

Abstract
The X Chromosome plays an important role in human development and disease. However, functional genomic and disease association studies of X genes greatly lag behind autosomal gene studies, in part owing to the unique biology of X-Chromosome inactivation (XCI). Because of XCI, most genes are only expressed from one allele. Yet, ∼30% of X genes “escape” XCI and are transcribed from both alleles, many only in a proportion of the population. Such interindividual differences are likely to be disease relevant, particularly for sex-biased disorders. To understand the functional biology for X-linked genes, we developed X-Chromosome inactivation for RNA-seq (XCIR), a novel approach to identify escape genes using bulk RNA-seq data. Our method, available as an R package, is more powerful than alternative approaches and is computationally efficient to handle large population-scale data sets. Using annotated XCI states, we examined the contribution of X-linked genes to the disease heritability in the United Kingdom Biobank data set. We show that escape and variable escape genes explain the largest proportion of X heritability, which is in large part attributable to X genes with Y homology. Finally, we investigated the role of each XCI state in sex-biased diseases and found that although XY homologous gene pairs have a larger overall effect size, enrichment for variable escape genes is significantly increased in female-biased diseases. Our results, for the first time, quantitate the importance of variable escape genes for the etiology of sex-biased disease, and our pipeline allows analysis of larger data sets for a broad range of phenotypes.
The human X Chromosome harbors approximately 1000 genes that perform diverse functions and play key roles in human development and disease etiology (Ross et al. 2005; Zhang et al. 2011; Khramtsova et al. 2019; Sidorenko et al. 2019). Yet, the study of X-linked genes in disease genetics and functional genomics greatly lags compared with those on autosomes (Accounting for sex in the genome 2017). Several analytical challenges arise from the unique biology of X, including chromosome copy number differences between males and females and X-Chromosome inactivation (XCI) in XX females. To balance dosage between sexes, XCI transcriptionally silences one X in female cells (Xi), and most genes only express the active X (Xa) allele. However, ∼10% of X-linked genes consistently escape XCI and are expressed from both the Xa and Xi. Moreover, as many as 30% of human X genes show variable XCI escape and show inter- and intra-individual differences; that is, they escape XCI in a subset of individuals or tissues within an individual but remain X-inactivated in others (Carrel and Willard 2005; Carrel and Brown 2017; Tukiainen et al. 2017; Garieri et al. 2018). As most genes that escape or variably escape XCI lack functionally equivalent Y homologs, Xi expression can result in dosage imbalance between the sexes. A role for XCI escape is emerging for sex-biased traits and disorders (Tukiainen et al. 2014; Wang et al. 2016; Dunford et al. 2017; Souyris et al. 2018; Harris et al. 2019; Natri et al. 2019; Syrett et al. 2019; Hagen et al. 2020; Mousavi et al. 2020; Foresta et al. 2021). For example, XCI escape in immune cells, owing to altered XCI maintenance that results in partial gene reactivation, may in part explain the severe sex bias in autoimmune disorders such as systemic lupus erythematosus (male:female ratio 1:9) (Wang et al. 2016; Souyris et al. 2018; Syrett et al. 2019; Yu et al. 2021). Nonetheless, a more complete understanding of the role that XCI escape plays in disorders, particularly those that are sex biased, has yet to be fully appreciated.
To better understand the biology of X-linked genes and unveil functional and clinical consequences of XCI escape, a critical first step is to identify genes that escape XCI in a particular tissue or disease state. The chromosome-wide analysis is complicated by the random nature of XCI in early development, as the determination of Xa and Xi Chromosomes differs from cell to cell (cellular mosaicism). Single-cell RNA-seq circumvents complications of XCI mosaicism and allows monoallelic/biallelic XCI state assessments (Tukiainen et al. 2017). However, most single-cell RNA-seq data sets are limited to a very small number of individuals (Garieri et al. 2018; Katsir and Linial 2019), making them inadequate for dissecting subtle inter-individual differences.
One approach to identify escape genes from samples with mosaic XCI is to quantify allele-specific expression (ASE) using heterozygous transcribed SNPs to compare the expression ratio between two alleles (Cotton et al. 2013; Larson et al. 2017). ASE levels from silenced genes mirror the level of XCI mosaicism in the sample, whereas escape genes deviate from this pattern and are expressed more evenly between the two alleles. ASE-based methods allow the assessment of XCI states of genes in individual samples and can maximize the utility of existing bulk RNA-seq data.
In this paper, we seek to develop a new statistical model that improves ASE analysis for the identification of XCI escape genes from bulk RNA-seq data sets, use this method to identify genes that escape and variably escape from XCI, and evaluate the impact of identified escape genes on sex-biased traits.
Results
Method overview
Here, we give a brief outline of the statistical framework underlying our XCI state inference model. A more thorough description is given in the Methods section. The XCIR approach, similar to other XCI states inference methods, uses a two-step strategy. We first estimate the degree of XCI skewing, or how much cellular mosaicism deviates from 50:50. XCI skewing is assessed using the ASE ratio observed for a prespecified set of commonly X-inactivated genes, which reflects the fraction of cells in a sample that express a particular X Chromosome. For this purpose, we use a training set of 177 commonly silenced X genes, as defined by Cotton et al. (2013). Subsequently, the XCI state for each X-linked gene that includes a transcribed informative SNP (or SNPs) at sufficient read depth is assessed by whether its ASE is more balanced than the estimated sample skewing.
