

Abstract
The pathologically form of amyloid (Aβ) peptide is shown to be toxic to the mitochondria and implicates this organelle in the progression and pathogenesis of Alzheimer’s disease(AD). Mitochondria are dynamic structures constantly undergoing fission and fusion, and altering their shape and size while traveling through neurons. Mitochondrial fission (Drp1, Fis1) and fusion (OPA1, Mfn1 and Mfn2) proteins are balanced in healthy neuronal cells. Glia maturation factor (GMF), a neuroinflammatory protein isolated and cloned in our laboratory plays an important role in the pathogenesis of AD. We hypothesized that GMF, a brain-localized inflammatory protein promotes oxidative stress mediated disruption of mitochondrial dynamics by alterations in mitochondrial fission and fusion proteins which eventually leads to apoptosis in the Aβ (1-42) treated human neuroblastoma (SH-SY5Y) cells. TheSH-SY5Y cells were incubated with GMF and Aβ (1-42) peptide, and mitochondrial fission and fusion proteins were analyzed by immunofluorescence, western blotting and Co-immunoprecipitation. We report that SH-SY5Y cells incubated with GMF and Aβ (1-42) promotes mitochondrial fragmentation, by potentiating oxidative stress, mitophagy and shifts in the Bax/Bcl2 expression and release of cytochrome-c, and eventual apoptosis. In the present study, we show that GMF and Aβ treatments significantly upregulates fission proteins and downregulates fusion proteins. The study shows that extracellular GMF is an important inflammatory mediator that mediates mitochondrial dynamics by altering the balance in fission and fusion proteins and amplifies similar effects promoted by Aβ. Upregulated GMF in the presence of Aβ could be an additional risk factor for AD and their synergistic actions need to be explored as a potential therapeutic target to suppress the progression of AD.
Keywords: Alzheimer’s disease, Amyloid-beta, Glia maturation factor, Mitochondrial dynamics, SH-SY5Y cells
Introduction
The pathological hallmarks of AD are senile plaques, neurofibrillary tangles (NFTs) along with loss of neurons and synapse in the AD brains [1,2]. Previous, studies have shown that amyloid beta (Aβ 1-42) peptide, the major component of senile plaques is toxic to the neuronal cells and is considered to have a causative role in the development and progression of AD [3]. Mitochondria are highly dynamic organelles with morphology and numbers regulated by fission and fusion proteins [4]. Excessive mitochondrial fission-fusion process leads to impaired mitochondrial function and neuronal death in AD [5,6]. Mitochondrial fission and fusion process is essential for the growth and division of immature cells to provide them with adequate numbers of mitochondria for effective bioenergetics. Mitochondrial fusion is an essential process for the complementation of damaged mitochondria and to rescue mitochondria to maximize the oxidative capacity in response to toxic stress to cells [7]. An equilibrium between mitochondrial fission and fusion events need to be maintained for the health of the neuronal cells. Mitochondrial fusion helps to maintain tubular mitochondrial network and optimal mitochondrial functions. Interconnected mitochondrial network facilitate transfer of mitochondrial membrane potential from oxygen rich region to regions of high demand [8]. Neurons are highly susceptible to mitochondrial dysfunction because of their high metabolic rate and complex morphology [9,10]. Therefore, maintaining healthy mitochondrial dynamics by balancing the levels of fission and fusion proteins will help in preventing mitochondrial dysfunction as seen in neurodegenerative disorders such as AD. Excessive mitochondrial fragmentation leads to the defective cellular metabolism, oxidative stress, mitochondrial dysfunction, neuronal dysfunction and neuronal cell death following Aβ-induced toxicity as seen in SH-SY5Y cells. Fission is initiated by cytosolic Dynamin-1 like protein (Drp1) is recruited from the cytosol to the mitochondria by Mid49, Mid51, and Mff [11]. However, fusion between outer mitochondrial membranes is mediated by dynamin family member proteins called Mfn1 and Mfn2. Fusion between mitochondrial inner membranes is mediated by a single dynamin family member protein called fusion protein, Optic atrophy protein 1(OPA1).
Following mitochondrial fragmentation and morphological changes, outer mitochondrial membrane permeabilization occurs through homo-oligomerization of Bax/Bak and opening of mitochondrial outer membrane mega pore, which allows release of cytochrome c (cyt c) from inner mitochondrial membrane into the cytoplasm and eventually apoptotic death [12]. The anti-apoptotic protein Bcl-2 inhibits homo-oligomerization of Bax/Bcl-2. Previous study has shown that Bcl-2 is downregulated in AD [13]. The progression of AD is characterized by activation of glial cells that contributes to local inflammation and oxidative stress resulting in mitochondrial dysfunction and neuronal damage [14-17].
Glia maturation factor(GMF), a neuroinflammatory protein containing 141 amino acids and having total molecular mass of (17 kDa), was discovered, isolated, cloned and sequenced from bovine brain for the first time in our laboratory [18-20]. GMF is one of the major intracellular inflammatory molecules expressed in glia and neuronal cells [21-23]. However, the role of GMF in mitochondrial dysfunction, especially in AD is not known. In the present study, we show that incubation of SH-SY5Y cells with Aβ led to the mitochondrial abnormalities including impaired mitochondrial dynamics and mitochondrial fragmentation with alterations in the equilibrium between fission and fusion proteins that is synergistically regulated by GMF.
Material Methods
Cell lines and culture condition
Human neuroblastoma SH-SY5Y cell line (Sigma Aldrich, ST Louis MO) was grown in (Sigma Ham’s F12: EMEM (1:1) medium supplemented with 10% fetal bovine serum (FBS: Sigma Aldrich, St. Louis, MO), 2mM Glutamine, 1% Non-Essential Amino Acids (NEAA), Penicillin (10 U/ml) and streptomycin (10 U/ml GIBCO Life Technology, Grand Island, NY).The cells were seeded at a density of 5×105 in a 75-cm2 tissue culture flask (Corning, New York, NY) and incubated at 37 °C under saturated humidity in 5% CO2/95% air.
Incubation of SH-SY5Y cells with GMF and Aβ (1-42)
After overnight seeding of SH-SY5Y cells in 96 well plate the cells were incubated with varying doses (5, 10 and 20 μM)of soluble form of Aβ (1-42) or were stimulated with varying concentrations of GMF (50, 100 and 200 ng/ml). In subsequent experiments, SH-SY5Y cells were grown to (75% to 85%) confluency and incubated for up to 24 h with 10 μM of Aβ (1-42) or were stimulated with optimum concentrations of GMF (100 ng/ml). Cells were trypsinized and collected for western blotting and seeded for immunofluorescence staining. Cell lysates were prepared for western blotting analysis and protein concentration of the cell lysates were quantified by using bicinchoninic acid assay (BCA) protein assay kit (Thermo Scientific, Waltham, MA) as per the manufacturer’s instructions.
MTT assay for cell viability
The cell viability of SH-SY5Y cells was determined by MTT (3-4, 5-dimethylthiazolyl-2)-2, 5-diphenyl tetrazolium bromide) dye-uptake method as described before [24,25]. The cell viability was measured based on the conversion of MTT to a purple formazan product by the mitochondrial dehydrogenase. MTT was dissolved at a concentration of 5 mg/ml in phosphate-buffered saline (PBS) and the cells were then pre-incubated with, the MTT solution (50 μl/well). Following the 2 h incubation period at 37 °C, the medium was decanted and the formazan precipitates were solubilized with acidified isopropanol (0.04–0.1 N HCl in absolute isopropanol). The absorbance was measured on a microplate reader (Molecular Devices; Sunnyvale, CA) at a wavelength of 570 nm with background subtraction at 630 nm. The solvent for MTT was employed as blank and results expressed as percentage of control.
Total antioxidant assay
SH-SY5Y cells (1 x 10 5cells/ml) were seeded in a 6well plate followed by incubation with GMF (100 ng/ml) and Aβ (1-42) 10 μM for 24 h. After treatment, cells were washed three times in DPBS and centrifuged at 2,000 g for 10 min at 4 °C. Cells were lysed at 4 °C with radio-immunoprecipitation assay (RIPA) cell lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% deoxycholate) containing protease inhibitors (Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitor cocktails (Cell Signaling Technology, Danvers, MA). Cell lysates were used for analysis of cellular glutathione peroxidase (GPX) (kit no. 703102) and superoxide dismutase (SOD) (kit no. 706002) according to the manufacturer’s instructions (Cayman Chemical Ann Arbor, MI). The activity of GPx was calculated as micromoles of glutathione (GSH) formed per milligram protein, using a molar extinction coefficient of 13.6 × 103/M/cm. The SOD results were expressed as units per milligram protein.
