

Abstract
Polyamines are known to play a significant role in cancer progression and treatment using difluoromethylornithine (DFMO), an inhibitor of polyamine biosynthesis, has shown some clinical promise. Interestingly, while DFMO is directly cytostatic in vitro, recent work has suggested that it achieves its anti-tumor efficacy in vivo by enhancing adaptive anti-tumor immune responses. Based on these data, we hypothesized that DFMO might act as an immune sensitizer to increase tumor responsiveness to checkpoint blockade. To test this hypothesis, we treated tumors with DFMO, in either the presence or absence of additional PD-1 blockade, and subsequently analyzed their immunological and therapeutic responses. Our data demonstrates that treatment with DFMO significantly enhances both the viability and activation status of intratumoral CD8+ T cells, most likely through an indirect mechanism. When combined with PD-1 blockade, this increased viability resulted in unique pro-inflammatory cytokine profiles and transcriptomes within the tumor microenvironment and improved therapeutic outcomes. Taken together, these data suggest that DFMO might represent a potential immunomodulatory agent that can enhance current PD-1-based checkpoint therapies.
Keywords: DFMO, polyamines, immunotherapy, PD-1, checkpoint
Introduction
Polyamines are chemically simple organic cations that have numerous functions in cellular biology including the regulation of DNA and RNA stability, protein translation, and cell cycle progression [1]. Due to these pleiotropic roles, it is unsurprising that polyamine metabolism is almost ubiquitously dysregulated in cancer and expression of numerous oncogenes are known to modulate cellular polyamine content [2–5]. Because of this, a significant amount of work has focused on developing polyamine-modulating drugs for use as cancer therapeutics [5–8].
Historically, the lead candidate for polyamine-deprivation-based therapies has been difluoromethylornithine (DFMO), a suicide inhibitor of ornithine decarboxylase (ODC) [6] which has shown modest anti-tumor efficacy in numerous preclinical models [9–12]. Interestingly, while DFMO’s anti-cancer potential has been extensively studied, the mechanisms mediating its observed anti-tumor efficacy in vivo remain unclear. In the absence of exogenous polyamines DFMO is potently cytostatic in vitro [13], however, a poorly characterized polyamine transport system exists which is able to effectively compensate for stalls in production by directly importing exogenous polyamines [1, 4]. Additionally, free polyamines are relatively abundant in vivo [14, 15], and DFMO monotherapy has been shown to have only modest effects on intratumoral polyamine levels [16, 17]. Furthermore, recent work has unexpectedly shown that DFMO’s anti-cancer effects are dependent on immunocompetency, with the drug’s therapeutic efficacy completely abrogated upon depletion of CD8+ T cells or in Rag−/− mice [18, 19]. These data suggest that – in contrast to its direct cytostatic mechanisms in vitro – the anti-tumor effects of DFMO in vivo may be primarily due to alterations of anti-tumor immunity. Several potential mechanisms through which DFMO might modulate immune responses have been suggested, including inhibition of Arginase 1 activity in tumor-associated macrophages (TAM) and/or myeloid-derived suppressor cells (MDSC) [18–20]. Regardless of the mechanism at hand, the breadth of DFMO’s immunomodulatory function(s) and how to best advance this novel mechanistic insight into the clinic remains unclear.
Here, we provide evidence that DFMO improves intratumoral CD8+ T cell viability and displays enhancement of α-PD-1 checkpoint blockade in multiple preclinical models. These data support the existing literature suggesting that DFMO acts as an immunomodulatory agent in vivo and presents a clear path for improved clinical translation.
Materials and Methods:
Mouse models:
Female C57Bl/6 mice aged six to ten weeks were subcutaneously seeded with 400,000 tumor cells in 50μL of cold PBS above the fourth mammary fat pad. Mice were only considered for treatment after forming palpable tumors by day of treatment (5 days post seeding B16/F10 tumors; 9 days post seeding LLC-A9F1 tumors). For survival experiments, mice were euthanized upon tumor size reaching 15mm in any direction. For molecular analysis experiments, all groups were euthanized upon the first mouse reaching euthanasia criteria in any group and tumors were excised, partitioned into halves, and weighed for downstream analysis (described below). For mice receiving α-PD-1 checkpoint blockade, 100μg of α-PD-1 monoclonal antibody (clone RMP 1–14) in 50μL of PBS was injected intraperitoneally every four days. DFMO treated mice received 1.5% w/v DFMO hydrochloride supplemented water ad libitum. For the depletion study, the following antibodies were injected intraperitoneally every four days: GR1 (NIMP-R14 100μg), NK1.1 (RK136, 300μg), CD8 (53–6.7, 100μg), CD4 (GK1.5, 100μg). All depleting and blocking antibodies used in this study were obtained from BioXcell (West Lebanon, NH).
Cytokine analysis:
Tumor portions were recorded for weight, crushed over a 40μm mesh strainer, and rinsed with 5mL of PBS. Resulting cell suspensions were centrifuged at 750 RCF for 5 minutes. 1mL of supernatants were collected for each sample. Supernatants were then clarified by three cycles of centrifugation at 15,000 RCF for 5 minutes per cycle. Samples were frozen at −80°C and shipped to Eve technologies (Calgary, CA) for analysis (Mouse Cytokine Array / Chemokine Array 31-Plex (MD31)).
Cell lines and culture reagents:
The B16/F10 cell line used for murine models was a CRISPR scramble control from the original ATCC cell bank. The BSC40 and Jurkat cell lines were purchased from ATCC (Manassas, VA). LLC-A9F1 cells were a gift from Dr. Mark Rubinstein at the Medical University of South Carolina. Jurkat cells were maintained in RPMI (Corning, Corning, NY) supplemented with 10% FBS (VWR, Radnor, PA) and 1x penicillin/streptomycin/glutamine. For Jurkat activation experiments, cells were treated with 5ng/mL PMA and 500ng/mL Ionomycin for 2 hours in RPMI, washed with PBS, and plated in fresh RPMI. All other cell lines were maintained in DMEM (Corning, Corning, NY) supplemented with 10% FBS (VWR, Radnor, PA) and 1x penicillin/streptomycin/glutamine (Corning, Corning, NY). Cultures were checked quarterly for mycoplasma contamination by qRT-PCR.
Splenocyte isolation and T cell enrichment:
Splenocyte cultures used for ex vivo assays were harvested from tumor-naïve female C57Bl/6 mice ten weeks of age. Spleens were harvested and crushed over a 70μM single-cell strainer in PBS, and plated in RPMI containing 100IU/mL IL-2 and 50μM ß-mercaptoethanol in 6-well plates coated with αCD3 for 24 hours prior for activation and enrichment of T cells. Media was exchanged after 48 hours with fresh RPMI+IL-2+BME. Cells were re-plated 24 hours after media exchange for experimental use.
Cell viability and count assays:
Cell viability was measured via CellTiter 96® Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) according to manufacturer’s recommendations. Cell counting was performed using cell aliquots mixed at a 1:1 dilution with Trypan blue and taking an average of 4 quadrants in a hemocytometer.
