

Abstract
Functional amyloids are highly organized protein/peptide structures that inter alia promote biofilm formation in different bacteria. One such example is provided by a family of 20–45 residue-long peptides called phenol-soluble modulins (PSMs) from Staphylococcus aureus. External components such as eukaryotic host proteins, which alter self-assembly of bacterial amyloids, can affect the biofilm matrix. Here, we studied the effect of the highly prevalent human plasma protein fibrinogen (Fg) on fibrillation of PSMs. Fg inhibits or suppresses fibrillation of most PSMs tested (PSMα1, PSMβ1, and PSMβ2) except for PSMα3, whose already rapid aggregation is accelerated even further by Fg but leads to amorphous β-rich aggregates rather than fibrils. Fg also induces PSMβ2 to form amorphous aggregates and diverts PSMα1 into off-pathway oligomers which consist of both Fg and PSMα1 and cannot seed fibrillation. Peptide arrays showed that Fg bound to the N-terminus of PSMα1, while it bound to the entire length of PSMα3 (except the C terminus) and to the C-termini of PSMβ1 and PSMβ2. The latter peptides are all positively charged, while Fg is negatively charged at physiological pH. The positive charges complement Fg’s net negative charge of −7.6 at pH 7.4. Fg’s ability to inhibit PSM fibrillation reveals a potential host-defense mechanism to prevent bacterial biofilm growth and infections in the human body.
Introduction
Many bacteria form biofilms where individual cells are part of a multicellular community encased in a shared extracellular matrix. Biofilms are notoriously tolerant to antibiotics, and biofilm infections are therefore difficult to treat. These infections often occur in association with biomedical implants, such as cardiac valves, prosthetic joints, vascular grafts, and implanted catheters.1,2 Staphylococci, particularly Staphylococcus aureus and Staphylococcus epidermidis, are the culprits of most nosocomial biofilm infections. They secrete high levels of small (20–45 residue) α-helical amphipathic peptides called phenol-soluble modulins (PSM).3 PSMs show cytolytic activity against white blood cells,4 and their surfactant-like properties can promote biofilm dispersion.5 Remarkably, they can also self-assemble into highly ordered amyloid structures, which may contribute to the establishment, integrity, and maturation of the biofilm,6 although the role and significance of the amyloid species remain contested.7 PSM peptides fall into three groups, namely, α-type (PSMα1-4, 20–25 amino acids), β-type (PSMβ1-2, 43–45 amino acids), and finally the separately encoded δ-toxin with the same size as α-type.8 Almost all α-type PSMs are net neutral or positively charged with an α-helical structure, whereas PSMβ2 is net negatively charged, contains three α-helical regions (of which α-helix2 and α-helix3 form a “v-like” structure), and shows lower lytic potential.9,10 PSMα3 is the most cytotoxic; moreover, this peptide assembles into a cross-α architecture consisting of α-helix peptides stacked perpendicular to the fibril axis, in striking contrast to conventional cross-β fibrils.11 Expression of all three types of PSMs is directly regulated by the quorum-sensing system, an accessory gene regulator (Agr) that controls secretion of several exotoxins from S. aureus during aggressive infections.12,13 PSMα peptides and the similarly sized δ-toxin are generally expressed at high levels whereas PSMβ peptides are produced to a much smaller extent.3
Biofilm formation is influenced by many components which accumulate in the biofilm matrix, such as polysaccharides, proteins, extracellular DNA (eDNA), bacterial biosurfactants like rhamnolipids, and outer membrane components such as lipopolysaccharides.14,15 Host components can also play a role, in particular blood plasma proteins.16,17 The 340 kDa protein fibrinogen (Fg) is the third-most abundant plasma protein (2–4 mg/mL). Fg has a negative net charge with an isoelectric point of 5.8 and is composed of Aα, Bβ, and γ chains linked together with disulfide bridges.18 Besides its central role in blood coagulation, it shows chaperone properties (preventing, e.g., thermal aggregation of citrate synthase19) and is able to suppress amyloid formation of various proteins. Thus, Fg inhibits fibrillation of the insulin-B chain by binding to prefibrillar intermediates.20 Similarly, fibrillation of the functional amyloid protein CsgA from Escherichia coli is also inhibited by Fg through interactions with early nuclei and more mature fibrils.21 Nevertheless, these anti-aggregatory properties are not an unconditional advantage. Fg binding to the Alzheimer peptide Aβ leads to abnormal blood clots that are resistant to degradation by plasmin and through co-deposition of Fg and Aβ leads to inflammation and permeability of blood vessels.22,23 The molecular basis for this is interactions of Aβ with the C-terminal region of the Fg-β chain (β384-393), which induce oligomerization of Fg; these oligomers can be processed by thrombin to form abnormal clots but are protected against cleavage by plasmin.24,25 Fg is also involved in biofilm formation through binding to bacteria, mediating the adherence of bacteria to endothelial cells or avoiding the immune system.17,26 In S. aureus, Fg-binding proteins (FnBPA and FnBPB) promote the adherence of bacteria to Fg, elastin, and fibronectin. The two proteins’ Fg-binding regions are located in the N2 and N3 subdomains of the N-terminal A domain.27,28 For coagulase-positive S. aureus, fibrin is also the main matrix component in vivo. S. aureus secretes two proteins, namely, coagulase (Coa) and von Willebrand factor–binding protein (vWbp), which activate prothrombin and staphylothrombin. These two proteases subsequently cleave Fg to form fibrin that fibrillates to form a fibrin network, essentially forming a blood clot around the bacteria.28,29 Thus, Fg is involved in many different protein–protein interactions in the S. aureus biofilm, indicating great versatility.
Here, we expand this versatility by demonstrating Fg interactions with another class of biofilm components, namely, the PSMs. We investigate how Fg impacts PSM amyloid formation using PSMα1 and PSMα3 along with PSMβ1 and PSMβ2 as model peptides (PSM sequences are provided in Figure 1A along with the structure of Fg). We excluded two PSM peptides PSMα2 and δ-toxin, which previous studies had shown to be very poor aggregators.30 Our results reveal a spectrum of effects. Fg strongly inhibits PSMα1 by redirecting monomers to off-pathway oligomers while promoting PSMα3 fibrillation. In contrast, it only had a rather modest inhibitory effect on PSMβ fibrillation. Fg increases the α-helical content of the PSM peptides and removes the seeding capacity of the resulting aggregates. Cationic residues in the PSM sequences are major drivers of the interaction with Fg. Taken together, our data indicate that Fg can modulate the function of PSM peptides in the S. aureus biofilm matrix through its impact on their aggregation behavior.
Figure 1.

(A) Sequences of PSM peptides used in the present study along with the structure of Fg. (B–E): Kinetics of PSM amyloid formation in presence and absence of Fg, monitored by ThT fluorescence. The data were fitted with a secondary nucleation unseeded model using the Amylofit web server (summarized in Table 1). The concentration of Fg (mg/mL) is indicated in the panels. (F) Relative lag time of fibrillation and (G) relative ThT-end point levels in presence of different [Fg], in both cases, normalized to the values for Fg-free PSMs.