Determination of Adenosine Triphosphate (ATP) Levels
ATP concentration was determined by using the ATP assay kit of Abcam (Cat No. ab83355, Cambridge MA). SH-SY5Y cells were grown in T25 cm2 cell culture flasks (1x 106) and after the incubation period with the GMF and Aβ, the cells were collected and washed with cold DPBS. Cell lysates were prepared from treated cells and mitochondrial fraction was isolated and the samples were used to measure ATP level according to the manufacturer instructions. Values of ATP activity levels were determined by using ATP standard curve.
Cell lysis and subcellular fractionation
SH-SY5Y cells were seeded in T25 cm2 cell culture flasks and incubated with GMF and Aβ peptide for 24 h, thereafter cells were washed with DPBS and adherent cells were detached from the culture flask, cells were collected and centrifuged at 2000 g for 10 min at 4 °C. Cells were lysed at 4 °C with RIPA cell lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% deoxycholate) containing protease inhibitors (Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitor cocktails (Cell Signaling Technology, Danvers, MA). The cell lysate were used for western blotting analysis, Mitochondrial and cytosolic fractions were separated by using the Abcam (Cat no; ab 110170, Cambridge, MA) mitochondria isolation kit according to the manufacturer’s instruction and protein concentration was estimated by the BCA protein assay kit.
Western blotting and co-Immunoprecipitation
Western blotting was performed as detailed previously from our laboratory [25] and co-immunoprecipitation (Co-IP) according to our published method[26]. The mitochondrial fraction was isolated by using mitochondrial isolation kit (Abcam) containing protease and phosphatase inhibitor cocktails. Equal amount of protein (ranging from 20-35 μg) were loaded on4-12% NuPAGE Tris-Glycine gradient gel (Invitrogen, Life technologies, Carlsbad, CA). The separated proteins were transferred onto PVDF membrane by wet protein transfer system (Life technologies). After blocking with 5% nonfat milk or bovine serum albumin (BSA) in Tris-buffered saline-containing 0.1% of Tween 20 (TBST) for 1 h, the membranes were then incubated with primary antibody at 4 °C overnight. The membranes were washed followed by incubation with HRP-conjugated secondary antibodies for 1 h at room temperature. Bound proteins were visualized using ECL prime western blotting detection reagent (Super Signal West Pico PLUS Substrate (Cat No. 34580; Thermo Scientific). Blots were stripped and re-probed for β-actin or COX4. Densitometry was performed by using the ChemiDoc-It2 Imaging System (UVP LLC, Upland, CA) analysis software and the semi quantitative analysis of bands were performed with ImageJ analysis software (Version 1.49; NIH, USA). B Means ± SEM was calculated from the data for statistical and graphical representation. Primary antibody sources are as follows; anti-OPA1(1:500), anti-Mfn1 (1:500), anti-Mfn2 (1:500), anti-Fis1 (1:500), anti-Mff (1:500), anti-bcl2 (1:700), (Santa Cruz, SCBT, Dallas, TX), anti-Drp1 (1;1000), anti-cytc (1:500), anti-COX4(1:1000), anti-P62 (Abcam) (1:1000), ant-p-Drp1 (1:700), anti-bax (1:500), anti-caspase3 (1:500), anti-caspase,9 (1:500), anti-LC3B (Cell Signaling) (1:700),β- actin(Sigma). For Co-IPs, 200 μg protein samples from mitochondrial fractions were incubated with 5 μg anti-Drp1 antibody overnight. After the addition of protein A/G-Sepharose (Santa Cruz Biotechnology), the mixture was incubated for 2 h at 4 °C. The beads were isolated by centrifugation, washed three times with lysis buffer, and eluted by heating at 100 ºC in 4X LDS sample buffer. The associated protein in immunoprecipitates was examined by western blot analysis with anti-Fis1 or anti-Mff antibody.
Immunofluorescence staining and confocal microscopy
Immunofluorescence staining was performed as detailed previously from our lab [27]. SH-SY5Y cells were seeded on glass coverslips pre-coated with Poly-L-Lysine (Sigma Aldrich). The cells were fixed with 4% paraformaldehyde for 1 h and permeabilized by incubation in PBS/0.1% Triton X-100 (PBST) for 15 min. Then the cells were rinsed three times in PBS, blocked with 5% normal goat serum (NGS) for 30 min and finally incubated for 24 h at 4 °C with anti-Tom20 (1:500), anti-OPA1 (1:100 dilution), anti-Mfn1 and anti-Mfn2 (1:150), and cyt c (1:500), Drp1, Fis1(1:150) primary antibody. Cells were washed three times at room temperature followed by incubation with proper Alexa Fluor 488/568 goat anti-mouse/rabbit/goat secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature. After washes, sections were mounted and cover slipped in Vectashield mounting medium with 4-6-diamidino-2-phenylindole (H-1200; Vector Laboratories, Burlingame, Inc, CA, USA). Cells were examined on a Leica TCP SP8 laser scanning confocal microscope with a 405-nm diode laser and tunable super continuum white light laser using 63X oil immersion objective (Molecular cytology core facility, University of Missouri, Columbia, MO).
Statistical analysis
The results were analyzed using GraphPad InStat 3 statistical software. Mean ± SEM was calculated and analyzed using One-way Analysis of Variance (ANOVA) followed by Tukey-Kramer multiple comparison tests to determine statistically significant differences between the groups. Results were expressed as mean ± SEM for four experiments in each group. *p values < 0.05 were considered as significant.
Results
Cell Viability of SH-SY5Y is affected by treatment with GMF and Aβ.
SH-SY5Y cells were treated with GMF (50,100 and 200 ng/ml) and Aβ (5, 10 and20 μM) for 24 h and the cell viability determined using the MTT assay. Results showed significant reduction in cell viability represented as reduction in mitochondrial activity demonstrated by the MTT reduction assay when compared to untreated cells. (Fig. 1a, b) Results showed that GMF and Aβ treatment caused significant dose-dependent cell death but does not seem to cause cell necrosis. Based on these results, a dose of 100 ng/ml GMF and 10μM Aβ were used in subsequent experiments.
Fig. 1.
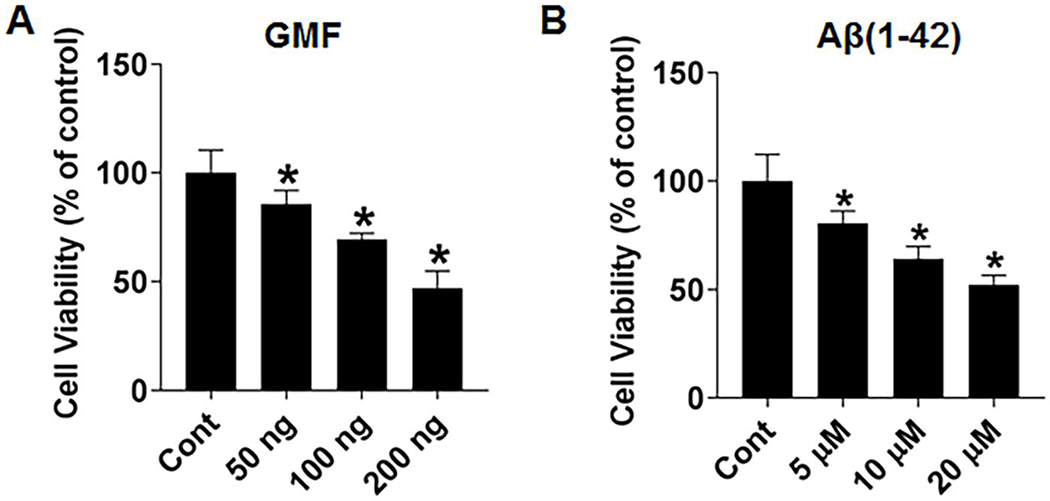
Effect of GMF and Aβ (1-42) on SH-SY5Y cell viability and proliferation. (a) Effect of dose of GMF (0, 50, 100 and 200 ng/ml) and (b) Effect of concentration of Aβ (1-42) (0, 5, 10 and 20, μM). Cell viability/Cytotoxicity was determined by the MTT assay. Higher dose of GMF and Aβ significantly reduced the cell viability when incubated for 24 h. Values are expressed as mean ± SE (n=6), *P< 0.05 Vs untreated control group.