Flow cytometry analysis:
Tumor portions were recorded for weight, crushed over a 40μm mesh strainer, and rinsed with 5mL of PBS. Resulting cell suspensions were centrifuged at 750 RCF for 5 minutes. Cell pellets were isolated for flow cytometry. Cells were stained using standard flow cytometry methods with the following antibodies: CD8 (clone 53–6.7), PD-1 (clone J43), CD25 (clone PC61), CD3 (clone 17A2), CD45 (clone 30-F11), CD69 (clone H1.2F3), CD4 (clone RM4–5), I-A[b] (clone AF6–120.1), Ly6G (clone 1A8), Ly6C (clone AL-21 ), CD11b (clone M1/70), CD11c (clone HL3). All antibodies used for flow cytometry were obtained from BD Biosciences (San Jose, CA)
RNAseq Sample Generation:
Tumor portions were immediately stored in RNAlater (Thermo Fisher Scientific, Waltham, MA) after excision, weighed, and then crushed over a 40μm mesh strainer. Resulting cell suspensions were centrifuged at 750 RCF for 5 minutes. Cell pellets were then processed through Qiagen’s RNeasy mini kit using the workflow with QIAshredder spin columns (Qiagen, Germantown, MD) to isolate RNA. RNA quality and concentration were determined by analysis on an Agilent 2100 bioanalyzer. RNAseq was conducted on samples by Novogene’s mRNA-seq service (Novogene, Sacramento, CA).
Statistical Methods and RNAseq Processing:
All statistical analyses were performed in SPSS, with each specific test notated within figure legends. All error bars indicate SEM unless otherwise specified for all figures. Downstream analyses of the RNAseq data were performed on the transcript counts provided by Novogene using RStudio and Rsubread package, and were filtered to only consider genes with CPM > 0.7 (~8–12 raw reads) in at least 5 samples. PCA was performed using ggbiplot package. Unsupervised hierarchical clustering was performed using edgeR and gplot packages. Enrichment analyses were performed in Cytoscape [21] using the ClueGO plugin [22]. RNAseq data is available upon request.
Results
DFMO treatment enhances checkpoint blockade in both α-PD-1 susceptible and α-PD-1 refractory tumor models
One of the most clinically impactful breakthroughs in modern cancer immunology is the discovery of the various immune checkpoints, such as the PD-1/PDL1 pathway [23–25]. These pathways inhibit localized immune function by causing long-lasting T cell exhaustion and represent a major mechanism of tumor immune evasion [26]. Critically, while checkpoint blockade therapy can potently regress established tumors in both preclinical models and human patients [27–29], its efficacy is dependent on the local inflammatory state. Since previous work had suggested that DFMO exerts therapeutic efficacy by modulating anti-tumor immunity [18–20] we hypothesized that it may potentiate α-PD-1 treatment.
To test for possible therapeutic benefits of combining DFMO and α-PD-1 we first examined treatment in a Lewis Lung Carcinoma (LLC-A9F1) model, which is partially responsive to α-PD-1 monotherapy. C57Bl/6 mice were injected subcutaneously (SQ) with 4×105 LLC-A9F1 cells and tumors allowed to establish for nine days. Mice were then separated into four groups and given either: no treatment (mock), drinking water supplemented with 1.5% DFMO (w/v), intraperitoneal (IP) injections of α-PD-1 blocking antibody, or both drinking water supplemented with 1.5% DFMO (w/v) and IP injections of α-PD-1 blocking antibody (combo) (Fig1A). Tumor burden was then measured twice weekly and animals euthanized when their tumors reached 15mm in any direction. Consistent with previous reports [11, 30–32], both DFMO and α-PD-1 monotherapies caused minor delays in tumor growth but limited tumor regression (0/10 mice in the DFMO group and 1/10 mice in the PD-1 blockade group) (Fig 1B). In contrast, animals treated with both DFMO and α-PD-1 displayed both significantly reduced tumor burden in partially responding animals (6/10 mice) as well as true tumor regression leading to complete phenotypic tumor eradication (no palpable tumors for 14+ days) in complete responders (4/10 mice) (Fig 1B and Fig 1C). This reduction also resulted in significantly improved overall survival suggesting that the combination of DFMO and PD-1 blockade improves therapeutic response compared to either as monotherapies(Fig 1D).
Figure 1: DFMO enhances PD-1 blockade in both partially checkpoint-responsive and refractory tumors.
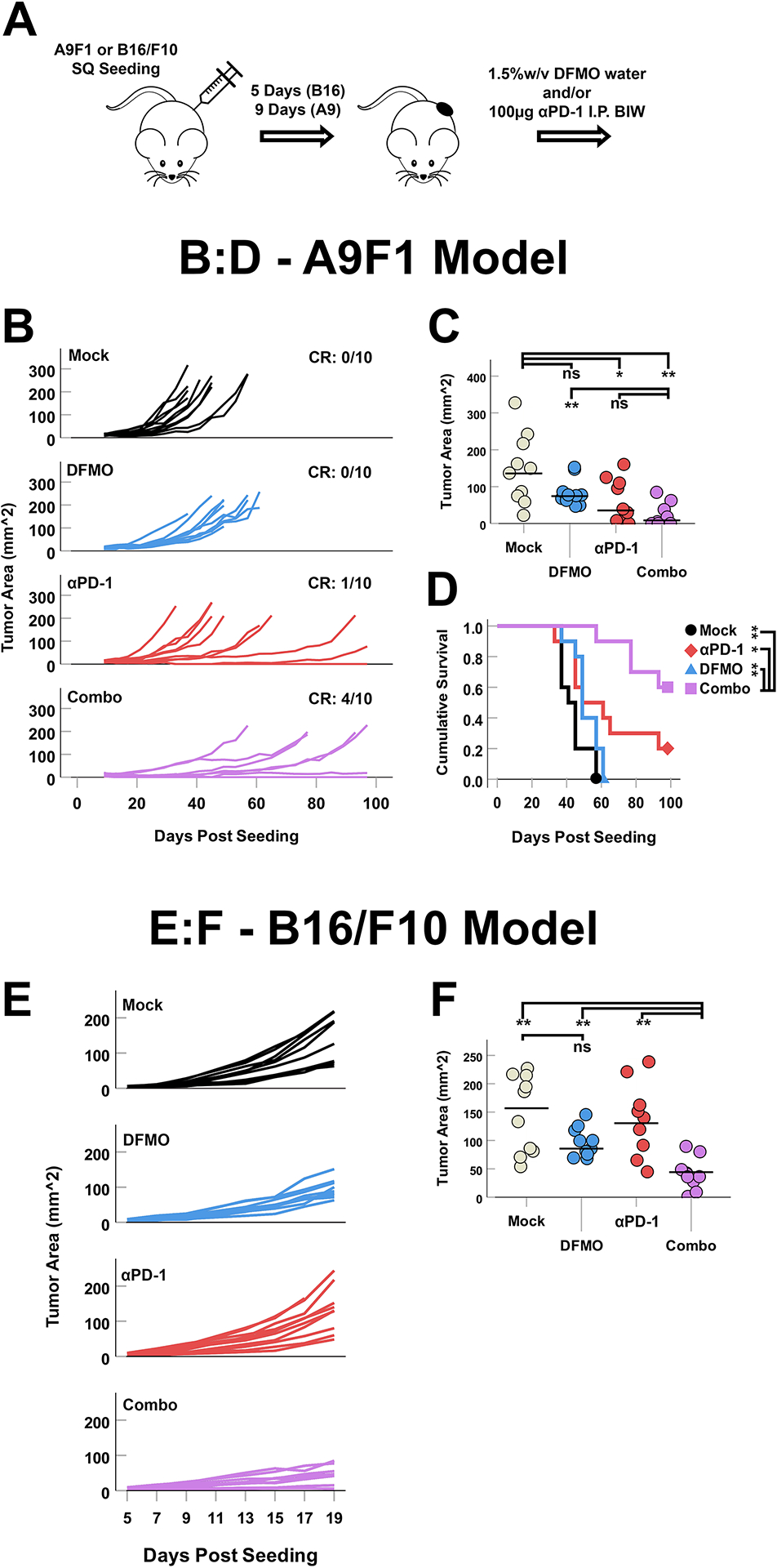
