

In this study, Simoneau et al. investigated why PARPi is more effective than other DNA-damaging drugs when used to treat BRCA1/2-deficient tumors. They show that PARPi induces DSBs progressively through trans-cell-cycle ssDNA gaps, and BRCA1/2-deficient cells fail to slow down and repair DSBs over multiple cell cycles, explaining the unique efficacy of PARPi in BRCA1/2-deficient cells.
Keywords: BRCA, DNA damage, PARP inhibitor, cell cycle, replication
Abstract
PARP inhibitor (PARPi) is widely used to treat BRCA1/2-deficient tumors, but why PARPi is more effective than other DNA-damaging drugs is unclear. Here, we show that PARPi generates DNA double-strand breaks (DSBs) predominantly in a trans cell cycle manner. During the first S phase after PARPi exposure, PARPi induces single-stranded DNA (ssDNA) gaps behind DNA replication forks. By trapping PARP on DNA, PARPi prevents the completion of gap repair until the next S phase, leading to collisions of replication forks with ssDNA gaps and a surge of DSBs. In the second S phase, BRCA1/2-deficient cells are unable to suppress origin firing through ATR, resulting in continuous DNA synthesis and more DSBs. Furthermore, BRCA1/2-deficient cells cannot recruit RAD51 to repair collapsed forks. Thus, PARPi induces DSBs progressively through trans cell cycle ssDNA gaps, and BRCA1/2-deficient cells fail to slow down and repair DSBs over multiple cell cycles, explaining the unique efficacy of PARPi in BRCA1/2-deficient cells.
In the past decade, the use of inhibitors of poly(ADP-ribose) polymerase (PARP) in the clinic has significantly improved the treatment of breast, ovarian, prostate, and pancreatic cancer patients. In particular, PARP inhibitors (PARPis) have shown efficacy in patients carrying mutations in BRCA1 and BRCA2 tumor suppressor genes (O'Connor 2015; Lord and Ashworth 2017). Both BRCA1 and BRCA2 encode proteins important for DNA repair and suppression of genomic instability (Roy et al. 2012; Venkitaraman 2014). Specifically, both BRCA1 and BRCA2 proteins are critical for homologous recombination (HR), a pathway that repairs DNA double-strand breaks (DSBs) (Chen et al. 2018). BRCA1 and BRCA2 are also important for protecting stalled DNA replication forks from nucleolytic degradation, thereby suppressing genomic instability during DNA replication (Schlacher et al. 2011, 2012). Cancer cells lacking functional BRCA1 and BRCA2 proteins are highly sensitive to PARPi, suggesting that the HR defects of cancer cells confer PARPi sensitivity (McCabe et al. 2006). PARP1, the primary target of PARPi, is involved in multiple DNA repair pathways, including base excision repair (BER), HR, alternative nonhomologous end joining (alt-NHEJ), and others (Dantzer et al. 1999; Robert et al. 2009; Li and Yu 2013). The efficacy of PARPi in HR-defective cancer cells provides an example of “synthetic lethality”: PARP is not essential in HR-proficient cells, but it becomes indispensable in HR-deficient cells because cells cannot tolerate the loss of both HR and PARP-mediated DNA repair (O'Connor 2015; Lord and Ashworth 2017). Although PARPi is already widely used in the clinic and the concept of synthetic lethality has been proven by a large body of studies, how exactly PARPi kills BRCA1/2-deficient cancer cells is still not fully understood. Furthermore, whether and how other oncogenic events in cancer cells can confer PARPi sensitivity and modulate the PARPi response is still far from completely clear (D'Andrea 2018; Pilié et al. 2019). These limitations in our understanding of the mechanism of action for PARPi present an obstacle to improving the use of PARPi in the clinic.
A number of models have been proposed to explain how PARPi selectively kills BRCA1/2-deficient cancer cells. PARP1 plays an important role in BER-mediated repair of DNA single-strand breaks (SSBs) (Dantzer et al. 1999). Inhibition of PARP1 by PARPi results in a surge of SSBs in DNA, which are subsequently converted to DSBs by DNA replication forks during S phase (Bryant et al. 2005; Farmer et al. 2005). The inability of BRCA1/2-deficient cancer cells to repair DSBs may underlie their sensitivity to PARPi (Lord et al. 2008; Helleday 2011). Studies comparing different clinical PARPis reveal that the ability of PARPis to trap PARP1 on DNA is important for their efficacy in cancer cells (Murai et al. 2012; Pommier et al. 2016). This concept is further supported by recent reports that defects in RNase H2 and ALC1 render cells hypersensitive to PARPi by increasing PARP trapping (Zimmermann et al. 2018; Blessing et al. 2020; Hewitt et al. 2020; Juhász et al. 2020; Verma et al. 2021). PARPi also accelerates replication forks (Maya-Mendoza et al. 2018). In the presence of PARPi, speeding replication forks are unable to stall properly in BRCA1-deficient cells, which may reduce the ability of replication forks to cope with stress and increase DSBs. In addition, PARPi induces more chromatin bridges and multinucleation in BRCA1/2-deficient cells during mitosis, inducing cell death (Schoonen et al. 2017). Finally, two recent studies suggested that PARPi induces ssDNA gaps in BRCA-deficient cells, and that replication gaps, rather than DSBs, underlie BRCA-deficiency and therapeutic response (Panzarino et al. 2020; Cong et al. 2021). All these models provided important insights into how PARPi preferentially kills BRCA1/2-deficient cancer cells. However, it should be noted that many other chemotherapeutics also interfere with DNA replication and induce replication-associated DNA damage, but their effects on cancer cells are not identical to those of PARPi (Cheung-Ong et al. 2013). It remains unclear whether and how PARPi induces DNA damage in a way distinct from other chemotherapeutics.
In addition to inducing DNA damage, PARPi also affects the normal function and stress response of replication forks. In cells treated with the topoisomerase 1 (TOP1) inhibitor camptothecin (CPT), replication forks increasingly undergo fork reversal, a process promoting fork recovery at DNA lesions (Ray Chaudhuri et al. 2012). In this context, PARPi suppresses fork reversal by alleviating the PARP-mediated inhibition of RECQ1, a helicase that resolves reversed forks (Berti et al. 2013). We recently showed that loss of CARM1, which stimulates PARP1 at replication forks, increases Primase and DNA-directed polymerase (PrimPol)-mediated repriming at stressed forks and single-stranded DNA (ssDNA) gaps (Genois et al. 2020). Even in the absence of DNA-damaging agents, poly(ADP-ribose) (PAR) is detected at replication forks, suggesting that PARP1 is active during unperturbed DNA replication (Hanzlikova et al. 2018). PARPi inhibits the repair of unligated Okazaki fragments, which was speculated to generate DSBs indirectly (van Wietmarschen and Nussenzweig 2018). While loss of PARP1 functions at replication forks clearly affects replication and the stress response, whether it contributes to the efficacy of PARPi in BRCA1/2-deficient cancer cells is still not known.
In this study, we investigated how PARPi induces DNA damage during consecutive cell cycles. To our surprise, PARPi induces a robust DNA damage response in the second but not the first S phase. During the first S phase, PARPi induces a transient cell cycle delay, and cells continue to complete the cell cycle. During the second S phase, however, a surge of DSBs is detected, leading to a profound cell cycle delay. Importantly, we found that PARPi induces ssDNA gaps behind replication forks in the first S phase in a PrimPol-dependent manner. Although cells activate postreplication repair mechanisms to fill the gaps, gap repair cannot complete in the presence of PARPi until the next S phase, generating DSBs in a trans cell cycle manner. In the second S phase, BRCA1/2-deficient cells fail to activate ATR and suppress firing of replication origins, resulting in continuous DNA synthesis and increased fork collapse. Furthermore, BRCA1/2-deficient cells are unable to recruit RAD51 to repair collapsed forks. Thus, PARPi has a unique ability to induce DSBs progressively in a trans cell cycle manner, and loss of BRCA1/2 exacerbates DSB accumulation over multiple cell cycles and simultaneously disrupts repair, rendering BRCA1/2-deficient cells particularly susceptible to PARPi. These findings provide a molecular basis for improving the use of PARPi in cancer therapy.
Results
PARPi causes a significant delay in the second cell cycle
To understand how PARPi induces DNA damage, we first investigated when PARPi exerts its cytotoxic effects during the cell cycle. To follow the cell cycle status of individual cells in asynchronously growing cell populations, we stained the chromatin-bound PCNA (a component of DNA replication fork) and DNA in U2OS cells with antibody and DAPI, respectively (Supplemental Fig. S1A). In addition, we pulse-labeled newly synthesized DNA with EdU (Supplemental Fig. S1A). Cells undergoing DNA replication displayed high levels of chromatin-bound PCNA and became EdU-positive (EdU+) right after pulse-labeling. Based on PCNA and DNA staining intensities, subpopulations of cells in G1, S, and G2 phases were identified (Supplemental Fig. S1A). Furthermore, based on the DNA contents of PCNA-positive cells, replicating cells were divided into early, mid, and late S-phase subpopulations (Supplemental Fig. S1A). Importantly, when we followed EdU+ cells over time, they gradually moved across various cell subpopulations at different cell cycle stages (Supplemental Fig. S1A), allowing us to monitor how the cells that were initially in S phase progressed through the cell cycle.
Using the strategy above, we analyzed how S-phase cells progress through two consecutive cell cycles in the presence or absence of PARPi. Treatment of asynchronously growing U2OS cells with 2 or 10 µM of the PARPi olaparib for 4 h efficiently reduced baseline PAR levels (Supplemental Fig. S1B). In the absence of olaparib, most of the EdU+ cells completed the first cell cycle and entered the second S phase by 24 h (Fig. 1A; Supplemental Fig. S1C). By 32 h, a significant fraction of the EdU+ cells completed the second S phase and entered G2. In the presence of olaparib, the EdU+ cells displayed a transient delay in the first cell cycle (Fig. 1A; Supplemental Fig. S1C, see 4 to 14 h). Nonetheless, most of the EdU+ cells entered the second S phase by 24 h (Fig. 1A; Supplemental Fig. S1C). Notably, however, the olaparib-treated EdU+ cells progressed through the second S phase much more slowly than control cells (Fig. 1A; Supplemental Fig. S1C). Even after 48 h of olaparib treatment, most of the EdU+ cells were still in the second S phase (Fig. 1A). Significant delays of the second S phase were observed in cells treated with 2 or 10 µM of olaparib (Supplemental Fig. S1C). In addition, two other PARPis, veliparib and talazoparib, also induced significant delays in the second S phase (Supplemental Fig. S1D,E). Of note, talazoparib was more potent than olaparib and veliparib in inducing a delay in the second cell cycle, suggesting that the ability of PARPis to trap PARP on DNA is important for their cell cycle effects. Together, these results reveal that PARPi primarily exerts its cell cycle effects in the second but not the first S phase.
Figure 1.

