

Abstract
Causation has multiple distinct meanings in genetics. One reason for this is meaning slippage between two concepts of the gene: Mendelian and molecular. Another reason is that a variety of genetic methods address different kinds of causal relationships. Some genetic studies address causes of traits in individuals, which can only be assessed when single genes follow predictable inheritance patterns that reliably cause a trait. A second sense concerns the causes of trait differences within a population. Whereas some single genes can be said to cause population-level differences, most often these claims concern the effects of many genes. Polygenic traits can be understood using heritability estimates, which estimate the relative influences of genetic and environmental differences to trait differences within a population. Attempts to understand the molecular mechanisms underlying polygenic traits have been developed, although causal inference based on these results remains controversial. Genetic variation has also recently been leveraged as a randomizing factor to identify environmental causes of trait differences. This technique—Mendelian randomization—offers some solutions to traditional epidemiological challenges, although it is limited to the study of environments with known genetic influences.
The meaning of cause in genetics is complicated in many ways. First, the way in which causation is understood differs depending on one's philosophical commitments. The “true nature” or best representation of causation is still discussed heavily in the philosophy of science, and there are distinct ways in which the term is used among both scientists and philosophers. Second, there are multiple senses of what it is to be genetic, or what counts as a gene, and different interpretations of both of these has led to confusion in debates about genetic causation.1
Thexs section Two Senses of the Gene considers two senses of the term “genetic,” and Causal Contribution and Difference Making covers two broad senses of genetic causation. The sections Single-Gene Traits, Polygenic Traits: Family-Based Studies, Polygenic Traits: Genomic Approaches, and Using Genes to Identify Environmental Causes then relate the ideas from Two Senses of the Gene and Causal Contribution and Difference Making to contemporary fields of genetic research.
TWO SENSES OF THE GENE
Empirical work demonstrates a plurality of gene concepts in use within scientific communities (Stotz et al. 2004). Whereas some, such as Keller (2000), have argued that this disarray provides conceptual value, most others believe that such plurality has led to conceptual confusion, stalling fruitful debate (Griffiths and Neumann-Held 1999; Griffiths 2001; Moss 2001). A useful development has been to think of the gene concept dichotomously. Whereas there are significant variations in how this dichotomy is expressed, it can be roughly amalgamated into two distinct notions: the Mendelian gene and the molecular gene. These two concepts play different explanatory roles and are suited to different scientific contexts (Griffiths and Stotz 2006, 2007).
Historically, genes were first understood in the Mendelian sense as inherited “factors” determining different traits. These factors, which were used to predict the outcomes of breeding experiments, remained unobserved theoretical entities. Each factor comes in alternative forms, now termed alleles. Whereas today alleles are thought of as variations that occur at a particular location in the genome (loci), Mendelian genes are identified in reference to their effects rather than their underlying physical basis, which for a long time was unknown (Griffiths and Stotz 2013). Although it is known today that trait differences are inherited due to DNA transmission, the Mendelian gene as it is used today need not refer to any particular DNA sequence(s), so long as it serves as a statistically valid predictor of phenotypes within a population. Thus, the Mendelian gene is synonymous with the classical gene of transmission genetics, and use of the term can continue to “black-box” the developmental role of DNA and its products. This gene concept is closely related to what Moss (2001) has termed “gene-P” (P for phenotype or prediction).
A second gene concept emerged with the discovery of the structure and function of DNA, and the advent of molecular genetics. A molecular gene is a sequence of DNA that has the potential to act as a transcriptional unit. Thus, the molecular gene serves as a developmental resource for “gene products” such as RNA, which in some cases leads to protein synthesis (Waters 1994). This concept roughly corresponds to Moss's (2001) “gene-D” (D for developmental resource) and what Dawkins (1976) terms a cistron (a term originating with the molecular biologist Seymour Benzer). These sequences sometimes appear clearly as a section in the genome where the beginning and end of the genes are marked by start and stop codons, which are identifiable nucleotide sequences used by enzymes, RNA polymerase, and ribosomes to commence and cease the transcription and translation process, respectively. Such a clear delineation, however, is not always the case, as often the specification of the linear sequence of a particular gene product derives from disparate sections of the genome. Additionally, the regulated synthesis of a polypeptide needs more than a coding region alone (Neumann-Held 2001).
In some instances, the molecular gene illuminates some of the developmental processes that had been “black-boxed” by the Mendelian gene. For instance, Menkes disease is an early-onset copper transport deficiency with serious physiological and developmental symptoms. The X-linked molecular gene associated with the disease was eventually isolated as mutations in ATP7A, a locus on the X chromosome sub-band Xq13.3, corresponding to an 8.5 kb transcript coding for a 1500 amino acid protein (Tumer et al. 1992). This began with linkage studies using family resemblance data, which capitalize on the genotype–phenotype mapping that epitomizes the Mendelian gene (Horn et al. 1984). This was followed by physical chromosomal mapping based on translocations and inversions (Verga et al. 1991), until the molecular ATP7A gene was eventually identified and cloned as part of the human genome project (Tumer et al. 1992).
However, there is imperfect overlap between the Mendelian and molecular gene concept. As Griffiths and Stotz (2013) point out, only a small percentage of the human genome corresponds to coding sequences that define molecular genes, yet the remaining sequences, such as those that perform regulatory functions, can be identified as components of Mendelian genes when they exert predictable phenotypic effects. One example of this is Lmbr1, a DNA sequence known to produce abnormal limb development via its regulatory influence on the sonic hedgehog's shh gene (Lettice et al. 2002 cited in Griffiths and Stotz 2013). Additionally, single molecular genes that are distributed across the genome (described above) can segregate independently as distinct Mendelian alleles.
As gene concepts vary, so too does the concept of the “environment.” The environment of a single molecular gene includes the internal environment of an individual as well as encompassing intra- and extracellular structures that can influence DNA expression such as proteins and RNA. Some evolutionary accounts also include other sequences of DNA as part of the environment of a gene (Sterelny and Kitcher 1988; Haig 2012). However, the influence of other DNA sequences on molecular gene expression is more commonly referred to as epistasis, or gene–gene interactions, and have been studied in genetics for over a century (Hollander 1955).
Mendelian genes on the other hand are inferred by examining family resemblances and transmission of phenotypes. Understanding the environment of these genes is often focused on factors outside of the organism, termed postnatal environmental effects, which include things like climate, nutrition, education, and socioeconomic status (Bazzett 2008). Contemporary geneticists also recognize the importance of within-organismal environmental factors or prenatal environmental effects, such as early embryonic conditions and the uterine environment, as nongenetic influences on physiological and behavioral differences (Hochberg et al. 2011).
Epigenetic effects also challenge traditional notions of “gene” and “environment,” although the transgenerational potential of these influences appears limited in human populations (Heard and Martienssen 2014). A further challenge to the gene/environment dichotomy is stochastic effects. These occur from the molecular level influencing gene expression (Tikhodeyev and Shcherbakova 2019) to chance events altering developmental trajectories (Smith 2011). Whereas they are thought to (in some cases substantially) influence phenotypic variation, they are often not regarded as “genetic” or “environmental” factors in the usual meaning of the term, although the quantitative genetics label of “nonshared environment”—into which they fall—confuses this.
An addition to the complications with defining and categorizing factors into “genetic” and “environmental” is the complexity with which genes and environments interact throughout development. In many cases, the environment an individual is exposed to is causally influenced by a parental genotype and/or their own genotype. These cases are discussed in the sections Polygenic Traits: Family-Based Studies and Using Genes to Identify Environmental Causes.
CAUSAL CONTRIBUTION AND DIFFERENCE MAKING
Every trait has a genetic underpinning in a trivial sense. For a physiological attribute, a psychological proclivity, or a behavior, to develop some stretches of DNA are expressed, transcribed into RNA, and, via a complex network of regulation, translated into polypeptides, forming proteins that form the building blocks for traits. Both DNA and a developmental environment are necessary for traits to develop. In this sense, not only is everything genetically caused, but also environmentally caused, what Kitcher (2001) terms the “interactionist consensus.”2
Take for example the claim that “curly hair is genetic.” For any individual to possess curly hair, the expression of genes in an environment are required, making genes and the environment causal contributors due to their necessary influence. As this is true for all traits, it does not say anything interesting about curly hair. Instead of merely considering causal contribution or necessity, a more useful sense of genetic causation involves difference making. What is it that makes a difference to hair texture? Is hair curly rather than straight because of differences in genes or differences in environments?
A popular contemporary difference making account of causation is the interventionist account (Woodward 2003). This theory of causation defines causes and effects in parallel to contemporary methods used to infer causality in experimental science. Causes and effects are treated as variables (X) and (Y), which can take on different values (x1, x2,…xn). X causes Y if an intervention on X—a manipulation that changes its value, say from x1 to x2—results in a subsequent change to the value of Y, for example, from y1 to y2. In this example, X is a deterministic cause of Y, but the interventionist account can similarly handle probabilistic causes. X can make a difference to Y if an intervention on X, changing its value, changes the probability distribution of Y. In both scenarios, X makes a difference to Y, and thus fulfils the criteria as a cause of Y.
Under a strict philosophical account, an intervention is a special kind of manipulation, which changes the value of X without accidentally altering the value of any other variables in the system. In this way, any observed changes to Y, as well as any downstream effects of Y, can be ascribed to changes to X alone, unhampered by confounding variables. Whereas precise direct genetic modifications are looking increasingly promising with the advent of new technologies such as CRISPR, in current scientific practice strict interventions are still often not possible (Chakrabarti et al. 2019). Manipulating a target variable is often done indirectly via the manipulation of a causally upstream variable such as inducing mutations using radiation exposure (Bedell et al. 1997). Other experimental manipulations are what Woodward (2008) calls “fat handed,” wherein an attempted manipulation of variable X inevitably changes the value of other variables (W, V, etc.). These manipulations can trigger mechanisms that influence the effects of interest, confounding experimental results. Moreover, in science and particularly human genetic research, cause and effect must often be inferred without carrying out any manipulations. Therefore, it is important to understand that the interventionist account does not set out to comprehensively describe the methods of causal ascertainment used by scientists; instead, it provides a way of specifying what it means to be a cause. In other words, this account defines what a causal relationship is rather than proscribing the means to investigate it. Because causes are understood using the criteria of “idealized” manipulations, causal relationships can be identified even when no manipulation is practically possible. This corresponds to the idea that some experiments, observational studies, and other methods of causal inference often set out to identify what would have happened if we were able to manipulate the cause in question.
