

Abstract
DNA lesions arise from a combination of physiological/metabolic sources and exogenous environmental influences. When left unrepaired, these alterations accumulate in the cells and can give rise to mutations that change the function of important proteins (i.e. tumor suppressors, oncoproteins), or cause chromosomal rearrangements (i.e. gene fusions) that also result in the deregulation of key cellular molecules. Progressive acquisition of such genetic changes promotes uncontrolled cell proliferation and evasion of cell death, and hence plays a key role in carcinogenesis. Another less-studied consequence of DNA damage accumulating in the host genome is the integration of oncogenic DNA viruses such as Human papillomavirus, Merkel cell polyomavirus, and Hepatitis B virus. This critical step of viral-induced carcinogenesis is thought to be particularly facilitated by DNA breaks in both viral and host genomes. Therefore, the impact of DNA damage on carcinogenesis is magnified in the case of such oncoviruses via the additional effect of increasing integration frequency. In this review, we briefly present the various endogenous and exogenous factors that cause different types of DNA damage. Next, we discuss the contribution of these lesions in cancer development. Finally, we examine the amplified effect of DNA damage in viral-induced oncogenesis and summarize the limited data existing in the literature related to DNA damage-induced viral integration. To conclude, additional research is needed to assess the DNA damage pathways involved in the transition from viral infection to cancer. Discovering that a certain DNA damaging agent increases the likelihood of viral integration will enable the development of prophylactic and therapeutic strategies designed specifically to prevent such integration, with an ultimate goal of reducing or eliminating these viral-induced malignancies.
Keywords: DNA lesions, DNA damaging agents, mutations, genomic instability, carcinogenesis, oncoviruses, viral-induced malignancies, viral integration
Introduction
DNA is the biological template needed for an organism to develop, function, grow, and reproduce. Its integrity and stability are therefore vital to life. However, due to its dynamic nature, this macromolecule is constantly subjected to several alterations. In fact, it has been estimated that each cell of the human body receives approximately 70,000 DNA lesions per day [1,2]. These aberrations arise from physiological or metabolic sources, as well as exogenous environmental influences [3,4]. Depending on the source or cause of DNA damage, the lesions can range from simple base changes to more complex alterations including single- or double-stranded breaks [5]. To avoid detrimental consequences to cellular functions and hence survival, life has evolved several systems that maintain genetic stability under strict control. Indeed, in addition to the proofreading activity of the DNA polymerases that correct mis-incorporated bases during replication, cells possess various DNA repair mechanisms to restore the damaged molecule [6,7]. Unfortunately, not all DNA lesions are efficiently repaired in an error-free manner, leading to the acquisition and accumulation of many mutations which can ultimately contribute to several diseases, including cancer [8,9]. The relevance of DNA damage in carcinogenesis became particularly apparent when it was recognized that almost all carcinogenic agents are also mutagenic, causing changes in the DNA sequence [10]. In recent years, DNA damage and repair processes have also received a special attention in the case of oncogenic viruses, since the process of malignant transformation by several of these viruses, including Hepatitis B virus (HBV), Merkel cell polyomavirus (MCV) and Human papillomavirus (HPV), is likely dependent on DNA damage promoting viral integration into human genome [11,12]. In this paper, we review the various endogenous and exogenous factors that cause different types of DNA damage and discuss the contribution of these lesions to cancer induction. Finally, we shed the light on the amplified effect of DNA damage in viral-induced carcinogenesis.
Sources and types of DNA damage
Based on the origin of the insult, DNA damage is classified into two main groups, namely endogenous and exogenous DNA damage. Endogenous DNA lesions are caused by cellular metabolic processes such as oxidation, hydrolysis, alkylation, and polymerase incorporation errors, whereas exogenous sources of DNA damage include environmental factors such as IR, UV radiation, and various chemical agents. Here we present a brief overview of the main endogenous and exogenous agents causing different types of DNA damage (summarized in Table 1).
Table 1.
Endogenous and exogenous sources of DNA damage, types of DNA lesions produced, repair mechanisms activated, and consequences on the DNA structure and processes
| Damaging event | Major forms of lesion | Repair mechanisms | Consequences on DNA | |
|---|---|---|---|---|
| Endogenous factors | Oxidation (ROS) | Oxidative base modifications | Base excision repair | Serve as miscoding template causing mutagenesis; helix distortion |
| SSBs | Single strand break repair or double strand break repair pathways | Collapse of DNA and RNA polymerase complexes; recombination events | ||
| Alkylation (SAM) | Methylated bases | Direct reversal; base excision repair; mismatch repair | G→A and T→C mutations; Inhibition of DNA replication | |
| Hydrolysis | AP sites | Base excision repair; lesion bypass | Misincorporation of bases; DNA or RNA polymerase block | |
| Deaminated bases (uracil) | DNA glycosylase & base excision repair | Changes in coding properties causing mutagenesis | ||
| Polymerase incorporation errors | Base substitutions, insertions or deletions | Mismatch repair | Mutagenic outcomes and genomic instability; fork collapse | |
| Exogenous factors | Ionizing radiation | DSBs | Non-homologous end-joining; Homologous recombination | Mutagenesis, chromosomal translocation and rearrangements; genomic instability |
| SSBs | AP endonucleases | Polymerase block | ||
| Single base alterations (mostly oxidation) | Base excision repair | |||
| Ultraviolet radiation | CPDs and 6-4PPs | Nucleotide excision repair | Mutagenicity (C to T and T to C transversions and CC to TT transition mutations); helix distortion; error-prone bypass | |
| Chemical agents (aromatic amines, alkylating agents, PAHs, reactive electrophiles, natural toxins, chemotherapeutic agents) | Base lesions and DNA adducts | Nucleotide excision repair; direct reversal; base excision repair; mismatch repair | Base substitutions; frameshift mutations; replication and transcription block; destabilization and breakage of DNA; helix distortion | |
| Intrastrand or interstrand crosslinks | Nucleotide excision repair; homologous recombination | Structure disruption; inhibition of DNA replication and transcription |
Endogenous DNA damage
Oxidative DNA damage
One of the most prominent sources of DNA damage is reactive oxygen species (ROS). These chemically reactive molecules containing oxygen are produced endogenously as common byproducts of aerobic cellular respiration, and are also derived from catabolic oxidases, anabolic processes, and peroxisomal metabolism. ROS can also be induced by various exogenous sources such as UV light, ionizing radiation, diet, stress, pathogens, and smoking [13,14]. At low or moderate levels, these free radicals play essential physiological roles in intracellular cell signaling and homeostasis, cell death, immune defense against pathogens, and induction of mitogenic response. However, in excess, they can cause oxidative damage to the biological macromolecules, severely compromising cell health and contributing to disease development [15-17]. For this reason, cells are equipped with several systems to protect the cellular components and mitigate the deleterious effects of ROS. For instance, aerobic respiration is confined to the mitochondrial compartment, thereby protecting the other cellular elements. Furthermore, the DNA is wrapped around complexes of histones, giving the chromosome a more compact shape, thereby protecting it from oxidizing effects. Most importantly, redox homeostasis is maintained by two arms of antioxidant defense machineries: enzymatic components (i.e. superoxide dismutase, catalase, glutathione peroxidase, glutathione S-transferase) and non-enzymatic, low molecular weight compounds (i.e. glutathione, vitamin A, vitamin C, vitamin E) [3,18,19]. When the balance between ROS production and antioxidant defense mechanisms are disrupted, oxidative stress occurs, causing damage to the DNA, proteins, and lipids.
Approximately 100 different oxidative DNA base lesions can be generated by excessive production of ROS [20-22]. Some of the most common and biologically significant oxidative base modifications include 7,8 dihydro-8-oxoguanine (8-oxoG), thymine glycol (TG), 8-hydroxyadenine, 5-hydroxymethyluracil, 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine (commonly called FaPys), and cyclopurines [23,24]. Depending on their chemical composition, these base lesions can either serve as a miscoding template causing mutagenesis or they can block DNA and RNA polymerase complexes thereby inhibiting DNA replication and transcription. For example, 8-oxoG pairs incorrectly with adenine instead of cytosine, causing G→T transversions and thereby adding to the mutational load [25,26]. On the other hand, the “bulky” adducts such as cyclopurines cause DNA helix distortion and therefore block DNA or RNA polymerase progression [27,28]. As for 5-hydroxymethyluracil, the oxidation of the methyl group of thymine hinders the binding of AP-1 transcription factors to DNA [29,30].
In addition to oxidation of the DNA bases, ROS radicals also cause single-strand DNA breaks (SSBs). It is estimated that oxidative stress generates around 2,300 SSBs per cell per hour [3]. This highly common type of DNA damage involves the breakage of the phosphodiester backbone in one of the DNA strands and often harbors single nucleotide losses or non-conventional damaged termini such as 3’-phosphate, 3’-phosphoglycolate, or 5’-hydroxyl groups. Consequences of such damage are collapse of DNA or RNA polymerase complexes, as well as recombination events during replication. Consequently, if SSBs are not repaired rapidly, they can lead to cell death, chromosomal aberrations, and genetic mutations [23,31]. While oxidized bases are commonly repaired by the base excision repair (BER) mechanism, DNA breaks are repaired by the single strand break repair (SSBR) or double strand break repair (DSBR) pathways [32-34].
Finally, oxidative damage to other cellular macromolecules such as lipids also produce additional forms of DNA damage. In fact, lipid peroxidation (LPO) products are responsible for about 1 adduct per 106-107 DNA bases [35-37]. For instance, malondialdehyde (MDA), generated as one of the final products of polyunsaturated fatty acid peroxidation, can interact with DNA bases to form mutagenic bulky adducts such as pyrimido[1,2α]purin-10(3H)-one (M1G), or induce the generation of a covalent interstrand crosslink. Both of these generated lesions create structural alterations in the DNA molecule, thereby inhibiting replication or transcription machinery of the cells [38,39]. Another major lipid peroxidation-derived aldehyde, 4-hydroxy-2-nonenals (4-HNE), can also react with the DNA to form four diastereomeric 1,N2-γ-hydroxypropano adducts of deoxyguanosine (HNE-dG), preferentially at the mutational hotspot codon 249 of the human p53 gene, and therefore these LPO products have been associated with p53 mutation-related cancers [40,41]. Removal of the different types of LPO-induced adducts from the DNA molecule is achieved by several repair systems, including direct reversal, BER, nucleotide excision repair (NER), and recombination [42].
Alkylating DNA damage
Besides ROS, several other small reactive molecules endogenously present in living cells can cause DNA damage. S-adenosyl-L-methionine (SAM), commonly used as a reactive methyl group donor by methyl transferases, is involved in DNA methylation reactions crucial for gene regulation [43]. However, like any reactive compound, this alkylating agent can spontaneously modify DNA bases to generate harmful adducts. It is estimated that up to 4,000 7-methylguanine, 600 3-methyladenine and 10-30 O6-methylguanine residues are formed per cell per day in mammals. SAM also produces two other minor mutagenic methyl lesions, 3-methylthymine and 3-methylcytosine [44,45]. While SAM is the most prominent type of alkylating agent, other examples of endogenous compounds that induce methylated DNA lesions include nitrosated bile salts, betaine, and choline [46]. Furthermore, alkylating DNA damage can be originated by exogenous sources such as tobacco smoke, environmental pollution, and diet (such as nitrate- or nitrite-containing food) (discussed later in Section II-B-3b) [45,47].
7-methylguanine residues are relatively harmless since they do not cause any alterations in the coding specificity of the guanine base. However, the destabilization of the glycosidic bond due to N-7 substitution on guanine can result in a spontaneous cleavage generating a mutagenic apurinic (AP) site and imidazole ring opening, which interferes with DNA replication [48,49]. Similarly, 3-methyladenine is partly cytotoxic due to its ability to inhibit DNA synthesis [50,51]. In contrast, O6-methylguanine and the related residues O4-methylthymine and O4-ethylthymine residues mispair during DNA replication, causing G→A and T→C transition mutations, respectively. Therefore, if left unrepaired, these methylated DNA bases are a major source of mutagenic and genotoxic lesions [52-54]. Methylated bases are typically repaired by one of two main pathways: direct reversal by O6-methylguanine-DNA methyltransferases and the BER pathway. In cases of abnormal base pairing with other residues, mismatch repair (MMR) may also be triggered [55-58].
Hydrolytic DNA damage
Endogenous DNA damage can also result from hydrolytic processes occurring under physiological conditions. As a molecule within an aqueous milieu, the covalent structure of the DNA is unstable and subject to hydrolytic reactions. DNA hydrolysis can result in spontaneous formation of apurinic/apyrimidinic (AP) sites, or deamination of individual bases [59].
Abasic or AP sites are generated in the DNA when water molecules attack and cleave the N-glycosidic bond between the bases and the sugar phosphate backbone. The generation of these DNA lesions can further be induced by ROS radicals or alkylating agents [60]. AP sites are also produced as an intermediate in the BER pathway when a DNA glycosylase removes the damaged base [61,62]. In humans, abasic sites are one of the most common types of endogenous DNA lesions, with an estimated 10,000 AP sites created per cell per day [63,64]. Because they lack the instructional information, AP sites can cause mutations due to misincorporation of bases (preferentially adenine) opposite the non-coding abasic site during semi-conservative replication [65,66]. Furthermore, they can block DNA or RNA polymerases and inhibit DNA replication and transcription respectively [67]. Due to their unstable nature, abasic sites are also often converted into SSBs from a β-elimination reaction [68,69]. The BER pathway is the major mechanism for the repair of AP sites, while translesion DNA synthesis, also referred to as lesion bypass, is used as a secondary defense mechanism against these abasic lesions [70].