PARPi induces a robust DNA damage response in the second S phase. (A) The effects of PARPi in two consecutive cell cycles. U2OS cells were pulse-labeled with 5 µM EdU for 15 min and released in the presence or absence of 10 µM olaparib. Cells were pre-extracted with detergent at indicated time points before immunofluorescence analysis of PCNA and EdU detection by click chemistry. Individual cells are plotted according to DNA content (x-axis), PCNA intensity (y-axis), and EdU positivity (colored in red). EdU-labeled cells were classified into G1, early S, mid S, late S, and G2 subpopulations (see Materials and Methods; Supplemental Fig. S1A), and the fractions of EdU-labeled cells in the subpopulations are shown in the stacked bar charts. (B) PARPi induces a more robust ATR response in the second S phase. U2OS cells were treated with 10 µM Olaparib for the indicated durations. Levels of the indicated proteins were analyzed by western blot. See Supplemental Figure S2A for quantifications. (C) PARPi triggers a more robust G2/M checkpoint after the second S phase. Cells were pulse-labeled with 2 µM EdU for 15 min and released in 0, 2, or 10 µM olaparib. To prevent cells from going through the first or second mitosis, 150 ng/mL nocodazole was added 0 or 24 h after EdU labeling, and cells were collected 24 h later. The relative levels of p-H3 S10-positive and EdU+ cells in PARPi-treated samples are normalized to DMSO samples. Error bars indicate SD of two independent experiments. Significance was calculated with a two-tailed Student's t-test. (*) P-value < 0.05, (**) P-value < 0.01. (D–F) PARPi induces a more robust DNA damage response in the second S phase. Cells were treated with 10 µM olaparib for the indicated durations, and S-phase cells were pulse-labeled with 10 µM EdU during the last 15 min. Foci of γH2AX (D), p-RPA32 S33 (E), or RAD51 (F) were analyzed by immunofluorescence. The total fluorescence of foci (D,E) or number of foci (F) in S-phase cells were quantified. (Red bar) Median intensity or focus number. More than 100 S-phase cells were analyzed in each sample (n > 100). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (n.s.) P-value > 0.05. (G) The entry into the second S phase is required for the robust DNA damage response after PARPi treatment. Cells were pulse-labeled with 2 µM EdU for 15 min and then treated with DMSO, 10 µM olaparib, 1 µM palbociclib, or olaparib and palbociclib for 32 h. Mean γH2AX focus intensity of EdU+ cells was quantified. (Red bar) Median intensity. More than 150 EdU+ cells were analyzed in each sample (n > 150). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (*) P-value < 0.05.
A previous study suggested that BrdU labeling of DNA increases sister chromatid exchange (Dillehay et al. 1983), raising the possibility that EdU-labeled DNA may interfere with DNA replication in the second S phase and affect the PARPi response. To test this possibility, we labeled a fraction of the genome with EdU in the first S phase and then allowed cells to progress into the second S phase (Supplemental Fig. S1F). In the second S phase, cells were exposed to PARPi or DMSO, and replication tracts were analyzed by sequential CldU/IdU labeling and DNA fiber assay. Replication fork instability was scored by uneven fork progression, which is reflected by high CldU/IdU or IdU/CldU ratios (Maya-Mendoza et al. 2018). Indeed, fork unevenness was increased in EdU-labeled replication tracts (Supplemental Fig. S1F), suggesting that EdU increases fork instability in the second S phase. However, PARPi exposure in the second S phase did not further increase fork instability in EdU-labeled tracts, ruling out the possibility that the effects of PARPi in the second S phase are caused by EdU labeling.
PARPi induces a more robust DNA damage response in the second S phase
To understand why PARPi-treated cells slow down in the second S phase, we asked whether the ATR checkpoint pathway is activated during this period. Chk1 and RPA32, two substrates of ATR, were phosphorylated at low levels during the first cell cycle after PARPi exposure (Fig. 1B). After cells entered the second cell cycle, the levels of phosphorylated Chk1 (p-Chk1 S317) and RPA32 (p-RPA32 S33) were significantly increased in the cell population (Fig. 1B; Supplemental Fig. S2A). To compare the PARPi-induced G2/M checkpoint responses in two consecutive cell cycles, we used nocodazole to block mitosis in the first or second cell cycle and measured the levels of mitotic cells positive for phosphorylated histone H3 Ser10 (Fig. 1C). At either 2 or 10 µM, olaparib induced a more significant reduction in mitotic cells in the second cell cycle than in the first. Thus, PARPi-treated cells slow down in the second cell cycle because of a checkpoint response.
The robust checkpoint effects in the second cell cycle indicate that PARPi induces a stronger DNA damage response in the second S phase. Indeed, PARPi induced higher levels of γH2AX in individual cells in the second S phase (Fig. 1D; Supplemental Fig. S2B). Consistent with the western data, PARPi also increased p-RPA32 S33 immunofluorescence in individual cells in the second S phase (Fig. 1E; Supplemental Fig. S2C). RAD51 is known to form nuclear foci at DSBs and collapsed replication forks (Petermann et al. 2010). While only baseline RAD51 foci were detected during the first S phase after PARPi exposure, a robust induction of RAD51 foci was observed in the second S phase (Fig. 1F; Supplemental Fig. S2D). These results support the idea that PARPi induces more replication stress and/or DSBs in the second S phase.
PARPi may induce more replication stress and/or DSBs in the second S phase simply because cells are exposed to PARPi for a longer period. To test whether the entry into the second S phase is important for the robust damage response, we arrested the PARPi-treated EdU+ cells in the first G2 with the CDK1 inhibitor RO-3306 (Supplemental Fig. S2E), or in the second G1 with the CDK4/6 inhibitor palbociclib (Fig. 1G; Supplemental Fig. S2F). After 24 h of PARPi exposure, the EdU+ cells arrested in the first G2 or the second G1 displayed much lower levels of γH2AX than those entered into the second S phase. These results suggest that PARPi does not simply induce DNA damage over time but requires successive rounds of DNA replication to exert the full extent of its cytotoxicity. Furthermore, these results raise the possibility that certain PARPi-induced DNA structures affect DNA replication differently in the first and second S phases.
PARPi induces ssDNA gaps behind replication forks
To identify the DNA structures induced by PARPi that may affect the second S phase, we considered the possibility of ssDNA gaps behind replication forks for two reasons. First, PARPi inhibits the repair of unligated Okazaki fragments (Hanzlikova et al. 2018), which may leave ssDNA gaps on the lagging strand. Second, PARPi suppresses fork reversal by allowing RECQ1 to resolve reversed forks (Berti et al. 2013), which may increase the use of PrimPol at stressed forks and PrimPol-generated ssDNA gaps on the leading strand (Fig. 2A; Quinet et al. 2020). If PARPi increases ssDNA gaps on both leading and lagging strands, both daughter arms of replication forks would be more susceptible to cleavage by the S1 nuclease, which specifically cuts ssDNA, leading to shortening of nascent DNA tracts after S1 digestion. To test this possibility, we performed DNA fiber assay in the presence and absence of PARPi. Nascent DNA was sequentially labeled with CldU and IdU, and only the IdU labeling was done in the presence or absence of PARPi (Fig. 2B). Furthermore, DNA fibers were treated with the S1 nuclease before the IdU/CldU ratio of replication tracts was measured. Because the S1 nuclease specifically cleaves ssDNA, the IdU-labeled DNA should be shortened and the IdU/CldU ratio should be reduced if PARPi induces ssDNA gaps in nascent DNA. Indeed, the IdU/CldU ratio was significantly reduced by S1 after cells were treated with PARPi (Fig. 2C,D, lanes 2,4 of each panel), demonstrating that PARPi induces ssDNA gaps in nascent DNA.
Figure 2.

PARPi generates persistent ssDNA gaps behind replication forks. (A) A schematic showing the possible effects of RECQ1 and PrimPol on stressed replication forks. (B) Experimental design for C and D. Olaparib was used at 10 µM. Cells were permeabilized and treated with or without S1 nuclease for 30 min before DNA fiber analysis. (C,D) PARPi generates ssDNA gaps behind replication forks in a PrimPol- and RECQ1-dependent manner. More than 125 CldU/IdU double-positive replication tracts were analyzed in each sample (n > 125). (Red bar) Median IdU/CldU ratio. Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (*) P-value < 0.05, (n.s.) P-value > 0.05. (E,F) Analysis of proteins at and behind replication forks. HEK293T cells were treated DMSO or 10 µM olaparib for 60 min, and nascent DNA was labeled with 10 µM EdU during the last 20 min. Cells were either processed for isolation of proteins on nascent DNA (iPOND) to capture proteins at progressing forks (Fork) or were chased with thymidine (T-chase) for 45 min to capture proteins on postreplicative DNA. Levels of the indicated proteins in input cell extracts and iPOND samples were analyzed by western blot. (G) PARPi and RAD51i block the repair of PARPi-induced ssDNA gaps. U2OS were treated as in B except cells were incubated in DMSO, 10 µM olaparib, or 50 µM RI-1 (RAD51i) for 4 h before S1 nuclease digestion. (Red bar) Median IdU/CldU ratio. More than 125 CldU/IdU double-positive replication tracts were analyzed in each sample (n > 125). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (H,I) PARPi-induced ssDNA gaps persist into the second S phase. (H) U2OS cells were treated as in B except cells were incubated in DMSO or 10 µM olaparib for 0 or 24 h before S1 nuclease digestion. (Red bar) Median IdU/CldU ratio. (I) U2OS were treated as in B except cells were incubated in DMSO or 10 µM olaparib for 6 h, then in 30 µM VdU with or without 10 µM olaparib for 16 h prior to S1 nuclease digestion. To exclude rereplicated fibers, only CldU+ IdU+ VdU− fibers were analyzed. (H,I) More than 125 CldU/IdU double-positive replication tracts were analyzed in each sample (n > 125). Significance was determined with a Mann–Whitney U-test. (****) P-value < 0.0001, (n.s.) P-value > 0.05. (J) Effects of PARPi exposure in the first or second cell cycle on fork stability in the second S phase. Cells were labeled with EdU for 90 min in the absence or presence of 10 µM olaparib, and then given 26.5 h to progress to the second cell cycle. Cells were either not exposed to PARPi throughout the time course, exposed to PARPi during the last 4 h, or exposed to PARPi throughout the time course. At the end of the time course, cells were analyzed by CldU/IdU labeling (20 min each) and DNA fiber assay. The longer/shorter ratio (CldU/IdU or IdU/CldU) of replication tracts in EdU+ fibers was determined. The first two samples are also shown in Supplemental Fig. S1F. Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (n.s.) P-value > 0.05.
Next, we asked how PARPi-induced ssDNA gaps are generated. To test whether PrimPol is responsible for the formation of ssDNA gaps, we analyzed PrimPol knockdown cells with the S1 nuclease assay (Fig. 2C). If loss of PrimPol reduces the formation of ssDNA gaps on the leading strand, the S1 nuclease will not be able to cut one of the daughter arms of replication forks efficiently, and nascent DNA tracts would not be shortened by S1 in the DNA fiber assay. In PrimPol-depleted cells, the IdU/CldU ratio of replication tracts was reduced by PARPi even in the absence of S1 nuclease (Fig. 2C, lanes 5,7; Supplemental Fig. S3A–C), suggesting that stressed forks rely on PrimPol to progress efficiently when fork reversal is suppressed by PARPi. Importantly, the IdU/CldU ratio of replication tracts in PARPi-treated PrimPol knockdown cells was not further reduced by the S1 nuclease (Fig. 2C, lanes 7,8; Supplemental Fig. S3B,C), showing that S1 cannot cut both daughter arms of replication forks in the absence of PrimPol. Knockdown of RECQ1 in PARPi-treated cells is expected to stabilize reversed forks and reduce the use of PrimPol (Fig. 2A). Similar to that in PrimPol-depleted cells, the IdU/CldU ratio of replication tracts in PARPi-treated RECQ1 knockdown cells was not reduced by the S1 nucleases (Fig. 2D, lanes 7,8; Supplemental Fig. S3D–F). Together, these results suggest that PARPi induces ssDNA gaps behind replication forks at least in part by reducing reversed forks and promoting PrimPol-mediated repriming (Fig. 2A).