(A) Syngeneic C57Bl/6 mice were injected SQ with 4×105 LLC-A9F1 or B16/F10 cells and tumors were allowed to establish for nine and five days, respectively. Animals were then either mock treated or treated with: DFMO, α-PD-1, or a combination (n = 10 per group). (B) Growth curves of individual LLC-A9F1 tumors over time. CR denoted complete phenotypic response (no palpable tumors for 14+ days). (C) Area of individual LLC-A9F1 tumors 33 days after tumor implantation (day of first tumor reaching predetermined end-point criteria). Statistical significance was determined using unpaired Student’s t-test (*=p<0.05, **=p<0.005). (D) Overall survival of LLC-A9F1 seeded animals treated as indicated. Statistical significance was determined using the Breslow test for survival (* = p<0.05, ** = p<0.005). (E) Growth curves of individual B16/F10 tumors over time. (F) Area of individual B16/F10 tumors 19 days after tumor implantation (day of first tumor reaching predetermined end-point criteria), after which all mice were sacrificed for downstream analyses. Statistical significance was determined using unpaired Student’s t-test (*=p<0.05, ** = p<0.005).
Our previous results were obtained in an LLC-A9F1 model, which has been shown to be partially responsive to α-PD-1 treatments [31]. Many forms of cancer, however, are initially resistant to checkpoint-based monotherapy. To test whether DFMO could also function as a sensitizer for non-responsive tumors, we asked whether its combination with PD-1 blockade would enhance efficacy in the normally non-responsive B16/F10 melanoma model. C57Bl/6 mice were subcutaneously seeded with 4×105 B16/F10 cells, separated into four cohorts (mock, DFMO, α-PD-1, and combo) and treated as above. Tumor progression was then measured for 21 days to assess acute therapeutic response. Consistent with previous findings [33], treatment of B16/F10 tumor-bearing mice with DFMO resulted in a reproducible but relatively minor delay in tumor growth while PD-1-monotherapy was completely ineffective (Fig 1E and Fig 1F). While curative events were noted in this experiment, mice in the combination group displayed significantly delayed tumor growth and reduced overall tumor burden at day 19 compared to either single treatment alone suggesting that DFMO treatment can sensitize tumors to α-PD-1 therapy. Taken together, these data suggest that DFMO potentiates PD-1 blockade in susceptible settings and additionally may act as a sensitizer for some PD-1 blockade resistant tumors.
DFMO monotherapy creates broad but modest pro-inflammatory changes that are aggravated by addition of α-PD-1
In order to identify potential mechanisms mediating the therapeutic benefit of using combinatory DFMO and α-PD-1 therapy, we next sought to interrogate global changes to immunity within treated tumors. To accomplish this, the B16/F10 tumors from the previous experiment were excised 21 days after the initiation of treatment and processed for downstream analyses.
RNAseq-based interrogation of whole-tumor transcriptomes indicated broad changes in the overall immune signature (Fig 2). Principal component analysis of the RNAseq dataset demonstrated three distinct clusters corresponding to tumors treated with: Mock or α-PD-1, DFMO, and the combinatory group (Fig 2A). Unsupervised hierarchical clustering of the top 500 most variable transcripts across samples also demonstrated some sample clustering dependent on treatment, with particular separation of the combination group from the other cohorts (Fig 2B). Within the clustering analysis, four transcript clusters unique to both DFMO monotherapy and combinatory groups were identified, with an additional six clusters exclusive to the DFMO/α-PD-1 combinatory group. Gene ontology enrichment analysis of differentially expressed genes (DEGs) identified by comparing the combination and mock cohorts identified the most significant changes occurring within the general immune compartment (Fig 2C). These enriched pathways included more general immune activation (such as regulation of the immune system and immune responses) as well as pathways specific to leukocyte activation and function (such as leukocyte migration, leukocyte mediated cytotoxicity, and immune effector processes) (see supplemental digital content 1 – Fig S1). Of note, these immune signatures were not apparent in comparisons between the mock and DFMO and no DEGs at all could be identified between the mock and α-PD-1 cohorts (criteria of FDR = 0.05, log2FC = 1).
Figure 2: Combination therapy with DFMO and anti-PD-1 induces unique transcriptomes within the tumor microenvironment with functional implications.
