

Summary
The emergence of megascale single-cell multiplex tissue imaging (MTI) datasets necessitates reproducible, scalable, and robust tools for cell phenotyping and spatial analysis. We developed open-source, graphics processing unit (GPU)-accelerated tools for intensity normalization, phenotyping, and microenvironment characterization. We deploy the toolkit on a human breast cancer (BC) tissue microarray stained by cyclic immunofluorescence and present the first cross-validation of breast cancer cell phenotypes derived by using two different MTI platforms. Finally, we demonstrate an integrative phenotypic and spatial analysis revealing BC subtype-specific features.
Keywords: multiplex tissue imaging, GPU data science, breast cancer, systems biology, image analytics
Graphical abstract
Highlights
-
•
Multiplex tissue imaging (MTI) data require a reliable analytical toolset
-
•
GPU acceleration of single-cell analyses enables rapid iteration and interaction
-
•
Inter-MTI platform validation of breast cancer (BC) cell types indicates consensus
-
•
Spatial features of tissues discriminate between BC clinical subtypes
Motivation
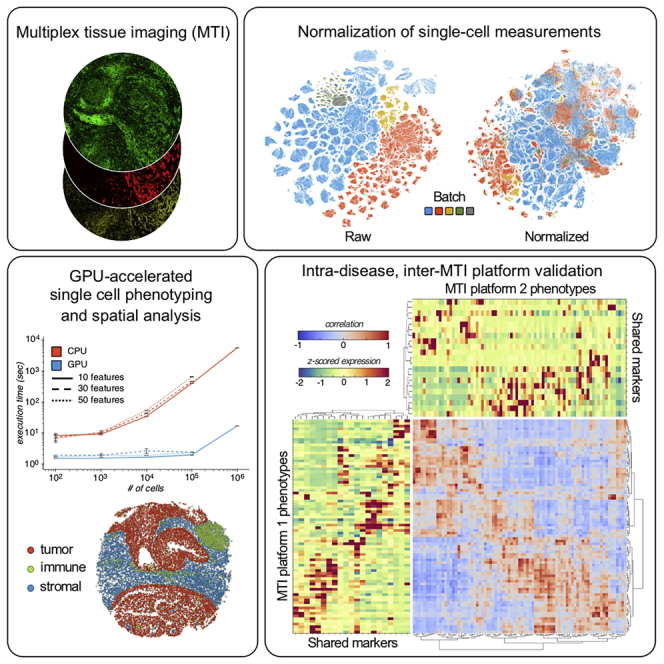
Multiplex tissue imaging (MTI) is transforming biology by linking biomarker expression with spatial context. As the number of MTI platforms and spatial tissue atlases increases, so too must the capacity of our methods to ingest and derive insight from these data. Many of the methods used in MTI analysis are crossovers from other domains like flow cytometry and single-cell genomics, do not scale well, and might be statistically unfit for atlas-level MTI data, constituting a crucial bottleneck in the search for the next generation of clinically relevant biomarkers. Here, we address this bottleneck with a computationally efficient and platform-agnostic MTI data analysis workflow and its proof-of-concept application to human breast cancer (BC) tissues.
Burlingame et al. describe a GPU-accelerated workflow for normalization, phenotyping, and spatial analysis of single-cell multiplex tissue imaging (MTI) data. This workflow is deployed on breast cancer (BC) tissues to derive a cell type dictionary, which is validated between MTI platforms. Tissue architecture is used to discriminate between BC subtypes.
Introduction
MTI methods like cyclic immunofluorescence (CyCIF) (Lin et al., 2018; Eng et al., 2020), CO-Detection by indEXing (CODEX) (Goltsev et al., 2018), multiplex immunohistochemistry (mIHC) (Tsujikawa et al., 2017), imaging mass cytometry (IMC) (Giesen et al., 2014), and multiplex ion beam imaging (Angelo et al., 2014) enable measurements of the expression and spatial distribution of tens of markers in tissues, and have facilitated our understanding of the interactions and relationships among distinct cell types in diverse tissue microenvironments. Nevertheless, for MTI to reach its full potential as a research paradigm, numerous computational challenges must be overcome, including (1) reproducible normalization of single-cell intensity measurements to enable intra- and inter-sample comparisons; (2) robust cell phenotyping at megascale to enable comparison—and soon compilation—of MTI datasets from different platforms; and (3) the development of insightful spatial features to characterize the microenvironment of the tissue or disease of interest, and so enable discrimination between tissues that vary over important clinical parameters.
The number of single-cell phenotyping algorithms has rapidly proliferated alongside the development of single-cell cytometry platforms (Weber and Robinson, 2016; Liu et al., 2019). With so many options from which to choose, the appropriate choice of algorithm depends on the questions one hopes to ask of the data being considered. For instance, prior biological knowledge can be leveraged by supervised or semi-supervised algorithms to bias identification toward known cell types of interest. In contrast, unsupervised algorithms identify cell types by leveraging only the internal data structure, making them the algorithms of choice in discovery-based studies where relatively little is known about the underlying biology. Among the unsupervised algorithms, PhenoGraph (Levine et al., 2015) and FlowSOM (Van Gassen et al., 2015) stand out for their abilities to precisely identify known cell types with high cluster coherence—i.e., high or low inter- or intra-cluster variance—and without the incorporation of prior biological knowledge (Liu et al., 2019). One notable tradeoff between these two CPU-executed algorithms is runtime: FlowSOM has a considerably faster runtime than PhenoGraph (Weber and Robinson, 2016). The relatively slow runtime of CPU-executed PhenoGraph motivated our GPU-accelerated implementation.
To address these challenges, we present (1) a broadened application of our data-intrinsic normalization method (Chang et al., 2020), which leverages the mutually exclusive expression pattern of marker pairs in MTI stain panels to estimate normalization factors without subjective and time-consuming manual gating; (2) a distributed and graphics processing unit (GPU)-accelerated implementation of PhenoGraph (Levine et al., 2015), the popular graph-based algorithm for subpopulation detection in high-dimensional single-cell data; and (3) an integrative analysis using this toolkit on 1.3 million cells from a 180-sample, pan-subtype human BC tissue microarray (TMA) dataset (Figure S1) stained by CyCIF using a marker panel that characterizes tumor, immune, and stromal compartments (Figure 1A, STAR Methods). Through consideration of both tissue composition and architecture, we identify features independent from hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) expression that discriminate between the canonical BC subtypes.
Figure 1.

GPU-accelerated analysis of single-cell phenotypes across BC clinical subtypes
(A) Overview of CyCIF analysis workflow. Once TMA cores are stained by CyCIF, cells are segmented and cell mean intensities are extracted, normalized, then used to define cell phenotypes for analyses of tissue composition and architecture. Box 1 shows an example of RESTORE normalization (Chang et al., 2020) of single-cell Ecad intensities when using mutually exclusive expression of CD68 to derive a normalization factor for Ecad. See Figure S2 for additional normalization details. Box 2 shows benchmarking results for CPU and GPU implementations of PhenoGraph for phenotyping of simulated single-cell datasets. Compared with the legacy CPU implementation, our GPU implementation of PhenoGraph is orders of magnitude faster at scale. Points and error bars show the mean and standard deviation of three replicate executions, respectively. See also Figure S3. Box 3 shows the spatial layout of high-dimensional cell phenotypes in a representative tissue core.
(B) t-SNE embedding of full single-cell CyCIF dataset colored by cell phenotype metacluster. See also Figure S3.
(C) Hierarchical clustering of PhenoGraph clusters and CyCIF markers. The color scale represents the Z-scored marker expression. The scatterplot displays how each BC subtype is composed, where point size represents the percentage of that BC subtype that is composed of that cluster. The bar plot represents that absolute number of cells belonging to each cluster and BC subtype. See also Figure S4.