GMF treatment potentiates alterations in mitochondrial dynamics by targeting fission protein levels in Aβ peptide treated SH--SY5Y cell line
Mitochondrial adaptor proteins Fis1 and Mff for Drp1, located on the outer mitochondrial membrane are involved in mitochondrial fission by binding to Drp1[28]. To examine the role of pro-inflammatory GMF on mitochondrial fission dynamics in Aβ- induced cytotoxicity in SH-SH5Y cells, we studied the association of the adapter protein Mff with the mitochondrial fission protein Drp1 by triple-labeling immunofluorescence staining and confocal microscopy. Co-localization of the fission proteins. (Fig. 2a, b) shows that GMF treatment promoted higher expression and mitochondrial localization of Drp1 and Mff proteins. Further, we performed western blotting and quantitative analysis in mitochondrial fraction to determine the fission proteins levels. (Fig. 2b). Results showed that the level of these fission proteins Drp1, Fis1, Mff were significantly elevated in GMF treated group compared with control (cont) group.
Fig. 2.
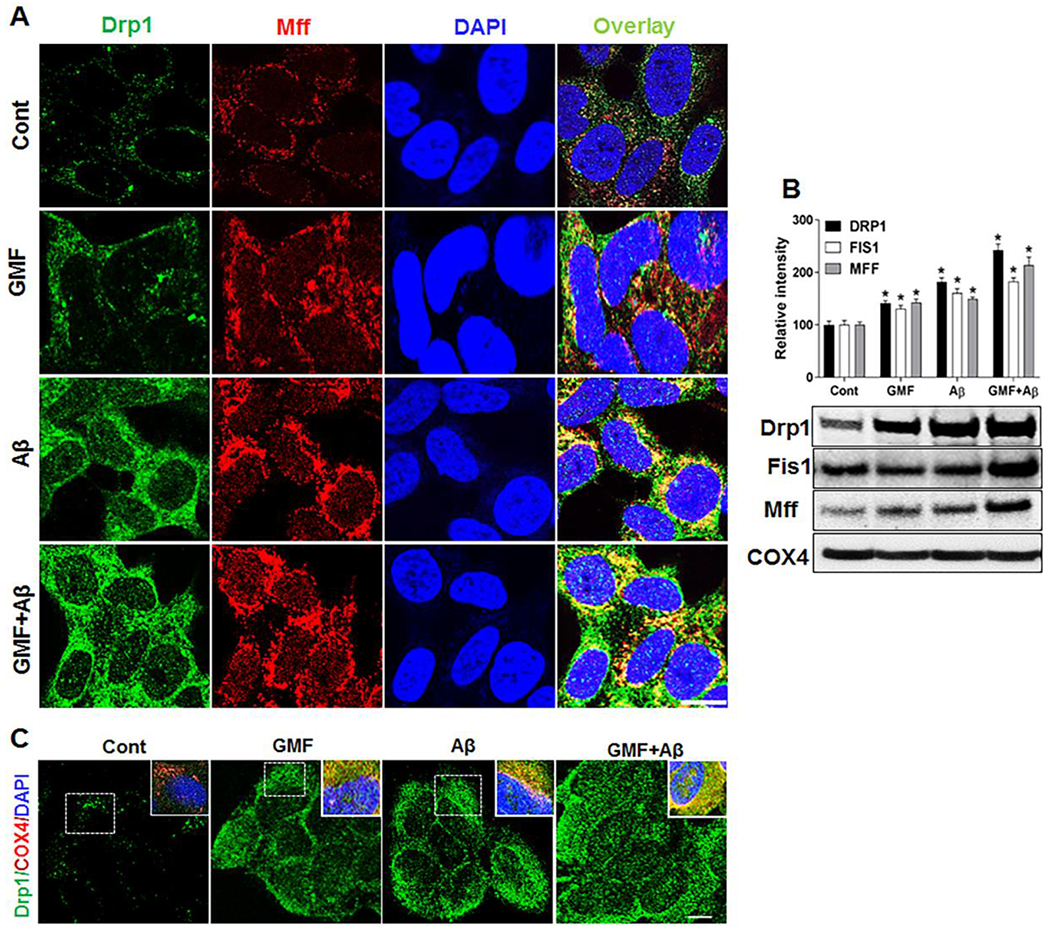
GMF increases the expression of mitochondrial fission proteins. Representative triple-labelling confocal microscopy images (a)showing staining of mitochondrial fission proteins Drp1 (green), Mff (red) and DAP1 (blue) scale bar=20 μm. Inserts are zoomed images of the boxed areas. Co-localization of fission protein Drp1 (green) along with the mitochondrial COX4 (red) staining shown in (c). Western blotting and quantitative analysis of mitochondrial fission protein, Drp1, Fis1 and Mff were performed using mitochondrial fraction from whole cell lysate. GMF treated group show higher expression of fission protein compared with control group shown in (b). Values are expressed as mean ± SE (n=3). *P<0.05 versus untreated control group.
GMF regulates Drp1 activity in the interaction of Drp1 with Fis1 and Mff in Aβ treated SH-SY5Y cells
Previous studies showed that Drp1 activity is regulated by several posttranslational modifications, including phosphorylation of Drp1 on Ser S616 (S616required for mitochondrial fission) and phosphorylation of Drp1 on Ser S637 (S637- inhibits mitochondrial fission process) [29,30].(Fig, 3a) showed that Drp1-S616 level was higher and Drp1-S637 level was significantly lower in GMF treated group compared with control (cont) group, suggesting that GMF promoted mitochondrial fission. Furthermore, we performed co-immunoprecipitation (Co-IPs) in mitochondrial protein fraction with Drp1 antibody and then separately blotted with ant-Fis1 or anti-Mff antibody, as shows in (Fig. 3b, c). The interactions and complex formation between Drp1-Fis1 and Drp1-Mff were significantly increased in GMF or Aβ treated group compared with control (cont) group. These results demonstrate that GMF promotes mitochondrial fission in neuronal cells.
Fig. 3.
