

Abstract
Mounting evidence supports immune dysfunction in psychiatric conditions such as post-traumatic stress disorder (PTSD). The association of immunomodulatory mechanisms with PTSD-relevant behavior and physiology is not well understood. Communication between neurons and microglia, resident immune cells of the central nervous system, is crucial for optimal regulation of behavior and physiology. In this regard, the fractalkine CX3CL1, secreted from neurons and its target, the microglial CX3CR1 receptor represent a primary neuron-microglia inter-regulatory system important for synaptic plasticity and function. The current study investigated the impact of CX3CR1 deficiency on behaviors relevant to PTSD, such as fear acquisition and memory, acoustic startle response and anxiety-like behavior. Morphological analysis of microglia and neuronal activation within PTSD-relevant forebrain nuclei regulating stress and fear behaviors was also conducted. CX3CR1-deficient (CX3CR1−/−) mice elicited increased fear acquisition as well as reinstatement of fear as compared to wild type (CX3CR1+/+) mice. Conditioned fear and extinction were not significantly different between genotypes. No significant differences were observed in unconditioned acoustic startle response between genotypes. CX3CR1−/− mice showed reduced anxiety-like behaviors as compared with CX3CR1+/+ mice. Morphological assessment of microglia showed region-selective effects of CX3CR1 deficiency, primarily within hypothalamic and cortical areas. Lastly, CX3CR1−/− mice elicited elevated neuronal activity in the PVN and the ventral tegmental-interpeduncular area following reinstatement of fear. Collectively, our data suggest that impaired CX3CR1 function may evoke region-selective alterations in forebrain circuits regulating stress, anxiety and fear, impacting behaviors relevant to disorders such as PTSD.
Keywords: CX3CR1, Fear, Startle, Anxiety, Microglia, PTSD
1. Introduction
Posttraumatic stress disorder (PTSD) is a debilitating, trauma-evoked psychiatric illness with a life-time prevalence of about 7.8% in adult Americans (National comorbidity survey replication NCS-R) (Kessler et al., 2005). PTSD patients have impaired processing of fear leading to re-experiencing, avoidance and hyperarousal symptoms to trauma reminders. Neurobiological studies support altered function of forebrain limbic regions regulating stress and emotion, such as the medial prefrontal cortex (mPFC), hippocampus and the amygdala in PTSD (Pitman et al., 2012). Investigation of potential regulatory mechanisms that may influence forebrain limbic function is crucial to understanding PTSD pathophysiology.
There is mounting evidence for altered immune function in PTSD (Lindqvist et al., 2014; Michopoulos et al., 2017; Neigh and Ali, 2016), although the association of immunomodulatory mechanisms with PTSD-relevant behavior and physiology is not well understood. Microglia, resident immune cells of the central nervous system (CNS) have gained attention in recent years as active participants in the regulation of neuronal activity and function (Tremblay et al., 2011; Wohleb, 2016). Various means of communication between neurons and microglia have been identified in recent years. Neurons are able to alter microglia activity via chemokine release, neurotransmitters as well as purine signaling (Eyo and Wu, 2013). Conversely, microglia are capable of altering neuronal behavior as well, via direct physical contact and paracrine signals (Eyo and Wu, 2013). The fractalkine CX3CL1, an exclusive ligand secreted from neurons and its target, the microglial CX3CR1 represent a primary neuron-microglia inter-regulatory system in the central nervous system (Ransohoff and El Khoury, 2016). CX3CL1–CX3CR1 signaling has been associated with the modulation of cytokine release, stress responsivity, and behavior (Rogers et al., 2011, Paolicelli et al., 2014, Winkler et al., 2017). Previous studies have shown a requirement of the CX3CR1 receptor in regulation of microglial activation, accordingly CX3CR1 deficiency leads to dysregulation in cognitive function and synaptic plasticity (Rogers et al., 2011, Reshef et al., 2014). We hypothesized that impaired CX3CR1 function would be associated with microglial alterations in forebrain limbic areas impacting behaviors relevant to PTSD such as fear, anxiety and startle reactivity as well as, modulate neuronal activity in stress-regulatory circuits. To test this hypothesis, we investigated CX3CR1-deficient mice in a fear conditioning paradigm and measured acquisition, extinction and reinstatement of fear. Testing fear reinstatement is relevant to the return of fear and relapse that is often observed in psychiatric disorders (Goode and Maren, 2014; Vervliet et al., 2013). Mice were also screened for acoustic startle reactivity and anxiety-like behavior. In addition, morphological measures of microglial and neuronal activation were assessed at baseline and following behavioral testing in several forebrain sub-regions regulating stress and fear associated behaviors.
Our data revealed significant enhancement of fear acquisition and reinstatement in CX3CR1 deficient mice, as well as, reduced anxiety-like behavior, with no significant effects on acoustic startle behavior. CX3CR1 deficiency was accompanied by altered morphological activation of microglia and enhanced neuronal activity in selective forebrain sub-regions. Collectively, these findings suggest an important role of microglia-neuron signaling in modulating forebrain stress and fear regulatory circuits impacting behaviors relevant to disorders such as PTSD.
2. Methods
2.1. Animals
Male, wild-type (CX3CR1+/+) and CX3CR1−/− mice on a C57BL/6J background were obtained from Jackson Laboratories (Bar Harbor, Maine, USA). Mice were housed in pairs in a climate-controlled vivarium (temperature averages 23 ± 4 °C, humidity averages 30 ± 6%). All behavioral tests were performed during the 12 h light cycle between 8 am and 2 pm. Study protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Cincinnati, in a vivarium accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All behavioral tests were performed with mice between 11 and 13 weeks of age. CX3CR1−/− and wild-type mice underwent behavioral testing for anxiety (elevated zero maze), startle and fear conditioning (see Fig. 1 schematic). This layout was adopted for consistency with rodent models of PTSD where these behavioral tests are often conducted in the same animal. A separate cohort was tested for open field (OF) and light dark box (LDB) to avoid consecutive testing for anxiety-like behavior in multiple tests. For morphological assessment of microglia, neuronal activation and cytokine measurement under baseline conditions, tissue was collected from mice left undisturbed under home cage conditions.
Fig. 1.
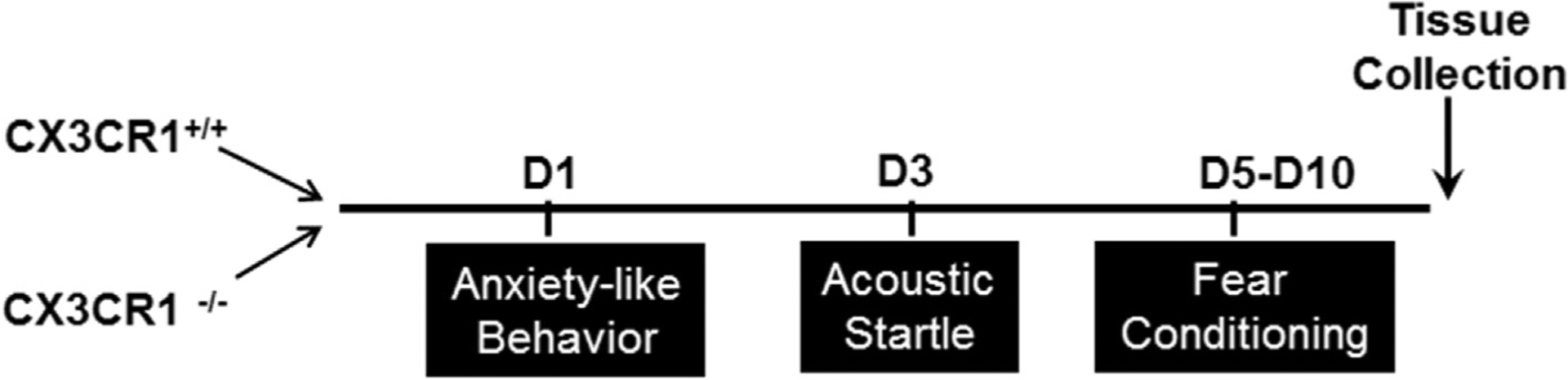
Schematic representation of experimental layout. Wild type (CX3CR1+/+) and CX3CR1-deficient (CX3CR1−/−) mice were subjected to behavioral testing for anxiety-like behavior (elevated zero maze) on Day 1 (D1) followed by acoustic startle (D3) and then underwent a contextual fear conditioning paradigm (acquisition (D5) conditioned fear testing (D6) extinction (D6–D9) and fear reinstatement (D10). A separate cohort was subjected to anxiety-like behavior testing in the open field and light dark box to avoid repeated testing for anxiety-like behavior using different anxiety tests. Brains were collected for morphological assessment of cFos-like immunoreactivity and microglial activation.