The second type of hydrolytic process that causes endogenous DNA damage is called spontaneous deamination. During this reaction, water molecules attack and replace exocyclic amine groups of the individual bases, converting cytosine, adenine, guanine, and 5-methylcytosine into uracil, hypoxanthine, xanthine, and thymine, respectively [71]. In addition, the rates of base deamination may be stimulated by environmental exposure to UV radiation, intercalating agents, nitrous acid, and sodium bisulfite [72-75]. Cytosine and its homologue 5-methyl cytosine are the preferential targets of hydrolytic deamination. Around 100-500 cytosines are deaminated to uracil per cell each day, whereas 5-methylcytosine is deaminated to thymine 3-4 times more rapidly than cytosine [3,71,76]. Since hydrolytic deamination introduces changes in the coding properties of the original bases, it is considered a major source of spontaneous mutagenesis in humans. For instance, cytosine deamination causes the alteration of original C:G base pairing into a U:A base pair in the first round of replication, which then results in a CG→TA mutation, a feature that underlies the common cancer-associated C/T mutational signature. On the other hand, deamination of 5-methylcytosine results in a G:T base pairing, causing GC→AT transition at the CpG sequences [77-80]. While deaminated cytosine is excised from the DNA by uracil-DNA glycosylase generating an AP site that is efficiently corrected via the BER pathway, the lesion caused by 5-methylcytosine deamination is a substrate for the thymine-DNA glycosylase or the relatively slower mismatch repair (MMR) process [81-84].
Damage from polymerase incorporation errors
Faulty DNA replication is another source of endogenous DNA damage. With every DNA replication, 3×109 nucleotide bases are copied by DNA polymerases in humans. These enzymes also fill the gaps generated during several DNA repair mechanisms such as BER, NER, and MMR [85]. While some DNA polymerases (particularly δ and ε) synthesize DNA at a high fidelity, others copy bases at a lower fidelity during DNA replication or repair processes. It is estimated that despite a highly evolved replication apparatus, base incorporation errors occur at a frequency of 10-6 to 10-8 per cell per generation [86-88]. Replication errors that escape the proofreading activity of replicative polymerases result in inaccurate base substitutions and single base insertion/deletion errors in the newly synthesized DNA strand. If these errors are not quickly and accurately repaired by the MMR pathway, they become incorporated into both strands during the next replication cycle, thereby leading to a change of DNA sequence and mutagenic outcomes [88,89]. Additionally, replicative polymerases can mis-incorporate uracil instead of thymine opposite adenine in the DNA due to alterations in the concentration of the nucleotide pool within the cell’s environment. Although U:A base pairing follows “normal” Watson-Crick geometry and does not cause any distortion, it still requires repair by uracil-DNA glycosylase since the methyl group of thymine is essential to bind with DNA-binding proteins in the major grove of the DNA [90-92]. Finally, replication errors can also accumulate from strand slippage events, particularly in repetitive sequences such as microsatellites (stretches of 2-6 nucleotide repeats), since nearby repetitive bases can stabilize the incorrect base pairing and allow DNA replication to resume, causing insertion and deletions of nucleotides that can potentially change the reading frame [23,93,94]. Worse, replication through microsatellite sequences or transcription-derived R-loops can lead to replicative stress, leading to fork collapse, double-strand DNA breaks (DSBs), and genomic instability [95-98]. Cells have evolved several checkpoint pathways that examine the internal and external cues and respond to potential failures. The S phase checkpoint in particular constitutes the surveillance mechanism that ensures successful DNA replication, preventing genomic instability upon replication stress [99,100].
Exogenous DNA damage
Ionizing radiation
Ionizing radiation, composed of alpha, beta, gamma, neutrons, and X-rays, is considered a major physical agent with DNA-damaging effects and is ubiquitous in our environment. Common sources of exposure to IR involve cosmic rays from outer space, radioactive materials in rocks and soil, and medical devices used for diagnosis and therapy [101,102]. IR can cause damage to the DNA by its direct interaction with the molecule, thereby disrupting the physical structure of the molecule. This type of damage accounts for 30-40% of IR-induced lesions. The remainder of the IR-induced DNA lesions are caused indirectly via radiolysis of water molecules generating free radicals that in turn cause oxidative damage to the DNA [103-105]. Hence, depending on the type and dose of exposure, IR can induce a spectrum of DNA damage lesions ranging from single base alterations (such as oxidation, alkylation, deamination, AP sites) to DNA-protein crosslinks, DNA-DNA crosslinks, SSBs and DSBs. For instance, studies have shown that each Gy of gamma radiation generates approximately 850 pyrimidine lesions, 450 purine lesions, 1,000 SSBs and 20-40 DSBs per cell [106-109].
Because ROS radicals account for about 60-70% of the IR-induced DNA damage, the base lesions produced by IR are, as expected, very similar to those generated by oxidative stress discussed earlier (see Section II-A-1). In contrast, SSBs induced by IR have a unique signature containing 3’ phosphate or 3’-phosphoglycolate ends rather than the usual 3’-OH ends at the DNA breaks. These modified ends are typically processed and repaired by AP endonucleases, polynucleotide kinases/phosphatases, and tyrosyl DNA phosphodiesterases [110-113]. Finally, DSBs, usually induced in clusters by IR, are considered to be the most important and dangerous of these lesions due to their high cytotoxic and mutagenic ability. Generated from multiple damaged sites that are closely positioned on both DNA strands, they lack an undamaged complementary strand that could be used as a template during repair [114,115]. Consequently, these DSBs are usually repaired without a template via non-homologous end-joining (NHEJ), resulting in deletion/insertion mutations as well as chromosomal translocation and rearrangements at the repair junctions. In those cases where they can seek a repair template such as sister chromatids during S/G2 phases or homologous chromosomes, DSBs can also be repaired by homologous recombination (HR). Although this repair mechanism is more accurate, translocations, inversions, deletions, and large-scale loss of heterozygosity can still occur from the associated crossover events [116-119].
Ultraviolet radiation
Exposure to ultraviolet radiation is another common environmental health hazard that causes DNA damage and alters the genomic integrity of an organism. Based on the range of wavelength, UV radiation is classified into three categories: UV-A (315-400 nm), UV-B (290-315 nm), and UV-C (280-100 nm). While UV-C is absorbed by the ozone layer, UV-A and UV-B are able to penetrate the Earth’s atmosphere and therefore comprise the primary damaging components of the solar UV spectrum, posing a great concern to humans [120].
The DNA molecule is most susceptible to UV damage at its relatively flexible areas. In fact, the p53 gene is considered to be one of the hot spots for UV-induced lesions [121]. This non-ionizing radiation causes two major types of DNA lesions, cyclobutane pyrimidine dimers (CPDs), and pyrimidine (6-4)-pyrimidone photoproducts (6-4PPs), at a rate of 105 DNA photolesions per hour in an exposed cell [122]. CPDs are produced when two adjacent pyrimidine bases become covalently linked to create a cyclic ring structure. On the hand, 6-4PPs are generated from a noncyclic covalent bond formed between the 5’ end of C6 and 3’ end of C4 of adjacent pyrimidines via spontaneous rearrangement of the unstable intermediates, oxetane (when the 3’-end is thymine) or azetidine (when the 3’-end is cytosine) [120,123,124]. 6-4PP adducts can also undergo isomerization to their Dewar form following exposure to another light photon from UV-A or UV-B, while reverting back to the conventional 6-4PP structure when exposed to UV-C radiation [125,126]. Although 6-4PPs make up around 1/3rd of the total UV-induced DNA modifications, they have a greater potential to cause mutagenesis [23]. CPDs and 6-4PPs are usually repaired by the specialized UV-induced DNA damage repair system called NER; but if left unrepaired, they result in cytotoxicity and mutagenicity [127]. Both of these bulky dimers distort the structure of the DNA helix, introducing bends that can hinder transcription and replication, requiring translesion polymerases to bypass them, thereby contributing to mutagenic load [128]. The most common mutations induced by pyrimidine dimers include C to T and T to C transversions followed by the characteristic tandem CC to TT transition mutations [129,130]. In addition to its direct effect on pyrimidine bases, UV radiation can also induce modifications to the DNA purine bases but to a very low extent (1×10-5 in native DNA). For instance, photoproducts such as adenine dimer and Pörschke photoproduct (an adenine residue that has undergone photocycloaddition reactions with adjacent adenine or thymine) have been reported following UV-B radiation [131,132]. Furthermore, UV radiation can also cause damage to the DNA indirectly through generation of ROS particles by photosensitizing reactions. These free radicals oxidize the DNA bases such as guanine causing G-T transversions as discussed earlier in the “oxidative DNA damage” section. Other oxidation products induced upon exposure of DNA to UV radiation include 8-oxo-Ade, 2,6-diamino-4-hydroxy-5-formamidoguanine (FapyGua), FapyAde, and oxazolone [120,133,134]. DNA strand breakages and DNA-protein and DNA interstrand crosslinks have also been documented in UV-exposed cells [135-138].
Exogenous DNA-damaging agents
In addition to ionizing radiation and UV radiation, in our daily lives we are exposed to numerous extrinsic DNA-damaging agents that create a massive diversity of DNA adducts. Exogenous sources of these agents include industrial and environmental chemicals, dietary products (including food preservatives and additives), and pharmaceutical agents. Due to the space restriction and scope of this manuscript, we will briefly discuss about the most important extrinsic DNA-damaging agents.
Aromatic amines
Aromatic amines, such as 2-aminofluorene and its acetylated derivative N-acetyl-2-aminofluorene, are found in cigarette smoke, fuel, coal, industrial dyes, synthetic chemical insecticides, and high temperature cooking. These aminofluorenes are activated by the P450 monooxygenase system into carcinogenic ester and sulfate alkylating agents, which attack the C8 position of guanine to cause C8-guanine lesions. If not repaired by the NER pathway, these adducts ultimately lead to base substitutions and frameshift mutations [139-144].
Exogenous alkylating agents
In addition to the endogenous alkylators naturally occurring in the cells (discussed in Section II-A-2), several alkylating agents are produced exogenously from diet components, tobacco smoke, industrial processing, biomass burning, and chemotherapeutic agents. As discussed earlier, alkylating agents react with great affinity to the highly nucleophilic base ring nitrogens and spontaneously modify DNA bases to generate methylated DNA lesions, including modified adenine (at N1, N3, N6 and N7), guanine (N1, N2, N3, N7 and O6), cytosine (N3, N4 and O2), thymine (N3, O2 and O4). Moreover, bifunctional alkylating agents can also cause formation of intrastrand or interstrand crosslinks that adversely impair replication or transcription [3,145-148].
Polycyclic aromatic hydrocarbons
Polycyclic aromatic hydrocarbons (PAHs) are potent atmospheric pollutants present in cigarette smoke, car exhaust fumes, charboiled food, and incomplete combustion of organic matter and fossil fuels. Examples of PAHs include naphthalene, anthracene, pyrene, 1-hydroxypyrene, 1-nitropyrene, dibenzo[a,l]pyrene, and benzo(a)pyrene, with the latter being the most studied [149,150]. Composed of carbon compounds with two or more aromatic rings, PAHs are activated by cytochrome P450 system as well as photo-oxidation, one electron oxidation, multiple ring-oxidation, and nitrogen-reduction pathways, to generate reactive intermediates that cause damage to the DNA [151-153]. Activated benzo(a)pyrene produces the carcinogen (+)-anti-BPDE [(+)- 7,8-hydroxy-9α, 10α-epoxy-7,8,9,10-tetrahydrobenzo(α)pyrene], and the (+)-anti-BPDE and the (-)-anti-BPDE intermediates, which intercalate into the DNA and bind to guanine at the N2 exocyclic position to form BPDE-N2-dG DNA adducts. If left unrepaired by NER and BER pathways, these lesions lead to G/T transversion mutations and harmfully affect replication, resulting in cancer development [154-157]. Indeed, many PAH-related mutation signatures have been reported in the tumor suppressor gene p53 [158-160].
Other reactive electrophiles
There are several other prominent reactive electrophiles that cause DNA damage and induce carcinogenesis. For example, N-nitrosamine, encountered by humans from tobacco smoke and preserved meats, is a potent carcinogen that has been linked with esophageal, gastric, and nasopharyngeal cancers. Formed by a reaction between nitrates or nitrites and certain amines, this compound alkylates N-7 of guanine, leading to destabilization and increased breakage of DNA, thereby causing toxic and mutagenic effects [161-164]. 4-nitroquinoline 1-oxide is another reactive electrophile naturally occurring in the environment, with mutagenic properties in oral carcinogenesis. This compound is activated to 4-acetoxyaminoquinoline 1-oxide which causes the formation of C8-guanine, N2-guanine, and N6-adenine covalent adducts, as well as oxidative DNA lesions such as 8-hydroxyguanine [165-167]. Another remarkable electrophilic compound is the estrogen hormone, commonly used in hormonal replacement therapy. Prolonged use of this hormone has been reported to increase risks of breast cancer due to its DNA damaging nature. Upon its hydroxylation by the P-450 1BI enzyme complex, estrogen is converted into reactive catechol estrogens, which either become oxidized to semiquinones and quinones that react with purines at the N3 and N7 positions, or they generate free radicals, ultimately leading to AP sites and strand breakages [168-171].