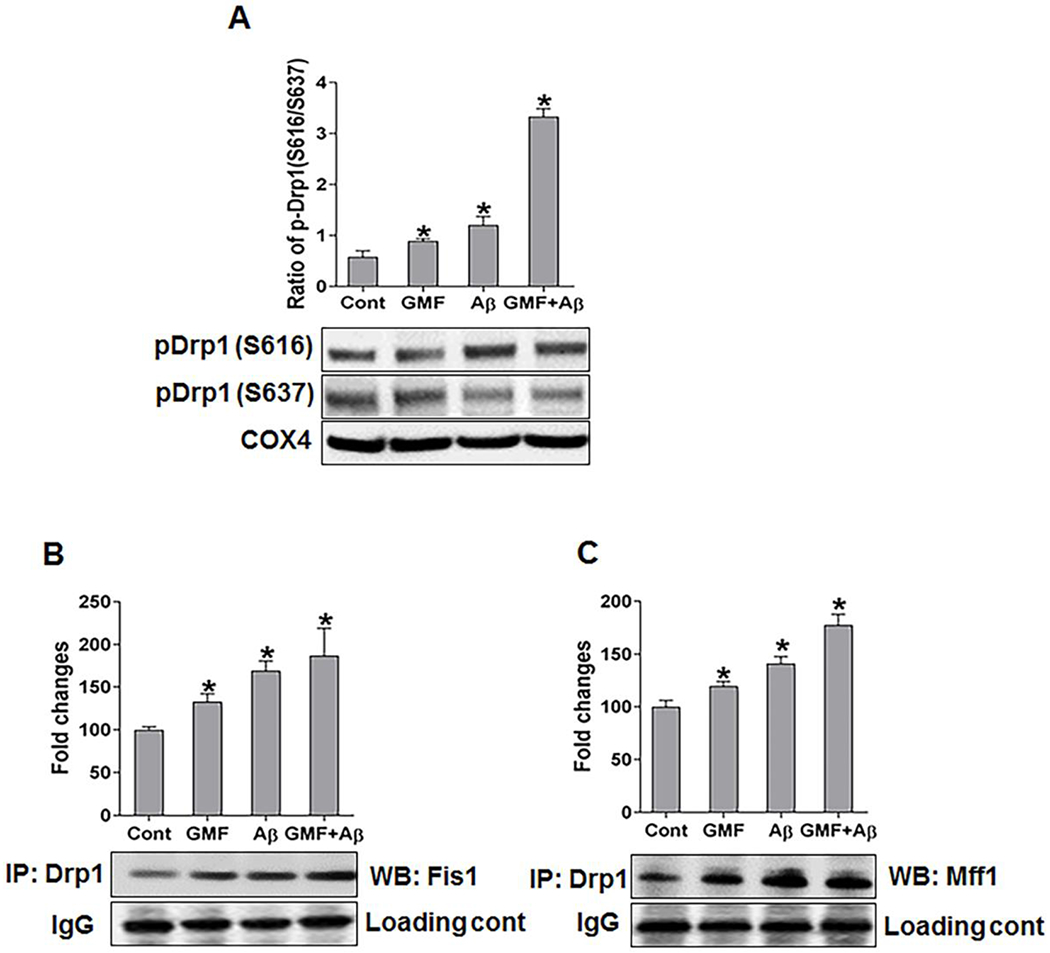
Effect of GMF on mitochondrial targeting of fission proteins and their interactions. (a) Western blotting and quantitative analysis of mitochondrial fractions from whole cell lysate were determined by using the indicated antibodies along with mitochondrial fission protein. pDrp1 (S616) blot shows that the Ser 616 is necessary for the fission event and pDrp1 (S637) blot shows that Ser 637 residue is necessary for the suppression of fission. Cells treated with GMF showed higher expression of pDrp1 (S616) and lower expression of pDrp1 (S637) compared with control group. COX4 is used as loading control for the mitochondrial protein. (b, c) Mitochondrial protein samples were also examined by immunoprecipitation (IP) with anti-Drp1 followed by western blotting and detection with anti-Fis1 or anti-Mff interaction. IgG was used as loading control for IPs. Values are expressed as mean ± SE (n=3). *P < 0.05versus untreated control group.
GMF suppresses mitochondrial fusion protein levels in Aβ treated SH-SY5Y cells
The fusion protein, Optic atrophy protein 1(OPA1), located in the inner mitochondrial membrane along with Mfn1 and Mfn2 located on outer membrane of mitochondria; key regulators of mitochondrial dynamics were analyzed by western blotting and triple-labeled confocal immunofluorescence staining. (Fig 4a, b) demonstrate that mitochondrial expression and localization of OPA1 and Mfn1 were drastically decreased in GMF treated group compared with control (cont) group. Furthermore, we investigated the levels of these fusion proteins by western blotting and quantitative analysis in mitochondrial fractions obtained from whole cell lysates. Results (Fig 4b) show a remarkable decrease in the level of fusion proteins in GMF treated group compared with control (cont) group. This result shows that GMF exerts its effect by creating an imbalance between fission and fusion proteins by suppression of mitochondrial fusion protein and promoting the elevation of fission proteins.
Fig. 4.
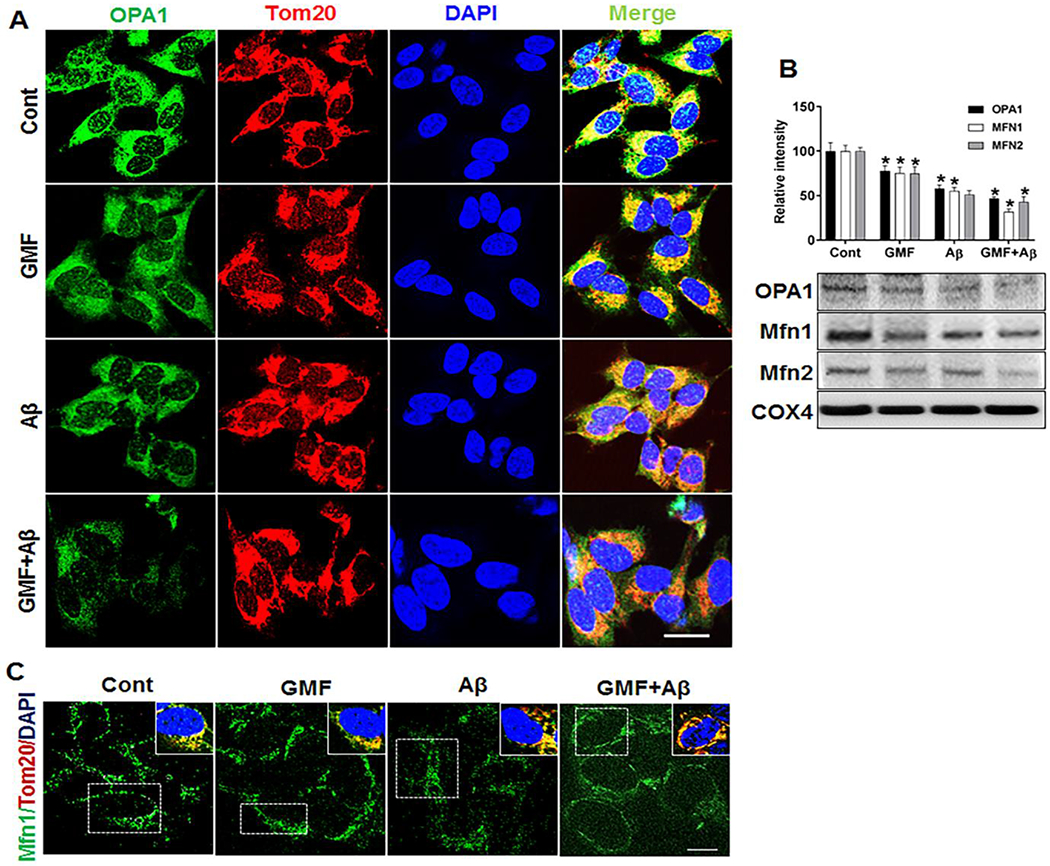
Effect of GMF on mitochondria by targeting the fusion proteins. (a, c) Representative triple-labelling confocal microscopy images showing staining of mitochondrial fusion proteins, OPA1 and Mfn1 after 24 incubation with Aβ and GMF. Inserts in (c) are the zoomed images of the boxed areas showing the co-localization of mitochondrial protein (Tom20, red) with fusion protein MFN1 (green). Nuclei were counter stained with DAPI (blue) scale bar =20 μm. (b) Western blot and quantitative analysis of mitochondrial fusion proteins OPA1, Mfn1 &Mfn2 were performed using mitochondrial fractions from cell lysate. GMF treatment significantly reduced the expression of mitochondrial fusion protein (OPA1, Mfn1&Mfn2) compared with cont. Values are expressed as mean ± SE of determination from each group (n=3). *P < 0.05 versus normal control group
GMF alters mitochondrial dynamics by promoting mitochondrial fragmentations
We next investigated the effect of GMF and Aβ on mitochondrial morphology as shown in (Fig.5a, b). Representative confocal images showed that mitochondria was extensively fragmented in SH-SY5Y cells following treatment with GMF. For clear mitochondrial localization and improved quantitative analysis, confocal images of mitochondrial translocase of outer membrane (Tom20) fluorescence was followed alongside parameters of threshold, filter, and binarized using ImageJ software. Quantitative analysis of fragmented mitochondrial segments using the parameters for total mitochondrial fragmentation in GMF or Aβ treated groups showed extensive fragmentation compared with control group. These results support the role of GMF in mitochondrial fragmentation in promoting Aβ induced toxicity in SH-SY5Y cells.
Fig. 5.