Results
Fg Shows Different Effects on Fibrillation of Different PSM Peptides
To investigate how Fg affects PSM fibrillation, we incubated 0.2 mg/mL of each peptide with 0–1 mg/mL Fg in the presence of an amyloid reporter dye ThioflavinT (ThT) (normalized data in Figure 1B–E and raw data in Figure S1A–D) and monitored how this affected the time course of fibrillation of the four PSM peptides. In the absence of Fg, all four peptides show a conventional sigmoidal ThT fluorescence growth curve, in which, there is a lag phase of 0.1–10 h followed by a growth phase until a certain plateau level is reached and there is no longer any net increase in fibril mass. Fg increased the lag time of fibrillation of PSMα1, PSMβ1, and PSMβ2 but had an opposite effect on PSMα3 fibrillation. As little as 0.25 mg/mL Fg strongly inhibited PSMα1 fibrillation, increasing the lag time ∼threefold (Figure 1E). The end-point ThT-end levels also decreased (Figure 1F). PSMα3 aggregates unusually rapidly (compared to the other PSM peptides and aggregating proteins in general) with a ∼10 min lag time and a stable plateau after ∼2 h. Fg not only abolished the lag time completely but also reduced the end-level ThT emission ∼twofold (Figure 1F). PSMβ1- and PSMβ2-aggregation curves were both inhibited by Fg in a dose-dependent manner, particularly PSMβ2, where 1 mg/mL Fg essentially abolished the ThT signal (Figure 1E,F). Fg alone did not show any signs of fibrillation at 0.5 and 1 mg/mL (data not shown).
To explore which aspects of fibrillation were affected by Fg, kinetic ThT-curves were fitted using the webserver Amylofit (http://www.amylofit.ch.cam.ac.uk/fit)31 (Figure 1B–E). Amylofit allows us to investigate which aggregation mechanisms provide the best model for the measured data. For all four PSMs, the secondary nucleation unseeded mechanism has previously been shown to provide the best fit to the data to describe PSM aggregation in the absence of other components such as Fg.30 This mechanism is characterized by three rate constants, namely, that of nucleation (kn) to form new fibrils, elongation (k+) to extend existing fibrils from the two growing ends, and secondary nucleation (k2) to form new fibrils catalyzed by the surface of the growing fibril. According to the kinetic model behind Amylofit and the associated equations, individual values of these rate constants are not obtained from the analysis which instead provides composite rate constants k+kn and k+k2. We extend this approach to include Fg as follows: the set of ThT time curves for each PSM obtained at different [Fg] are fitted in a global fit, where we either allow k+kn or k+k2 to vary with [Fg] while restricting fitting of the other composite parameter to one global value.
For PSMα1, we obtained the best fit by varying the composite rate constant k+kn, while keeping k+k2 constant (Table 1). k+kn was markedly reduced by 0.25 mg/mL Fg, and higher [Fg] completely inhibited fibrillation (Supporting Information Figure S1A). Because k+k2 (and by inference k+ in the composite k+kn) is constant, we conclude that Fg inhibits PSMα1 fibrillation by reducing kn, that is, primary nucleation. Similar conclusions were made for PSMβ1. The effect of Fg on PSMβ2 was different; to obtain a better fit, we needed to keep k+kn constant while varying k+k2 with [Fg], indicating that secondary nucleation is being targeted. 1 mg/mL Fg completely suppressed fibrillation (Figure S1D). In contrast, Fg accelerated PSMα3 fibrillation through increased k+kn values. Consistent with the abolition of the lag phase by Fg (Figure S1B), we conclude that Fg promotes the nucleation phase. Because k+k2 is constant, increased k+kn values imply an increase in kn, which also agrees with the disappearance of the lag phase.
Table 1. Results from Analysis of ThT Time Curves for Fibrillation of PSM Peptides at Different Fg Concentrations (Figure 1) Using Amylofita.
|
k+kn (M–nc h–2) |
k+k2 (M–n2 h–2) | |||
|---|---|---|---|---|
| Fg (mg/mL) | PSMα1 | PSMα3 | PSMβ1 | PSMβ2 (k+k2) |
| 0 | 2.48 × 107 | 1.84 × 107 | 2.62 × 108 | 3.73 × 106 |
| 0.25 | 4.67 × 104 | 1.04 × 109 | 9.48 × 107 | 9.12 × 105 |
| 0.5 | 1.20 × 109 | 1.31 × 107 | 4.89 × 105 | |
| 1 | 1.45 × 109 | 1.82 × 106 | ||
| global parameters | k+k2 = 7.05 × 107 M–n2 h–2 | k+k2 = 2.51 × 1015 M–n2 h–2 | k+k2: 1.8 × 106 M–n2 h–2 | k+kn: 1.46 × 107 M–nc h–2 |
| nc = 2.55 | nc = 1.60 | nc: 2.55 | nc: 2.49 | |
| n2 = 1.41 | n2 = 2.07 | n2: 0.5 | n2: 0.536 | |
An aggregation model with secondary nucleation was used. Only the compound rate constant k+kn (PSMα1, PSMα3, and PSMβ1) or k+k2 (PSMβ2) was allowed to vary with [Fg], while other indicated parameters were fitted to a single value for each peptide.
Fg Only Alters the Secondary Structure of PSMα1 Aggregates
The secondary structure of PSM aggregates formed in the presence of Fg was evaluated by UV circular dichroism (far-UV CD, 260–190 nm) and attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) (amide I band at 1600–1700 cm–132). The samples were spun down to remove dimethyl sulfoxide (DMSO) and soluble Fg and PSMs. The Fg-free PSMα1 fibrils’ ATR-FTIR spectrum shows a characteristic amyloid peak at 1628 cm–1 and a peak at 1654 cm–1 (Figure 2A) attributed to the α-helical structure. Deconvolution of this spectrum indicates 73.6% β-strand and 26.4% α-helical content (Figures 2E, S2 and Table S1). Incubation of PSMα1 with 0.25 and 1 mg/mL Fg led to a decrease in the β-strand peak and an increase in the α-helical peak to 56.6 and 89%, respectively, consistent with the Fg’s α-helix structure.33 The presence of Fg in these aggregates was confirmed by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) (see below), and allowed us to estimate that aggregates formed in the presence of 1 mg/mL (2.9 μM) Fg and 0.2 mg/mL (88 μM) PSMα1 (when resuspended in the same volume) consist of ∼60 μM PSMα1 and 2.2 μM Fg, that is, essentially the same molar ratio of 30 PSMα1:1 Fg in both cases with a ∼70% aggregation level. To better clarify the secondary structure of these aggregates, we used circular dichroism (CD). Fg is a helix-rich protein containing 62% α-helix, 31% parallel β-strands, and 7% turns and other structures according to the deconvolution of the CD curve using BeStSel. Fg-free PSMα1 fibrils contain β-sheets with a (red-shifted) minimum at around 222 nm (Figure 2F); however, 1 mg/mL Fg led to Fg–PSMα1 co-aggregates consisting of 11% β-strands (mainly parallel) and 89% α-helix. A simple combination of the β-sheet and α-helix content of PSMα1 and Fg mixed in a 0.2:1 mass ratio should lead to 38% β-sheet, 56% α-helix, and 6% turns. This discrepancy suggests that the interaction of PSMα1 with Fg leads to co-aggregates structurally different from the individual components.
Figure 2.

Secondary structure analysis of the PSM aggregates incubated in absence and presence of fibrinogen by using CD and FTIR. (A) PSMα1, (B) PSMα3, (C) PSMβ1, and (D) PSMβ2. Legends in panel (A) also apply to panels (B–D). (E) FTIR analysis of the secondary structure of PSM peptides in presence of different concentrations of fibrinogen based on deconvolution of peaks. (F) CD of different PSM fibrils and aggregates formed in presence of 1 mg/mL Fg.
In contrast to the co-aggregative behavior of Fg and PSMα1, only a small fraction of Fg (∼4, 12, and 21% of total Fg at 1 mg/mL) was incorporated into aggregates formed by 0.2 mg/mL PSMα3, PSMβ1, and PSMβ2, respectively. Therefore, the signals for these co-aggregates are largely derived from the PSM peptides themselves. CD curves of Fg-free PSMα3 fibrils reveal an α-helix structure with two minima at 210 and 226 nm, consistent with its reported cross-α structure.34 Aggregates formed in the presence of Fg show the same helicity by CD (Figure 2F) and FTIR spectra (Figure 2B) are relatively unchanged, indicating that Fg does not lead to changes in the secondary structure of PSMα3 aggregates.
