 s−1) and did not affect the faster trans-cleavage rates (Figure S3C). Together, these results demonstrate that the Cas13a RNase is rapidly activated after binding of the target, which may be cleaved more slowly than trans-substrates, suggesting the former is not generally required for the latter.
Cas13a: Trans-RNase
We explored the effect of substrate length and crRNA form on Cas13a trans-cleavage. The longer U20 trans-substrate was cleaved somewhat more efficiently than shorter ones (Figure S2C). For one crRNA-target pair, direct dependence of product formation on target concentration revealed apparent turnover rates kapp of 0.065 s−1 and 0.89 s−1 for 10 and 100 nM U10 trans-substrate, respectively, over 2 hr (Figure 3A), suggesting KM >100 nM and kcat/KM may be as low as 7–9 × 106 M−1 s−1. Again, crRNA precursors were considerably more potent in activating targeted trans-cleavage than mature forms, an effect observed for several crRNA-target pairs, as well as when used to activate Cas13a K1082A (Figures 3B, 3C, and S2D–S2H; Table S3), a crRNA-processing deficient variant (East-Seletsky et al., 2017). These results indicate that higher trans-RNase activity resulting from crRNA precursors does not require processing to occur.
Figure 3.

Characterization of target-activated Cas13a trans-RNase
(A–C) Varied Target RNA-1 and trans-RNA U10 were reacted with WT (A, B) or processing mutant K1082A (C) Cas13a bound with mature (A) or both mature and precursor (B, C) crRNA-1. (Insets) Lines show linear fit to yield kapp (see Table S3). (D–F) Trans-cleavage using Cas13a bound with mature (D, E) or precursor (F) crRNA-1, activated with 10 pM Target-1, then reacted with varied trans-RNA. Lines show linear fit of time course of U20 cleavage to determine V0 (D) or V0 dependence on substrates U5 and U10 substrates (E) or hyperbolic fit of all others (E − F) to yield kinetic constants (Table 1). (G–I) Target-activated Cas13a RNP cleaves RNaseAlert, untethering quencher (Q) from fluorescein (F). Cas13a bound to mature (H, I) or precursor (I) crRNA-1 was activated by 10 pM Target RNA-1, then reacted with substrate, and fluorescence was recorded continuously (H). Lines show linear fit to determine V0 (H, Inset) or hyperbolic fit of V0 dependence on substrate (I) to yield kinetic constants (Table 1). Data in panels (A–C and H) represent mean ± SD and in (E, F, I) represent value ±standard error (SE). See also Figures S2 and S3 and Tables S3 and S4.
Kinetic constants for non-target RNA trans-cleavage were determined by pre-reacting RNP with target, then measuring initial velocities (V0) for trans-cleavage (Figure 3D). Using the mature crRNA-Target-1 activating pair, V0 for U5 and U10 increased linearly without signs of approaching saturation, indicating a KM >1 μM (Figure 3E); kinetic constants estimated from direct dependence of V0 on substrate concentration (Figure S3D; Tables S1 and S4) gave kcat/KM of 0.33 x 107 M−1 s−1 and 1.0 x 107 M−1 s−1 and lower limit estimates for kcat of 3.3 s−1 and 12 s−1, respectively. A similar kcat/KM was obtained for U10 using four-fold higher RNP (Figure S3J), validating the use of target concentration as a proxy for that of activated enzyme, further indicating the Kd of target binding to RNP is < 2.5 nM. Saturation kinetics observed for U20 enabled determination for KM of 760 nM and kcat of 17 s−1 (Figure 3E), yielding a kcat/KM of 2.2 x 107 M−1 s−1 (Table 1), corroborated by analysis at substrate concentrations below KM (Figure S3D; Table S4). Together, these results confirm that post-binding trans-cleavage rates are considerably greater than those measured for target cleavage.
Table 1.
Kinetic constants for trans-cleavage of nuclei acids by crRNA-target-activated Cas enzymes
| Enzyme | crRNA (form) | Target | Trans-substratea | kcat (s−1) | KM (nM) | kcat/KM (M−1 s−1) | Figure |
|---|---|---|---|---|---|---|---|
| Cas13a | 1 (mature) | 1 | U5b | >3.3 | >1000 | 3.3 (±0.2) x 106 | 3E |
| U10b | >12 | >1000 | 1.03 (±0.03) x 107 | 3E | |||
| U20 | 16.7 (±0.9) | 760 (±70) | 2.2 (±0.2) x 107 | 3E | |||
| RNaseAlert | 27.1 (±0.6) | 294 (±16) | 9.2 (±0.5) x 107 | 3I | |||
| 1 (precursor) | 1 | U5 | 164 (±7) | 2100 (±120) | 7.9 (±0.5) x 107 | 3F | |
| U10 | 360 (±50) | 3400 (±600) | 1.1 (±0.2) x 108 | 3F | |||
| U20 | 290 (±8) | 1390 (±60) | 2.1 (±0.1) x 108 | 3F | |||
| RNaseAlert | 25.8 (±0.7) | 330 (±20) | 7.8 (±0.5) x 107 | 3I | |||
| 3b (mature) | 3 | U10 | 320 (±10) | 2230 (±90) | 1.43 (±0.08) x 108 | S3G | |
| U20 | 273 (±6) | 1500 (±50) | 1.82 (±0.07) x 108 | S3G | |||
| RNaseAlert | 17.1 (±0.3) | 101 (±6) | 1.7 (±0.1) x 108 | S3I | |||
| 3 (precursor) | 3 | U10 | 740 (±40) | 3500 (±200) | 2.1 (±0.2) x 108 | S3H | |
| U20 | 360 (±30) | 1100 (±100) | 3.2 (±0.4) x 108 | S3H | |||
| RNaseAlert | 24.9 (±0.7) | 220 (±20) | 1.1 (±0.1) x 108 | S3I | |||
| Cas12a | 1 (mature) | 1 | C10 | 2.77 (±0.05) | 28 (±2) | 9.8 (±0.7) x 107 | 5D |
| C20 | 2.26 (±0.08) | 28 (±4) | 8.0 (±1.0) x 107 | S4C | |||
| DNaseAlert | 1.30 (±0.03) | 26 (±2) | 5.1 (±0.4) x 107 | 5E | |||
| dsDNA | 0.044 (±0.001) | 13 (±1) | 3.5 (±0.7) x 106 | 5F | |||
| U20 | 0.118 (±0.005) | 133 (±17) | 8.9 (±1.1) x 105 | 5G | |||
| IS2404 | C10 | 1.32 (±0.04) | 49 (±4) | 2.7 (±0.2) x 107 | 5D | ||
| 2 (mature) | 2 | C10 | 3.83 (±0.18) | 35 (±5) | 1.1 (±0.2) x 108 | 5D | |
| C20 | 2.28 (±0.05) | 18 (±2) | 1.3 (±0.1) x 108 | S4C | |||
| DNaseAlert | 1.02 (±0.03) | 23 (±2) | 4.4 (±0.5) x 107 | 5E | |||
| dsDNA | 0.054 (±0.002) | 16 (±2) | 3.3 (±0.4) x 106 | 5F | |||
| U20 | 0.338 (±0.008) | 108 (±8) | 3.2 (±0.2) x 106 | 5G |
Trans-substrate: C10 and C20 are ssDNA; U5, U10 and U20 are ssRNA.
Estimates based on direct dependence of V0 on substrate concentration through entire concentration range.
Trans-cleavage of U10 and U20 activated by a mature crRNA-target pair (crRNA-Target-3) eliciting greater trans-RNase activity showed its higher efficiency arises mainly from increased kcat (Tables S1 and S4; Figures S3G and S3H). Again, precursor crRNA stimulated a more active Cas13a trans-RNase than mature, processed forms for two different crRNA-target pairs (Figures 3E, 3F, S3D, S3E, S3G, and S3H; Tables S1 and S4). For the most active Cas13a trans-RNases, kcat and KM were together highest for U10 (Table 1): kcat values were as high as 360–740 s−1. The close agreement between kinetic constants obtained from steady-state analysis and endpoint measurements (Table S3) demonstrates trans-activity is maintained for at least 1 hr at 37°C. As expected for a cellular RNA degraded by trans-RNase activity in vitro (East-Seletsky et al., 2016), tRNA competed for U20 cleavage (Figure S3K).
Finally, trans-cleavage of RNaseAlert (Figures 3G–3I) exhibited saturation kinetics for all crRNA-target pairs tested and reduced kcat and KM compared to those for polyuridine substrates (Table 1). Moreover, for Cas13a activated by mature or precursor crRNA, or by different crRNA-target pairs, kinetic constants for RNaseAlert varied only slightly: KM ranged between 101 and 330 nM, kcat ranged from 17–35 s−1, and kcat/KM spanned 0.78–1.1 x 108 M−1 s−1 (Figures 3I and S3I; Table 1). Estimates of kcat/KM for RNaseAlert from V0 at low substrate concentrations corroborated values obtained by Michaelis-Menten analysis (Figures S3F and S3I; Table S4). These results indicate that the use of RNaseAlert does not record the full catalytic potential of Cas13a, nor does it reflect differences in catalytic activity that may arise from the crRNA processed form or the activating crRNA-target combination.
Cas12a: target cleavage and trans-nuclease activation
To develop a Cas12a-based diagnostic, we sought to better understand steps leading to activation of its RuvC domain by performing kinetic analysis of LbCas12a crRNA-guided target and trans-cleavage. We used the substrate-capture approach to measure cleavage of dsDNA targets containing a dual-labeled TS (Figure 4A) to record double-stranded cleavage, since Cas12a proceeds with NTS first, and cleavage of ssDNA trans-substrates labeled at opposite ends (Figure 4B). As expected, both target and trans-cleavage require complementarity between crRNA spacer and target and presence of divalent cations.
Figure 4.