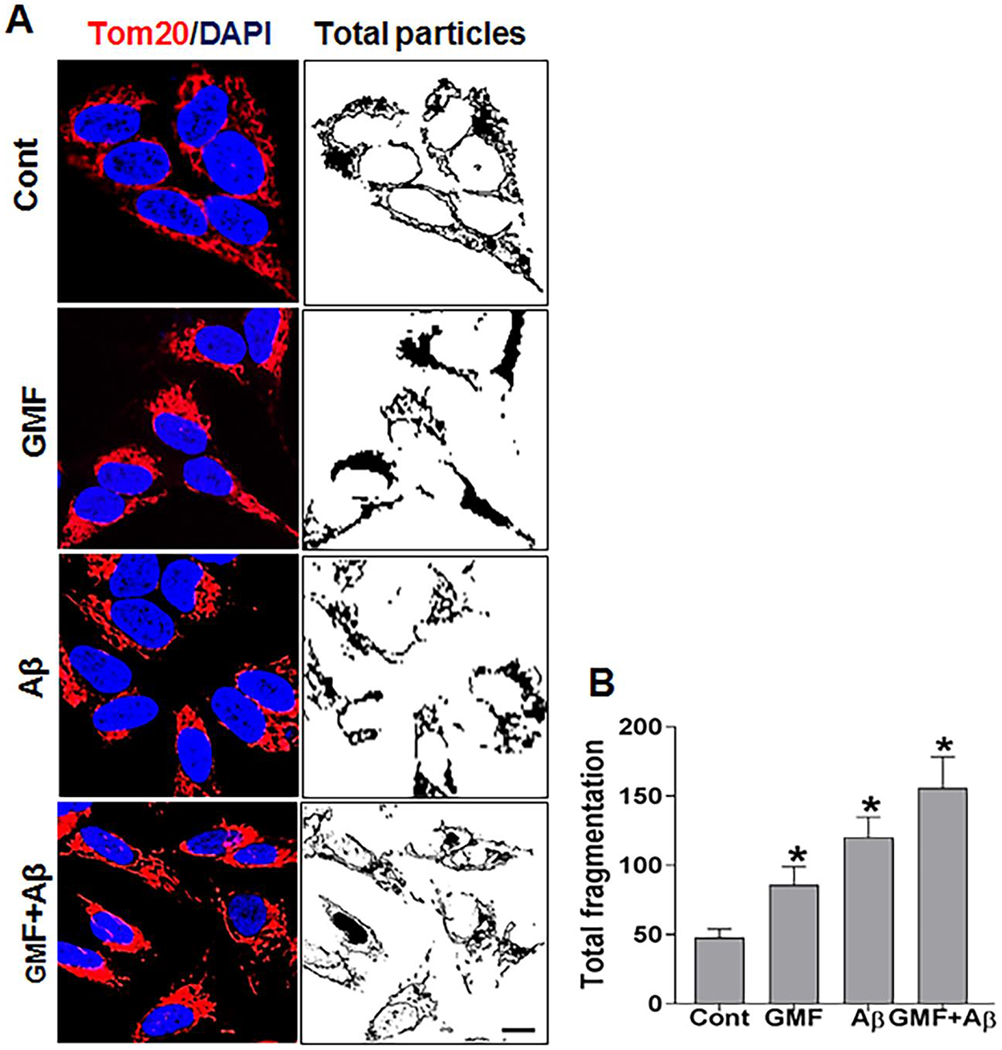
Effect of GMF on mitochondrial dynamics. (a) Representative confocal microscopy images of SH-SY5Y cells showing mitochondria with anti-Tom20 antibody (red). Nuclei were counterstained with DAPI (blue). Acquired images of Tom20 fluorescent staining were threshold filtered (median, 2. 0 pixel), and binarized for image views and quantitative analysis was done for each group. Mitochondrial fragmentation was analyzed (b) Total mitochondrial fragmentation in each case was calculated as the percentage of the particle normalized with total mitochondrial area. Data are represented as mean ± SE (n=3) in each group. Scale bar=5μm. *P < 0.05 versus normal control group.
GMF promotes mitochondrial autophagy (mitophagy) and mitochondrial dysfunction
Mitochondrial dysfunction following extensive fragmentation is an early and causal event in neurodegenerative disorder [31]. Mitochondrial degradation by autophagy (mitophagy) process has long been proposed to remove damaged and dysfunctional mitochondria, which has been implicated in the progression of AD [32,33]. We determined mitophagy in mitochondrial fraction by monitoring the expression levels of LC3B and the sequestosome protein P62well-known marker of autophagy. (Fig 6a, b) shows increased levels of LC3B and P62 in GMF treated group compared with control group. Furthermore, we investigated the level of Bax/Bcl2, (Fig 6c).GMF treated group showed higher level of Bax/Bcl2 compared with control group. Finally, we quantified the levels of ATP production as a measure of mitochondrial health on the isolated mitochondrial fractions as shown in (Fig 6d). GMF or Aβ treatment significantly depleted the level of ATP compared with control. Our results revealed that GMF treatment enhances mitophagy and disturbs the balance between Bax/Bcl2 that causes ATP deficiency.
Fig. 6.
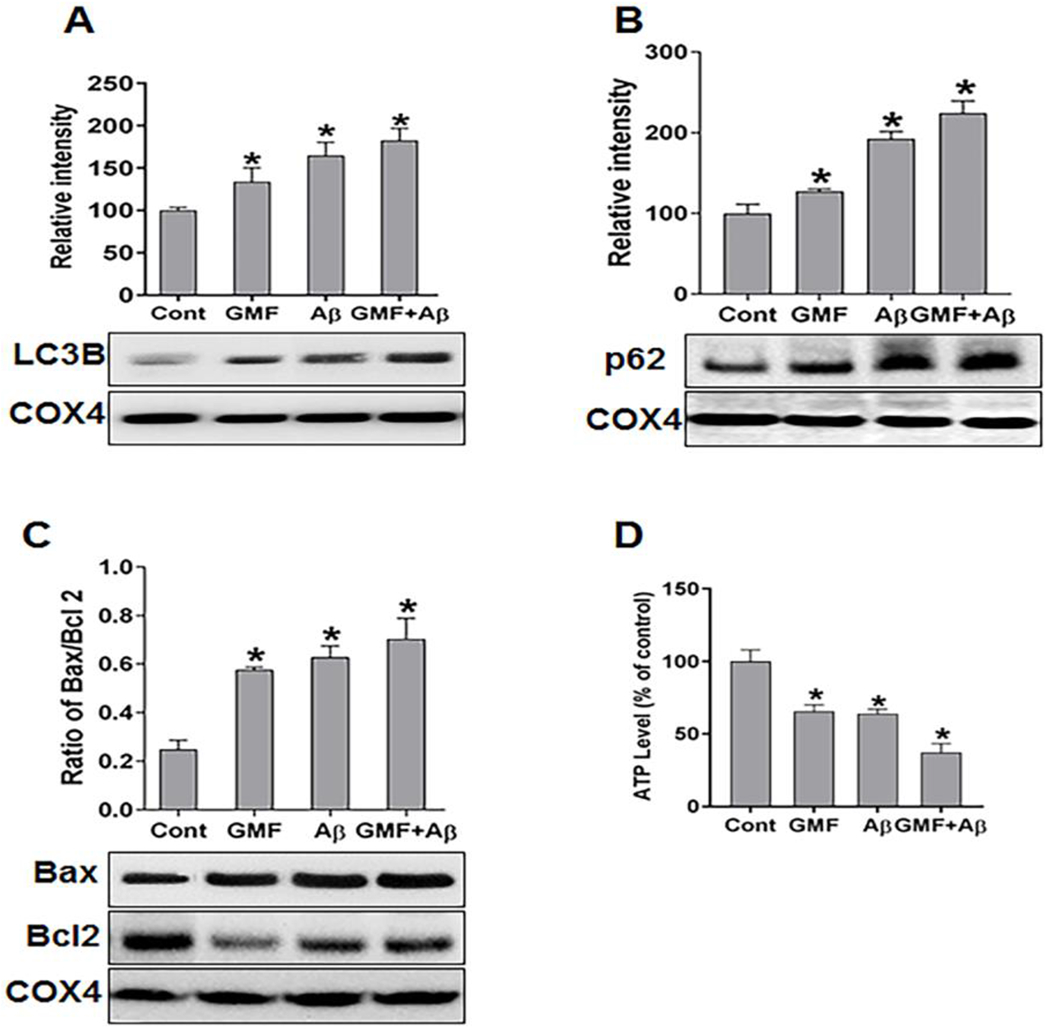
GMF induces mitochondrial dysfunction by ATP depletion and mitophagy (a, b) western blotting and quantitative analysis of mitophagy markers LC3B and P62sequestosome in mitochondrial fractions, COX4 is used as loading control for mitochondrial protein. GMF treated cells showed higher expression of LC3B and P62 compared with control group. (c) Further, we calculated the ratio of Bax/Bcl2 protein expression. GMF treatment showed elevation in the ratio of Bax/Bcl2 compared with control. The levels of ATP production were tested by using mitochondrial fraction to evaluate the mitochondrial function. (d) Shows the ATP levels in control and GMF treated group. Data were expressed as means ± SE (n=3). *p<0.05 versus control.