Considerations of “what would have happened” are a common tool for understanding difference making in philosophical accounts of causation. A counterfactual theory of causation stipulates that X causes Y if, had X not occurred, Y would also have not occurred (Lewis 1973). Interventionist causation can be understood in counterfactual terms: X not occurring corresponds to a case where an intervention on X changes the value of X, say from present to absent, which results in a change to the value of Y from occurring to not occurring.
Counterfactual accounts often address individual-level causation (also called “token” or “actual” causation), wherein one aims to understand what makes a difference in a particular instance. For example, “would my hair still be curly if I had different genes?” is a counterfactual claim pertaining to an individual in a single particular instance. In individual cases, the values of the cause variable include the actual value (the genes that I actually possess), and some other possible value used as a counterfactual contrast. How to specify the counterfactual value is a general problem for counterfactual accounts, and many have proposed complicated solutions (for example, see Menzies 2004). In genetics, this is no exception. For complicated traits involving many genes, it is unclear how many genes would be different in a counterfactual scenario and precisely how they would differ. Some philosophers have suggested that counterfactuals be specified by the “normal” alternative to the actual situation (Hitchcock and Knobe 2009). In genetics, this would correspond to the most prevalent allelic variations in the population. But this becomes complicated when there are multiple “normal” and prevalent alternatives, such as observed in the human ABO blood group system. This approach also relies on an assessment of the population most relevant to the individual in question. Whereas the B allele is dominant in Asian populations, it is relatively uncommon among Caucasians (Dean 2005). For an individual, this raises the question: Should you counterfactually consider the most prevalent allele in the local geographic population or in the wider demographic one or perhaps the population containing individuals who are most similar to the token individual in other genetic respects? The answer is unclear. It is for this reason that individual-level causal claims in genetics are rarely made, although some exceptions are discussed in the section Single-Gene Traits.
Instead, most genetic causal explanations involve “type” causation, which refers to general causal relationships with multiple instantiations such as: Do differences in genes cause differences in hair texture? Or more specifically, do differences in gene G cause differences in hair texture within the general population?
SINGLE-GENE TRAITS
Genetic influence is traditionally divided into single-gene or qualitative traits and polygenic or quantitative traits. This distinction is somewhat artificial, as shall become clear in this section. For simplicity I shall refer to single-gene traits throughout this section, beginning with a seemingly textbook example, and gradually show the complexities involved in the cases that undermine this terminology.
Single-gene traits are sometimes referred to as Mendelian traits,3 as the influence of single genes can be observed to follow Mendel's principles of inheritance. The presence or absence of these traits are thought to occur because of differences in a single section of DNA. Menkes disease, described in the section Two Senses of the Gene, is an example of a trait caused by a single gene, which was first conceptualized using a Mendelian gene concept, and is now thought of in the molecular sense, referring to a single identifiable locus that specifies a gene product. The genes associated with these traits are often protein coded, although some single-gene traits are known to be caused by mutations in regulatory genes, corresponding to a Mendelian gene concept (Kondo et al. 2002). Single-gene traits often present a case in which the molecular and Mendelian gene concepts overlap, as sections of DNA that are inherited in discrete units following predictable patterns are also responsible for the production of a discrete trait. These traits are often studied from a molecular angle to understand the mechanisms and possible intervention pathways leading to trait development.
An idealized or textbook version of a single-gene trait is one where a gene (G) takes just a few values corresponding to different genotypes at a single locus, and these different values correspond to distinct (often qualitatively so) values of a trait (T). The ABO blood group is an example of this: it has three alleles that in combination correspond to six genotypes. These genotypes result in four possible blood groups, which specifically correspond to distinct phenotypes, red blood cell antigens (Fig. 1). This kind of mapping is a form of specificity, a feature of causal relationships related to perceived explanatory depth (Woodward 2010). However, many apparently single-gene traits do not have this precisely specific relationship. Instead of mapping neatly to a single trait, the effects of single genes are often pleiotropic, influencing many distinct phenotypes. Whereas mutations in ATP7A are understood as characterizing a single disease (Menkes), the disease is characterized by a variety of symptoms, such as growth retardation, kinky hair, tissue abnormality, and neurological impairment. Even in the textbook blood group example, the specificity of the mapping between genotype and phenotype is imperfect, as the A and B blood groups are multiply realized by different genotypes (Fig. 1). Additionally, there appear to be pleiotropic effects of these blood type genes, with O genotypes associated with stomach ulcers and lower levels of blood-clotting proteins, and A genotypes associated with gastric cancer (Dean 2005).
Figure 1.
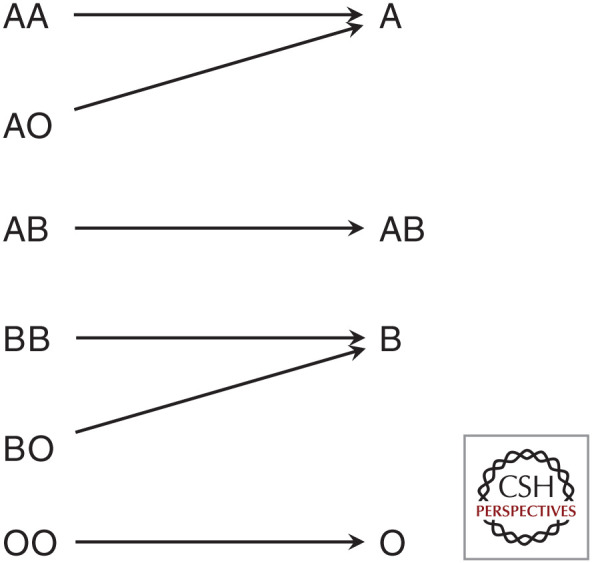
The ABO blood group is often used to show a specific relationship between genotype and phenotype, although two blood types (A and B) are multiply realized by two different genotypes (A, AO and B, BO).
As indicated above, specificity can also be disrupted when the same trait is multiply realized by multiple genes. Osteogenesis imperfecta (OI) is a connective tissue disorder caused by mutations in two genes on different chromosomes: COL1A1 (chromosome 17) and COL1A2 (chromosome 7). This means that neither mutation in COL1A1 or COL1A2 is necessary for OI to occur, although both are sufficient. However, both genes fulfil the difference-making criteria under an interventionist account of causation, as described in the section Causal Contribution and Difference Making. An intervention on either gene, changing it from a wild- to mutant-type, would make a difference to the occurrence of OI (although as I describe below, this difference is not deterministic). Like Menkes disease and many other single-gene traits, OI also demonstrates pleiotropy, with mutations in the genes causing bone fragility, blue sclera, thin skin, and hearing loss (Fig. 2; Byers 1994).
Figure 2.

An unspecific causal relationship between genotype and phenotype. (A) Osteogenesis imperfect (OI) is multiply realized by two different genes: COL1A1 and COL1A2. (B) When effects of these genes are characterized by symptoms, both COL1A1 and COL1A2 are pleiotropic for the same set of traits.
OI shows a further problematic assumption about single-gene traits. It is often assumed that qualitative traits arising from single genes are highly penetrant, meaning that individuals with the associated genes inevitably develop the trait. However, many single-gene traits such as OI show incomplete penetrance, meaning that some individuals with identical COL1A1 or COL1A2 mutations do not present with the disease at all. This shows that the genes involved in qualitative traits are best thought of in terms of probabilistic, as opposed to deterministic, causation, as discussed in the section Causal Contribution and Difference Making. Penetrance can be influenced in a number of ways. For traits characterized by a gene mutation, the mutation type and location can influence the penetrance and expressivity (variation in how the trait is expressed) of the trait. Some mutations may reduce the amount of a protein produced by a gene sequence, while others result in a disrupted molecule. The penetrance of other traits is influenced by the genetic background of the individual that may harbor other susceptibility genes, meaning they are not single-gene traits after all. Niemi et al. (2018) found that the penetrance of neurodevelopmental disorders thought to be monogenic could in part be explained by commonly inherited genetic variation. Last, more broad environmental factors, such as diet and lifestyle, can influence the penetrance of a single-gene trait. This has been observed for many genes involved in cancer (Shawky 2014).
The causal implications of penetrance and expressivity can be understood in terms of the invariance and stability of the causal relationship. When a causal relationship is maintained across a large number of different background conditions, it is deemed to be stable. All other things being equal, the more stable a causal relationship, the more causal explanatory power that relationship is thought to have (Woodward 2010). Traits that vary in their penetrance or expressivity due to environmental perturbations or differences in genetic background exhibit limited stability.
Invariance refers to how a causal relationship is maintained across changes to the values of the causal variable and is also thought to relate to causal explanation (Woodward 2010). When different types of mutations are considered as different values of the cause, then they can be mapped to different values of the effect characterized by penetrance or expressivity. In some cases, penetrance indicates an invariant causal relationship between mutation type and trait, as different mutation values map to different degrees of trait penetrance. To return to OI, mutations that disrupt the carboxy terminal of the corresponding collagen molecule are associated with higher penetrance and expressivity compared to amino-terminal disruptions. This is because alterations at this end of the molecule are more disruptive during protein folding, resulting in overfolded proteins with limited functionality (Arnold and Fertala 2013).
Some single-gene traits have the unique feature of being used to explain both group differences and individual phenotypes. When mutations in a single gene are known causes of a trait, the relevant counterfactual for a causal claim is clear; it is the absence of what is usually a single-gene mutation or a reversion to a “wild-type” genotype. Wild types for single-gene traits, particularly disease traits, are generally ubiquitous and homogenous across populations, which avoids the need to appeal to norms as discussed in the section Causal Contribution and Difference Making. Individuals with a mutated form of ATP7A can easily imagine a counterfactual scenario in which they had a functional copy of the gene and thus no disease. This corresponds with the causal claim that in an individual's particular case, ATP7A is the cause of Menkes disease.4
For type- or group-level explanations, the causal claim takes the following form: differences in gene G (values g1, g2,…gn) cause differences in trait T (values t1, t2,…tn) across many populations. This kind of claim is rooted in a body of research that has observed associations between individuals with genotypic values at a particular loci (g1, g2,…gn) with individuals with differences in trait values (t1, t2,…tn) within a population. When an association between G and T is found, then causation between genes and the trait is inferred because the gene appears to be a difference maker in the population(s) studied. In many of these studies, environments are trivially important in the sense that they are necessary for trait development; however, variation in the environment does make a difference as to whether the trait is present or absent (although see the discussion of penetrance above). Another way of thinking about this is that the relationship between gene and phenotype is relatively stable across a large number of environmental backgrounds. A consequence of this is that under a difference-making account of causation, the causal role of environments is marginal for single-gene traits.