Cas12a cleavage of target DNA and activation of trans-nuclease
(A) Cas12a cleavage of dual-labeled target dsDNA. Components tested on Fluorescent Target DNA-1 containing protospacer specified by crRNA-1 but not −2. (B) Target-activated Cas12a cleavage of dual-labeled trans-ssDNA. Components tested on substrate C10. (C and D) Single-turnover target cleavage using varied Cas12a-crRNA-1 reacted with 1.0 nM Fluorescent Target DNA-1 (C) or 5.0 nM Cas12a-crRNA-1 reacted with varied Fluorescent Target DNA-1 (D). Lines show single-exponential fit to yield kobs. (E) Simultaneously recorded single-turnover target and multiple-turnover trans-cleavage using dual-labeled dsDNA target tagged with Alexa 488 (F1) and trans-ssDNA tagged with Alexa 594 (F2). 5.0 nM Cas12a-crRNA-1 was reacted with a mixture containing 1.0 nM Alexa 488-labeled Target DNA-1 and varied Alexa 594-labeled C10. Lines show single-exponential (target cleavage, top) or sequential double-exponential (trans-cleavage, bottom) fit to yield kobs. (F) Multiple-turnover cleavage of dual-labeled non-target trans-dsDNA. 5.0 nM Cas12a-crRNA-2 was reacted with mixtures containing 1.0 nM unlabeled Target DNA-2 and varied Fluorescent Target DNA-1 at 37°C. Lines show sequential double-exponential fit to yield a lag phase followed by accumulation of product (kobs 0.017–0.018 s−1). (Inset) Similar experiment of shorter duration. In all panels data represent mean ± SD. See also Figures S4 and S5.
Target cleavage was performed under single-turnover conditions using mature crRNA-Cas12a in excess over target. At 37°C, cleavage proceeded with kobs of 0.023 s–1 (Figure S4A) and two-fold more slowly (kobs 0.012 s-1) at 25°C (Figure 4C), the temperature chosen for further studies, at a rate independent of RNP concentration above 2.5 nM. Similar cleavage rates independent of target concentration spanning 0.5 to 2.0 nM were observed (Figure 4D). Unlike Cas13a, target cleavage by precursor crRNA-Cas12a forms, containing additional 15- and 6-nt 5′ to the processing site, were no different (Figure S4B). These results suggest target binds rapidly to RNP with a dissociation constant considerably <2 nM, and its cleavage is rate-limited by a post-binding step, in agreement with studies demonstrating slow unwinding of target strands and formation of an R-loop preceding cleavage of bound target DNA in AsCas12a (Strohkendl et al., 2018).
To test whether the appearance of trans-nucleolytic activity is also limited by a post-target binding step, target and trans-cleavages were monitored simultaneously by reacting RNP with mixtures of target and trans-substrate labeled with distinguishable Alexa tags (Figure 4E). Target cleavage rates were similar to those measured above and were not affected by the presence of trans-substrates, and trans-cleavage exhibited a lag phase of a similar duration as target cleavage (kobs 0.011–0.024 s−1). After this lag, trans-cleavage accelerated during the steady-state phases as the substratewas increased from 25 to 100 nM, indicating trans-ssDNA cleavage operates below its maximal velocity under these conditions. Together, these results indicate activation of Cas12a trans-activity is also rate-limited by post-target binding steps, suggesting its initiation is coupled to target cleavage.
Target-activated dsDNA trans-cleavage (see below) was then recorded by reacting RNP with mixtures of unlabeled target and dual-labeled trans-dsDNA lacking the crRNA-specified protospacer (Figure 4F). Because dsDNA trans-cleavage was considerably slower than ssDNA trans-cleavage, the reaction temperature was raised to 37°C to shorten the time course. Products of dsDNA trans-cleavage also accumulated after a lag likely reflecting rate-limiting target cleavage. However, for the first minutes after this lag, initial cleavage rates were indistinguishable over substrate concentrations of 25–100 nM (Figure 4F, inset), indicating dsDNA trans-cleavage operates at low, albeit maximal, and velocity under these conditions. These results indicate both ss and dsDNA trans-cleavage are kinetically coupled to steps involved in target cleavage.
Cas12a: trans-nuclease activities
We measured trans-cleavage rates of substrates varying in length and composition. At least three-fold greater activity was observed against 10- or 20-nt substrates compared to 5-nt substrates (Figure 5A), and 10-nt ssDNA pyrimidines were at most two-fold more efficiently cleaved than those composed of purines (Figure 5B). Likewise, trans-DNase activity of Acidaminococcus sp. BV3L6 Cas12a (AsCas12a) showed little or no base preference (Figure S5A). Trans-activity arising from varying C10 and target concentrations (Figure 5C), corresponded to kapp of 0.64 s−1 and 2.01 s−1 for 10 and 100 nM substrate, respectively, suggesting a KM of 31 nM, kcat of 2.6 s−1, and kcat/KM of 8.4 x 107 M−1 s−1. Again, RNP assembled from either mature or precursor crRNA displayed indistinguishable trans-cleavage (Figure S5B).
Figure 5.

Characterization of target activated Cas12a trans-nuclease
(A–C) Cas12a, crRNA-1, and varied Target DNA-1 tested with trans-substrates consisting of (A) single or multiple repeats of TTATT, (B) differing sequence, or (C) varied concentrations of trans-substrate C10 where linear fit was used to estimate kinetic constants (Inset). Data are represented as mean ± SD. (D–G) (Top) Cas12a RNP was pre-reacted with target, then reacted with varied trans-substrate used in each panel. (Middle) Linear fit of time courses to determine V0. Examples shown: (D) Cas12a-crRNA-1, 10 pM Target DNA-1, substrate C10; (E) Cas12a-crRNA-2, 10 pM Target DNA-2, DNaseAlert substrate; (F) Cas12a-crRNA-2, 1.0 nM Target DNA-2, non-target dsDNA substrate (Fluorescent Target DNA-1); (G) Cas12a-crRNA-1, 1.0 nM Target DNA-1, RNA substrate U20. (Bottom) Hyperbolic fit of V0 (symbols) to yield kinetic constants (Table 1); data are represented as value ±SE. See also Figures S4 and S5.
Though the Cas12a RuvC trans-nuclease has been widely described as a ssDNase, Cas12a has been reported to also cleave dsDNA and ssRNA in trans (Chen et al., 2017; Fu et al., 2019; Fuchs et al., 2019; Li et al., 2018; Murugan et al., 2020). We observed multi-turnover, divalent cation-dependent trans-cleavage of other nucleic acid substrates, including DNaseAlert (Figure S5C), widely used to monitor ssDNase trans-activity, non-target dsDNA (Figure S5D), and ribonucleotide U20 (Figure S5E), demonstrating the substrate nucleobase indifference of the Cas12a RuvC nuclease.
To gain insight into the mechanism of trans-cleavage and the potential physiological relevance of the different substrates, we measured steady-state rates of trans-cleavage. Kinetic constants were determined by pre-reacting RNP with target, then measuring V0 of trans-cleavage (Figures 5D–5G). Cas12a activated by two different crRNA-target pairs cleaved both C10 and C20 with kinetic constants varying only little: kcat of 2.3–2.8 s−1, KM of 18–35 nM, and kcat/KM of 0.8–1.3 x 108 M−1 s−1 (Figures 5D and S4C; Table 1). Similar constants were obtained for IS2404, a 1299-bp fragment of DNA from the pathogenic bacterium M. ulcerans containing protospacers specified by crRNA-1, -2, and -3. Since these values are comparable to those estimated from 2 hr endpoints (Figure 5C), trans-activity persists over several hours at 37°C. Furthermore, these KM values account for the observed post-lag acceleration of trans-cleavage (Figure 4E). Kinetic constants remained largely unchanged when RNP concentration was increased four-fold (Figure S4D), further indicating Kd is < 2 nM and target concentration can serve as a proxy for that of activated enzymes. Kinetic constants for cleavage of polycytidine were comparable to those for DNaseAlert determined via a parallel approach for both crRNA-target pairs (Figures 5E and S4E; Table 1). Increasing ionic strength with additional salt or including the heparin polyanion decreased cleavage efficiency of both C10 and DNaseAlert by increasing KM with minimal effect on kcat (Figures S4F and S4G). Together, these results demonstrate steady-state trans-cleavage of ssDNA and DNaseAlert (kcat 1–3 s−1) is considerably faster than the rate-limiting step in target cleavage.
Multi-turnover trans-cleavage of dsDNA by Cas12a (Figure 5F) activated by two crRNA-target pairs was considerably less efficient than ssDNA, with kcat/KM of 3.3–3.5 x 106 M−1 s−1, primarily due to 60–70-fold reduction of kcat to 0.04–0.05 s−1 and reduction of KM to 13–16 nM (Table 1). Besides illustrating the strong structural preference for single-stranded trans-substrates, these results account for the approach of Cas12a to a low, albeit maximal, velocity for trans-dsDNA cleavage (Figure 4F). Finally, kcat for multi-turnover ssRNA trans-cleavage (Figure 5G) by Cas12a activated by both crRNA-target pairs was only 1/10th that for ssDNA and, combined with higher KM, resulted in considerably lower catalytic efficiencies, with kcat/KM of 0.9–3.2 x 106 M−1 s−1 (Table 1). Thus, steady-state analysis indicates trans-nucleolytic activity of the Cas12a RuvC domain may be described as a preferential DNase structurally selective for single-stranded substrates.
Cas13a and Cas12a: quantitative, amplification-free Detection of Pathogen Nucleic Acids
To illustrate their utility, we applied the findings to improve assay sensitivity to enable amplification-free detection of nucleic acid targets. Having shown that the RNP-target affinity was sufficiently high—Kd is considerably less than typical assay RNP concentrations—we predicted multiple protospacers could be bound by RNP assembled from pools of multiple crRNA species, generating trans-cleavage activity greater than that of single target-RNP. We screened 21 Cas13a crRNA-target pairs corresponding to different regions of hepatitis C viral RNA, assessing apparent turnover rates from endpoint values (Figure 6A). Of these, we identified 13 non-overlapping crRNA, which, when pooled, increased the apparent trans-cleavage rate 10-fold and enhanced the sensitivity for detection of pooled RNA targets by a factor of 6- to 7-fold using U20 or RNaseAlert trans-substrates (Figure 6B; Table S5). We estimate if such targets were encoded within intact viral genomes, then the analytical limit of detection (LOD) by the pooled crRNA, 29 fM (based on 13 pooled targets), would translate to 2 fM of viral genomes.