2.2. Behavioral tests
2.2.1. Anxiety-like behaviors
2.2.1.1. Elevated zero maze (EZM).
Animals were tested on the EZM following previous studies (Braun et al., 2011) with modifications. The maze (Stoelting Co., Wood Dale, IL) was a circular track divided in four quadrants of equal lengths with two opposing open quadrants with 1 cm high clear acrylic curbs to prevent falls and two opposing closed quadrants with black acrylic walls 20 cm in height. Dim halogen lamp lighting (35 lux on the open quadrant) was used during testing. Mice were placed in a closed quadrant and allowed to explore for 5 min, during which they were video-taped with an overhead video camera. The track and walls were cleaned with 10% ethanol between trials. Time spent in the open and closed quadrants, latency to enter the open quadrant and distance travelled were scored offline using TopScan software (CleverSys. Inc., Reston, VA).
2.2.1.2. Open field test (OFT).
Mice were tested in the OFT following previously described procedures (Vollmer et al., 2013). Briefly, the arena (19×19×8 in) consisted of a border area, a center (50% of the total area), and a center of center (40% of the center area) evenly illuminated at 20 lux. Mice were placed in the corner of the arena and behavior was recorded for 5 min. The arena was cleaned with 10% ethanol between each animal. Videos were scored for time spent in the center and border areas, total distance traveled, and latency to enter the center area.
2.2.1.3. Light/dark box (LDB).
Behavior in the LDB was tested following previous procedure (Costall et al., 1989) with modifications. The light dark box consists of a light and dark compartment each measuring 14 × 8 × 14 inches. Illumination in the light arena was 850 lux and 0 lux in the dark arena. Mice were placed in the dark compartment and behavior was recorded for 5 min. The arena was cleaned with 10% ethanol between each trial. Videos were recorded for offline scoring. Time spent in different compartments, latency to enter the light compartment and vertical exploration (rearing) in the light arena were scored and analyzed.
2.2.2. Acoustic startle
Startle response to an acoustic stimulus was measured using previously published procedures (Schmeltzer et al., 2015). The SR-Lab acoustic response system (San Diego Instruments, San Diego, California, USA) was used. The apparatus included ventilated, soundproof chambers measuring approximately 52 cm × 52 cm × 76 cm and contained an enclosure to keep the mouse over the sensor. The enclosure was of sufficient size to restrict but not restrain the animal and allowed it to turn around. The chambers were equipped with a fan delivering background noise at 68 dB The chambers were calibrated using the SR-LAB standardization unit (San Diego Instruments, San Diego, CA), which transmits a precise series of pulses to the sensor located on each enclosure, allowing each chamber to be adjusted to the same read-out value for an identical stimulus prior to testing. To minimize any effect of measurement differences between the chambers, an equal number of CX3CR1+/+ and −/− mice were tested in each chamber. The chambers and enclosures were cleaned between mice with 10% ethanol.
Mice were habituated in the chamber for 10 min. The test consisted of 30 trials. The acoustic stimulus was a 40 ms, 108 dB burst of white noise emitted at intervals determined semi-randomly by computer with inter-stimulus intervals were between 3 and 30 s with a minimum step between interval lengths of 3 s. Movement inside the tube was detected by a piezoelectric accelerometer below the frame. For each trial, measurements were taken at 1 ms intervals for a response window of 150 ms following the startle stimulus using National Instruments Data Acquisition Software (San Diego Instruments, San Diego, CA). The maximum voltage change (Vmax) within the recording window over the averaged baseline (5 ms) just prior to stimulus was used for data. Mean startle (vmax) for the entire session was also compared between genotypes.
2.2.3. Contextual fear conditioning
All mice underwent a contextual conditioning paradigm to investigate fear acquisition, conditioned fear, extinction and reinstatement following previously published procedures (McGuire et al., 2010; Vollmer et al., 2013) with modifications. Operant chambers housed in sound attenuated isolation cabinets were used (Clever Sys Inc.). The floors of the chambers consisted of stainless steel grid bars that delivered scrambled electric shocks. The grid, floor trays and chamber walls were wiped with 10% ethanol and allowed to dry completely. Each animal was acclimated to the chamber for 5 min, then received 3 shocks of 0.5 mA intensity, 1 s duration administered 1 min apart. The animals were placed in the chamber the next 4 days and recorded for 5 min without shocks to measure fear conditioning and extinction. On Day 6 mice were placed in the chamber for 5 min then received a single 0.5 mA, 1 s shock and behavior was recorded for another 5 min. Freezing, defined as complete lack of movement except respiration, was measured on Day 1 (acquisition), Days 2–5 (conditioned fear and extinction) and Day 6 (reinstatement) using the Freeze Scan software (Clever Sys Inc.).
2.3. Immunohistochemistry
Mice were perfused transcardially with 4% paraformaldehyde and brains were removed. Brains were cut at 30 μm on a sliding microtome and the resulting sections were stored in cryoprotectant (0.1 M phosphate buffer, 30% sucrose, 1% polyvinylpyrrolidone, and 30% ethylene glycol) at −20 °C until processed for immunohisto-chemistry. Primary antibodies used were anti-ionized calcium binding adapter molecule (IBA-1) (1:1000, Synaptic Systems Inc., cat/ 3 234-003, Germany) and c-Fos (1:1000, Santa Cruz Biotechnology, cat/ 10415, Santa Cruz, CA). Slices were transferred to 50 mM KPBS (pH 7.2; 40 mM potassium phosphate dibasic, 10 mM potassium phosphate monobasic, and 0.9% sodium chloride) and rinsed five times for 5 min. at RT. Sections were transferred to 0.3% H2O2 in KPBS and incubated for 10 min at RT. Slices were rinsed five times for 5 min. in KPBS and transferred to blocking solution [50 mM KPBS, 0.5% bovine serum albumin (BSA), and 0.4% Triton X-100] for 1 h at RT. Slices were incubated overnight at 4 °C in primary antibody diluted in blocking solution. The following day, sections were rinsed (five times for 5 min) in KPBS and incubated in secondary antibody (Cy-3 anti-rabbit; Jackson Immunoresearch) diluted (1:500) in 50 mM KPBS plus 0.5% BSA for 1hr at RT on a shaker in the dark. Tissue was rinsed five times for 5 min. in KPB, mounted onto UltraStick micro slides (Gold Seal Products, cat. 3039, Portsmouth, NH) and coverslipped with Gelvatol.
2.3.1. Imaging, quantification and analysis
Immunolabeled sections were imaged using the AxioImager ZI microscope (Zeiss) equipped with apotome (z-stack) imaging capability (Axiocam MRm camera and AxioVision Release 4.6 software; Zeiss). Processing for IBA-1 and cfos imaging and quantification was performed following previously published procedures from our lab (Schmeltzer et al., 2015; Vollmer et al., 2016). Briefly, for IBA-1 labeled tissue, Z-stacks were acquired using the 20× air objective lens at 568 nm. Images were analyzed using Image J software (NIH open access). For Iba-1postitive microglial cells, soma perimeter was analyzed using the ImageJ software tool “Freehand line” and recorded using the “Analyze and Measure” option. To determine branch length and number of endpoints a skeleton analysis was performed using the Image J “skeleton analysis” and “skeletonize” plug-ins. Images were enhanced to make processes visible and converted into binary images. The “despeckle” option was used to remove background staining. Then images were skeletonized and analyzed to quantify length and number of endpoints for each branch.
For c-Fos analysis, images were acquired in regions demonstrating cfos immunoreactive cells. The regions were delineated using characteristics of each nucleus taken from the atlas of Paxinos and Watson (1998), and quantified at a similar distance from bregma within all animals. Image magnifications were chosen in order to quantify the number of immunoreactive nuclei in one uni-lateral image from each section. If possible, at least four images per region of interest per mouse were collected. To quantify the number of immunoreactive nuclei the Image-J “cell counter” tool was used by an investigator blind to experimental group. Cell counts for each section were averaged for each animal and individual means averaged to derive group means.
2.4. Measurement of cytokines
Cytokine concentrations were measured using the Bio-Plex® Mouse Cytokine Assays (Bio-Rad, USA) as described previously by us (Vollmer et al., 2016). Briefly, mice were sacrificed via decapitation and snap frozen brains were stored at −80 °C until dissection. Forebrain sub-region tissue samples were micro-dissected from 2.3 mm slices on a cryostat (−20 °C) using atlas coordinates. Samples were homogenized in a dounce homogenizer with a tight fitting pestle in 500 μl of PBS supplemented with a cOmplete ULTRA protease inhibitor cocktail tablet (Roche). Samples were then centrifuged at 14,000 RPM for 15 min at 4 °C and the supernatant was collected for cytokine analysis.