Natural toxins
Aflatoxins (B1, B2, G1, G2, and M1), produced by pathogenic fungi such as Aspergillus flavus and Aspergillus parasiticus, have been associated with increased risk of liver cancer [172-174]. Humans are exposed to these genotoxic and carcinogenic compounds from contaminated cereals, oilseeds, spices, tree nuts, and milk products [172]. Following dietary or inhalation exposure, the most prevalent and potent aflatoxin B1 (AFB1) is activated by the P450 complex into aflatoxin B1-8,9-epoxide which alkylates guanine to generate an AFB1-N7-guanine adduct. The latter weakens the glycosidic bond and causes depurination or is further hydrolyzed to form AFB1-formamidopyrimidine, which causes less DNA distortion but still blocks replication with a greater G/T transversion potential [175-177].
Chemotherapeutic agents
Chemotherapeutic agents are intentionally employed in clinical settings to treat patients suffering from different types of cancer. Several of these anti-cancer drugs exert their effect through DNA damage, including cisplatin or cis-diamminedichloroplatinum (II), carboplatin, oxaliplatin, 5-fluorouracil (5-FU), methotrexate, temozolomide, etoposide, and doxorubicin [178]. For instance, upon cell entry, cisplatin is hydrolyzed to an electrophile that attacks nucleophilic centers of the DNA to form interstrand and intrastrand crosslinks, disrupting the structure of the DNA and thereby interfering with DNA replication and transcription [179,180]. On the other hand, 5-FU and methotrexate impair DNA replication through different mechanisms. These anti-metabolites substitute for the natural nucleotides during replication or promote nucleotide pool imbalances that cause arrest in chromosomal duplication [178,181-183]. As for temozolomide, this drug acts as an alkylator and reacts with the DNA to form O6 methylated guanine adducts causing cytotoxic and mutagenic outcomes [184,185]. Finally, agents such as etoposide and doxorubicin act by inhibiting the superhelical relaxation activity of topoisomerases, which leads to protein-DNA adduct formation and DSBs [186-188]. Collectively, the extent of the DNA damage induced by these chemotherapeutic agents overwhelm the cell’s DNA repair ability leading to the activation of cell death responses.
Implications of DNA damage in carcinogenesis
DNA damage is implicated in the development of several human diseases including Alzheimer’s disease, coronary artery disease, diabetes, chronic obstructive pulmonary disease, and cancer [189,190]. The relevance of DNA damage in carcinogenesis became particularly apparent when it was recognized that almost all carcinogenic agents are also mutagenic, causing changes in the DNA sequence [10,191]. Indeed, studies have shown that the mutation rates in the cancer genome increase when cells are exposed to substantial exogenous DNA damaging agents such as radiation, tobacco smoke, and aflatoxins, which are associated with skin, lung, and liver cancers, respectively [192-197]. These DNA aberrations include substitutions, insertions, deletions of small or large fragments of DNA, genomic amplification, and rearrangements [198,199]. While some of these aberrations act as passenger mutations that do not directly drive carcinogenesis, others, defined as driver mutations, play a key role in cancer initiation and progression [199-201].
A multitude of acquired alterations have been identified in protein-coding genes that lead to the activation of oncogenes or to the loss of tumor suppressor functions in various types of cancer. One well-characterized example is the set of TP53 mutations, which are extremely widespread in human sporadic cancers, occurring at rates that range from 38%-50% in ovarian, esophageal, colorectal, head and neck, and lung cancers to about 5% in leukemia, sarcoma, malignant melanoma, and cervical cancer [202]. Missense mutations in this gene give rise to mutant p53 proteins that lose their tumor suppressive functions, which drastically disrupt the nature of the p53 pathway, promoting invasion, metastasis and chemoresistance [203]. RB1, another tumor suppressor gene which plays a key role in regulating cell cycle, is also commonly found to be mutated in various cancers, including retinoblastoma, osteosarcoma, and small cell lung cancer. The loss of function of this gene results in increased cell proliferation and a failure in terminal differentiation [204-206]. Cancer development can also be promoted via mutations that activate proto-oncogenes such as secreted growth factors (e.g. PDGF), cell surface tyrosine kinase receptors (e.g. EGFR), signal transduction G-proteins (e.g. RAS), and nuclear transcription factors (e.g. MYC), leading to the stimulation of cell proliferation and expansion of the transformed cell population [207,208].
Another common class of genomic aberrations in cancer includes chromosomal rearrangements and gene fusions, also resulting in the deregulation of key cellular proteins. One of the most prominent example of reciprocal chromosomal translocation is the t(9;22) Philadelphia translocation observed in chronic myeloid leukemia. This translocation of the proto-oncogene ABL1 located on chromosome 9 to the BCR gene on chromosome 22 results in the BCR-ABL1 fusion gene, which encodes a BCR-ABL1 protein with enhanced tyrosine kinase activity that promotes uncontrolled cell proliferation in the absence of growth factors [209-211]. Another well studied chromosomal translocation is the t(14;18) in follicular lymphoma, which leads to the overexpression of anti-apoptotic protein BCL2, that provides extended survival to B-cells [212,213]. Although gene fusions are most frequently found in hematological malignancies, they have also been identified in epithelial tumors such as fusion of TMPRSS2 to ERG or ETV1 in prostate cancer, EML4-ALK gene fusion in non-small-cell lung cancer, and RAF kinase pathway gene fusion in gastric and prostate cancer [214]. Needless to say, DSBs are prerequisites for these chromosomal translocations since they facilitate the swapping of chromosomal arms between heterologous chromosomes [215].
In addition to carcinogens acting directing on the DNA sequence and causing mutagenesis, DNA repair processes are also involved in the induction of many permanent DNA sequence changes accountable for oncogenesis. Under normal circumstances, DNA repair processes correct the deleterious DNA lesions thereby suppressing mutagenesis. However, when DNA repair mechanisms are defective, genome instability arises, increasing mutation rates and leading to cellular transformation [216-218]. Inhibitions of proteins involved in DNA repair pathways have been linked to increased risks of various cancers. For instance, hereditary non-polyposis colon cancer is caused by defective MMR [219,220], and a large proportion of breast and ovarian cancers are caused by mutations in the BRCA1 and BRCA2 genes, which are crucial for the process of DSB repair by HR [221-223]. Other inherited human diseases of DNA repair with cancer susceptibility syndromes include Xeroderma pigmentosum, Ataxia-telangiectasia, Bloom’s and Werner’s syndromes, and Li-Fraumeni-syndrome [8].
According to the multistep model of cancer development, a lifetime exposure to endogenous and exogenous DNA damaging agents accounts for the progressive accumulation of driver mutations that give rise to a clonal cell population with an advantage in proliferation [224,225]. Indeed, unlike the non-cancerous somatic cells where genomic integrity is well maintained and cell divisions are strictly controlled, the pre-cancerous cells are prone to a high frequency of genomic changes and alterations due to defects in the regulation of surveillance mechanisms, as well as in the DNA damage checkpoint, DNA repair machinery and mitotic checkpoint pathways [226]. This process, defined as genomic instability, is an enabling characteristic and a driving force of tumorigenesis [227,228]. Ranging from single nucleotide changes to gross chromosomal alterations, genomic instability is classified into 3 types based on the level of disruption. Nucleotide instability is the increased frequency of a single or a few nucleotide changes (i.e., insertions, deletions, and substitutions); microsatellite instability includes deletions or expansion of short nucleotide repeats called microsatellites; and the most prominent form of genomic instability, chromosomal instability, is characterized by chromosomal abnormalities such as aneuploidy, amplifications, deletions, translocations, and inversions [229,230].
As mentioned earlier, in hereditary cancers or cancer-predisposing syndromes, including hereditary breast and ovarian cancer, hereditary non-polyposis colon cancer, xeroderma pigmentosum, and MYH-associated polyposis, genomic instability is caused by mutations in DNA repair genes, supporting the mutator phenotype hypothesis [231]. The latter is based on the idea that genomic instability, present in precancerous cells due to mutations in caretaker genes that maintain genomic stability, drives tumorigenesis by increasing the rate of spon0taneous mutations [232-234]. On the other hand, genomic instability in sporadic cancers is supported by the oncogene-induced DNA replication stress model in which mutations in oncogenes, checkpoint regulating genes, and tumor suppressors that drive proliferation are responsible for DNA replication stress causing DNA damage and genomic instability in the early cancer stages [235-237]. In any case, unrepaired DNA damage therefore plays a culprit role in cancer development and aggressiveness due to the detrimental consequences of genetic alterations and genomic instability. In the next section, we discuss how the DNA damage response mechanisms activated by DNA lesions are exploited by oncogenic DNA viruses to promote viral integration and cause carcinogenesis.
Role of DNA damage in DNA oncovirus integration and carcinogenesis
Approximately 20%-25% of all human cancers is caused by DNA oncogenic viruses such as HBV, MCV, and HPV [238]. However, infection with these oncogenic viruses does not necessarily cause cancer development. In fact, progression to cancer rarely occurs following infection with an oncogenic virus [239,240]. Several mechanisms underlying the ability of DNA oncogenic viruses to cause cellular transformation have been reported, with unrepaired DNA damage being the most common and overlapping feature of these oncoviruses to promote tumor development [12,241]. During viral infections, the host cells generally activate their surveillance mechanisms to detect and repair DNA damage in order to maintain genomic integrity. Many viruses also stimulate DNA damage signaling pathways, either directly by virus infection itself or indirectly via the expression of viral proteins, to ensure an S-phase-like replication environment, preventing apoptosis and promoting episomal maintenance [242]. However, when DNA damage accumulates in the host genome and the activated DNA damage repair system is not sufficient to repair the lesions, one side-effect is the integration of the viral episome into the host genome, which can trigger cancer development in the infected cells.
Integration of the viral genome is a crucial event in the process of malignant transformation for several oncogenic viruses. For example, integration of the HBV episome into the genome of hepatocytes has been reported in over 85%-90% of HBV-related hepatocellular carcinoma cases [243-245]. HBV integration leads to overexpression of cellular cancer-related genes, such as telomerase reverse transcriptase (TERT), mixed-lineage leukemia 4 (MLL4), and CCNE1 encoding cyclin E1 [246]. Furthermore, integrated HBV sequences encoding HBx and/or truncated envelope pre-S2/S proteins induce genomic instability and cell cycle deregulation by interfering with NER, repressing p53-mediated gene transcription and inactivating p53-dependent tumor suppression, as well as transactivating several cellular genes and signaling pathways linked to carcinogenesis [247-252]. Overall, HBV integration promotes further genetic alterations such as chromosomal deletions, translocations, fusion of transcripts and amplification of DNA, which lead to activation of oncogenes and depletion of tumor suppressor genes, promoting the development of hepatocellular carcinoma [253,254].
While HBV integration contributes to carcinogenesis by stimulating both cellular and viral responses, integration of HPV into the host genome solely activates viral mechanisms that promote malignant transformation. Integration of HPV into the host genome typically results in the disruption and loss of the viral negative regulator E2, allowing persistent over-expression of the two viral oncoproteins, E6 and E7 [255-258]. The E6 oncoprotein causes evasion of apoptosis and perturbation of cell cycle control by forming a complex with p53 and targeting it to proteasomal degradation. On the other hand, the E7 oncoprotein stimulates unrestrained replication and cell division by binding to and stimulating the degradation of Rb, leading to the inappropriate release of the E2F transcription factor. Therefore, when these viral oncoproteins are over-expressed following viral integration, the HPV-infected cells undergo uncontrolled cellular proliferation and survival, and consequently develop HPV-induced malignancies [259-261]. It is estimated that about 90% of HPV-positive cervical cancer cases contain HPV integrated into the host genome, supporting the idea that development of cervical cancer in HPV-infected women is tightly linked to the integration status of the virus [262-264]. The frequency of HPV integration in other anogenital cancers is not as well documented, with one study reporting integrated HPV in nearly 80% of anal cancer cases [265]. In the case of HPV-positive head and neck carcinomas, viral integration has been detected at lower rates, with many tumors having either episomal or mixed episomal and integrated viral DNA [266-268].
As for MCV, integration of this viral episome into the human genome has been noted in 70%-80% of Merkel cell carcinoma tumors [269,270]. While the exact role of MCV integration in Merkel cell carcinoma development requires further study, it has been documented that the integrated viral genome almost always contains mutations that truncate the C-terminal DNA binding and helicase domains of large tumor (T) antigen, preventing auto-activation of integrated virus replication, which would be detrimental for cell survival [271-273].