PARPi prevents complete repair of postreplicative ssDNA gaps
To follow the fate of PARPi-induced ssDNA gaps, we used the isolation of proteins on nascent DNA (iPOND) assay to test whether certain DNA repair proteins are recruited to these gaps in postreplicative DNA (Sirbu et al. 2012). The nascent DNA at progressing replication forks was pulse-labeled with EdU in the presence and absence of PARPi, and the proteins associated with forks were captured by iPOND and detected by western blot (Fig. 2E). Furthermore, the proteins on postreplicative DNA were also captured after a 45-min thymidine chase and detected by western blot. As expected, replication fork proteins PCNA and RPA70 were detected at forks but not on postreplicative DNA (Fig. 2E). In contrast, RAD51 was detected on postreplicative DNA in a PARPi-induced manner (Fig. 2E). The binding of RAD51 to postreplicative DNA is dependent upon PrimPol and RECQ1 (Fig. 2F; Supplemental Fig. S3G), suggesting that RAD51 is recruited to ssDNA gaps. Of note, we cannot exclude the possibility that slow fork progression contributes to the reduction of RAD51 on DNA in PrimPol- and RECQ1-depleted cells (Fig. 2C,D). Consistent with the recruitment of RAD51 to DNA, chromatin-bound RAD51 was slightly increased after 4 h of PARPi treatment (Supplemental Fig. S3H). While PARPi-induced RAD51 binding to DNA was detected by iPOND and chromatin fractionation, no increase of RAD51 foci was observed in the first cell cycle (Fig. 1F). Chromatin-bound RAD51 was further increased after 24 h of PARPi treatment (Supplemental Fig. S3H), which correlates with the formation of RAD51 foci in the second S phase. A previous study showed that replication-associated RAD51 foci are only detectable after prolonged replication stress and fork collapse (Petermann et al. 2010). Thus, the RAD51 detected by iPOND and chromatin fractionation in the first S phase likely reflects the low levels of RAD51 recruited to ssDNA gaps, whereas the RAD51 foci in the second S phase are likely caused by the high levels of RAD51 at collapsed forks.
To test the effects of PARPi and RAD51 on the repair of PARPi-induced ssDNA gaps, we designed a two-step experiment to follow the clearance of ssDNA gaps (Fig. 2G). In the first step, nascent DNA was sequentially labeled with CldU and IdU, and the IdU labeling was done in the presence or absence of PARPi. This step allows PARPi to induce ssDNA gaps in IdU-labeled DNA. In the second step, IdU was removed from the media, and cells were given 4 h to repair ssDNA gaps. To assess the impacts of PARPi and RAD51 on repair, cells were treated with DMSO, PARPi, or the RAD51 inhibitor (RAD51i) RI-1 during the 4 h (Budke et al. 2012). At the end of this experiment, the levels of remaining ssDNA gaps in IdU-labeled DNA were determined with the S1 nuclease assay. When cells were treated with DMSO during the repair period, IdU-labeled DNA was not shortened by S1 (Fig. 2G, lanes 3,4), showing that ssDNA gaps were fully repaired. In contrast, IdU-labeled DNA was significantly shortened by S1 when cells were incubated in PARPi (Fig. 2G, lanes 5,6) or RAD51i (Fig. 2G, lanes 7,8) during the repair period. Thus, when PARPi is continuously present or RAD51 is inhibited, PARPi-induced ssDNA gaps are not completely repaired. These results suggest that RAD51 promotes the repair of postreplicative ssDNA gaps, possibly through template switching between sister chromatids (Vanoli et al. 2010). Moreover, in the presence of PARPi, the repair of ssDNA gaps cannot complete even when RAD51 is functional, suggesting that the trapping of PARP by PARPi may prevent the complete filling of ssDNA gaps. We noted that only RAD51, but not RPA, was detected at postreplicative ssDNA gaps in the presence of PARPi (Fig. 2E). This is possibly because a RAD51-containing but RPA-free intermediate is stuck by trapped PARP during gap repair.
PARPi-induced ssDNA gaps persist into the second S phase
Given that PARPi blocked the repair of PARPi-induced ssDNA gaps 4 h postreplication (Fig. 2G), we asked whether PARPi-induced ssDNA gaps can persist into the second S phase in the presence of PARPi. To test this possibility, we performed the two-step assay above and kept cells in PARPi for 24 h to allow them to enter the second S phase (Fig. 2H). As shown in Figure 1A, most of the PARPi-treated EdU+ cells have entered the second S phase in 24 h. Remarkably, even after 24 h of PARPi exposure, IdU-labeled DNA was still significantly shortened by S1 (Fig. 2H, lanes 7,8), suggesting that PARPi-induced ssDNA gaps can persist into the second cell cycle. The shortening of IdU-labeled DNA by S1 was reduced in the second cell cycle compared with that right after IdU labeling (Fig. 2H), indicating that some ssDNA gaps were repaired over time. Nonetheless, significant levels of ssDNA gaps were clearly present in IdU-labeled DNA in the second cell cycle.
To exclude the possibility that the ssDNA gaps in IdU-labeled DNA detected in the second cell cycle are generated during the second round of DNA synthesis, we treated cells with PARPi and arrested them in the second G1 with palbociclib (Supplemental Fig. S3I). Even without the second S phase, IdU-label DNA was still shortened by S1 after 24 h of PARPi treatment (Supplemental Fig. S3I, lanes 7,8). To test more directly whether the PARPi-induced ssDNA gaps persist into the second S phase, we carried out CldU-IdU labeling and PARPi treatment as above and added VdU to cells after they finished the first S phase (6 h after CldU/IdU labeling) (Fig. 2I). After 24 h of PARPi treatment, we performed the S1 nuclease assay and specifically analyzed the CldU+ IdU+ VdU− DNA fibers. These CldU+ IdU+ VdU− fibers may come from unreplicated DNA regions of cells in the second S phase or cells that have not reached the second S phase. Because most of the EdU+ cells have reached early S phase after 24 h in PARPi (Fig. 1A), we infer that the majority of the CldU+ IdU+ VdU− fibers are from cells in the second S phase. The IdU-labeled DNA in the CldU+ IdU+ VdU− fibers was significantly shortened by S1 (Fig. 2I; Supplemental Fig. S3J), suggesting that the PARPi-induced ssDNA gaps from the first S phase persist into the second S phase without another round of DNA synthesis.
To directly test whether the PARPi exposure in the first S phase affects fork stability in the second S phase, we labeled a fraction of the genome with EdU in the first S phase and then allowed cells to progress into the second S phase (Fig. 2J). In one sample, cells were not exposed to PARPi. In another sample, cells were treated with PARPi in the second S phase. In the third sample, cells were exposed to PARPi during EdU labeling in the first S phase and kept in PARPi until the second S phase. All samples were analyzed by CldU/IdU labeling and DNA fiber assay in the second S phase. Strikingly, fork unevenness was increased in EdU-labeled tracts only when PARPi was added in the first S phase (Fig. 2J), showing that the PARPi exposure during EdU labeling in the first S phase is critical for the fork instability in EdU-labeled DNA in the second S phase.
Persistent ssDNA gaps cause DSBs in a trans cell cycle manner
The persistence of PARPi-induced ssDNA gaps into the second S phase will inevitably increase collisions of replication forks with ssDNA gaps, leading to fork collapse and one-ended DSBs. In addition, resection of DSBs at collapsed forks will increase ssDNA. To test this possibility, we performed native BrdU staining to measure the exposure of ssDNA in cells treated or untreated with PARPi (Fig. 3A). In cells treated with PARPi for 8 h, a modest increase of ssDNA was observed (Fig. 3A; Supplemental Fig. S4A,B). After 24 h of PARPi treatment, the levels of ssDNA were drastically increased (Fig. 3A; Supplemental Fig. S4A,B), showing that PARPi induces much more ssDNA in the second S phase. Knockdown of PrimPol did not affect the progression of PARPi-treated cells through the second S phase but reduced PARPi-induced ssDNA (Fig. 3B; Supplemental Fig. S4C,D), linking the formation of ssDNA gaps to the induction of ssDNA in the second S phase. Furthermore, knockdown of CtIP or treatment with mirin, an inhibitor of the MRE11 nuclease, suppressed the induction of ssDNA in the second S phase (Fig. 3B; Supplemental Fig. S4D,E), suggesting that ssDNA is generated in a resection-dependent manner. It should be noted that DNA synthesis is drastically reduced in PARPi-treated cells in the second S phase (see Fig. 5C), slowing the induction of ssDNA gaps. Therefore, most of the effects of ssDNA gaps in the second S phase are likely attributed to the ssDNA gaps generated in the first S phase, supporting the idea that persistent ssDNA gaps give rise to collapsed forks and resected DNA ends in the second S phase.
Figure 3.

ssDNA gaps induce a surge of DSBs and ssDNA in the second S phase. (A) PARPi induces a surge of ssDNA in the second S phase. U2OS cells were labeled with 20 µM BrdU and treated with DMSO or 10 µM olaparib as indicated. BrdU-labeled ssDNA and PCNA were analyzed by immunofluorescence under a nondenaturing condition. The BrdU intensity of S-phase cells (PCNA+) was quantified. (Red bar) Median intensity. At least 200 S-phase cells were analyzed in each sample (n ≥ 200). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (n.s.) P-value > 0.05. (B) PARPi-induced ssDNA formation in the second S phase requires PrimPol and MRE11 nuclease activity. Cells were transfected with control and PrimPol siRNAs for 24 h and then treated as in A. Mirin (50 µM) was added where indicated. (Red bar) Median intensity. At least 200 S-phase cells were analyzed in each sample (n ≥ 200). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (C–F) PARPi-induced DNA damage responses in the second S phase require PrimPol. U2OS cells were transfected with control or PrimPol siRNA for 48 h. Cells were then incubated with DMSO or 10 µM olaparib for 24 h, or DMSO for 20 h and 10 µM olaparib for 4 h, and S-phase cells were labeled with 10 µM EdU during the last 15 min. Foci of p-RAP32 S33 (C), RAD51 (D), and γH2AX (E) were analyzed by immunofluorescence. The total foci intensity (C,E) or focus number (D) of S-phase cells were quantified. (Red bar) Median intensity (C,E) and focus number (D). More than 100 S-phase cells were analyzed in each sample (n > 100). (F) Median γH2AX intensities of three independent experiments normalized to the siCTRL 4-h time point are compiled. Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (**) P-value < 0.01, (n.s.) P-value > 0.05.
Figure 5.

BRCA1-deficient cells fail to suppress DNA synthesis upon PARPi treatment. (A,B) BRCA1-deficient cells fail to slow down in the second S phase after PARPi treatment. (A) U2OS cells were transfected with control or BRCA1 siRNA for 48 h. Cells were pulse-labeled with 2 µM EdU for 15 min and then released in the presence or absence of 10 µM olaparib. The fractions of EdU-labeled cells in the subpopulations are shown in the stacked bar charts. (B) UWB1 and UWB1 + B1 cells were pulse-labeled with 2 µM EdU for 15 min and released in the presence or absence of 2 µM olaparib. (C–E) Analysis of DNA synthesis following PARP inhibition in cells lacking PARP1 (C), BRCA1 (D), or BRCA2 (E). U2OS cells were transfected with the indicated siRNA for 48 h, treated with DMSO or 10 µM olaparib for 24 h, and nascent DNA was labeled with 10 µM EdU during the last 15 min. (Top panel) Cells were plotted according to PCNA intensity (y-axis), DAPI intensity (x-axis), and mean EdU intensity (color gradient). (Bottom panel) The mean EdU intensities of cell subpopulations are shown. More than 50 cells were analyzed in each subpopulation (n > 50). Error bars indicate SEM.