Figure 6.

Pooling of crRNA enables sensitive cas-directed detection of pathogen nucleic acids
(A) Trans-RNase activity of Cas13a bound to individual precursor crRNA, then reacted with target RNA and trans-RNA U10. Filled symbols indicate 13 crRNA selected for pooling. (B) Trans-RNase activity of Cas13a bound to single or 13 pooled crRNA, then reacted with 13 pooled target RNA and U20 (left) or RNaseAlert (right). (Insets) Linear fit to yield kapp (Table S5). (C) Trans-DNase activity of Cas12a bound to crRNA-1, then reacted with IS2404 or genomic DNA and trans-ssDNA C10. Genomic DNA isolated from M. ulcerans (Mu) contains IS2404 protospacers targeted by crRNA-1, whereas genomic DNA isolated from M. tuberculosis (Mt), E. coli (Ec), and humans (Hs) do not. (D) Trans-DNase activity of components tested on C10. Protospacer in target IS2404 complementary to crRNA-1 abuts a PAM, whereas that of crRNA-4 does not. (E) Trans-DNase activity of Cas12a bound to individual crRNA, then reacted with IS2404, which contains protospacers for each crRNA, and C10. Colors indicate 20 crRNA selected for sub-pooling. Activity of Cas12a-crRNA-4 is indicated by black symbol. (F–G) Trans-DNase activity of Cas12a bound to crRNA pools added cumulatively (F) or together (G), then reacted with IS2404 and C10. Linear fits were used to calculate kapp (see Table S5). (H) IS2404 in DNA samples isolated from 38 patient skin swabs quantified by Cas12a trans-activity (y axis) or by qPCR (x axis). Color-coded patient qPCR status was determined from DNA extracted initially at test site. Solid line, detailed in Inset, indicates linear fit to yield kapp (Table S5); gray shading indicates 95% confidence interval. In all panels data represent mean ± SD. See also Figure S6 and Table S5.
We then developed a sensitive Cas12a-based assay to detect M. ulcerans, the causative agent of Buruli ulcer disease (Johnson et al., 2005), which harbors multiple copies of IS2404. Cas12a trans-cleavage was activated by DNA isolated from M. ulcerans, but not from the related bacterium M. tuberculosis, which lacks IS2404, or from E. coli or humans as they do not have a crRNA-complementary protospacer and an adjacent PAM (Figures 6C and 6D). We screened 39 crRNA designed to target protospacers adjacent to PAM sequences within IS2404 for trans-nuclease activity (Figure 6E). Perhaps worth further study, higher activity was observed for several crRNA guiding Cas12a to protospacers within 5′ AND-3′ untranslated regions of the IS2404 transposase gene (Figure S6A). Four sub-pools from the 20 highest potency crRNA, excluding those demonstrating pairwise interference (Figures S6B–S6D), cumulatively increased trans-activity of Cas12a (Figure 6F). The use of 20-pooled crRNA increased kapp and enhanced detection of IS2404, for which an analytical LOD of 310 aM was determined (Figure 6G; Table S5). By calibrating the trans-activity of Cas12a loaded with the 20-pooled crRNA against IS2404, we quantified the concentration of IS2404 in DNA extracted from wound swabs of 38 patients suspected to be infected with M. ulcerans, whose disease status was originally ascertained using IS2404 qPCR at the field hospital where samples were collected (Figure 6H). Linear analysis indicated Cas12a detects ∼25% of qPCR-identified IS2404 segments in DNA isolates. Thus, sensitive and quantitative detection of nucleic acid targets by Cas enzymes may be achieved by combining multiple crRNA.
Discussion
Cas13a: target RNA cleavage and trans-nuclease activation
Target RNA binding to Cas13a RNP is rapid, achieving equilibrium within seconds, and does not limit its cleavage rate. We estimate the Kd for initial encounter complex formation is < 1 nM, approximately 20-fold lower than measured for LbuCas13a in the presence of tRNA (Tambe et al., 2018), a potential competitive inhibitor that decreases trans-cleavage efficiency by approximately 100-fold. We observed Cas13a target cleavage to be considerably slower than steady-state trans-cleavage, indicating constraints in this particular target or some other post-binding step limit its cleavage. It remains unknown whether target cleavage proceeds in cis via intra-molecular cleavage by the same Cas13a to which it is bound or in trans via inter-molecular cleavage by a separate activated Cas13a RNP, as in the LbuCas13a dimeric complex (Liu et al., 2017). Though 5′-extended targets may not readily access the RNase site, longer targets might be more efficiently cleaved in cis if not physically constrained. Trans-cleavage commences seconds after enzyme encounters target, indicating HEPN nuclease activation is rapid. Trans-cleavage rates were approximately five to ten times faster than target cleavage using Cas13a bound to either mature and precursor crRNA, indicating that activation of HEPN trans-nuclease activity does not generally require target cleavage nor does cleavage of targets in cis pose a rate-limiting barrier to trans-activity. Target cleavage rates remained unchanged in the presence of up to 100 nM trans-ssRNA, conditions whereby Cas13a can undergo multiple rounds of trans-cleavage, indicating trans-substrates may only weakly competitively inhibit target cleavage due to their high KM values.
Our steady-state analysis of Cas13a trans-cleavage revealed several features of the activated catalytic state of the HEPN nuclease. First, catalytic efficiencies triggered by different crRNA-target pairs varied over a 15–30-fold range, mainly due to variations in kcat, indicating different conformational states are sampled by Cas13a depending on crRNA-target duplex sequence. Notably, this variability was not observed for trans-cleavage of RNaseAlert, suggesting it probes a subset of the conformational states. Second, addition of nucleotides 5′ to the protospacer had little or no effect on HEPN trans-nuclease activity. Third, precursor crRNA possessing additional nucleotides 5′ to its processing site stimulate 2- to 10-fold higher catalytic efficiency than mature forms on polyuridine trans-substrates (but not RNaseAlert) for both the wild-type and crRNA-processing deficient variant of Cas13a. These increases were associated with increased kcat. Fourth, the agreement between kinetic constants from endpoint and steady-state readings indicates the activated conformational state is maintained at high levels for at least 1 hr at 37°C. Together, these results suggest the determinants for allosteric activation of the Cas13a HEPN nuclease extend beyond the complex specificity landscape demonstrated for base pairs involved in target binding (Tambe et al., 2018).
Though variable, estimates for the HEPN trans-nuclease catalytic efficiency converge on a value of ∼108 M−1 s−1, providing a lower-limit bimolecular rate constant for trans-substrate binding to the activated enzyme, a value within two orders of magnitude of the diffusion-controlled limit. In the most highly activated forms of Cas13a, kcat of several hundred per sec were observed, which is very high compared to the trans-nuclease activity observed for Cas12a, but within the range for other ribonucleases, including pancreatic ribonucleases in the RNase A family (EC 4.1.1.18): kcat of 323 s−1 for bovine (Moussaoui et al., 2007) and 2317 s−1 for human (Rehman et al., 2011) enzymes. For Cas13a, kcat for polyuridine trans-substrates was as much as 30 times higher than for RNaseAlert, indicating once bound, the former are more rapidly cleaved and released than the latter, and the use of RNaseAlert does not accurately record the full catalytic potential of Cas13a and may be suboptimal for diagnostics.
Spacer regions of type VI systems show complementarity to DNA, but not RNA viruses (Smargon et al., 2017; Yan et al., 2018), suggesting Cas13a is activated by viral transcripts made during infection and targets destruction of nascent transcripts (Hampton et al., 2020; Varble and Marraffini, 2019). This hypothesis is strongly supported by in vivo experiments demonstrating Cas13a-mediated transcript destruction suffices to disrupt phage infection (Koonin and Zhang, 2017; Meeske et al., 2019). Our observation that trans-RNase activity is triggered immediately and proceeds rapidly likely reflects the requirement for this mode of interference to outcompete the rapid viral transcription. The slow rate of target cleavage exposes the relatively long half-life of the ternary complex, which prevents translation of the transcript to which the enzyme is bound and enables the enzyme to undergo multiple rounds of trans-cleavage before releasing the target sequence, presumably deactivating nuclease activity. Further experiments are needed to determine the effect, if any, of Cas13a trans-RNAse activity when expressed in eukaryotic cells.
Cas12a: target DNA cleavage and trans-nuclease activation
Our results indicate dsDNA target binding to LbCas12a RNP is rapid, likely equilibrating within seconds, and are consistent with the bimolecular constant measured for AsCas12a binding to target (Strohkendl et al., 2018). Binding is tight, with a Kd of <5 nM for initial encounter complex formation, in agreement with the values for AsCas12a (Strohkendl et al., 2018)and LbCas12a (Jeon et al., 2018). Despite rapid binding, target cleavage is slow, consistent with previous measurements with Lb and AsCas12a (Jeon et al., 2018; Knott et al., 2019; Strohkendl et al., 2018). Given the steady-state rate of ssDNA trans-cleavage is almost 100-fold faster, our results agree with a model whereby NTS and TS are rapidly cleaved following slow rate-limiting strand unwinding and R-loop formation (Stella et al., 2018; Strohkendl et al., 2018).
Our results show trans-nuclease activity proceeds via two kinetically resolvable steps. The first appears as a lag in trans-cleaved product accumulation at a rate corresponding to target cleavage, comparable to TS cleavage rates and PAM-distal target fragments release measured for AsCas12a (Strohkendl et al., 2018), indicating they likely represent the same steps in orthogonal enzymes. The second step represents trans-substrate binding and cleavage itself, which is considerably faster for ssDNA trans-substrates than for dsDNA. We conclude that trans-nuclease activity acquisition accompanies late steps of target DNA destruction, including TS cleavage and, perhaps, PAM-distal fragment release. Cas12a remains tightly bound to the PAM-proximal target fragment (Singh et al., 2018), locked into a conformational state capable of engaging in multiple rounds of trans-cleavage for 2 hr or longer at 37°C.