GMF treatment stimulates apoptosis as determined by mitochondrial cyt c release
Next, we examined the mitochondrial permeability by following the release and cytosolic distribution of cyt c in response to GMF. We determined both mitochondrial and cytosolic levels of cyt c by western blotting analysis. As shown in (Fig 7a, b) GMF treated group showed significantly higher level of cyt c in cytosolic fraction and reduced level in mitochondrial fraction compared to control group. Also, we analyzed the release of cyt c from mitochondrial membrane to cytosolic region by immunofluorescence staining. (Fig 7c) Representative images by confocal microscopy showed the higher expression and release of cytc in cytosolic region and we observed intense translocation of cytc from mitochondrial membrane to cytosolic region in GMF treated group compared with control group. Release of cyt c from mitochondria to the cytosol, which then activates the effector caspases resulting in apoptotic neuronal death in SH-SY5Y cells, was explored by western blotting analysis. (Fig 7d) shows the GMF mediated activation of caspase-9 and final executioner of apoptosis marker caspase-3. Combined incubation of GMF and Aβ showed higher level of capsase-9 and caspase-3 as compared with GMF or Aβ treated group.
Fig. 7.
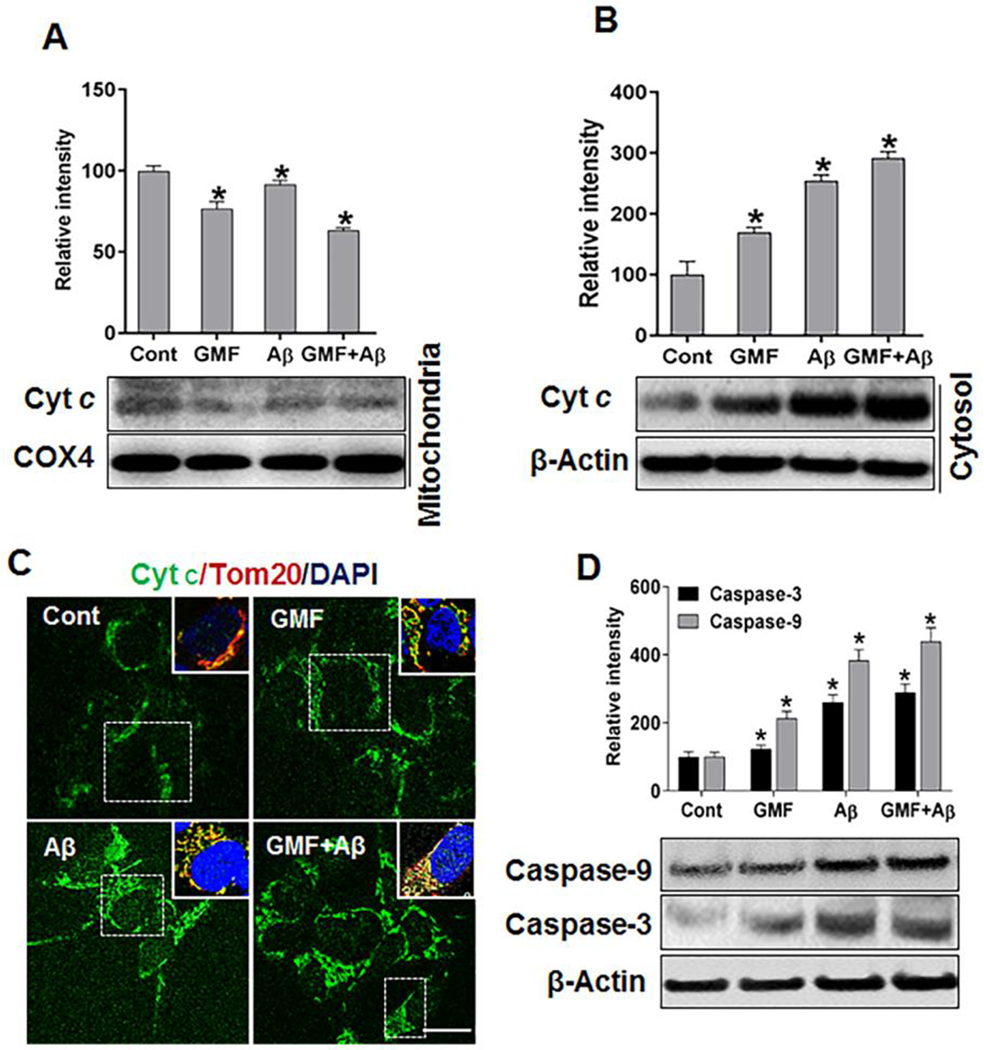
Effect of GMF treatment on the release of mitochondrial cytochrome c to cytosolic region and on apoptosis in Aβ- treated SH-SY5Y cells. (a) The levels of cytochrome c (cyt c) determined by western blotting and quantitative analysis showed reduced levels of expression of cyt c in mitochondrial fraction compared to control group. However, (b) cytosolic fraction showed more released from mitochondria to cytosol in GMF treated group compared with control group. (c) Representative images of immunofluorescence staining of cytc. Inserts are zooms of the boxed area showing the mitochondrial cyt c (green) and Tom20 (red) and nuclei were stained with DAPI (blue) scale bar =20 μm. (d) Western blotting and quantitative analyses of caspase-9 and caspase-3 in 10 μM, Aβ- treated SH-SY5Y cells using cytosolic protein. GMF treated group showed elevated level of caspase-9 and caspase-3 compared with control group. Data were represented by mean ± SE (n=3) in each group. *p<0.05 versus untreated control.
Glia maturation factor induced oxidative stress augments Aβ -mediated neurotoxicity
Mitochondria are a major source of oxidants and cause of oxidative stress due to uncontrolled electron leakage from the electron transport chain that leads to the massive production of superoxide anion and disruption of cellular antioxidant system. Abundant evidence demonstrates that mitochondrial dysfunction and oxidative stress are strongly involved in pathology of AD [15,34]. (Fig 8a, b) shows the levels of antioxidant enzyme superoxide dismutase and glutathione peroxidase (SOD and GPX) following GMF treatment in SH-SY5Y cells. Results show that GMF treatment significantly decreases the level of these antioxidant enzymes compared with control group. Furthermore, we performed immunofluorescence staining of the lipid peroxidation product,4-hydroxynonenal (4-HNE), a marker for oxidative damage and the specific oxidized marker of DNA damage,8-Hydroxydeoxyguanosine(8-OHdG)(Fig 8c). From the representative triple labelling immunofluorescence staining of 8-OHdG, Tom20 with DAPI mitochondrial oxidative DNA damage was found to be substantially greater in GMF treated group as compared with control group. Higher concentrations of 8-OHdG within cells is a measure of increased oxidative stress. Representative confocal immunofluorescence staining of lipid peroxidation marker 4-HNE (Fig 8d) and DAPI showed higher expression of 4-HNE in GMF treated group compared with cont group.
Fig. 8.