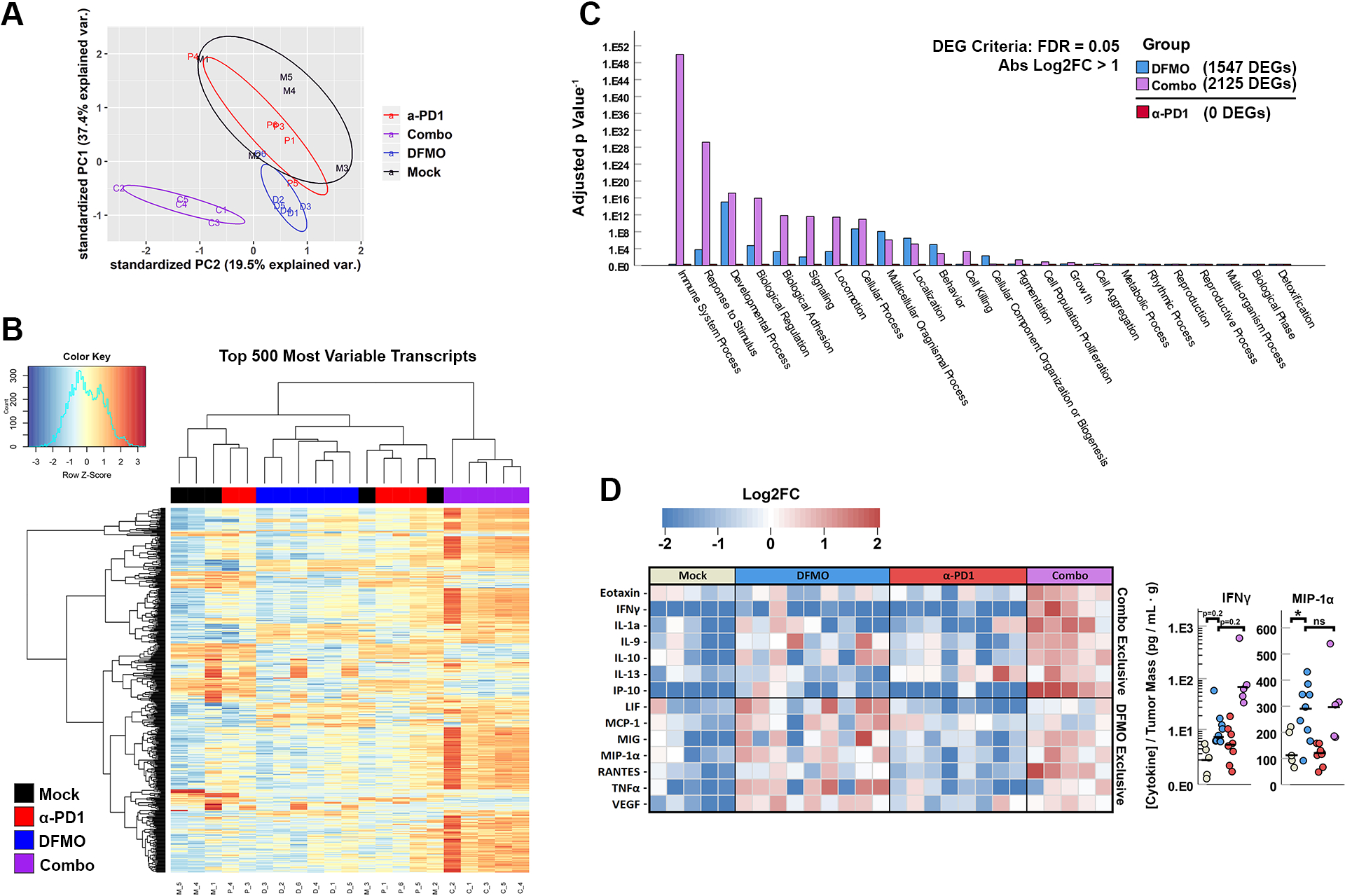
Syngeneic C57Bl/6 mice were injected SQ with 4×105 B16/F10 cells and tumors were allowed to establish for five days. Mice were then either mock treated or treated with DFMO, α-PD-1, or a combination (n = 5 per group). 19 days post tumor implantation, tumors were harvested and total RNA subjected to RNAseq analysis. (A) log2FC principal component analysis of CPM reads from each sample library showing unique clustering of combination treated tumors. (B) Heat map displaying an unsupervised hierarchical clustering of log2CPM reads of the top 500 most variable transcripts between the mock, single-agent, or combination cohorts. (C) Gene ontology pathway enrichment analysis of differentially expressed genes (FDR = 0.05, log2FC > 1) in DFMO, α-PD-1, and DFMO/α-PD-1 compared to mock within 24 daughter terms of GO’s Biological Processes. (D) Heatmap depicting log2FC in concentration of cytokines compared to collective averages within tumor supernatants of B16/F10 tumor bearing mice in 4 treatment cohorts. Cytokines were grouped for those induced uniquely by combination treatment (top section, all cytokines p < 0.05 in DFMO vs. Mock, p > 0.05 in DFMO vs. Combo) or by DFMO treatment alone (bottom section, all cytokines p < 0.05 in Combo vs. Mock, p > 0.05 in DFMO vs. Mock). Statistical significance was determined using unpaired Student’s t-test (ns = no significance, * = p<0.05, ** = p<0.005).
To supplement our global RNA profiling, we also assessed the overall cytokine expression within treated tumors (Fig 2D). Consistent with combination therapy enhancing overall immune activity, tumors from mice treated with both DFMO and α-PD-1 displayed increased levels of several immune modulating cytokines including: IFNγ, IL-9, IL-10, IL-13 and RANTES. Interestingly, despite no obvious immune signature in the RNAseq data, tumors from mice treated with DFMO also demonstrated significant changes to cytokine profiles including increases in numerous cytokines associated with innate immunity, such as: LIF, IL-1α, TNFα, MIP-1α, and MCP-1.
Our previous results suggested that the combination of DFMO and α-PD-1 therapy reduced tumor growth by altering intratumoral immune activity. To determine which immune populations may be mechanistically responsible for the combination treatment’s enhanced anti-tumor efficacy, we therefore conducted an immune depletion study to test the efficacy of combined DFMO and α-PD-1 treatment in the absence of specific immune subsets. Mice were again seeded with subcutaneous B16/F10 tumors and placed under DFMO/α-PD-1 combination treatment. Animals were then separated into six cohorts and given depleting antibodies against CD8, NK1.1, GR1, CD4, or isotype controls. Animals were then assessed for tumor growth and euthanized as above. The results indicated that the ability of DFMO/α-PD-1 therapy to delay tumor growth was largely lost in mice depleted of CD8+ T cells (Fig 3). No other depleted cohort showed a significant reduction in tumor control at day 19, however, NK1.1 depletion displayed a strong trend towards reduced efficacy (p=0.06) suggesting the possible additional involvement of natural killer (NK) cells.
Figure 3: Anti-tumor effects of DFMO/α-PD-1 combination therapy is primarily mediated by CD8+ T cells.