The FTIR spectra of fibrils of PSMβ1 and PSMβ2 both show peaks at 1630/1629 cm–1 (amyloid) and a shoulder at 1660/1657 cm–1 (β-turns), indicative of β-intermolecular sheets packed into a characteristic amyloid structure. This is confirmed by a deep minimum at around 221 nm in CD curves of fibrils of PSMβ1 and PSMβ2 (Figure 2F) (the CD curve shows a small shoulder at around 204–206 nm for PSMβ1 although deconvolution indicated mainly a β-sheet structure). For PSMβ1, there is no significant change in the fibril structure by Fg, whereas incubation of PSMβ2 with 0.25–1 mg/mL Fg leads to a shift in maximum of the peak from 1660 to 1650 cm–1, which translates to an increase in α-helical content to 92–98% (Figures 2C,E, S2 and Table S1). These data are nicely consistent with our previously mentioned ThT curves in which Fg only retarded PSMβ1 fibrillation to a certain extent but completely inhibited fibrillation of PSMβ2 (Figure S1C,D). The CD curves of PSMβ1 and PSMβ2 in presence of Fg both show a lower signal intensity. For PSMβ2, this is attributed to inhibition of fibril formation whereas for PSMβ1, we observed difficulties in dispensing from pipette tips, indicative of a pronounced stickiness of the PSMβ1 aggregates, especially at 1 mg/mL Fg (Figure S2K).
Fg Induces Non-amyloid Structures in PSM Aggregates, Particularly PSMα1
We complemented these spectroscopic data with transmission electron microscopy (TEM) images of end-point fibrillation samples, which turned out to be highly consistent with the ThT data. Fg-free PSMα1 shows high amounts of rod-like fibrils, but 1 mg/mL Fg completely shifted this to small spherical (probably oligomeric) species of diameter ∼25 nm (Figure 3), consistent with the disappearance of ThT signals. Although PSMβ1 formed fibrils both in the absence and presence of Fg, those formed with 1 mg/mL Fg were slightly thicker and stacked together. Based on this, we attribute the small decline in the ThT signal to a combination of precipitation and occlusion of ThT binding sites. PSMβ2 forms rod-like fibrils, which in the presence of Fg change to a network of amorphous aggregates, consistent with the near-total loss of the ThT signal. PSMα3 formed fibrils in the presence and absence of Fg, but Fg led to clumping into largely amorphous aggregates (Figure 3), indicating a dense network (which presumably is formed quickly, hence abolishing the lag time of fibrillation). On its own, Fg did not show any increase in ThT fluorescence over time or form any insoluble aggregates.
Figure 3.

Transmission electron microscopy images of PSM aggregates formed in the presence and absence of 1 mg/mL fibrinogen. All scale bars are 200 nm except for “PSMβ2 + Fg” and “PSMα3 + Fg” (500 nm). The yellow circles highlight spherical oligomers of ca. 25 nm diameter formed by PSMα1 in the presence of Fg.
Fg Co-aggregates with PSMα1 and PSMβ2 Seed Poorly
Next, we investigated whether PSM aggregates formed in the presence of Fg can act as seeds in fibrillation. Previous studies on the PSM seeding experiment indicated that different PSM fibrils show different seeding behaviors.30 Specifically, PSMα1 fibrils strongly seed fibrillation whereas other fibrils show weaker effects.30 We selected PSM aggregates formed in the presence of 1 mg/mL Fg, removed soluble Fg and PSMs by centrifugation, and then incubated the resuspended aggregates with fresh peptides at 10% (mass/mass) seed concentration without shaking. Seeds of Fg-free PSMα1 fibrils completely abolished the lag phase (Figure 4A). In contrast, aggregates formed with Fg inhibit the fibrillation, leading to a prolonged lag phase and a slower growth (elongation) phase. Thus, the oligomeric species formed in the presence of Fg (Figure 3) directly inhibit aggregation, acting as off-pathway oligomers similar to (although more effectively than) α-synuclein oligomers.35 This is consistent with Fg’s inhibitory role toward PSMα1 aggregation and the lack of fibrils seen by TEM. However, both normal PSMα3 fibrils and Fg-induced aggregates only have a modest effect on the lag phase (Figure 4B). A low seeding capacity of PSMα3 fibrils is observed in other studies.30 PSMβ1 seeds with and without Fg both accelerate PSMβ1 fibrillation (Figure 4C), while only Fg-free PSMβ2 seeds accelerate PSMβ2 fibrillation (Figure 4D). All these data are nicely consistent with the ThT and TEM data, given that 1 mg/mL Fg essentially completely abolishes PSMβ2 aggregation (Figure 1F).
Figure 4.

Fibrillation of 0.2 mg/mL PSM peptides in presence of 10% (mass/mass) preformed fibrils. The seeds were prepared by incubation of PSM peptides in presence and absence of 1 mg/mL fibrinogen. (A) PSMα1. (B) PSMα3, (C) PSMβ1, and (D) PSMβ2.
We used SDS–PAGE to quantify how large a fraction of Fg and PSM was incorporated into the aggregates when 0.2 mg/mL PSM was incubated with 1 mg/mL Fg. The pelleted aggregates were depolymerized in reducing sample buffer and boiled for 5 min before loading. The SDS–PAGE gel reveals the characteristic three Fg chains (α, β, and γ chains). Band intensity was quantified by densitometry and converted to protein mass using standard curves (Figure S3). The Fg–PSMα1 pellet contains ∼75% of total Fg and 67% of PSMα1, highlighting the strong interaction of Fg with PSMα1 (∼60 μM PSMα1 and 2.2 μM Fg, i.e., a molar PSMα1/Fg ratio of 27.3:1). The weaker bands at around 20 kDa (seen more clearly in the supernatant of Fg-PSMα3) might be a degradation product from the α-chain of Fg as reported for incubation of Aβ42 with Fg.25 For the three other PSMs, Fg is mostly found in the supernatant fraction (88, 79, and 99.5% for PSMβ1, PSMβ2, and PSMα3, respectively), leading to aggregate molar ratios (PSM/Fg) of 54:1, 11.6:1, and 123:1 for the three PSMs. The appearance of monomeric PSM bands obtained from PSM aggregates indicates that the fibrils of PSMα1 and PSMβ1 are sufficiently unstable to dissociate in SDS–PAGE.
Only Net Positively Charged PSM-Derived Peptides Interact with Fg
To gain insight into the interaction of PSMs with Fg, Alexa Fluor-488-labeled Fg was incubated with a 384-spot peptide microarray consisting of different parts of the PSM sequences. Note that this array technology in practice is limited to peptides of maximum 14-residue length, and we decided to restrict ourselves to 10-residue peptides to focus on local sequence effects. Accordingly, each spot consists of a 10-residue peptide sequence displaced one residue forward in the PSM sequence (i.e., with a 9-residue overlap with the next spot). The signal intensities of the first six spots of PSMα1 (spanning residues 1–15) are clearly higher than the last part (Figure 5A). These peptides have higher charge and hydrophobicity than the next six residues. When all 12 spots are considered, binding intensity is correlated with charge and hydrophobicity with p-values of 2.94 × 10–5 and 0.001, respectively. Thus, the greater the positive charge, the greater the binding, while hydrophobicity correlates more weakly with binding. A similar correlation was seen with PSMα3, for which almost all peptides except the last two ones (which have net charges of 0 and −1, respectively) interacted with Fg (Figure 5B) (p-values of 0.0007 and 0.11 for charge and hydrophobicity, respectively). This is perhaps not surprising given Fg’s overall negative charge at physiological pH (with an isoelectric point of 5.8).
Figure 5.