Although the mechanism of these viral integrations has not been well defined, none of these DNA oncoviruses encode genes that produce integrase enzymatic activity proteins similar to those encoded by the human immunodeficiency virus [274]. Instead, the integrative process of these oncogenic viruses is thought to be linked to the extent of DNA damage existing in the host cells. According to this model, viral integration requires DSBs in both the host DNA and in the circular viral episome, following which the recruitment of DNA damage repair complexes ensures the accessibility of ligases that can reconnect the recombined host and viral sequences, creating the perfect microenvironment for viral integration [12,275]. While DSBs can be directly caused by exogenous DNA damaging agents and IR as discussed earlier, spontaneous DSBs also occur at a rate of 1 DSB per 108 bp during normal cellular processes [276]. Furthermore, it has been reported that about 1% of single strand DNA lesions such as SSBs, AP sites, oxidation products 8-oxoG and TG, and alkylation products, are converted to 50 DSBs per cell per cell cycle during the S phase (a rate of 1 DSB per 108 bp) [277]. Indeed, if left unresolved, SSBs cause the collapse of a replication fork, leading to DSB formation; while unrepaired base damages in the template strand cause stalling of replication fork, leading to the formation of nicks and gaps in DNA that generate DSBs (reviewed in [116]). Consequently, these simultaneous breaks in the phosphate backbones of the two complementary DNA strands serve as integration sites for oncogenic DNA viruses (Figure 1). In support of this hypothesis, environmental conditions known to cause DNA damage have been epidemiologically associated with increased incidence of viral-induced carcinogenesis. For instance, exposure to UV and possibly ionizing radiation is a risk factor for MCV-induced Merkel cell carcinoma [278-280]. In the case of HBV-related hepatocellular carcinoma, environmental risk factors include aflatoxin B1 exposure, alcohol consumption and smoking [281-286]. As for HPV-mediated malignancies, smoking, long term use of oral contraceptives, high parity, inflammation, and co-infections with STD-associated pathogens Chlamydia trachomatis and Neisseria gonorrhoeae are considered as non-viral risk factors [287-294]. Several publications have also reported increased oxidative stress and subsequent DNA damage in patients with cervical carcinoma compared to normal population (reviewed in [295]). In addition to these observational and epidemiological studies, some pieces of evidence have also mechanistically linked DNA damage to a higher integration frequency of these oncogenic DNA viruses. For example, the integration frequency of HBV into the host genome has been shown to be increased in the presence of DNA strand breaks induced by oxidative stress [296]. In the case of HPV, Winder et al. (2007) have demonstrated that generation of DSBs due to Ku70 depletion in cervical keratinocyte cell line W12 containing HPV16 episomes results in the integration of viral DNA into the host genome [297]. Our lab has also shown that chronic oxidative stress, induced either by the exogenous agent L-Buthionine-sulfoximine or by the HPV E6* protein, increases the frequency of integration of HPV16 into the genome of cervical keratinocytes [275]. Furthermore, the activity of DNA-dependent protein kinase, an important enzyme involved in DSB repair via the NHEJ pathway, has been found to be significantly lower in patients with cervical cancer, compared to a healthy population [298]. It was recently demonstrated that this disruption of the NHEJ pathway is mediated by the HPV16 E7 oncoprotein and often results in a reciprocal increase in microhomology-mediated end-joining [299]. Supporting this observation, integration breakpoints in cervical and oropharyngeal cancers have been found to be enriched with micro-homologous sequence between the HPV and human genomes, indicating the involvement of microhomology-mediated DNA repair pathways in the process of HPV integration [266,300]. Collectively, these data support the idea that DNA lesions can serve as sites for viral integration and that inducing DNA damage dramatically increases viral integration frequency.
Figure 1.
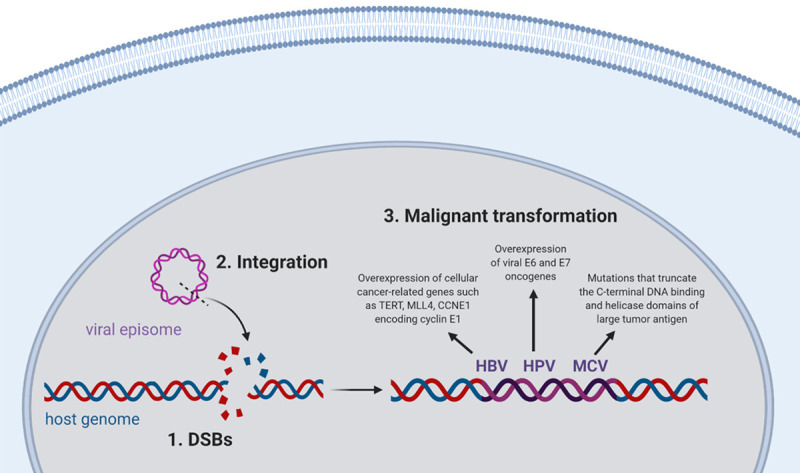
DNA lesions serve as integration sites for oncogenic DNA viruses and promote malignant transformation of the infected cells. Created with BioRender.com.
Conclusions
To summarize, the DNA molecule is subject to continuous damage from a combination of endogenous and exogenous sources. Endogenous DNA lesions are caused by cellular metabolic and physiological processes such as oxidation, hydrolysis, alkylation, and polymerase incorporation errors, whereas exogenous sources of DNA damage include exposure to environmental factors such as IR, UV radiation, chemical agents (aromatic amines, alkylating agents, PAHs, reactive electrophiles, aflatoxins) as well as chemotherapeutic agents. When left unrepaired, DNA damage accumulates in the cells and gives rise to mutations that change the function of important genes (i.e. activation of oncogenes, loss of tumor suppressor functions), or causes chromosomal rearrangements and gene fusions that also result in the deregulation of key cellular proteins. Progressive acquisition of such genetic mutations promotes uncontrolled cell proliferation and evasion of cell death, and hence plays a key role in cancer initiation and progression. Another less-studied consequence of DNA damage accumulating in the host genome is the integration of oncogenic DNA viruses. This critical step of malignant transformation by HBV, MCV and HPV is thought to be particularly facilitated by DSBs in both viral and host genome. Therefore, the impact of DNA damage on carcinogenesis is magnified in the case of these oncoviruses via the additional effect of increasing integration frequency.
In this paper, we also reviewed the limited data connecting DNA damage and repair mechanisms with viral oncogenesis through viral integration. As discussed, several epidemiological pieces of evidence point in support of the idea that DNA damaging agents are risk factors for viral integration and subsequent carcinogenesis, yet mechanistically only a few studies have examined the connection of these DNA damaging agents with viral oncogenesis. Therefore, a critical evaluation should be undertaken to further assess the etiology of virus-mediated carcinogenesis and identify the DNA damage pathways involved in the progression from viral infection to cancer development. In particular, the role of these non-viral factors in viral integration needs to be more clearly elucidated, with the ultimate goal of reducing or eradicating these viral-mediated malignancies that account for about 20% of human cancers worldwide. Indeed, demonstrating that a certain DNA damaging agent increases the likelihood of viral-host integration and magnifies the process of carcinogenesis will enable the development of both preventative and therapeutic strategies designed specifically to intercept the critical step of malignant transformation by the oncogenic DNA viruses. For instance, the current epidemiological data support the implementation of precautionary measures such as HBV vaccination programs in regions with high AFB1 exposure as well as abstinence from smoking and alcohol to prevent hepatocellular carcinoma, or cessation of smoking and limitation of oral contraceptive for HPV management. More importantly, research on the molecular mechanisms involved in viral integration will allow the development of more effective treatment approaches to eradicate virus-based carcinogenesis. Of particular interest, molecules such as poly(ADP-ribose) polymerase-1 (PARP-1) that protect DNA strand breaks and act as anti-recombinogenic factors need to be the focus of future studies, since these guardians of genomic integrity have the potential to prevent viral integration [296].
Disclosure of conflict of interest
None.
References
- 1.Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168:644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 3.Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58:235–263. doi: 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saul RL, Ames BN. Background levels of DNA damage in the population. Basic Life Sci. 1986;38:529–535. doi: 10.1007/978-1-4615-9462-8_55. [DOI] [PubMed] [Google Scholar]
- 5.Martin LJ. DNA damage and repair: relevance to mechanisms of neurodegeneration. J Neuropathol Exp Neurol. 2008;67:377–387. doi: 10.1097/NEN.0b013e31816ff780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eker AP, Quayle C, Chaves I, van der Horst GT. DNA repair in mammalian cells: direct DNA damage reversal: elegant solutions for nasty problems. Cell Mol Life Sci. 2009;66:968–980. doi: 10.1007/s00018-009-8735-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bębenek A, Ziuzia-Graczyk I. Fidelity of DNA replication-a matter of proofreading. Curr Genet. 2018;64:985–996. doi: 10.1007/s00294-018-0820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiesmüller L, Ford JM, Schiestl RH. DNA damage, Repair, and diseases. J Biomed Biotechnol. 2002;2:45. doi: 10.1155/S1110724302001985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nature Reviews Cancer. 2016;16:35–42. doi: 10.1038/nrc.2015.4. [DOI] [PubMed] [Google Scholar]
- 11.Williams VM, Filippova M, Soto U, Duerksen-Hughes PJ. HPV-DNA integration and carcinogenesis: putative roles for inflammation and oxidative stress. Future Virology. 2010;6:45–57. doi: 10.2217/fvl.10.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Williams V, Filippova M, Filippov V, Duerksen-Hughes P. Viral carcinogenesis: factors inducing DNA damage and virus integration. Cancers (Basel) 2014;6:2155–2186. doi: 10.3390/cancers6042155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henle ES, Linn S. Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. 1997;272:19095–19098. doi: 10.1074/jbc.272.31.19095. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94:329–354. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, Bitto A. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763. doi: 10.1155/2017/8416763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 17.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 18.Matés JM, Pérez-Gómez C, Núñez de Castro I. Antioxidant enzymes and human diseases. Clin Biochem. 1999;32:595–603. doi: 10.1016/s0009-9120(99)00075-2. [DOI] [PubMed] [Google Scholar]
- 19.Matés JM, Sánchez-Jiménez F. Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci. 1999;4:D339–345. doi: 10.2741/mates. [DOI] [PubMed] [Google Scholar]
- 20.Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;53:137–180. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free Radic Biol Med. 1991;10:225–242. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- 22.Cadet J, Douki T, Ravanat JL. Oxidatively generated base damage to cellular DNA. Free Radic Biol Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 23.Tiwari V, Wilson DM. DNA damage and associated DNA repair defects in disease and premature aging. Am J Hum Genet. 2019;105:237–257. doi: 10.1016/j.ajhg.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takao K. Aging and the accumulation of oxidative damage to DNA. J Clin Biochem Nutr. 2004;34:51–60. [Google Scholar]
- 25.Fleming AM, Burrows CJ. 8-Oxo-7,8-dihydroguanine, friend and foe: epigenetic-like regulator versus initiator of mutagenesis. DNA Repair (Amst) 2017;56:75–83. doi: 10.1016/j.dnarep.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakabeppu Y. Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int J Mol Sci. 2014;15:12543–12557. doi: 10.3390/ijms150712543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks PJ. The cyclopurine deoxynucleosides: DNA repair, biological effects, mechanistic insights, and unanswered questions. Free Radic Biol Med. 2017;107:90–100. doi: 10.1016/j.freeradbiomed.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 28.Jaruga P, Dizdaroglu M. 8,5’-Cyclopurine-2’-deoxynucleosides in DNA: mechanisms of formation, measurement, repair and biological effects. DNA Repair. 2008;7:1413–1425. doi: 10.1016/j.dnarep.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Rogstad DK, Liu P, Burdzy A, Lin SS, Sowers LC. Endogenous DNA lesions can inhibit the binding of the AP-1 (c-Jun) transcription factor. Biochemistry. 2002;41:8093–8102. doi: 10.1021/bi012180a. [DOI] [PubMed] [Google Scholar]
- 30.Berney M, McGouran JF. Methods for detection of cytosine and thymine modifications in DNA. Nat Rev Chem. 2018;2:332–348. [Google Scholar]
- 31.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 32.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 33.Abbotts R, Wilson DM 3rd. Coordination of DNA single strand break repair. Free Radic Biol Med. 2017;107:228–244. doi: 10.1016/j.freeradbiomed.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blair IA. DNA adducts with lipid peroxidation products. J Biol Chem. 2008;283:15545–15549. doi: 10.1074/jbc.R700051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gentile F, Arcaro A, Pizzimenti S, Daga M, Cetrangolo GP, Dianzani C, Lepore A, Graf M, Ames PRJ, Barrera G. DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017;4:103–137. doi: 10.3934/genet.2017.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Łuczaj W, Skrzydlewska E. DNA damage caused by lipid peroxidation products. Cell Mol Biol Lett. 2003;8:391–413. [PubMed] [Google Scholar]