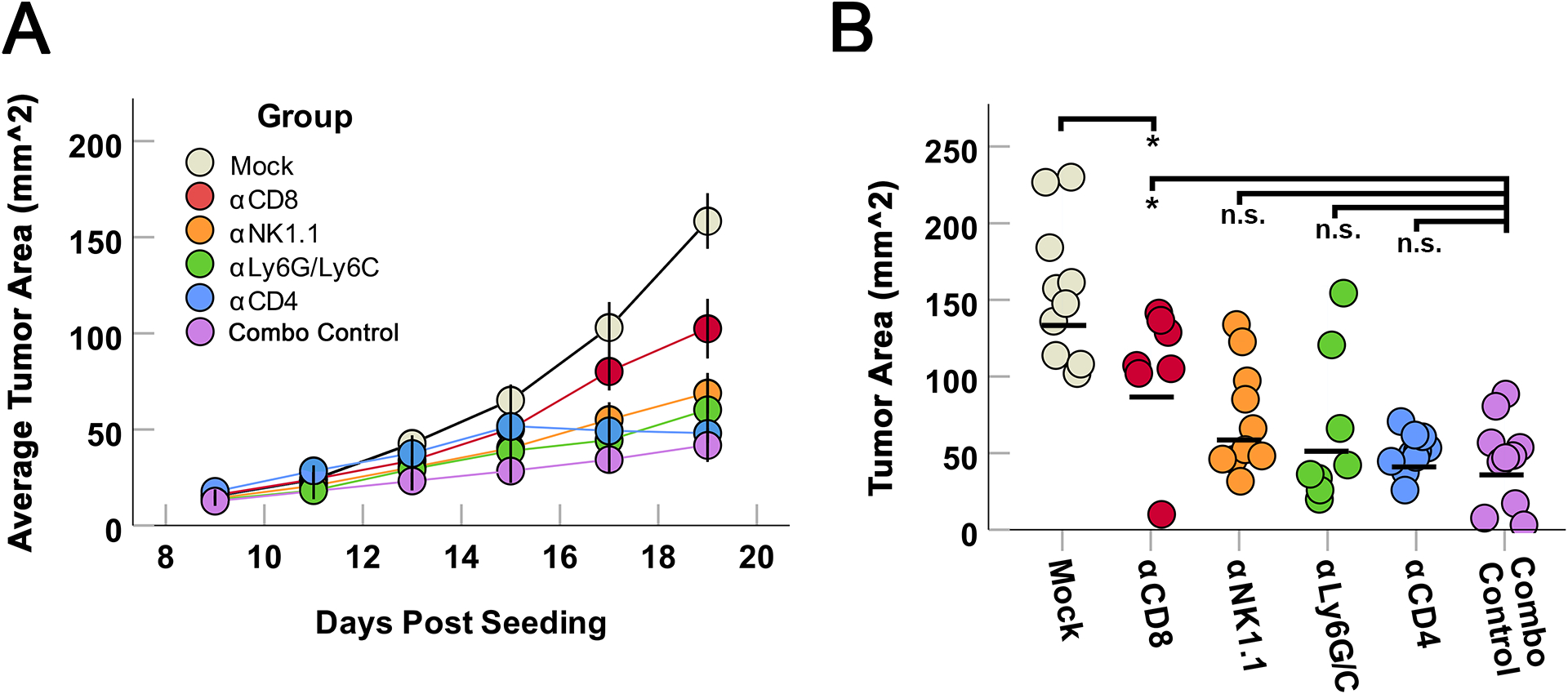
Mice were SQ seeded with B16/F10 cells as previously and subsequently treated with DFMO/α-PD-1 therapy as previously described. Beginning on day 5 and continuing throughout the experiment, groups of mice were also given twice-weekly intraperitoneal injections of the indicated immune cell depletion antibodies (α-CD4, α-CD8, α-NK1.1, or α-GR1) (n=10/group) twice weekly. (A) Average growth of tumors within each cohort over time. (B) Area of individual tumors 19 days after tumor implantation (the day the first mock tumor reached predetermined end-point criteria). Statistical significance was determined using unpaired Student’s t-test (*=p<0.05, ** = p<0.005).
DFMO combination therapy enhances the survival and activity of intratumoral CD8+ T cells
To determine how DFMO might influence the intratumoral CD8+ T cell compartment, we next sought to identify phenotypic changes within this population following either DFMO, α-PD-1, or DFMO/α-PD-1 therapy. Mice were subcutaneously seeded with B16/F10 cells as above and subsequently treated with either saline, DFMO, α-PD-1, or DFMO/ α-PD-1. Tumors were then excised 21 days after the initiation of treatment and tumor infiltrating lymphocytes (TIL) analyzed using flow cytometry (Fig 4A). This analysis indicated that mice treated with either DFMO or combination therapy displayed a strong trend towards increased total CD45+ infiltrates. Within the CD8+ T cell compartment, we observed that neither DFMO nor combination therapy increased the average percentage of CD8+ T cells within the tumor. However, a significant increase in the percentage of viable CD8+ T cells was observed within tumors from DFMO and DFMO/α-PD-1 combination groups. DFMO/α-PD-1 treatment was also associated with an increase expression of PD-1 on CD8+ TIL, suggesting possible enhanced activation or exhaustion.
Figure 4: DFMO/anti-PD-1 combination therapy potently alters tumoral CD8+ T cell viability through an indirect mechanism.
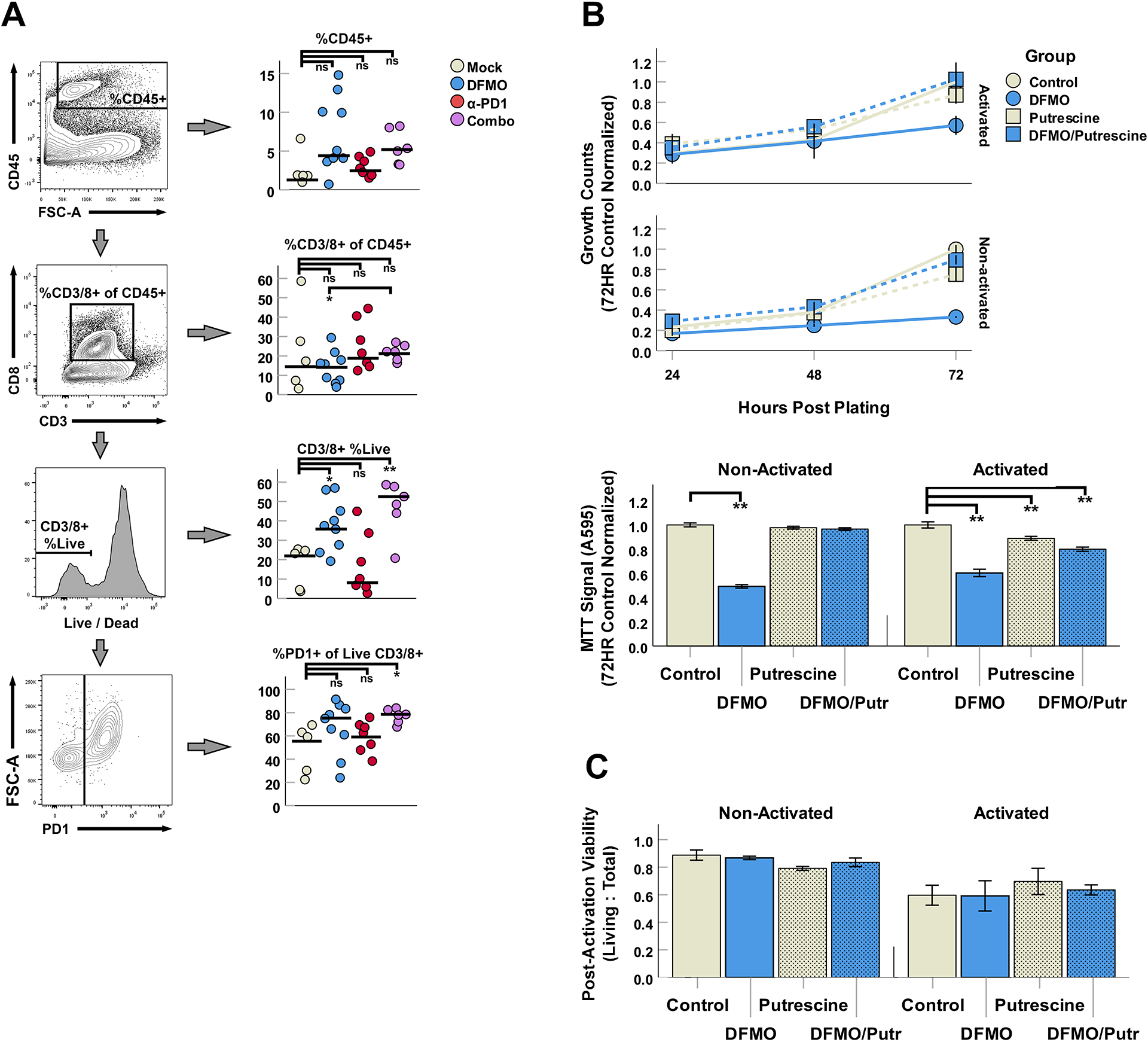
(A) Phenotypic flow cytometry analysis of CD8+ T cell populations within B16/F10 tumors in 4 treatment cohorts. (B) In vitro analyses of PMA/ionomycin activated T cell (Jurkat cell line) proliferation in 10mM DFMO and/or 80μM putrescine over 72 hours (top; total cell counts normalized to 72HR controls) and MTT signal at 72 hours post-treatment (bottom; absorbance (A595) normalized to control). (C) Quantification of activation-induced cell death (AICD) via trypan blue counts of Jurkat cultures 24 hours post activation. Values represent ratio of Living (Trypan−) to Total cells in each culture condition.