Results
GPU-accelerated BC cell type identification at megascale
For robust intensity normalization method for multiplexed imaging (RESTORE) normalization (Chang et al., 2020) of each TMA core, we leverage the fact that tumor, immune, and stromal cells exhibit mutually exclusive expression of cell-type-specific markers, and use a graph-based clustering to define positive and negative cells and normalization factors (Figure S2A, Table S1, see subsubsection “single-cell intensity normalization”). When the raw expression vectors of all cells across TMAs are embedded by t-stochastic neighbor embedding (t-SNE) (van der Maaten and Hinton, 2008), cells are segregated on the basis of TMA source (Figure S2B, left), mainly because of batch effect and in part because of subtype bias within TMAs (Figures S1B and S1C). Afters normalization, shared cell types between TMAs are co-embedded (Figure S2B, right) and cell expression of immune, tumor, and stromal markers is segregated (Figure S2C), a validation of the normalization process.
To define cell types among the 1.3 million cells in the normalized feature table, we first attempted to use the central processing unit (CPU)-based version of the widely used algorithm PhenoGraph (Levine et al., 2015), but found it to be inefficient at this scale. To overcome this computational bottleneck, we re-implemented PhenoGraph to be executable on GPUs. Using the Python libraries RAPIDS (Raschka et al., 2020) and CuPy (Okuta et al., 2017) to parallelize and accelerate several of PhenoGraph's computations (see subsubsection ”single-cell phenotyping algorithm selection”), we observed multiple orders of magnitude improvement in the algorithm's speed without sacrificing clustering quality (Figure 1A, box 2). Our GPU implementation of PhenoGraph is competitive with FlowSOM in terms of execution time (Figure S3A), which until now was one of the primary motivations for choosing FlowSOM over PhenoGraph (Weber and Robinson, 2016; Liu et al., 2019). Moreover, to ensure that our GPU implementation of PhenoGraph produced results consistent with the CPU implementation, we benchmarked each by using synthetic datasets and detected no significant difference between CPU and GPU clustering modularity (Figures S3B–S3D).
Our PhenoGraph implementation identified diverse tumor, immune, and stromal cell types across tissues and BC subtypes (Figures 1B and 1C). To define phenotypes shared across tissues, metaclusters of similar phenotypes were aggregated on the basis of the hierarchical clustering of phenotypes based on their mean marker expression. Although tissues from all BC subtypes contained similar populations of immune, stromal, and endothelial cells, differences between BC subtypes were largely driven by variable tumor cells expression of luminal and basal cytokeratins, HER2, and the HRs estrogen receptor (ER) and PgR, as previously reported (Jackson et al., 2020).
Identified cell phenotypes are robust against noise and subsampling
To assess PhenoGraph clustering robustness to sampling shift, PhenoGraph clusters were derived by using random subsets of tissues of varying cardinality, from to of all tissues, and compared the Z-scored mean marker intensities of the PhenoGraph-derived clusters from the full reference dataset and each subset by using pairwise Pearson's correlation. Even with heavy subsampling, the median of the maximum correlations between matching clusters from reference-to-sample comparisons held at 0.75 (Figure S4A), indicating that we are defining a robust core set of cell phenotypes. Indeed, the major variation between reference and subsample clusters appeared to be sample-specific tumor cell phenotypes from the tissues held out from each subsample (Figures S4B and S4C), suggesting that PhenoGraph defines robust cell phenotypes that are shared across tissues and is capable of detecting new phenotypes as they are added to the dataset.
We further assessed the robustness of our identified cell phenotypes by applying noise to normalization factors for each marker for each core (Figure S4D) and found that phenotypes are identified as they are sampled and are robust to minor variation in normalization factors. We simulated measurement noise by multiplying the normalized cell intensity vectors for each tissue and marker by a scaling factor drawn uniformly at random from the range [0.8,1.2] and compared the Z-scored mean marker intensities of the PhenoGraph-derived clusters from the clean reference and noisy datasets by using pairwise Pearson's correlation. Even with these significant perturbations to the intensity profiles of cells, the median of the maximum correlations between matching clean and noisy clusters held above 0.8 (Figure S4D), indicating cluster robustness to differentials in preanalytical variables like tissue fixation or autofluorescence which can affect measured IF intensity across a TMA.
We acknowledge that segmentation is a critical pre-processing step that can affect downstream analyses. Instead of comparing various segmentation methods and downstream analysis results, we demonstrated robustness of cell types with 20% random noise added to each marker in each core alternatively. This variation implicitly reflects the mean intensity variation of each marker expected using different segmentation methods.
Inter-MTI platform benchmarking of cell phenotypes suggests basis for inter-MTI platform data integration
With the growth of MTI in the cancer research and translational communities, there is an acute need for robust and integrative analyses of MTI data across platforms and cohorts (Rozenblatt-Rosen et al., 2020). In a step toward addressing that need, we validated our identified cell types through comparison with a recently published survey of BC by IMC (Jackson et al., 2020). Although the total number of cells from each BC subtype varied between the Basel (IMC) and Oregon Health and Science University (OHSU) (CyCIF) cohorts (Figure 2A), there was substantial overlap between the marker panels used for each MTI platform (Figure 2B). By aligning the cell phenotypes independently detected by PhenoGraph in each cohort (Figure 2C), we found highly correlated clusters for stromal, immune, basal, and proliferating cell types, among others (Figure 2D), suggesting that shared cell types could be matched across cohorts and MTI platforms, a necessary step for data integration. We note that differences between cohort cell types might reflect the differences between cohort composition with respect to BC subtype. Consistent between cohorts and platforms, tumor cells differed more between samples than did immune, stromal, and endothelial cells (Figures S3E–S3H).
Figure 2.

Cross-validation of BC cell phenotypes between MTI platforms reveals commonalities between two BC cohorts
(A) Cellular ratio highlighting compositional differences between Basel (Jackson et al., 2020) and OHSU (this work) cohorts with respect to BC subtype.
(B) The intersection of the IMC and CyCIF marker panels used to stain tissues from the Basel and OHSU cohorts, respectively.
(C) PhenoGraph cluster matching between Basel and OHSU cohorts. Using only the intersecting markers, we independently clustered cells from each cohort by using PhenoGraph with the same parameterization, then cohort clusters were pairwise correlated and hierarchically clustered on the basis of the resulting correlation structure. We identified highly correlated clusters between cohorts, including those corresponding to epithelial, immune, stromal, endothelial, and proliferating cell populations.
(D) Maximum Pearson's correlation corresponding to inter-cohort cluster matches. Lines in boxes indicate the medians, and whiskers indicate data within 1.5 interquartile range of the upper and lower quartiles. Outliers are shown as distinct points.
Integrative analysis reveals BC subtype-specific features of tissue composition and architecture
Although BC is appreciated as a genetically and morphologically heterogeneous disease, its clinical subtyping is based on the expression of relatively few markers, in particular, tumor cell expression of the hormone receptors for estrogen and progesterone (ER and PgR, respectively, or HR, collectively), and human epidermal growth factor 2 (HER2), which is insufficient to explain differences in treatment response within each subtype (Prat et al., 2015). Recent studies using MTI to interrogate intact BC tissues have found that the spatial contexture of the BC microenvironment can improve our ability to predict clinical outcome (Jackson et al., 2020; Keren et al., 2018). However, because these studies have focused on either disease risk or a single BC clinical subtype, here we focused on compositional and spatial features that differentiate between subtypes.