If PARPi primarily induces ssDNA gaps behind replication forks in the first S phase and these gaps cause fork collapse in the second S phase, one would expect that ssDNA gaps are required for the robust DNA damage response in the second S phase. Indeed, knockdown of PrimPol significantly reduced the induction of p-RPA32 after 24 h of PARPi treatment (Fig. 3C), implying that ssDNA gaps are important for the ATR response in the second S phase. Furthermore, knockdown of PrimPol prevented the induction of RAD51 foci after 24 h (Fig. 3D), suggesting that ssDNA gaps generate collapsed forks in the second S phase. Finally, depletion of PrimPol did not affect the induction of γH2AX by PARPi after 4 h but prevented the further increase of γH2AX after 24 h (Fig. 3E,F), indicating that ssDNA gaps are converted to DSBs in the second S phase. Collectively, these results suggest that PARPi-induced and PrimPol-generated ssDNA gaps give rise to DSBs in a trans cell cycle manner.
BRCA1/2-deficient cells fail to repair PARPi-induced DSBs in the second S phase
The ability of PARPi to induce ssDNA gaps and collapsed forks raises a question as to how BRCA1/2-deficient cells respond to these types of DNA damage. To investigate effects of PARPi in BRCA1/2-deficient cells, we performed the S1 nuclease assay on BRCA1 knockdown and control cells in the presence or absence of PARPi (Fig. 4A; see Fig. 6A for BRCA1 knockdown). In the absence of PARPi, knockdown of BRCA1 increased the shortening of IdU-labeled DNA by S1 (Fig. 4A, lanes 5,6), suggesting that BRCA1 suppresses the accumulation of ssDNA gaps in nascent DNA. However, in the presence of PARPi, IdU-labeled DNA was similarly shortened by S1 in BRCA1 knockdown and control cells (Fig. 4A, lanes 3,4,7,8), suggesting that BRCA1 loss does not increase PARPi-induced ssDNA gaps. Similar observations were made in the BRCA1-deficient ovarian cancer cell line UWB1 and its derivative UWB1+B1, which is complemented with wild-type BRCA1 (Fig. 4B). Of note, iPOND analysis revealed that the binding of RAD51 to PARPi-induced ssDNA gaps was dependent on BRCA1 (Supplemental Fig. S5A). Thus, BRCA1 enables the loading of RAD51 to postreplicative ssDNA gaps to promote repair, explaining why BRCA1 loss increases ssDNA gaps. However, in the presence of PARPi, the trapping of PARP by PARPi prevents complete repair of ssDNA gaps even when BRCA1 is functional, making the levels of PARPi-induced ssDNA gaps similar in BRCA1-proficient and -deficient cells.
Figure 4.

BRCA1-deficient cells are defective for RAD51-mediated repair in the second S phase. (A,B) BRCA1 loss does not affect the induction of ssDNA gaps by PARPi. U2OS cells transfected with control or BRCA1 siRNA (A) or UWB1 and UWB1+B1 cells (B) were labeled with CldU and IdU in the presence or absence of 10 µM olaparib and incubated with or without S1 nuclease for 30 min. (Red bar) Median IdU/CldU ratio. More than 125 CldU/IdU double-positive replication tracts were analyzed in each sample (n > 125). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (*) P-value < 0.05, (n.s.) P-value > 0.05. (C,D) PARPi induces BRCA1 and RAD51 foci in the second S phase. U2OS cells were transfected with control, BRCA1, or BRCA2 siRNA for 48 h. Cells were then treated with DMSO or 10 µM olaparib for 24 h, and S-phase cells were labeled with 10 µM EdU during the last 15 min. Foci of BRCA1 (C) and RAD51 (D) were analyzed by immunofluorescence, and the number of foci in S-phase cells were quantified. (Red bar) Median focus number. More than 60 S-phase cells were analyzed in each sample (n > 60). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (E) RAD51 is required for the repair of PARPi-induced DSBs in the second S phase. Cells were treated with DMSO or 10 µM olaparib for 24 h, and S-phase cells were labeled with 10 µM EdU during the last 15 min. The number of γH2AX foci in S-phase cells was quantified. (Red bar) Median focus number. More than 60 S-phase cells were analyzed in each sample (n > 60). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (F) PARPi preferentially induces DSBs in BRCA1-deficient cells in the second S phase. U2OS cells were transfected with control or BRCA1 siRNA for 72 h. Cells were either untreated or treated with 10 µM olaparib during the last 8 or 24 h and analyzed by neutral comet assay. Box plots represent the tail moment of comets. More than 100 cells were analyzed in each sample (n > 100). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (G) PARPi induces more DSBs in BRCA1-deficient cancer cells over multiple cell cycles. UWB1 and UWB1 + B1 cell lines were treated with 10 µM olaparib for 48 h and processed for a neutral comet assay. More than 100 cells were analyzed in each sample (n > 100). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001, (**) P-value < 0.01.
Figure 6.

Levels of origin firing and overall DNA synthesis are determinants of PARPi sensitivity. (A) BRCA1/2-deficient cells are defective for ATR activation in the second S phase. Cells were treated with DMSO or 10 µM olaparib for 24 h. Levels of the indicated proteins were analyzed by western blot. (B) BRCA1-deficient cells are unable to suppress origin firing in the second S phase. Cells were treated with 10 µM olaparib for the indicated durations and analyzed by CldU/IdU labeling (20 min each) and DNA combing. More than 320 CldU/IdU double-positive replication tracts were analyzed for inter-origin distance in each sample (n > 320). Significance was determined with a two-tailed Student's t-test. (****) P-value < 0.0001, (n.s.) P-value > 0.05. (C) BRCA1-deficient UWB1 cells fail to activate the G2/M checkpoint in the second S phase. UWB1 and UWB1+B1 cells were analyzed as in Figure 1C. Error bars indicate SD of two independent experiments. Significance was calculated with a two-tailed Student's t-test. (*) P-value < 0.05, (**) P-value < 0.01, (n.s.) P-value > 0.05. (D) ATR is critical for suppressing DNA damage in the second S phase. U2OS cells were treated with 10 µM olaparib for the indicated durations. VE-821 (ATRi; 10 µM) was added during the last 4 h as indicated, and S-phase cells were labeled with 10 µM EdU during the last 15 min. The total intensity of γH2AX foci in S-phase cells was quantified. (Red bar) Median intensity. More than 200 S-phase cells were analyzed in each sample (n > 200). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (E) Inhibition of origin firing suppresses the induction of DNA damage by ATRi in the second S phase. U2OS cells were treated with DMSO or 10 µM olaparib for 24 h. VE-821 (ATRi; 10 µM) and/or XL-413 (CDC7i; 5 µM) were added during the last 4 h, and S-phase cells were labeled with 10 µM EdU during the last 15 min. The mean intensity of γH2AX in S-phase cells was quantified. (Red bar) Median intensity. More than 200 S-phase cells were analyzed in each sample (n > 200). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (F) ATRi enhances PARPi sensitivity by increasing origin firing. U2OS cells were treated with increasing concentrations of VE-821 (ATRi), 1 µM olaparib (PARPi), and 1 µM XL-413 (CDC7i) as indicated for 6 d. Cell viability was determined with CellTiter Glo. Three technical replicates were analyzed in each sample (n = 3). Significance was determined with a two-tailed Student's t-test. Error bar indicates standard deviation. (*) P-value < 0.01. (G) Inhibition of origin firing suppresses PARPi-induced DNA damage in BRCA1-deficient cells. U2OS cells were transfected with BRCA1 siRNA for 48 h and then treated with DMSO or 10 µM olaparib for 24 h in the presence or absence of 1 µM XL-413. S-phase cells were labeled with EdU during the last 15 min. The mean γH2AX intensity in S-phase cells was quantified. (Red bar) Median intensity. More than 200 S-phase cells were analyzed in each sample (n > 200). Significance was determined with a Mann–Whitney U test. (****) P-value < 0.0001. (H) The ability to suppress overall DNA synthesis in the second S phase correlates with PARPi resistance in cancer cell lines. The indicated cell lines were treated with DMSO or 10 µM olaparib for 24 h, and S-phase cells were labeled with 10 µM EdU in the last 15 min. PCNA levels in EdU+ cells were then quantified by immunofluorescence and normalized to the untreated DMSO control of each cell line. At least 200 cells were analyzed in each condition (n ≥ 200). Error bars indicate SD of three independent experiments. Significance was calculated with a one-way ANOVA with the Tukey procedure. (*) P-value < 0.05, (**) P-value < 0.01, (n.s.) P-value > 0.05.
Next, we compared the responses of BRCA1-proficient and -deficient cells to PARPi in the second cell cycle. In BRCA1-proficient cells, the levels of BRCA1 foci were significantly increased after 24 h of PARPi treatment (Fig. 4C; Supplemental Fig. S5B,C), suggesting that BRCA1 is recruited to PARPi-induced DSBs in the second S phase. Knockdown of BRCA1 drastically reduced RAD51 foci in the second S phase (Fig. 4D; Supplemental Fig. S5D), showing that BRCA1 is required for RAD51 loading at collapsed forks (Feng and Zhang 2012). Similarly, knockdown of BRCA2 also suppressed RAD51 foci in the second S phase (Fig. 4D; see Fig. 6A for BRCA2 knockdown; Supplemental Fig. S5D), confirming that both BRCA1 and BRCA2 are required for RAD51 loading in this context. In BRCA1 knockdown cells, the induction of ssDNA by PARPi in the second S phase was reduced (Supplemental Fig. S5E–G), indicating compromised DNA end resection at collapsed forks. Furthermore, knockdown of RAD51 led to an increase of PARPi-induced γH2AX foci in the second S phase (Fig. 4E; Supplemental Fig. S5H), showing that RAD51 is required for the repair of PARPi-induced DSBs. Thus, in response to PARPi treatment, both BRCA1 and BRCA2 are important for the loading of RAD51 to collapsed forks and DSB repair in the second S phase.
To directly measure the impacts of BRCA1 loss on the levels of DSBs in PARPi-treated cells, we used a neutral comet assay to quantify the PARPi-induced DSBs in the first and second S phases (Fig. 4F). Knockdown of BRCA1 did not affect DSB levels in the first S phase but significantly increased DSBs in the second S phase (Fig. 4F). Furthermore, PARPi induced higher levels of DSBs in UWB1 cells than in UWB1+B1 cells after multiple cell cycles (Fig. 4G), lending further support to the notion that BRCA1/2-deficient cells are sensitive to PARPi at least in part because they accumulate more DSBs in response to the trans cell cycle effects of PARPi.
BRCA1/2-deficient cells fail to suppress DNA synthesis in the second S phase
While analyzing the effects of BRCA1 loss on the repair of PARPi-induced DSBs in the second S phase, we noted that BRCA1-deficient cells did not progress through the second S phase in the same way as control cells. Consistent with Figure 1A, EdU+ control cells displayed a significant delay in the second S phase after PARPi treatment (Fig. 5A). In marked contrast, BRCA1 knockdown progressed through the second S phase quite efficiently (Fig. 5A). Similar observations were made in UWB1 and UWB1+B1 cells (Fig. 5B). These results reveal that BRCA1-deficient cells continue to replicate DNA in the second S phase after PARPi treatment despite that they accumulate more DSBs than BRCA1-proficieint cells.