2.5. Data analysis and statistics
Data are shown as mean ± SEM and were analyzed by two-factorial ANOVA, Student t test, or non-parametric test where applicable. Normality was tested formally for all data and met assumptions of the statistical tests being used. Fear conditioning expressed as percent freezing were analyzed by one- or two way repeated measures ANOVA using genotype as the between subject variable- and time as the repeated measure. Acoustic startle response over time blocks were analyzed by two-way repeated measures ANOVA using genotype and time as factors. Bonferroni’s post hoc analysis was applied where main effects were significant. Measures on the EZM, LDB and OF tests were assessed by two tailed student t tests except for latency and rearing frequency data in the EZM and LDB tests, respectively, that was analyzed by non-parametric Mann Whitney U test for group differences. Quantified microglia morphological measures were analyzed by two-way ANOVA using genotype and testing (basal versus post behavior test) as factors. cFos data was analyzed by two-tailed student t test. Statistical significance was taken as p < 0.05. Prism 6.0 software was used for statistical analysis (GraphPad Software, Inc., La Jolla, CA).
3. Results
3.1. Morphological alterations in microglia within forebrain limbic sub-regions in CX3CR1−/− and CX3CR1+/+ mice at baseline and post behavior
Alterations in soma size and process length were assessed in forebrain limbic subdivisions including the basolateral subdivision of the amygdala (BLA), the pre-frontal cortex (PFC), hippocampal dentate gyrus (DG), and paraventricular nucleus of the hypothalamus (PVN) (Fig. 2). Regional microglia were assessed at basal (homecage) conditions in the absence of any immunologic or stress challenges, and post-behavior testing. Significant differences were observed in microglial soma perimeter and microglial process length that were dependent on genotype as well as testing, and were region-selective. Consistently, CX3CR1−/− mice had increased soma perimeter and process length compared to wild-type mice under basal conditions (see Fig. 2 images and data). Interestingly, exposure to behavioral testing produced region-specific alterations in these parameters. In wild type mice, behavioral manipulations resulted in a significant increase in soma perimeter specifically in the PFC (Fig. 2G) and the PVN (Fig. 2O), an effect that was not observed in CX3CR1−/− mice. Two way ANOVA revealed a significant genotype, testing and genotype × testing interaction in the PFC [genotype (F1,31 = 250.5; p < 0.05), testing (F1,31 = 22.07; p < 0.05), genotype × testing (F1,31 = 68.45; p < 0.05) and PVN [genotype (F1,34 = 712.4; p < 0.05), testing (F1,31 = 114.6; p < 0.05), genotype × testing (F1,31 = 205.9; p < 0.05). Post-hoc analyses revealed increased soma perimeter in CX3CR1−/− mice vs +/+ mice at baseline and increased soma perimeter post-behavior only in CX3CR1+/+ mice. A significant effect of genotype but no effect of testing, or a genotype × testing interaction was observed for the BLA (Fig. 2C) [genotype (F1,33 = 36.74; p < 0.05), testing (F1,33 = 3.585; p > 0.05), genotype × testing (F1,33 = 1.286; p > 0.05)], and DG (Fig. 2K) [genotype (F1,33 = 57.16; p < 0.05), testing (F1,33 = 2.352; p > 0.05), genotype × testing (F1,33 = 0.638; p > 0.05). Post-hoc analyses revealed significant increase in soma perimeter in CX3CR1−/− mice compared to CX3CR1+/+ only under basal conditions. Exposure to behavioral testing also led to a significant increase in process length in both genotypes within all tested subregions, however this effect was absent in CX3CR1−/− mice only within the PVN. For process length, two way ANOVA revealed a significant effect of genotype (F1,34 = 18.95; p < 0.05), testing (F1,34 = 48.92; p < 0.05), and a genotype × test interaction (F1,34 = 10.98; p < 0.05) for the PVN (Fig. 2P). Post-hoc analyses revealed significant increase in branch length in CX3CR1−/− mice vs wild type under basal condition and a significant test-evoked increase in branch length in the wild type mice that was blunted in CX3CR1−/− mice. For the other regions, a significant effect of test and genotype × test interaction was observed, with no effect of genotype: PFC (Fig. 2H): test (F1,20 = 191.3; p < 0.05), genotype × test (F1,20 = 4.678; p < 0.05), genotype (F1,20 = 2.715; p > 0.05), BLA (Fig. 2D) test (F1,33 = 48.87; p < 0.05), genotype × test (F1,33 = 7.46; p < 0.05), genotype (F1,33 = 4.096; p > 0.05), and DG (Fig. 2L) test (F1,33 = 48.19; p < 0.05), genotype × test (F1,33 = 4.48; p < 0.05), genotype (F1,20 = 2.34; p > 0.05). Post hoc analyses revealed significantly greater branch length in CX3CR1−/− mice at baseline (PFC, BLA). Testing-evoked increases in branch length in these areas were similar between genotypes.
Fig. 2.
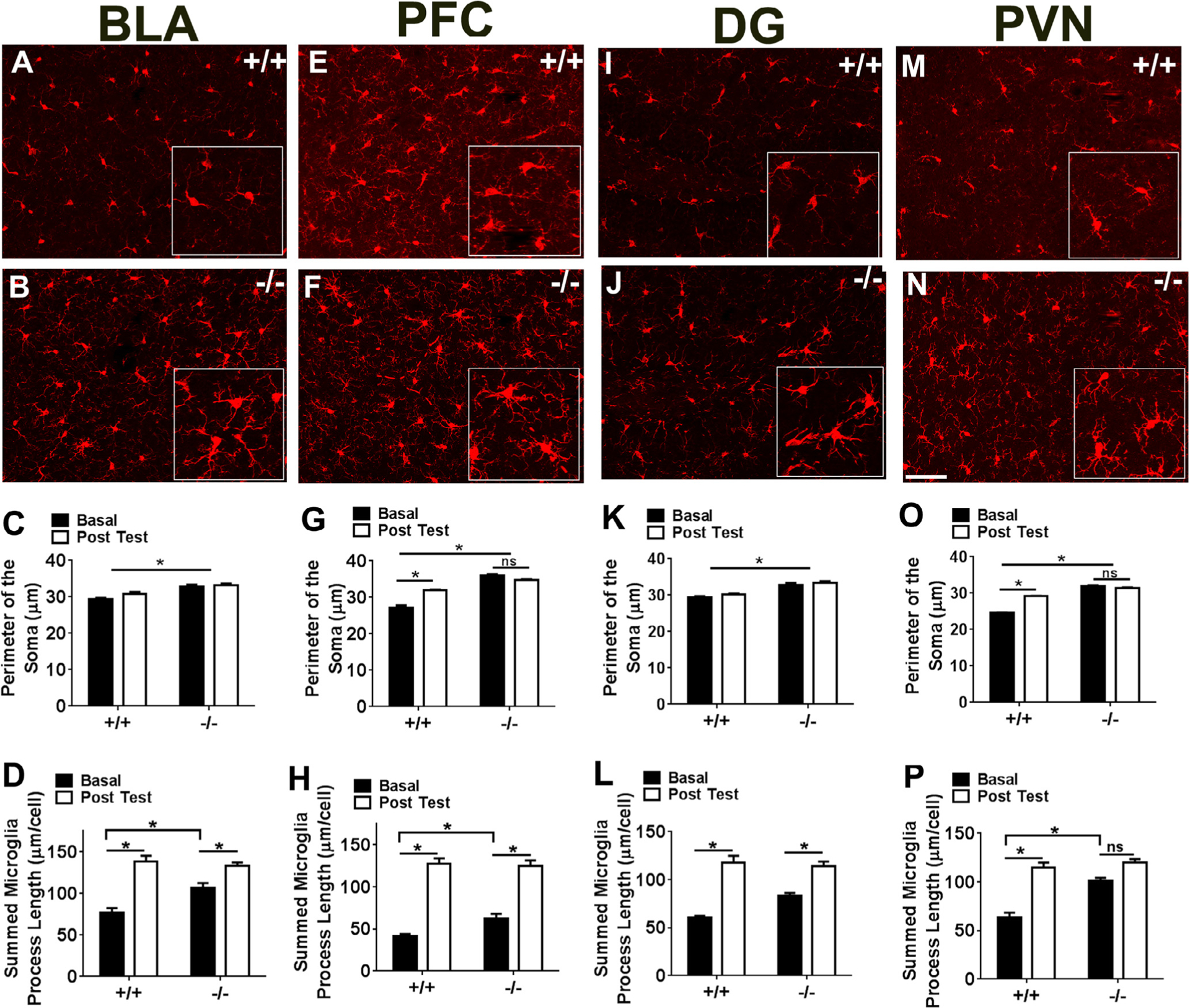
Morphological analysis of microglia in CX3CR1 deficient (−/−) and wild type (+/+) mice within forebrain limbic sub-regions. Images show IBA-1 (ionized calcium binding adaptor molecule 1) stained microglia within the basolateral amygdala, BLA, (A, B), pre-frontal cortex, PFC (E, F), dentate gyrus, DG (I, J), and hypothalamic paraventricular nucleus, PVN (M, N). Insets show magnified area within the image. Bar graphs show quantified soma perimeter (C,G,K, O) and branch length (D,H,L,P) within these areas under basal, home cage conditions (basal) and following completion of behavioral testing (post test). Data are represented as mean +/− sem; *p < 0.05 versus basal +/+ or basal −/− as depicted by lines. (n = 6–12 mice per group; ns = not significant).