At the composition level, we first considered the tumor cell differentiation states of BC subtypes through their expression of luminal and basal cytokeratins (CKs). Although CK+ cells in HR−/HER2+, HR+/HER2−, and HR+/HER2+ tissues primarily expressed luminal CKs 19, 8, and 7, CK+ cells in triple negative (TN) tissues exhibited significantly greater differentiation state heterogeneity (Figure 3A), as they expressed many different combinations of luminal and basal cytokeratins (Figure 3B). This differentiation state heterogeneity is consistent with the genetic and histological heterogeneity of triple-negative BC (TNBC) described in other studies (Haupt et al., 2010; Bianchini et al., 2016). We next determined the composition of each tissue core with respect to the cell metaclusters we defined above. Hierarchical clustering of cores on the basis of their cell metacluster densities highlighted the broad variability of cellular composition within and between BC subtypes (Figures 3C and 3D). When the cell metaclusters were further aggregated into immune, stromal, and tumor cell types (see subsubsection “aggregation of immune, stromal, and tumor cell phenotypes”), we found the HR+HER2− tissues to have lower overall immune cell density than the other BC subtypes, and no differences in stromal or tumor cell density between subtypes (Figure 3E).
Figure 3.

BC subtypes are differentiated by single-cell composition
(A) Epithelial differentiation heterogeneity across BC subtypes. Box plot displaying CK expression heterogeneity across BC subtypes, where each box represents the distribution of tissue cores from a BC subtype, and each core is summarized on the basis of the entropy of the distribution of CK+ cell types contained within it. Lines in boxes indicate the medians, and whiskers indicate data within 1.5 interquartile range of the upper and lower quartiles. Outliers are shown as distinct points. Groupwise comparisons were made by using one-way ANOVA with pairwise Tukey post-hoc test (TN, ; HR+HER2−, ; HR−HER2+, ; HR+HER2+, ). ∗p < 0.001 for all TN comparisons with other BC subtypes.
(B) UpSet plot summarizing the distribution of CK+ cell types across BC subtypes, considering each CK alone (left margin) or in combination (upper margin).
(C) Cell phenotype density across tissue cores. Bar plot where each bar represents a TMA core, the full bar height represents its total cell density, and each colored segment represents the density of a particular cell metacluster. Bars are hierarchically clustered on the basis of cell metacluster densities. Each bar is labeled with its corresponding subtype, stage, and grade, if a label is available. The inset brackets indicate (1) cores with abundant H3K27me3+ tumor cells, which could indicate a mechanism of HR repression in some TN and HR−/HER2+ tissues (Chen et al., 2016); (2) cores with abundant infiltrating B cells (TIL-B), consistent with association found between TIL-B and high-grade, HR− BC (Garaud et al., 2019); and (3) cores with relatively low immune density, consistent with the finding that HR+/HER− tissues are immunologically cold compared with TN and HER2+ tissues (Ali et al., 2015; Wimberly et al., 2015).
(D) A selection of representative tissue cores.
(E) The immune, stromal, and tumor densities of tissue cores from each BC subtype. Lines in boxes indicate the medians, and whiskers indicate data within 1.5 interquartile range of the upper and lower quartiles. Groupwise comparisons were made by using one-way Welch ANOVA and Games-Howell post-hoc test. ∗p < 0.034, ∗∗p < 0.035, ∗∗∗p < 0.079 (TN, ; HR+HER2−, ; HR−HER2+, ; HR+HER2+, ).
Recognizing that cell density measurements fail to capture the organization of cells in each tissue, we next characterized the spatial architectures of BC subtypes by building cell neighborhood graphs for each tissue. Given the recent evidence that the quantity and diversity of BC tumor cell interactions with other cell types can inform disease outcome (Jackson et al., 2020; Keren et al., 2018), we first identified the neighboring cells to each tumor cell and compared the composition of tumor cell neighborhoods across BC subtypes (Figure 4A). Although most tumor cell interactions (70%–80%) are with other tumor cells typical of their BC subtype, we observed increased tumor-stromal interaction in the HR+HER2− subtype. When considering tissues that contain an appreciable population of stromal cells (tissues comprising at least 25% stromal cells), we confirmed that there was significantly more stromal mixing with tumor cells in HR+HER2− tissues (Figure 4B). Importantly, stromal mixing can vary widely between tissues in spite of their similar stromal densities (Figure 4C), highlighting the importance of the spatial context that is preserved in intact tissues. Given that malignant epithelial cells can suppress fibroblast maturation and thus promote fibroblast aromatase activity (Bulun et al., 2012), ER+HER2− tumors likely favor more proximal fibroblasts as a source of growth-inducing estrogen than other BC subtypes, and might even act to maintain tumor microenvironments with high stromal mixing (Brechbuhl et al., 2017).
Figure 4.

BC cellular composition belies tumor-stromal interaction and tumor architecture
(A) Stacked bar plots displaying the proportion of tumor cell 10-nearest neighbors for each BC subtype. Each colored bar segment represents the proportion of tumor cell neighbors that are composed of the corresponding cell metacluster.
(B) Bar plot representation of tissue core stromal mixing, where only cores with greater than 0.25 stromal fraction are shown. Cores are ordered on the basis of increasing stromal mixing. Inset shows box plot comparing stromal mixing over BC subtype. For inset, lines in boxes indicate the medians, and whiskers indicate data within 1.5 interquartile range of the upper and lower quartiles. Outliers are shown as distinct points. Groupwise comparisons were made by using one-way Welch ANOVA and Games-Howell post-hoc test. ∗p < 0.001 for all HR+HER2− comparisons (TN, ; HR+HER2−, ; HR−HER2+, ; HR+HER2+, ).
(C) Scatterplot displaying stromal density versus stromal mixing and images of representative cores with similar stromal density but different stromal mixing.
(D) Overview of tumor architecture characterization. A spatial graph is defined over tumor cells, where each tumor cell is connected to others within a 65-μm radius from its centroid, and the closeness centrality is then measured over this graph. Each core is then summarized as a histogram of centrality values.
(E) Hierarchical clustering of cores on the basis of their tumor closeness centrality histograms. The upper and lower outset tumor graphs correspond to the right and left tissue core images in Figure 4C, respectively. Scale bar in tumor graphs represents 150 μm.
(F) Comparison of tumor closeness centrality between BC subtypes. Lines in boxes indicate the medians, and whiskers indicate data within 1.5 interquartile range of the upper and lower quartiles. Outliers are shown as distinct points. Groupwise comparisons of mean tumor closeness centrality were made by using one-way Welch ANOVA and Games-Howell post-hoc test. ∗p = 0.016, ∗∗p = 0.0099, ∗∗∗p = 0.0010 (TN, ; HR+HER2−,; HR−HER2+, ; HR+HER2+, ).
We reasoned that differences in tumor-stromal interaction might translate into detectable differences between BC subtypes based on their tumor architectures alone. To characterize the tumor architecture of each tissue, we constructed tumor architecture graphs over which we computed the closeness centrality for each tumor cell, which quantifies the relative closeness of that tumor cell to all other tumor cells in the tissue (Figure 4D). Consistent with the stromal mixing trend observed above, HR+HER2− tissues had a mean tumor closeness centrality significantly greater than tissues from the other BC subtypes (Figures 4E and 4F), which is in part a reflection of HR+HER2− tumor cell nests tending to be separated by narrower streams of stromal cells than tumor nests in tissues from other BC subtypes (Figure 4C). In summary, by analyzing BC tissues with spatially resolved MTI, we have identified inter-cell phenotype (stromal mixing) and intra-cell phenotype (tumor closeness centrality) interactions that can be leveraged to help discriminate between canonical BC subtypes on a basis other than receptor expression.