Cas12a trans-cleavage can be described in terms of a Michaelis-Menten enzyme, in agreement with previous reports (Chen et al., 2017; Knott et al., 2019). Kinetic constants for Cas12a trans-cleavage activated by two different crRNA-target combinations are similar, suggesting they represent general kinetic features for LbCas12a activated by fully matched pairs. The LbCas12a kcat of ∼2 s−1 for polycytidine trans-substrates is similar to the NTS cleavage rate (>4 s−1) of the Cas9 RuvC domain (Gong et al., 2018) but is smaller than rates exhibited by other DNases, such as the single-strand-specific, non-sequence-specific endonuclease S1 (kcat of 42 s−1) (Li et al., 2000) or the Cas13a trans-RNase (see above). Nevertheless, the low-turnover Cas12a trans-activity may be of physiological importance considering the double-strand-, site-specific endonuclease EcoRI, a component of the bacterial modification system providing defense against invading viruses, cleaves targets with kcat of 0.063 s−1 (Modrich and Zabel, 1976). The kcat/KM ratio of ∼108 M−1 s−1 for Cas12a trans-cleavage represents a bimolecular rate constant lower limit estimate for trans-substrate binding to the enzyme ternary complex, suggesting target DNA binding and cleavage serve as the main allosteric switch activating trans-nuclease activity. Finally, we show kinetic properties for trans-cleavage of DNaseAlert are like those for polycytidine substrates, demonstrating that its cleavage can serve as a generalized marker for activation of the Cas12a trans-nuclease.
We interpret trans-cleavage activities of Cas12a against different substrates in context of a proposed two-Mg2+ mechanism, whereby catalytic residues and the scissile phosphate jointly coordinate metal ions roughly in line with the phosphoribosyl backbone on opposite sides of target bases (Yang et al., 2006). Lack of trans-nucleobase preference by both Lb and AsCas12a suggests most contacts between enzyme and substrate, as well as DNA target (Swarts et al., 2017), are mediated through the phosphoribosyl backbone. Inhibition of trans-cleavage by polyanionic heparin suggests non-specific electrostatic interactions help steer trans-substrates into the catalytic site. Preference for trans-substrates containing deoxyribose suggests the RNA 2′ OH interferes with the positioning of the metal ions. LbCas12a prefers single-stranded trans-substrates, even though the attack of the phosphoribosyl backbone by RuvC nucleases does not require gross distortion of the phosphate backbone or base unstacking (Yang, 2011), similar to the structural preference displayed by other RuvC domains characterized to date, including Cas9, as well as the bacterial Holliday junction-resolving endonuclease RuvC itself (Górecka et al., 2019). Low-level Cas12a trans-activity against dsDNA likely represents cleavage of denatured single-stranded ends of duplexes and may represent the same low-level nicking of dsDNA observed by others (Murugan et al., 2020) on localized regions of dynamically melted DNA strands providing transient access for cleavage. Structural selectivity for single-strands and base indifference are likely to be general properties of the Cas12a RuvC nuclease, reflecting both that dsDNA target cleavage requires R-loop formation and specificity for target cleavage is governed not by the catalytic site itself but rather by sequences involved in crRNA-target hybridization, enabling Cas12a to act as a generic nuclease against any number of invading protospacers.
Experimental data probing the physiological role of Cas12a, including trans-nuclease activity, is currently lacking, though both nuclease activities play essential roles in interference with target cleavage degrading infectious dsDNA, similar to most CRISPR systems including Cas9, and trans-activity targeting ssDNA species in transcription bubbles or during rolling circle replication (Hampton et al., 2020; Varble and Marraffini, 2019). Trans-nuclease activity of ssDNA is conserved among members of type V systems (Yan et al., 2019) and is also key to type III systems, supporting this hypothesis. Our observation that target cleavage is a prerequisite for trans-nuclease activation may reflect the relative importance of the two viral interference activities. Based on the relatively low efficiencies for cleavage of dsDNA and RNA in trans, we hypothesize these activities are of little physiological significance. While Cas12a trans-ssDNase activity appears to have no consequences in the context of genome editing (Wei et al., 2021), similar non-specific ssDNA cleaving Cas enzymes were recently shown to drive host mutations (Mo et al., 2021).
Cas13a and Cas12a: quantification of pathogen nucleic acids in clinical specimens
We show that for both Cas13a and Cas12a, different crRNA generate widely varying trans-activities. From kinetic analysis, we improved sensitivity of Cas13a for target RNA by replacing mature crRNA with precursor forms activating higher trans-activity. Further, we capitalized on the high affinities of RNP for targets to develop a crRNA pooling strategy that enables targeting of up to twenty sequences to improve the sensitivity of both Cas enzymes. This strategy drives the analytical LOD for Cas13a down to the low femtomolar range (for viral genome equivalents) and Cas12a to the attomolar range. Moreover, we could quantifiably detect pathogen DNA in clinical specimens from patients experiencing Buruli ulcer disease—a devastating, neglected tropical disease that often goes undetected due to the lack of diagnostics appropriate for low-resource settings (Siegmund et al., 2007; Yotsu et al., 2018). Further experiments are needed to ascertain how well Cas12a-based detection of IS2404 correlates with clinical features and outcomes and what role it may play as a diagnostic.
The COVID-19 pandemic highlighted the importance of having multiple diagnostic options for disease management, especially during outbreaks. Point-of-care solutions, including antigen tests (LOD ∼200 aM), are often more appropriate than highly sensitive RT-qPCR (LOD ∼2 aM) (Larremore et al., 2020). Already a large number of reports have demonstrated detection of SARS-CoV-2 using Cas enzymes. In most cases NAATs were used to pre-amplify target sequences, and it is difficult to imagine how these chemistries could be used at the point-of-care without employing costly closed assay architectures to limit cross-contamination. Our approach of tuning assay conditions to optimally exploit the kinetic properties of Cas enzymes drops the LOD of non-NAAT amplified reactions to be compatible with SARS-CoV-2 nasopharyngeal samples (20 aM–20 fM), among other diseases (Nouri et al., 2021). While our paper was under review, a similar strategy was used to detect SARS-CoV-2 RNA in extracted patient samples (Fozouni et al., 2021). Synergizing with recent advances in Cas-based diagnostics for low-resource settings, including lyophilization to avoid cold chain requirements (Curti et al., 2021; Lee et al., 2020), this new class of diagnostics may meet the World Health Organization's ASSURED criteria of being affordable, sensitive, specific, user-friendly, robust, equipment-free, and deliverable to end-users.
Conclusions
We developed a quantitative experimental approach to uncover key kinetic details of the steps in activation of nucleolytic activities and potential physiological roles of representative type V and VI Cas enzymes. This approach may be used to investigate the catalytic properties of other Cas enzymes, as well as other nucleases. Further, we applied these findings to design a novel approach for quantitative diagnostic applications using Cas enzymes and anticipate these findings may also guide engineering of Cas enzymes for gene editing.
Limitations of the study
Given the wide range of trans-cleavage activities displayed by different Cas-crRNA RNP on a given target (Figures 6A and 6E), we anticipate that kinetic constants, which we have determined for representative Cas enzyme-crRNA-target combinations (Tables S1, S3, and S4), will likewise vary considerably. Though we have shown that analytical sensitivity for both Cas12a- and Cas13a-based assays is increased by crRNA pooling (Figure 6, Table S5), we have not tested whether signals are generated in both assays from different but related pathogen nucleic acids. As expected, individual Cas-crRNA RNP are specific for their intended targets (see Figures 2A and 2B for Cas13a and Figures 4A, 4B, 6C, S5D, and S5E for Cas12a), and we do not anticipate that crRNA pooling will have any effect on specificity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Mycobacterium ulcerans NCTC 10417 | ATCC | Cat#ATCC 19432 |
| Biological samples | ||
| Human skin swabs (Negative and positive for M. ulcerans) | Centre de Dépistage et de Traitement de l'Ulcère de Buruli, Pobè, Benin | https://ilepfederation.org/centre-de-depistage-et-de-traitement-de-la-lepre-et-de-lulcere-de-buruli-raoul-et-madeleine-follereau-de-pobe/ |
| Human genomic DNA | Promega | Cat#G304A |
| Chemicals, peptides, and recombinant proteins | ||
| LbCas12a | NEB | Cat#M0653T |
| Recombinant Cas12a from Acidaminococcus sp. BV3L6 | IDT | AsCas12a |
| Recombinant Cas13a from Leptotrichia buccalis, wild-type | East-Seletsky et al., 2017 | LbuCas13a |
| Recombinant Cas13a from Leptotrichia buccalis, processing mutant K1082A | East-Seletsky et al., 2017 | LbuCas13a K1082A |
| DNase I | NEB | Cat#M0303S |
| RNase A | IDT | Cat#11-02-01-02 |
| Dynabeads™ MyOne™ Streptavidin C1 | ThermoFisher | Cat#65002 |
| Critical commercial assays | ||
| MagMAX™ Total Nucleic Acid Isolation Kit | ThermoFisher | Cat#AM1840 |
| Qubit™ 1X dsDNA HS Assay Kit | ThermoFisher | Cat#Q33231 |
| TaqMan Gene Expression Master Mix | Life Technologies | Cat# 4369106 |
| Oligonucleotides | ||
| ssRNA oligos | Synthego | See Table S1 |
| DNA oligos | IDT | See Table S1 |
| IS2404 | IDT | See Table S1 |
| RNaseAlert Substrate | IDT | Cat#11-04-03-03 |
| DNaseAlert Substrate | IDT | Cat#11-04-03-04 |
| Software and algorithms | ||
| Prism 8 | GraphPad | https://www.graphpad.com/ |
| Excel | Microsoft | https://www.microsoft.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Damian Madan (damian.madan@ghlabs.org).
Material availability
This study did not generate new unique reagents.
Experimental models and subject details
Patient samples
Collection and use of human samples used in this study was approved by Le Ministre de la Santé, Bénin. Swabs were collected from open wounds of patients suspected of Buruli ulcer disease at Centre de Dépistage et de Traitement de l'Ulcère de Buruli, in Pobé, Bénin. At the field hospital collection site, swabs were rehydrated in 2.0 mL water, 400 μL was processed for qPCR to ascertain presence of IS2404 as described (Marion et al., 2014), and the remaining sample was stored at -20°C.