One unique contribution of XCIR is that it includes rigorous statistical models to improve XCI skewing estimation. Although simple in concept, sample skewing estimation may be confounded by the presence of sequencing errors, which can make a homozygous variant appear heterozygous. Additionally, some commonly silenced genes may escape XCI in a given individual or tissue, making mosaicism appear more balanced than it truly is. Both artifacts can severely bias the estimation of sample skewing and affect the downstream inference of the XCI states. Using a mixture of beta-binomials, we jointly modeled three possible scenarios for each of the training set genes: (1) the gene is truly silenced and the ASE ratio calculated from the heterozygous SNPs accurately reflects sample skewing; (2) the identified heterozygous SNPs for some genes are in fact homozygous and reflect sequencing errors; or (3) some training set genes escape XCI in a given sample. An Akaike information criterion (AIC)–based model selection procedure was developed to select the mixture components that best fit the data. Finally, to infer the XCI state for each candidate gene, we compared each candidate gene's ASE with the sample skewing. Genes with more balanced ASE than the inferred sample skewing are deemed XCI escape genes.
XCIR was compared with two other published approaches. The Xi-threshold approach was originally developed for cDNA hybridization to SNP arrays (Cotton et al. 2013). This method estimates sample skewing as described above and focuses analysis on sufficiently skewed samples (>25:75). Using these skewed samples, the method defines escape genes as those with Xi expression levels >10% of Xa expression levels. To compare performance with XCIR, it was necessary to extend the method for RNA-seq data. In our extension, we estimated sample skewing by replacing the allelic expression intensity from SNP array with allele-specific expression (read counts) from RNA-seq, selected skewed samples, and identified escape genes using the previously established Xi expression level threshold.
An additional method, BayesMix, was developed by Larson et al. (2017) to identify escape genes from RNA-seq data. The method also identifies significantly skewed samples and uses them to identify XCI escape genes. Subsequently, the method uses a cluster model to analyze all X-linked genes. The model implicitly assumes that Xa and Xi expression is equal for escape genes. Based upon the assumption, BayesMix identifies a cluster of genes with a mean ASE ratio of 50:50, representing the set of escape genes, and another cluster with a more skewed ASE ratio representing the set of silenced genes. Each gene is assigned a posterior probability of escaping XCI (PPE). Genes with PPE > 50% are deemed escape genes. The assumption of equal Xa/Xi expression levels for escape genes, however, is not supported, as Xi expression of most escape genes are <30% of Xa expression levels (Carrel and Willard 2005; Tukiainen et al. 2017) and thus may lead to incorrect classification of escape genes.
The determination of escape genes from Xi-threshold and BayesMix approaches requires a predetermined cutoff on Xi expression or the PPE. Although both methods recommend default cutoffs, using them can lead to high type 1 errors and prevent power comparisons. Therefore, to allow comparison with XCIR, we recalibrated cutoffs so that all methods have the same type 1 error. Specifically, we analyze each gene in the training set using BayesMix and Xi-threshold methods, estimate the PPE and Xi expression level for the commonly silenced genes, and use them as an empirical distribution to determine cutoffs in order to get well-controlled type 1 errors.
Simulations to validate XCIR
We first performed simulations to evaluate sample skewing estimates. The simulation parameters for read depths were based upon the estimates obtained in the Geuvadis data set (Lappalainen et al. 2013). A complete description of the simulation settings is presented in the Methods section and Supplemental Methods. For a complete list of simulation settings, see Supplemental Table S1.
Briefly, we simulated samples at XCI skewing mean μ = (0.15, 0.25, 0.35) and variance σ2 = (4 × 10−8, 2 × 10−4, 1 × 10−3), where the skewing mean represents the true level of mosaicism in an individual, and the variance represents how much variability there is in the observed ASE around the skewing mean. We then compared estimates of the skewing from XCIR, BayesMix, and Xi-threshold. Finally, we obtain predicted XCI states for all genes in the test set and computed type 1 error and power across methods.
Overall, the XCIR method gave much more accurate estimates of the sample skewing compared with the BayesMix model (Fig. 1A). The mean squared error of Xi-threshold's and BayesMix's skewing estimates is on average 6.7 and 7.4 times that of XCIR, respectively. In bulk RNA-seq, the presence of sequencing errors in some training genes may decrease average ASE, and this can bias the estimates of the skewing toward zero (Supplemental Fig. S1A). On the other hand, if the training set contains genes that escape XCI in the sample, the skewing estimate can be biased toward 0.5 (Supplemental Fig. S2A). In our simulations, we considered the possibility of both artifacts with one-third of the samples containing sequencing errors and one-third containing genes that escape XCI in the training set. Xi-threshold and BayesMix do not account for these sources of bias, leading to the decreased accuracy.
Figure 1.

Comparison of skewing and XCI state estimates in XCIR, BayesMix, and Xi-threshold for different XCI skewing means (μ) and variances (σ2) of the true skewing. (A) Skewing estimates. The dashed line indicates the true mean. (B) Type 1 error. The dashed line indicates the significance threshold, 0.05. (C) Rescaled power. The Xi-threshold and BayesMix posterior probabilities of escape cutoffs are adjusted until a type 1 error of 5% is achieved. Power is then computed at the recalibrated thresholds for all three methods. Scaled power of zero for BayesMix indicates that a type 1 error of 0.05 or less in the training set can only be achieved using a very high PPE threshold, thus classifying every gene as silenced.
Simulation of the type 1 error and power for XCIR, Xi-threshold, and BayesMix
In addition to assessing XCI skewing, we also performed simulations to evaluate the ability of the three models to correctly identify XCI states in samples with varied XCI skewing. We simulated 100 silenced genes as well as 100 escape genes with different levels of Xi expression in order to compare the type 1 error and power for detecting XCI escape genes.
XCIR has a well-controlled type 1 error across all nine scenarios with different skewing and variance parameters (Fig. 1B). In contrast, using default cutoffs for the Xi-threshold (the Xi expression >10% of the Xa expression) and BayesMix (PPE > 50%) models, the type 1 error depends on the sample skewing and becomes extremely high for less-skewed samples. For example, at a significance threshold of α = .05 when the sample skewing mean is 0.35, and its variance is 1 × 10−3, the type 1 errors for Xi-threshold and BayesMix are 35% and 27%, respectively (4.7% for XCIR).