- 38.Marnett LJ. Lipid peroxidation-DNA damage by malondialdehyde. Mutat Res. 1999;424:83–95. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- 39.Jeong YC, Nakamura J, Upton PB, Swenberg JA. Pyrimido[1,2-a] -purin-10(3H)-one, M1G, is less prone to artifact than base oxidation. Nucleic Acids Res. 2005;33:6426–6434. doi: 10.1093/nar/gki944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong H, Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol. 2015;4:193–199. doi: 10.1016/j.redox.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choudhury S, Pan J, Amin S, Chung FL, Roy R. Repair kinetics of trans-4-hydroxynonenal-induced cyclic 1, N2-propanodeoxyguanine DNA adducts by human cell nuclear extracts. Biochemistry. 2004;43:7514–7521. doi: 10.1021/bi049877r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winczura A, Zdżalik D, Tudek B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radic Res. 2012;46:442–459. doi: 10.3109/10715762.2012.658516. [DOI] [PubMed] [Google Scholar]
- 43.Lin H. S-Adenosylmethionine-dependent alkylation reactions: when are radical reactions used? Bioorg Chem. 2011;39:161–170. doi: 10.1016/j.bioorg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rydberg B, Lindahl T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982;1:211–216. doi: 10.1002/j.1460-2075.1982.tb01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 46.O’Driscoll M, Macpherson P, Xu YZ, Karran P. The cytotoxicity of DNA carboxymethylation and methylation by the model carboxymethylating agent azaserine in human cells. Carcinogenesis. 1999;20:1855–1862. doi: 10.1093/carcin/20.9.1855. [DOI] [PubMed] [Google Scholar]
- 47.Pegg AE. DNA repair and carcinogenesis by alkylating agents. In: Cooper CS, Grover PL, editors. Chemical Carcinogenesis and Mutagenesis II. Berlin, Heidelberg: Springer Berlin Heidelberg; 1990. pp. 103–131. [Google Scholar]
- 48.Barbarella G, Tugnoli V, Zambianchi M. Imidazole ring opening of 7-methylguanosine at physiological pH. Nucleosides & Nucleotides. 1991;10:1759–1769. [Google Scholar]
- 49.Tudek B, Boiteux S, Laval J. Biological properties of imidazole ring-opened N7-methylguanine in M13mp18 phage DNA. Nucleic Acids Res. 1992;20:3079–3084. doi: 10.1093/nar/20.12.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Settles S, Wang RW, Fronza G, Gold B. Effect of N3-Methyladenine and an isosteric stable analogue on DNA polymerization. J Nucleic Acids. 2010;2010:426505. doi: 10.4061/2010/426505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, Woodgate R. Eukaryotic Y-family polymerases bypass a 3-methyl-2’-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Research. 2008;36:2152–2162. doi: 10.1093/nar/gkn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerchman LL, Ludlum DB. The properties of O6-methylguanine in templates for RNA polymerase. Biochimica et Biophysica Acta (BBA)-Nucleic Biochim Biophys Acta. 1973;308:310–316. doi: 10.1016/0005-2787(73)90160-3. [DOI] [PubMed] [Google Scholar]
- 53.Abbott PJ, Saffhill R. DNA synthesis with methylated poly(dC-dG) templates. Evidence for a competitive nature to miscoding by O6-methylguanine. Biochim Biophys Acta. 1979;562:51–61. doi: 10.1016/0005-2787(79)90125-4. [DOI] [PubMed] [Google Scholar]
- 54.Ezerskyte M, Paredes JA, Malvezzi S, Burns JA, Margison GP, Olsson M, Scicchitano DA, Dreij K. O6-methylguanine-induced transcriptional mutagenesis reduces p53 tumor-suppressor function. Proc Natl Acad Sci U S A. 2018;115:4731–4736. doi: 10.1073/pnas.1721764115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zak P, Kleibl K, Laval F. Repair of O6-methylguanine and O4-methylthymine by the human and rat O6-methylguanine-DNA methyltransferases. J Biol Chem. 1994;269:730–733. [PubMed] [Google Scholar]
- 56.Ye N, Holmquist GP, O’Connor TR. Heterogeneous repair of N-methylpurines at the nucleotide level in normal human cells. J Mol Biol. 1998;284:269–285. doi: 10.1006/jmbi.1998.2138. [DOI] [PubMed] [Google Scholar]
- 57.Huang JC, Hsu DS, Kazantsev A, Sancar A. Substrate spectrum of human excinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc Natl Acad Sci U S A. 1994;91:12213–12217. doi: 10.1073/pnas.91.25.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci U S A. 1993;90:6424–6428. doi: 10.1073/pnas.90.14.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shapiro R. Damage to DNA Caused by Hydrolysis. In: Seeberg E, Kleppe K, editors. Chromosome Damage and Repair. New York, NY: Springer US; 1981. pp. 3–18. [Google Scholar]
- 60.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59:2522. [PubMed] [Google Scholar]
- 61.Nilsen L, Forstrøm RJ, Bjørås M, Alseth I. AP endonuclease independent repair of abasic sites in Schizosaccharomyces pombe. Nucleic Acids Res. 2012;40:2000–2009. doi: 10.1093/nar/gkr933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim YJ, Wilson DM 3rd. Overview of base excision repair biochemistry. Curr Mol Pharmacol. 2012;5:3–13. doi: 10.2174/1874467211205010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu ZJ, Martínez Cuesta S, van Delft P, Balasubramanian S. Sequencing abasic sites in DNA at single-nucleotide resolution. Nature Chemistry. 2019;11:629–637. doi: 10.1038/s41557-019-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 65.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 66.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 67.Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in saccharomyces cerevisiae. DNA Repair (Amst) 2004;3:1–12. doi: 10.1016/j.dnarep.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 68.Simonelli V, Narciso L, Dogliotti E, Fortini P. Base excision repair intermediates are mutagenic in mammalian cells. Nucleic Acids Res. 2005;33:4404–4411. doi: 10.1093/nar/gki749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bailly V, Verly WG. Possible roles of beta-elimination and delta-elimination reactions in the repair of DNA containing AP (apurinic/apyrimidinic) sites in mammalian cells. Biochem J. 1988;253:553–559. doi: 10.1042/bj2530553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Otterlei M, Kavli B, Standal R, Skjelbred C, Bharati S, Krokan HE. Repair of chromosomal abasic sites in vivo involves at least three different repair pathways. EMBO J. 2000;19:5542–5551. doi: 10.1093/emboj/19.20.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yonekura S, Nakamura N, Yonei S, Zhang-Akiyama QM. Generation, biological consequences and repair mechanisms of cytosine deamination in DNA. J Radiat Res. 2009;50:19–26. doi: 10.1269/jrr.08080. [DOI] [PubMed] [Google Scholar]
- 72.Peng W, Shaw BR. Accelerated deamination of cytosine residues in UV-induced cyclobutane pyrimidine dimers leads to CC-->TT transitions. Biochemistry. 1996;35:10172–10181. doi: 10.1021/bi960001x. [DOI] [PubMed] [Google Scholar]
- 73.Moyer R, Briley D, Johnsen A, Stewart U, Shaw BR. Echinomycin, a bis-intercalating agent, induces C-->T mutations via cytosine deamination. Mutat Res. 1993;288:291–300. doi: 10.1016/0027-5107(93)90097-y. [DOI] [PubMed] [Google Scholar]
- 74.Hayatsu H. Discovery of bisulfite-mediated cytosine conversion to uracil, the key reaction for DNA methylation analysis--a personal account. Proceedings of the Japan Academy. Proc Jpn Acad Ser B Phys Biol Sci. 2008;84:321–330. doi: 10.2183/pjab/84.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frankel AD, Duncan BK, Hartman PE. Nitrous acid damage to duplex deoxyribonucleic acid: distinction between deamination of cytosine residues and a novel mutational lesion. J Bacteriol. 1980;142:335–338. doi: 10.1128/jb.142.1.335-338.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 77.Sassa A, Kanemaru Y, Kamoshita N, Honma M, Yasui M. Mutagenic consequences of cytosine alterations site-specifically embedded in the human genome. Genes Environ. 2016;38:17–17. doi: 10.1186/s41021-016-0045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen JC, Rideout WM 3rd, Jones PA. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994;22:972–976. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schärer OD, Kawate T, Gallinari P, Jiricny J, Verdine GL. Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc Natl Acad Sci U S A. 1997;94:4878–4883. doi: 10.1073/pnas.94.10.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu X, Meng FL. Generation of genomic alteration from cytidine deamination. In: Zhang Y, editor. Chromosome Translocation. Singapore: Springer Singapore; 2018. pp. 49–64. [DOI] [PubMed] [Google Scholar]
- 81.Waters TR, Swann PF. Kinetics of the action of thymine DNA glycosylase. J Biol Chem. 1998;273:20007–20014. doi: 10.1074/jbc.273.32.20007. [DOI] [PubMed] [Google Scholar]
- 82.Wiebauer K, Jiricny J. Mismatch-specific thymine DNA glycosylase and DNA polymerase beta mediate the correction of G.T mispairs in nuclear extracts from human cells. Proc Natl Acad Sci U S A. 1990;87:5842. doi: 10.1073/pnas.87.15.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krokan HE, Drabløs F, Slupphaug G. Uracil in DNA - occurrence, consequences and repair. Oncogene. 2002;21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 84.Drohat AC, Coey CT. Role of base excision “repair” enzymes in erasing epigenetic marks from DNA. Chem Rev. 2016;116:12711–12729. doi: 10.1021/acs.chemrev.6b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pavlov YI, Shcherbakova PV, Rogozin IB. Roles of DNA polymerases in replication, repair, and recombination in eukaryotes. In: Jeon KW, editor. International Review of Cytology. Academic Press; 2006. pp. 41–132. [DOI] [PubMed] [Google Scholar]
- 86.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 88.Kunkel TA. Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol. 2009;74:91–101. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Preston BD, Albertson TM, Herr AJ. DNA replication fidelity and cancer. Semin Cancer Biol. 2010;20:281–293. doi: 10.1016/j.semcancer.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andersen S, Heine T, Sneve R, König I, Krokan HE, Epe B, Nilsen H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis. 2005;26:547–555. doi: 10.1093/carcin/bgh347. [DOI] [PubMed] [Google Scholar]
- 91.Vértessy BG, Tóth J. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc Chem Res. 2009;42:97–106. doi: 10.1021/ar800114w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, Chabes A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011;39:1360–1371. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Viguera E, Canceill D, Ehrlich SD. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001;20:2587–2595. doi: 10.1093/emboj/20.10.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hile SE, Eckert KA. DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Res. 2007;36:688–696. doi: 10.1093/nar/gkm1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kaushal S, Freudenreich CH. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer. 2019;58:270–283. doi: 10.1002/gcc.22721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Patel DR, Weiss RS. A tough row to hoe: when replication forks encounter DNA damage. Biochem Soc Trans. 2018;46:1643–1651. doi: 10.1042/BST20180308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- 98.Richard P, Manley JL. R loops and links to human disease. J Mol Biol. 2017;429:3168–3180. doi: 10.1016/j.jmb.2016.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hustedt N, Gasser SM, Shimada K. Replication checkpoint: tuning and coordination of replication forks in s phase. Genes. 2013;4:388–434. doi: 10.3390/genes4030388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goldman M. Ionizing radiation and its risks. West J Med. 1982;137:540–547. [PMC free article] [PubMed] [Google Scholar]
- 102.Donya M, Radford M, ElGuindy A, Firmin D, Yacoub MH. Radiation in medicine: origins, risks and aspirations. Glob Cardiol Sci Pract. 2014;2014:437–448. doi: 10.5339/gcsp.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Desouky O, Ding N, Zhou G. Targeted and non-targeted effects of ionizing radiation. J Radiat Res. 2015;8:247–254. [Google Scholar]
- 104.Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327:48–60. doi: 10.1016/j.canlet.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Santivasi WL, Xia F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid Redox Signal. 2013;21:251–259. doi: 10.1089/ars.2013.5668. [DOI] [PubMed] [Google Scholar]
- 106.Reynolds P, Botchway SW, Parker AW, O’Neill P. Spatiotemporal dynamics of DNA repair proteins following laser microbeam induced DNA damage - when is a DSB not a DSB? Mutat Res. 2013;756:14–20. doi: 10.1016/j.mrgentox.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leatherbarrow EL, Harper JV, Cucinotta FA, O’Neill P. Induction and quantification of gamma-H2AX foci following low and high LET-irradiation. Int J Radiat Biol. 2006;82:111–118. doi: 10.1080/09553000600599783. [DOI] [PubMed] [Google Scholar]
- 108.Cadet J, Douki T, Ravanat JL. Oxidatively generated damage to the guanine moiety of DNA: mechanistic aspects and formation in cells. Acc Chem Res. 2008;41:1075–1083. doi: 10.1021/ar700245e. [DOI] [PubMed] [Google Scholar]
- 109.Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 110.Henner WD, Grunberg SM, Haseltine WA. Sites and structure of gamma radiation-induced DNA strand breaks. J Biol Chem. 1982;257:11750–11754. [PubMed] [Google Scholar]
- 111.Henner WD, Rodriguez LO, Hecht SM, Haseltine WA. gamma Ray induced deoxyribonucleic acid strand breaks. 3’ Glycolate termini. J Biol Chem. 1983;258:711–713. [PubMed] [Google Scholar]
- 112.Price A. The repair of ionising radiation-induced damage to DNA. Semin Cancer Biol. 1993;4:61–71. [PubMed] [Google Scholar]