There were no significant differences in regional microglia cell number between genotypes (data not shown).
3.2. Concentration of pro-inflammatory cytokine, interleukin 1β (IL-1β) in forebrain limbic sub-regions is not altered by CX3CR1 deficiency
IL-1β concentration within the BLA, PVN, PFC and DG was not significantly different between CX3CR1+/+ and CX3CR1−/− mice in tissue collected from home cage mice under basal conditions (Table 1). Other pro-inflammatory cytokines, interleukin 6 (IL-6) and tumor necrosis factor α (TNF α) were not detectable at baseline within these areas using the BioPlex assay.
Table 1.
IL-1β concentrations within forebrain limbic sub-regions in wild type and CX3CR1-deficient mice at basal conditions.
| Region | Wild Type | CX3CR1−/− |
|---|---|---|
| BLA | 0.098 ± 0.015 | 0.06 ± 0.008 |
| PFC | 0.10 ± 0.04 | 0.06 ± 0.015 |
| DG | 0.095 ± 0.027 | 0.049 ± 0.011 |
| PVN | 0.049 ± 0.01 | 0.049 ± 0.006 |
Data represent mean ± sem of IL-1β levels detected in each region using BioPlex assay.
Tissue was collected from home cage housed mice.
Note: Other pro-inflammatory cytokines (IL-6 and TNF-a) were not detectable within these areas in the same tissue homogenates (n = 5/group).
3.3. Enhanced fear acquisition and potentiated fear reinstatement in CX3CR1 deficient mice
Given microglial alterations under basal conditions within brain areas regulating stress, fear and memory, CX3CR1−/− mice were tested in a contextual fear-conditioning paradigm. As shown, CX3CR1−/− mice showed enhanced acquisition of fear evidenced by increased post-shock freezing during the training phase (Fig. 3A). A two way repeated measures ANOVA revealed a significant effect of training trials (F3,66 = 29.86; p < 0.05), a significant genotype × time interaction (F3,66 = 3.104; p < 0.05) and a trending effect of genotype (F1,22 = 3.476; p = 0.07). Post hoc analysis revealed a significant difference between genotypes for conditioning trial 2 (p < 0.05). When exposed to the shock context 24 h post training, both genotypes demonstrated a similar magnitude of conditioned freezing (Day 1, Fig. 3B). Exposure to context on subsequent days (extinction training) did not reveal statistically significant differences between genotypes on days 2–4. On day 5, both groups were tested for fear reinstatement following a single reminder shock. CX3CR1−/− mice elicited significantly higher post-reminder shock freezing as compared with CX3CR1+/+ mice (Fig. 3C and D). Repeated measures ANOVA revealed a significant effect of genotype (F1,20 = 4.971; p < 0.05), time (F5,100 = 10.49; p < 0.05) and a genotype × time interaction (F5,100 = 3.76; p < 0.05). Mean post reminder shock freezing in CX3CR1−/− mice was significantly higher than CX3CR1+/+ mice (Fig. 3D). One-way ANOVA revealed significant main effect (F3,40 = 12.37; p < 0.05). Post hoc analyses revealed a significant difference in freezing between genotypes post reminder shock.
Fig. 3.
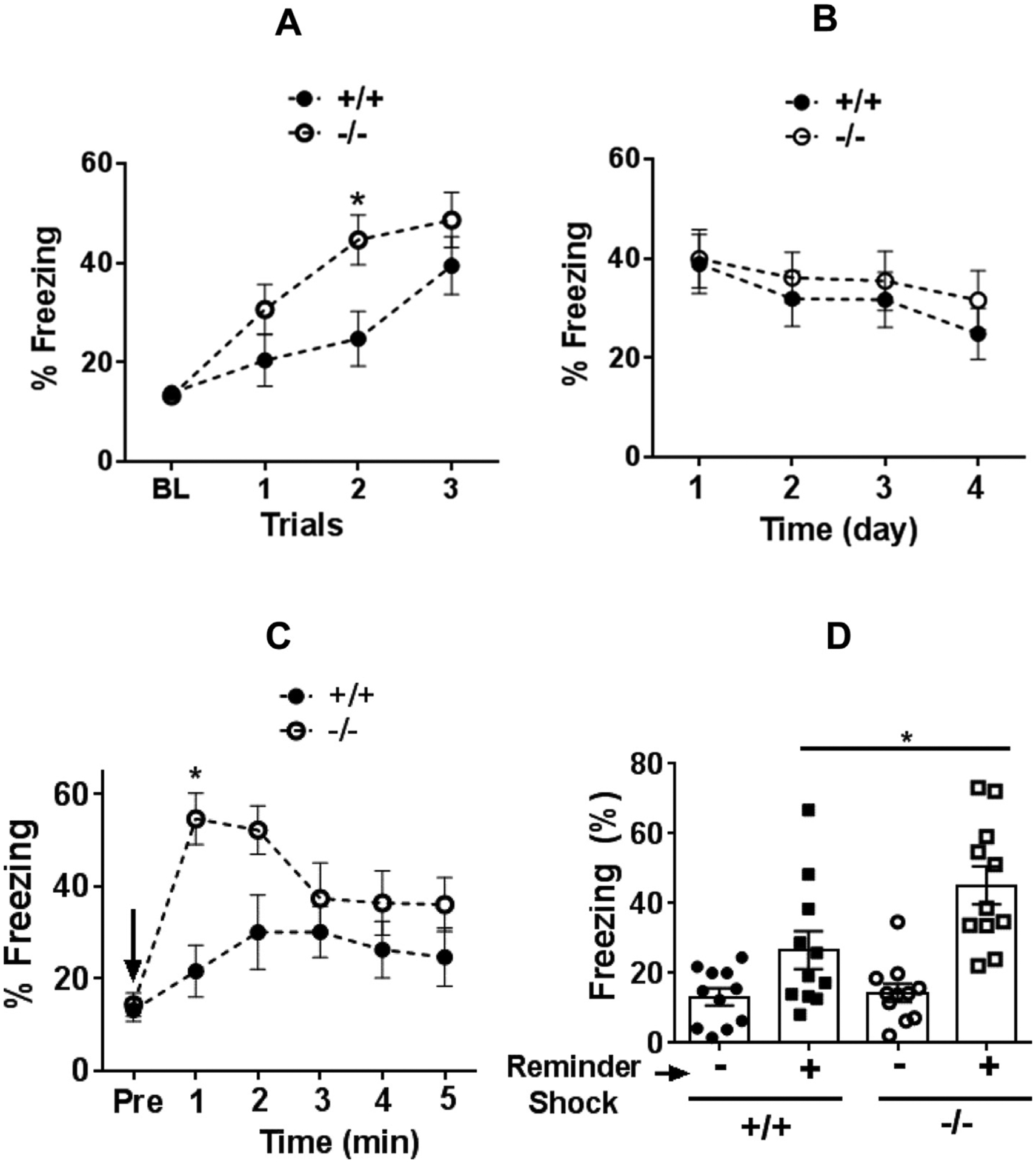
Freezing response in CX3CR1-deficient (−/−) and wild type (+/+) mice in a contextual fear conditioning paradigm to assess fear acquisition (A), conditioned fear and extinction (B) and reinstatement of fear (C,D). CX3CR1−/− mice showed significantly higher freezing during training trials (A). Baseline pre-shock freezing (BL) was not significantly different between genotypes. (B) No significant difference was observed during testing for conditioned freezing 24 h post-training (day 1) or extinction training (days 2–4). (C) Minute-by minute freezing during reinstatement following a single reminder shock (arrow). Pre = pre-shock freezing prior to reminder shock. Significantly higher freezing was observed in CX3CR1−/− mice. (D) Mean freezing following reminder shock was significantly higher in CX3CR1−/− mice than wild type (+/+) mice. Data are shown as mean ± sem (n = 12 mice/group) *p < 0.05 versus +/+ mice.
3.4. Acoustic startle responsivity is not impacted by CX3CR1 deficiency
Exposure of CX3CR1+/+ and −/− mice to unexpected bouts of a 108 dB acoustic stimulus did not produce statistically significant differences in startle amplitude between genotypes (Fig. 4). A two way repeated measures ANOVA revealed a significant effect of trial block (F5,201 = 11.59; p < 0.05 Fig. 4A), but no significant difference in genotype (p > 0.05) or genotype × time interaction (p > 0.05 vs +/+). Mean startle amplitude was also not significantly different between groups (p > 0.05 vs +/+, Fig. 4B).