Discussion
This work is motivated by an understanding that the spatial context of the tumor microenvironment in intact cancer tissues enables a more granular definition of disease, and we hope for the design of more personalized and effective treatments. With spatially resolved MTI, our analysis makes clear that the cellular composition of BC tissue can belie important aspects of its spatial architecture. Ongoing work involves validating these findings in a cohort with more extensive clinical annotation to assess their significance to disease outcome between and within BC subtypes. Although the BC cell phenotypes and architectural features we have derived will be assets to future BC studies, our generic toolkit can be used alone or integrated with existing toolkits (Schapiro et al., 2017) to improve the efficiency and reproducibility of analytics for any single-cell measurement platform.
Although we demonstrated the computational efficiency of GPU-implemented PhenoGraph, graph-based clustering methods and other extensions could benefit from our implementation, and the CPU-to-GPU software translation approach, in general. For instance, some components of PhenoGraph (e.g., constructing a k-nearest neighbor graph) are also implemented as part of Seurat (Butler et al., 2018), a popular tool for single-cell RNA sequencing analysis. Thus, our approach could help extended applications like Seurat scale up for the megascale and greater meta-analyses of the near future.
Limitations of the study
In this study, we limit our scope to the identification of a robust BC cell phenotype dictionary and its validation against a BC cell phenotype dictionary defined by using another MTI platform. As such, we focus on methodological details, validation, and demonstration of reproducible, scalable, and robust data analysis. Although the clinical metadata for the described data were highly fragmented and limited the application scope of the study, we envision that the identified BC cell phenotypes we derive and robustly cross-validate here will be useful to future MTI-based studies that have more comprehensive clinical metadata at their disposal.
Although we acknowledge that segmentation is a critical pre-processing step that can affect downstream analyses, we feel that that a broad comparison against state-of-the-art segmentation methods is beyond the scope of the current study. Alternatively, we demonstrated robustness of cell types with variation added to each marker in each core. This variation implicitly reflects the mean intensity variation of each marker expected when using different segmentation methods, indicating that the methods we propose here are also robust to differences in segmentation approach.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CD20 (clone EP459Y) | Abcam | Cat#ab198941 |
| Rabbit anti-CD4 (clone EPR6855) | Abcam | Cat#ab196147 |
| Rabbit anti-CD44 (clone EPR1013Y) | Abcam | Cat#ab216647 |
| Rabbit anti-CD45 (clone EP322Y) | Abcam | Cat#ab214437 |
| Mouse anti-CD68 (clone KP1) | Biolegend | Cat#916104 |
| Mouse anti-FOXP3 (clone 206D) | Biolegend | Cat#320102 |
| Rabbit anti-Granzyme B (GRNZB) (clone EPR20129-217) | Abcam | Cat#ab219803 |
| Rabbit anti-Cytokeratin 5 (CK5) (clone EP1601Y) | Abcam | Cat#ab193894 |
| Mouse anti-Cytokeratin 14 (CK14) (clone LL002) | Abcam | Cat#ab212547 |
| Rabbit anti-Cytokeratin 17 (CK17) (clone EP1623) | Abcam | Cat#ab185032 |
| Rabbit anti-Cytokeratin 7 (CK7) (clone EPR1619Y) | Abcam | Cat#ab185048 |
| Rabbit anti-Cytokeratin 8 (CK8) (clone EP1628Y) | Abcam | Cat#ab192467 |
| Mouse anti-Cytokeratin 19 (CK19) (clone A53-B/A2) | Biolegend | Cat#628502 |
| Rabbit anti-E Cadherin (Ecad) (clone EP700Y) | Abcam | Cat#ab201499 |
| Rabbit anti-Androgen Receptor (AR) (polyclonal) | Sigma-Aldrich | Cat#06-680-AF555 |
| Rabbit anti-Estrogen Receptor (ER) (clone EPR4097) | Abcam | Cat#ab205851 |
| Rabbit anti-Progesterone Receptor (PgR) (clone YR85) | Abcam | Cat#ab199455 |
| Mouse anti-HER2 (clone 3B5) | Santa Cruz | Cat#sc-33684 |
| Mouse anti-aSMA (clone 3B5) | Santa Cruz | Cat#sc-32251 |
| Rabbit anti-CD31 (clone EPR3094) | Abcam | Cat#ab218582 |
| Rabbit anti-Vimentin (Vim) (clone D21H3) | Cell Signaling Technology | Cat#9854 |
| Rabbit anti-Collagen I (ColI) (clone EPR7785) | Abcam | Cat#ab215969 |
| Mouse anti-Collagen IV (ColIV) (clone 1042) | ThermoFisher | Cat#51-9871-82 |
| Mouse anti-Lamin A/C (LamA/C) (clone 4C11) | Sigma-Aldrich | Cat#SAB4200236 |
| Rabbit anti-Lamin B1 (LamB1) (clone EPR8985(B)) | Abcam | Cat#ab194106 |
| Rabbit anti-Lamin B2 (LamB2) (clone EPR9701(B)) | Abcam | Cat#ab200427 |
| Rabbit anti-H3K4me3 (clone C42D8) | Cell Signaling Technology | Cat#11960 |
| Rabbit anti-H3K27me3 (clone C36B11) | Cell Signaling Technology | Cat#5499 |
| Mouse anti-Podoplanin (PDPN) (polyclonal) | Biolegend | Cat#916606 |
| Rabbit anti-Cleaved PARP (cPARP) (clone D64E10) | Cell Signaling Technology | Cat#6894 |
| Rabbit anti-gH2AX (clone EP854(2)Y) | Abcam | Cat#ab195189 |
| Rabbit anti-Ki67 (clone D3B5) | Cell Signaling Technology | Cat#12075 |
| Mouse anti-PCNA (clone PC10 | Cell Signaling Technology | Cat#8580 |
| Rabbit anti-pHH3 (clone D2C8) | Cell Signaling Technology | Cat#3465 |
| Rabbit anti-p-S6 (clone D57.2.2E) | Cell Signaling Technology | Cat#3985 |
| Biological samples | ||
| Breast cancer tissue array | US Biomax Inc. | BR1201a |
| Breast cancer tissue array | US Biomax Inc. | BR1506 |
| Breast cancer tissue array | US Biomax Inc. | Her2B |
| Breast cancer tissue array | Dowsett et al., 2008 | T-ATAC-4A |
| Chemicals, peptides, and recombinant proteins | ||
| SlowFade Gold Antifade Mountant with DAPI | Life Technologies | Cat#S36938 |
| Normal Goat Serum (NGS) | Vector Laboratories | Cat#S-1000 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat#A7906 |
| Phosphate Buffered Saline (PBS) | Fisher Scientific | Cat#BP39920 |
| Target Retrieval Solution, pH 9 | Agilent | Cat#S236784-2 |
| Deposited data | ||
| Single-cell feature data (OHSU CyCIF) | This paper | 10.5281/zenodo.4908899 |
| Single-cell feature data (Basel IMC) | Jackson et al., 2020 | 10.5281/zenodo.3518284 |
| Software and algorithms | ||
| PhenoGraph | Levine et al., 2015 | https://github.com/dpeerlab/phenograph |
| RESTORE | Chang et al., 2020 | https://gitlab.com/Chang_Lab/cycif_int_norm/ |
| Code for analysis and figure generation | This paper | https://gitlab.com/eburling/BCTMA |
| CuPy | Okuta et al., 2017 | https://cupy.dev/ |
| SciPy | Virtanen et al., 2020 | https://www.scipy.org/ |
| Grapheno | This paper | https://gitlab.com/eburling/grapheno |
| NetworkX | Hagberg et al., 2008 | https://networkx.org/ |
| Pingouin | Vallat, 2018 | https://pingouin-stats.org/ |
| Seaborn | Waskom and the seaborn development team, 2020 | https://seaborn.pydata.org/ |
| Holoviews | Rudiger et al., 2019 | http://holoviews.org/ |
| Bokeh | Bokeh Development Team, 2020 | https://docs.bokeh.org |
| Matplotlib | Hunter, 2007 | https://matplotlib.org/ |
| Napari | Sofroniew et al., 2020 | https://napari.org/ |
| RAPIDS | Raschka et al., 2020 | https://rapids.ai/ |
| Dask | Dask Development Team, 2016 | https://dask.org/ |
| Other | ||
| 24x50 mm rectangular #1½ cover glass | Corning | Cat#2980-245 |
| 24x30 mm rectangular #1½ cover glass | Corning | Cat#2980-243 |
| Slide chambers | Bio-Rad | Cat#SLF0601 |
| Tabletop incubator | Clinical Scientific Equipment Inc. | No. 100 |
| Hybridization incubator | Robbins Scientific | Model 1000 |
| Decloaking chamber | Biocare Medical | Cat#DC2012 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Young Hwan Chang (chanyo@ohsu.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The single-cell feature data have been deposited on Zenodo and are publicly available as of the date of publication. The DOI is listed in the key resources table. The code use for analysis and figure generation is publicly available at https://gitlab.com/eburling/BCTMA. The GPU-accelerated implementation of PhenoGraph is publically available at https://gitlab.com/eburling/grapheno. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Acquisition of breast cancer tissue microarrays (TMAs)
The tissues used in this study are a compilation of multiple TMAs: BR1201a-SG48 (US Biomax Inc., https://www.biomax.us/BR1201a), BR1506-A019 (US Biomax Inc., https://www.biomax.us/tissue-arrays/Breast/BR1506), Her2B-K154 (US Biomax Inc., https://www.biomax.us/tissue-arrays/Breast/Her2B), and the TransATAC TMAs T-ATAC-4A-Left and T-ATAC-4A-Right (Dowsett et al., 2008). All tissues that were successfully stained and imaged were included in the study, representing 180 tissue cores from 128 patients.