Single-gene traits are often subject to the “gene for” rhetoric, which is sometimes interpreted as entailing genetic determinism, which is the thesis that this gene causes the trait in all background conditions. Phenylketonuria (PKU) is often used as a counterexample to this claim, as environmental modifications (a low phenylalanine diet) alter disease presentation. In this case, genotype interacts with the dietary environment, and both genes and environments are considered to be difference makers. One can imagine that an intervention on either the PAH gene, or on an individual's diet will make a difference to the absence, presence, and severity of the disease. This can be interpreted as an extreme case of penetrance being influenced by environmental factors, showing the instability of the causal relationship between genotype and phenotype.
A more charitable interpretation of “gene for” claims is that single genes cause these kinds of traits across a range of normal background conditions (Okasha 2009). Thus, the causal relationship between gene and phenotype can be assessed as relatively stable, given a set of normal background conditions. However, as discussed above, variation in the penetrance and expressivity of genotypes across a range of environmental conditions and differences in genetic backgrounds undermines this assumption.
POLYGENIC TRAITS: FAMILY-BASED STUDIES
Most traits are influenced by multiple genes, termed polygenic traits. Often these traits are quantitative, forming a continuous distribution within populations, or in other words, the effect variable takes on many different values. Differences in trait values are the products of differences in many genes as well as environmental factors. The key scientific aim for the study of quantitative traits has traditionally been to understand the degree to which genes and environments influence trait differences. This is the crux of the “nature–nurture” debate and has historically been studied using heritability estimates.
Heritability is a statistical parameter that seeks to partition the relative contribution of genetic and environmental variation to population-level differences. To obtain a heritability estimate, phenotypic variance (VP) is decomposed into the sum of environmental variance (VE) and genetic variance (VG) (Equation 1),5 which itself can be further subdivided into additive (VA) and nonadditive genetic variance (Equation 2). Dominance variance (VD) arises from the interactions of different alleles at the same locus, while epistasis variance (VI) occurs when alleles at one locus influence the expression of alleles at another. The remaining genetic variance—that which contributes without influencing other loci or alleles—is additive genetic variance. VA is the only kind of genetic variance that responds to artificial and natural selection, as VD and VI are context dependent; they depend upon particular combinations of genes for their expression. VD effects are not transmitted from parents to offspring, whereas VA effects are predictably inherited between generations based on principles of inheritance and genetic similarity (Knopik et al. 2017). For this reason, only VA is used to reliably predict evolutionary adaptation (Nagylaki 1992). Broad heritability (H2) is the proportion of phenotypic variance that can be accounted for by all genetic differences (Equation 3), and narrow heritability (h2) concerns the proportion accounted for by additive genetic variance alone (Equation 4) (Nagylaki 1992).6
| (1) |
| (2) |
| (3) |
| (4) |
Equations 3 and 4 result in a heritability estimate between 0 and 1, with a high H2 or h2, indicating that trait differences are largely due to (additive) genetic differences, and a low H2 or h2, suggesting that trait differences are largely due to environmental differences.
In human genetic research, genetic differences are inferred by identifying family resemblances, which are due to the probabilities of inheriting the same sections of DNA. Because of this, the Mendelian gene concept is used in heritability studies. This is assessed by using phenotypic data from individuals sharing a common family environment, with varying degrees of genetic relatedness, such as twins, siblings, parents, and adopted children. Twins are of particular interest to geneticists, as they provide a natural experiment: monozygotic (identical [MZ]) twins share 100% of their segregated genes and are twice as genetically similar7 than dizygotic (fraternal [DZ]) twins (Knopik et al. 2017). This means that if MZ twin pairs have greater phenotypic similarities compared to DZ twins, those similarities can be attributed to genetics, given a number of assumptions. For example, it is assumed in these cases that both MZ and DZ twin pairs experience the same degree of environmental differences between them. This premise has been contested by some (Kendler et al. 1993; Horwitz et al. 2003), although supported by others, for instance by studying twin pairs with mistaken zygosity (Scarr and Carter-Saltzman 1979; Borkenau et al. 2002). More details of family-based designs can be found in Thapar and Rice (2021), McAdams et al. (2021), and Hwang et al. (2021).
As twins develop in the same family environment, a distinction is made between the common family environment, which is shared by siblings (C), and the nonshared environment (E), which is specific to an individual. Combined with additive genetic variance (A), these components make up phenotypic variance in the ACE model (Equation 5). Twins studies are used to infer narrow heritability, as the effects of additive genetic variation (A) are estimated (although see the Appendix for a complication with this estimate).
| (5) |
Statistical associations between genetic factors and phenotypes derived from twin and family studies are often used to infer causal relationships (for some references to these kinds of causal claims, see Lynch and Bourrat 2017, p. 15). However, there is an ongoing debate as to whether these associations truly demonstrate causality. Some authors contest that heritability relates to any form of causality (Sarkar 1998), while others offer specific, population-level causal interpretations (Tabery 2014; Lynch and Bourrat 2017). Under the latter interpretations, genetic (or environmental) differences make a difference to trait differences, and so are considered to cause those trait differences within a population. For example, hair-curliness has a heritability between 85% and 95% (Medland et al. 2009). This means that most of the trait variance in hair-curliness can be accounted for by differences in genes within a population, indicating that genes are primarily the difference makers to this trait. As such, hair-curliness is considered to be largely genetically caused.
The population relativity of heritability also entails that the statistic cannot be used to infer causal relationships in individuals. For instance, it makes no sense to say that the curliness of my hair is 85% caused by genes, and 15% by the environment.
Populations are an important feature of genetic causation, particularly with regard to heritability. Different populations vary in terms of the constituent individuals and their genes as well as the environments in which they develop. This means that genes could have more or less of a causal influence on a trait in one population compared to another. Genetic variation may account for a large amount of trait variation in one population but not in another if they differ with respect to difference-making genetic variants. For instance, different geographical populations differ in the degree and variability of hair-curliness (Hrdy 1973; Loussouarn et al. 2007). Similarly, hair color is highly variable in European populations, with heritability estimated between 0.77 and 0.92 (Sulem et al. 2007; Lin et al. 2015). This is because European populations are more diverse with respect to several pigment-related genes compared to populations of individuals of African or Asian descent (Rees 2003).
Heritability can also vary across populations if there are consistent environmental differences between them. This is because individuals in different populations develop in different environments, and some environments make bigger differences to the trait of interest than others. Different populations could differ in the degree of environmental influence on hair-curliness through cosmetic means (artificial straightening or curling), depending upon the aesthetic preferences of each community. This shows differences in the degree of both genetic and environmental variation for hair-curliness per population, meaning that heritability estimates for this trait will differ depending upon the population studied.
The population relativity of genetic causal explanations has been leveraged as a criticism of genetic research, particularly investigations of heritability (Lewontin 1974; Bateson 2001; Rutter 2002; West-Eberhard 2003). This is related to the problem of gene–environment interaction (G×E), which occurs when the genetic influence on a trait is modifiable depending on the environment (and vice versa), undermining the additivity assumption in Equations 1, 2, and 5. When G×E occurs, genes make a different degree and type of difference in different populations, making it problematic to make general claims about the genetic causation of certain traits (for a more in-depth discussion of this problem, see Tabery 2014). Although theoretically troublesome, many researchers are skeptical that G×E is responsible for much phenotypic variance in human populations (Duncan and Keller 2011; Dick et al. 2015).
For example, early findings suggested that the heritability of childhood IQ varies with respect to socioeconomic status (SES). Turkheimer et al. (2003) found that in low SES families, the heritability of IQ was significantly lower compared to high SES families, suggesting that shared environmental differences played more or less of a causal role in IQ differences depending upon the population. However, this finding has failed to replicate in other large, quantitative genetic studies (Figlio et al. 2017), and a meta-analysis of this effect showed moderate effects in U.S. populations and no effect in European and Australian populations (Tucker-Drob and Bates 2016). Further, a more recent study by Allegrini et al. (2020), using genome-wide polygenic scores (discussed in the section Polygenic Traits: Genomic Approaches) and a range of environmental measures, found no contribution of G×E toward educational attainment. Other recent attempts to replicate G×E findings have similarly failed, such as Caspi et al.’s (2003) finding of an interaction between life stress and a single polymorphism for depression (Border et al. 2019; Dick et al. 2015).
However, Lewontin (1974) initially argued that G×Es remain problematic for ideas about genetic causation even when there are no realized influences on phenotypic variation. Lewontin's concern is that even when no G×Es are identified in studied populations (such as in Fig. 3A–C), they may exist in other unobserved or hypothetical unrealized populations (such as in Fig. 3D). This, he believes, restricts the explanatory utility of heritability estimates. He concludes that general causal claims about the relative contributions of genes and the environment cannot be made by appealing to heritability estimates, as they are relevant only to the populations observed within the corresponding study. Instead, Lewontin advocates for the use of reaction norms, a visual representation of the relative influences of genes and environment on phenotypes (Fig. 3).
Figure 3.

Norms of reaction that show the phenotypic distribution (y-axis) of difference genotypes (G1, G2, G3) across different environments (E1, E2). (A) A reaction norm where most phenotypic variance is accounted for by genetic variance, (B) where most phenotypic variance is accounted for by genetic variance, (C) where both genetic and environmental variance contribute to phenotypic variance additively, and (D) where genetic and environmental variance interact statistically, a case of G×E. The effects of G on P are relatively stable in A, although not in B, C, or D, as the values of P corresponding to values of G are altered. The effects of G on P are relatively invariant in A, B, and C, although not in D, as there is a functional causal relationship between G and P in these first three, although not the latter.