To evaluate power with controlled false-positive rates, we recalibrated the cutoffs used in the Xi-threshold and BayesMix models such that both methods have a controlled type 1 error on the training set with (Fig. 1C). Power is consistently higher in XCIR compared with the other approaches. All methods are adversely affected by decreased sample skewing and increased variance of ASEs across commonly silenced genes in the training set; however, XCIR retains higher power than the other approaches even for samples with less skewing or large variance. We further compared type 1 errors and power in samples with only sequencing or only training errors (Supplemental Figs. S1, S2), in order to separately assess their impact on the performance of different methods. Finally, we allowed sequencing errors to vary across genes (Supplemental Fig. S3). XCIR outperforms other approaches under these scenarios as well.
Validation using single-cell-derived cell line mixing experiments
To complement our simulations, we evaluated the methods using experimental data from mixes of single-cell-derived clonal lines isolated from the lymphoblastoid cell line (LCL) GM07345. Cell lines derived from a single-cell all have the same Xa/Xi assignment; that is, they are nonrandomly X inactivated and hence allow direct assessment of XCI status, as any gene that escapes XCI will show biallelic expression. To reflect different levels of XCI skewing, we experimentally generated mixes of two single-cell-derived clonal lines with different Xa/Xi assignments (Supplemental Fig. S4). For each mixed sample, we performed RNA-seq and evaluated ASE (see Methods) (Supplemental Methods). This approach allows us to estimate the type 1 error and power empirically, as escape genes are inferred in the mixes, and the accuracy of results can be evaluated using the XCI states observed in the nonrandomly inactivated single-cell-derived lines. We applied the XCIR, BayesMix, and Xi-threshold methods to the data. For BayesMix, we considered both flat and informative prior for single-sample analysis.
The XCI status for each informative, well-expressed gene was confirmed by the RNA-seq data from the nonrandomly inactivated single-cell-derived lines. Eighty-three genes are expressed from only one X and were deemed X-inactivated, and 28 genes were expressed from both Xs and therefore escape XCI (Supplemental Table S2). Of the silenced genes, 57 (68%) were part of the training set of commonly silenced genes (Balaton et al. 2015).
Evaluations based upon the clonal cell line mixes are concordant with the simulation experiment, with XCIR outperforming existing approaches. XCIR has a well-controlled type 1 error in all four mixes, including the less-skewed 60:40 and 70:30 mixes (Fig. 2A). Based upon the default cutoff values, both the Xi-threshold and BayesMix methods have very high type 1 errors in all samples, particularly those that are less skewed. Using single-sample analysis with the suggested flat prior, the observed type 1 error rate for BayesMix lies between 22% and 40%. Similarly, the type 1 error rate for Xi-threshold lies between 7% and 37%.
Figure 2.
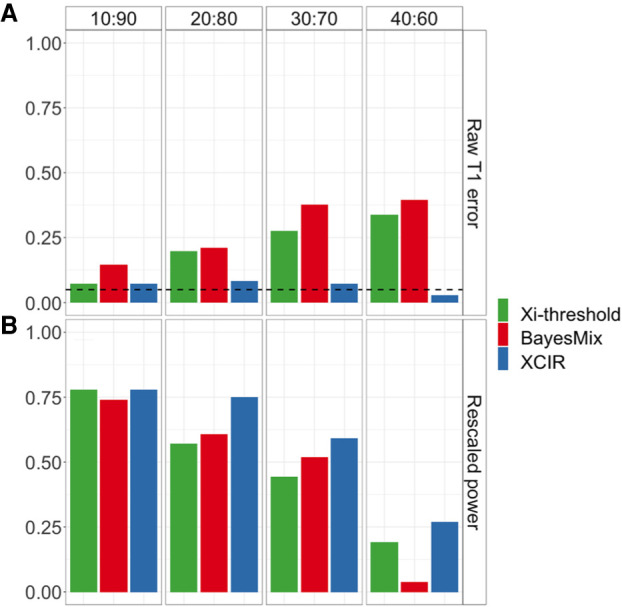
XCI inference in single-cell clone mixing experiment. The proportion of each single-cell clonal line in the mixed sample is indicated at the top of the panel and is equivalent to the true skewing of each sample. (A) Raw type 1 error. (B) Rescaled power at the empirical threshold for 5% type 1 error.
We recalibrated the cutoffs used to call escape genes and compared the power of Xi-threshold and BayesMix with XCIR (Fig. 2B). The empirical power was calculated by the fraction of the 25 XCI escape genes that was correctly called by each method. For the most skewed samples, the identification of escape genes becomes the easiest, and the power for the three methods is comparable. For samples with skewing 90:10, all methods can identify ∼75% of the escape genes that are sufficiently expressed and covered by enough reads. On the other hand, as XCI skewing decreases, XCIR outperforms the existing approaches. For more balanced mixes (i.e., 60:40, 70:30, or 80:20), the power for XCIR is much higher than the competing approaches. For example, in the sample with 80:20 skewing, the power for BayesMix is 60% compared with 75% for XCIR. As some of the commonly silenced genes escape in the sample, they may bias the empirical null distribution for the Xi expression level used to calibrate the cutoffs, and the resulting thresholds were overly stringent. Likewise, the posterior probability of escape cutoffs is affected by escape genes in the training set, which can lead to overly conservative cutoffs (Supplemental Table S3).