Method details
Enzymes, RNA, and DNA
Recombinant LbCas12a and DNAase I were purchased from New England Biolabs (cat. no. M0653T, M0303S). Recombinant Cas12a from Acidaminococcus sp. BV3L6 (AsCas12a) was a gift from Integrated DNA Technologies (IDT). RNase A was purchased from IDT (cat. no. 11-02-01-02). Wild-type and crRNA-processing variant LbuCas13a were produced recombinantly and purified as described (East-Seletsky et al., 2017). RNA was purchased from Synthego. Synthetic DNA was purchased from IDT. IS2404 consists of a synthetic gBlock spanning 1299 bp of the repeat region present in M. ulcerans. RNaseAlert™ and DNaseAlert™ were purchased from IDT (cat. no. 11-04-03-03 and 11-04-03-04). Nucleic acid sequences are provided in Table S1. crRNA and the cognate targets they specify are designated by matching numbers: for example, the 20-nt spacer of crRNA-1 is complementary to the 20-nt protospacer of Target-1. Human genomic DNA was purchased from Promega (cat. no. G304A). Bacterial genomic DNA from E. coli and M. ulcerans NCTC 10417 (ATCC cat. no. 19432) was isolated using the MagMAX™ Total Nucleic Acid Isolation Kit (ThermoFisher, cat. no. AM1840) and quantified using the Qubit™ 1X dsDNA HS Assay Kit (ThermoFisher, cat. no. Q33231).
Substrate-capture and product quantification
Nuclease reactions described below were terminated by diluting reactions into a salt-EDTA quench solution composed of 1.0 M NaCl, 10 mM Tris-Cl, pH 7.5, 10 mM EDTA, and 0.01% Tween-20, unless otherwise indicated. Uncleaved biotin-labeled substrate was removed by addition of Dynabeads™ MyOne™ Streptavidin C1 (ThermoFisher Scientific, cat. no. 65002) diluted in salt-EDTA quench solution for at least 15 min at room temperature with end-over-end rocking. Bead quantities used in each reaction were scaled according to the amount of biotinylated substrate to be captured, based on the binding capacity of the beads (∼500 pmol of ss oligo per mg of beads). Supernatants were removed from capture beads collected by magnetization and read on a BioTeK Synergy H1 fluorescence microplate reader at λex 490 nm and λem 525 nm for Alexa Fluor™ 488 (Alexa488) and λex 584 nm and λem 616 nm for Alexa Fluor™ 594 (Alexa594). When necessary, the concentration of product was calculated by converting fluorescence values (RFU) to molarity by comparison to linear standard curves of un-treated substrate performed in parallel.
Cas13a nuclease assays
Cleavage reactions were conducted in Cas13a assay buffer composed of 20 mM 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), pH 6.8, 50 mM KCl, 5.0 mM MgCl2, 100 μg/mL BSA, 0.01% Igepal CA-630, and 5% glycerol (East-Seletsky et al., 2017) in microtiter plates at final concentrations of reactants indicated (see Method Table). Assays performed in an alternative buffer lacking glycerol and KCl provided results qualitatively similar to the ones presented throughout this report; however, an approximately 10-fold reduction in catalytic efficiency for trans-cleavage of polyuridine and RNaseAlert by Cas13a activated by cRNA-Target-1 was observed (data not shown). For all experiments, 100 nM RNP was formed by incubating 200 nM Cas13a with 100 nM crRNA for at least 15 min at room temperature in buffer lacking Mg2+. Cleavage reactions were then performed at 37°C unless otherwise indicated. EDTA was pre-mixed with fluorescence substrates when testing the Mg2+-dependence of reactions. Reactions were terminated and processed as described above unless otherwise indicated.
In endpoint target-cleavage experiments (Figure 2A), RNP was incubated with dual fluorescent-biotin-labeled target DNA, and uncleaved substrate was removed by magnetic bead capture. In all endpoint trans-substrate cleavage experiments (Figures 2B, 3A–3C, S2, 6A, and 6B), RNP was incubated with unlabeled target RNA for at least 15 min at 37°C in buffer lacking Mg2+, then fluorescent trans-substrate was added in buffer containing Mg2+. For assays using dual fluorescent-biotin-labeled substrates (Figures 2B, 3A–3C, S2B–S2H, 6A, and 6B), uncleaved substrate was removed by magnetic bead capture. For assays using RNaseAlert trans-substrate (Figure S2A), reactions were diluted into the salt-EDTA quench solution, and fluorescein fluorescence was recorded directly (λex 490 nm and λem 525 nm).
In transient-state target-cleavage experiments (Figures 2C–2E, S3A, and S3B), RNP was mixed with fluorescent dsDNA target in buffer containing Mg2+ to initiate time courses. Control reactions, consisting of enzyme reactions lacking crRNA, were conducted in parallel. Transient-state experiments in which target and trans-substrate cleavage were measured simultaneously (Figures 2G and S3C) were conducted by mixing RNP with a solution containing a dual fluorescent-biotin-labeled RNA target (labeled with Alexa 488) and a dual fluorescent-biotin-labeled trans-substrate (labeled with Alexa 594) in a buffer containing Mg2+ to initiate time courses. Control reactions, consisting of enzyme reactions lacking crRNA, were conducted in parallel. At successive time points, cleavage reactions were terminated by dilution into the salt-EDTA quench solution or a denaturing solution containing 6.0 M guanidinium chloride and EDTA (Figure S3B) prior to removal of uncleaved substrate by magnetic bead capture.
In steady-state kinetics experiments (Figures 3D–3F, 3H, 3I, and S3D–S3K), RNP was incubated with RNA target in buffer lacking Mg2+ for at least 15 min, then mixed with varying amounts of fluorescent trans-substrate in buffer containing Mg2+ to initiate time courses, which were terminated by diluting reactions at successive time points into the salt-EDTA quench solution. Control reactions, consisting of enzyme reactions lacking crRNA, were conducted in parallel. Assays using dual fluorescent-biotin-labeled substrates (Figures 3D–3F, S3D, S3E, S3G, S3H, S3J, and S3K), uncleaved biotin-labeled trans-substrate was removed by magnetic bead capture. For cleavage of RNaseAlert trans-substrate (Figures 3H, 3I, S3F, and S3I), fluorescein fluorescence was measured continuously over several hours. When necessary, the concentration of cleaved product was determined by converting fluorescence values to molarity using a linear standard curve generated from the fluorescence values of cleavage reactions that had reached completion. The effect of tRNA was tested by including yeast tRNA (Sigma-Aldrich: cat. no. R5636) during the trans-cleavage phase of the reaction (Figure S3K).
Cas12a nuclease assays
Cleavage reactions were conducted in Cas12a assay buffer composed of 10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1.0 mM Tris(2-carboxyethyl)phosphine hydrochloride, 50 μg/mL BSA, and 0.01% Igepal CA-630 in microtiter plates at final concentrations of reactants indicated (see Method Table). Where indicated (Figures S4F and S4G), assays were performed in an alternative buffer composed of Cas12a assay buffer to which 5% glycerol and 100 mM KCl were included (Chen et al., 2017). For all experiments, 100 nM RNP was formed by incubating 100 or 200 nM Cas12a with 100 nM crRNA for at least 15 min at room temperature. Cleavage reactions were then performed at 37°C unless otherwise indicated. EDTA was pre-mixed with fluorescence substrates when testing the Mg2+-dependence of reactions.
In endpoint target-cleavage experiments (Figure 4A), RNP was incubated with dual fluorescent-biotin-labeled target DNA, and uncleaved substrate was removed by magnetic bead capture. In all endpoint trans-substrate cleavage experiments (Figures 4B, 5A–5C, 6C–6H, S5, and S6), RNP was incubated with unlabeled target DNA for at least 15 min at 37°C, then fluorescent trans-substrate was added. For assays using dual fluorescent-biotin-labeled trans-substrates (Figures 4B, 5A–5C, 6C–6H, S5A, S5B, S5D, S5E, and S6), uncleaved substrate was removed by magnetic bead capture. Low and high concentrations of target tested in Figure 6C are 1.0 and 10 pM of IS2404 or 0.26 and 2.6 ng of genomic DNA: assuming 250 copies of IS2404 per genome (Phillips et al., 2005), these amounts of M. ulcerans contain 1.0 and 10 pM of protospacers in 20 μL reactions. For assays using DNaseAlert trans-substrate (Figure S5C), reactions were diluted into the salt-EDTA quench solution, and HEX fluorescence was recorded directly (λex 535 nm and λem 563 nm).
In transient-state target-cleavage experiments (Figures 4C, 4D, S4A, and S4B), RNP was mixed with dual fluorescent-biotin-labeled dsDNA target to initiate time courses. Control reactions, consisting of reactions lacking crRNA (Figures 4C, 4D, and S4B) or RNP mixed with a solution of the target DNA containing EDTA (Figure S4A), were conducted in parallel. For transient-state experiments in which target and trans-substrate cleavage were measured simultaneously (Figure 4E), RNP was mixed with a solution containing a dual fluorescent-biotin-labeled dsDNA target (labeled with Alexa 488) and a dual fluorescent-biotin-labeled ssDNA trans-substrate (labeled with Alexa 594) to initiate time courses. For transient-state experiments in which cleavage of a dsDNA trans-substrate was measured (Figure 4F), RNP was mixed with a solution containing unlabeled dsDNA target and dual fluorescent-biotin-labeled non-target dsDNA trans-substrate to initiate time courses. For both types of experiments, control reactions consisted of RNP mixed with a solution of targets and trans-substrates containing EDTA. At successive time points, cleavage reactions were terminated by dilution into a denaturing quench solution composed of 90% formamide and 20 mM EDTA prior to removal of uncleaved substrates by magnetic bead capture.