Fig. 4.
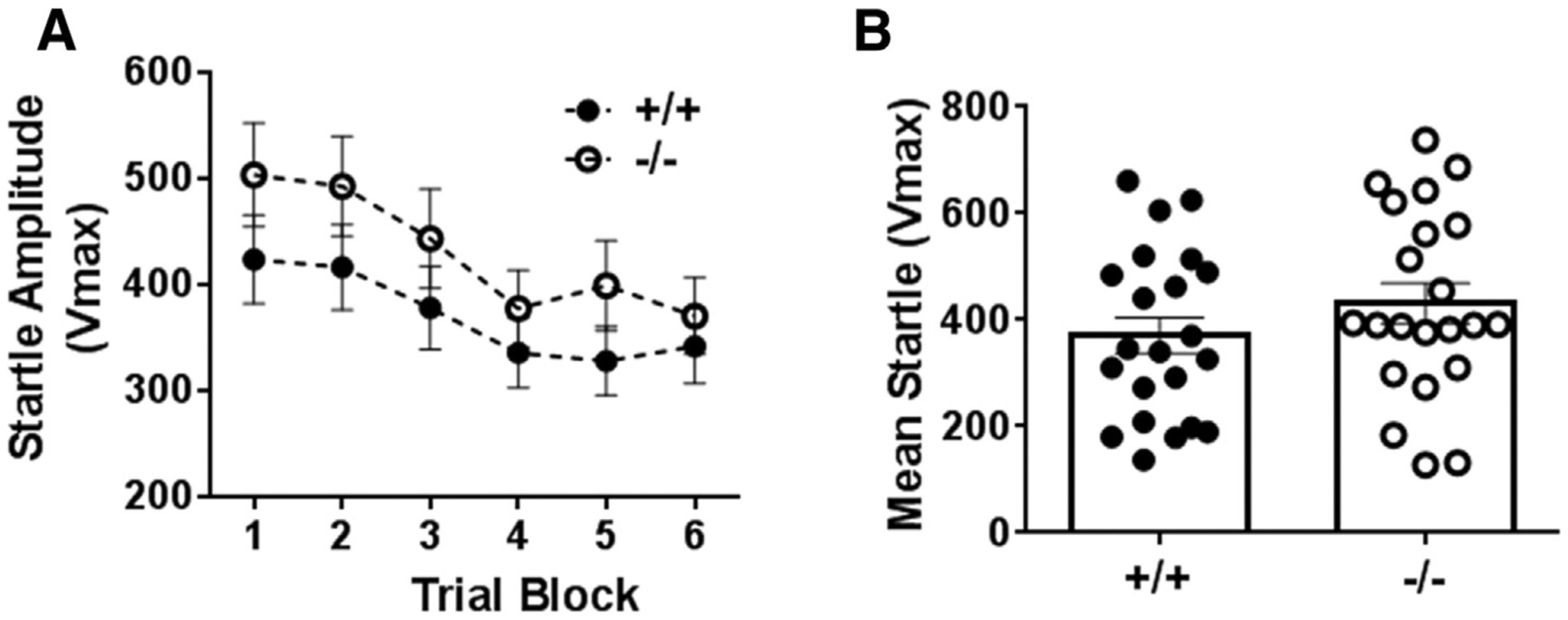
Acoustic startle response in CX3CR1−/− and +/+ mice over trial blocks to assess peak startle and habituation (A). No significant differences were observed between genotypes. Mean startle amplitude averaged over trial blocks revealed no differences between groups (B). Data are shown as mean ± sem (n = 12 mice/group).
3.5. Attenuated anxiety-like behaviors in CX3CR1 deficient mice
CX3CR1-deficient mice were screened in three separate tests for anxiety-like behavior. As shown in Fig. 5, attenuation of anxiety-like behavior was observed in the elevated zero maze (EZM) and light dark box (LDB) tests but not in the open field test (OFT). When tested in the EZM, CX3CR1−/− mice spent a significantly higher amount of time in the open quadrants of the zero maze (t22 = 2.881; p < 0.05 vs +/+ mice; Fig. 5A). We also observed significantly reduced latency to enter the open quadrant [median latencies 292.0 (+/+) and 29.50 (−/−)] and the distributions were significantly different (Mann Whitney U = 0.50; exact p = 0.0001; Fig. 5B). No genotype differences in motor activity were observed (p > 0.05; Fig. 5C). In the LDB, no significant genotype differences were observed in time spent in the light compartment (p > 0.05, Fig. 5D). However, CX3CR1−/− elicited significantly reduced latency to enter the light compartment (t18 = 3.662; p < 0.05 vs +/+ mice; Fig. 5E), and demonstrated a significantly higher rearing frequency (vertical exploration) in the lighted area (median rearing frequency were 0.50 (+/+) and 7.5 (−/−) and the distributions were significantly different (Mann Whitney U = 13.5; exact p = 0.0039; Fig. 5F). Exposure of +/+ and −/− mice to the open field revealed no significant differences in time spent in the center zone (p < 0.05, Fig. 4G), or latency to enter the center zone (p > 0.05, Fig. 5H). A significant reduction in motor activity was observed in CX3CR1−/− mice as revealed by unpaired t test (t17 = 3.418; p < 0.05 vs +/+ mice; Fig. 5I).
Fig. 5.
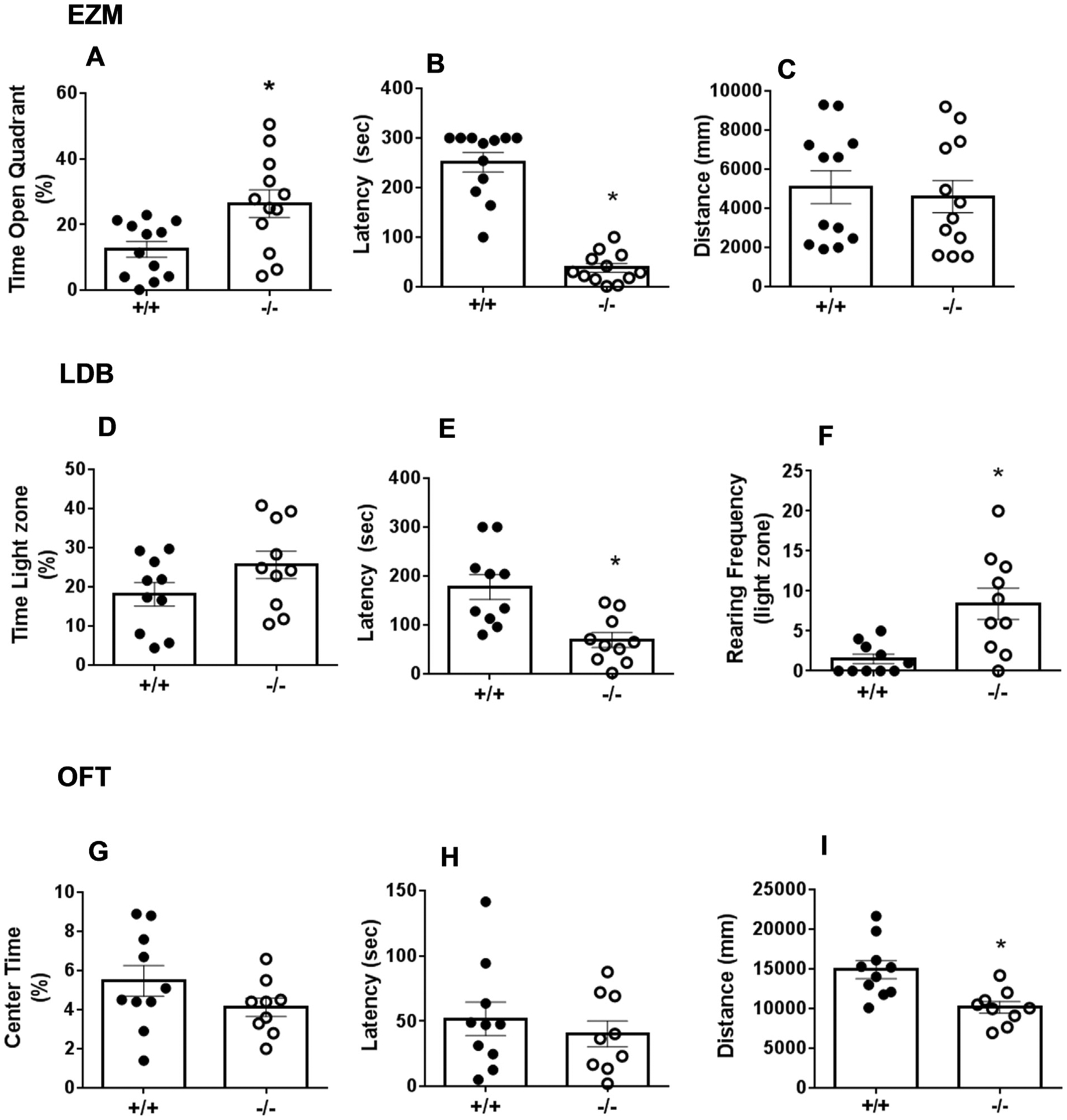
Anxiety-like behavior in CX3CR1−/− and +/+ mice assessed in the elevated zero maze, EZM (panels A–C), light dark box, LDB (panels D–F) and open field test, OFT (panels G-I). In the EZM: CX3CR1−/− mice elicited significantly increased open quadrant time (A) and reduced latency to enter open quadrant (B), while motor activity was not significantly different (C). In the light dark box time spent in the light zone was not significantly different (D), however, CX3CR1−/− mice showed significantly reduced latency to enter the light zone (E), and significantly increased rearing in the light zone (F). In the open field no significant differences were observed in center time duration (G) or latency to enter center field (H). CX3CR1−/− mice showed a significant reduction in motor activity as compared with +/+ mice (I). Data represented are mean ± sem (n = 10–12 mice/group) *p < 0.05 versus +/+ mice.