Interaction of Alexa Fluor-488-labeled Fg with different PSM peptide sequences immobilized on a peptide array. Signal intensity for Fg binding to sequences from (A) PSMα1, (B) PSMα3, (C) PSMβ1, and (D) PSMβ2. For each spot, the numbers on the x-axis give the residue position in the intact PSM sequence, corresponding to the starting residue in the spot’s 10-mer peptide.
The interactions with PSMβ1 and PSMβ2 were significantly weaker. Only 3–5 C-terminal peptides (the only peptides with a net positive charge, here +1) interacted with Fg (Figure 5C,D), underlining the importance of charge in the interaction with Fg.
Ala-Scan of PSM-Derived Peptides Highlight the Role of Lys Residues in the Interaction with Fg
We extended our peptide-array analysis using an Ala scan of each residue in the PSM sequences to probe the importance of each residue in the interaction with labeled Fg. The PSM sequences were divided into blocks of 10 residues, each of which was subsequently replaced with Ala (see Figure 6). In the PSMα1 sequence, this scan identified M1, I3, G5, K9, and K21 as the most important interacting residues, whose substitution by Ala reduced the Fg interaction (Figure 6A).
Figure 6.
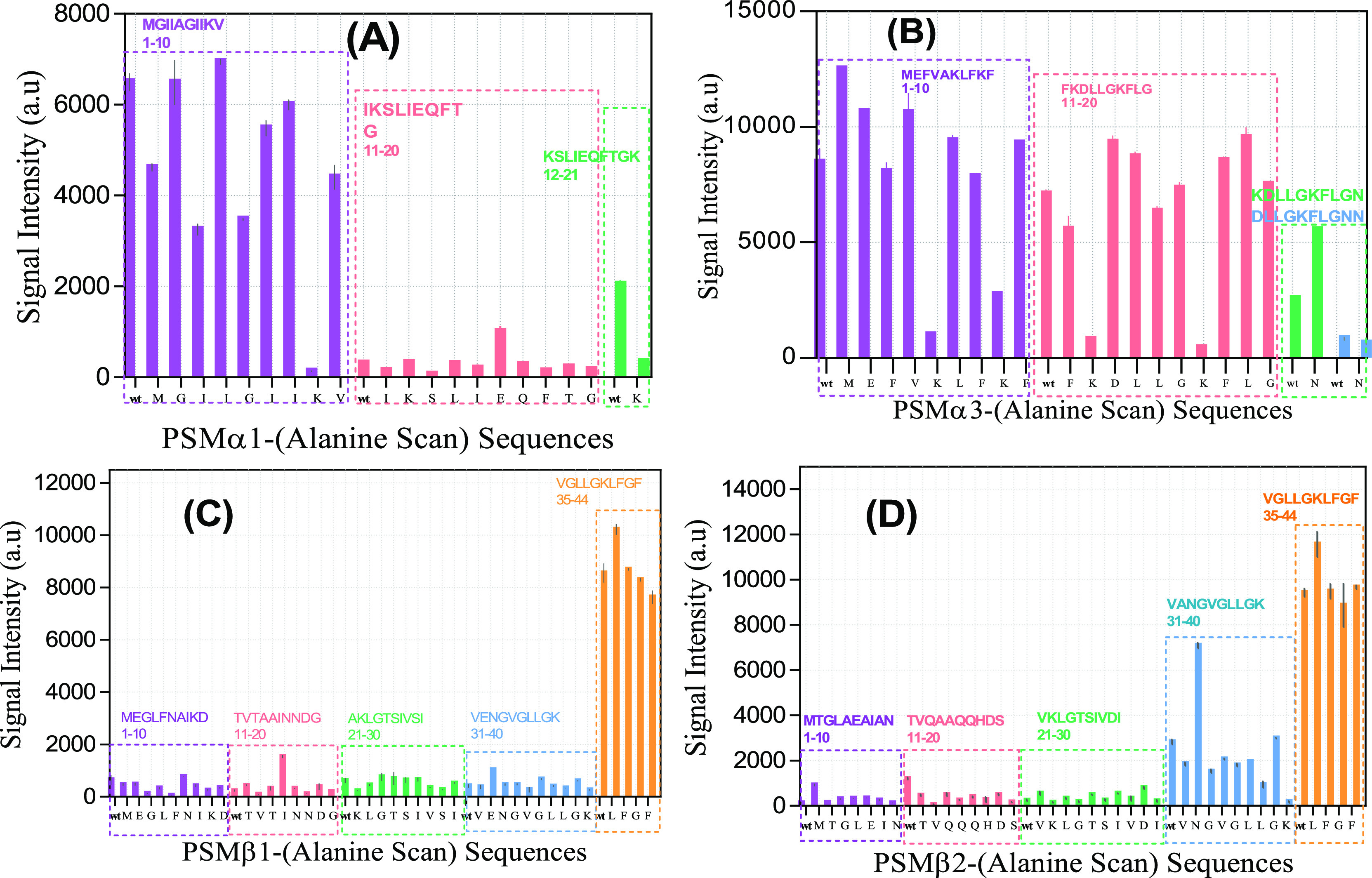
Signal intensities from interaction of labeled fibrinogen with peptide-array peptides involving Ala scans of different PSMs. Signal intensity for Fg binding to sequences from (A) PSMα1, (B) PSMα3, (C) PSMβ1, and (D) PSMβ2. The peptides were divided into 10-residue peptides (each with their own color); in each peptide, positions were individually replaced by Ala from left to right. “wt” is the initial peptide before starting the Ala scan. Each letter on the x-axis shows the residue that is replaced by Ala.
By far the greatest attenuation was seen with K9A and K21A, emphasizing the importance of charge. Weak interactions in wt residues 10–20 were not improved by Ala scanning. Similar to PSMα1, the most significant decrease in signal intensity for PSMα3 was related to the four Lys-to-Ala mutations at positions 6, 9, 12, and 17, again emphasizing the importance of charge (Figure 6B). PSMα3 mutants K9A, K12A, and K17A were still able to form amyloid fibrils,34 indicating that these residues were available for contacts with other components and providing a possible binding interface for fibrinogen (Figure 7). Ala-scanning of PSMβ1 and PSMβ2 did not turn up any major changes except the mutation N33A in PSMβ2, which increased binding intensity 2–3 fold (Figure 6C,D). For all Ala scans, charge is more important than hydrophobicity in the interaction with Fg (Table S2).
Figure 7.
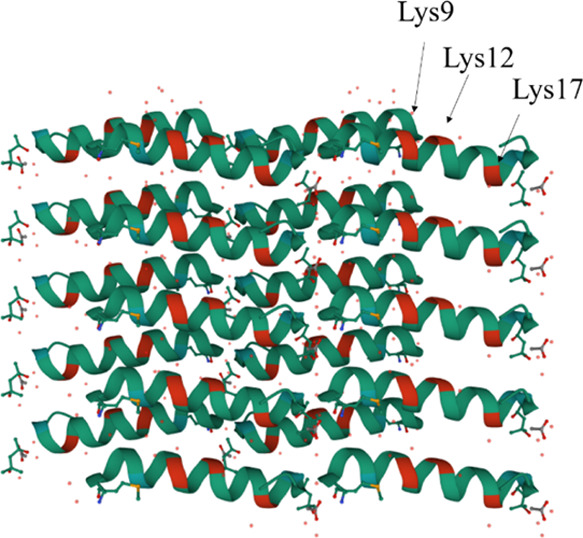
Crystal structure of PSMα3 (PDB 5I55) showing Lys 9, 12, and 17 in red as potential binding sites for fibrinogen.