Lewontin's criticism concerns the extrapolation of heritability results and has been termed the locality objection (Sesardic 2005). Two important features of causal relationships—stability and invariance8 introduced in the section Single-Gene Traits—help to shed light on Lewontin's concerns about genetic causality. Recall that stability refers to how often a causal relationship is maintained across various background conditions and corresponds to how well an association between particular genotype values and corresponding phenotype values continue to associate across environmental changes. This is nicely shown by a norm of reaction, suggesting that reaction norms can provide a useful assessment tool for indicating explanatory depth. In Figure 3, the relationship between G and P is relatively stable in A, where there is relatively high heritability, and no gene–environment interaction. That is, the value G1 maps to roughly the same P value (represented on the y-axis), despite changes in the background environment (x-axis). In Figure 3B–D, the relationship between G and P is relatively unstable as it changes depending upon the environment. Conversely, the relationship between E and P is relatively stable across changes in G in Figure 3B where the heritability is low, although not in A, C, or D.
Invariance refers to how a causal relationship is maintained across changes to the values of the causal variable, rather than the background conditions. For instance, an invariant relationship between E and P would occur if, when changes are made to E, corresponding changes to the value of P can be expected, often following a predictable functional relationship. For example, if E represented nutrient load and P plant height, then it would be expected that changes to E (increasing or decreasing nutrient load) would correspond to a functional change in P (increased or decreased growth). Instead of the actual genotype and phenotype values needing to be maintained, as with stability, invariance occurs when there is a functional causal relationship maintained between the two variables.
For heritability and reaction norms, the invariance of the relationship between G and P is represented by how variations in G map to variations in P. In Figure 3, the relationship between G and P is invariant in A, B, and C, but not D. This is because a functional relationship can be seen in A, where the same values of G map to P irrespective of the environment, and in B and C, where the values of G map to different values of P in different environments, although the differences between genotypes remain the same between environments. In D, however, no functional relationship is maintained across the different values of G and P.
Both stability and invariance capture how easily the relationship between genes and phenotypes can be altered and are not restricted to the study of genetic causation. For instance, randomized controlled trials (RCTs), considered the gold standard for causal research, study causal relationships between treatment and outcome across different demographic, physiological, and genetic backgrounds. Only when a treatment produces the desired outcome across many different backgrounds is the relationship between treatment and outcome considered causal. This means that Lewontin's in principle criticism is not restricted to genetic causation but would apply to any cases where stability and invariance are low. Like with clinical and other scientific research, genetic causal relationships that are reliably maintained across a sufficient amount of variation in genetic, demographic, and environmental backgrounds can be thought of as good genetic causal explanations. As noted above, most human quantitative traits fit this paradigm, with even those initially thought to be subject to G×Es maintaining relatively stable genetic effects across a range of backgrounds.
A second criticism of heritability questions the independence of genes and environments. Correlations between particular genotypes and environments (rGEs) can occur when an individual's genotype causally influences the environment in which they develop, or when there is a common cause of an individual's genotype and of their environment. rGEs are commonly divided into three different types: active, evocative (also called reactive), and passive (Plomin et al. 1977; Scarr and McCartney 1983) and are further addressed in McAdams et al. (2021) and Thapar and Rice (2021).
For example, children who inherit genes contributing to intelligence are also more likely to develop in a more stimulating parental environment compared to others. This is a passive rGE, where children “inherit” environments that are correlated with their genetics. One way these correlations can occur is through “genetic nurture,” also termed “dynastic effects,” where the parental genotype influences the child's phenotype via the parental phenotypes’ impact on the child's environment (Fig. 4). At a population level, genes and environments are correlated, although when this is not explicitly detected the phenotypic variance derived from both will be attributed only to genes. Evidence for passive rGEs comes from studies of adopted subjects—where passive correlations between genes and the environment have been severed—or by examining the influence of noninherited parental genes determined by molecular means. In adoptive subjects, behaviors related to depression, anxiety, and anger have been found to be influenced by the (nonbiological) adoptive parents’ behaviors, which themselves are thought to be in part genetically influenced (Rhoades et al. 2011; Grabow et al. 2017). Molecular studies have identified the influence of nontransmitted parental alleles on offspring's educational attainment. This suggests a genetic nurture effect, through the influence of noninherited parental alleles (Kong et al. 2018), although the effects of inherited parental alleles, expressed in both parents and children, are also likely to be at play (Trejo and Domingue 2018).
Figure 4.
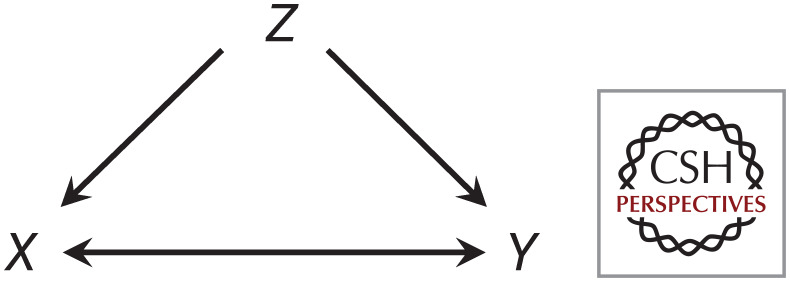
The possible causal relationships that may underlie an association between two variables. If X and Y are associated within a population, it may be that X causes Y, Y causes X, or both are caused by another variable, Z.
Active rGEs occur when an individual with a particular genotype is more likely to put themselves in a certain environment. That is, an individual's genotype influences their phenotype via the environment (Fig. 4). For example, children with particular genes may be more likely to spend more time in the library and actively participate in class, thus seeking out an environment containing more intellectual resources. There is good evidence that children actively shape their environment (Ambert 1997), and significant heritabilities have been found for environmental variables such as SES, television viewing, quality of social support, and family warmth9 (Plomin and Bergeman 1991). In an adolescent twin study, heritable life events were found to be correlated with depressive symptoms, suggesting an environmental influence on later behaviors indicative of active rGEs (Silberg et al. 1999). Molecular genetic studies support this, finding associations between polygenic variation and many of the above environmental exposures, which are correlated with later outcomes such as educational attainment, schizophrenia, and body-mass index (Krapohl et al. 2017).
Evocative rGEs occur when other people differentially interact with an individual based on their phenotype, which is an expression of a particular genotype. For example, parents and teachers are thought to differentially respond to children who behave differently due to genotypic differences. This could include teachers encouraging and supporting gifted children, furthering their intellectual development via the provision of an enriched environment and resulting in a multiplying effect of genotype on the phenotype. Conversely, special attention may be paid to struggling children who are intellectually delayed for genotypic reasons. Successful interventions on these children may allow them to “catch up” and, as such, the evocative rGE may have a “canceling out” effect on the child's genetic influence on phenotype. Like active rGEs, evocative rGEs arise when an individual's genotype causes them to experience a particular kind of environment (Fig. 4).
Because of their identical underlying causal structure, active and evocative rGEs are difficult to empirically disentangle. Differences in stressful life events, as well as socioeconomic, educational, and occupational status, have all been shown to have some genetic influences (Rutter and Silberg 2002), although this may be due to active or evocative explanations. Adoption studies have shed some light on the possibility of evocative rGE. For example, Ge et al. (1996) and O'Connor et al. (1998) both found that adoptive children with biological parents with antisocial behavior disorder were more likely to experience hostile parenting in their adoptive families, suggesting a biologically elicited parenting response. Molecular genetic methods can also be used to investigate evocative rGE hypotheses. Avinun and Hariri (2019) found that polygenic risk scores for obesity predicted early life stress, which in turn predicted depressive symptoms in adulthood. This finding coheres with an evocative explanation, whereby individuals with a genetic predisposition toward obesity suffer from depression due to bullying and mistreatment as a reaction to their weight. Similarly, Sallis et al. (2020) found an association between polygenic risk scores for schizophrenia and trauma exposure in childhood or adolescence, suggesting that certain genotypes put individuals at risk of evoking particular environmental influences including domestic violence, sexual abuse, and emotional and physical cruelty.
Debate remains about how to interpret rGEs, particularly active and reactive forms. Some argue that active rGEs are simply a “natural manifestation of the genotype,” and thus any resulting variance should be attributed to genetics. Others believe that these kinds of indirect heritable effects should be treated as a separate source of variance (for a comprehensive discussion of this debate, see Lynch 2017). Accepting reactive rGEs as caused by genetic differences leads to some uncomfortable consequences when one begins to consider their application in prejudicial societies. If a genetic basis of skin color is the cause societal prejudice against the skin color phenotype leading to impoverished environments, then any resulting effects, such as correlated low IQ, would be attributed to genetics. Thinking about genetic causation in this way seems to have, as Block (1995, p. 116) puts it, “…nothing to do with our ordinary socially important ideas of causation and is often violently in conflict with them.” An alternative approach is to argue that the attribution of traits to genes versus the environment depends upon the phenotype being studied (Lynch 2017). Whereas this approach might more appropriately cohere with intuitive ideas about causation, it could also lead to inconsistent interpretations within genetics as a field of study.
This interpretative point also highlights the context dependency of rGE effects. In a society without racial prejudice, rGEs relating to IQ and other phenotypes would be eliminated. Active and reactive rGEs rely upon the provision of particular environments for genotypes to elicit corresponding active or reactive effects. To take another example, children with a particular genetic predisposition for seeking out stimulating environments will only be at an advantage when those environments are available to them. This highlights the potential relationship between active and reactive rGEs and G×Es, as indirect genetic effects in many cases will only occur in certain environmental contexts.
POLYGENIC TRAITS: GENOMIC APPROACHES
More recently, scientists have moved from trying to understanding how much genes and environments make a difference, to trying to identify which stretches of DNA make a difference. Instead of genetic causal inference using family resemblances, genome-wide association studies (GWASs) examine genetic variants associated with trait differences. Commonly, these variants are single-nucleotide polymorphisms (SNPs), which are single base-pair differences that tend to vary among individuals within the general population. Associated traits are studied in two ways: First, as a binary variable, such as the presence or absence of a trait, where participants are selected using a case-control design. Alternatively, for polygenic traits, populations of individuals that vary continuously with respect to the trait of interest and SNP variants associated with quantitative trait differences are studied.