- 113.El-Khamisy SF, Hartsuiker E, Caldecott KW. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst) 2007;6:1485–1495. doi: 10.1016/j.dnarep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 114.Sutherland BM, Bennett PV, Sidorkina O, Laval J. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc Natl Acad Sci U S A. 2000;97:103–108. doi: 10.1073/pnas.97.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nickoloff JA, Sharma N, Taylor L. Clustered DNA double-strand breaks: biological effects and relevance to cancer radiotherapy. Genes. 2020;11:99. doi: 10.3390/genes11010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cannan WJ, Pederson DS. Mechanisms and consequences of double-strand DNA break formation in chromatin. J Cell Physiol. 2016;231:3–14. doi: 10.1002/jcp.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kavanagh JN, Redmond KM, Schettino G, Prise KM. DNA double strand break repair: a radiation perspective. Antioxid Redox Signal. 2013;18:2458–2472. doi: 10.1089/ars.2012.5151. [DOI] [PubMed] [Google Scholar]
- 118.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rodgers K, McVey M. Error-prone repair of DNA double-strand breaks. J Cell Physiol. 2016;231:15–24. doi: 10.1002/jcp.25053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rastogi RP, Richa , Kumar A, Tyagi MB, Sinha RP. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980. doi: 10.4061/2010/592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Benjamin CL, Ullrich SE, Kripke ML, Ananthaswamy HN. p53 tumor suppressor gene: a critical molecular target for UV induction and prevention of skin cancer†. Photochem Photobiol. 2008;84:55–62. doi: 10.1111/j.1751-1097.2007.00213.x. [DOI] [PubMed] [Google Scholar]
- 122.Cadet J, Douki T. Formation of UV-induced DNA damage contributing to skin cancer development. Photochem Photobiol Sci. 2018;17:1816–1841. doi: 10.1039/c7pp00395a. [DOI] [PubMed] [Google Scholar]
- 123.Mullenders LHF. Solar UV damage to cellular DNA: from mechanisms to biological effects. Photochem Photobiol Sci. 2018;17:1842–1852. doi: 10.1039/c8pp00182k. [DOI] [PubMed] [Google Scholar]
- 124.de Lima-Bessa KM, Armelini MG, Chiganças V, Jacysyn JF, Amarante-Mendes GP, Sarasin A, Menck CF. CPDs and 6-4PPs play different roles in UV-induced cell death in normal and NER-deficient human cells. DNA Repair (Amst) 2008;7:303–312. doi: 10.1016/j.dnarep.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 125.Taylor JS, Lu HF, Kotyk JJ. Quantitative conversion of the (6-4) photoproduct of tpdc to its dewar valence isomer upon exposure to simulated sunlight. Photochem Photobiol. 1990;51:161–167. doi: 10.1111/j.1751-1097.1990.tb01698.x. [DOI] [PubMed] [Google Scholar]
- 126.Mitchell DL, Jen J, Cleaver JE. Relative induction of cyclobutane dimers and cytosine photohydrates in dna irradiated in vitro and in vivo with ultraviolet-C and ultraviolet-B light. Photochem Photobiol. 1991;54:741–746. doi: 10.1111/j.1751-1097.1991.tb02084.x. [DOI] [PubMed] [Google Scholar]
- 127.Yu SL, Lee SK. Ultraviolet radiation: DNA damage, repair, and human disorders. Molecular and Cellular Toxicology. 2017;13:21–28. [Google Scholar]
- 128.Yang W. Surviving the sun: repair and bypass of DNA UV lesions. Protein Sci. 2011;20:1781–1789. doi: 10.1002/pro.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brash DE. UV signature mutations. Photochem Photobiol. 2015;91:15–26. doi: 10.1111/php.12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rochette PJ, Therrien JP, Drouin R, Perdiz D, Bastien N, Drobetsky EA, Sage E. UVA-induced cyclobutane pyrimidine dimers form predominantly at thymine-thymine dipyrimidines and correlate with the mutation spectrum in rodent cells. Nucleic Acids Res. 2003;31:2786–2794. doi: 10.1093/nar/gkg402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Duker NJ, Gallagher PE. Purine photoproducts. Photochem Photobiol. 1988;48:35–39. [PubMed] [Google Scholar]
- 132.Kumar S, Sharma ND, Davies RJH, Phillipson DW, McCloskey JA. The isolation and characterisation of a new type of dimeric adenine photoproduct in UV-irradiated deoxyadenylates. Nucleic Acids Res. 1987;15:1199–1216. doi: 10.1093/nar/15.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Markiewicz E, Idowu OC. DNA damage in human skin and the capacities of natural compounds to modulate the bystander signalling. Open Biol. 2019;9:190208–190208. doi: 10.1098/rsob.190208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Doetsch PW, Zasatawny TH, Martin AM, Dizdaroglu M. Monomeric base damage products from adenine, guanine, and thymine induced by exposure of DNA to ultraviolet radiation. Biochemistry. 1995;34:737–742. doi: 10.1021/bi00003a005. [DOI] [PubMed] [Google Scholar]
- 135.Brem R, Li F, Montaner B, Reelfs O, Karran P. DNA breakage and cell cycle checkpoint abrogation induced by a therapeutic thiopurine and UVA radiation. Oncogene. 2010;29:3953–3963. doi: 10.1038/onc.2010.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Brem R, Daehn I, Karran P. Efficient DNA interstrand crosslinking by 6-thioguanine and UVA radiation. DNA Repair. 2011;10:869–876. doi: 10.1016/j.dnarep.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 137.Gueranger Q, Kia A, Frith D, Karran P. Crosslinking of DNA repair and replication proteins to DNA in cells treated with 6-thioguanine and UVA. Nucleic Acids Res. 2011;39:5057–5066. doi: 10.1093/nar/gkr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Labiuk SL, Delbaere LT, Lee JS. Gamma and ultraviolet radiation cause DNA crosslinking in the presence of metal ions at high pH. Photochem Photobiol. 2001;73:579–584. doi: 10.1562/0031-8655(2001)073<0579:gaurcd>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 139.Skipper PL, Kim MY, Sun HLP, Wogan GN, Tannenbaum SR. Monocyclic aromatic amines as potential human carcinogens: old is new again. Carcinogenesis. 2010;31:50–58. doi: 10.1093/carcin/bgp267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Neumann HG. The role of DNA damage in chemical carcinogenesis of aromatic amines. J Cancer Res Clin Oncol. 1986;112:100–106. doi: 10.1007/BF00404390. [DOI] [PubMed] [Google Scholar]
- 141.Hammons GJ, Milton D, Stepps K, Guengerich FP, Tukey RH, Kadlubar FF. Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis. 1997;18:851–854. doi: 10.1093/carcin/18.4.851. [DOI] [PubMed] [Google Scholar]
- 142.Kriek E. Fifty years of research on N-acetyl-2-aminofluorene, one of the most versatile compounds in experimental cancer research. J Cancer Res Clin Oncol. 1992;118:481–489. doi: 10.1007/BF01225261. [DOI] [PubMed] [Google Scholar]
- 143.Mah MC, Maher VM, Thomas H, Reid TM, King CM, McCormick JJ. Mutations induced by aminofluorene-DNA adducts during replication in human cells. Carcinogenesis. 1989;10:2321–2328. doi: 10.1093/carcin/10.12.2321. [DOI] [PubMed] [Google Scholar]
- 144.Heflich RH, Neft RE. Genetic toxicity of 2-acetylaminofluorene, 2-aminofluorene and some of their metabolites and model metabolites. Mutat Res. 1994;318:73–114. doi: 10.1016/0165-1110(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 145.Brookes P, Lawley PD. Alkylating agents. Br Med Bull. 1964;20:91–95. doi: 10.1093/oxfordjournals.bmb.a070323. [DOI] [PubMed] [Google Scholar]
- 146.Singer B, Kuśmierek JT. Chemical mutagenesis. Annu Rev Biochem. 1982;51:655–693. doi: 10.1146/annurev.bi.51.070182.003255. [DOI] [PubMed] [Google Scholar]
- 147.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 148.Kondo N, Takahashi A, Ono K, Ohnishi T. DNA damage induced by alkylating agents and repair pathways. J Nucleic Acids. 2010;2010:543531. doi: 10.4061/2010/543531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lawal AT. Polycyclic aromatic hydrocarbons. A review. Cogent Environmental Science. 2017 [Google Scholar]
- 150.Abdel-Shafy HI, Mansour MSM. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egyptian Journal of Petroleum. 2016;25:107–123. [Google Scholar]
- 151.Yu H. Environmental carcinogenic polycyclic aromatic hydrocarbons: photochemistry and phototoxicity. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2002;20:149–183. doi: 10.1081/GNC-120016203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ewa B, Danuta MŠ. Polycyclic aromatic hydrocarbons and PAH-related DNA adducts. J Appl Genet. 2017;58:321–330. doi: 10.1007/s13353-016-0380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Melendez-Colon VJ, Luch A, Seidel A, Baird WM. Cancer initiation by polycyclic aromatic hydrocarbons results from formation of stable DNA adducts rather than apurinic sites. Carcinogenesis. 1999;20:1885–1891. doi: 10.1093/carcin/20.10.1885. [DOI] [PubMed] [Google Scholar]
- 154.Phillips DH. Fifty years of benzo(a)pyrene. Nature. 1983;303:468–472. doi: 10.1038/303468a0. [DOI] [PubMed] [Google Scholar]
- 155.Meehan T, Wolfe AR, Negrete GR, Song Q. Benzo[a]pyrene diol epoxide-DNA cis adduct formation through a trans chlorohydrin intermediate. Proc Natl Acad Sci U S A. 1997;94:1749–1754. doi: 10.1073/pnas.94.5.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cosman Balhorn M, De Los Santos C, Fiala R, Hingerty B, Singh S, Ibanez V, Margulis L, Live D, Geacintov N, Broyde S, Patel D. Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a] pyrene diol epoxide and DNA. Proc Natl Acad Sci U S A. 1992;89:1914–1918. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kozack R, Seo KY, Jelinsky SA, Loechler EL. Toward an understanding of the role of DNA adduct conformation in defining mutagenic mechanism based on studies of the major adduct (formed at N2-dG) of the potent environmental carcinogen, benzo[a] pyrene. Mutat Res. 2000;450:41–59. doi: 10.1016/s0027-5107(00)00015-4. [DOI] [PubMed] [Google Scholar]
- 158.Mordukhovich I, Rossner P, Terry Mary B, Santella R, Zhang YJ, Hibshoosh H, Memeo L, Mansukhani M, Long CM, Garbowski G, Agrawal M, Gaudet Mia M, Steck Susan E, Sagiv Sharon K, Eng Sybil M, Teitelbaum Susan L, Neugut Alfred I, Conway-Dorsey K, Gammon Marilie D. Associations between polycyclic aromatic hydrocarbon-related exposures and p53 mutations in breast tumors. Environ Health Perspect. 2010;118:511–518. doi: 10.1289/ehp.0901233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Park JH, Shen Y, Field J, Penning TM. PAH o-quinone-mediated mutations of p53 correlate with 8-oxo-dGuo formation. Cancer Res. 2006;66:833. [Google Scholar]
- 160.Park JH, Gelhaus S, Vedantam S, Oliva AL, Batra A, Blair IA, Troxel AB, Field J, Penning TM. The pattern of p53 mutations caused by PAH o-quinones is driven by 8-oxo-dGuo formation while the spectrum of mutations is determined by biological selection for dominance. Chem Res Toxicol. 2008;21:1039–1049. doi: 10.1021/tx700404a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jakszyn P, Gonzalez CA. Nitrosamine and related food intake and gastric and oesophageal cancer risk: a systematic review of the epidemiological evidence. World J Gastroenterol. 2006;12:4296–4303. doi: 10.3748/wjg.v12.i27.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Song P, Wu L, Guan W. Dietary nitrates, nitrites, and nitrosamines intake and the risk of gastric cancer: a meta-analysis. Nutrients. 2015;7:9872–9895. doi: 10.3390/nu7125505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stuff JE, Goh ET, Barrera SL, Bondy ML, Forman MR. N-nitroso compounds: assessing agreement between food frequency questionnaires and 7-day food records. J Am Diet Assoc. 2009;109:1179–1183. doi: 10.1016/j.jada.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Swann PF, Magee PN. Nitrosamine-induced carcinogenesis. The alklylation of nucleic acids of the rat by N-methyl-N-nitrosourea, dimethylnitrosamine, dimethyl sulphate and methyl methanesulphonate. Biochem J. 1968;110:39–47. doi: 10.1042/bj1100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Galiègue-Zouitina S, Bailleul B, Loucheux-Lefebvre MH. Adducts from in vivo action of the carcinogen 4-hydroxyaminoquinoline 1-oxide in rats and from in vitro reaction of 4-acetoxyaminoquinoline 1-oxide with DNA and polynucleotides. Cancer Res. 1985;45:520. [PubMed] [Google Scholar]
- 166.Kanojia D, Vaidya MM. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol. 2006;42:655–667. doi: 10.1016/j.oraloncology.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 167.Fronza G, Campomenosi P, Iannone R, Abbondandolo A. The 4-nitroquinoline 1-oxide mutational spectrum in single stranded DNA is characterized by guanine to pyrimidine transversions. Nucleic Acids Res. 1992;20:1283–1287. doi: 10.1093/nar/20.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Caldon CE. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol. 2014;4:106. doi: 10.3389/fonc.2014.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 170.Yasuda MT, Sakakibara H, Shimoi K. Estrogen-and stress-induced DNA damage in breast cancer and chemoprevention with dietary flavonoid. Genes Environ. 2017;39:10. doi: 10.1186/s41021-016-0071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Miller K. Estrogen and DNA damage: the silent source of breast cancer? J Natl Cancer Inst. 2003;95:100–102. doi: 10.1093/jnci/95.2.100. [DOI] [PubMed] [Google Scholar]
- 172.Kumar P, Mahato DK, Kamle M, Mohanta TK, Kang SG. Aflatoxins: a global concern for food safety, human health and their management. Front Microbiol. 2017;7:2170. doi: 10.3389/fmicb.2016.02170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Barrett JR. Liver cancer and aflatoxin: new information from the Kenyan outbreak. Environ Health Perspect. 2005;113:A837–A838. [Google Scholar]