Application to Geuvadis RNA-seq data
To further quantify XCI states in multiple samples using a population-scale data set, we applied the pipeline to the Geuvadis data set, which contains RNA-seq data for 217 female LCLs. DNA genotypes for the same set of individuals were obtained from The 1000 Genomes Project phase 3 (The 1000 Genomes Project Consortium 2015).
Determining XCI states with XCIR
Given that ASE-based methods such as XCIR have maximal power to detect escape genes in relatively skewed samples, we restricted our analysis to the 136 samples with skewing greater than 25:75. As a result, although the full data set contains 351 genes with at least one heterozygous SNP covered at a sufficient depth, only the 215 genes that could be scored in at least 10 of the 136 skewed samples were used for the final classification. The Geuvadis samples were part of The 1000 Genomes Project that included DNA-sequence genotypes and phased haplotypes. Although the default input for our XCIR analysis takes the read depths of a single heterozygous SNP, aggregating reads from multiple heterozygous SNPs on phased haplotypes can potentially improve power. In simulations, we show that using phasing information can increase the accuracy of XCI state predictions, especially in less-skewed samples with an average increase of 11% (Supplemental Fig. S5). Specifically, in Geuvadis samples, using haplotype information increases the average per-gene read depth by 48% from 81 to 120.
We classified genes using previously established criteria (Carrel and Willard 2005): Escape genes were expressed from the inactive X in >75% of individuals; genes that escape in <25% of the individuals were deemed X-inactivated; and those that escape in 25%–75% of the individuals were classified as variable escape genes. Following these criteria, 165 (76.7%) genes were found to be X-inactivated, 20 (9.3%) were predicted to escape XCI, and 30 (14%) showed variable XCI escape in the data set.
As described above, XCIR includes features to identify if the observed ASE ratio for a SNP is owing to sequencing errors or the inclusion of an escape gene within the commonly silenced training set genes. Subsequently, the likelihood of each scenario can be quantified, and the model that best fits the data was used to infer XCI states. Applying the method, we noted that 28% of the samples were fitted with a two- or three-component mixture model, which emphasizes the necessity of correcting for both confounding errors.
Using XCIR's ability to reliably identify inter-individual variability, we refined the previous classification of X-linked genes (Supplemental Table S4, S5). Compared with the consensus (Balaton et al. 2015), we identified five new genes variably escaping XCI that were previously classified as strictly X-inactivated (EOLA2, DMD, EMD, OTUD5, SASH3). Finally, our results confirm known escapers as we did not reclassify any of the escape genes identified in all prior studies.
Although the underlying XCI landscape for all Geuvadis samples is not known, results were confirmed with multiple lines of evidence. Because of their presence on both the X and Y Chromosomes, pseudoautosomal region (PAR) genes are expected to escape XCI. Indeed, the 10 PAR1 genes with sufficient read counts were predicted as escape XCI in >80% of the subjects. Furthermore, of the 124 training set genes that could be scored in this data set, only three escape in >25% of the subjects (SEPTIN6, STK26, and MAP7D2). It is notable, however, that other studies also classified these three genes as variably escaping XCI (Cotton et al. 2013; Balaton et al. 2015), which confirms both our findings and the necessity to account for potential errors in the training list. Although such findings could support refining the list of training set genes, they perhaps, more importantly, underscore that the tools integrated into XCIR have tremendous merit for defining the XCI landscape in larger data sets and cell types in which XCI states are less well described.
Differential gene expression for X-linked genes
We also examined expression between sexes using the 244 male lines included in the Geuvadis project. Transcript abundance was quantified using kallisto (Bray et al. 2016), and differential expression analysis was performed with limma (Ritchie et al. 2015). Using a false-discovery rate threshold of 0.05, we found 33 differentially expressed genes on the X Chromosome.
Because most escape genes lack functionally equivalent Y homologs, we sought to examine whether escape genes are more likely to be differentially expressed between sexes. We asked if genes with significant differential expression were those that escape in the most individuals. Our analyses were focused on the 215 X-linked genes for which XCI calls were available in at least 10 skewed samples (Fig. 3). As expected, we found that genes predicted to escape XCI are up-regulated in females and show a significant difference in expression versus males. When a Y homolog exists, such as for genes in the PAR, there is expression from both sex chromosomes. Although most of these genes fail to show significantly increased expression in females, XCIR consistently predicted a very high percentage of escape for the 10 PAR1 genes available in the data set (Fig. 3, triangles). Although differential gene expression analysis cannot reliably assess individual-level XCI states, XCIR can be successfully applied to infer XCI states of genes in each individual, provided the presence of an informative expressed SNP. Moreover, we found that five of these PAR1 genes show male-biased expression, including ZBED1, which was significantly differentially expressed. This observation is consistent with a previous report that escape from XCI in these genes is only partial and can lead to higher expression on the Xa and Y than on the Xi (Gershoni and Pietrokovski 2017; Tukiainen et al. 2017).
Figure 3.
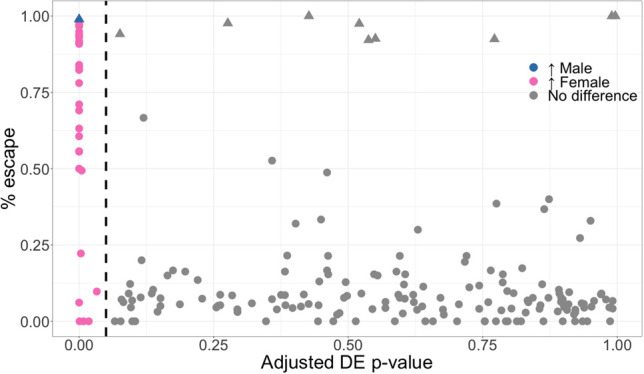
Genes that escape XCI are differentially expressed. Adjusted differential expression (DE) P-values as reported by limma for 215 genes against the frequency of samples that escape XCI as predicted by XCIR in Geuvadis. The dashed line indicates the 5% significance cutoff, and the significantly differentially expressed genes are colored based on the sex in which increased expression is observed. Escape genes are mostly female-biased, reflecting expression from both X copies. PAR genes (triangles) are correctly identified as escaping XCI despite most of them not being significantly differentially expressed, as the expression on the Y is similar or higher to that on the Xi.