SNPs do not correspond to a molecular gene concept, as they correspond to base-pair variation in just a single nucleotide, and so on their own are not responsible for a gene product. To put this in perspective, the molecular gene ATP7A, which codes for a copper transport protein and is responsible for Menkes disease, spans over 150,000 base pairs (Tumer et al. 1992). SNPs can fall within coding and nonregions of DNA, and many SNPs may be present within a single molecular gene.
However, SNPs can act as difference makers to and thus causes of a trait. For example, the SNP rs671 is located on the molecular gene ALDH2, which produces a protein involved in alcohol metabolism.10 An rs671 allele, particularly found in East Asian populations, produces an inactive subunit in the protein that corresponds to ALDH2, resulting in individuals who cannot (when homozygous for the allele) or with limited ability to (when heterozygous) metabolize acetaldehyde (Yoshida et al. 1984). This is akin to a Mendelian gene, which is defined in reference to observably inherited effects.
Most SNPs occur in noncoding regions of DNA (Barreiro et al. 2008). They may have no phenotypic effects or effect sizes too small to be detectable through the study of inheritance patterns, meaning that they neither qualify as Mendelian nor molecular genes. However, even noncoding alterations to DNA can make a difference to phenotypic expression via the regulation of coding sequences. SNPs with strong phenotypic associations are sometimes used as markers for potential molecular candidate genes. For example, 11 SNPs have been recently identified as having a strong association with squamous cell carcinoma, with seven of these occurring within known pigment-related genes (Chahal et al. 2016; Ioannidis et al. 2018).
For quantitative traits traditionally studied using heritability methods, some SNP associations have been found. However, the associations between individual SNPs and trait differences account for only a small percentage of genetic influenced estimated by heritability. Human height has a heritability of ∼80% when estimated using the family-based study designs described in the section Polygenic Traits: Family-Based Studies (Visscher 2008). A SNP, for example, rs1042725, which is a SNP located in the HMGA2 gene, accounts for ∼0.3% of height variation (Weedon et al. 2007). Added together, the effects of ∼50 significantly associated SNPs in genome-wide association studies initially accounted for just ∼5% of height or 1/16th of heritability (Visscher 2008).
These results differ dramatically compared to heritability estimates from family-based studies. For other traits such as intelligence, or many psychological disorders, the gap between SNP heritability and family-based heritability is similarly immense (Turkheimer 2011; Schaffner 2016; Matthews and Turkheimer 2019). This problem, termed “missing heritability” (Manolio et al. 2009), could be due to a number of factors, such as parental effects (Eichler et al. 2010) or limitations with current molecular genetic approaches, such as the inability to detect variants with penetrance too low to statistically associate with a trait and/or genetic variants too rare to be captured in a GWAS using common SNPs (Maher 2008; Manolio et al. 2009; Zuk et al. 2014).
To combat the problem of SNPs with a low statistical association, Yang et al. (2010) estimated the heritability of height by estimating the variance in height that could be explained by the aggregate effects of large numbers of common SNPs. As an aggregate, much more phenotypic variance was explained by these SNPs, yielding a heritability of ∼45%. Many of these SNPs would not meet the significance threshold for explaining variance in height individually. This suggests that a significant amount of missing heritability is due to genetic variants with small effect sizes.
Aggregates of weighted SNP associations with particular traits found in a given individual can be used to form genome-wide polygenic scores (GPSs), sometimes also termed polygenic risk scores. GPSs are calculated using population data from GWASs, which give an indication of the degree of phenotypic variance the summation of effects of particular gene variants are associated with. Individuals can obtain a unique score based on the type and number of SNP associations found in their genome. GPS thus represents a move from population-level explanations used in SNP heritability to individual-level explanations. They also offer a further abstraction away from the gene concept. As Matthews and Turkheimer (2019) point out, DNA variants that were already divorced from the molecular gene concept are amalgamated into theoretical statistical variables and used as predictive factors. Most do not consider GWASs or GPSs as methods that address causal relationships, with researchers in this field explicitly reporting on associations and addressing issues of prediction, although some still tend to use causal language (e.g., Marigorta and Navarro 2013; Plomin and von Stumm 2018). Some of the conditions under which causal claims about GPSs are justified are outlined in Dudbridge (2021).
Many GWASs include only common SNPs, that is, those that have a minor allele frequency (MAF) (the frequency that the second most common allele occurs within a population), greater than 5% (Marouli et al. 2017). Studies that have included rare (MAF <1%) and low-frequency (1<MAF<5%) SNPs have recovered additional phenotypic variance (e.g., for height, see Wood et al. 2014; Marouli et al. 2017), and the promise of whole-genome studies suggests that for some traits, the missing heritability gap will one day be closed (Yang et al. 2015; Wainschtein et al. 2019).
USING GENES TO IDENTIFY ENVIRONMENTAL CAUSES
The above research describes efforts to identify genetic influences and, in some cases, candidate genes as causes. Another focus, largely in the field of epidemiology, has been to identify candidate environmental variables termed exposures. Whereas heritability studies estimate the relative influence of the environment on trait differences, epidemiologists strive to identify particular environmental variables of interest, akin to a candidate gene approach for the “nurture” side of the debate.
A classic challenge in epidemiology is inferring causal relationships from correlational data. When an exposure (environment) is associated with a trait (outcome), it may be causal, or there may be reverse causation; the trait may increase the likelihood of that particular variable. For example, an association between alcohol and high blood pressure could mean that alcohol consumption causes increases in blood pressure, or that individuals increase their alcohol intake in response to high blood pressure. Associations between environments and traits may also be confounded, where some other unknown variable causes both the trait and the associated exposure, for instance, if both high blood pressure and alcohol consumption shared a common cause (Fig. 5). To circumvent these issues, exposures must be randomly assigned, such as in RCTs. However, for many exposures, these kinds of study designs are not possible.
Figure 5.

The different possible forms of gene–environment correlation. (A) No correlation between genes and environment occurs, and both influence phenotype independently. (B) Genes causally influence environments, corresponding to active and reactive cases of gene–environment correlation. (C) Parental genes causes both the child's genotype and the child's environment (via genetic nurture), correlating the child's genes and environment.
Mendelian randomization (MR) is a method of causal inference that uses existing genetic variation as a “randomizing” factor for exposures associated with health outcomes, which in some cases include environmental factors such as the propensity to drink alcohol (Chen et al. 2008) or to smoke (Bjørngaard et al. 2013). These environmental factors are causally influenced by known genes, which differ within populations, and, as such, genetic differences result in systematically different environmental exposures. When these kinds of exposures are known to have impacts on health, MR studies can be used to investigate the causal links between environmental variables and health outcomes. Note that for MR studies, the Mendelian gene concept is used, as genes are defined in reference to their effects. However, the term “Mendelian” does not reference this gene concept, rather it references Mendel's laws of independent assortment and segregation, which are capitalized on as a way of randomizing environmental exposures in a population.
Because an individual's genome is determined at conception, it cannot be altered by the outcome (e.g., a disease) of interest, or by the associated intermediate trait (such as smoking or drinking alcohol), which the genetic variants influence in an MR study. This rules out reverse causation from outcome to genome and from environment to genome. It is also assumed that there are no factors that act as common causes of both genes and outcome traits or of genes and environments of interest. This makes these types of genes useful instrumental variables for exposures (environments) of interest, enabling researchers to make causal claims about the relationships between particular environments and other (outcome) traits (Smith and Ebrahim 2003; Davies et al. 2018, see also Richmond 2021).
MR studies can be used to demonstrate the difference-making nature of particular environmental variables to traits often but not always related to disease. The effect variables can be discrete, such as the absence or presence of disease or continuous, such as blood pressure. The causal variables of interest (exposures) are generally grouped into discrete categories based on the associated genetic variants. For instance, alcohol intake is categorized into three values based the associated genotype for ALDH2: a gene related to alcohol consumption. These are low intake (homozygous recessive), intermediate intake (heterozygote), and high intake (homozygote dominant) (Chen et al. 2008).
When associations are made between the instrumental genotype and trait (outcome), and it is assumed that this association is only due to genetic influences that act via the intermediate environmental variable (exposure), it not only demonstrates the corresponding trait risk for individuals with those genes, but also for individuals with different genotypes who are exposed to the same environmental variable. Type causal claims are then made about the influence of the environmental variable on the trait of interest within a population.
Notably, the observed indirect effects of genotypes on environmental exposures reflected in these types of MR studies can be used to identify causal pathways and explain GWAS results. When a genetic variant is associated with a trait in using GWAS methods, it is usually assumed that these associations are due to genetic influences on internal, biological pathways. However, MR studies have demonstrated that single gene variants can have significant phenotypic effects via environmental exposures. This is important as components of GWAS results may reflect the contribution of modifiable causal influences that can be leveraged for public health benefits (Gage et al. 2016).
Curiously, these kinds of MR associations correspond to some types of gene–environment correlation (rGE), whereby genetic effects on phenotype are mediated by an intermediate environmental variable. Thus, MR can show how indirect environmentally mediated effects are likely to contribute to heritabilities estimated by both family-based and molecular methods. As discussed in the section Polygenic Traits: Family-Based Studies, rGEs are traditionally regarded as controversial and difficult to causally interpret in quantitative and behavioral genetics. Philosophical discussions of rGEs suggest that biases in causal reasoning and differing notions of the concepts of “genetic” and “environmental” as well as different ideas about the phenotype of interest can influence their interpretation and account for disagreement among scholars (Lynch 2017; Lynch and Bourrat 2017). This controversy does not currently exist in MR studies, suggesting something interesting about the particular types of rGEs included. One possibility is that the corresponding rGEs examined in these studies are incredibly stable. That is, the relationships between genotype and environment in MR studies persist across a large range of other environmental and genetic background factors as there is a negligible effect of G×E (Wang et al. 2019). This is consistent with the fact that, as mentioned in the section Polygenic Traits: Family-Based Studies, controversial (often hypothetical) examples of rGE such as those related to racial discrimination reflect situations where the indirect genetic effect is sensitive to environmental context; that is, they are subject to Lewontin's locality (G×E) objection if counterfactual environments are considered.