3.6. Increased c-Fos in stress and fear regulatory circuits in CX3CR1-deficient mice
To investigate brain regions and circuits that might be associated with behavioral effects of CX3CR1 deficiency, we measured cfos immunoreactivity, a measure of neuronal activation at basal conditions and following reinstatement in CX3CR1−/− and CX3CR1+/+ mice. Assessment of cfos staining revealed genotype differences that were highly region-selective. As expected, c-fos counts were low under basal conditions, however CX3CR1−/− mice had significantly higher cell counts within the PVN and the PFC (Table 2). Following reinstatement, compared to CX3CR1+/+ mice, increased number of cfos-positive cells were observed in CX3CR1−/− mice within the PVN (Fig. 6A–C). We also observed increased c-fos counts within the ventral tegmental area (VTA) and adjacent interpeduncular nucleus (IPN) area post reinstatement (quantified as VTA-IPN) (Fig. 6D–F). Unpaired t test revealed significantly higher cfos counts in deficient mice within the PVN (t22 = 4.034; p < 0.05 vs +/+ mice; Fig. 6C), and the VTA-IPN (t22 = 4.375; p < 0.05 vs +/+ mice; Fig. 6F).
Table 2.
cFos-immunoreactive cells within sub regions modulating stress and fear behaviors in wild type and CX3CR1-deficient mice at basal conditions.
| Region | Wild Type | CX3CR1−/− |
|---|---|---|
| PVN | 1.04 ± 0.28 | 11.50 ± 1.58* |
| PFC | 2.3 ± 0.35 | 6.5 ± 1.7* |
| DG | 19.88 ± 4.176 | 12.25 ± 1.605 |
| BLA | ND | ND |
Data represent mean ± sem of immunopositive cells counted in each region. Tissue was collected from home cage housed mice.
ND = Immunopositive cells were not detected in this area.
PVN = paraventricular nucleus of the hypothalamus, PFC = prefrontal cortex, DG = dentate gyrus, BLA = basolateral amygdala.
p < 0.05 versus Wild type mice, n = 6/group.
Fig. 6.
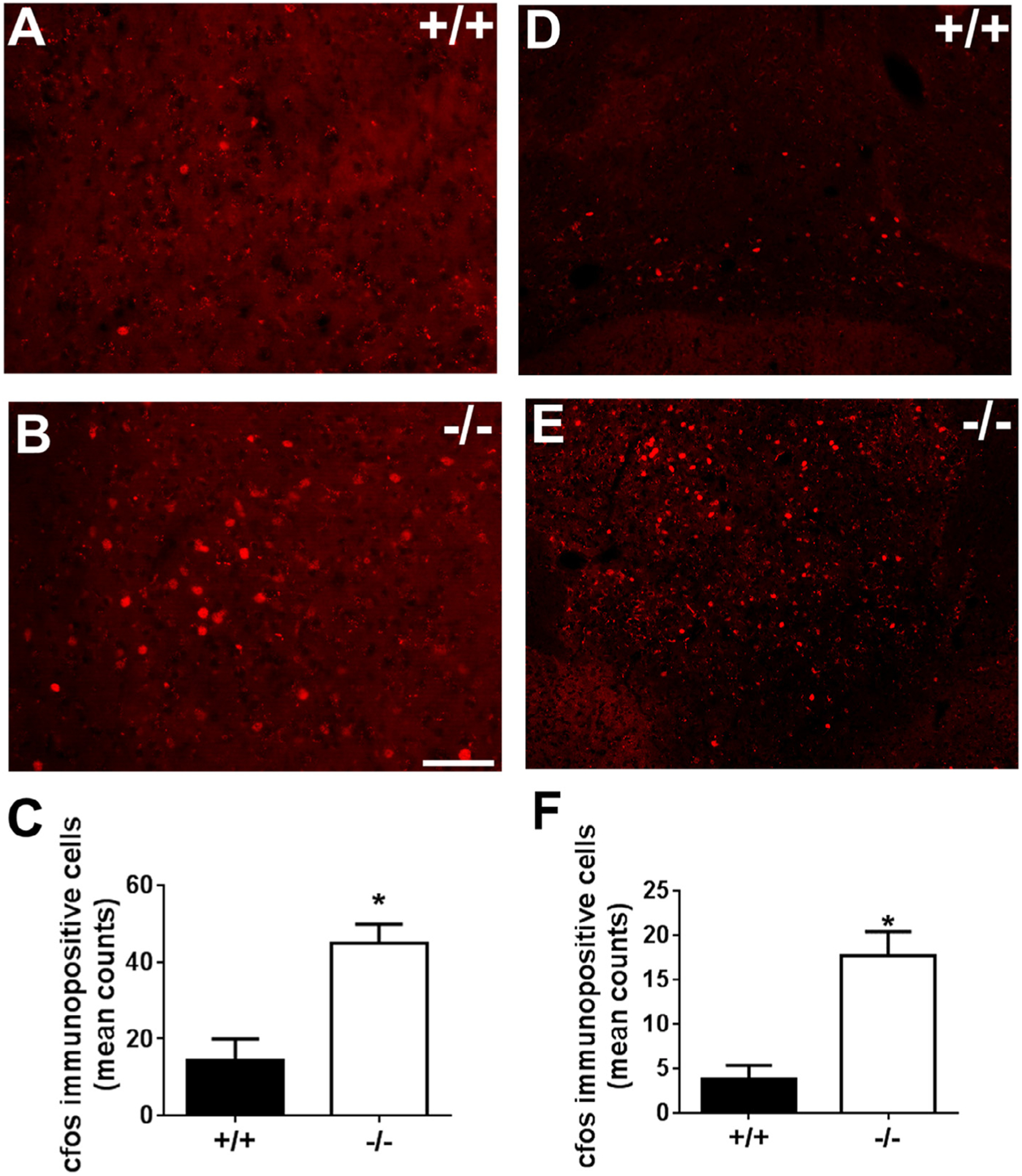
cfos-like immunoreactivity in the paraventricular nucleus (PVN) and the ventral tegmental -interpeduncular area (VTA-IPN) of CX3CR1 deficient (−/−) and wild type (+/+) mice. Top panels show representative images of fluorescent cfos immunoreactive nuclei from PVN (A,B) and VTA-IPN (D,E) of +/+ and −/− mice. Bottom panels show bar graphs of quantified data. Significant increase in cfos immunoreactive cells was observed in CX3CR1−/− mice in the PVN (C), and VTA-IPN (F). (*p < 0.05 versus +/+). Data are shown as mean + SEM, n = 6–7/group.
4. Discussion
The present study tested the hypothesis that chemokine signaling may be required in modulating fear, startle and anxiety-like behaviors that are pertinent to PTSD. Our data demonstrated a role of the chemokine receptor, CX3CR1 in regulating fear acquisition and reinstatement potentially through altered microglial-neuronal activity in stress and fear regulatory circuits. Genetic disruption of CX3CR1 impacts anxiety-related behaviors but may not regulate unconditioned sensorimotor startle responses.