Heritability analysis of UK Biobank phenotypes
We next sought to determine whether XCI state influences X-linked disease heritability. Disease heritability of X genes was estimated by extending LD score regression. We first annotated the entire X Chromosome by XCI states based upon our analysis of Geuvadis data and quantified the heritability for 319 self-reported phenotypes in pooled (while adjusting for sex as a covariate) and sex-specific GWAS of the UK Biobank (Bycroft et al. 2018). Annotations and heritability analysis results are available in Supplemental Table S6. We focus on variants with MAF > 0.01 in the analysis. Although association results from low-frequency variants in unbalanced case control studies should generally be interpreted with caution, we ensured that the SLE analysis results are well behaved by observing the quantile–quantile and Manhattan plot (Supplemental Figs. S6, S7).
To examine the contribution of inactive and escape genes, we partitioned heritability across XCI states. Enrichment is defined as the ratio of the proportion of heritability explained over the proportion of SNPs for a specific functional category. Overall, in the self-reported phenotypes, the analysis of traits in combined sexes indicates that the heritabilities from variable escape and escape genes are significantly enriched (mean enrichment of 5× and 3.8×) relative to silenced genes (1.3×) and intergenic SNPs (0.5×). These enrichment patterns are also observed when analyzing each sex separately (Fig. 4).
Figure 4.

Genes that escape and variably escape XCI are enriched for heritability. (A) Distribution of heritability enrichment for all self-reported phenotypes available in females (240 phenotypes), males (218 phenotypes), or both (280 phenotypes). Heritability was independently assessed for each XCI state: escape (E), variably escape (VE), and silenced (S). X genes with Y homologs that escape or variably escape are differentiated (Y homolog). (B) Lupus heritability enrichment measured in females (392 cases, 182,316 controls).
Notably, we observe that heritability is particularly enriched in escape or variable escape genes with functional Y homologs, including SNPs associated with genes in the PARs (9.5×). Such enrichment may reflect evolutionary constraints on the dosage of these genes (Slavney et al. 2016). This is supported by previous observations that these genes are widely expressed in multiple cell types and function broadly to regulate targets throughout the genome (Bellott et al. 2014). We noted slightly weaker enrichment for escape genes compared with variable escape genes. Constitutive escape genes are expected to have consistent biallelic expression across all females. Therefore, there is little inter-individual differences in the escape status, and the escape genes do not explain the inter-individual phenotypic differences. On the other hand, escape of a typically silenced gene (i.e., variable escape) will lead to more dramatic expression changes between individuals, which is likely to affect phenotypes (Fig. 4A).
Although alterations in the XY-homologous genes may lead to broad effects that underlie their role in many disease phenotypes, the more modest overall enrichment observed in escape and variable escape genes that lack functional Y homologs could reflect significant enrichment in a subset of diseases. Moreover, as escape and variable escape genes without Y homologs can result in gene dosage imbalance between males and females, we hypothesize that heritability at these gene loci may be enriched in disorders with a female bias. Indeed, for systemic lupus erythematosus, an autoimmune disease that predominantly affects females, we find that silenced genes explain virtually none of the heritability observed in females, whereas genes that escape or variably escape XCI lacking Y homologs are significantly enriched for heritability (3.2× and 7.7×, respectively) (Fig. 4B).
To more broadly examine this hypothesis, we identified sex-biased phenotypes from the UK Biobank with a female/male ratio greater than two among affected individuals. In this subset of 51 phenotypes, we observe a significantly higher enrichment of heritability for variable escape genes than for the 154 traits with a balanced sex ratio (with P-value = 0.007 based upon permutation testing; see Methods) (see Supplemental Methods). Importantly, for both female- and male-biased diseases (Table 1), escape and variable escape genes are the only XCI states that show significant enrichment in these sex-biased diseases, strongly supporting a role for XCI escape in many female-biased diseases (Khramtsova et al. 2019).
Table 1.
Permutation tests to assess heritability enrichment differences between sex-biased diseases and nonbiased phenotypes for each XCI state

Computational advantages
Despite having a more complex statistical model than some existing approaches, XCIR remains computationally efficient. Analysis of the full Geuvadis data set is performed under 1 min using a standard computer server (with Intel Xeon CPU E5-2680 v2 and 128GB RAM), including reading the data, fitting all mixture models for the skewing estimates, and classifying X-linked genes. In comparison, using BayesMix, the estimation of the sample skewing alone exceeded 2 h, and the PPE could not be computed on a single core. Likewise, the analysis of 720 simulated samples with 107 genes each takes <2 min with XCIR. BayesMix required massive parallelization, and the processing of a subset of 36 samples takes >2 h. Our method is implemented in an R package with examples and full documentation available on Bioconductor and GitHub. The source code is also uploaded as part of the Supplemental Material. The improvements in speed and ease of use brought by XCIR will allow further analysis of existing and future large-scale data sets by other researchers in the research community and present an opportunity for the systematic inclusion of X in all genomics studies.
Discussion
In this article, we have proposed and validated a novel and principled statistical method to identify XCI escape genes from RNA-seq data. The approach has multiple advantages. First, XCIR effectively models genotype errors, as well as misclassification of X-inactivated training set genes owing to inter-individual heterogeneity in XCI states. This model-based inference for XCI states allows accurate control of type 1 errors and yields much higher power than alternative approaches. As the study of XCI escape extends to new tissue types, grows to larger sample sizes, and incorporates samples from individuals with particular disorders/traits, the set of commonly silenced genes may be less accurate, as many of them were inferred from smaller cohorts of LCLs or fibroblasts. Thus, we expect that the model can be even more useful to infer XCI states in other tissues.