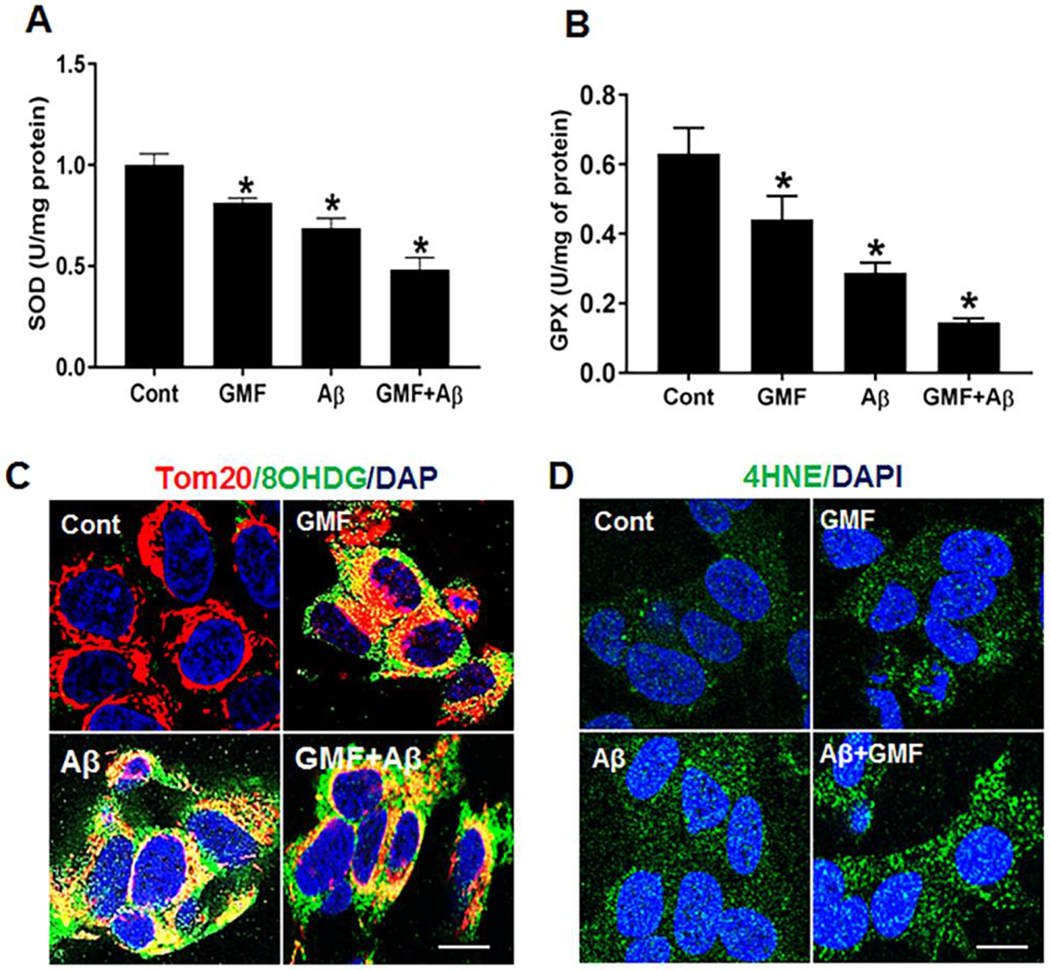
GMF enhances the cytotoxicity through inhibition of activities of the antioxidant enzymes. (a, b) showed the activities of antioxidant enzymes (SOD, GPX) levels, GMF treated cells showed downregulation of these enzymes compared with control group. However, markers of oxidative stress showed enhancement in GMF treated group. (c, d) Representative confocal immunofluorescence staining of oxidative damage markers of oxidative DNA damage (8-OHdG) and marker for lipid peroxidation (4-HNE) showed higher expression in GMF treated group compared with control group. Data are represented as mean ± SE (n=3). Scale bar 50 μm, *p<0.05 vs untreated control group.
Discussion
In the present study we show that GMF in conjunction with the neurotoxic Aβ causes alterations in fission (Drp1, Fis1) and fusion (OPA1, Mfn1 and mfn2) proteins in neuroblastoma SH-SY5Y cells. We demonstrate for the first time that GMF was able to disrupt mitochondrial dynamics by altering the expression of fission and fusion proteins, along with oxidative stress and apoptosis. We found a disruption in cellular homeostasis with major reduction in cellular energy production and extensive oxidative stress following GMF treatment that induced mitochondrial dysfunction and initiation of neuronal dysfunction and neurodegeneration. Preservation of mitochondrial structure and morphology has been widely associated with the balance of mitochondrial fission and fusion protein levels [5,35,31]. Defects in mitochondrial fission and fusion proteins alter mitochondrial morphology that subsequently affects mitochondrial mobility and transmission of energy signals along the length of the neurons.
Excessive mitochondrial fragmentation and mitochondrial dysfunction in neurodegenerative disorders associated with morphological changes is brought about by permeabilization of outer mitochondrial membrane. The initiation of oligomerization of Bax and Bak to form mega pores releases cytochrome c into the cytoplasm [12]. In the present study, we report that SH-SY5Y cells incubated with GMF for 24 h promotes mitochondrial abnormalities, excessive mitochondrial fragmentation, and alterations in mitochondrial dynamics towards more fission rather than fusion events. Besides oxidative stress GMF treatment, also causes oxidative DNA damage, apoptosis, mitochondrial fragmentation and disruption in mitophagy. Our studies highlight an important role of GMF in disruption of mitochondrial dynamics. Mitochondria are important organelles in the nervous system that are essential to fulfil the high-energy demand for proper neuronal function. Unlike other cell types, neurons cannot switch to glycolysis when energy source is depleted [36,37]. Mitochondria transport materials through a dynamic process of mitochondrial fission and fusion events that provide energy across the length of neurons. Impaired mitochondrial function, low levels of ATP and structural defects in mitochondria are observed in AD patients [6,38,39], suggesting that the brain is under high-energy stress in AD pathogenesis. Incubation of human neuroblastoma SH-SY5Y cells with GMF leads to the activation and phosphorylation of Drp1. Our data demonstrate increased activation and association of Drp1 with the mitochondria, which is facilitated through binding to Fis1 causing excessive mitochondrial fragmentation in GMF treated SH-SY5Y cells compared to control. On the effect of GMF on mitochondrial fusion proteins, we found reduced level of fusion proteins OPA1, Mfn1 and Mfn2 in GMF treated SHS-SY5Y cells compared with control group. The anti-apoptotic protein Bcl-2 which is downregulated in AD brain is a central regulator of mitochondrial apoptosis and prevents the homo-oligomerization of Bax and Bak [13]. Overall, GMF disrupts mitochondrial homeostasis, through oxidative DNA damage and induces mitochondrial fragmentation and disruption of mitochondrial dynamics with impaired mitophagy and Bcl-2- mediated apoptotic cell death.
Impaired oxidative phosphorylation and disrupted ATP synthesis have detrimental effect evident in neurodegenerative disease. It has been known for a long time that in aged brain there is a continuous reduction of ATP level and continuous decline in bioenergetics capacity. Also ATP levels and its synthesis are impaired in the brain of AD patients [39,38]. It is suggested that the AD brain is under high-energy demand and reduced mitochondrial enzyme activities in AD accompanies neurodegeneration and neurophathogical conditions [40]. Our results showed that GMF enhanced Aβ toxicity by altering mitochondrial dynamics with significant reduction in ATP synthesis.
It is becoming increasingly clear that oxidative stress and mitochondrial dysfunction are early events in neurodegenerative diseases like AD and play an important role in the neuro- pathology of AD [41,42]. Previous studies demonstrated that treatment of neuronal cell with Aβ caused oxidative stress by inhibition of respiratory chain electron transport and induction of ROS through leakage of excessive electrons from respiratory chain [1,3]. In line with these our current results show that GMF treatment amplifies Aβ (1-42) neurotoxicity by significantly decreasing the levels of antioxidant enzymes and enhanced ROS production and increased lipid peroxidation; all of which lead to alterations in fission and fusion proteins, mitochondrial fragmentation and dysfunction and ultimate neuronal death.
Conclusions
GMF is brain specific, biologically active neuroinflammatory protein that mediates disruption of mitochondrial dynamics by altering the balance in levels of mitochondrial fission and fusion proteins. Our current findings reveal that GMF disrupts mitochondrial dynamics through excessive fragmentation brought about by increased ROS generation, oxidative DNA damage, altered mitophagy and Bax/Bcl-2mediated apoptotic neuronal cell death.
Acknowledgements
This work was supported by National Institutes of Health Grants NS073670, AG048205and Veterans Affairs Merit Award I01BX002477 to AZ.