Whereas MR provides strong evidence for causal explanations exploiting genetic properties, it is not without its limitations. Determining a causal relationship between an exposure and a trait depends on there being identifiable candidate genes for exposures of interest, and so many environmental variables cannot currently be studied using these methods. The genetic effect must also be large enough to produce reliable associations stably across populations. Finally, the genetic effect on the disease of interest must occur only through the exposure of interest and not via other pathways if the gene is pleiotropic, otherwise the gene can act as a common cause of both the exposure of interest and the disease trait. Details of MR are further discussed in Richmond (2021), Dudbridge (2021), Sanderson (2021), and Hwang et al. (2021).
CONCLUDING REMARKS
Genes can be understood as factors that are transmitted between generations: Mendelian genes, and as stretches of DNA responsible for a gene product, molecular genes. For single-gene traits, the Mendelian and molecular gene concepts can overlap. In quantitative genetics, which studies polygenic traits, the Mendelian gene concept has been traditionally used, although molecular techniques such as GWASs indicate a move toward the use of DNA markers to identify molecular genes.
Causation can be usefully understood in genetics as corresponding to difference makers, factors that determine or, more commonly, raise the probability of their effects. There are two types of difference-making causal claims that can be made within genetics. The first considers the causes of the trait for a given individual. This type of causation makes sense when considering single-gene traits, where counterfactual reasoning is possible with a high degree of certainty. It may also be appropriate in MR studies for the same reasons, when a single environmental variable is identified as causal. Individual causal reasoning has been employed by some geneticists when discussing GPS, although this kind of causal talk is controversial.
The second type of causation is “type” causal claims. Type causal claims can be made about the influences of single genes and isolated environmental exposures. In the former case, they can be deterministic or probabilistic, and in the latter case, they are generally probabilistic. Type causal claims also apply where differences in genes account for differences in trait values within a population. For polygenic traits, where a large number of genes are involved and most candidate genes are unknown, only population-relative “type” causal claims make sense. The exact values of genetic and environmental variables are unknown for polygenic traits, and the degree to which genes causally influence a trait is relative to how much environmental influence there is on that trait. These influences depend upon the degree and type of genetic and environmental variation in the population under study, making causal claims of this nature population relative. When a relationship between genetic and environmental variation and trait variation can be reliably observed across multiple populations and contexts (stability), there are good grounds for more general causal claims to be made about the relative influences on that trait. This is true for single-gene traits, polygenic traits, and those influenced by environmental exposures.
ACKNOWLEDGMENTS
I thank Paul Griffiths and Davide Serpico for comments on an earlier version of this paper and the editors of this volume for their many helpful comments on early drafts. This research was generously supported by the Sir John Templeton Foundation (Genetics and Human Agency initiative).
Footnotes
Editors: George Davey Smith, Rebecca Richmond, and Jean-Baptiste Pingault
Additional Perspectives on Combining Human Genetics and Causal Inference to Understand Human Disease and Development available at www.perspectivesinmedicine.org
Additionally, the way that people interpret genetic information is influenced by biases and aspects of our cognition. This is true for both laypeople and professionals who are educated about genetics. (For a review, see Lynch et al. 2019.)
Note that the term interaction here differs from interaction as discussed in the context of gene–environment interactions and gene–gene interactions (see sections Single-Gene Traits and Polygenic Traits: Family Based Studies). Interaction in Kitcher's (2001) sense simply refers to both genes and the environment being necessary components for a phenotype.
See Online Mendelian Inheritance in Man (www.omim.org) for a comprehensive and up-to-date catalog of known single-gene traits.
This of course assumes that the disease presentation in each individual is not multiply realized by the presence of another gene mutation with similar effects. Whereas there is never absolute certainty when counterfactually reasoning in individual single-gene cases, given the rarity of these kinds of mutations, it is highly unlikely that a given individual would possess two different mutations overdetermining the same trait. Thus, these inferences can be made with a high degree of certainty.
An error term is sometimes included in Equations 1 and 2 or is otherwise incorporated into the VE term.
In humans, VD and VI do not appear to influence phenotypic variance to a great degree for most quantitative traits (Hivert et al. 2020), meaning that h2 and H2 estimates should generally converge.
See the Appendix for a discussion of the genetic similarity among twins, and the appropriate use of broad and narrow heritability in these studies.
Stability and invariance are just two of many features that philosophers have discussed as being important to causality and causal explanation. For a discussion on the limitations of these features, see Strand and Oftedal (2019) and Oftedal (2020).
Although these estimates themselves may be in part due to gene–environment correlations.
Alcohol is metabolized first into acetaldehyde, which is then further metabolized and excreted from the body. The gene product of ALDH2 aids in this second process.
APPENDIX: HERITABILITY AND TWIN STUDIES
The ACE model used in twin research includes the effects of additive genetic variance, and shared and nonshared environments (Equation 5). This assumes that genetic similarly between twins is due solely to additive genetic variance, corresponding to narrow heritability (Equation 4). Heritability is estimated by comparing the correlations found between monozygotic twins (rMZ) and dizygotic twins (rDZ), with similarities attributed to both additive heritable factors and shared environment (Knopik et al. 2017, p. 351):
Although h2 is assumed in the equations above, twin studies do not exactly estimate broad or narrow heritability. Twins share 100% of their DNA: additive (VA), dominant, and epistatic (VD+I). DZ twins are expected to share approximately 50% of VA, but only 25% of VD and VI, due to the probability of DZ twins inheriting the same two interacting alleles from their parents (in the case of dominance, these are both at the same loci, and in the case of epistasis, these are at different loci) (Falconer and MacKay 1996), so that:
When MZ and DZ twins are compared, the resulting heritability estimate is somewhere in between broad and narrow (Falconer and MacKay 1996, p. 174):
REFERENCES
*Reference is also in this collection.
- Allegrini AG, Karhunen V, Coleman JRI, Selzam S, Rimfeld K, von Stumm S, Pingault JB, Plomin R. 2020. Multivariable G-E interplay in the prediction of educational achievement. PLoS Genet 16: e1009153. 10.1371/journal.pgen.1009153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambert AM. 1997. Parents, children, and adolescents: interactive relationships and development in context. Haworth, New York. [Google Scholar]
- Arnold WV, Fertala A. 2013. Skeletal diseases caused by mutations that affect collagen structure and function. Int J Biochem Cell Biol 45: 1556–1567. 10.1016/j.biocel.2013.05.017 [DOI] [PubMed] [Google Scholar]
- Avinun R, Hariri AR. 2019. A polygenic score for body mass index is associated with depressive symptoms via early life stress: evidence for gene–environment correlation. J Psychiatr Res 118: 9–13. 10.1016/j.jpsychires.2019.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. 2008. Natural selection has driven population differentiation in modern humans. Nat Genet 40: 340–345. 10.1038/ng.78 [DOI] [PubMed] [Google Scholar]
- Bateson P. 2001. Behavioural development and Darwinian evolution. In Cycles of contingency: developmental systems and evolution (ed. Oyama S, et al. ), pp. 149–166. MIT Press, Cambridge, MA. [Google Scholar]
- Bazzett TJ. 2008. An introduction to behavior genetics. Sinauer, Sunderland, MA. [Google Scholar]
- Bedell MA, Jenkins NA, Copeland NG. 1997. Mouse models of human disease. Part I: Techniques and resources for genetic analysis in mice. Genes Dev 11: 1–10 10.1101/gad.11.1.1 [DOI] [PubMed] [Google Scholar]
- Bjørngaard JH, Gunnell D, Elvestad MB, Smith GD, Skorpen F, Krokan H, Vatten L, Romundstad P. 2013. The causal role of smoking in anxiety and depression: a Mendelian randomization analysis of the HUNT study. Psychol Med 43: 711–719. 10.1017/S0033291712001274 [DOI] [PubMed] [Google Scholar]
- Block N. 1995. How heritability misleads about race. Cognition 56: 99–128. 10.1016/0010-0277(95)00678-R [DOI] [PubMed] [Google Scholar]
- Border R, Johnson EC, Evans LM, Smolen A, Berley N, Sullivan PF, Keller MC. 2019. No support for historical candidate gene or candidate gene-by-interaction hypotheses for major depression across multiple large samples. Am J Psychiatry 176: 376–387. 10.1176/appi.ajp.2018.18070881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkenau P, Riemann R, Angleitner A, Spinath FM. 2002. Similarity of childhood experiences and personality resemblance in monozygotic and dizygotic twins: a test of the equal environments assumption. Pers Individ Dif 33: 261–269. 10.1016/S0191-8869(01)00150-7 [DOI] [Google Scholar]
- Byers PH. 1994. Osteogenesis imperfecta. In Connective tissue and its heritable disorders: molecular, genetic, and medical aspects (ed. Royce PM, Steinmann B), pp. 317–350. Wiley, New York. [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. 2003. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301: 386–389. [DOI] [PubMed] [Google Scholar]