Second, unlike Xi-threshold and BayesMix (Cotton et al. 2013; Larson et al. 2017), XCIR does not rely on an arbitrary threshold for the inference of XCI states. XCIR does not need recalibration, a process that is particularly sensitive to the contamination of the training set of commonly silenced genes with escape genes. Critically, approaches that require such recalibration typically do not account for this source of error.
Third, the method is computationally efficient and publicly available as an R package. It can be applied to the broadly available bulk RNA-seq data, which maximizes the utility of the existing and future data sets regardless of their size.
For the BayesMix approach (Larson et al. 2017), the investigators suggested the use of informative priors to improve the analysis. When the informative prior is used, the prior knowledge tends to dominate the analysis. Although no type 1 errors were made, the power is consistently lower for less-skewed samples. The method with informative prior is only more powerful than XCIR for very balanced samples, in which the observed ASE is highly uninformative for XCI state inference. As the analysis of XCI escape genes in other tissues remains limited, the usefulness of prior information in the detection of XCI escape genes remains to be examined.
Lower performance in samples with more balanced mosaicism and less skewing has been reported with other methods (Cotton et al. 2013; Larson et al. 2017). Despite the loss of power, we showed that type 1 error is well controlled at all skewing levels. Furthermore, with bulk RNA-seq, higher sample sizes somewhat compensate for the reduced number of skewed samples as ∼25% of normal individuals are skewed >75:25 (Amos-Landgraf et al. 2006). This means that single-subject predictions, such as in the context of personalized medicine, may not always be feasible, depending on the mosaicism in a particular patient. Single-cell RNA-seq may provide an answer for such cases, although such analyses are complicated by intraindividual heterogeneity among cells that could reflect XCI state differences or transcription states (e.g., transcriptional bursting). Moreover, until single-cell RNA-seq is routinely performed for large population data sets, it will likely miss much of the inter-individual differences highlighted by XCIR. In this case, a combined approach that uses bulk RNA-seq and XCIR on large disease data sets in conjunction with single-cell RNA-seq (e.g., Tukiainen et al. 2017; Garieri et al. 2018; Katsir and Linial 2019) on less-skewed samples will be helpful to fully explore the genetics of X-linked disease. In addition, such combined approaches using single-cell RNA-seq and XCIR may be further extended to incorporate covariates, for example, age or cell type heterogeneity to improve the modeling of bulk-RNA-seq.
Finally, similar to other expression-based approaches, XCIR may be influenced by the presence of eQTL regulatory variants, which may also contribute allelic imbalance. In the estimation of skewing, multiple genes are used, and hence, the eQTL effects may be canceled out across genes and have negligible effects on the estimates. On the level of individual XCI calls, eQTLs may influence ASE. Yet, in simulations, we show that under realistic conditions, the effect of regulatory variants is limited and that by regressing the allelic expression over the eQTL genotypes and using the residuals as input for XCIR, we are able to recover most of the lost accuracy (Supplemental Table S7). Importantly, how and whether cis-regulatory variants influence XCI escape have never been addressed but may be revealed by such analyses.
To our knowledge, this work is the first to characterize the importance of each class of XCI state to the heritability of hundreds of traits, including male- and female-biased diseases. Although sex biases can be explained by both hormonal and/or sex chromosomal differences, the heritability enrichment observed here is seen not only in comparisons between males and females but also in male-only and female-only analyses and therefore strongly points to chromosomal differences. Our analyses reveal an overall high contribution from SNPs in the escape and variable escape regions. Mechanistically, these data may support genetic influences on XCI escape. Additionally, large contributions from escape and variable escape genes that retain Y homologs are consistent with current knowledge of the biology of these XY pairs (Bellott et al. 2014; Cortez et al. 2014), and their comparison for the enrichment of each XCI class between sex-biased and nonbiased phenotypes revealed a significant increase of enrichment for the variable escape genes in female-biased diseases. These results may suggest that overexpression of some X-linked genes, owing to XCI escape in a subset of females, plays a previously unappreciated role in diseases that merits further examination. We showed that XCI variable escape genes, despite being critically understudied because of technical complexity, play important roles in the regulation of many phenotypes. Our annotated list of escape and variable escape genes come from our XCI evaluation in LCLs with results that are largely concordant with other surveys from multiple tissue/cell types. As shown by Tukiainen et al. (2017), <6% of genes show tissue-specific escape. Therefore, we expect our enrichment analysis results will remain as studies are broadened to consider additional tissues. As part of future work, it is important to incorporate GTEx data and elucidate the causal roles of tissue-specific escape genes on diseases.
In conclusion, we have developed the methodology needed to establish XCI status for population-sized data sets. We have tested our pipeline on simulated and real data sets and have implemented it into an intuitive, well-documented, and freely available software. Using our approach, we reclassified X-linked genes, highlighting their increased importance to female-biased diseases. We anticipate that applications of our method to other biobank-scale data sets will lead to further characterization of the role of specific genes and mutations in a broad range of phenotypes.
Methods
In this section, we describe in detail the XCIR model, whereas the methodological details for the simulation evaluation, the experimental validation, the analysis of the Geuvadis data, and heritability analysis of the UK Biobank data set can be found in the Supplemental Methods.