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Mattson MP (2004) Pathways towards and away from Alzheimer's disease. Nature 430 (7000):631–639. doi: 10.1038/nature02621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thangavel R, Kempuraj D, Zaheer S, Raikwar S, Ahmed ME, Selvakumar GP, Iyer SS, Zaheer A (2017) Glia Maturation Factor and Mitochondrial Uncoupling Proteins 2 and 4 Expression in the Temporal Cortex of Alzheimer’s Disease Brain. Front Aging Neurosci 9:150. doi: 10.3389/fnagi.2017.00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qu M, Zhou Z, Xu S, Chen C, Yu Z, Wang D (2011) Mortalin overexpression attenuates beta-amyloid-induced neurotoxicity in SH-SY5Y cells. Brain Res 1368:336–345. doi: 10.1016/j.brainres.2010.10.068 [DOI] [PubMed] [Google Scholar]
- 4.Itoh K, Nakamura K, Iijima M, Sesaki H (2013) Mitochondrial dynamics in neurodegeneration. Trends Cell Biol 23 (2):64–71. doi: 10.1016/j.tcb.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu Y, Wang R, Dong Y, Tucker D, Zhao N, Ahmed ME, Zhu L, Liu TC, Cohen RM, Zhang Q (2017) Low-level laser therapy for beta amyloid toxicity in rat hippocampus. Neurobiol Aging 49:165–182. doi: 10.1016/j.neurobiolaging.2016.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cadonic C, Sabbir MG, Albensi BC (2016) Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol Neurobiol 53 (9):6078–6090. doi: 10.1007/s12035-015-9515-5 [DOI] [PubMed] [Google Scholar]
- 7.Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337 (6098):1062–1065. doi: 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skulachev VP (2001) Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci 26 (1):23–29 [DOI] [PubMed] [Google Scholar]
- 9.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26 (2):207–210. doi: 10.1038/79936 [DOI] [PubMed] [Google Scholar]
- 10.Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R (2007) Mitochondrial bioenergetics and structural network organization. J Cell Sci 120 (Pt 5):838–848. doi: 10.1242/jcs.03381 [DOI] [PubMed] [Google Scholar]
- 11.Elgass K, Pakay J, Ryan MT, Palmer CS (2013) Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta 1833 (1):150–161. doi: 10.1016/j.bbamcr.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, Blomgren K, Kroemer G (2009) Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 10 (7):481–494. doi: 10.1038/nrn2665 [DOI] [PubMed] [Google Scholar]
- 13.Satou T, Cummings BJ, Cotman CW (1995) Immunoreactivity for Bcl-2 protein within neurons in the Alzheimer’s disease brain increases with disease severity. Brain Res 697 (1-2):35–43 [DOI] [PubMed] [Google Scholar]
- 14.Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR (2009) An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J Alzheimers Dis 16 (4):741–761. doi: 10.3233/JAD-2009-0972 [DOI] [PubMed] [Google Scholar]
- 15.Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G (2010) Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta 1802 (1):2–10. doi: 10.1016/j.bbadis.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 16.Steardo L Jr., Bronzuoli MR, Iacomino A, Esposito G, Steardo L, Scuderi C (2015) Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front Neurosci 9:259. doi: 10.3389/fnins.2015.00259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verkhratsky A, Zorec R, Rodriguez JJ, Parpura V (2016) Astroglia dynamics in ageing and Alzheimer’s disease. Curr Opin Pharmacol 26:74–79. doi: 10.1016/j.coph.2015.09.011 [DOI] [PubMed] [Google Scholar]
- 18.Kaplan R, Zaheer A, Jaye M, Lim R (1991) Molecular cloning and expression of biologically active human glia maturation factor-beta. J Neurochem 57 (2):483–490 [DOI] [PubMed] [Google Scholar]
- 19.Lim R, Zaheer A, Lane WS (1990) Complete amino acid sequence of bovine glia maturation factor beta. Proc Natl Acad Sci U S A 87 (14):5233–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaheer A, Fink BD, Lim R (1993) Expression of glia maturation factor beta mRNA and protein in rat organs and cells. J Neurochem 60 (3):914–920 [DOI] [PubMed] [Google Scholar]
- 21.Choudhury A, Marks DL, Proctor KM, Gould GW, Pagano RE (2006) Regulation of caveolar endocytosis by syntaxin 6-dependent delivery of membrane components to the cell surface. Nat Cell Biol 8 (4):317–328. doi: 10.1038/ncb1380 [DOI] [PubMed] [Google Scholar]
- 22.Aerbajinai W, Liu L, Zhu J, Kumkhaek C, Chin K, Rodgers GP (2016) Glia Maturation Factor-gamma Regulates Monocyte Migration through Modulation of beta1-Integrin. J Biol Chem 291 (16):8549–8564. doi: 10.1074/jbc.M115.674200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Afeseh Ngwa H, Kanthasamy A, Anantharam V, Song C, Witte T, Houk R, Kanthasamy AG (2009) Vanadium induces dopaminergic neurotoxicity via protein kinase Cdelta dependent oxidative signaling mechanisms: relevance to etiopathogenesis of Parkinson’s disease. Toxicol Appl Pharmacol 240 (2):273–285. doi: 10.1016/j.taap.2009.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E (2000) Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res 6 (4):1205–1218 [PubMed] [Google Scholar]
- 25.Selvakumar GP, Iyer SS, Kempuraj D, Raju M, Thangavel R, Saeed D, Ahmed ME, Zahoor H, Raikwar SP, Zaheer S, Zaheer A (2018) Glia Maturation Factor Dependent Inhibition of Mitochondrial PGC-1alpha Triggers Oxidative Stress-Mediated Apoptosis in N27 Rat Dopaminergic Neuronal Cells. Mol Neurobiol. doi: 10.1007/s12035-018-0882-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmed ME, Dong Y, Lu Y, Tucker D, Wang R, Zhang Q (2017) Beneficial Effects of a CaMKIIalpha Inhibitor TatCN21 Peptide in Global Cerebral Ischemia. J Mol Neurosci 61 (1):42–51. doi: 10.1007/s12031-016-0830-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kempuraj D, Selvakumar GP, Thangavel R, Ahmed ME, Zaheer S, Kumar KK, Yelam A, Kaur H, Dubova I, Raikwar SP, Iyer SS, Zaheer A (2018) Glia Maturation Factor and Mast Cell-Dependent Expression of Inflammatory Mediators and Proteinase Activated Receptor-2 in Neuroinflammation. Journal of Alzheimer’s disease : JAD 66 (3):1117–1129. doi: 10.3233/JAD-180786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT (2013) Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem 288 (38):27584–27593. doi: 10.1074/jbc.M113.479873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 105 (41):15803–15808. doi: 10.1073/pnas.0808249105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8 (10):939–944. doi: 10.1038/sj.embor.7401062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9 (7):505–518. doi: 10.1038/nrn2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baloyannis SJ (2006) Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis 9 (2):119–126 [DOI] [PubMed] [Google Scholar]
- 33.Kim I, Rodriguez-Enriquez S, Lemasters JJ (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462 (2):245–253. doi: 10.1016/j.abb.2007.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moreira PI (2012) Alzheimer’s disease and diabetes: an integrative view of the role of mitochondria, oxidative stress, and insulin. J Alzheimers Dis 30 Suppl 2:S199–215. doi: 10.3233/JAD-2011-111127 [DOI] [PubMed] [Google Scholar]
- 35.Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnaune-Pelloquin L, Davezac N, Mils V, Miquel MC, Rojo M, Belenguer P (2016) Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis 90:3–19. doi: 10.1016/j.nbd.2015.10.011 [DOI] [PubMed] [Google Scholar]
- 36.Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125 (7):1241–1252. doi: 10.1016/j.cell.2006.06.010 [DOI] [PubMed] [Google Scholar]
- 37.Hoppins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048 [DOI] [PubMed] [Google Scholar]
- 38.Pettegrew JW, Panchalingam K, Klunk WE, McClure RJ, Muenz LR (1994) Alterations of cerebral metabolism in probable Alzheimer’s disease: a preliminary study. Neurobiol Aging 15 (1):117–132 [DOI] [PubMed] [Google Scholar]
- 39.Terni B, Boada J, Portero-Otin M, Pamplona R, Ferrer I (2010) Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol 20 (1):222–233. doi: 10.1111/j.1750-3639.2009.00266.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullivan PG, Brown MR (2005) Mitochondrial aging and dysfunction in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry 29 (3):407–410. doi: 10.1016/j.pnpbp.2004.12.007 [DOI] [PubMed] [Google Scholar]
- 41.Freund-Levi Y, Vedin I, Hjorth E, Basun H, Faxen Irving G, Schultzberg M, Eriksdotter M, Palmblad J, Vessby B, Wahlund LO, Cederholm T, Basu S (2014) Effects of supplementation with omega-3 fatty acids on oxidative stress and inflammation in patients with Alzheimer’s disease: the OmegAD study. J Alzheimers Dis 42 (3):823–831. doi: 10.3233/JAD-132042 [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842 (8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]