Discussion
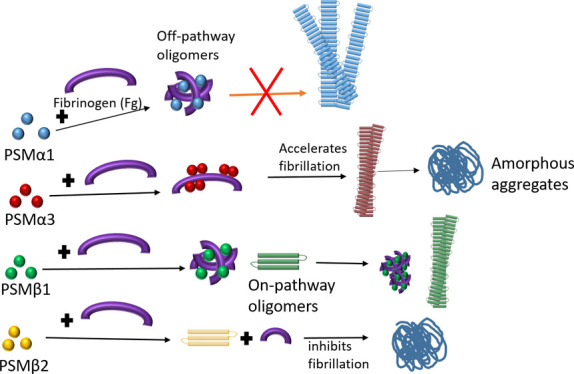
In this study, we have investigated the effect of the human plasma protein Fg on the fibrillation of four PSM peptides. Fibrillation of these peptides can promote biofilm formation in S. aureus, leading to antibiotic resistance and increased infectivity.6,8 As we summarize in Figure 8, the ThT fluorescence, CD, and ATR-FTIR data demonstrate various effects of Fg on different PSMs. Fg inhibits the fibrillation of PSMs except for PSMα3, whose fibrillation is accelerated, with an effect that is saturated by 0.25 mg/mL Fg. However, the final structure of PSM-Fg aggregates is different. Fg induces round oligomers with PSMα1, amorphous aggregates with PSMβ2, slightly thicker fibrils with PSMβ1, and a dense network of mostly amorphous (although seeding competent) aggregates with PSMα3. Fg also induces oligomerization in other amyloidogenic proteins such as CsgA, insulin B chain, β-amyloid, and yeast prion protein Sup35 (NM).19−21,24 However, the specific effect varies with protein. Thus, the functional amyloid CsgA formed 35–45 nm oligomers21 while Aβ42 was redirected to form co-oligomers with the D-fragment of Fg.24 Fg binds to prefibrillar intermediates of the insulin B chain to halt further fibril growth and alter the spectroscopic signature of the aggregates20 and also binds to Sup35 (NM) at early stages of fibrillation to form amorphous deposits.19
Figure 8.
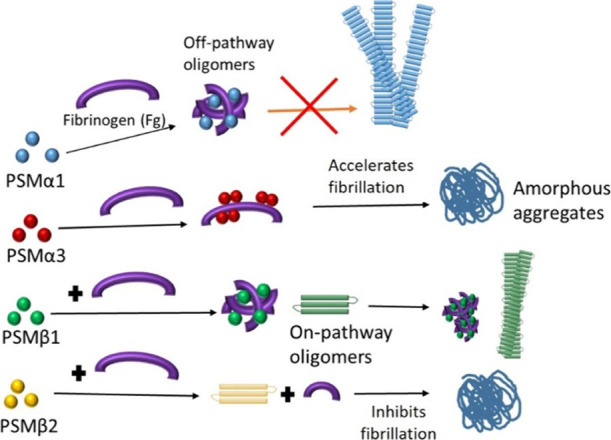
Schematic representation of different pathways of aggregation, resulting from incubation of PSMs with human fibrinogen.
The lack of aromatic residues in PSM peptides precluded the use of absorption to estimate concentrations of PSMs in aggregates. Instead, we quantitated concentration using SDS–PAGE and discovered that PSMα1 co-aggregates to a much greater extent with Fg than other PSMs, indicating particularly strong and stable interactions between PSMα1 and Fg. Neither aggregates of PSMα1 and PSMβ2 formed with Fg could seed fibrillation, showing that they are not bona fide fibrils, and in the case of PSMα1, aggregates directly inhibit fibrillation. In contrast, aggregates of PSMα3 and PSMβ1 formed with Fg both seed fibrillation. The effect was particularly striking for PSMβ1 which does not aggregate efficiently on its own. However, for both peptides, preformed fibrils are known to accelerate aggregation and thus perpetuate their inherent amyloid structure.30 The incorporation of the predominantly α-helical protein Fg into the non-fibrillar aggregates formed with PSMα1 and PSMβ2 is also confirmed by their high α-helical content and corresponding lower amount of β-sheet. The more modest (10% or less) incorporation of Fg into aggregates with PSMα3 and PSMβ1 also leads to significantly smaller changes in the secondary structure.
Molecular Basis of Fg Interactions with PSMs: Lys Ridges and Chaperone Activity?
Binding of Fg generally inhibits the nucleation step of fibrillation. This is seen both for CsgA and insulin B chain where Fg markedly inhibits the nucleation phase where early intermediates are formed. Using Amylofit, we could fit the PSM ThT aggregation curves with a secondary nucleation unseeded model previously used to describe aggregation of disease-related amyloids like α-synuclein,36 Aβ,37 and insulin.38 However, the mechanistic impact of Fg on PSM fibrillation is not uniform. Fg markedly decreased PSMα1’s rate constant of nucleation (kn) and to a smaller extent that of PSMβ1. For PSMβ2, the decrease in k+k2 (but insignificant impact on k+kn) indicates inhibition of secondary nucleation, that is, Fg impedes the ability of PSMβ2 to form fibrils on the side of the fibrils, perhaps by blocking access to these sites. This may be similar to the insulin B chain where Fg blocks fibril growth by surrounding prefibrillar intermediates.20 Unlike the other PSMs, Fg accelerated PSMα3 fibrillation by abolishing the lag phase. Given that PSMα3 forms a different amyloid structure known as cross-α with very rapid fibrillation kinetics,11 it is possible that Fg increases the local PSMα3 concentration by interacting temporarily with it through coiled-coil interactions or other templating phenomena. Fg did not change the secondary structure of PSMα3 fibrils, indicating that it favors cross-α fibril formation. This likely occurs through contacts with Lys residues which are known to be accessible in the PSMα3 structure and whose mutation in peptide arrays strongly reduces Fg binding although their mutation does not prevent PSMα3 fibril formation.34 These Lys residues are generally solvent-accessible and constitute a potential binding ridge for Fg contacts, for example, for initial templating of fibrillation, cfr. Figure 7. Only a small fraction of Fg formed stable and pelletable complexes with PSMα3, confirming a catalytic role of Fg in PSMα3 fibrillation.
Although PSMα1 and PSMα3 have almost the same length (21 vs 22aa), PSMα1 fibrils form a cross-β structure in contrast to PSMα3’s cross-α structure, but Fg shifts PSMα1 to α-helical oligomers. PSMα1’s strongest Fg interaction is found in the N-terminal residues, which are the most hydrophobic and the most amyloidogenic part of the peptide (Figure S4). This reveals a potential chaperoning behavior of Fg with an affinity for hydrophobic regions.39 The C-terminal region of the α-chain in human Fg is reported to have chaperone-like activity,19,40 and different chaperones are known to inhibit fibrillation of amyloidogenic proteins like α-synuclein and Aβ.41−43
Biological Perspectives
It has been reported that macromolecules like eDNA, polysaccharide intercellular adhesin (PIA), heparin, and lipopolysaccharides promote and stabilize biofilm formation by themselves or through enhancing amyloid formation.14,15,44,45 It is a remarkable feature that most of these macromolecules have a negative net charge at physiological pH, emphasizing negative charge as a general motif contributing to amyloid formation and biofilm strengthening.
The biofilm matrix composition is highly dependent on the environment and on precursors which are available for matrix formation. In vivo, the biofilm matrix of S. aureus is dominated by fibrin as the secretion of coagulases activates prothrombin, which leads to formation of a fibrin pseudocapsule around the bacteria. The interaction between Fg and PSMs may therefore not only affect biofilm formation via its effect on amyloid formation but also via its effect on fibrin formation. To the best of our knowledge, the effect of PSMs on fibrin formation in S. aureus biofilms remains unexplored. However, our study reveals that apart from PSMα3, Fg actually inhibits the fibrillation of PSMs, which implies an inhibitory effect on biofilm formation in view of PSM amyloid contributions to this process.6 On the other hand, fibrin produced by Fg promotes biofilm formation, potentially suggesting two antagonistic effects of Fg on biofilm formation.