- Chahal HS, Lin Y, Ransohoff KJ, Hinds DA, Wu J, Dai HJ, Qureshi AA, Li WQ, Kraft P, Tang JY, et al. 2016. Genome-wide association study identifies novel susceptibility loci for cutaneous squamous cell carcinoma. Nat Commun 7: 12048. 10.1038/ncomms12048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti AM, Henser-Brownhill T, Monserrat J, Poetsch AR, Luscombe Nm, Scaffidi P. 2019. Target-specific precision of CRISPR-mediated genome editing. Mol Cell 73: 699–713.e6. 10.1016/j.molcel.2018.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Smith GD, Harbord RM, Lewis SJ. 2008. Alcohol intake and blood pressure: a systematic review implementing a Mendelian randomization approach. PLoS Med 5: e52. 10.1371/journal.pmed.0050052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies NM, Holmes, MV, Smith GD. 2018. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362: k601. 10.1136/bmj.k601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawkins R. 1976. The selfish gene. Oxford University Press, New York. [Google Scholar]
- Dean L. 2005. The ABO blood group. National Center for Biotechnology Information, Bethesda, MD. www.ncbi.nlm.nih.gov/books/NBK2267 [PubMed] [Google Scholar]
- Dick DM, Agrawal A, Keller MC, Adkins A, Aliev F, Monroe S, Hewitt JK, Kendler KS, Sher KJ. 2015. Candidate gene–environment interaction research: reflections and recommendations. Perspect Psychol Sci 10: 37–59. 10.1177/1745691614556682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Dudbridge F. 2021. Polygenic Mendelian randomization. Cold Spring Harb Perspect Med 10.1101/cshperspect.a039586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan LE, Keller MC. 2011. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry 168: 1041–1049. 10.1176/appi.ajp.2011.11020191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. 2010. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 11: 446–450. 10.1038/nrg2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. 1996. Introduction to quantitative genetics, 4th ed. Longman, Harlow, UK. [Google Scholar]
- Figlio DN, Freese J, Karbownik K, Roth J. 2017. Socioeconomic status and genetic influences on cognitive development. Proc Natl Acad Sci 114: 13441–13446. 10.1073/pnas.1708491114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage SH, Smith GD, Ware JJ, Flint J, Munafo MR. 2016. G=E: what GWAS can tell us about the environment. PLoS Genet 12: e1005765. 10.1371/journal.pgen.1005765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Conger RD, Cadoret RJ, Neiderhiser JM, Yates W, Troughton E, Stewart MA. 1996. The developmental interface between nature and nurture: a mutual influence model of child antisocial behavior and parent behaviors. Dev Psychol 32: 574–589. 10.1037/0012-1649.32.4.574 [DOI] [Google Scholar]
- Grabow AP, Khurana A, Natsuaki MN, Neiderhiser JM, Harold GT, Shaw DS, Ganiban JM, Reiss D, Leve LD. 2017. Using an adoption biological family design to examine associations between maternal trauma, maternal depressive symptoms, and child internalizing and externalizing behaviors. Dev Psychopathol 29:1707–1720. 10.1017/S0954579417001341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths PE. 2001. Genetic information: a metaphor in search of a theory. Philos Sci 68: 394–412. 10.1086/392891 [DOI] [Google Scholar]
- Griffiths PE, Neumann-Held EM. 1999. The many faces of the gene. Bioscience 49: 656–662. 10.2307/1313441 [DOI] [Google Scholar]
- Griffiths PE, Stotz K. 2006. Genes in the postgenomic era. Theor Med Bioeth 27: 499–521. 10.1007/s11017-006-9020-y [DOI] [PubMed] [Google Scholar]
- Griffiths PE, Stotz K. 2007. Gene. In Cambridge companion to the philosophy of biology (ed. Hull D, Ruse M), pp. 85–102. Cambridge University Press, Cambridge. [Google Scholar]
- Griffiths PE, Stotz K. 2013. Genetics and philosophy: an introduction. Cambridge University Press, Cambridge. [Google Scholar]
- Haig D. 2012. The strategic gene. Biol Philos 27: 461–479. 10.1007/s10539-012-9315-5 [DOI] [Google Scholar]
- Heard E, Martienssen RA. 2014. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157: 95–109. 10.1016/j.cell.2014.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock C, Knobe J. 2009. Cause and norm. J Philos 106: 587–612. 10.5840/jphil20091061128 [DOI] [Google Scholar]
- Hivert V, Sidorenko J, Rohart F, Goddard ME, Yang J, Wray NR, Yengo L, Visscher PM. 2020. Estimation of non-additive genetic variance in human complex traits from a large sample of unrelated individuals. bioRxiv 10.1101/2020.11.09.375501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg ZE, Feil R, Constancia M, Fraga M, Junien, C, Carel JC, Boileau P, Le Bouc Y, Deal CL, Lillycopr K, et al. 2011. Child health, developmental plasticity, and epigenetic programming. Endocr Rev 32: 159–224. 10.1210/er.2009-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander WF. 1955. Epistasis and hypostasis. J Hered 46: 222–225. 10.1093/oxfordjournals.jhered.a106562 [DOI] [Google Scholar]
- Horn N, Stene J, Móllekar AM, Friedrich U. 1984. Linkage studies in Menkes’ disease: the Xg blood group system and C-banding of the X chromosome. Ann Hum Genet 48: 161–172. 10.1111/j.1469-1809.1984.tb01011.x [DOI] [PubMed] [Google Scholar]
- Horwitz AV, Videon TM, Schmitz MF, Davis D. 2003. Rethinking twins and environments: possible social sources for assumed genetic influences in twin research. J Health Soc Behav 44: 111–129. 10.2307/1519802 [DOI] [PubMed] [Google Scholar]
- Hrdy D.1973. Quantitative hair form variation in seven populations. Am J Phys Anthropol 39: 7–17. 10.1002/ajpa.1330390103 [DOI] [PubMed] [Google Scholar]
- *.Hwang LD, Davies NM, Warrington NM, Evans DM. 2021. Integrating family-based and Mendelian randomization designs. Cold Spring Harb Perspect Med 10.1101/cshperspect.a039503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis NM, Wang W, Furlotte NA, Hinds DA, Bustamante CD, Jorgenson E, Asgari MM, Whittemore AS. 2018. Gene expression imputation identifies candidate genes and susceptibility loci associated with cutaneous squamous cell carcinoma. Nat Commun 9: 4264. 10.1038/s41467-018-06149-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller EF. 2000. The century of the gene. Harvard University Press, Cambridge. [Google Scholar]
- Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. 1993. A test of the equal-environment assumption in twin studies of psychiatric illness. Behav Genet 23: 21–27. 10.1007/BF01067551 [DOI] [PubMed] [Google Scholar]
- Kitcher P. 2001. Battling the undead: how (and how not) to resist genetic determinism. In Thinking about evolution: historical, philosophical, and political perspectives (ed. Singh RS, Krimbas K, Paul DB, Beatty J), pp. 396–414. Cambridge University Press, Cambridge. [Google Scholar]
- Knopik VS, Neiderhiser JM, DeFries JC, Plomin R. 2017. Behavioral genetics, 7th ed.Macmillan Learning, New York. [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, Ferreira de Lima RLL, Daack-Hirsch S, Sander A, et al. 2002. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet 32: 285–289. 10.1038/ng985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Frigge ML, Vilhjalmsson BJ, Young AI, Thorgeirsson TE, Benonisdottir S, Oddsson A, Halldorsson BV, Masson G, et al. 2018. The nature of nurture: effects of parental genotypes. Science 359: 424–428. 10.1126/science.aan6877 [DOI] [PubMed] [Google Scholar]
- Krapohl E, Hannigan LJ, Pingault JB, Patel H, Kadeva N, Curtis C, Breen G, Newhouse SJ, Eley TC, O'Reilly PF, et al. 2017. Widespread covariation of early environmental exposures and trait-associated polygenic variation. Proc Natl Acad Sci 114: 11727–11732. 10.1073/pnas.1707178114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D. 1973. Causation. J Philos 70: 556–567. 10.2307/2025310 [DOI] [Google Scholar]
- Lewontin RC. 1974. The analysis of variance and the analysis of causes. Am J Hum Genet 26: 400–411. [PMC free article] [PubMed] [Google Scholar]
- Lin BD, Mbarek H, Willemsen G, Dolan CV, Fedko IO, Abdellaoui A, De Geus EJ, Boomsma DI, Hottenga JJ. 2015. Heritability and genome-wide association studies for hair color in a Dutch twin family based sample. Genes 6: 559–576. 10.3390/genes6030559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G, Garcel A, Lozano I, Coullaudin C, Porter C, Panhard S, Saint-Léger D, de La Mettrie, R. 2007. Worldwide diversity of hair curliness: a new method of assessment. Int J Dermatol 46: 2–6. 10.1111/j.1365-4632.2007.03453.x [DOI] [PubMed] [Google Scholar]
- Lynch KE. 2017. Heritability and causal reasoning. Biol Philos 32: 25–49. 10.1007/s10539-016-9535-1 [DOI] [Google Scholar]
- Lynch KE, Bourrat P. 2017. Interpreting heritability causally. Philos Sci 84: 14–34. 10.1086/688933 [DOI] [Google Scholar]
- Lynch KE, Morandini JS, Dar-Nimrod I, Griffiths PE. 2019. Causal reasoning about human behavior genetics: synthesis and future directions. Behav Genet 49: 221–234. 10.1007/s10519-018-9909-z [DOI] [PubMed] [Google Scholar]
- Maher B. 2008. Personal genomes: the case of the missing heritability. Nature 456: 18–21. 10.1038/456018a [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthey MI, Ramos EM, Cardon LR, Chakravarti A, et al. 2009. Finding the missing heritability of complex diseases. Nature 461: 747–753. 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigorta UM, Navarro A. 2013. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet 9: e1003566. 10.1371/journal.pgen.1003566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, Fine RS, Lu Y, Schurmann C, Highland HM, et al. 2017. Rare and low-frequency coding variants alter human adult height. Nature 542: 186–190. 10.1038/nature21039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews LJ, Turkheimer E. 2019. Across the great divide: pluralism and the hunt for missing heritability. Synthese 10.1007/s11229-019-02205-w [DOI] [Google Scholar]
- *.McAdams TA, Rijsdijk FV, Zavos HMS, Pingault JB. 2021. Twins and causal inference: leveraging nature's experiment. Cold Spring Harb Perspect Med 10.1101/cshperspect.a039552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medland SE, Zhu G, Martin NG. 2009. Estimating the heritability of hair curliness in twins of European ancestry. Twin Res Hum Genet 12: 514–518. 10.1375/twin.12.5.514 [DOI] [PubMed] [Google Scholar]