Next, we further analyzed how DNA synthesis is affected by PARPi in the second S phase by monitoring chromatin-bound PCNA and EdU incorporation. After 24 h of PARPi treatment, EdU incorporation was drastically reduced in cells transfected with control siRNA (Fig. 5C). Furthermore, the levels of chromatin-bound PCNA were also decreased, especially in mid- and late-S-phase cells (Fig. 5C). Knockdown of PARP1 largely reversed the effects of PARPi on EdU and PCNA (Fig. 5C; Supplemental Fig. S6A) and suppressed the induction of γH2AX (Supplemental Fig. S6B), confirming that these are on-target effects of PARPi. Importantly, knockdown of BRCA1 and BRCA2 also significantly reversed the effects of PARPi on EdU and PCNA (Fig. 5D,E), showing that BRCA1/2-deficient cells cannot suppress overall DNA synthesis efficiently. CPT traps TOP1 at DNA nicks ahead of replication forks (Pommier et al. 2010), leading to fork collapse similar to that in PARPi-treated cells in the second S phase. Reminiscent of the effects of a 24-h PARPi exposure, a 4-h CPT treatment drastically reduced EdU incorporation and chromatin-bound PCNA in control cells (Supplemental Fig. S6C). In contrast, BRCA1 knockdown cells only displayed modest reductions in EdU incorporation and chromatin-bound PCNA after CPT exposure (Supplemental Fig. S6C). Together, these results suggest that BRCA1/2-deficient cells fail to suppress overall DNA synthesis upon collisions of replications forks with ssDNA gaps.
BRCA1/2-deficient cells fail to activate ATR in the second S phase
Because ATR is important for suppressing DNA synthesis after DNA damage and it is robustly activated by PARPi in the second S phase (Fig. 1C), we asked whether BRCA1/2-deficient cells are defective for ATR activation in the second cell cycle after PARPi treatment. Knockdown of either BRCA1 or BRCA2 reduced the PARPi-induced p-Chk1 (Fig. 6A), showing that both BRCA1 and BRCA2 are indeed required for ATR activation. Notably, depletion of BRCA1 but not BRCA2 also reduced the PARPi-induced p-RPA32 (Fig. 6A). This result suggests that BRCA1 may promote ATR activation by generating ssDNA, whereas BRCA2 may facilitate Chk1 activation after ssDNA formation (Huen et al. 2010; Roy et al. 2012). The ATR-Chk1 pathway inhibits DNA synthesis by suppressing firing of replication origins (Costanzo et al. 2003). To test whether cells suppress origin firing in the second S phase after PARPi treatment, we performed DNA combing assay and measured the inter-origin distance and newly fired origins (Fig. 6B; Supplemental Fig. S7A). A 24-h PARPi treatment significantly reduced newly fired origins and increased the inter-origin distance in control cells but not BRCA1 knockdown cells (Fig. 6B; Supplemental Fig. S7A), showing that BRCA1-proficient, but not BRCA1-deficient, cells suppressed origin firing. Of note, PARPi increased replication fork speed in control cells but not BRCA1 knockdown cells (Supplemental Fig. S7B). Although fork speed was increased by PARPi in control cells, the overall DNA synthesis in these cells was reduced (Fig. 5C), showing that the effects of origin suppression outweigh fork speeding. In addition to suppressing origin firing, ATR and Chk1 also promote the G2/M checkpoint arrest after DNA damage. The PARPi-induced G2/M arrest in the second cell cycle was more robust in UWB1+B1 cells than in UWB1 cells (Fig. 6C), further supporting the idea that BRCA1/2-deficient cells are unable to slow down in the second cell cycle after PARPi treatment due to their defects in ATR activation.
Origin firing and overall DNA synthesis in the second S phase are determinants of PARPi sensitivity
The defect of BRCA1-deficient cells in suppressing origin firing in the second S phase after PARPi treatment raises the possibility that excessive origin firing is a determinant of PARPi sensitivity. Given that BRCA1/2-deficient cells are defective for ATR activation, we used ATR inhibitor (ATRi) VE-821 to recapitulate the defect of origin suppression in BRCA1/2-deficient cells. We treated cells with PARPi for 4 or 24 h and exposed cells to ATRi during the last 4 h in PARPi (Fig. 6D). ATRi increased the levels of PARPi-induced γH2AX at both 4 and 24 h, but the effects of ATRi were significantly stronger after 24 h (Fig. 6D). This result suggests that ATR activation is more important in the second S phase than in the first S phase.
To investigate whether the effects of ATRi in the second S phase are attributed to increased origin firing, we used the CDC7 inhibitor (CDC7i) XL-413 to repress replication initiation (Fig. 6E; Rainey et al. 2017). The ATRi-induced γH2AX in the second S phase was significantly reduced by CDC7i (Fig. 6E). Using CPT to mimic the effects of PARPi in the second S phase, we confirmed that ATRi induces DNA damage in a CDC7-dependent manner (Supplemental Fig. S7C). Furthermore, ATRi increased the PARPi sensitivity of cells in a concentration-dependent manner, which was partially reversed by CDC7i (Fig. 6F). These results suggest that unrestricted origin firing is an important determinant of PARPi sensitivity.
Finally, we asked whether the unrestricted origin firing in BRCA1/2-deficient cells is a contributor to their PARPi sensitivity. As expected, treatment of BRCA1 knockdown cells with PARPi for 24 h induced a drastic increase of γH2AX (Fig. 6G). Adding CDC7i in the second S phase reduced the induction of γH2AX (Fig. 6G), suggesting that the excessive origin firing in BRCA1-deficient cells indeed contributes to the cytotoxic effects of PARPi in the second S phase. To test whether the inability of BRCA1-deficient cancer cells to suppress overall DNA synthesis in the second S phase is relevant to their PARPi sensitivity, we analyzed UWB1, UWB1+B1, and three UWB1-derived PARPi-resistant cell lines (SYr12, SYr13, and SYr14) (Yazinski et al. 2017). We treated cells with PARPi for 24 h and used the levels of chromatin-bound PCNA to monitor overall DNA synthesis (Fig. 6H). In UWB1+B cells, the levels of chromatin-bound PCNA were significantly reduced. In contrast, UWB1 cells did not display a reduction in chromatin-bound PCNA, confirming their defect in suppressing DNA synthesis. Importantly, the PARPi-induced reduction in chromatin-bound PCNA was restored in all three BRCA1-deficient, PARPi-resistant cell lines (Fig. 6H), showing that these cells regain the ability to suppress DNA synthesis in the second S phase. These results strongly suggest that the inability of BRCA1/2-deficient cells to suppress origin firing and overall DNA synthesis in the second cell cycle is a key determinant of their sensitivity to PARPi.
Discussion
Compared with many other cancer drugs that damage DNA or inhibit DNA replication, PARPi induces DNA damage more slowly. Even in actively proliferating cells, it takes many hours and sometimes days for PARPi-induced DNA damage to become readily detectable (Michelena et al. 2018; Ryu et al. 2019). Although DNA replication is clearly required for PARPi to induce DNA damage, it is unclear why the time required to detect significant PARPi-induced DNA damage is usually longer than one S phase. In this study, we found that PARPi induces DSBs in a trans cell cycle manner, providing a plausible explanation for the slow induction of DNA damage by PARPi (Fig. 7A). We show that PARPi induces ssDNA gaps behind replication forks. Inhibition of PARP1 allows RECQ1 to resolve reversed forks and channel them into the PrimPol-mediated repriming pathway, giving rise to ssDNA gaps on the leading strand (Berti et al. 2013; Quinet et al. 2020). PARPi may also interfere with maturation of Okazaki fragments, generating ssDNA gaps on the lagging strand (Hanzlikova et al. 2018). The presence of PARPi-induced ssDNA gaps on both leading and lagging strands allows the S1 nuclease to break replication tracts efficiently. Notably, we only detected low levels of PARPi-induced DSBs in the first S phase, suggesting that PARPi-induced ssDNA gaps are not converted to DSBs efficiently during this period. Although PARPi may induce some SSBs and ssDNA gaps ahead of replication forks by inhibiting BER, most of the PARPi-induced ssDNA gaps are likely generated during replication and distributed behind forks in the first S phase, which would explain why the S-phase cells exposed to PARPi can complete the first cell cycle with only a transient delay.
Figure 7.
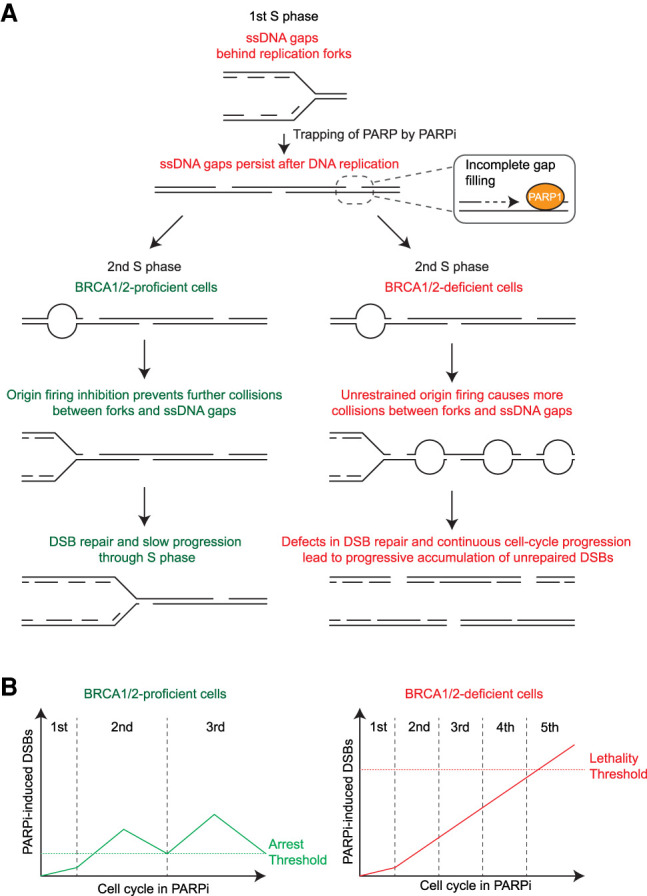
Models for the trans cell cycle effects of PARPi in BRCA1-proficient and -deficient cells. (A) A model for the effects of PARPi in BRCA1/2-proficient and -deficient cells during two consecutive S phases. During each S phase, PARPi induces ssDNA gaps behind DNA replication forks. The trapping of PARP by PARPi prevents complete filling of these gaps, allowing them to persist into the next cell cycle. During the next round of DNA replication, the ssDNA gaps generated in the previous cell cycle collide with replication forks, leading to fork collapse and a surge of DSBs. BRCA1/2-proficient cells activate ATR to suppress replication origin firing and recruit RAD51 to repair collapsed forks. In contrast, BRCA1/2-deficient cells fail to suppress origin firing and repair collapsed forks, leading to more DSBs. The unique ability of PARPi to induce DSBs in a trans cell cycle manner explains why PARPi generates more DSBs in BRCA1/2-deficeint cells than in BRCA1/2-proficient cells. (B) A model for the effects of PARPi in BRCA1/2-proficient and -deficient cells over multiple cell cycles. The ssDNA gaps generated in each S phase are converted to DSBs in the next S phase. Because BRCA1/2-proficient cells slow down and repair DSBs in each cell cycle, DSB levels will not reach the threshold for cell death, and cells will continue to proliferate after DSBs are repaired. In contrast, BRCA1/2-deficient cells fail to slow down and repair in each cell cycle, which allows PARPi-induced DSBs to accumulate progressively over multiple cell cycles. These repeating trans cell cycle effects of PARPi eventually push DSB levels over the lethal threshold and kill BRCA1/2-deficient cancer cells.