In steady-state kinetic experiments (Figures 5D–5G and S4C–S4G), RNP was incubated with dsDNA target for at least 15 min, then mixed with varying amounts of fluorescent trans-substrate to initiate time courses. Control reactions, consisting of enzyme reactions lacking target DNA, were conducted in parallel unless indicated otherwise. For cleavage of dual fluorescent-biotin-labeled trans-substrates (Figures 5D, 5F, 5G, S4C, S4D, and S4F), time courses were terminated by diluting reactions at successive time points into the salt-EDTA quench solution prior to removal of uncleaved substrate by magnetic bead capture. For cleavage of the fluorescent dsDNA serving as both target and trans-substrate (Figure 5F), RNP was mixed with varying amounts of the fluorescent dsDNA to initiate time courses; control reactions, consisting of enzyme reactions lacking crRNA, were conducted in parallel. For cleavage of DNaseAlert trans-substrate, HEX fluorescence was measured continuously over several hours after initiation of time courses (Figures 5E and S4G) or after quenching at intervals with the salt-EDTA solution (Figure S4E), and cleaved product was quantified when need as described above for RNaseAlert reactions. The effect of heparin was tested by including sodium heparin (MP Biomedicals: cat. no 101931) during the trans-cleavage phase of the reaction (Figures S4F and S4G).
Fluorophore quantification and data analysis
Fluorescence readings of supernatants from which uncleaved substrates were removed by magnetic beads were compared to standard curves derived from titrations of starting solutions of fluorescent substrates to derive estimates of fluorophore concentrations (molarity). No adjustments were made to values to account for changes in fluorescence due to nuclease digestion of substrates (see below, Figure S1 and Table S2). Analytical limits of detection (LOD) and 95% confidence intervals were calculated using unweighted four-parameter logistic fit of raw fluorescence values of assay supernatants (Holstein et al., 2015) from selected experiments (Table S5).
In transient-state kinetic experiments, time-dependent changes in fluorescence reporter levels were processed as follows. Control fluorescence values collected in parallel were subtracted and converted to molarity, and the resulting values, F(t), were subjected to ordinary least-squares fitting in GraphPad Prism v8.2 (GraphPad Software, Inc.). The single-exponential equation is given by:
where t is time, F0 and Ff are initial and final fluorescent reporter values, respectively, and kobs is the apparent first-order rate constant. The double-exponential (sequential) equation is given by:
where kobs1 and kobs2 represent the apparent first-order rate constants for the two sequential steps (Fierke and Hammes, 1995).
In steady-state kinetic experiments, linear fitting of control-corrected reporter values was performed to obtain initial velocities (V0). Fitting of the substrate (S) dependence of V0 to the hyperbolic Michaelis-Menten equation (Michaelis and Menten, 1913):
was performed to obtain Vmax and Michaelis-Menten constant KM. The constant kcat was obtained by dividing Vmax by the concentration of activated enzyme, which is given by the target concentration, assuming target is fully bound by RNP, an assumption validated in experiments in which RNP was increased four-fold (see Figure S3J for Cas13a and Figures S4C and S4D for Cas12a). In some cases, no approach to saturation was observed (e.g. U5 and U10 cleavage by Cas13a activated by mature crRNA-Target-1 in Figure 3E), indicating KM ≫ highest [S] tested. In these cases, a lower limit estimate for KM was set at the highest substrate concentration tested and linear fitting of the substrate-dependence of V0 yielded slopes that, when divided by activated enzyme concentrations, yielded estimates for kcat/KM, and hence, kcat. The validity of all Cas13a trans-kinetic constants was corroborated by similarly analyzing rate data at low substrate concentrations (Table S4).
Steady-state kinetic constants were also estimated from endpoint experiments, as follows. For a given trans-substrate concentration, under conditions in which substrate conversion is low, V0 is maintained throughout the experiment, and product increases linearly with time. Under such conditions, trans-product increases in direct proportion to target concentration (which serves as proxy for activated enzyme), and an apparent turnover rate (kapp) can be determined from the constant of proportionality (the slope) by taking into account the duration of the reaction. Since kapp represents kcat ([S]/([S] + KM), kcat and KM may be estimated from the ratio of kapp calculated from two widely differing trans-substrate concentrations. For instance, for Cas12a activated by crRNA-Target-1 (Figure 5C, inset), kapp in the presence of 100 nM substrate C10 was 2.01 (±0.02) s−1, which is less than ten times the value obtained using 1/10th that concentration of substrate, which was 0.64 (±0.02) s−1 for 10 nM C10, suggesting the KM value for C10 lies between 10 and 100 nM. Indeed, from the ratio of kapp values, an estimate of KM equal to 31 nM is obtained, and, substituting this value back into the Michaelis-Menten equation, yields estimates for kcat of 2.6 s−1 and kcat/KM of 8.4 x 107 M−1 s−1, which agree reasonably well with steady-state analysis, which gave kcat of 2.8 s−1, KM of 28 nM and kcat/KM as 9.8 x 107 M−1 s−1 (Table 1). For Cas13a activated by mature crRNA-Target-1, based on slope values, the kapp of 0.89 (±0.01) s−1 for 100 nM U10 is approximately 10-times that in the presence of 1/10th the concentration of trans-substrate, corresponding to 0.065 (±0.001) s−1, using the crRNA-Target-1 pair (Figure 3A, inset). These results may be explained by an enzyme operating well below KM, and assuming KM ≫ 100 nM (born out in steady-state analysis), kapp values suggest kcat/KM > 7–9 x 106 M−1 s−1 and kcat > 0.7 s−1, both consistent with steady-state analysis (Table 1). Similar analysis was performed for other Cas13a crRNA-target combinations (Figures 3B, 3C, and S2D–S2H; Table S3).
Nuclease digestion of nucleic acid reporter probes
Solutions containing 1.0 μM reporter nucleic acids and DNase I (4 units) or RNase A (0.02 milliunits) were incubated for 2 hr at 37°C in buffer composed of 10 mM Tris-HCl, pH 7.6, 2.5 mM MgCl2, and 0.5 mM CaCl2, or Cas13a assay buffer, respectively. Digests were then diluted into salt-EDTA quench solution, and fluorescence was recorded and normalized to that of untreated material (Figure S1 and Table S2). Since fluorescence output of DNase I-cleaved trans-substrate G9C increases seven-fold (Table S2) and fluorescence signals were not corrected for this effect, product released using this substrate is likely overrepresented.
Creation of crRNA pools
A total of 21 crRNA designed against protospacers in hepatitis C viral RNA were tested individually with Cas13a on varying concentrations of their cognate target RNA and 100 nM trans-substrate U10. Based on the resulting trans-RNase activities, 13 non-overlapping crRNA were chosen for pooling (Figure 6A), including crRNA-3 to −5, 1-2, 2-2, −6 to −10, 11-2, 12-2, and 13. Trans-activity of Cas13a bound with crRNA-3 or pooled crRNA (containing 77 pM of each crRNA) was tested on varied pooled target RNA (Figure 6B), consisting of equimolar concentrations of Target RNA-3 to −5, -1a, -2a, −6 to −10, −11, −12, and 13.
A total of 39 crRNA designed to target protospacers containing adjacent PAM sequences within IS2404 were tested individually with Cas12a on 10 pM IS2404 and trans-substrate C10 without pre-incubation of RNP and target (Figure 6E). Interference between equipotent pairs of Cas12a-crRNA complexes targeting adjacent or overlapping protospacers was investigated by measuring trans-activity of individual or paired crRNA (Figure S6), the results of which were interpreted as follows. If two different Cas12a-crRNA bind to and cleave target DNA at equal rates, then the resulting trans-activity of the paired crRNA should be at least 50% of the sum of individual activities, which was observed for every pair tested. If the two Cas12a-crRNA complexes act completely independently, then the resulting trans-activity of the pair should be 100% of the sum, which was observed for crRNA pairs targeting distantly separated protospacers. For interfering Cas12a-crRNA complexes, paired trans-activity will be considerably less than 100%, which was observed for complexes whose targeted footprints overlapped in a non-favorable relative orientation. The lower activity crRNA of an interfering pair was then excluded from consideration. Of the highest potency crRNA remaining, 20 were chosen for four sets of sub-pools (A – D), including crRNA-1 to −3, −5, and −7 to −22 (Figure 6E). Cumulative inclusion of sub-pools resulted in enhanced trans-activity that increased with diminishing magnitude reflecting the decreasing potency of the added sub-pools (Figure 6F). Cas12a bound with 20 pooled crRNA containing each crRNA at 1/20th the total crRNA concentration was used for sensitive detection of IS2404 (Figures 6G and 6H).
Analysis of patient samples
Rehydrated swabs samples were shipped on dry ice prior to subsequent DNA extraction from 200 μL of material using the QIAamp DNA Microbiome Kit (Qiagen, cat. no. 51704). The resulting DNA (50 μL) was quantified by Qubit. qPCR with TaqMan Gene Expression Master Mix (Life Technologies cat. no. 4369106) using primers and probes specific for IS2404 (Table S1) was performed on 2.0 μL of extracted material in triplicate using IS2404 as DNA standard (Figure 6H). Trans-activity of Cas12a was measured on 3.0 μL of extracted material in triplicate in 20 μL reactions composed of Cas12a, crRNA (pool of 20), trans-substrate C20 in assay buffer supplemented with BSA (Figure 6H). Target DNA in samples was determined by interpolation to standard curves generated from a dilution series of IS2404 spanning 3.0–200 fM. From both qPCR and Cas12a, concentration of IS2404 is stated with reference to the 50 μL extract.