In summary, human Fg interacts in a number of different ways with different PSMs from S. aureus. Fg inhibits fibrillation of PSMα1, PSMβ1, and PSMβ2 but induces fibrillation in PSMα3. Of note, Fg drives PSMα1 to off-pathway oligomers and aggregates that contain both Fg and PSMα1 and lack the capacity to seed fibrillation of PSMα1. PSMα1 fibrils strongly seed the fibrillation of other PSMs and even drive fibrillation of PSMα2 and δ-toxin that do not fibrillate on their own.30 Consequently, Fg’s inhibition of PSMα1 fibrillation can in itself effectively halt the fibrillation of other PSMs. We propose that by inhibiting PSM fibrillation, Fg can modulate biofilm formation of S. aureus. This may be a host defense mechanism to reduce bacterial infection which can be overruled by the bacterial ability to bind to host cells through contact with fibrinogen and thus congregate to form a biofilm.
Materials and Methods
Materials
Synthetic N-terminally formylated PSM peptides were purchased from GenScript at >95% purity. Fg from human plasma, Alexa Fluor 488-labeled Fg, hexafluoroisopropanol (HFIP), trifluoroacetic acid, and other chemicals were from Sigma-Aldrich (St. Louis, MO).
PSM Peptide Pretreatment
To dissolve any preformed aggregates and optimize reproducibility, the synthetic peptides were dissolved in HFIP/TFA (1:1) to 5–6 mg/mL and then sonicated for 2 min (10 s on and 10 s off) with a QSonica Sonicator (Q500, Newtown, CT, USA) at 20% amplitude. After 1 h at room temperature in a chemical hood, remaining solvent was evaporated completely in a speed vac.
Fibrillation Kinetics Assays Using a Plate Reader
PSM peptides were dissolved to 20 mg/mL in DMSO, diluted in Milli-Q water to 1 mg/mL, spun down (5 min, 10,000 rpm or 6700g) to remove aggregates, and kept on ice. The supernatant was diluted to 0.2 mg/mL in filtered sodium phosphate buffer (containing 47.5 mM Na2HPO4 and 2.5 mM NaH2PO4) pH 8, mixed with 200 μM ThT (from stock of 14 mM in Milli-Q water), and incubated at 37 °C in a 96-well plate (Nunc, Thermo Fisher Scientific, Roskilde, Denmark). The final DMSO concentration was always 1% (vol/vol). Fg was dissolved in phosphate-buffered saline (PBS) at 10 mg/mL and directly added to the wells to reach the desired concentration. Crystal clear sealing tape (Hampton Research, Aliso Viejo, CA, USA) was used to cover the plate and prevent evaporation of solvent. ThT fluorescence was measured every 5 min using excitation at 438 nm and emission at 480 nm. Before each cycle, the plate was shaken at the 300 rpm orbital mode for 15 s. Due to PSMα3’s very rapid fibrillation kinetics, measurements were carried out in the quiescent mode using 2 min cycles. All measurement were carried out in triplicate. Note that due to batch variations in peptide synthesis, there are minor differences in the specific fibrillation kinetics compared to the previously reported data.30
Seeding Experiments
To prepare the seeds, the fibrils and aggregates formed in the presence of 1 mg/mL Fg were spun down (13,000 rpm or 11,340g for 10 min) and resuspended in the same amount of Milli-Q water and then sonicated for 2 min (20 s on and 10 s off) with a QSonica Sonicator (Q500, Newtown, CT, USA) using 20% amplitude. 0.2 mg/mL pretreated peptides (prepared and incubated the same as in sections 2–3 but under quiescent condition) were incubated in presence of 10% (mass) of different seeds (normal fibrils of each peptide and aggregates formed in presence of Fg). ThT-emission time profiles were recorded in a GENios Pro fluorescence plate reader (Tecan, Mänerdorf, Switzerland). The data were fitted using the Amylofit web server (http://www.amylofit.ch.cam.ac.uk/fit). When fitting ThT curves recorded at different Fg concentrations, the PSM concentration is fixed at its experimental value, and the reaction orders of primary nucleation (nc), secondary nucleation (n2), and k+k2 were globally fitted to obtain rate constants (k+kn) for each ThT curve recorded at different [Fg] (except for PSMβ2, where k+k2 was fitted to individual [Fg]). Here, kn is the rate constant for formation of nuclei from monomers, whereas k+ is the rate constant of elongation in which the fibril grows by addition of monomer to fibril ends, and k2 is the rate constant of formation of secondary nuclei on fibrils.31
CD Spectroscopy
Fibrillated samples were centrifuged (15 min at 13,000 rpm or 11,340g) to remove DMSO after which the pellet was resuspended in the same volume of Milli-Q water. Far-UV CD spectra of samples in a 0.1 mm quartz crystal cuvette were recorded from 190 to 260 nm with a step size of 1 nm on a Chirascan CD spectrophotometer (Applied Photophysics, Surrey, UK). Spectra were deconvoluted using the BeStSel web server.46
Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy
To remove DMSO and soluble Fg, samples were spun down (15 min at 13,000 rpm or 11,340g) and the pellets were resuspended in the same volume of Milli-Q water. 2 μL of samples were dried in a Golden Gate Single Reflection Diamond ATR cell (Specac, Kent, UK) under a stream of nitrogen, and the FTIR spectra were monitored on a Tensor 27 FTIR spectrophotometer (Bruker, Billerica, Massachusetts). Baseline correlation and atmospheric compensation considering CO2 and H2O were conducted with OPUS 5.5 software (Bruker, Billerica, Massachusetts). Deconvolution of peaks was carried out in Origin.
SDS–PAGE of PSMs Incubated with Fg
Aggregates formed in the presence and absence of Fg were spun down (15 min at 13,000 rpm or 11,340g) and resuspended in the same amount of Milli-Q water, after which 20 μL of both supernatants and pellet was separately mixed with sample buffer and boiled for 5 min. Due to their small size, the PSM peptides migrate in the dye front. Therefore, bromophenol blue was omitted from the loading buffer to quantify peptide bands. 10 μL of samples loaded on SDS–PAGE (15% polyacrylamide gel) and tris/tricine buffer containing 0.1 M tris and 0.02 M HCl with pH 8.9 as anode buffer, while 0.1 M tris, 0.1 M tricine, and 0.1% SDS with pH 8.25 as cathode buffer was used to obtain sharper peptide bands.47 A voltage of 150 V was applied on the gel for around 3 h and a prestained protein ladder was used to follow the running process.
Transmission Electron Microscopy
PSMs samples fibrillated in the presence or absence of 1 mg/mL Fg were stained with phosphotungstic acid, and images were recorded on a JEM 1010 electron microscope as described.48
Celluspots Peptide Microarrays
A peptide microarray displaying all the PSM sequences was prepared as described49 and blocked using 25 mL of 3% (w/v) whey protein in tris buffer saline containing 0.1% Tween-20 (TBS-T) in a falcon tube on a rolling board overnight at 4 °C. The peptide microarray was washed three times with PBS and incubated with 0.05 mg/mL Alexa Fluor 488-labeled Fg in PBS for 4 h on a rolling table at room temperature and then washed three times with TBS-T to remove free Fg. Subsequently, the array was air-dried for 1 h in a dark place (to protect from photobleaching) without touching the surface and scanned with a Typhoon Trio scanner (GE Life Sciences, Pittsburgh, PA).
Acknowledgments
We thank Maria Andreasen and Masihuz Zaman for ongoing discussions and collaborations on PSM.
Glossary
Abbreviations
- Fg
fibrinogen
- PSM
phenol soluble modulin
- CD
circular dichroism
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02333.