- Menzies P. 2004. Difference making in context. In Causation and counterfactuals (ed. Collins JD, Hall NE, Paul LA), pp. 139–180. MIT Press, Cambridge, MA. [Google Scholar]
- Moss L. 2001. Deconstructing the gene and reconstructing molecular developmental systems. In Cycles of contingency: developmental systems and evolution (ed. Oyama S, Griffiths PE, Gray RD), pp. 85–97. MIT Press, Cambridge, MA. [Google Scholar]
- Nagylaki T. 1992. Introduction to theoretical population genetics. Springer, Berlin. [Google Scholar]
- Neumann-Held EM. 2001. Let's talk about genes: the process molecular gene concept and its context. In Cycles of contingency: developmental systems and evolution (ed. Oyama S, Griffiths PE, Gray RD), pp. 69–84. MIT Press, Cambridge, MA. [Google Scholar]
- Niemi MEK, Martin HC, Rice DL, Gallone G, Gordon S, Kelemen M, McAloney K, McRae J, Radford EJ, Yu S, et al. 2018. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562: 268–271. 10.1038/s41586-018-0566-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor TG, Deater-Deckard K, Fulker D, Rutter M, Plomin R. 1998. Genotype–environment correlations in late childhood and early adolescence: antisocial behavioral problems and coercive parenting. Dev Psychol 34: 970–981. 10.1037/0012-1649.34.5.970 [DOI] [PubMed] [Google Scholar]
- Oftedal G. 2020. Problems with using stability, specificity, and proportionality as criteria for evaluating strength of scientific causal explanations: commentary on Lynch et al. (2019). Biol Philos 35: 26. 10.1007/s10539-020-9739-2 [DOI] [Google Scholar]
- Okasha S. 2009. Causation in biology. In The Oxford handbook of causation (ed. Menzies P, Beebee H, Hitchcock C), pp. 707–725. Oxford University Press, Oxford. [Google Scholar]
- Plomin R, Bergeman CS. 1991. The nature of nurture: genetic influence on “environmental” measures. Behav Brain Sci 14: 373–386. 10.1017/S0140525X00070278 [DOI] [Google Scholar]
- Plomin R, von Stumm S. 2018. The new genetics of intelligence. Nat Rev Genet 19: 148–159. 10.1038/nrg.2017.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, Loehlin JC. 1977. Genotype-environment interaction and correlation in the analysis of human behavior. Psychol Bull 84: 309–322. 10.1037/0033-2909.84.2.309 [DOI] [PubMed] [Google Scholar]
- Rees JL. 2003. Genetics of hair and skin color. Annu Rev Genet 37: 67–90. 10.1146/annurev.genet.37.110801.143233 [DOI] [PubMed] [Google Scholar]
- Rhoades KA, Leve LD, Harold GT, Neiderhiser JM, Shaw DS, Reiss D. 2011. Longitudinal pathways from marital hostility to child anger during toddlerhood: genetic susceptibility and indirect effects via harsh parenting. J Fam Psychol 25: 282–291. 10.1037/a0022886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Richmond R. 2021. Mendelian randomization: concepts and scope. Cold Spring Harb Perspect Med 10.1101/cshperspect.a040501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter M. 2002. Nature nurture and development: from evangelism through science toward policy and practice. Child Dev 73: 1–21. 10.1111/1467-8624.00388 [DOI] [PubMed] [Google Scholar]
- Rutter M, Silberg J. 2002. Gene–environment interplay in relation to emotional and behavioral disturbance. Annu Rev Psychol 53: 463–490. 10.1146/annurev.psych.53.100901.135223 [DOI] [PubMed] [Google Scholar]
- Sallis HM, Croft J, Havdahl A, Jones HJ, Dunn EC, Smith GD, Zammit S, Munafò MR. 2020. Genetic liability to schizophrenia is associated with exposure to traumatic events in childhood. Psychol Med 10.1017/S0033291720000537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Sanderson E. 2021. Multivariable Mendelian randomization and mediation. Cold Spring Harb Perspect Med 10.1101/cshperspect.a038984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S. 1998. Genetics and reductionism. Cambridge University Press, Cambridge. [Google Scholar]
- Scarr S, Carter-Saltzman L. 1979. Twin method: defense of a critical assumption. Behav Genet 9: 527–542. 10.1007/BF01067349 [DOI] [PubMed] [Google Scholar]
- Scarr S, McCartney K. 1983. How people make their own environments: a theory of genotype environment effects. Child Dev 54: 424–435. [DOI] [PubMed] [Google Scholar]
- Schaffner KF. 2016. Behaving: what's genetic, what's not, and why should we care? Oxford University Press, Oxford. [Google Scholar]
- Sesardic N. 2005. Making sense of heritability. Cambridge University Press, Cambridge. [Google Scholar]
- Shawky RM. 2014. Reduced penetrance in human inherited disease. Egypt J Med Hum Genet 15: 103–111. 10.1016/j.ejmhg.2014.01.003 [DOI] [Google Scholar]
- Silberg J, Pickles A, Rutter M, Hewitt J, Simonoff E, Maes H, Carbonneau R, Mur Relle L, Foley D, Eaves L. 1999. The influence of genetic factors and life stress on depression among adolescent girls. Arch Gen Psychiatry 56: 225–232. 10.1001/archpsyc.56.3.225 [DOI] [PubMed] [Google Scholar]
- Smith GD. 2011. Epidemiology, epigenetics and the “gloomy prospect”: embracing randomness in population health research and practice. Int J Epidemiol 40: 537–562. 10.1093/ije/dyr117 [DOI] [PubMed] [Google Scholar]
- Smith GD, Ebrahim S. 2003. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32: 1–22. 10.1093/ije/dyg070 [DOI] [PubMed] [Google Scholar]
- Sterelny K, Kitcher P. 1988. The return of the gene. J Philos 85: 339–361. 10.2307/2026953 [DOI] [Google Scholar]
- Stotz K, Griffiths PE, Knight R. 2004. How biologists conceptualize genes: an empirical study. Stud Hist Philos Sci C 35: 647–673. [Google Scholar]
- Strand A, Oftedal G. 2019. Restricted causal relevance. Br J Philos Sci 70: 431–457. 10.1093/bjps/axx034 [DOI] [Google Scholar]
- Sulem P, Gudbjartsson DF, Stacey SN, Helgason A, Rafnar T, Magnusson KP, Manolescu A, Karason A, Palsson A, Thorleifsson G, et al. 2007. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat Genet 39: 1443–1452. 10.1038/ng.2007.13 [DOI] [PubMed] [Google Scholar]
- Tabery J. 2014. Beyond versus: the struggle to understand the interaction of nature and nurture. MIT Press, Cambridge, MA. [Google Scholar]
- *.Thapar A, Rice F. 2021. Family-based designs that disentangle inherited factors from pre-and postnatal environmental exposures: in vitro fertilization, discordant sibling pairs, maternal versus paternal comparisons, and adoption designs. Cold Spring Harb Perspect Med 10.1101/cshperspect.a038877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhodeyev ON, Shcherbakova OV. 2019. The problem of non-shared environment in behavioral genetics. Behav Genet 49: 259–269. 10.1007/s10519-019-09950-1 [DOI] [PubMed] [Google Scholar]
- Trejo S, Domingue BW. 2018. Genetic nature or genetic nurture? Introducing social genetic parameters to quantify bias in polygenic score analyses. Biodemography Soc Biol 64: 187–215, 10.1080/19485565.2019.1681257 [DOI] [PubMed] [Google Scholar]
- Tucker-Drob EM, Bates TC. 2016. Large cross-national differences in gene × socioeconomic status interaction on intelligence. Psychol Sci 27: 138–149. 10.1177/0956797615612727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumer Z, Chelly J, Tommerup N, Ishikawa-Brush Y, Tonnesen T, Monaco AP, Horn N. 1992. Characterization of a 1.0 Mb YAC contig spanning two chromosome breakpoints related to Menkes disease. Hum Mol Genet 1: 483–489. 10.1093/hmg/1.7.483 [DOI] [PubMed] [Google Scholar]
- Turkheimer E. 2011. Still missing. Res Hum Devel 8: 227–241. [Google Scholar]
- Turkheimer E, Haley A, Waldron M, D'Onofrio B, Gottesman II. 2003. Socioeconomic status modifies heritability of IQ in young children. Psychol Sci 14: 623–628. 10.1046/j.0956-7976.2003.psci_1475.x [DOI] [PubMed] [Google Scholar]
- Verga V, Hall BK, Wang SR, Johnson S, Higgins JV, Glover TW. 1991. Localization of the translocation breakpoint in a female with Menkes syndrome to Xq13. 2-q13. 3 proximal to PGK-1. Am J Hum Genet 48: 1133. [PMC free article] [PubMed] [Google Scholar]
- Visscher PM. 2008. Sizing up human height variation. Nat Genet 40: 489–490. 10.1038/ng0508-489 [DOI] [PubMed] [Google Scholar]
- Wainschtein P, Jain DP, Yengo L, Zheng Z. 2019. Recovery of trait heritability from whole genome sequence data. bioRxiv 10.1101/588020 [DOI] [Google Scholar]
- Wang H, Zhang F, Zeng J, Wu Y, Kemper KE, Xue A, Zhang M, Powell JE, Goddard ME, Wray NR, et al. 2019. Genotype-by-environment interactions inferred from genetic effects on phenotypic variability in the UK Biobank. Sci Adv 5: eaaw3538. 10.1126/sciadv.aaw3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CK. 1994. Genes made molecular. Philos Sci 61: 163–185. 10.1086/289794 [DOI] [Google Scholar]
- Weedon MN, Lettre G, Freathy RM, Lindgren CM, Voight BF, Perry JR, Elliott KS, Hackett R, Guiducci C, Shields B, Zeggini E. 2007. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet 39: 1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Eberhard MJ. 2003. Developmental plasticity and evolution. Oxford University Press, Oxford. [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Kutalik Z, Amin N, et al. 2014. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet 39: 1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward J. 2003. Making things happen: a theory of causal explanation. Oxford University Press, New York. [Google Scholar]
- Woodward J. 2008. Invariance, modularity, and all that. In Nancy Cartwright's philosophy of science (ed. Hartman S, Hoefer C, Bovens L), pp. 198–237. Taylor & Francis, New York. [Google Scholar]
- Woodward J. 2010. Causation in biology: stability, specificity, and the choice of levels of explanation. Biol Philos 25: 287–318. 10.1007/s10539-010-9200-z [DOI] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. 2010. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42: 565–569. 10.1038/ng.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH, Robinson MR, Perry JR, Nolte IM, van Vliet-Ostaptchouk JV, et al. 2015. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet 47: 1114–1120. 10.1038/ng.3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Huang IY, Ikawa M. 1984. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. Proc Natl Acad Sci 81: 258–261. 10.1073/pnas.81.1.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, Daly MJ, Neale BM, Sunyaev SR, Lander ES. 2014. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci 111: E455–E464. 10.1073/pnas.1322563111 [DOI] [PMC free article] [PubMed] [Google Scholar]