Once generated behind replication forks, PARPi-induced ssDNA gaps can persist for a long time in the presence of PARPi (Fig. 7A). This is likely due to the trapping of PARP1 at ssDNA gaps (Murai et al. 2012). Although cells can activate postreplication repair mechanisms such as translesion synthesis (TLS) and template switching to fill the gaps (Daigaku et al. 2010; García-Rodríguez et al. 2018; Nayak et al. 2020), the trapping of PARP at these gaps may prevent the completion of gap filling. This idea is consistent with the previous model that the trapping of PARP1/2 by PARPi is critical for the effects of PARPi in cancer cells. It should be noted that trapping of PARP1/2 is not particularly toxic in the first S phase or the subsequent G1. The toxicity of trapped PARP1/2 only becomes evident when cells enter the second S phase. It is plausible that the replication forks initiated in the second S phase collide with the PARP1/2 trapped in the first cell cycle, leading to fork collapse and DSBs. Consistent with this idea, there is a significant increase of γH2AX, BRCA1, p-RPA32, and RAD51 foci and DSBs in the second S phase after PARPi treatment. PrimPol is required for DSB formation in the second but not the first S phase, suggesting that ssDNA gaps primarily give rise to DSBs during the second round of DNA replication. The collisions of replication forks with PARPi-induced ssDNA gaps in the second S phase are reminiscent of the previous model in which replication forks collapse at PARPi-induced SSBs (Bryant et al. 2005; Farmer et al. 2005). However, our data suggest that in the first S phase PARPi primarily induces ssDNA gaps behind replication forks rather than SSBs ahead of forks. Furthermore, collisions of forks and gaps primarily occur during the second round of DNA replication when newly generated forks run into gaps. Our model that PARPi induces persistent ssDNA gaps and fork–gap collisions in a trans cell cycle manner explains why PARPi induces DNA damage slowly. It is conceivable that PARPi induces ssDNA gaps in every S phase during prolonged PARPi treatment, and these ssDNA gaps are converted to DSBs in a trans cell cycle manner (Fig. 7B). Cells with a proficient ATR checkpoint and HR pathway will slow down in each cell cycle and repair DSBs, keeping DNA damage at tolerable levels. In contrast, cancer cells defective for the ATR checkpoint and HR will not slow down and repair DSBs in each cell cycle, leading to progressive accumulation of DSBs and cell death over multiple cell cycles.
Our studies also provide insights into why PARPi selectively kills BRCA1/2-deficient cells (Fig. 7A,B). BRCA1/2 knockdown cells are unable to recruit RAD51 to PARPi-induced collapsed forks in the second S phase, compromising repair of DSBs. The requirement of BRCA1/2 for repairing PARPi-induced DSBs is consistent with the model proposed to explain the synthetic lethal relationship between PARP inhibition and BRCA1/2 loss (Helleday 2011). However, our data emphasize that BRCA1/2 primarily executes its repair function in the second but not the first S phase after PARPi treatment. The results of this study also reveal that BRCA1/2-deficient cells fail to activate the ATR pathway and suppress origin firing in the second S phase. In the absence of proper ATR activation, BRCA1/2-deficient cells continue to fire origins and synthesize DNA despite the presence of PARPi-induced ssDNA gaps, which will inevitably lead to more fork-gap collisions and DSBs (Fig. 7A). Most importantly, during prolonged PARPi treatment, BRCA1/2-deficient cells are expected to experience the same problems in every S phase starting from the second cell cycle (Fig. 7B). The inability of BRCA1/2-deficient cancer cells to slow down and repair in each S phase will result in progressive accumulation of DSBs over multiple cell cycles, which eventually kills the cancer cells.
Our model is consistent with a recent report that PARPi-induced ssDNA gaps underlie the therapeutic response of BRCA1/2-deficient cancers (Panzarino et al. 2020). However, our data suggest that the DSBs resulting from fork-gap collisions, rather than ssDNA gaps themselves, drive the killing of BRCA1/2-deficient cells by PARPi. Another recent study suggested that PARPi induces more ssDNA gaps in BRCA1-deficient cells than in BRCA1-proficient cells (Cong et al. 2021). Even in the absence of PARPi, higher levels of ssDNA gaps were detected in BRCA2-deficient cells (Somyajit et al. 2021). Although we also observed more ssDNA gaps in BRCA1-deficient cells, we did not detect a significant difference in the PARPi-induced ssDNA gaps between BRCA1-proficient and -deficient cells. Our results suggest that the sensitivity of BRCA1/2-deficient cells to PARPi primarily stems from their checkpoint and repair defects in response to trans cell cycle gaps, rather than the levels of ssDNA gaps induced by PARPi. Nonetheless, we do not exclude the possibility that PARPi-induced ssDNA gaps are differently processed in BRCA1/2-proficient and -deficient cells, which may affect how these gaps generate DSBs in the next cell cycle. It should be noted that, although BRCA1 and RAD51 are involved in the repair of postreplicative ssDNA gaps, depletion of BRCA2 and RAD51 did not reduce mitotic DNA synthesis (MiDAS) (Bhowmick et al. 2016), suggesting that the ssDNA gaps in BRCA1/2-deficient cells can be repaired by a BRCA-independent mechanism in mitosis. Therefore, BRCA1/2-deficient cells may not generate trans cell cycle ssDNA gaps efficiently without PARPi.
Finally, the results of this study may help improve the use of PARPi in cancer therapy. Our data suggest that the level of origin firing is an important determinant of PARPi sensitivity and that ATRi can enhance PARPi sensitivity by increasing origin firing. These findings support the notion that combining ATR, Chk1, and WEE1 inhibitors with PARPi will increase origin firing and help the treatment of tumors that respond to PARPi alone modestly or tumors that acquire PARPi resistance (Kim et al. 2017, 2020; Yazinski et al. 2017; Hill et al. 2018; Fang et al. 2019; Parmar et al. 2019). Because excessive origin firing matters more in the second S phase than in the first S phases after PARPi treatment, sequential therapy with PARPi and ATRi may be beneficial (Fang et al. 2019). In addition, our data reveal that multiple BRCA1-deficient, PARPi-resistant cell lines regain the ability to suppress overall DNA synthesis in the second S phase after PARPi treatment, strongly suggesting that the inability of BRCA1/2-deficient cells to slow down S phase is a key component of their “BRCAness” and a cause of PARPi sensitivity. Recent studies showed that a DNA fiber assay can be applied to patient-derived organoids or circulating tumor cells to monitor the replication stress response, which may be used to predict the response of patients to PARPi (Yazinski et al. 2017; Hill et al. 2018). However, the current assays rely on the replication inhibitor hydroxyurea (HU) rather than PARPi, leaving the possibility that they do not fully reflect the PARPi response of tumors. The findings of this study suggest that we can follow the PARPi response of tumors by analyzing the ability of tumor cells to suppress origin firing and overall DNA synthesis in the second cell cycle after PARPi treatment, providing a means to functionally detect the BRCAness or the loss of it in tumors. This approach may help establish a clinically useful assay to monitor the BRCAness of tumors and guide the use of PARPi in mono and combination therapies. To fully understand the mechanism of action for PARPi, it will be important to test the revised model from this work in preclinical and clinical studies and compare it with other models.
Materials and methods
Cell lines
U2OS and HEK293T cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 2 mM L-glutamine. The ovarian cancer cell line UWB1.249 and its derivatives, UWB1+B1, SYr12, SYr13, and SYr14 were cultured in 1:1 Roswell Park Memorial Institute (RPMI) 1640–mammary epithelial cell growth medium (MEGM) supplemented with 3% FBS and 1% penicillin/streptomycin. UWB1+B1 cells were maintained with 200 µg/mL G-418 and SYr12, SYr13, and SYr14 cells were cultured with 1 µM olaparib. All cell lines were incubated at 37°C with 5% CO2. Inhibitors used are listed in Supplemental Table S1.
RNA interference
Cell transfection was carried out by reverse transfection method with RNAiMAX and 4 nM silencer select siRNAs. Unless otherwise stated, experiments involving transfected cells were initiated 48 h after transfection. siRNA sequences are listed in Supplemental Table S2.
Immunofluorescence
Cells were cultured on #1.5 22 × 22-mm coverslips in six-well plates. Samples were pre-extracted for 10 min at 4°C in phosphate-buffered saline (PBS) + 0.5% Triton X-100 and fixed for 10 min at room temperature in 3% paraformaldehyde and 2% sucrose. For experiments with p-H3 S10 staining, cells were not pre-extracted but were permeabilized with PBS + 0.25% Triton X-100 for 10 min at 4°C after paraformaldehyde fixation. For experiments involving PCNA staining, cells were further fixed with 100% methanol for 10 min at −20°C and allowed to dry at room temperature. Cells were washed with PBS between each step. Samples were then incubated for 1 h at room temperature in blocking solution consisting of PBS with 0.05% Tween-20 (PBS-T) and 1% milk. In BrdU-labeling experiments, 2% bovine serum albumin was used instead of milk for blocking and subsequent incubation steps. Primary antibody staining was carried out for 1 h at 37°C at a 1:500 concentration for most antibodies, except for RAD51 (1:100) and BrdU (1:50) antibodies. Samples were washed three times for 5 min in PBS-T and incubated for 1 h at room temperature with 1:500 dilution of secondary antibodies conjugated with AlexaFluor 488 (γH2AX, RAD51, RPA32-S33P, and BrdU) or AlexaFluor 568 (PCNA, BRCA1, and H3S10P). Cells were washed three times with PBS-T and where applicable, EdU was then stained by click chemistry. To this end, samples were incubated for 30 min at room temperature in PBS with 2 mM CuSO4, 10 mM sodium ascorbate, and 1 µM picolyl-azide AF647. Coverslips were then washed once with PBS and stained with 1 µg/mL DAPI in PBS for 10 min at room temperature. Coverslips were washed once with PBS, allowed to dry at room temperature, and mounted on slides with ProLong Gold. Images were acquired using NIS elements software with a Nikon i90 microscope.
S1 nuclease DNA fiber assay
Cells were labeled for the indicated time with 50 µM CldU, washed twice with prewarmed media, and incubated in fresh warm media containing 100 µM IdU for the indicated time. In conditions where cells were further incubated before S1 nuclease treatment, cells were washed twice with prewarmed media before incubation with fresh warm media. S1 nuclease treatment was then carried out according to previously published protocols (Quinet et al. 2017). Cells were washed with PBS and permeabilized by incubating 10 min at room temperature with CSK100 buffer (10 mM MOPS at pH 7, 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5% Triton X-100). Cells were washed once with PBS and once with S1 nuclease buffer (30 mM sodium acetate at pH 4.6, 10 mM zinc acetate, 5% glycerol, 50 mM NaCl) before incubation for 30 min at 37°C in 1 mL of S1 nuclease buffer ± 20 U of S1 nuclease. S1 nuclease buffer was replaced with PBS + 0.1% BSA, and cells were collected using a cell lifter. Cell suspensions containing ∼3000 cells (in 3 µL) were then dropped onto clean microscope slides and allowed to sit for 2 min. DNA fiber lysis buffer (7 µL; 200 mM Tris-HCl at pH 7.4, 0.5% SDS, 50 mM EDTA) was added, and slides were incubated for 8 min at room temperautre before tilting at a 15° angle to let liquid droplets slowly travel down slides, thereby stretching DNA fibers. Slides were then fixed in 3:1 methanol/acetic acid solution for 5 min at −20°C and allowed to dry overnight. DNA fibers were denatured in 2.5 M HCl for 45 min and washed five times for 1 min with PBS. For experiments involving VdU staining, slides were then incubated for 4 h at 37°C with PBS + 10 µM AF488 tetrazine and then washed once with PBS. Slides were blocked for 30 min at room temperature in PBS-T containing 2% BSA. Slides were then incubated with 1:25 mouse anti-BrdU (B44) and 1:100 rat anti-BrdU (BU1/75) antibodies in PBS-T + 2% BSA for 1 h at 37°C in a humid chamber. Samples were washed three times for 5 min in PBS-T before incubation for 1 h at 37°C in a humid chamber with antimouse and antirat secondary antibodies conjugated to AlexaFluor 488 (or mouse AlexaFluor 647 in the VdU experiments) and Cy3, respectively. Slides were washed three times for 5 min in PBS-T and mounted with ProLong Gold. Images were captured using NIS elements software with a Nikon i90 microscope and analyzed using Fiji software.