Kinetics of PSM amyloid formation in presence and absence of fibrinogen monitored by ThT fluorescence; FTIR analysis of the secondary structure of PSMs peptides in presence of different concentrations of fibrinogen; deconvolution results from FTIR of PSMs incubated with Fg; SDS–PAGE of the pellet and supernatants of aggregates formed in the presence of 1 mg/mL Fg densitometrically quantified using ImageJ; p-values of multiple regression analysis of the correlation between signal intensity of alanine scans from peptide arrays and the two parameters charge and hydrophobicity, and computational prediction of aggregation propensity of different PSMs based on Rosetta energies that predict amyloid propensity in hexapeptides (PDF)
Accession Codes
Accession codes—PSMα1: A9JX05. PSMα3: A9JX07. PSMβ1: H9BRQ3. PSMβ2: H9BRQ4. Fibrinogen: α chain P02671, β chain P02675, and γ chain P02679.
Author Contributions
Z.N. and D.E.O. designed experiments. Z.N., A.F., and J.N. carried out experiments. V.S. and K.S. provided reagents. Z.N., R.L.M., and D.E.O. analyzed the data. Z.N., R.L.M., and D.E.O. wrote and edited the article and D.E.O. acquired funding.
This work was supported by the Danish Research Foundation|Technical Sciences (grant no. 6111-00241B) and the Danish Research Foundation|Natural Sciences (grant no. 8021-00208B), both to D.E.O.
The authors declare no competing financial interest.
Supplementary Material
References
- Otto M. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Ann. Rev. Med. 2013, 64, 175–188. 10.1146/annurev-med-042711-140023. [DOI] [PubMed] [Google Scholar]
- Otto M.Staphylococcal Biofilms in Gram−Positive Pathogens, 3rd edition. Fischetti V. A., Novick R. P., Ferretti J. F., Portnoy D. A., Braunstein M., Rood J. I., eds.; ASM Press: Washington, DC, 2019; pp 699−711. [Google Scholar]
- Joo H.-S.; Chatterjee S. S.; Villaruz A. E.; Dickey S. W.; Tan V. Y.; Chen Y.; Sturdevant D. E.; Ricklefs S. M.; Otto M. Mechanism of gene regulation by a Staphylococcus aureus toxin. mBio 2016, 7, e01579 10.1128/mbio.01579-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le K. Y.; Dastgheyb S.; Ho T. V.; Otto M. Molecular determinants of staphylococcal biofilm dispersal and structuring. Front. Cell. Infect. Microbiol. 2014, 4, 167. 10.3389/fcimb.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy S.; Joo H.-S.; Duong A. C.; Bach T.-H. L.; Tan V. Y.; Chatterjee S. S.; Cheung G. Y. C.; Otto M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 1281–1286. 10.1073/pnas.1115006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz K.; Syed A. K.; Stephenson R. E.; Rickard A. H.; Boles B. R. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012, 8, e1002744 10.1371/journal.ppat.1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le K. Y.; Villaruz A. E.; Zheng Y.; He L.; Fisher E. L.; Nguyen T. H.; Ho T. V.; Yeh A. J.; Joo H.-S.; Cheung G. Y. C.; Otto M. Role of Phenol-Soluble Modulins in Staphylococcus epidermidis Biofilm Formation and Infection of Indwelling Medical Devices. J. Mol. Biol. 2019, 431, 3015–3027. 10.1016/j.jmb.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung G. Y. C.; Joo H.-S.; Chatterjee S. S.; Otto M. Phenol-soluble modulins–critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. 10.1111/1574-6976.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towle K. M.; Lohans C. T.; Miskolzie M.; Acedo J. Z.; van Belkum M. J.; Vederas J. C. Solution structures of phenol-soluble modulins α1, α3, and β2, virulence factors from Staphylococcus aureus. Biochemistry 2016, 55, 4798–4806. 10.1021/acs.biochem.6b00615. [DOI] [PubMed] [Google Scholar]
- Laabei M.; Jamieson W. D.; Yang Y.; Van Den Elsen J.; Jenkins A. T. A. Investigating the lytic activity and structural properties of Staphylococcus aureus phenol soluble modulin (PSM) peptide toxins. Biochim. Biophys. Acta Biomembr. 2014, 1838, 3153–3161. 10.1016/j.bbamem.2014.08.026. [DOI] [PubMed] [Google Scholar]
- Tayeb-Fligelman E.; Tabachnikov O.; Moshe A.; Goldshmidt-Tran O.; Sawaya M. R.; Coquelle N.; Colletier J.-P.; Landau M. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science 2017, 355, 831–833. 10.1126/science.aaf4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick R. P.; Geisinger E. Quorum sensing in staphylococci. Annu. Rev. Genet. 2008, 42, 541–564. 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- Matsumoto M.; Nakagawa S.; Zhang L.; Nakamura Y.; Villaruz A. E.; Otto M.; Wolz C.; Inohara N.; Núñez G. Interaction between Staphylococcus Agr virulence and neutrophils regulates pathogen expansion in the skin. Cell Host Microbe 2021, 29, 930. 10.1016/j.chom.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz K.; Ganesan M.; Payne D. E.; Solomon M. J.; Boles B. R. Extracellular DNA facilitates the formation of functional amyloids in S taphylococcus aureus biofilms. Mol. Microbiol. 2016, 99, 123–134. 10.1111/mmi.13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najarzadeh Z.; Pedersen J. N.; Christiansen G.; Shojaosadati S. A.; Pedersen J. S.; Otzen D. E. Bacterial amphiphiles as amyloid inducers: Effect of Rhamnolipid and Lipopolysaccharide on FapC fibrillation. Biochim. Biophys. Acta Protein Proteonomics 2019, 1867, 140263. 10.1016/j.bbapap.2019.140263. [DOI] [PubMed] [Google Scholar]
- Akiyama H.; Ueda M.; Kanzaki H.; Tada J.; Jirô Arata J. Biofilm formation of Staphylococcus aureus strains isolated from impetigo and furuncle: role of fibrinogen and fibrin. J. Dermatol. Sci. 1997, 16, 2–10. 10.1016/s0923-1811(97)00611-7. [DOI] [PubMed] [Google Scholar]
- Pickering A. C.; Vitry P.; Prystopiuk V.; Garcia B.; Höök M.; Schoenebeck J.; Geoghegan J. A.; Dufrêne Y. F.; Fitzgerald J. R. Host-specialized fibrinogen-binding by a bacterial surface protein promotes biofilm formation and innate immune evasion. PLoS Pathog. 2019, 15, e1007816 10.1371/journal.ppat.1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman J. M.; Pandi L.; Sawaya M. R.; Riley M.; Doolittle R. F. Crystal structure of human fibrinogen. Biochemistry 2009, 48, 3877–3886. 10.1021/bi802205g. [DOI] [PubMed] [Google Scholar]
- Tang H.; Fu Y.; Cui Y.; He Y.; Zeng X.; Ploplis V. A.; Castellino F. J.; Luo Y. Fibrinogen has chaperone-like activity. Biochem. Biophys. Res. Commun. 2009, 378, 662–667. 10.1016/j.bbrc.2008.11.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N.; Akai T.; Inoue R.; Sugiyama M.; Tamura A.; Chatani E. Structural insights into the inhibition of amyloid fibril formation by fibrinogen via interaction with prefibrillar intermediates. Biochemistry 2019, 58, 2769–2781. 10.1021/acs.biochem.9b00439. [DOI] [PubMed] [Google Scholar]
- Swasthi H. M.; Bhasne K.; Mahapatra S.; Mukhopadhyay S. Human fibrinogen inhibits amyloid assembly of biofilm-forming CsgA. Biochemistry 2018, 57, 6270–6273. 10.1021/acs.biochem.8b00841. [DOI] [PubMed] [Google Scholar]
- Rybarczyk B. J.; Lawrence S. O.; Simpson-Haidaris P. J. Matrix-fibrinogen enhances wound closure by increasing both cell proliferation and migration. Blood 2003, 102, 4035–4043. 10.1182/blood-2003-03-0822. [DOI] [PubMed] [Google Scholar]