Quantification and statistical analysis
Data in Figures 2, 3, 4, 5, 6, and S1–S6 were processed and visualized using Microsoft Excel and GraphPad Prism 8. The number of replicate wells analyzed for each data point in all experiment is indicated in the Method Table. The model used for least-squares fitting of data points in each experiment is indicated in figure legends and the procedures (see above).
| Method table. Cleavage reaction conditionsa | |||||||
|---|---|---|---|---|---|---|---|
| Figure | Replicates | Cas (nM) | crRNA (nM) | Target | Trans-substrate (nM) | Time (hr) | Other (when indicated) |
| 2A | 3 | 5.0 | 2.5 | 1.0 nM | None | 0.5 | 50 mM EDTA |
| 2B | 3 | 2.0 | 1.0 | 10 pM | 100 | 1.0 | 25 mM EDTA |
| 2C and 2D | 2 | Vary | Vary | 1.0 nM | None | Vary | |
| 2E | 2 | 10 | 5.0 | Vary | None | Vary | |
| 2G and S3C | 1 | 10 | 5.0 | 1.0 nM | Vary | Vary | |
| 3A | 3 | 2.0 | 1.0 | Vary | 10, 100 | 2.0 | |
| 3B and 3C | 3 | 2.0 | 1.0 | Vary | 100 | 1.0 | |
| 3D–3F, S3D, S3E, S3G, and S3H | 1 | 2.0 | 1.0 | 10 pM | Vary | Vary | |
| 3H, 3I, S3F, and S3I | 3 | 2.0 | 1.0 | 10 pM | Vary | Vary | |
| 4A | 3 | 5.0 | 2.5 | 1.0 nM | None | 0.5 | 50 mM EDTA |
| 4B and 6D | 3 | 1.0 | 1.0 | 10 pM | 100 | 1.0 | 50 mM EDTA |
| 4C | 2 | Vary | Vary | 1.0 nM | None | Vary | |
| 4D | 2 | 10 | 5.0 | Vary | None | Vary | |
| 4E | 2 | 5.0 | 5.0 | 1.0 nM | Vary | Vary | |
| 4F | 2 | 5.0 | 5.0 | 1.0 nM | Vary | Vary | |
| 5A, 5B, and 6C | 3 | 1.0 | 1.0 | Vary | 100 | 2.0 | |
| 5C | 3 | 1.0 | 1.0 | Vary | 10, 100 | 2.0 | |
| 5D | 1 | 1.0 | 1.0 | 10 pM | Vary | Vary | |
| 5E | 3 | 1.0 | 1.0 | 10 pM | Vary | Vary | |
| 5F | 1 | 2.0 | 1.0 | 1.0 nM | Vary | Vary | |
| 5G | 1 | 2.0 | 2.0 | 1.0 nM | Vary | Vary | |
| 6A and 6B | 3 | 2.0 | 1.0 | Vary | 100 | 1.0 | |
| 6E and S6A | 4–13 | 1.0 | 1.0 | 10 pM | 100 | 1.0 | |
| 6F | 3 | 1.0 | 1.0 | 10 pM | 10 | 2.0 | |
| 6G | 3 | 1.0 | 1.0 | Vary | 10 | 2.0 | |
| 6H | 3 | 1.0 | 1.0 | Vary | 100 | 2.0 | 0.5 mg/mL BSA |
| S2A | 3 | 2.0 | 1.0 | 100 pM | 83 | 0.5 | |
| S2B | 3 | 1.0 | 0.5 | 500 pM | 100 | 1.0 | |
| S2C | 3 | 2.0 | 1.0 | 10, 100 pM | 100 | 2.0 | |
| S2D–S2H | 3 | 2.0 | 1.0 | Vary | 100 | 1.0 | |
| S3A | 3 | 5.0 | 2.5 | 1.0 nM | None | Vary | |
| S3B | 3 | Vary | Vary | 1.0 nM | None | Vary | |
| S3J | 1 | 8.0 | 4.0 | 10 pM | Vary | Vary | |
| S3K | 1 | 2.0 | 1.0 | 100 pM | Vary | Vary | 100 μg/mL yeast tRNA |
| S4A | 3 | 5.0 | 5.0 | 1.0 nM | None | Vary | 50 mM EDTA |
| S4B | 3 | 10 | 5.0 | 1.0 nM | None | Vary | |
| S4C | 1 | 1.0 | 1.0 | 10 pM | Vary | Vary | |
| S4D | 1 | 4.0 | 4.0 | 10 pM | Vary | Vary | |
| S4E | 1 | 1.0 | 1.0 | 100 pM | Vary | Vary | |
| S4F | 1 | 5.0 | 6.26 | 100 pM | Vary | Vary | 50 μg/mL heparin |
| S4G | 3 | 5.0 | 6.25 | 100 pM | Vary | Vary | 100 μg/mL heparin |
| S5A | 3 | 1.0 | 1.0 | 2.5, 5.0, 10 pM | 100 | 2.0 | |
| S5B | 3 | 1.0 | 1.0 | Vary | 100 | 0.5 | |
| S5C | 3 | 2.0 | 2.0 | 1.0 nM | 83 | 0.25 | |
| S5D | 3 | 1.0 | 1.0 | 1.0 nM | 100 | 1.0 | 50 mM EDTA |
| S5E | 3 | 2.0 | 2.0 | 1.0 nM | 100 | 1.0 | 50 mM EDTA |
| S6B and S6D | 3 | 1.0 | 1.0 | 10 pM | 100 | 1.0 | |
a Cleavage reactions performed in the number of wells for each point in the indicated figures. Concentrations are given for the components during the cleavage phase of the reactions for the time indicated.
Acknowledgments
The authors thank Brittney Thornton for technical assistance and Ted Baughman, Joshua Bishop, Shannon Kuyper, Akos Somoskovi, David Bell, and Robert Jenison for valuable discussions. E.A.N., N.P., P.J.Y.L., Z.I., R.M.K., I.P, A-L.M.L.N, and D.M. were supported by the Intellectual Ventures Global Good Fund. J.A.D. was supported by NIH grant nos. RM1HG009490 and U01AI142817-02, the William M. Keck Foundation, the National Multiple Sclerosis Society, NSF grant no. 1817593, the Paul Allen Frontiers Group, and the Howard Hughes Medical Institute. G.J.K. was supported by an NHMRC Investigator Grant (EL1, 1175568) and previously by an American Australian Association Fellowship. E.M. was supported by the Raoul Follereau Fondation.
Author contributions
Conceptualization, E.A.N., A.-L.M.L.N., and D.M.; Methodology, E.A.N., A-L.M.L.N., and D.M.; Investigation, E.A.N., N.P., P.J.Y.L., Z.I., R.M.K., and I.P.; Resources, E.M., G.J.K., and J.A.D.; Writing- Original Draft, E.A.N.; Writing – Review and Editing, A.-L.M.L.N., D.M, G.J.K., and J.A.D.
Declaration of interests
Some methods described herein have patents pending on which E.A.N., P.Y.J.L., G.J.N., J.A.D, A.-L.M.L.N, and D.M are inventors. The Regents of the University of California have patents issued and pending for CRISPR technologies on which G.J.K. and J.A.D. are inventors. J.A.D. is a cofounder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, and Mammoth Biosciences; is a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, and Inari; is a director at Johnson & Johnson; and has research projects sponsored by Biogen and Pfizer.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102996.
Contributor Information
Eric A. Nalefski, Email: eric.nalefski@ghlabs.org.
Damian Madan, Email: damian.madan@ghlabs.org.
Supplemental information
Data and code availability
The published article includes all datasets generated or analyzed during this study.
References
- Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B., Shmakov S., Makarova K.S., Semenova E., Minakhin L. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2015;353:1–23. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanzadeh J. Preventing PCR amplification carryover contamination in a clinical laboratory. Ann Clin Lab Sci. 2004;34:389–396. [PubMed] [Google Scholar]
- Bissonnette L., Bergeron M. Next revolution in the molecular theranostics of infectious diseases: microfabricated systems for personalized medicine. Expert Rev Mol Diagn. 2006;6:433–450. doi: 10.1586/14737159.6.3.433. [DOI] [PubMed] [Google Scholar]
- Borst A., Box A.T.A., Fluit A.C. False-positive results and contamination in nucleic acid amplification assays: suggestions for a prevent and destroy strategy. Eur J Clin Microbiol Infect Dis. 2004;4:289–299. doi: 10.1007/s10096-004-1100-1. [DOI] [PubMed] [Google Scholar]
- Chen J.S., Ma E., Harrington L.B., Tian X., Doudna J.A. CRISPR-Cas12a target binding unleashes single-stranded DNase activity. Science. 2017;360:436–439. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cofsky J.C., Karandur D., Huang C.J., Witte I.P., Kuriyan J., Doudna J.A. CRISPR-Cas12a exploits R-loop asymmetry to form double strand breaks. eLife. 2020;9:e55143. doi: 10.7554/eLife.55143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curti L.A., Primost I., Valla S., Alegre D.I., Perglione C.O., Repizo G.D., Lara J., Parcerisa I., Palacios A., Llases M.E. Evaluation of a lyophilized CRISPR-cas12 assay for a sensitive, specific, and rapid detection of SARS-CoV-2. Viruses. 2021;13:420. doi: 10.3390/v13030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H.D., Tjian R., Doudna J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A., O’Connell M.R., Burstein D., Knott G.J., Doudna J.A. RNA targeting by functionally orthogonal type VI-A CRISPR-cas enzymes. Mol. Cell. 2017;66:373–383.e3. doi: 10.1016/j.molcel.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierke C.A., Hammes G.G. Transient kinetic approaches to enzyme mechanisms. Methods Enzymol. 1995;249:3–37. doi: 10.1016/0076-6879(95)49029-9. [DOI] [PubMed] [Google Scholar]
- Fozouni P., Son S., Díaz de León Derby M., Knott G.J., Gray C.N., D’Ambrosio M.V., Zhao C., Switz N.A., Kumar G.R., Stephens S.I. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell. 2021;184:323–333.e9. doi: 10.1016/j.cell.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B.X.H., Smith J.D., Fuchs R.T., Mabuchi M., Curcuru J., Robb G.B., Fire A.Z. Target-dependent nickase activities of the CRISPR–cas nucleases Cpf1 and Cas9. Nat. Microbiol. 2019;4:888–897. doi: 10.1038/s41564-019-0382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs R.T., Curcuru J., Mabuchi M., Yourik P., Robb G.B. Cas12a trans-cleavage can be modulated in vitro and is active on ssDNA, dsDNA, and RNA. bioRxiv. 2019:600890. doi: 10.1101/600890. [DOI] [Google Scholar]
- Gong S., Yu H.H., Johnson K.A., Taylor D.W. DNA unwinding is the primary determinant of CRISPR-cas9 activity. Cell Rep. 2018;22:359–371. doi: 10.1016/j.celrep.2017.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Górecka K.M., Krepl M., Szlachcic A., Poznański J., Šponer J., Nowotny M. RuvC uses dynamic probing of the Holliday junction to achieve sequence specificity and efficient resolution. Nat. Commun. 2019;10:4102. doi: 10.1038/s41467-019-11900-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton H.G., Watson B.N.J., Fineran P.C. The arms race between bacteria and their phage foes. Nature. 2020;577:327–336. doi: 10.1038/s41586-019-1894-8. [DOI] [PubMed] [Google Scholar]
- Holstein C.A., Griffin M., Hong J., Sampson P.D. Statistical method for determining and comparing limits of detection of bioassays. Anal. Chem. 2015;87:9795–9801. doi: 10.1021/acs.analchem.5b02082. [DOI] [PubMed] [Google Scholar]