EdU-labeled fiber assays
Nascent DNA of U2OS cells was labeled with 2 µM EdU for 90 min, and cells were then washed twice before releasing into media without EdU for 26.5 h. In the PARPi-treated conditions, 10 µM olaparib was added either 4 h before collecting samples or throughout the experiment including during EdU labeling. Forty minutes prior to collecting samples, cells were labeled with 50 µM CldU for 20 min, washed twice with prewarmed media, and subsequently labeled for 20 min with 100 µM IdU. Importantly, in the PARPi-treated conditions, 10 µM olaparib was added during CldU and IdU labeling as well as in the media used for washes. Cells were then trypsinized and resuspended at ∼1000 cells/µL in PBS. Three microliters of cell suspension was dropped onto slides and allowed to settle for 3 min. Seven microliters of lysis buffer (200 mM Tris-HCl at pH 7.4, 0.5% SDS, 50 mM EDTA) was then gently added, and cell lysis was allowed to occur for 3 min at room temperature. Slides were then tilted at a 15° angle to let cell lysates slowly flow down the length of slides and stretch DNA fibers. Immunofluorescence was then carried out as described in the S1 nuclease assay with few modifications. EdU was biotinylated prior to blocking by incubating slides in PBS with 2 mM CuSO4, 10 mM sodium ascorbate, and 1 µM biotin azide (Click chemistry tools 1265) for 1 h at room temperature. Biotinylated EdU was detected with a rabbit anti-biotin antibody (Abcam ab53494). Antirabbit AlexaFluor 488, antimouse AlexaFluor 647, and antirat Cy3-conjugated secondary antibodies were used.
DNA combing
Cells were labeled with 50 µM CldU for 20 min, washed twice with prewarmed media, and subsequently labeled for 20 min with 100 µM IdU. Cells were trypsinized, collected, and resuspended in PBS. DNA extraction for DNA combing was carried out with a Fiber Prep DNA extraction kit (Genome Vision) following the manufacturer's guidelines. Extracted DNA fibers were combed on CombiSlips silanized coverslips with the the FiberComb Molecular Combing System (Genome Vision). Immunofluorescence was performed based on previously published procedures (Bianco et al. 2012; Gallo et al. 2016). Coverslips were baked for 2 h at 60°C and then successively incubated for 5 min in 70%, 90%, and 100% ethanol before being allowed to dry at room temperature. DNA was denatured by dipping coverslips in 1M NaOH solution for 20 min at room temperature. Coverslips were then washed five times for 1 min in PBS before proceeding to immunodetection of CldU and IdU using a similar protocol as for fiber assays. ssDNA was subsequently detected by incubating for 1 h at 37°C in a humid chamber with 1:100 dilution of a mouse IgG 2a anti-ssDNA antibody, followed by three 5-min PBS-T washes and a 1-h incubation at 37°C with antimouse IgG 2a secondary antibody conjugated to AlexaFluor 647. Coverslips were further washed three times for 5 min with PBS-T and mounted on slides with ProLong Gold. Images were captured using NIS elements software with a Nikon i90 microscope and analyzed for inter-origin distance and fork speed using Fiji software.
Isolation of proteins on nascent DNA (iPOND)
One hundred million HEK293T cells were treated with 10 µM olaparib or an equal volume of DMSO for 40 min. To label nascent DNA, 10 µM EdU was added, and cells were incubated for an additional 20 min. Fork samples were immediately fixed with 1% formaldehyde in PBS. Thymidine chase samples were washed once with prewarmed media and incubated for 45 min with fresh media containing 10 µM thymidine and 10 µM olaparib or an equal volume of DMSO prior to fixation. Formaldehyde was then quenched with 0.125 M glycine, and cells were collected by scraping with a cell lifter. Cells were washed with PBS and permeabilized in PBS + 0.25% Triton X-100 for 30 min at room temperature. EdU was then labeled with biotin by click chemistry by resuspending cells in reaction buffer (1× PBS, 2 mM CuSO4, 10 mM sodium ascorbate, 1 µM biotin azide) and incubating for 2 h at room temperature. Cells were washed with PBS, resuspended in 1 mL lysis buffer (100 mM HEPES pH 8, 1% SDS), and sonicated with a 4710 series ultrasonic homogenizer (Cole-Parmer) at setting 3 three times for 30 sec at 4°C interspersed with 1-min incubations on ice. Lysates were spun at 13,000 rpm for 10 min at room temperature, and supernatant was collected and quantified using a Pierce BCA protein assay kit (Thermo Fisher Scientific 23227). Protein concentrations were normalized and diluted 1:1 in 100 mM HEPES (pH 8) to reduce SDS concentration to 0.5%. A prewashed streptavidin agarose beads slurry (100 µL; Millipore Sigma 69203-3) was added to each sample, and bead-lysate mixtures were incubated with rotation overnight at 4°C. Beads were then successively washed for 5 min on a rotating platform with lysis buffer, low-salt wash buffer (20 mM Tris at pH 8, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100), high-salt wash buffer (20 mM Tris at pH 8, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100), 50 mM Tris (pH8), and 1% SDS. Beads were then resuspended in 2× sample buffer (100 mM Tris at pH 6.8, 12% glycerol, 3.5% SDS, 0.2M DTT), boiled for 30 min, and processed for immunoblotting.
Chromatin fractionation
Cells were trypsinized, washed with PBS, and incubated for 5 min at 4°C in hypotonic buffer (10 mM HEPES at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X-100, 10 mM NaF, protease inhibitor cocktail [Millipore Sigma P8340]).The cell suspension was centrifuged at 1300g at 4°C, and the supernatant (S1) was transferred to a new tube, while the pellet (P1) was further washed twice with hypotonic buffer. The pellet (P1) was then resuspended in hypertonic buffer (3 mM EDTA, 0.2 mM EGTA, 10 mM NaF, 1 mM DTT, protease inhibitor cocktail) and incubated for 30 min at 4°C.The supernatant (S1) was centrifuged at 15,700g for 15 min at 4°C, and the supernatant was transferred to a new tube with an equivalent volume of 2× sample buffer (100 mM Tris at pH 6.8, 12% glycerol, 3.5% SDS, 0.2M DTT). The pellet (P1) was centrifuged at 1700g for 4 min at 4°C, and the supernatant (S2) was transferred to a new tube with an equivalent volume of 2× sample buffer prior to combining with supernatant S1 (soluble fraction).The pellet (P1) was washed twice with hypertonic buffer, and the remaining pellet was resuspended in 2× sample buffer (chromatin fraction).
Immunoblots
Cells were resuspended and lysed in lysis buffer (100 mM Tris at pH 6.8, 1% SDS), sonicated for 10 sec with a 4710 series ultrasonic homogenizer (Cole-Parmer), and boiled for 3 min. Protein concentrations were normalized using a Pierce BCA protein assay kit (Thermo Fisher Scientific 23227) and mixed 1:1 with 2× SDS-PAGE loading buffer (100 mM Tris at pH 6.8, 12% glycerol, 3.5% SDS, 0.2 M DTT). Samples were boiled for 3 min, loaded on Bolt Bis-Tris Plus 3%–12% gels, and run at 100 V for 90 min. Proteins were transferred onto PVDF membranes using a CBS Scientific electrophoretic blotting liquid transfer system (EBX-700) for 2 h at 100 V. Membranes were then blocked in Tris-buffered saline with 0.05% Tween-20 (TBS-T) and 5% milk for 30 min at room temperature. Membranes were then immunoblotted with primary antibodies overnight at 4°C with mild shaking. Most antibodies were used at a 1:1000 concentration in TBS-T 5% milk, except for RAD51 (1:200), H3 (1:40,000), RECQ1 (1:10,000), and GAPDH (1:20,000). Antibodies are listed in Supplemental Table S3. The PrimPol antibody was kindly provided by the Méndez laboratory. Membranes were washed with TBS-T and incubated for 1 h at room temperature with secondary antibodies conjugated to horseradish peroxidase. Membranes were washed four times for 10 min with TBS-T and an enhanced chemiluminescence (ECL Bio-Rad 1705061) substrate was applied. Signals were detected using a Chemidoc imaging system (Bio-Rad) with ImageLab v6.0.1. software.
Neutral comet assay
Neutral comet assays were performed according to the Trevigen protocol. Cells were trypsinized and resuspended in PBS at a density of 3 × 105 cells/mL. Cells were mixed at a 1:10 v:v ratio with molten 1% low melting point agarose (Trevigen) at 37°C, and 50 µL of cell-agarose solution was dropped onto Flare slides (Trevigen). The agarose was allowed to solidify at 4°C, and slides were soaked in ice-cold Comet lysis buffer (Trevigen) and incubated at 4°C overnight. Slides were then incubated for 30 min at 4°C in cold neutral electrophoresis buffer (100 mM Tris, 300 mM sodium acetate at pH 9.0). Slides were carefully immersed in neutral electrophoresis buffer in a Thermo Scientific Owl EasyCast B1A mini gel electrophoresis system gel tank and electrophoresis was carried out at 15 V (1 V/cm) for 1 h at 4°C. Slides were sequentially incubated for 30 min in DNA precipitation solution (1 M ammonium sulfate, 95% ethanol) and 70% EtOH. Slides were dried and 200 µL of SYBR Gold DNA staining solution (1:10,000 SYBR Gold, 10 mM Tris at pH 7.5, 1 mM EDTA) was dropped onto each sample. Slides were incubated 30 min at room temperature, washed twice with H2O, and completely dried before mounting with ProLong Gold. Images were captured using NIS elements software with a Nikon i90 microscope and tail moments were analyzed with the OpenComet software integrated in Fiji (Gyori et al. 2014).
Cell viability assay
Cell viability was assessed with a CellTiter-Glo cell viability assay (Promega). Cells (in 50 µL) were seeded in 96-well flat-bottom plates at a density of 400 cells/well and incubated overnight. The next day, 50 µL of media containing the appropriate concentration of drugs was added to each well, and plates were incubated for an additional 6 d. Plates were then equilibrated for 30 min to room temperature and 50 µL of CellTiter-Glo reagent was added to each well, after which plates were incubated for 15 min at room temperature. Luminescence of each was then measured with a Perkin Elmer EnVision 2103 multilabel plate reader. Viability was calculated as the luminescence signal ratio of treated versus untreated samples.
Image analysis and data processing
Cell microscopy images were analyzed with MATLAB R2020a software together with the Image Processing Toolbox and Bioformats 6.4.0 package (Linkert et al. 2010). Cells were segmented by adaptive thresholding of the DAPI channel, which was then used to derive mean and total intensities of every channel for each cell. In experiments where the number of foci or total foci intensity was analyzed, a mask was created by applying a threshold on a Laplacian of Gaussian-filtered image of the corresponding channel. The mask was then used to quantify the number of elements (i.e., foci) in each cell and determine the total intensity in the original image of masked elements in each cell. These data were then compiled into a table where the values for each cell could easily be accessed.