- Vidal B.; Serrano A. L.; Tjwa M.; Suelves M.; Ardite E.; De Mori R.; Baeza-Raja B.; Martinez de Lagran M.; Lafuste P.; Ruiz-Bonilla V.; Jardi M.; Gherardi R.; Christov C.; Dierssen M.; Carmeliet P.; Degen J. L.; Dewerchin M.; Munoz-Canoves P. Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn H. J.; Zamolodchikov D.; Cortes-Canteli M.; Norris E. H.; Glickman J. F.; Strickland S. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 21812–21817. 10.1073/pnas.1010373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamolodchikov D.; Berk-Rauch H. E.; Oren D. A.; Stor D. S.; Singh P. K.; Kawasaki M.; Aso K.; Strickland S.; Ahn H. J. Biochemical and structural analysis of the interaction between β-amyloid and fibrinogen. Blood 2016, 128, 1144–1151. 10.1182/blood-2016-03-705228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung A. L.; Krishnan M.; Jaffe E. A.; Fischetti V. A. Fibrinogen acts as a bridging molecule in the adherence of Staphylococcus aureus to cultured human endothelial cells. J. Clin. Invest. 1991, 87, 2236–2245. 10.1172/jci115259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. A.; Monk I. R.; O’Gara J. P.; Foster T. J. Subdomains N2N3 of fibronectin binding protein A mediate Staphylococcus aureus biofilm formation and adherence to fibrinogen using distinct mechanisms. J. Bacteriol. 2013, 195, 2675–2683. 10.1128/jb.02128-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane F. M.; Loughman A.; Valtulina V.; Brennan M.; Speziale P.; Foster T. J. Fibrinogen and elastin bind to the same region within the A domain of fibronectin binding protein A, an MSCRAMM of Staphylococcus aureus. Mol. Microbiol. 2007, 63, 711–723. 10.1111/j.1365-2958.2006.05552.x. [DOI] [PubMed] [Google Scholar]
- Thomer L.; Schneewind O.; Missiakas D. Pathogenesis of Staphylococcus aureus bloodstream infections. Annu. Rev. Phytopathol. 2016, 11, 343–364. 10.1146/annurev-pathol-012615-044351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman M.; Andreasen M. Cross-talk between individual phenol soluble modulins in S. aureus biofilm enables rapid and efficient amyloid formation. eLife 2020, 9, e59776 10.7554/elife.59776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisl G.; Kirkegaard J. B.; Arosio P.; Michaels T. C. T.; Vendruscolo M.; Dobson C. M.; Linse S.; Knowles T. P. J. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 2016, 11, 252–272. 10.1038/nprot.2016.010. [DOI] [PubMed] [Google Scholar]
- Moran S. D.; Zanni M. T. How to get insight into amyloid structure and formation from infrared spectroscopy. J. Phys. Chem. Lett. 2014, 5, 1984–1993. 10.1021/jz500794d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinov R. I.; Faizullin D. A.; Zuev Y. F.; Weisel J. W. The α-helix to β-sheet transition in stretched and compressed hydrated fibrin clots. Biophys. J. 2012, 103, 1020–1027. 10.1016/j.bpj.2012.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayeb-Fligelman E.; Salinas N.; Tabachnikov O.; Landau M. Staphylococcus aureus PSMα3 Cross-α Fibril Polymorphism and Determinants of Cytotoxicity. Structure 2020, 28, 301–313.e6. 10.1016/j.str.2019.12.006. [DOI] [PubMed] [Google Scholar]
- Lorenzen N.; Nielsen S. B.; Buell A. K.; Kaspersen J. D.; Arosio P.; Vad B. S.; Paslawski W.; Christiansen G.; Valnickova-Hansen Z.; Andreasen M.; Enghild J. J.; Pedersen J. S.; Dobson C. M.; Knowles T. P. J.; Otzen D. E. The role of stable α-synuclein oligomers in the molecular events underlying amyloid formation. J. Am. Chem. Soc. 2014, 136, 3859–3868. 10.1021/ja411577t. [DOI] [PubMed] [Google Scholar]
- Gaspar R.; Meisl G.; Buell A. K.; Young L.; Kaminski C. F.; Knowles T. P.; Sparr E.; Linse S. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q. Rev. Biophys. 2017, 50, e6 10.1017/S0033583516000172. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 9758–9763. 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foderà V.; Librizzi F.; Groenning M.; Van De Weert M.; Leone M. Secondary nucleation and accessible surface in insulin amyloid fibril formation. J. Phys. Chem. B 2008, 112, 3853–3858. 10.1021/jp710131u. [DOI] [PubMed] [Google Scholar]
- Rüdiger S.; Germeroth L.; Schneider-Mergener J.; Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997, 16, 1501–1507. 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Fu Y.; Zhan S.; Luo Y. αEC, the C-terminal extension of fibrinogen, has chaperone-like activity. Biochemistry 2009, 48, 3967–3976. 10.1021/bi900015n. [DOI] [PubMed] [Google Scholar]
- Huang C.; Cheng H.; Hao S.; Zhou H.; Zhang X.; Gao J.; Sun Q.-H.; Hu H.; Wang C.-c. Heat shock protein 70 inhibits α-synuclein fibril formation via interactions with diverse intermediates. J. Mol. Biol. 2006, 364, 323–336. 10.1016/j.jmb.2006.08.062. [DOI] [PubMed] [Google Scholar]
- Willander H.; Presto J.; Askarieh G.; Biverstål H.; Frohm B.; Knight S. D.; Johansson J.; Linse S. BRICHOS domains efficiently delay fibrillation of amyloid β-peptide. J. Biol. Chem. 2012, 287, 31608–31617. 10.1074/jbc.m112.393157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P.; Michaels T. C.; Linse S.; Månsson C.; Emanuelsson C.; Presto J.; Johansson J.; Vendruscolo M.; Dobson C. M.; Knowles T. P. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016, 7, 10948. 10.1038/ncomms10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holubová M.; Lobaz V.; Loukotová L.; Rabyk M.; Hromádková J.; Trhlíková O.; Pechrová Z.; Groborz O.; Štěpánek P.; Hrubý M. Does polysaccharide glycogen behave as a promoter of amyloid fibril formation at physiologically relevant concentrations?. Soft Matter 2021, 17, 1628–1641. 10.1039/d0sm01884h. [DOI] [PubMed] [Google Scholar]
- Chen X. e.; Ling P.; Duan R.; Zhang T. Effects of heparosan and heparin on the adhesion and biofilm formation of several bacteria in vitro. Carbohydr. Polym. 2012, 88, 1288–1292. 10.1016/j.carbpol.2012.02.006. [DOI] [Google Scholar]
- Micsonai A.; Wien F.; Bulyáki É.; Kun J.; Moussong É.; Lee Y.-H.; Goto Y.; Réfrégiers M.; Kardos J. BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acid Res. 2018, 46, W315–W322. 10.1093/nar/gky497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger H. Tricine–sds-page. Nat. Protoc. 2006, 1, 16. 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- Pedersen J. N.; Jiang Z.; Christiansen G.; Lee J. C.; Pedersen J. S.; Otzen D. E. Lysophospholipids induce fibrillation of the repeat domain of PMEL17 through intermediate core-shell structures. Biochim. Biophys. Acta Protein Proteonomics 2019, 1867, 519–528. 10.1016/j.bbapap.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleem A.; Christiansen G.; Madsen D. J.; Maric H.; Strømgaard K.; Bryers J. D.; Daggett V.; Meyer R. L.; Otzen D. E. Protein engineering reveals mechanisms of functional amyloid formation in Pseudomonas aeruginosa biofilms. J. Mol. Biol. 2018, 430, 3751–3763. 10.1016/j.jmb.2018.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.