Method details
Preparation and cyclic immunofluorescence (CyCIF) staining of tissues
Formalin-fixed paraffin-embedded (FFPE) human tissues were received mounted on adhesive slides. The slides were baked overnight in an oven at 55°C (Robbin Scientific, Model 1000) and an additional 30 minutes at 65°C (Clinical Scientific Equipment, NO. 100). Tissues were deparaffinized with xylene and rehydrated with graded ethanol baths. Two step antigen retrieval was performed in the Decloaking Chamber (Biocare Medical) using the following settings: set point 1 (SP1), 125°C, 30 seconds; SP2: 90°C, 30 seconds; SP limit: 10°C. Slides were further incubated in hot Target Retrieval Solution, pH 9 (Agilent, S236784-2) for 15 minutes. Slides were then washed in two brief changes of diH2O (2 seconds) and once for 5 minutes in 1x phosphate buffered saline (PBS), pH 7.4 (Fisher, BP39920). Sections were blocked in 10% normal goat serum (NGS, Vector S-1000), 1% bovine serum albumin (BSA, Sigma A7906) in PBS for 30 minutes at 20°C in a humid chamber, followed by PBS washes. Primary antibodies were diluted in 5% NGS, 1% BSA in 1x PBS and applied overnight at 4°C in a humid chamber, covered with plastic coverslips (Bio-Rad, SLF0601). Following overnight incubation, tissues were washed 3 x 10 min in 1x PBS. Coverslips (Corning; 2980-243 or 2980-245) were mounted in Slowfade Gold plus DAPI mounting media (Life Technologies, S36938).
Fluorescence microscopy
Fluorescently stained slides were scanned on the Zeiss AxioScan.Z1 (Zeiss, Germany) with a Colibri 7 light source (Zeiss). The filter cubes used for image collection were DAPI (Zeiss 96 HE), Alexa Fluor 488 (AF488, Zeiss 38 HE), AF555 (Zeiss 43 HE), AF647 (Zeiss 50) and AF750 (Chroma 49007 ET Cy7). The exposure time was determined individually for each slide and stain to ensure good dynamic range but not saturation. Full tissue scans were taken with the 20x objective (Plan-Apochromat 0.8NA WD=0.55, Zeiss) and stitching was performed in Zen Blue image acquisition software (Zeiss).
Quenching fluorescence signal
After successful scanning, slides were soaked in 1x PBS for 10–30 minutes in a glass Coplin jar, waiting until glass coverslip slid off without agitation. Quenching solution containing 20 mM sodium hydroxide (NaOH) and 3% hydrogen peroxide (H2O2) in 1x PBS was freshly prepared from stock solutions of 5 M NaOH and 30% H2O2, and each slide placed in 10 ml quenching solution. Slides were quenched under incandescent light, for 30 minutes for FFPE tissue slides. Slides were then removed from chamber with forceps and washed 3 x 2 min in 1x PBS. The next round of primary antibodies was applied, diluted in blocking buffer as previously described, and imaging and quenching were repeated over ten rounds for FFPE tissue slides.
Cell segmentation and mean intensity extraction
Cell segmentation and mean intensity extraction were performed as previously described (Eng et al., 2020). The nuclei and cells segmentation are performed using mathematical morphology. The process starts by segmenting the nuclei:
-
1
The DAPI image contrast is equalized using contrast-limited adaptive histogram equalization to remove illumination and staining irregularities.
-
2
The equalized DAPI image is cleaned by removing noise and artifacts as well as flattening the texture using an alternative sequential filter (alternation of opening and closing with structuring elements of increasing size).
-
3
A white top-hat filter is applied to separate the nuclei from the remaining background.
-
4
Area openings and closings (opening/closing based on the surface instead of a structuring element) are performed to flatten nuclei texture.
-
5
An ultimate opening is employed to find nuclei centers.
-
6
Nuclei centers are used as seeds in a watershed algorithm applied on the Sobel gradient of the original image, which provides the final nuclei segmentation.
-
7
For cell segmentation, nuclear segmentation masks are used as seeds in another watershed algorithm applied on a gradients combination of the markers CD44, CD45, CK7, CK19, and E-cadherin.
Mean intensities for each cell were extracted from the biologically-relevant compartment for each marker, i.e. mean intensities for markers with known nuclear (cytoplasmic) localization were extracted from nuclear (cytoplasmic) segmentation masks (Table S1). Cytoplasmic segmentation masks were computed by subtracting nuclear segmentation masks from full cell body segmentation masks.
Single-cell intensity normalization
Normalization factors for single-cell mean intensities were computed as previously described (Chang et al., 2020) using the putative mutually-exclusive marker pairs in Table S1. Normalization factors are computed for each pair of reference and mutually-exclusive markers, and the median of these factors is used to normalize each raw single-cell mean intensity vector for each CyCIF marker and each TMA core. Raw intensities were normalized using the equation:
| (Equation 1) |
where and are the normalized and raw single-cell mean intensity vectors for CyCIF marker i for all cells in tissue core j, respectively, and is the corresponding normalization factor determined as described above. Therefore, cells with a normalized intensity greater than 1 are considered to be above the background intensity level.
Single-cell phenotyping algorithm selection
PhenoGraph was used to define breast cancer cell types in a related study (Jackson et al., 2020), so we opted to use PhenoGraph in the current study to enable a fair comparison of the cell types defined between studies. Furthermore, the relatively slow runtime of CPU-executed PhenoGraph motivated our GPU-accelerated implementation.