Model-based inference for XCI skewing
For a gene g in a female individual, let N be the total number of reads, and N1 and N2 be the number of reads mapped to each haplotype. When haplotype information is available, N1 and N2 represent the total read count from haplotypes 1 and 2 summed across heterozygous SNPs. If haplotype information is not available, N1 and N2 may represent the read counts from the most highly expressed SNP within the transcribed region. Let Na, Ni be the number of reads expressed from Xa and Xi, respectively. Given that the bulk RNA-seq data consist of a mosaic of cells with different Xa/Xi assignment, we further denote the pairs Na1, Na2 and Ni1, Ni2 as the number of reads on the active and inactive chromosomes that also belong to the first and second haplotypes, respectively. For convenience, we assume that X1 is the least expressed haplotype such that, for any gene, N1 ≤ N2. The relationship between different quantities is given by
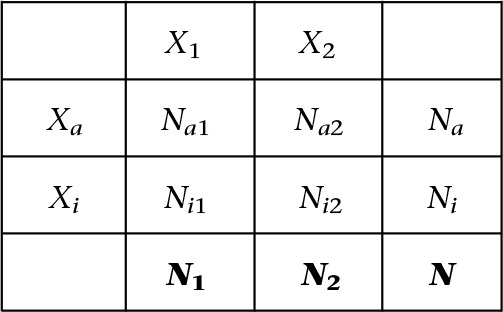
where only N1, N2, and N are observed (shown in bold in the bottom row). The read counts Na1, Na2, Ni1, Ni2 are not directly observed in bulk RNA-seq data and need to be statistically inferred. For genes that escape XCI, we expect Ni,1 > 0, Ni,2 > 0. The relationship between these read counts satisfies
A key parameter of interest for XCI inference is XCI skewing (denoted as f), which can be represented by the fraction of cells in which a given haplotype (e.g., the first haplotype) is actively expressed. The observed allelic expression and the number of reads from Xa and Xi satisfy
For genes that are silenced by XCI, E(Ni1) = 0, and the above equation reduces to
In theory, the sample skewing f can be estimated using the ratio of N1/N. Yet, it should be noted that the training set of commonly silenced genes may include genes that escape XCI in a particular sample. The contamination can be extensive as the original training set was obtained using relatively small data sets (Carrel and Willard 2005; Cotton et al. 2013, 2015). Variable escape genes that escape in a small fraction of individuals may be incorrectly included as a commonly silenced gene. The observed read counts may also be sequence errors.
To account for these potential artifacts and infer XCI states rigorously, we adopted a likelihood-based approach. If the observed read counts are owing to sequence errors (with probability perr), we assume that the read count follows Bin(N, πerr). If the read counts come from a silenced gene (with probability ps (1 − perr), and ps is the fraction of silenced genes), we assume that they follow a beta-binomial (BB) distribution, which allows for overdispersion (Pickrell et al. 2010), that is, BB(N, αs, βs). Finally, if the reads come from an escape gene (with probability (1 − ps)(1 − perr)), we assume that they follow BB(N, αe, βe).
Together, the observed read counts on haplotype 1 are assumed to follow the full mixture model (Mfull) below:
The model parameters for Mfull are estimated for each sample separately.
Determination of the mixture components using AIC
Although the full model Mfull incorporates all possibilities, it is of practical interest to determine for each sample if any commonly silenced gene escapes in the sample and if the reads contain sequencing errors. In addition, the downstream XCI inference would also benefit from a more parsimonious model with fewer mixture components, as the skewing estimates can be more accurate.
We determine the optimal number of mixture components using AIC based variable selection. We consider four possible submodels for the read count from one haplotype allele. Specifically, the three submodels that we consider along with Mfull are
The model with the smallest AIC will be selected. Based upon the selected model, the parameters of interest can be estimated using a maximum likelihood approach. The sample skewing estimate and its variance are given by
Inference of XCI escape states
To perform hypothesis testing and infer the XCI states, we compare the observed ASE of each gene to the sample skewing:
For a given gene g, the ASE ratio of an escape gene will always be greater or equal to that of an inactivated gene. Thus, we test the following one-sided hypothesis:
using the t-statistic
where is the skewing estimated in the first step, and is the observed ASE ratio for gene g.
Under H0, the variance for satisfies
The P-value can be approximated from normal distribution. We also calculate the exact P-value based upon the BB distribution as
The mid-p procedure was used in the above formula to account for discreteness in the exact P-values.
Software availability
Our method is implemented in an R package (R Core Team 2021) complete with examples and full documentation available on Bioconductor (http://bioconductor.org/packages/3.10/bioc/html/XCIR.html) and GitHub (https://github.com/SRenan/XCIR). The XCIR source code is also uploaded as Supplemental Code.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health (National Institute of General Medical Sciences) R01GM126479, the Lupus Research Alliance, and CURE funds from the Pennsylvania Department of Health.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275677.121.