In experiments where S-phase cells are shown, analyzed cells were selected by applying a threshold on PCNA or EdU mean intensity. For EdU pulse-labeling experiments (Figs. 1A, 5A,B; Supplemental Fig. S1B–D), cells were classified into five cell cycle phase (G1, early S, mid S, late-S, and G2) subpopulations based on PCNA mean intensity and DAPI total intensity. To this end, the mean PCNA intensity and total DAPI intensity of each cell were plotted and a threshold on PCNA intensity was applied to separate S-phase cells from G1 or G2 cells. S-phase cells were then split into three groups, corresponding to early, mid, and late S, by dividing the range of DAPI intensities into three equal bins (Supplemental Fig. S1A). Pie charts were generated by quantifying the number of EdU-positive cells in each cell cycle phase and dividing on the total number of EdU-positive cells.
Unless otherwise stated, each experiment was repeated three times. Statistical significance was determined in fiber assay and cell microscopy experiments with a Mann–Whitney U test of at least 100 individual fibers or cells. Statistical significance of viability assays was assessed with a two-tailed Student's t-test of triplicate experiments.
Supplementary Material
Acknowledgments
We thank Dr. A. Vindigni and members of the Zou, Dyson, Lan, and Elia laboratories for discussions. A.S. is the recipient of postdoctoral fellowships from the Canadian Institutes of Health Research and Fonds de Recherche du Québec-Santé. L.Z. is the James and Patricia Poitras Endowed Chair in Cancer Research. This work is supported by grants from the National Institutes of Health (CA240243, CA197779, and CA218856) and Gray Foundation to L.Z.
Author contributions: A.S. and L.Z. designed the project. A.S. and R.X. performed the experiments and data analysis. L.Z. supervised the experiments and data analysis. A.S. and L.Z. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348479.121.
Competing interest statement
The authors declare no competing interests.
References
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. 2013. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20: 347–354. 10.1038/nsmb.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick R, Minocherhomji S, Hickson ID. 2016. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol Cell 64: 1117–1126. 10.1016/j.molcel.2016.10.037 [DOI] [PubMed] [Google Scholar]
- Bianco JN, Poli J, Saksouk J, Bacal J, Silva MJ, Yoshida K, Lin YL, Tourrière H, Lengronne A, Pasero P. 2012. Analysis of DNA replication profiles in budding yeast and mammalian cells using DNA combing. Methods 57: 149–157. 10.1016/j.ymeth.2012.04.007 [DOI] [PubMed] [Google Scholar]
- Blessing C, Mandemaker IK, Gonzalez-Leal C, Preisser J, Schomburg A, Ladurner AG. 2020. The oncogenic helicase ALC1 regulates PARP inhibitor potency by trapping PARP2 at DNA breaks. Mol Cell 80: 862–875.e6. 10.1016/j.molcel.2020.10.009 [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. 2012. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res 40: 7347–7357. 10.1093/nar/gks353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Feng W, Lim PX, Kass EM, Jasin M. 2018. Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol 2: 313–336. 10.1146/annurev-cancerbio-030617-050502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Ong K, Giaever G, Nislow C. 2013. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol 20: 648–659. 10.1016/j.chembiol.2013.04.007 [DOI] [PubMed] [Google Scholar]
- Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, Krais J, VanderVere-Carozza PS, Pawelczak KS, Calvo J, et al. 2021. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell 81: 3128–3144.e7. 10.1016/j.molcel.2021.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. 2003. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11: 203–213. 10.1016/S1097-2765(02)00799-2 [DOI] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, Ulrich HD. 2010. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465: 951–955. 10.1038/nature09097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea AD. 2018. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 71: 172–176. 10.1016/j.dnarep.2018.08.021 [DOI] [PubMed] [Google Scholar]
- Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Ménissier-de Murcia J, de Murcia G. 1999. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie 81: 69–75. 10.1016/S0300-9084(99)80040-6 [DOI] [PubMed] [Google Scholar]
- Dillehay LE, Thompson LH, Minkler JL, Carrano AV. 1983. The relationship between sister-chromatid exchange and perturbations in DNA replication in mutant EM9 and normal CHO cells. Mutat Res 109: 283–296. 10.1016/0027-5107(83)90053-2 [DOI] [PubMed] [Google Scholar]
- Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, et al. 2019. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell 35: 851–867.e7. 10.1016/j.ccell.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang J. 2012. A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic Acids Res 40: 726–738. 10.1093/nar/gkr748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo D, Wang G, Yip CM, Brown GW. 2016. Analysis of replicating yeast chromosomes by DNA combing. Cold Spring Harb Protoc 2016: pdb.prot085118. 10.1101/pdb.prot085118 [DOI] [PubMed] [Google Scholar]
- García-Rodríguez N, Wong RP, Ulrich HD. 2018. The helicase Pif1 functions in the template switching pathway of DNA damage bypass. Nucleic Acids Res 46: 8347–8356. 10.1093/nar/gky648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genois MM, Gagne JP, Yasuhara T, Jackson J, Saxena S, Langelier MF, Ahel I, Bedford MT, Pascal JM, Vindigni A, et al. 2020. CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol Cell 81: 784–800.e8. 10.1016/j.molcel.2020.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV. 2014. OpenComet: an automated tool for comet assay image analysis. Redox Biol 2: 457–465. 10.1016/j.redox.2013.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW. 2018. The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol Cell 71: 319–331.e3. 10.1016/j.molcel.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T. 2011. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 5: 387–393. 10.1016/j.molonc.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G, Borel V, Segura-Bayona S, Takaki T, Ruis P, Bellelli R, Lehmann LC, Sommerova L, Vancevska A, Tomas-Loba A, et al. 2020. Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Mol Cell 81: 767–783.e11. 10.1016/j.molcel.2020.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ, Feltmate CM, Nucci MR, Swisher EM, Nguyen H, et al. 2018. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov 8: 1404–1421. 10.1158/2159-8290.CD-18-0474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Sy SM, Chen J. 2010. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol 11: 138–148. 10.1038/nrm2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhász S, Smith R, Schauer T, Spekhardt D, Mamar H, Zentout S, Chapuis C, Huet S, Timinszky G. 2020. The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci Adv 6: eabb8626. 10.1126/sciadv.abb8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. 2017. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res 23: 3097–3108. 10.1158/1078-0432.CCR-16-2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, Medvedev S, Kinose Y, Devins K, Verma P, et al. 2020. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 11: 3726. 10.1038/s41467-020-17127-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Yu X. 2013. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23: 693–704. 10.1016/j.ccr.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, Macdonald D, et al. 2010. Metadata matters: access to image data in the real world. J Cell Biol 189: 777–782. 10.1083/jcb.201004104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. 2017. PARP inhibitors: synthetic lethality in the clinic. Science 355: 1152–1158. 10.1126/science.aam7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. 2008. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 7: 2010–2019. 10.1016/j.dnarep.2008.08.014 [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. 2018. High speed of fork progression induces DNA replication stress and genomic instability. Nature 559: 279–284. 10.1038/s41586-018-0261-5 [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, et al. 2006. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66: 8109–8115. 10.1158/0008-5472.CAN-06-0140 [DOI] [PubMed] [Google Scholar]
- Michelena J, Lezaja A, Teloni F, Schmid T, Imhof R, Altmeyer M. 2018. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat Commun 9: 2678. 10.1038/s41467-018-05031-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. 2012. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72: 5588–5599. 10.1158/0008-5472.CAN-12-2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK, Cantor SB. 2020. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci Adv 6: eaaz7808. 10.1126/sciadv.aaz7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor MJ. 2015. Targeting the DNA damage response in cancer. Mol Cell 60: 547–560. 10.1016/j.molcel.2015.10.040 [DOI] [PubMed] [Google Scholar]
- Panzarino NJ, Krais JJ, Cong K, Peng M, Mosqueda M, Nayak SU, Bond SM, Calvo JA, Doshi MB, Bere M, et al. 2020. Replication gaps underlie BRCA-deficiency and therapy response. Cancer Res 81: 1388–1397. 10.1158/0008-5472.CAN-20-1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar K, Kochupurakkal BS, Lazaro JB, Wang ZC, Palakurthi S, Kirschmeier PT, Yang C, Sambel LA, Färkkilä A, Reznichenko E, et al. 2019. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin Cancer Res 25: 6127–6140. 10.1158/1078-0432.CCR-19-0448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37: 492–502. 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilié PG, Gay CM, Byers LA, O'Connor MJ, Yap TA. 2019. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res 25: 3759–3771. 10.1158/1078-0432.CCR-18-0968 [DOI] [PubMed] [Google Scholar]
- Pommier Y, Leo E, Zhang H, Marchand C. 2010. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 17: 421–433. 10.1016/j.chembiol.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, O'Connor MJ, de Bono J. 2016. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 8: 362ps17. 10.1126/scitranslmed.aaf9246 [DOI] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, Vindigni A. 2017. DNA fiber analysis: mind the gap!. Methods Enzymol 591: 55–82. 10.1016/bs.mie.2017.03.019 [DOI] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, González-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. 2020. PRIMPOL-mediated adaptive response suppresses replication fork reversal in BRCA-deficient cells. Mol Cell 77: 461–474.e9. 10.1016/j.molcel.2019.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey MD, Quachthithu H, Gaboriau D, Santocanale C. 2017. DNA replication dynamics and cellular responses to ATP competitive CDC7 kinase inhibitors. ACS Chem Biol 12: 1893–1902. 10.1021/acschembio.7b00117 [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. 2012. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19: 417–423. 10.1038/nsmb.2258 [DOI] [PubMed] [Google Scholar]
- Robert I, Dantzer F, Reina-San-Martin B. 2009. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J Exp Med 206: 1047–1056. 10.1084/jem.20082468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN. 2012. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 12: 68–78. 10.1038/nrc3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Kim HJ, Song JY, Hwang SG, Kim JS, Kim J, Bui THN, Choi HK, Ahn J. 2019. A small compound KJ-28d enhances the sensitivity of non-small cell lung cancer to radio- and chemotherapy. Int J Mol Sci 20: 6026. 10.3390/ijms20236026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542. 10.1016/j.cell.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22: 106–116. 10.1016/j.ccr.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonen PM, Talens F, Stok C, Gogola E, Heijink AM, Bouwman P, Foijer F, Tarsounas M, Blatter S, Jonkers J, et al. 2017. Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat Commun 8: 15981. 10.1038/ncomms15981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Cortez D. 2012. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc 7: 594–605. 10.1038/nprot.2012.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Spies J, Coscia F, Kirik U, Rask MB, Lee JH, Neelsen KJ, Mund A, Jensen LJ, Paull TT, et al. 2021. Homology-directed repair protects the replicating genome from metabolic assaults. Dev Cell 56: 461–477.e7. 10.1016/j.devcel.2021.01.011 [DOI] [PubMed] [Google Scholar]
- Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. 2010. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet 6: e1001205. 10.1371/journal.pgen.1001205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wietmarschen N, Nussenzweig A. 2018. Mechanism for synthetic lethality in BRCA-deficient cancers: no longer lagging behind. Mol Cell 71: 877–878. 10.1016/j.molcel.2018.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR. 2014. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 343: 1470–1475. 10.1126/science.1252230 [DOI] [PubMed] [Google Scholar]
- Verma P, Zhou Y, Cao Z, Deraska PV, Deb M, Arai E, Li W, Shao Y, Puentes L, Li Y, et al. 2021. ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat Cell Biol 23: 160–171. 10.1038/s41556-020-00624-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. 2017. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 31: 318–332. 10.1101/gad.290957.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaitė Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. 2018. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559: 285–289. 10.1038/s41586-018-0291-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.