GPU acceleration of PhenoGraph
Given a cell-by-feature dataframe, the PhenoGraph algorithm (Levine et al., 2015) consists of two primary steps: (1) defining a k-nearest neighbor graph over all cells that is then refined by computing the Jaccard similarity measure over graph edges, and (2) partitioning the graph into discrete cell phenotypes through optimization of partition modularity using the Louvain algorithm, such that cells in the same partition are more connected to each other than to cells of another partition. In the official version of PhenoGraph (https://github.com/dpeerlab/PhenoGraph), these steps are implemented using a combination of Python and C++ libraries that execute on CPU. In Figure 1A, we show that PhenoGraph execution time increases exponentially with increasing dataset size, taking approximately 3 hours to process a synthetic 1 million cell-by-10 feature dataset. Most MTI datasets measure tens of features, but CPU-based PhenoGraph was unable to fully process the 1 million cell-by-30- and 50-feature synthetic datasets in the 8 hours allotted for the experiment. We see such computational bottlenecks—which would be even further constricted when compiling multiple MTI or cytometry datasets—as a major obstacle to current studies and future meta-studies of high-dimensional MTI datasets, where rapid iteration will be essential to the validation of cross-platform data integration techniques.
Owing to recent advances in GPU computing and its ever-broadening adoption in machine learning research, there now exist accelerated GPU-based analogs of many Python scientific computing libraries (Raschka et al., 2020; Okuta et al., 2017), including those with which the CPU-based PhenoGraph is implemented. Some of these libraries even allow computation to be distributed across multiple GPUs (Raschka et al., 2020; Dask Development Team, 2016). We employed two of such libraries, CuPy (Okuta et al., 2017) and RAPIDS (Raschka et al., 2020), to accelerate each step of the PhenoGraph algorithm and enable distributed computing over multiple GPUs. For example, for a synthetic dataset containing 50,000 samples and 50 features, the GPU implementation realizes a 354-fold speed up in the graph building and refinement step (97.3 seconds for CPU vs. 0.275 seconds for GPU) and a 141-fold speed up in the Louvain partitioning step (11.2 seconds for CPU vs. 0.0795 seconds for GPU). With our GPU implementation, it is now possible to phenotype cells in megascale cytometry datasets in seconds-to-minutes rather than hours-to-days, and without subsampling. We have packaged our GPU implementation into a Python library called grapheno and have adopted the API from the official CPU implementation of PhenoGraph found at https://github.com/dpeerlab/phenograph. To run a simple clustering of synthetic data:
import cudf
import cuml
import grapheno
X, _ = cuml.make_blobs()
X = cudf.Dataframe.from_records(X)
communities, G, Q = grapheno.cluster(X)
In practice, X can be any single-cell dataframe with cells as rows and features as columns. For a dataframe with N cells, will be a vector of length N specifying the cluster label for each cell. G is a RAPIDS graph object representing the relationships between cells that were used for clustering. Q is the modularity score for the clustering result defined by . Installation instructions and full details about our implementation can be found at https://gitlab.com/eburling/grapheno.
Benchmarking CPU and GPU implementations of PhenoGraph
To ensure that our GPU implementation of PhenoGraph produced results consistent with the CPU implementation, we benchmarked each using synthetic datasets which varied in terms of number of samples and number of features (Figures S3B–S3D). Synthetic data were generated using the make_classification function from the RAPIDS library, using the settings n_classes=16, class_sep=4, and weights=list(np.random.dirichlet(np.ones(16)∗5.)) to randomly scale the proportion of samples in each class. Examples of clustering results for each implementation are shown in Figure S3B. To check for differences in clustering results, we compared the purity and modularity of clusters derived using either CPU or GPU implementations and the same parameterization (). Purity measures the percentage of correctly clustered objects—in this case the percentage of agreement between clustering results from each implementation—and is calculated as:
| (Equation 2) |
where N is the total number of objects, m is the number of clusters from the CPU implementation, is the i-th cluster from the CPU implementation, and is the cluster from the GPU implementation which has the maximum number of objects from the cluster . The CPU and GPU implementations yielded almost identical clustering results, i.e. inter-implementation cluster purity for all combinations of n_sample and n_features (Figure S3C). Modularity (Q) is the quantity optimized by the Louvain algorithm used by PhenoGraph to partition k-nearest neighbor graphs of cells into clusters of similar cells. As anticipated based on the tight agreement between cluster results between implementations, we detected no significant difference between CPU and GPU clustering modularity (Figure S3D). Very slight differences between clustering results correspond to a maximum of 0.003% of cells being mis-matched between implementations and can be attributed to the degeneracy of Louvain partitioning.
7.3.9. Single-cell phenotyping and metacluster annotation
Apart from the benchmarking experiment described in Figure 1A, we use only our GPU-based implementation of PhenoGraph throughout this work. Single-cell phenotypes were defined based on single-cell mean intensity for the 35-marker CyCIF panel (STAR Methods). Prior to application of PhenoGraph, data were 99.9th-percentile normalized and arcsin transformed (cofactor = 5). Following (Jackson et al., 2020), PhenoGraph was parameterized (k=40) to over-cluster the data and detect rare cell types. PhenoGraph clustering was followed by aggregation of phenotypes into metaclusters based on hierarchical clustering of phenotype mean marker intensities and to preserve known biological variation.
t-stochastic neighbor embedding (t-SNE)
To enable visualization, the full 35-feature single-cell dataset was reduced to 2 dimensions using the RAPIDS implementation of t-SNE (van der Maaten and Hinton, 2008) with default parameters except perplexity = 60. Prior to t-SNE processing, data were 99.9th-percentile normalized and arcsin transformed (cofactor = 5). Plots containing t-SNE embeddings of the full 1.3 million-cell dataset were created using Datashader (https://github.com/holoviz/datashader).
Cross-platform breast cancer cell phenotype validation
The imaging mass cytometry (IMC) dataset (Jackson et al., 2020) used for our cell phenotype validation experiment was retrieved from https://zenodo.org/record/3518284. To make a fair comparison between the OHSU (CyCIF) and Basel (IMC) datasets, we independently ran PhenoGraph on each using the same parameters (k=40, Louvain partitioning) and only the overlapping features between IMC and CyCIF stain panels (Figure 2B). The phenotypes derived from each platform were then cross-correlated to identify inter-platform phenotype matches. Using the clustermap function from seaborn (Waskom and the seaborn development team, 2020), the cross-correlation matrix was then hierarchically clustered with Ward linkage and used to sort the matching clusters between the two cohort heatmaps.
Epithelial differentiation heterogeneity
Cells from each core were labeled as positive for each cytokeratin if their mean intensity was greater than the normalization factor computed for that cytokeratin for that core. The plot from Figure 3B was generated using UpSetPlot (https://github.com/jnothman/UpSetPlot). The CK heterogeneity of each core was computed by measuring the Shannon entropy of its distribution of CK-expressing cells. The homogeneity of variances assumption was met, so comparison of the CK heterogeneity over BC subtypes was made using the pairwise_tukey function from pingouin (Vallat, 2018).
Aggregation of immune, stromal, and tumor cell phenotypes
To enable high-level comparison of cell phenotype distribution over BC subtypes (Figures 3E and 4), cell metaclusters were aggregated into immune, stromal, and tumor groups with the following metacluster membership:
-
•
immune = [B cell/T cell, B cell, T cell/macrophage, T cell, macrophage]
-
•
stromal = [CD44+ endothelial, PDPN+/aSMA+ stromal, ColI+ stromal, Lamin+/Vimentin+ stromal, ColI+/PDPN+ stromal, Vimentin+ stromal, Vimentin+ endothelial]
-
•
tumor = [Ecad+/CK low, CK5+, CK5+/CK14+, myoepithelial, CK+/H3K27me3+, epithelial low, CK14+, Ecad+/CK+, proliferating, apoptotic, H3K4me3+, HER2+/CK8+, ER+, HER2 low, HER2+/CK+, ER+/PgR+]
Cell phenotype density
The density of each of the 27 cell metaclusters in each tissue core was measured by counting the number of cells of each metacluster in the core, then dividing the count by the area of the convex hull defined by the cell centroids of the core. Tissue cores were then hierarchically clustered based on their z-scored cell metacluster densities using the clustermap function with Ward linkage from seaborn (Waskom and the seaborn development team, 2020). The homogeneity of variances assumption was not met, so comparisons of immune, stromal, and tumor cell densities over BC subtypes were made using the pairwise_gameshowell function from pingouin (Vallat, 2018).