- Jeon Y., Choi Y.H., Jang Y., Yu J., Goo J., Lee G., Jeong Y.K., Lee S.H., Kim I.S., Kim J.S. Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nat. Commun. 2018;9:2777. doi: 10.1038/s41467-018-05245-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P.D.R., Stinear T., Small P.L.C., Plushke G., Merritt R.W., Portaels F., Huygen K., Hayman J.A., Asiedu K. Buruli ulcer (M. ulcerans infection): new insights, new hope for disease control. PLoS Med. 2005;2:0282–0286. doi: 10.1371/journal.pmed.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott G.J., Thornton B.W., Lobba M.J., Liu J.J., Al-Shayeb B., Watters K.E., Doudna J.A. Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat. Struct. Mol. Biol. 2019;26:315–321. doi: 10.1038/s41594-019-0208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E.V., Zhang F. Coupling immunity and programmed cell suicide in prokaryotes: life-or-death choices. BioEssays. 2017;39:1–9. doi: 10.1002/bies.201600186. [DOI] [PubMed] [Google Scholar]
- Koonin E.V., Makarova K.S., Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017;37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larremore D.B., Wilder B., Lester E., Shehata S., Burke J.M., Hay J.A., Milind T., Mina M.J., Parker R. Test sensitivity is secondary to frequency and turnaround time for COVID-19 surveillance. medRxiv. 2020 doi: 10.1101/2020.06.22.20136309. [Preprint]. 2020.06.22.20136309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R.A., de Puig H., Nguyen P.Q., Angenent-mari N.M., Donghia N.M., McGee J.P., Dvorin J.D., Klaperich C.M., Pollock N.R., Collins J.J. Ultrasensitive CRISPR-based diagnostic for field-applicable detection of Plasmodium species in symptomatic and asymptomatic malaria. Proc. Natl. Acad. Sci. U S A. 2020:1–10. doi: 10.1073/pnas.2010196117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.J., Geyer R., Tan W. Using molecular beacons as a sensitive fluorescence assay for enzymatic cleavage of single-stranded DNA. Nucleic Acids Res. 2000;28:52e–552. doi: 10.1093/nar/28.11.e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.-Y., Cheng Q.-X., Wang J.-M., Li X.-Y., Zhang Z.-L., Gao S., Cao R.-B., Zhao G.-P., Wang J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018;4:20. doi: 10.1038/s41421-018-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Li S., Wang J., Liu G. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 2019;37:730–743. doi: 10.1016/j.tibtech.2018.12.005. [DOI] [PubMed] [Google Scholar]
- Liu L., Li X., Ma J., Wang M., Zhang X., Wang Y., Liu L., Li X., Ma J., Li Z. The molecular architecture for RNA-guided RNA cleavage by Cas13a the molecular architecture for RNA-guided RNA cleavage by Cas13a. Cell. 2017;170:714–720.e10. doi: 10.1016/j.cell.2017.06.050. [DOI] [PubMed] [Google Scholar]
- Marion E., Ganlonon L., Claco E., Blanchard S., Kempf M., Adeye A., Chauty A. Establishment of quantitative PCR (qPCR) and culture laboratory facilities in a field hospital in Benin: 1-year results. J. Clin. Microbiol. 2014;52:4398–4400. doi: 10.1128/JCM.02131-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske A.J., Nakandakari-Higa S., Marraffini L.A. Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature. 2019;570:241–245. doi: 10.1038/s41586-019-1257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis L., Menten M. Die kinetik der Invertinwirkung. Biochem. Z. 1913;49:333–369. [Google Scholar]
- Mo C.Y., Mathai J., Rostøl J.T., Varble A., Banh D.V., Marraffini L.A. Type III-A CRISPR immunity promotes mutagenesis of staphylococci. Nature. 2021;592:611–615. doi: 10.1038/s41586-021-03440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P., Zabel D. EcoRI endonuclease physical and catalytic properties of the homogeneous enzyme. J. Biol. Chem. 1976;251:5866–5874. [PubMed] [Google Scholar]
- Moussaoui M., Cuchillo C.M., Nogues M.V. A phosphate-binding subsite in bovine pancreatic ribonuclease A can be converted into a very efficient catalytic site. Protein Sci. 2007;1:99–109. doi: 10.1110/ps.062251707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugan K., Seetharam A., Severin A., Sashital D. CRISPR-Cas12a has widespread off-target and dsDNA-nicking effects. J. Biol. Chem. 2020;295:5538–5553. doi: 10.1074/jbc.RA120.012933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouri R., Tang Z., Dong M., Liu T., Kshirsagar A., Guan W. SCRISPR-based detection of SARS-CoV-2: a review from sample to result. Biosens. Bioelectron. 2021;178:113012. doi: 10.1016/j.bios.2021.113012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R., Horsfield C., Kuijper S., Lartey A., Tetteh I., Nyamekye B., Awuah P., Nyarko K.M., Lucas S., Kolk A.H.J. Sensitivity of PCR targeting the IS2404 insertion sequence of Mycobacterium ulcerans in an assay using punch biopsy specimens for diagnosis of Buruli ulcer. J. Clin. Microbiol. 2005;43:3650–3656. doi: 10.1128/JCM.43.8.3650-3656.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman M.T., Dey P., Hassan M.I., Ahman F., Batra J.K. Functional role of glutamine 28 and arginine 39 in double stranded RNA cleavage by human pancreatic ribonuclease. PLoS ONE. 2011;6::e17159. doi: 10.1371/journal.pone.0017159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S., Abudayyeh O.O., Makarova K.S., Wolf Y.I., Gootenberg J.S., Semenova E., Minakhin L., Joung J., Konermann S., Severinov K. Discovery and functional characterization of diverse class 2 CRISPR-cas systems. Mol. Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund V., Adjei O., Nitschke J., Thompson W., Klutse E., Herbinger K.H., Thompson R., van Vloten F., Racz P., Fleischer B. Dry reagent-based polymerase chain reaction compared with other laboratory methods available for the diagnosis of Buruli ulcer disease. Clin. Infect. Dis. 2007;45:68–75. doi: 10.1086/518604. [DOI] [PubMed] [Google Scholar]
- Singh D., Mallon J., Poddar A., Wang Y., Tippana R., Yang O., Bailey S., Ha T. Real-time observation of DNA target interrogation and product release by the RNA-guided endonuclease CRISPR Cpf1 (Cas12a) Proc. Natl. Acad. Sci. U S A. 2018;115:5444–5449. doi: 10.1073/pnas.1718686115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargon A.A., Cox D.B.T., Pyzocha N.K., Zheng K., Slaymaker I.M., Gootenberg J.S., Abudayyeh O.A., Essletzbichler P., Shmakov S., Makarova K.S. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol. Cell. 2017;65:618–630.e7. doi: 10.1016/j.molcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella S., Mesa P., Thomsen J., Moses M.E., Hatzakis N.S., Montoya G., Jensen S.B., Saligram B., Moses M.E., Hatzakis N.S. Conformational activation promotes CRISPR-cas12a catalysis and resetting of the endonuclease article conformational activation promotes CRISPR-cas12a catalysis and resetting of the endonuclease activity. Cell. 2018;175:1856–1871. doi: 10.1016/j.cell.2018.10.045. [DOI] [PubMed] [Google Scholar]
- Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohkendl I., Saifuddin F.A., Rybarski J.R., Finkelstein I.J., Russell R. Kinetic basis for DNA target specificity of CRISPR-cas12a. Mol. Cell. 2018;71:1–9. doi: 10.1016/j.molcel.2018.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts D.C., van der Oost J., Jinek M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-cas12a. Mol. Cell. 2017;66:221–233.e4. doi: 10.1016/j.molcel.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambe A., East-Seletsky A., Knott G.J., Doudna J.A., O’Connell M.R. RNA binding and HEPN-nuclease activation are decoupled in CRISPR-cas13a. Cell Rep. 2018;24:1025–1036. doi: 10.1016/j.celrep.2018.06.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varble A., Marraffini L.A. Three new Cs for CRISPR: collateral, communicate, cooperate. Trends Genet. 2019;35:446–456. doi: 10.1016/j.tig.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y., Zhou Y., Liu Y., Ying W., Lv R., Zhao Q., Zhou H., Zuo E., Sun Y., Yang H. Indiscriminate ssDNA cleavage activity of CRISPR-Cas12a induces no detectable off-target effects in mouse embryos. Protein Cell. 2021 doi: 10.1007/s13238-021-00824-z. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W.X., Chong S., Zhang H., Makarova K.S., Koonin E.V., Cheng D.R., Scott D.A., Yan W.X., Chong S., Zhang H. Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol. Cell. 2018;70:327–339. doi: 10.1016/j.molcel.2018.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W.X., Hunnewell P., Alfonse L.E., Carte J.M., Keston-Smith E., Sothiselvam S., Garrity A.J., Chong S., Makarova K.S., Koonin E.V. Functionally diverse type V CRISPR-Cas systems. Science. 2019;363:88–91. doi: 10.1126/science.aav7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Lee J.Y., Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Yotsu R.R., Suzuki K., Simmonds R.E., Bedimo R., Ablordey A., Yeboah-Manu D., Phillips R., Asiedu K. Buruli ulcer: a review of the current knowledge. Curr. Trop. Med. Rep. 2018;5:247–256. doi: 10.1007/s40475-018-0166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Van Der Oost J., Regev A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.