To assess whether DFMO might have a directly beneficial effect on T cell viability, we finally asked whether treatment with this drug would alter either T cell proliferation or viability in vitro (Fig 4B). Jurkat T cells were either left non-stimulated or activated for 2 hours with PMA/Ionomycin. Cells were subsequently treated with DFMO in either the presence or absence of exogenous putrescine (putrescine being the product of ODC – the enzyme inhibited by DFMO) to confirm if any direct biological changes were strictly attributable to polyamine starvation. The growth of each culture was then tracked for 72 hours after which total cell number and viability was determined. The results demonstrated that cultures of Jurkat cells containing DFMO grew significantly slower than mock treated cultures, with this effect expectedly eliminated in cultures that were rescued with exogenous putrescine. Cultures of Jurkats that were activated with PMA/Ionomycin displayed a significant decrease in cellular viability consistent with T cell activation induced cell death; however, this reduced cellular viability was not influenced by the presence of either DFMO or putrescine within non-activated samples, though a slight loss of MTT signal was observed in putrescine and DFMO + putrescine combination groups. Experiments with activated T cells from splenocyte cultures (IL-2, αCD3 plate coating) yielded similar results at 48 hours, with no obvious benefit induced by DFMO in viability or CD8+ T cell abundance (Fig S3).
Discussion
The recent observations of DFMO’s immunomodulatory nature has raised questions of the possible relevance of polyamine metabolism to immune response and function, and has offered opportunities to explore the use of DFMO as an immunotherapeutic agent. Here, our work demonstrates that DFMO potentiates intratumoral T cell survival and improves α-PD-1 checkpoint blockade in both α-PD-1 monotherapy-responsive and refractory cancer models.
The therapeutic efficacy of DFMO as a mono-agent in the LLC-A9F1 and B16/F10 melanoma models (Fig 1) was modest with a significant delay in tumor growth but marginal impact on survival. Though multiple groups have now demonstrated immunocompetency is required for DFMO to exert its anti-tumor effects [18, 19, 34], the majority of our results show relatively restrained changes to the overall immune signature following DFMO therapy. The changes that were observed were largely indicative of a functional shift within the innate immunity compartment, highlighted by increases in IL-1α, TNFα, and MIP-1α (Fig 2). Flow cytometry of TIL in B16/F10 tumors from DFMO treated mice also demonstrated only slight changes compared to mock, with a trending, but non-statistically significant, increase in total CD45+ cells and an appreciable increase in CD8+ T cell viability. No major changes were observed within the myeloid compartment save for a potential shift in MDSC phenotype from monocytic to granulocytic (see supplemental digital content 2 - Fig S2), a phenomenon of uncertain significance that has also been observed by a previous group [19]. These changes seemed to be lost entirely within the RNAseq dataset, where the determined DEGs were outside of terms associated with immune response and function. If the effects of DFMO are indeed more nuanced, observing the global transcriptomes of tumors (rather than only immune populations) may have been sufficient to obfuscate changes within the immune compartment, particularly with stringent DEG criteria.
Interestingly, despite its exciting clinical successes, PD-1 blockade monotherapy was often less effective than DFMO in the preclinical models used in this study. This therapy completely failed to induce tumor delay in a standard B16/F10 model and was associated with even more limited immunological changes compared to those of DFMO. Even in the ‘responsive’ LLC-A9F1 model, only 3/10 mice treated with PD-1 blockade outperformed animals treated with DFMO alone. Using the same stringency for DEG classification as the DFMO and DFMO/αPD-1 Combination cohorts, the RNAseq dataset within the PD-1 blockade cohorts failed to detect any DEGs compared to mock mice. The B16/F10 model being entirely refractory to αPD-1 therapy reflects this lack of observed change, though our analysis of bulk tumor RNA likely masks any more nuanced changes induced by αPD-1 mentioned above for the DFMO DEGs. However, given a large portion of immunological changes are observed in the DFMO/αPD-1 cohort that are not observed within the DFMO monotherapy cohort, the data suggests that the B16/F10 tumors do exploit PD-L1/PD-1 signaling, but it is secondary to some immunological barrier that is circumvented through the use of DFMO. Only once this primary immunosuppression is removed by DFMO does the αPD-1 therapy become effective within the B16/F10 model.
In contrast, combination therapy consistently displayed robust performance both in metrics of efficacy and analyses of immune function. The RNAseq of whole tumors demonstrated a marked association of DEGs within immune system processes, with the most significant changes occurring in Immune Response, Leukocyte Activation, Antigen Processing and Presentation, and Immune Effector Process daughter terms (see supplemental digital content 1 – Fig S1). Within immune infiltrates of tumors, combination treatment was characterized by an even greater proportion of viable CD8+ T cells (Fig 4) as well as a shift to a more conventional CD4+ T cell predominant population (see supplemental digital content 2 - Fig S2) that are not explained by a simple additive effect of α-PD-1 and DFMO. Evidence for enhancing therapeutic response was also seen within the cytokine datasets, with increases in pro-inflammatory cytokines such as IFNγ, RANTES, and IL-9.
Collectively, these results suggest that DFMO/α-PD-1 combination treatment is likely removing two sequential immunological barriers to mounting an effective immune response. In the first step, rather than directly acting on T cells, DFMO modulates the tumor microenvironment in a way that leads to increased CD8+ T cell survival. In the second step, the addition of PD-1-blocking reagents prevents the functional exhaustion of these surviving T cells leading to enhanced anti-tumor effects. This explanation is supported in part by our data indicating that αPD-1 had no effect on B16/F10 cells as a monotherapeutic despite the majority of T cells within the tumor bed being PD-1+ in all four cohorts, and that CD8+ T cell survival was potentiated in both DFMO and DFMO/αPD-1 combination groups (Fig 4A).