Competing interest statement
The authors declare no competing interests.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accounting for sex in the genome. 2017. Nat Med 23: 1243. 10.1038/nm.4445 [DOI] [PubMed] [Google Scholar]
- Amos-Landgraf JM, Cottle A, Plenge RM, Friez M, Schwartz CE, Longshore J, Willard HF. 2006. X chromosome-inactivation patterns of 1005 phenotypically unaffected females. Am J Hum Genet 79: 493–499. 10.1086/507565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaton BP, Cotton AM, Brown CJ. 2015. Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol Sex Differ 6: 35. 10.1186/s13293-015-0053-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho TJ, Koutseva N, Zaghlul S, Graves T, Rock S, et al. 2014. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508: 494–499. 10.1038/nature13206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, et al. 2018. The UK Biobank resource with deep phenotyping and genomic data. Nature 562: 203–209. 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L, Brown CJ. 2017. When the lyon(ized chromosome) roars: ongoing expression from an inactive X chromosome. Philos Trans R Soc Lond B Biol Sci 372: 20160355. 10.1098/rstb.2016.0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L, Willard HF. 2005. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434: 400–404. 10.1038/nature03479 [DOI] [PubMed] [Google Scholar]
- Cortez D, Marin R, Toledo-Flores D, Froidevaux L, Liechti A, Waters PD, Grützner F, Kaessmann H. 2014. Origins and functional evolution of Y chromosomes across mammals. Nature 508: 488–493. 10.1038/nature13151 [DOI] [PubMed] [Google Scholar]
- Cotton AM, Ge B, Light N, Adoue V, Pastinen T, Brown CJ. 2013. Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome Biol 14: R122. 10.1186/gb-2013-14-11-r122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton AM, Price EM, Jones MJ, Balaton BP, Kobor MS, Brown CJ. 2015. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum Mol Genet 24: 1528–1539. 10.1093/hmg/ddu564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunford A, Weinstock DM, Savova V, Schumacher SE, Cleary JP, Yoda A, Sullivan TJ, Hess JM, Gimelbrant AA, Beroukhim R, et al. 2017. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat Genet 49: 10–16. 10.1038/ng.3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foresta C, Rocca MS, Di Nisio A. 2021. Gender susceptibility to COVID-19: a review of the putative role of sex hormones and X chromosome. J Endocrinol Invest 44: 951–956. 10.1007/s40618-020-01383-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garieri M, Stamoulis G, Blanc X, Falconnet E, Ribaux P, Borel C, Santoni F, Antonarakis SE. 2018. Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts. Proc Natl Acad Sci 115: 13015–13020. 10.1073/pnas.1806811115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni M, Pietrokovski S. 2017. The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol 15: 7. 10.1186/s12915-017-0352-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen SH, Henseling F, Hennesen J, Savel H, Delahaye S, Richert L, Ziegler SM, Altfeld M. 2020. Heterogeneous escape from X chromosome inactivation results in sex differences in type I IFN responses at the single human pDC level. Cell Rep 33: 108485. 10.1016/j.celrep.2020.108485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris VM, Harley ITW, Kurien BT, Koelsch KA, Scofield RH. 2019. Lysosomal pH Is regulated in a sex dependent manner in immune cells expressing CXorf21. Front Immunol 10: 578. 10.3389/fimmu.2019.00578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsir KW, Linial M. 2019. Human genes escaping X-inactivation revealed by single cell expression data. BMC Genomics 20: 201. 10.1186/s12864-019-5507-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khramtsova EA, Davis LK, Stranger BE. 2019. The role of sex in the genomics of human complex traits. Nat Rev Genet 20: 173–190. 10.1038/s41576-018-0083-1 [DOI] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ’t Hoen PAC, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. 2013. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501: 506–511. 10.1038/nature12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson NB, Fogarty ZC, Larson MC, Kalli KR, Lawrenson K, Gayther S, Fridley BL, Goode EL, Winham SJ. 2017. An integrative approach to assess X-chromosome inactivation using allele-specific expression with applications to epithelial ovarian cancer. Genet Epidemiol 41: 898–914. 10.1002/gepi.22091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi MJ, Mahmoudi M, Ghotloo S. 2020. Escape from X chromosome inactivation and female bias of autoimmune diseases. Mol Med 26: 127. 10.1186/s10020-020-00256-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natri H, Garcia AR, Buetow KH, Trumble BC, Wilson MA. 2019. The pregnancy pickle: evolved immune compensation due to pregnancy underlies sex differences in human diseases. Trends Genet 35: 478–488. 10.1016/j.tig.2019.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772. 10.1038/nature08872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2021. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, et al. 2005. The DNA sequence of the human X chromosome. Nature 434: 325–337. 10.1038/nature03440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorenko J, Kassam I, Kemper KE, Zeng J, Lloyd-Jones LR, Montgomery GW, Gibson G, Metspalu A, Esko T, Yang J, et al. 2019. The effect of X-linked dosage compensation on complex trait variation. Nat Commun 10: 3009. 10.1038/s41467-019-10598-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavney A, Arbiza L, Clark AG, Keinan A. 2016. Strong constraint on human genes escaping X-inactivation is modulated by their expression level and breadth in both sexes. Mol Biol Evol 33: 384–393. 10.1093/molbev/msv225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, Pienkowski C, Chaumeil J, Mejia JE, Guery JC. 2018. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol 3: eaap8855. 10.1126/sciimmunol.aap8855 [DOI] [PubMed] [Google Scholar]
- Syrett CM, Paneru B, Sandoval-Heglund D, Wang J, Banerjee S, Sindhava V, Behrens EM, Atchison M, Anguera MC. 2019. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight 4: e126751. 10.1172/jci.insight.126751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen T, Pirinen M, Sarin AP, Ladenvall C, Kettunen J, Lehtimäki T, Lokki ML, Perola M, Sinisalo J, Vlachopoulou E, et al. 2014. Chromosome X-wide association study identifies loci for fasting insulin and height and evidence for incomplete dosage compensation. PLoS Genet 10: e1004127. 10.1371/journal.pgen.1004127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, Aguirre M, Gauthier L, Fleharty M, Kirby A, et al. 2017. Landscape of X chromosome inactivation across human tissues. Nature 550: 244–248. 10.1038/nature24265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Syrett CM, Kramer MC, Basu A, Atchison ML, Anguera MC. 2016. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc Natl Acad Sci 113: E2029–E2038. 10.1073/pnas.1520113113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Qi Y, Li R, Shi Q, Satpathy AT, Chang HY. 2021. B cell-specific XIST complex enforces X-inactivation and restrains atypical B cells. Cell 184: 1790–1803.e17. 10.1016/j.cell.2021.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Klein K, Sugathan A, Nassery N, Dombkowski A, Zanger UM, Waxman DJ. 2011. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. PLoS One 6: e23506. 10.1371/journal.pone.0023506 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.