- 174.Liu Y, Wu F. Global burden of aflatoxin-induced hepatocellular carcinoma: a risk assessment. Environ Health Perspect. 2010;118:818–824. doi: 10.1289/ehp.0901388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM. The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2002;99:6655–60. doi: 10.1073/pnas.102167699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bedard LL, Massey TE. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett. 2006;241:174–183. doi: 10.1016/j.canlet.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 177.Bailey EA, Iyer RS, Stone MP, Harris TM, Essigmann JM. Mutational properties of the primary aflatoxin B1-DNA adduct. Proc Natl Acad Sci U S A. 1996;93:1535–1539. doi: 10.1073/pnas.93.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 179.Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010:201367. doi: 10.4061/2010/201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hu J, Lieb JD, Sancar A, Adar S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc Natl Acad Sci U S A. 2016;113:11507. doi: 10.1073/pnas.1614430113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Li LS, Morales JC, Veigl M, Sedwick D, Greer S, Meyers M, Wagner M, Fishel R, Boothman DA. DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets. Br J Pharmacol. 2009;158:679–692. doi: 10.1111/j.1476-5381.2009.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Goulian M, Bleile B, Tseng BY. Methotrexate-induced misincorporation of uracil into DNA. Proc Natl Acad Sci U S A. 1980;77:1956. doi: 10.1073/pnas.77.4.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.De Angelis PM, Svendsrud DH, Kravik KL, Stokke T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Cancer. 2006;5:20–20. doi: 10.1186/1476-4598-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Strobel H, Baisch T, Fitzel R, Schilberg K, Siegelin MD, Karpel-Massler G, Debatin KM, Westhoff MA. Temozolomide and other alkylating agents in glioblastoma therapy. Biomedicines. 2019;7:69. doi: 10.3390/biomedicines7030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fan CH, Liu WL, Cao H, Wen C, Chen L, Jiang G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013;4:e876. doi: 10.1038/cddis.2013.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Delgado JL, Hsieh CM, Chan NL, Hiasa H. Topoisomerases as anticancer targets. Biochem J. 2018;475:373–398. doi: 10.1042/BCJ20160583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cuya SM, Bjornsti MA, van Waardenburg RCAM. DNA topoisomerase-targeting chemotherapeutics: what’s new? Cancer Chemother Pharmacol. 2017;80:1–14. doi: 10.1007/s00280-017-3334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nelson BC, Dizdaroglu M. Implications of DNA damage and DNA repair on human diseases. Mutagenesis. 2020;35:1–3. doi: 10.1093/mutage/gez048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Møller P, Stopper H, Collins AR. Measurement of DNA damage with the comet assay in high-prevalence diseases: current status and future directions. Mutagenesis. 2019;35:5–18. doi: 10.1093/mutage/gez018. [DOI] [PubMed] [Google Scholar]
- 191.Besaratinia A, Pfeifer GP. Investigating human cancer etiology by DNA lesion footprinting and mutagenicity analysis. Carcinogenesis. 2005;27:1526–1537. doi: 10.1093/carcin/bgi311. [DOI] [PubMed] [Google Scholar]
- 192.Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res. 2005;571:43–56. doi: 10.1016/j.mrfmmm.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 193.Pfeifer GP, You YH, Besaratinia A. Mutations induced by ultraviolet light. Mutat Res. 2005;571:19–31. doi: 10.1016/j.mrfmmm.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 194.Wogan GN. Aflatoxin as a human carcinogen. Hepatology. 1999;30:573–575. doi: 10.1002/hep.510300231. [DOI] [PubMed] [Google Scholar]
- 195.Yu MC, Yuan JM. Environmental factors and risk for hepatocellular carcinoma. Gastroenterology. 2004;127:S72–78. doi: 10.1016/j.gastro.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 196.Smith LE, Denissenko MF, Bennett WP, Li H, Amin S, Tang M, Pfeifer GP. Targeting of lung cancer mutational hotspots by polycyclic aromatic hydrocarbons. J Natl Cancer Inst. 2000;92:803–811. doi: 10.1093/jnci/92.10.803. [DOI] [PubMed] [Google Scholar]
- 197.Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 198.Chakravarthi BV, Nepal S, Varambally S. Genomic and epigenomic alterations in cancer. Am J Pathol. 2016;186:1724–1735. doi: 10.1016/j.ajpath.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Adjiri A. DNA mutations may not be the cause of cancer. Oncol Ther. 2017;5:85–101. doi: 10.1007/s40487-017-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Pon JR, Marra MA. Driver and passenger mutations in cancer. Annu Rev Pathol. 2015;10:25–50. doi: 10.1146/annurev-pathol-012414-040312. [DOI] [PubMed] [Google Scholar]
- 202.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26:199–212. doi: 10.1038/s41418-018-0246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bhateja P, Chiu M, Wildey G, Lipka MB, Fu P, Yang MCL, Ardeshir-Larijani F, Sharma N, Dowlati A. Retinoblastoma mutation predicts poor outcomes in advanced non small cell lung cancer. Cancer Med. 2019;8:1459–1466. doi: 10.1002/cam4.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357–363. doi: 10.1093/jnci/djh058. [DOI] [PubMed] [Google Scholar]
- 206.Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 2016;30:1492–1502. doi: 10.1101/gad.282145.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med. 2015;5:a006098. doi: 10.1101/cshperspect.a006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hartl M, Bister K. Oncogenes. In: Maloy S, Hughes K, editors. Brenner’s encyclopedia of genetics (Second Edition) San Diego: Academic Press; 2013. pp. 164–166. [Google Scholar]
- 209.Kang ZJ, Liu YF, Xu LZ, Long ZJ, Huang D, Yang Y, Liu B, Feng JX, Pan YJ, Yan JS, Liu Q. The Philadelphia chromosome in leukemogenesis. Chin J Cancer. 2016;35:48–48. doi: 10.1186/s40880-016-0108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Soverini S, Mancini M, Bavaro L, Cavo M, Martinelli G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17:49. doi: 10.1186/s12943-018-0780-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Deininger MW, Vieira SA, Parada Y, Banerji L, Lam EW, Peters G, Mahon FX, Köhler T, Goldman JM, Melo JV. Direct relation between BCR-ABL tyrosine kinase activity and cyclin D2 expression in lymphoblasts. Cancer Res. 2001;61:8005. [PubMed] [Google Scholar]
- 212.Kridel R, Sehn LH, Gascoyne RD. Pathogenesis of follicular lymphoma. J Clin Invest. 2012;122:3424–3431. doi: 10.1172/JCI63186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Rabkin CS, Hirt C, Janz S, Dölken G. t(14;18) translocations and risk of follicular lymphoma. J Natl Cancer Inst Monogr. 2008:48–51. doi: 10.1093/jncimonographs/lgn002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kumar-Sinha C, Kalyana-Sundaram S, Chinnaiyan AM. Landscape of gene fusions in epithelial cancers: seq and ye shall find. Genome Med. 2015;7:129. doi: 10.1186/s13073-015-0252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nambiar M, Raghavan SC. How does DNA break during chromosomal translocations? Nucleic Acids Res. 2011;39:5813–5825. doi: 10.1093/nar/gkr223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014;30:326–339. doi: 10.1016/j.tig.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 217.van Gent DC, Kanaar R. Exploiting DNA repair defects for novel cancer therapies. Mol Biol Cell. 2016;27:2145–2148. doi: 10.1091/mbc.E15-10-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Torgovnick A, Schumacher B. DNA repair mechanisms in cancer development and therapy. Front Genet. 2015;6:157–157. doi: 10.3389/fgene.2015.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Steinke V, Engel C, Büttner R, Schackert HK, Schmiegel WH, Propping P. Hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome. Dtsch Arztebl Int. 2013;110:32–38. doi: 10.3238/arztebl.2013.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Peltomäki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- 221.Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7:a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gorodetska I, Kozeretska I, Dubrovska A. BRCA genes: the role in genome stability, cancer stemness and therapy resistance. J Cancer. 2019;10:2109–2127. doi: 10.7150/jca.30410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Barrett JC, Wiseman RW. Cellular and molecular mechanisms of multistep carcinogenesis: relevance to carcinogen risk assessment. Environ Health Perspect. 1987;76:65–70. doi: 10.1289/ehp.877665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Barrett JC. Mechanisms of multistep carcinogenesis and carcinogen risk assessment. Environ Health Perspect. 1993;100:9–20. doi: 10.1289/ehp.931009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yao Y, Dai W. Genomic instability and cancer. J Carcinog Mutagen. 2014;5:1000165. doi: 10.4172/2157-2518.1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shen Z. Genomic instability and cancer: an introduction. J Mol Cell Biol. 2011;3:1–3. doi: 10.1093/jmcb/mjq057. [DOI] [PubMed] [Google Scholar]
- 228.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 229.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 230.Pikor L, Thu K, Vucic E, Lam W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013;32:341–352. doi: 10.1007/s10555-013-9429-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Moon JJ, Lu A, Moon C. Role of genomic instability in human carcinogenesis. Exp Biol Med (Maywood) 2019;244:227–240. doi: 10.1177/1535370219826031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075. [PubMed] [Google Scholar]
- 233.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 234.Loeb LA. A Mutator Phenotype in Cancer. Cancer Res. 2001;61:3230. [PubMed] [Google Scholar]
- 235.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 236.Sarni D, Kerem B. Oncogene-induced replication stress drives genome instability and tumorigenesis. Int J Mol Sci. 2017;18:1339. [Google Scholar]
- 237.Primo LMF, Teixeira LK. DNA replication stress: oncogenes in the spotlight. Genet Mol Biol. 2019;43(Suppl 1):e20190138. doi: 10.1590/1678-4685-GMB-2019-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Pagano JS, Blaser M, Buendia MA, Damania B, Khalili K, Raab-Traub N, Roizman B. Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol. 2004;14:453–471. doi: 10.1016/j.semcancer.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 239.McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochim Biophys Acta. 2008;1782:127–150. doi: 10.1016/j.bbadis.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. 2014;15:266–282. doi: 10.1016/j.chom.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kgatle MM, Spearman CW, Kalla AA, Hairwadzi HN. DNA oncogenic virus-induced oxidative stress, genomic damage, and aberrant epigenetic alterations. Oxid Med Cell Longev. 2017;2017:3179421. doi: 10.1155/2017/3179421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Weitzman MD, Lilley CE, Chaurushiya MS. Genomes in conflict: maintaining genome integrity during virus infection. Annu Rev Microbiol. 2010;64:61–81. doi: 10.1146/annurev.micro.112408.134016. [DOI] [PubMed] [Google Scholar]
- 243.Hai H, Tamori A, Kawada N. Role of hepatitis B virus DNA integration in human hepatocarcinogenesis. World J Gastroenterol. 2014;20:6236–6243. doi: 10.3748/wjg.v20.i20.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Shafritz DA, Shouval D, Sherman HI, Hadziyannis SJ, Kew MC. Integration of hepatitis B virus DNA into the genome of liver cells in chronic liver disease and hepatocellular carcinoma. Studies in percutaneous liver biopsies and post-mortem tissue specimens. N Engl J Med. 1981;305:1067–1073. doi: 10.1056/NEJM198110293051807. [DOI] [PubMed] [Google Scholar]
- 245.Brechot C, Pourcel C, Louise A, Rain B, Tiollais P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286:533–535. doi: 10.1038/286533a0. [DOI] [PubMed] [Google Scholar]
- 246.Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM, Poon RT, Fan ST, Chan KL, Gong Z, Hu Y, Lin Z, Wang G, Zhang Q, Barber TD, Chou WC, Aggarwal A, Hao K, Zhou W, Zhang C, Hardwick J, Buser C, Xu J, Kan Z, Dai H, Mao M, Reinhard C, Wang J, Luk JM. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 247.Jia L, Wang XW, Harris CC. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer. 1999;80:875–879. doi: 10.1002/(sici)1097-0215(19990315)80:6<875::aid-ijc13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 248.Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U S A. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Feitelson MA, Reis HMGPV, Tufan NL, Sun B, Pan J, Lian Z. Putative roles of hepatitis B x antigen in the pathogenesis of chronic liver disease. Cancer Lett. 2009;286:69–79. doi: 10.1016/j.canlet.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Tsai WL, Chung RT. Viral hepatocarcinogenesis. Oncogene. 2010;29:2309–2324. doi: 10.1038/onc.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Schlüter V, Meyer M, Hofschneider PH, Koshy R, Caselmann WH. Integrated hepatitis B virus X and 3’ truncated preS/S sequences derived from human hepatomas encode functionally active transactivators. Oncogene. 1994;9:3335–3344. [PubMed] [Google Scholar]