Additionally, the presence of αPD-1 increased IFNγ and RANTES only when combined with DFMO, and therapeutic efficacy was reduced upon depletion of CD8+ T cells. It is of note, however, that loss of CD8+ T cells was not sufficient to ablate therapy entirely, with α-CD8+ cohorts still performing significantly better than the mock group yet worse than the combination control. This may be explained when considering the NK cell depleted cohort was statistically borderline in terms of significance in tumor burden at this level of statistical power, and might be playing a secondary population of anti-tumor cytotoxic cells capable of producing IFNγ [35]. As in the RNAseq data, these data support the frequently proposed idea that PD-L1/PD-1 checkpoint is therapeutically irrelevant in immunologically barren tumors, but becomes a major obstacle to immune function once other forms of immune inhibition are lifted.
Our in vitro and ex vivo experiments (Fig 4B–C, supplemental digital content 3 - Fig S3) demonstrated that DFMO had no direct pro-survival or proliferative effects on Jurkat cells or primary splenic CD8+ T cells in states of both non-activation and activation, and expectedly induced cytostasis in the DFMO controls without exogenous putrescine [13]. In Jurkat DFMO cultures rescued with putrescine and the single putrescine controls, a slight decrease in MTT signal was observed for activated T cells though not living cell counts; a likely explanation for this may be mild mitochondrial toxicity induced by the degradation of putrescine by diamine oxidase, an enzyme naturally found within fetal bovine serum that produces H2O2, ammonia, and aminoaldehyde from putrescine [36]. Regardless, the primary purpose of these studies was to attempt to identify any possible benefit to T cell survival or proliferation induced by DFMO directly, specifically any potential off-target effects outside of ODC; clearly no beneficial differences were observed at 72 hours for Jurkats or 48 hours for activated splenic T cells, showing there is likely no gain of function by virtue of dosing with DFMO. These results further suggest that DFMO is acting as a preventative to loss-of-function in vivo, which would align with other studies indicating a possible DFMO induced disruption of immunosuppression within the myeloid compartment [19, 20].
The recently published work of Alexander et. al. also explored the use of DFMO as an immunomodulatory agent to potentiate response to checkpoint blockade, but in the context of total polyamine blockade by the addition of a polyamine transport inhibitor (PTI) [18]. Although the authors of this work observe clear therapy-induced disruptions of pathological M2 polarization and MDSC differentiation, and thoroughly interrogate functional changes within these populations, the in vivo performance of total polyamine blockade by the addition of a PTI to DFMO and α-PD-1 seems to be comparable to the efficacy observed in our work. This may be partially explained by a disparate dosing regimen of DFMO (1.5% w/v compared to 0.25% w/v), however, transport of exogenous polyamines would remain unimpeded regardless of DFMO concentration. As noted by this group and others, what remains to be determined is how much of DFMO’s immunomodulation can be attributed directly to a polyamine deficit, or rather if the observed effects on anti-tumor immunity stem from ODC-inhibition-induced alterations within the several metabolic pathways that are inextricably linked to polyamine biosynthesis [37–40].
Though additional pre-clinical testing to obtain a mechanistic understanding of the effects of DFMO/αPD-1 combination therapy (and how similar this mechanism is in relation to total polyamine blockade using a PTI/DFMO with αPD-1) is desirable, it is worth noting that the combination of DFMO/αPD-1 did not result in any obvious increase of toxicity compared to either monotherapy. Consistent with DFMO single-agent controls, the combination treatment cohorts exhibited loose stools and one case of possible ototoxicity, with both side effects documented in the literature [13]. Though mice were not weighed throughout treatment, no clear change or aggravation of weight loss was observed while handling mice for tumor measurements. We believe that this exceptionally mild toxicity of DFMO - even when clinically employed at multi-gram doses daily [13, 41] - further argues for investigation into its use as a combinatory therapeutic with checkpoint blockade, the latter being a therapy infamous for severe toxicity [42–44]. Lastly, the pre-existing FDA approval of DFMO for treatment of late stage African trypanosomiasis and hirsutism [45] poses a further significant advantage should efforts be taken to attempt clinical transition of this combination therapy.
In conclusion, while our work does not directly address the mechanism(s) through which the observed immunomodulation occurs beyond determining that these effects are CD8+ T cell/NK cell dependent, it adds additional support to previous studies implicating DFMO as an immunomodulatory agent capable of potentiating anti-cancer immunity. Our data demonstrates that DFMO could be a valuable addition to cancer immunotherapies even in the absence of a polyamine transport inhibitor.
Supplementary Material
Supplemental digital content 1: Figure S1 showing gene-ontology pathway enrichments from RNA seq data.
Supplemental digital content 2: Figure S2 showing complete immune phenotyping of treated tumors.
Supplemental digital content 3: Figure S3 showing effect of DFMO on activated splenic CD8+ T cell in terms of proliferation and viability over 48 hours.
Acknowledgements:
This work was supported by grants to Dr. Eric Bartee from: the NIH (NIAID-AI142387 and NCI-CA194090), and the ACS (RSG-17047-01-MPC). This work was also supported by the Hollings Cancer Center’s Support Grant (P30-CA138313).
References
- 1.Miller-Fleming L, et al. , Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. Journal of Molecular Biology, 2015. 427(21): p. 3389–3406. [DOI] [PubMed] [Google Scholar]
- 2.Arruabarrena-Aristorena A, Zabala-Letona A, and Carracedo A, Oil for the cancer engine: The cross-talk between oncogenic signaling and polyamine metabolism. Science Advances, 2018. 4(1): p. eaar2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bachmann AS and Geerts D, Polyamine synthesis as a target of MYC oncogenes. Journal of Biological Chemistry, 2018. 293(48): p. 18757–18769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casero RA, Murray Stewart T, and Pegg AE, Polyamine metabolism and cancer: treatments, challenges and opportunities. Nature Reviews Cancer, 2018. 18(11): p. 681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nowotarski SL, Woster PM, and Casero RA Jr., Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert reviews in molecular medicine, 2013. 15: p. e3–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bey P, et al. , Analogues of ornithine as inhibitors of ornithine decarboxylase. New deductions concerning the topography of the enzyme’s active site. J Med Chem, 1978. 21(1): p. 50–5. [DOI] [PubMed] [Google Scholar]
- 7.Luk GD and Casero RA, Polyamines in normal and cancer cells. Advances in Enzyme Regulation, 1987. 26: p. 91–105. [DOI] [PubMed] [Google Scholar]
- 8.Murray-Stewart TR, Woster PM, and Casero RA Jr., Targeting polyamine metabolism for cancer therapy and prevention. The Biochemical journal, 2016. 473(19): p. 2937–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowlin TL, et al. , Effects of Three Irreversible Inhibitors of Ornithine Decarboxylase on Macrophage-mediated Tumoricidal Activity and Antitumor Activity in B16F1 Tumor-bearing Mice. Cancer Research, 1990. 50(15): p. 4510. [PubMed] [Google Scholar]