Tumor cell neighborhood interactions
To characterize the microenvironments of tumor cells across BC subtypes, we identified the cell metaclusters of the 10 nearest cells within 65 μm (double the median of the minimum tumor-stromal distances across all tissue cores) of each tumor cell. Tumor cells were then split based on the BC subtype of the tissue from which they were derived, and counts for each metacluster were summed over all tumor cells such that each metacluster could be represented as a proportion of the total tumor neighborhood for each BC subtype.
To measure the extent of tumor-stromal cell interactions in each tissue core, we computed their stromal mixing scores, an adaptation of a previously described cell-cell mixing score (Keren et al., 2018). To focus on cores that had substantial stromal composition, we first selected cores which are comprised of at least 25% stromal cells and for each we defined a 10-nearest neighbor spatial graph over all cells in that core. Second, we removed edges between cells with an interaction distance greater than 65 μm. Finally, we computed the stromal mixing score for tissue core j as:
| (Equation 3) |
Tumor graph centrality
To characterize tumor architecture in each tissue core, we considered the spatial interactions between tumor cells only. To account for variation in tissue core diameter (Figure S1A) which would affect the scale of spatial graph characteristics, we subsampled large diameter cores to be equal in size to the smallest diameter cores by only considering cells within the 300 μm-radius circle drawn about the centroid of each core. With the spatially-subsampled cores, we first construct a 4-nearest neighbor spatial graph over all tumor cells in each core. Here we use rather than to construct a sparser graph since we are focusing on tumor cells only. Over this graph we compute the Wasserman-Faust closeness centrality of each cell using the closeness_centrality function from the Python package networkx (Hagberg et al., 2008). The Wasserman-Faust closeness centrality of cell u is computed as:
| (Equation 4) |
where is the shortest-path distance between cells v and u, n is the number of cells that can reach u, and N is the number of cells in the graph. For heatmap visualization, the distribution of tumor cell centrality for each core was max-normalized, converted into a 50-bin histogram over range = (0,1), then hierarchically clustered using the clustermap function from seaborn (Waskom and the seaborn development team, 2020) with Jensen-Shannon distance and average linkage.
Plotting and visualization
Unless otherwise noted, all plots were generated using Holoviews (Rudiger et al., 2019) with either the Bokeh (Bokeh Development Team, 2020) or matplotlib (Hunter, 2007) backends. Images of CyCIF-stained tissue cores were generated using napari (Sofroniew et al., 2020).
Computing hardware
The GPU-accelerated PhenoGraph implementation was developed and deployed on the NVIDIA V100 GPU with 32 GB memory, but the grapheno Python library can be compiled to work with any NVIDIA GPU.
Quantification and statistical analysis
For statistical analyses, a p value of less than 0.05 was considered significant. For the groupwise comparisons in Figure 3A, 3E, 4B, and 4F, we first tested the assumption of homogeneity of variances using the bartlett function from the Python package SciPy (Virtanen et al., 2020). When the assumption was (not) met, we made groupwise comparisons using one-way ANOVA with Tukey-HSD post-hoc test using the pairwise_tukey (one-way Welch ANOVA with Games-Howell post-hoc test using the pairwise_gameshowell) function from the Python package pingouin (Vallat, 2018). Additional statistical details can be found in the corresponding figure legends.
Acknowledgments
This work was supported in part by the National Cancer Institute (U54CA209988, U2CCA233280, U01 CA224012, R01 CA253860), CRUK-OHSU joint funding award, the OHSU Center for Spatial Systems Biomedicine, and a Biomedical Innovation Program Award from the Oregon Clinical and Translational Research Institute. We acknowledge expert technical assistance by staff in the Advanced Multiscale Microscopy Shared Resource, supported by the OHSU Knight Cancer Institute (NIH P30 CA069533) and the Office of the Senior Vice President for Research. Equipment purchases included support by the OHSU Center for Spatial Systems Biomedicine, the MJ Murdock Charitable Trust, and the Collins Foundation. We also acknowledge Mitchell Dowsett for his contribution of TMAs used in this study. The resources of the Exacloud high-performance computing environment developed jointly by OHSU and Intel and the technical support of the OHSU Advanced Computing Center are gratefully acknowledged. E.A.B. receives support from a scholar award provided by the ARCS (Achievement Rewards for College Scientists) Foundation Oregon.
Author contributions
Conceptualization, E.A.B., J.W.G., and Y.H.C.; methodology, E.A.B., J.E., G.T., K.C., J.W.G., and Y.H.C.; software, E.A.B., G.T., and Y.H.C.; validation, E.A.B., J.E., K.C., and Y.H.C.; formal analysis, E.A.B.; investigation, E.A.B. and J.E.; resources, K.C. and J.W.G.; data curation, E.A.B. and J.E.; writing – original draft, E.A.B. and Y.H.C.; Writing – review & editing, E.A.B., J.W.G., and Y.H.C.; visualization, E.A.B.; supervision, J.W.G. and Y.H.C.; project administration, J.W.G. and Y.H.C.; funding acquisition, E.A.B., Y.H.C., and J.W.G.
Declaration of interests
J.W.G. has licensed technologies to Abbott Diagnostics; has ownership positions in Convergent Genomics, Health Technology Innovations, Zorro Bio, and PDX Pharmaceuticals; serves as a paid consultant to New Leaf Ventures; has received research support from Thermo Fisher Scientific (formerly FEI), Zeiss, Miltenyi Biotech, Quantitative Imaging, Health Technology Innovations, and Micron Technologies; and owns stock in Abbott Diagnostics, AbbVie, Alphabet, Amazon, Amgen, Apple, General Electric, Gilead, Intel, Microsoft, Nvidia, and Zimmer Biomet.
Published: July 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100053.