- 252.Hildt E, Munz B, Saher G, Reifenberg K, Hofschneider PH. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002;21:525–535. doi: 10.1093/emboj/21.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Bonilla Guerrero R, Roberts LR. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J Hepatol. 2005;42:760–777. doi: 10.1016/j.jhep.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 254.Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007;252:157–170. doi: 10.1016/j.canlet.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 255.Oyervides-Muñoz MA, Pérez-Maya AA, Rodríguez-Gutiérrez HF, Gómez-Macias GS, Fajardo-Ramírez OR, Treviño V, Barrera-Saldaña HA, Garza-Rodríguez ML. Understanding the HPV integration and its progression to cervical cancer. Infect Genet Evol. 2018;61:134–144. doi: 10.1016/j.meegid.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 256.Peitsaro P, Johansson B, Syrjänen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol. 2002;40:886–891. doi: 10.1128/JCM.40.3.886-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 258.Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol. 1987;61:962–971. doi: 10.1128/jvi.61.4.962-971.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Yim EK, Park JS. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res Treat. 2005;37:319–324. doi: 10.4143/crt.2005.37.6.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Yeo-Teh NSL, Ito Y, Jha S. High-risk human papillomaviral oncogenes E6 and E7 target key cellular pathways to achieve oncogenesis. Int J Mol Sci. 2018;19:1706. doi: 10.3390/ijms19061706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Tomaić V. Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers (Basel) 2016. 2016;8:95. doi: 10.3390/cancers8100095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center & Research Institute at Christiana Care Health Services; HudsonAlpha Institute for Biotechnology; ILSbio, LLC; Indiana University School of Medicine; Institute of Human Virology; Institute for Systems Biology; International Genomics Consortium; Leidos Biomedical; Massachusetts General Hospital; McDonnell Genome Institute at Washington University; Medical College of Wisconsin; Medical University of South Carolina; Memorial Sloan Kettering Cancer Center; Montefiore Medical Center; NantOmics; National Cancer Institute; National Hospital, Abuja, Nigeria; National Human Genome Research Institute; National Institute of Environmental Health Sciences; National Institute on Deafness & Other Communication Disorders; Ontario Tumour Bank, London Health Sciences Centre; Ontario Tumour Bank, Ontario Institute for Cancer Research; Ontario Tumour Bank, The Ottawa Hospital; Oregon Health & Science University; Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center; SRA International; St Joseph’s Candler Health System; Eli & Edythe L. Broad Institute of Massachusetts Institute of Technology & Harvard University; Research Institute at Nationwide Children’s Hospital; Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University; University of Bergen; University of Texas MD Anderson Cancer Center; University of Abuja Teaching Hospital; University of Alabama at Birmingham; University of California, Irvine; University of California Santa Cruz; University of Kansas Medical Center; University of Lausanne; University of New Mexico Health Sciences Center; University of North Carolina at Chapel Hill; University of Oklahoma Health Sciences Center; University of Pittsburgh; University of São Paulo, Ribeir ão Preto Medical School; University of Southern California; University of Washington; University of Wisconsin School of Medicine & Public Health; Van Andel Research Institute; Washington University in St Louis. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–384. doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Cullen AP, Reid R, Campion M, Lörincz AT. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J Virol. 1991;65:606–612. doi: 10.1128/jvi.65.2.606-612.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Klaes R, Woerner SM, Ridder R, Wentzensen N, Duerst M, Schneider A, Lotz B, Melsheimer P, von Knebel Doeberitz M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999;59:6132. [PubMed] [Google Scholar]
- 265.Valmary-Degano S, Jacquin E, Prétet JL, Monnien F, Girardo B, Arbez-Gindre F, Joly M, Bosset JF, Kantelip B, Mougin C. Signature patterns of human papillomavirus type 16 in invasive anal carcinoma. Hum Pathol. 2013;44:992–1002. doi: 10.1016/j.humpath.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 266.Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, Lee S, Hadjipanayis AG, Ivanova EV, Wilkerson MD, Protopopov A, Yang L, Seth S, Song X, Tang J, Ren X, Zhang J, Pantazi A, Santoso N, Xu AW, Mahadeshwar H, Wheeler DA, Haddad RI, Jung J, Ojesina AI, Issaeva N, Yarbrough WG, Hayes DN, Grandis JR, El-Naggar AK, Meyerson M, Park PJ, Chin L, Seidman JG, Hammerman PS, Kucherlapati R. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014;111:15544–15549. doi: 10.1073/pnas.1416074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Olthof NC, Speel EJ, Kolligs J, Haesevoets A, Henfling M, Ramaekers FC, Preuss SF, Drebber U, Wieland U, Silling S, Lam WL, Vucic EA, Kremer B, Klussmann JP, Huebbers CU. Comprehensive analysis of HPV16 integration in OSCC reveals no significant impact of physical status on viral oncogene and virally disrupted human gene expression. PLoS One. 2014;9:e88718. doi: 10.1371/journal.pone.0088718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Vojtechova Z, Sabol I, Salakova M, Turek L, Grega M, Smahelova J, Vencalek O, Lukesova E, Klozar J, Tachezy R. Analysis of the integration of human papillomaviruses in head and neck tumours in relation to patients’ prognosis. Int J Cancer. 2016;138:386–395. doi: 10.1002/ijc.29712. [DOI] [PubMed] [Google Scholar]
- 269.Arora R, Chang Y, Moore PS. MCV and Merkel cell carcinoma: a molecular success story. Curr Opin Virol. 2012;2:489–498. doi: 10.1016/j.coviro.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, Chang Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105:16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, Moore PS, Becker JC. Merkel cell polyomavirus-infected merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84:7064. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Li J, Wang X, Diaz J, Tsang SH, Buck CB, You J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J Virol. 2013;87:9173–9188. doi: 10.1128/JVI.01216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Doolittle-Hall JM, Cunningham Glasspoole DL, Seaman WT, Webster-Cyriaque J. Meta-analysis of DNA tumor-viral integration site selection indicates a role for repeats, gene expression and epigenetics. Cancers (Basel) 2015;7:2217–2235. doi: 10.3390/cancers7040887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chen Wongworawat Y, Filippova M, Williams VM, Filippov V, Duerksen-Hughes PJ. Chronic oxidative stress increases the integration frequency of foreign DNA and human papillomavirus 16 in human keratinocytes. Am J Cancer Res. 2016;6:764–780. [PMC free article] [PubMed] [Google Scholar]
- 276.Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Lunder EJ, Stern RS. Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. N Engl J Med. 1998;339:1247–1248. doi: 10.1056/NEJM199810223391715. [DOI] [PubMed] [Google Scholar]
- 279.Laikova KV, Oberemok VV, Krasnodubets AM, Gal’chinsky NV, Useinov RZ, Novikov IA, Temirova ZZ, Gorlov MV, Shved NA, Kumeiko VV, Makalish TP, Bessalova EY, Fomochkina II, Esin AS, Volkov ME, Kubyshkin AV. Advances in the understanding of skin cancer: ultraviolet radiation, mutations, and antisense oligonucleotides as anticancer drugs. Molecules (Basel, Switzerland) 2019;24:1516. doi: 10.3390/molecules24081516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Mogha A, Fautrel A, Mouchet N, Guo N, Corre S, Adamski H, Watier E, Misery L, Galibert MD. Merkel cell polyomavirus small T antigen mRNA level is increased following in vivo UV-radiation. PLoS One. 2010;5:e11423. doi: 10.1371/journal.pone.0011423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Wu HC, Wang Q, Yang HI, Ahsan H, Tsai WY, Wang LY, Chen SY, Chen CJ, Santella RM. Aflatoxin B1 exposure, hepatitis B virus infection, and hepatocellular carcinoma in Taiwan. Cancer Epidemiol Biomarkers Prev. 2009;18:846–853. doi: 10.1158/1055-9965.EPI-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Wu HC, Santella R. The role of aflatoxins in hepatocellular carcinoma. Hepat Mon. 2012;12:e7238. doi: 10.5812/hepatmon.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Lin CW, Lin CC, Mo LR, Chang CY, Perng DS, Hsu CC, Lo GH, Chen YS, Yen YC, Hu JT, Yu ML, Lee PH, Lin JT, Yang SS. Heavy alcohol consumption increases the incidence of hepatocellular carcinoma in hepatitis B virus-related cirrhosis. J Hepatol. 2013;58:730–735. doi: 10.1016/j.jhep.2012.11.045. [DOI] [PubMed] [Google Scholar]
- 284.Shohdy KS, Abdel-Rahman O. Is smoking causally-associated with hepatitis B virus-related hepatocellular carcinoma? Ann Transl Med. 2019;7:S44–S44. doi: 10.21037/atm.2019.02.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wang YH, Chuang YH, Wu CF, Jan MC, Wu WJ, Lin CL, Liu CJ, Yang YC, Chen PJ, Lin SM, Tsai MH, Huang YW, Yu MW. Smoking and hepatitis B virus-related hepatocellular carcinoma risk: the mediating roles of viral load and alanine aminotransferase. Hepatology. 2019;69:1412–1425. doi: 10.1002/hep.30339. [DOI] [PubMed] [Google Scholar]
- 286.Li W, Deng R, Liu S, Wang K, Sun J. Hepatitis B virus-related hepatocellular carcinoma in the era of antiviral therapy: the emerging role of non-viral risk factors. Liver Int. 2020;40:2316–2325. doi: 10.1111/liv.14607. [DOI] [PubMed] [Google Scholar]
- 287.Almonte M, Albero G, Molano M, Carcamo C, García PJ, Pérez G. Risk factors for human papillomavirus exposure and co-factors for cervical cancer in Latin America and the Caribbean. Vaccine. 2008;26:L16–L36. doi: 10.1016/j.vaccine.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 288.Finan RR, Musharrafieh U, Almawi WY. Detection of Chlamydia trachomatis and herpes simplex virus type 1 or 2 in cervical samples in human papilloma virus (HPV)-positive and HPV-negative women. Clin Microbiol Infect. 2006;12:927–930. doi: 10.1111/j.1469-0691.2006.01479.x. [DOI] [PubMed] [Google Scholar]
- 289.Hawes SE, Kiviat NB. Are genital infections and inflammation cofactors in the pathogenesis of invasive cervical cancer? J Natl Cancer Inst. 2002;94:1592–1593. doi: 10.1093/jnci/94.21.1592. [DOI] [PubMed] [Google Scholar]
- 290.Deacon JM, Evans CD, Yule R, Desai M, Binns W, Taylor C, Peto J. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: a case-control study nested within the Manchester cohort. Br J Cancer. 2000;83:1565–1572. doi: 10.1054/bjoc.2000.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Plummer M, Herrero R, Franceschi S, Meijer CJ, Snijders P, Bosch FX, de Sanjosé S, Muñoz N. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case--control study. Cancer Causes Control. 2003;14:805–814. doi: 10.1023/b:caco.0000003811.98261.3e. [DOI] [PubMed] [Google Scholar]
- 292.Moreno V, Bosch FX, Muñoz N, Meijer CJ, Shah KV, Walboomers JM, Herrero R, Franceschi S. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet. 2002;359:1085–1092. doi: 10.1016/S0140-6736(02)08150-3. [DOI] [PubMed] [Google Scholar]
- 293.Appleby P, Beral V, Berrington de González A, Colin D, Franceschi S, Goodhill A, Green J, Peto J, Plummer M, Sweetland S. Cervical cancer and hormonal contraceptives: collaborative reanalysis of individual data for 16,573 women with cervical cancer and 35,509 women without cervical cancer from 24 epidemiological studies. Lancet. 2007;370:1609–1621. doi: 10.1016/S0140-6736(07)61684-5. [DOI] [PubMed] [Google Scholar]
- 294.Muñoz N, Franceschi S, Bosetti C, Moreno V, Herrero R, Smith JS, Shah KV, Meijer CJ, Bosch FX. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet. 2002;359:1093–1101. doi: 10.1016/S0140-6736(02)08151-5. [DOI] [PubMed] [Google Scholar]
- 295.Katerji M, Filippova M, Duerksen-Hughes P. Approaches and methods to measure oxidative stress in clinical samples: research applications in the cancer field. Oxid Med Cell Longev. 2019;2019:1279250. doi: 10.1155/2019/1279250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Dandri M, Burda MR, Bürkle A, Zuckerman DM, Will H, Rogler CE, Greten H, Petersen J. Increase in de novo HBV DNA integrations in response to oxidative DNA damage or inhibition of poly(ADP-ribosyl)ation. Hepatology. 2002;35:217–223. doi: 10.1053/jhep.2002.30203. [DOI] [PubMed] [Google Scholar]
- 297.Winder DM, Pett MR, Foster N, Shivji MK, Herdman MT, Stanley MA, Venkitaraman AR, Coleman N. An increase in DNA double-strand breaks, induced by Ku70 depletion, is associated with human papillomavirus 16 episome loss and de novo viral integration events. J Pathol. 2007;213:27–34. doi: 10.1002/path.2206. [DOI] [PubMed] [Google Scholar]
- 298.Someya M, Sakata KI, Matsumoto Y, Yamamoto H, Monobe M, Ikeda H, Ando K, Hosoi Y, Suzuki N, Hareyama M. The association of DNA-dependent protein kinase activity with chromosomal instability and risk of cancer. Carcinogenesis. 2005;27:117–122. doi: 10.1093/carcin/bgi175. [DOI] [PubMed] [Google Scholar]
- 299.Leeman JE, Li Y, Bell A, Hussain SS, Majumdar R, Rong-Mullins X, Blecua P, Damerla R, Narang H, Ravindran PT, Lee NY, Riaz N, Powell SN, Higginson DS. Human papillomavirus 16 promotes microhomology-mediated end-joining. Proc Natl Acad Sci U S A. 2019;116:21573. doi: 10.1073/pnas.1906120116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, Shen H, Zhang C, Liu H, Liu X, Zhao Y, Fang X, Li S, Chen W, Tang T, Fu A, Wang Z, Chen G, Gao Q, Li S, Xi L, Wang C, Liao S, Ma X, Wu P, Li K, Wang S, Zhou J, Wang J, Xu X, Wang H, Ma D. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nature Genetics. 2015;47:158–163. doi: 10.1038/ng.3178. [DOI] [PubMed] [Google Scholar]