- 10.Levin VA, et al. , Treatment of recurrent gliomas with eflornithine. J Natl Cancer Inst, 1992. 84(18): p. 1432–7. [DOI] [PubMed] [Google Scholar]
- 11.Paulsen JE and Lutzow-Holm C, In vivo growth inhibition of human colon carcinoma cells (HT-29) by all-trans-retinoic acid, difluoromethylornithine, and colon mitosis inhibitor, individually and in combination. Anticancer Res, 2000. 20(5b): p. 3485–9. [PubMed] [Google Scholar]
- 12.Green JE, et al. , 2-difluoromethylornithine and dehydroepiandrosterone inhibit mammary tumor progression but not mammary or prostate tumor initiation in C3(1)/SV40 T/t-antigen transgenic mice. Cancer Res, 2001. 61(20): p. 7449–55. [PubMed] [Google Scholar]
- 13.Meyskens FL and Gerner EW, Development of Difluoromethylornithine (DFMO) as a Chemoprevention Agent. Clinical Cancer Research, 1999. 5(5): p. 945. [PubMed] [Google Scholar]
- 14.Liu R, et al. , Determination of polyamines in human plasma by high-performance liquid chromatography coupled with Q-TOF mass spectrometry. Journal of Mass Spectrometry, 2012. 47(10): p. 1341–1346. [DOI] [PubMed] [Google Scholar]
- 15.Löser C, et al. , Polyamine Concentrations in Pancreatic Tissue, Serum, and Urine of Patients with Pancreatic Cancer. Pancreas, 1990. 5(2): p. 119–127. [DOI] [PubMed] [Google Scholar]
- 16.Muth A, et al. , Polyamine Transport Inhibitors: Design, Synthesis, and Combination Therapies with Difluoromethylornithine. Journal of Medicinal Chemistry, 2014. 57(2): p. 348–363. [DOI] [PubMed] [Google Scholar]
- 17.Samal K, et al. , AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. International Journal of Cancer, 2013. 133(6): p. 1323–1333. [DOI] [PubMed] [Google Scholar]
- 18.Alexander ET, et al. , A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget, 2017. 8(48): p. 84140–84152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye C, et al. , Targeting Ornithine Decarboxylase by α-Difluoromethylornithine Inhibits Tumor Growth by Impairing Myeloid-Derived Suppressor Cells. Journal of immunology (Baltimore, Md. : 1950), 2016. 196(2): p. 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geng Z, et al. , alpha-Difluoromethylornithine suppresses inflammatory arthritis by impairing myeloid-derived suppressor cells. Int Immunopharmacol, 2019. 71: p. 251–258. [DOI] [PubMed] [Google Scholar]
- 21.Shannon P, et al. , Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 2003. 13(11): p. 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bindea G, et al. , ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics (Oxford, England), 2009. 25(8): p. 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong J, et al. , Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. Journal for ImmunoTherapy of Cancer, 2018. 6(1): p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamid O, et al. , Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med, 2013. 369(2): p. 134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robert C, et al. , Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet, 2014. 384(9948): p. 1109–17. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X, et al. , Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Molecular Cancer, 2019. 18(1): p. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rausch MP and Hastings KT, Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application, in Cutaneous Melanoma: Etiology and Therapy, Ward WH and Farma JM, Editors. 2017, Codon Publications The Authors.: Brisbane (AU). [PubMed] [Google Scholar]
- 28.Azoury SC, Straughan DM, and Shukla V, Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety. Curr Cancer Drug Targets, 2015. 15(6): p. 452–62. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, et al. , Application of PD-1 Blockade in Cancer Immunotherapy. Computational and structural biotechnology journal, 2019. 17: p. 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gitto SB, et al. , Difluoromethylornithine Combined with a Polyamine Transport Inhibitor Is Effective against Gemcitabine Resistant Pancreatic Cancer. Molecular Pharmaceutics, 2018. 15(2): p. 369–376. [DOI] [PubMed] [Google Scholar]
- 31.Nagato T, et al. , Combinatorial immunotherapy of polyinosinic-polycytidylic acid and blockade of programmed death-ligand 1 induce effective CD8 T-cell responses against established tumors. Clin Cancer Res, 2014. 20(5): p. 1223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jun JY, et al. , Effects of polyamine depletion by α-difluoromethylornithine on in vitro and in vivo biological properties of 4T1 murine mammary cancer cells. Breast Cancer Research and Treatment, 2008. 107(1): p. 33–40. [DOI] [PubMed] [Google Scholar]
- 33.Kubota S, Ohsawa N, and Takaku F, Effects of DL-alpha-difluoromethylornithine on the growth and metastasis of B16 melanoma in vivo. Int J Cancer, 1987. 39(2): p. 244–7. [DOI] [PubMed] [Google Scholar]
- 34.Hayes CS, et al. , Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer immunology research, 2014. 2(3): p. 274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paolini R, et al. , NK cells and interferons. Cytokine & Growth Factor Reviews, 2015. 26(2): p. 113–120. [DOI] [PubMed] [Google Scholar]
- 36.Eero N and William W Wharton III, Diamine Oxidase Is Important in Assessment of Polyamine Effects on Hemopoietic Cell Proliferation in vitro. In Vitro Cellular & Developmental Biology, 1987. 23(4): p. 257–260. [DOI] [PubMed] [Google Scholar]
- 37.Lam S-K, et al. , Inhibition of ornithine decarboxylase 1 facilitates pegylated arginase treatment in lung adenocarcinoma xenograft models. Oncology reports, 2018. 40(4): p. 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selamnia M, et al. , Alpha-difluoromethylornithine (DFMO) as a potent arginase activity inhibitor in human colon carcinoma cells. Biochem Pharmacol, 1998. 55(8): p. 1241–5. [DOI] [PubMed] [Google Scholar]
- 39.Shantz LM, et al. , Regulation of S-adenosylmethionine decarboxylase activity by alterations in the intracellular polyamine content. The Biochemical journal, 1992. 288 ( Pt 2)(Pt 2): p. 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Witherspoon M, et al. , Unbiased metabolite profiling indicates that a diminished thymidine pool is the underlying mechanism of colon cancer chemoprevention by alpha-difluoromethylornithine. Cancer discovery, 2013. 3(9): p. 1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sholler GLS, et al. , Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Scientific Reports, 2018. 8(1): p. 14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marin-Acevedo JA, Chirila RM, and Dronca RS, Immune Checkpoint Inhibitor Toxicities. Mayo Clinic Proceedings, 2019. 94(7): p. 1321–1329. [DOI] [PubMed] [Google Scholar]
- 43.Spiers L, Coupe N, and Payne M, Toxicities associated with checkpoint inhibitors-an overview. Rheumatology (Oxford, England), 2019. 58(Suppl 7): p. vii7–vii16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puzanov I, et al. , Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. Journal for ImmunoTherapy of Cancer, 2017. 5(1): p. 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laukaitis CM and Gerner EW, DFMO: targeted risk reduction therapy for colorectal neoplasia. Best Pract Res Clin Gastroenterol, 2011. 25(4–5): p. 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content 1: Figure S1 showing gene-ontology pathway enrichments from RNA seq data.
Supplemental digital content 2: Figure S2 showing complete immune phenotyping of treated tumors.
Supplemental digital content 3: Figure S3 showing effect of DFMO on activated splenic CD8+ T cell in terms of proliferation and viability over 48 hours.