Supplemental information
References
- Ali H.R., Glont S.E., Blows F.M., Provenzano E., Dawson S.J., Liu B., Hiller L., Dunn J., Poole C.J., Bowden S., et al. Pd-l1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann. Oncol. 2015;26:14881493. doi: 10.1093/annonc/mdv192. [DOI] [PubMed] [Google Scholar]
- Angelo M., Bendall S.C., Finck R., Hale M.B., Hitzman C., Borowsky A.D., Levenson R.M., Lowe J.B., Liu S.D., Zhao S., et al. Multiplexed ion beam imaging (MIBI) of human breast tumors. Nat. Med. 2014;20:436442. doi: 10.1038/nm.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini G., Balko J.M., Mayer I.A., Sanders M.E., Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016;13:674690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokeh Development Team Bokeh: Python library for interactive visualization. 2020. https://bokeh.org/
- Brechbuhl H.M., Finlay-Schultz J., Yamamoto T.M., Gillen A.E., Cittelly D.M., Tan A.-C., Sams S.B., Pillai M.M., Elias A.D., Robinson W.A., et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. 2017;23:17101721. doi: 10.1158/1078-0432.CCR-15-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulun S.E., Chen D., Moy I., Brooks D.C., Zhao H. Aromatase, breast cancer and obesity: a complex interaction. Trends Endocrinology Metabolism: TEM. 2012;23:83–89. doi: 10.1016/j.tem.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.H., Chin K., Thibault G., Eng J., Burlingame E., Gray J.W. Restore: robust intensity normalization method for multiplexed imaging. Commun. Biol. 2020;3:1–9. doi: 10.1038/s42003-020-0828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Hu H., He L., Yu X., Liu X., Zhong R., Shu M. A novel subtype classification and risk of breast cancer by histone modification profiling. Breast Cancer Res. Treat. 2016;157:267–279. doi: 10.1007/s10549-016-3826-8. [DOI] [PubMed] [Google Scholar]
- Dask Development Team Dask: library for dynamic task scheduling. 2016. https://dask.org
- Dowsett M., Allred C., Knox J., Quinn E., Salter J., Wale C., Cuzick J., Houghton J., Williams N., Mallon E., et al. Relationship between quantitative estrogen and progesterone receptor expression and human epidermal growth factor receptor 2 (her-2) status with recurrence in the arimidex, tamoxifen, alone or in combination trial. J. Clin. Oncol. 2008;26:1059–1065. doi: 10.1200/JCO.2007.12.9437. [DOI] [PubMed] [Google Scholar]
- Eng J., Thibault G., Luoh S.-W., Gray J.W., Chang Y.H., Chin K. Methods in Molecular Biology. Springer; 2020. Cyclic multiplexed-immunofluorescence (cmIF), a highly multiplexed method for single-cell analysis; pp. 521–562. [DOI] [PubMed] [Google Scholar]
- Garaud S., Buisseret L., Solinas C., Gu-Trantien C., de Wind A., Van den Eynden G., Naveaux C., Lodewyckx J.-N., Boisson A., Duvillier H., et al. Tumor-infiltrating B cells signal functional humoral immune responses in breast cancer. JCI Insight. 2019;4 doi: 10.1172/jci.insight.129641. https://insight.jci.org/articles/view/129641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen C., Wang H.A.O., Schapiro D., Zivanovic N., Jacobs A., Hattendorf B., Schüffler P.J., Grolimund D., Buhmann J.M., Brandt S., et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods. 2014;11:417422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- Goltsev Y., Samusik N., Kennedy-Darling J., Bhate S., Hale M., Vazquez G., Black S., Nolan G.P. Deep profiling of mouse splenic architecture with codex multiplexed imaging. Cell. 2018;174:968–981.e15. doi: 10.1016/j.cell.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg A.A., Schult D.A., Swart P.J. In: Proceedings of the 7th Python in Science Conference’, Pasadena, CA USA. Varoquaux G., Vaught T., Millman J., editors. 2008. Exploring network structure, dynamics, and function using networkx; pp. 11–15. [Google Scholar]
- Haupt B., Ro J.Y., Schwartz M.R. Basal-like breast carcinoma: a phenotypically distinct entity. Arch. Pathol. Lab. Med. 2010;134:130133. doi: 10.5858/134.1.130. [DOI] [PubMed] [Google Scholar]
- Hunter J.D. Matplotlib: a 2d graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- Jackson H.W., Fischer J.R., Zanotelli V.R.T., Ali H.R., Mechera R., Soysal S.D., Moch H., Muenst S., Varga Z., Weber W.P., et al. The single-cell pathology landscape of breast cancer. Nature. 2020;578:615620. doi: 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- Keren L., Bosse M., Marquez D., Angoshtari R., Jain S., Varma S., Yang S.-R., Kurian A., Valen D.V., West R., et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. 2018;174:1373–1387.e19. doi: 10.1016/j.cell.2018.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.-a. D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.-R., Izar B., Wang S., Yapp C., Mei S., Shah P.M., Santagata S., Sorger P.K. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife. 2018;7:e31657. doi: 10.7554/eLife.31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Song W., Wong B.Y., Zhang T., Yu S., Lin G.N., Ding X. A comparison framework and guideline of clustering methods for mass cytometry data. Genome Biol. 2019;20:297. doi: 10.1186/s13059-019-1917-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuta R., Unno Y., Nishino D., Hido S., Loomis C. Proceedings of Workshop on Machine Learning Systems (LearningSys) in the Thirty-First Annual Conference on Neural Information Processing Systems (NeurIPS) 2017. CuPy: a NumPy-compatible library for NVIDIA GPU calculations; p. 7. [Google Scholar]
- Prat A., Pineda E., Adamo B., Galván P., Fernández A., Gaba L., Díez M., Viladot M., Arance A., Muñoz M. Clinical implications of the intrinsic molecular subtypes of breast cancer. The Breast. 2015;24:S26S35. doi: 10.1016/j.breast.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Raschka S., Patterson J., Nolet C. Machine learning in python: main developments and technology trends in data science, machine learning, and artificial intelligence. arXiv. 2020 arXiv:2002.04803. [Google Scholar]
- Rozenblatt-Rosen O., Regev A., Oberdoerffer P., Nawy T., Hupalowska A., Rood J.E., Ashenberg O., Cerami E., Coffey R.J., Demir E., et al. The human tumor atlas network: charting tumor transitions across space and time at single-cell resolution. Cell. 2020;181:236249. doi: 10.1016/j.cell.2020.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudiger P., Stevens J.-L., Bednar J.A., Nijholt B., Andrew B.,C., Tenner V., Mease J., Randelhoff A., maxalbert, Kaiser M., et al. holoviz/holoviews: version 1.12.7. 2019. [DOI]
- Schapiro D., Jackson H.W., Raghuraman S., Fischer J.R., Zanotelli V.R.T., Schulz D., Giesen C., Catena R., Varga Z., Bodenmiller B. histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods. 2017;14:873876. doi: 10.1038/nmeth.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew N., Evans K., Lambert T., Nunez-Iglesias J., Solak A.C., Yamauchi K., Buckley G., Tung T., Bokota G., Boone P., Freeman J., Har-Gil H., Royer L., Axelrod S., jakirkham, Dunham R., Vemuri P., Huang M., Hector Bryant, Rokem A., Kiggins J., van Kemenade H., Patterson H., Gay G., Perlman E., Bennett D., Gohlke C., Chopra B., de Siqueira A. napari/napari: 0.3.0. 2020. [DOI]
- Tsujikawa T., Kumar S., Borkar R.N., Azimi V., Thibault G., Chang Y.H., Balter A., Kawashima R., Choe G., Sauer D., et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 2017;19:203217. doi: 10.1016/j.celrep.2017.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat R. Pingouin: statistics in python. J. Open Source Softw. 2018;3:1026. [Google Scholar]
- van der Maaten L., Hinton G. Visualizing data using t-SNE. J. Machine Learn. Res. 2008;9:25792605. [Google Scholar]
- Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry. Part A: J. Int. Soc. Anal. Cytol. 2015;87:636645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., et al. SciPy 1.0 contributors SciPy 1.0: fundamental algorithms for scientific computing in python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskom M., seaborn development team seaborn: statistical data visualization. 2020. [DOI]
- Weber L.M., Robinson M.D. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry A. 2016;89:10841096. doi: 10.1002/cyto.a.23030. [DOI] [PubMed] [Google Scholar]
- Wimberly H., Brown J.R., Schalper K., Haack H., Silver M.R., Nixon C., Bossuyt V., Pusztai L., Lannin D.R., Rimm D.L. Pd-l1 expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol. Res. 2015;3:326332. doi: 10.1158/2326-6066.CIR-14-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The single-cell feature data have been deposited on Zenodo and are publicly available as of the date of publication. The DOI is listed in the key resources table. The code use for analysis and figure generation is publicly available at https://gitlab.com/eburling/BCTMA. The GPU-accelerated implementation of PhenoGraph is publically available at https://gitlab.com/eburling/grapheno. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.