To investigate regional effects of CX3CR1-deficiency, we assessed morphological alterations in microglia within several forebrain sub-regions such as the PFC, BLA, DG and PVN that are well-established sites for the stress and emotional regulation, and implicated in PTSD pathophysiology (Pitman et al., 2012, Stevens et al., 2013). Structural alterations in microglial morphology (increased soma perimeter and hyper-ramification) were observed in home-cage housed CX3CR1−/− mice as compared to CX3CR1+/+ mice within the PVN, BLA and PFC, while the dentate elicited increased soma perimeter. Hyper-ramification of microglia is often observed following non-pathological signals (experience-dependent modifications) such as alterations in neuronal activity, neurotransmitter fluctuations as opposed to the classical injury-evoked morphological changes (reviewed in Walker et al., 2013). Collectively, this is suggestive of an activated state of microglia in these regions under basal, tonic conditions. Previous studies have reported high CX3CL1 concentrations in the “non-perturbed” brain suggesting constitutive release of the chemokine for maintaining an optimal microglial milieu in the normal CNS (Biber et al., 2007). The absence of this physiological “brake” system in CX3CR1-deficient mice is expected to promote a sensitized microglial state. Previous studies have reported potentiated LPS-evoked microglial activation and enhanced pro-inflammatory responses in CX3CR1−/− mice (Cardona et al., 2006; Corona et al., 2010). In our study, altered microglial morphology within limbic sub-regions in CX3CR1−/− mice was not accompanied by elevated pro-inflammatory cytokines suggesting that at least within these nuclei cytokines were maintained within physiological concentrations. Exposure to behavioral testing produced changes in microglial morphology associated with CX3CR1 deficiency in a region-selective manner. Post-behavior increases in soma perimeter and process arborization were dependent on CX3CR1 only within selective limbic nuclei such as the mPFC and the PVN, but not the dentate and BLA. This suggests that adaptive changes in microglia and potentially microglia-neuron crosstalk evoked by behavioral manipulations may differ between limbic brain regions. Regional differences in stress-evoked microglial alterations have been reported previously (Tynan et al., 2010), an effect that may depend on the modality of stressors (Kopp et al., 2013). Similar to our observations, a previous study reported altered microglial responsivity within the PVN to chronic stress in CX3CR1−/− mice (Winkler et al., 2017). Impaired CX3CR1 function may induce a mild activation state of microglia within limbic nuclei under baseline conditions, that may be primed for sensitized responses to secondary challenges, in a region-specific manner.
Significant enhancement of fear acquisition and reinstatement was observed in CX3CR1−/− mice. Previous studies investigating the effect of CX3CR1 deficiency on fear learning and memory have yielded contradictory results. Increased freezing and improved learning and memory in CX3CR1−/− mice was reported (Reshef et al., 2014). In contrast, attenuated conditioned fear and learning deficits were associated with CX3CR1 deficiency (Rogers et al., 2011). In our paradigm, fear conditioning was preceded by exposure to anxiety and startle testing that may have led to potentiated fear acquisition. Despite higher fear acquisition, we did not observe significant differences in conditioned freezing or extinction. An interesting finding was the higher freezing in CX3CR-deficient mice during the reinstatement test, suggesting that the return of fear was potentiated in these mice. Higher fear acquisition (suggestive of a more aversive US, unconditioned stimulus experience) or the stress engendered by the unsignaled footshocks may have resulted in increased reinstatement in CX3CR1−/− mice. Stress systems overlap and interact with fear circuitry and stress has been known to facilitate reinstatement and relapse (Goode and Maren, 2014). Our cFos data strongly supports contributions of stress systems in observed fear behaviors. Significantly higher neuronal activation in the paraventricular nucleus (PVN) of the hypothalamus was observed in CX3CR1−/− mice at basal conditions and following reinstatement. The PVN plays a central role in stress response and regulation (Ulrich-Lai and Herman, 2009). Previous reports have shown cfos activation in PVN neurons following a physically aversive footshock, as well as following a behaviorally conditioned stimulus (Pezzone et al., 1992), suggesting an integrative role of the PVN in unconditioned and conditioned aversive stimuli. Significantly increased cfos staining in this area in CX3CR1−/− mice indicates modulation of stress regulatory pathways by CX3CR1 deficiency impacting fear acquisition and reinstatement. Mounting evidence suggests interaction between microglial systems and stress (Walker et al., 2013). Animals exposed to stressors such as restraint stress or chronic mild stress exhibit increased expression of the CX3CR1 receptor (Bollinger et al., 2016; Rossetti et al., 2016) suggesting recruitment of the receptor under stress conditions. Our data resonates with a recent study reporting higher neuronal activation in the PVN and increased stress response in CX3CR1−/− mice (Winkler et al., 2017). In addition to the PVN, increased cfos immunoreactivity was also observed in the ventral tegmental-interpeduncular area in CX3CR1 mice following reinstatement. Previous studies on fear reinstatement have reported reminder shock-induced elevation of cfos in VTA neurons projecting to and inhibiting the infralimbic prefrontal cortex resulting in reinstatement of an extinguished fear memory (Hitora-Imamura et al., 2015). The IPN plays an important role in convergence of forebrain limbic signals and relay to midbrain centers and has been implicated in regulation of fear and anxiety behaviors (McLaughlin et al., 2017; Thompson 1960). Collectively, our data suggests impaired microglia-neuron signaling (as in CX3CR1−/− mice) may modulate fear behavior via altered neuronal activity in selective circuits resulting in increased freezing during acquisition and reinstatement. Our observations highlight a role of extra-hippocampal sites and circuits that may be impacted by the CX3CR1 receptor to regulate fear-associated behaviors.
Attenuated anxiety-like behaviors were observed in CX3CR1−/− mice. Three separate anxiety paradigms were used for verification of genotype effects. In EZM and LDB tests, CX3CR1 mice elicited increased time in open quadrants as well as reduced latency to enter aversive open areas and light compartment consistent with increased exploration and attenuated anxiety-like behaviors. Thus, approach rather than avoidance behavior was more evident in the CX3CR1−/− mice than wild type mice. This is consistent with active coping behaviors recently reported in CX3CR1−/− mice under aversive conditions (Winkler et al., 2017). No significant differences in area preference but reduced motor activity was noted in CX3CR1−/− mice in the OFT. Motor activity in the OFT represented horizontal exploration and did not account for vertical activity, which may have accounted for these differences. Previous studies using the OFT and elevated plus maze (EPM) paradigm reported no significant differences in anxiety-like behavior or activity between CX3CR1−/− and CX3CR1+/+ mice (Hellwig et al., 2016; Rogers et al., 2011). It is apparent from our data that modality of the test may be a driving factor in these differences. Although the exact explanation for anxiolytic behavior in CX3CR1−/− mice is not evident, it may be an outcome of altered functioning of anxiety regulatory circuits. As reported previously, PFC-hippocampus connectivity encodes location, navigation, attention and decision making associated with anxiety testing in rodents (Zhan, 2015) and CX3CR1−/− mice have altered coherence within PFC circuits (Zhan et al., 2014). We observed microglial alterations and higher cFos immunopositive cell counts within the mPFC in CX3CR1 mice under basal conditions, although functional effects and association with anxiety-like behaviors remains to be investigated.
No significant genotype differences were observed in acoustic startle responses suggestive of normal sensorimotor function in CX3CR1−/− mice. The acoustic startle response is a fast reflexive response to a strong unexpected acoustic stimulus primarily mediated by a specific neural pathway involving auditory pathway converging at brain stem areas such as the cochlear nucleus and the reticular pontine nucleus which projects to the motor neurons in the spinal for regulating muscle responses. Our data shows that in contrast to forebrain regulated fear and anxiety-associated behaviors, unconditioned reflexive responses regulated by sensorimotor pathways are not impacted by CX3CR1 deficiency.
Implications of CX3CR1 deficiency
Previous studies have investigated the role of impaired CX3CL1/CX3CR1 function in neurodegenerative and neurotoxic conditions (Corona, et al., 2010; Cardona, et al., 2006). Association of CX3CR1 deficiency with increased fear behaviors as well as anxiolytic-like phenotype was observed in our study. Interestingly, recent studies have reported attenuated depression-like behavior, active coping and resiliency to chronic stress in CX3CR1−/− mice in conjunction with increased conditioned fear and elevated stress-evoked hypothalamic pituitary adrenal (HPA) responses (Reshef et al., 2014, Milior et al., 2016, Rimmerman et al., 2017, Winkler et al., 2017). Collectively, our observations suggest that CX3CR1 signaling may differentially regulate microglial-neuronal communication within discrete brain regions and circuits leading to variable and seemingly disparate phenotypes. In support, we observed differential, region-selective alterations in microglial and neuronal activation in CX3CR1−/− mice following behavioral testing. It would be important to consider regional differences when investigating microglia-neuron inter-regulatory mechanisms and their impact on behavior and physiology. In addition to previously reported hippocampal abnormalities (Rogers et al., 2011, Rimmerman et al., 2017), our findings provide further evidence supporting a potential role of CX3CR1 regulation and microglia-neuron crosstalk within hypothalamic and cortical circuits in regulation of fear and anxiety related behaviors. It will be important to conduct mechanistic studies to assess immune-stress interactions within these limbic sub-regions in CX3CR1 modulation of behavior. Although our study focused on specific nuclei, contributions of other brain areas/subregions should also be considered. In conjunction with mounting evidence supporting recruitment of the CX3CR1 receptor following stress our data suggests that impaired or deficient functioning of the receptor may lead to potentiated fear-associated behaviors following stress. Accumulating evidence from previous studies has supported a regulatory role and therapeutic potential of the CX3CR1 receptor in neurodegenerative conditions (D’Haese et al., 2012). Our data suggests that manipulation of the CX3CR1 pathway may offer therapeutic strategies for fear and anxiety associated disorders.
In conclusion, impaired CX3CR1 function may evoke alterations in fear and anxiety regulatory circuits regulating behaviors relevant to stress and trauma-evoked disorders such as PTSD.
Acknowledgment
Support from VA Merit Award grant (2I01BX001075) to Dr. Renu Sah is acknowledged. We would like to thank Ms Jennifer Schurdak for technical assistance.
References
- Biber K, Neumann H, Inoue K, Boddeke HWGM, 2007. Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 30, 596–602. [DOI] [PubMed] [Google Scholar]
- Bollinger JL, Bergeon Burns CM, Wellman CL, 2016. Differential effects of stress on microglial cell activation in male and female medial prefrontal cortex. Brain. Behav. Immun 52, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun AA, Skelton MR, Vorhees CV, Williams MT, 2011. Comparison of the elevated plus and elevated zero mazes in treated and untreated male Sprague-Dawley rats: effects of anxiolytic and anxiogenic agents. Pharmacol. Biochem. Behav 97, 406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee J-C, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM, 2006. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci 9, 917–924. [DOI] [PubMed] [Google Scholar]
- Corona AW, Huang Y, O’Connor JC, Dantzer R, Kelley KW, Popovich PG, Godbout JP, 2010. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J. Neuroinflammation 7, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costall B, Jones BJ, Kelly ME, Naylor RJ, Tomkins DM, 1989. Exploration of mice in a black and white test box: validation as a model of anxiety. Pharmacol. Biochem. Behav 32, 777–785. [DOI] [PubMed] [Google Scholar]
- D’Haese JG, Friess H, Ceyhan GO, 2012. Therapeutic potential of the chemokine-receptor duo fractalkine/CX3CR1: an update. Expert Opin Ther Targets 16, 613–618. [DOI] [PubMed] [Google Scholar]
- Eyo UB, Wu L-J, 2013. Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. 2013, 456857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode TD, Maren S, 2014. Animal models of fear relapse. ILAR J. 55, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwig S, Brioschi S, Dieni S, Frings L, Masuch A, Blank T, Biber K, 2016. Altered microglia morphology and higher resilience to stress-induced depression-like behavior in CX3CR1-deficient mice. Brain Behav. Immun 55, 126–137. [DOI] [PubMed] [Google Scholar]
- Hitora-Imamura N, Miura Y, Teshirogi C, Ikegaya Y, Matsuki N, Nomura H, 2015. Prefrontal dopamine regulates fear reinstatement through the downregulation of extinction circuits. Elife 4, e08274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE, 2005. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 62, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp BL, Wick D, Herman JP, 2013. Differential effects of homotypic vs. heterotypic chronic stress regimens on microglial activation in the prefrontal cortex. Physiol. Behav 122, 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist D, Wolkowitz OM, Mellon S, Yehuda R, Flory JD, Henn-Haase C, Bierer LM, Abu-Amara D, Coy M, Neylan TC, Makotkine I, Reus VI, Yan X, Taylor NM, Marmar CR, Dhabhar FS, 2014. Proinflammatory milieu in combat-related PTSD is independent of depression and early life stress. Brain. Behav. Immun 42, 81–88. [DOI] [PubMed] [Google Scholar]
- McGuire J, Herman JP, Horn PS, Sallee FR, Sah R, 2010. Enhanced fear recall and emotional arousal in rats recovering from chronic variable stress. Physiol. Behav 101, 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin I, Dani JA, De Biasi M, 2017. The medial habenula and interpeduncular nucleus circuitry is critical in addiction, anxiety, and mood regulation. J. Neurochem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michopoulos V, Powers A, Gillespie CF, Ressler KJ, Jovanovic T, 2017. Inflammation in fear- and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology 42, 254–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milior G, Lecours C, Samson L, Bisht K, Poggini S, Pagani F, Deflorio C, Lauro C, Alboni S, Limatola C, Branchi I, Tremblay M-E, Maggi L, 2016. Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav. Immun 55, 114–125. [DOI] [PubMed] [Google Scholar]
- Neigh GN, Ali FF, 2016. Co-morbidity of PTSD and immune system dysfunction: opportunities for treatment. Curr. Opin. Pharmacol 29, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bisht K, Tremblay MÈ, 2014. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell Neurosci 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C, 1998. The Mouse Brain in Stereotaxic Coordinates. Academic Press, San Diego, CA. [Google Scholar]
- Pezzone MA, Lee WS, Hoffman GE, Rabin BS, 1992. Induction of c-Fos immunoreactivity in the rat forebrain by conditioned and unconditioned aversive stimuli. Brain Res. 597, 41–50. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I, 2012. Biological studies of post-traumatic stress disorder. Nat. Rev. Neurosci 13, 769–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, El Khoury J, 2016. Microglia in health and disease. Cold Spring Harb. Perspect. Biol, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshef R, Kreisel T, Beroukhim Kay D, Yirmiya R, 2014. Microglia and their CX3CR1 signaling are involved in hippocampal- but not olfactory bulb-related memory and neurogenesis. Brain Behav. Immun 41, 239–250. [DOI] [PubMed] [Google Scholar]
- Rimmerman N, Schottlender N, Reshef R, Dan-Goor N, Yirmiya R, 2017. The hippocampal transcriptomic signature of stress resilience in mice with microglial fractalkine receptor (CX3CR1) deficiency. Brain Behav Immun. 61, 184–196. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, Weeber EJ, Bickford PC, Gemma C, 2011. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci 31, 16241–16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti AC, Papp M, Gruca P, Paladini MS, Racagni G, Riva MA, Molteni R, 2016. Stress-induced anhedonia is associated with the activation of the inflammatory system in the rat brain: restorative effect of pharmacological intervention. Pharmacol. Res 103, 1–12. [DOI] [PubMed] [Google Scholar]
- Schmeltzer SN, Vollmer LL, Rush JE, Weinert M, Dolgas CM, Sah R, 2015. History of chronic stress modifies acute stress-evoked fear memory and acoustic startle in male rats. Stress 18, 244–253. [DOI] [PubMed] [Google Scholar]
- Stevens JS, Jovanovic T, Fani N, Ely TD, Glover EM, Bradley B, Ressler KJ, 2013. Disrupted amygdala-prefrontal functional connectivity in civilian women with posttraumatic stress disorder. J. Psychiatr. Res 47, 1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R, 1960. Interpeduncular nucleus and avoidance conditioning in the rat. Science 132, 1551–1553. [DOI] [PubMed] [Google Scholar]
- Tremblay M-E, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A, 2011. The role of microglia in the healthy brain. J. Neurosci 31, 16064–16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, Day TA, Walker FR, 2010. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun. 24, 1058–1068. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai Y, Herman JP, 2009. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci 10, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervliet B, Craske MG, Hermans D, 2013. Fear extinction and relapse: state of the art. Annu. Rev. Clin. Psychol 9, 215–248. 10.1146/annurevclinpsy-050212-185542. [DOI] [PubMed] [Google Scholar]
- Vollmer LL, Ghosal S, Rush J, Sallee F, Herman J, Weinert M, Sah R, 2013. Attenuated stress-evoked anxiety, increased sucrose preference and delayed spatial learning in glucocorticoid-induced receptor-deficient mice. Genes. Brain. Behav 12, 241–249. 10.1111/j.1601-183X.2012.00867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer LL, Ghosal S, McGuire JL, Ahlbrand RL, Li K-Y, Santin JM, Ratliff-Rang CA, Patrone LGA, Rush J, Lewkowich IP, Herman JP, Putnam RW, Sah R, 2016. Microglial acid sensing regulates carbon dioxide-evoked fear. Biol. Psychiatry 80, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FR, Nilsson M, Jones K, 2013. Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr. Drug Targets 14, 1262–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler Z, Kuti D, Ferenczi S, Gulyás K, Polyák Á, Kovács KJ, 2017. Impaired microglia fractalkine signaling affects stress reaction and coping style in mice. Behav Brain Res. 334, 119–128. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, 2016. Neuron-microglia interactions in mental health disorders: “for better, and for worse”. Front. Immunol 7, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, 2015. Theta frequency prefrontal-hippocampal driving relationship during free exploration in mice. Neuroscience 300, 554–565. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT, 2014. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci 17, 400–406. [DOI] [PubMed] [Google Scholar]