

Abstract
Regulatory T cells (Tregs) control immune responses in autoimmune disease, transplantation, and enable antigen-specific tolerance induction in protein-replacement therapies. Tregs can exert a broad array of suppressive functions through their T cell receptor (TCR) in a tissue-directed and antigen-specific manner. This capacity can now be harnessed for tolerance induction by “redirecting” polyclonal Tregs to overcome low inherent precursor frequencies and simultaneously augment suppressive functions. With the use of hemophilia A as a model, we sought to engineer antigen-specific Tregs to suppress antibody formation against the soluble therapeutic protein factor (F)VIII in a major histocompatibility complex (MHC)-independent fashion. Surprisingly, high-affinity chimeric antigen receptor (CAR)-Treg engagement induced a robust effector phenotype that was distinct from the activation signature observed for endogenous thymic Tregs, which resulted in the loss of suppressive activity. Targeted mutations in the CD3ζ or CD28 signaling motifs or interleukin (IL)-10 overexpression were not sufficient to restore tolerance. In contrast, complexing TCR-based signaling with single-chain variable fragment (scFv) recognition to generate TCR fusion construct (TRuC)-Tregs delivered controlled antigen-specific signaling via engagement of the entire TCR complex, thereby directing functional suppression of the FVIII-specific antibody response. These data suggest that cellular therapies employing engineered receptor Tregs will require regulation of activation thresholds to maintain optimal suppressive function.
Keywords: Treg, CAR, scFv, FVIII, TRuC, engineered receptor, ITAM, hemophilia, tolerance, inhibitors
Graphical abstract
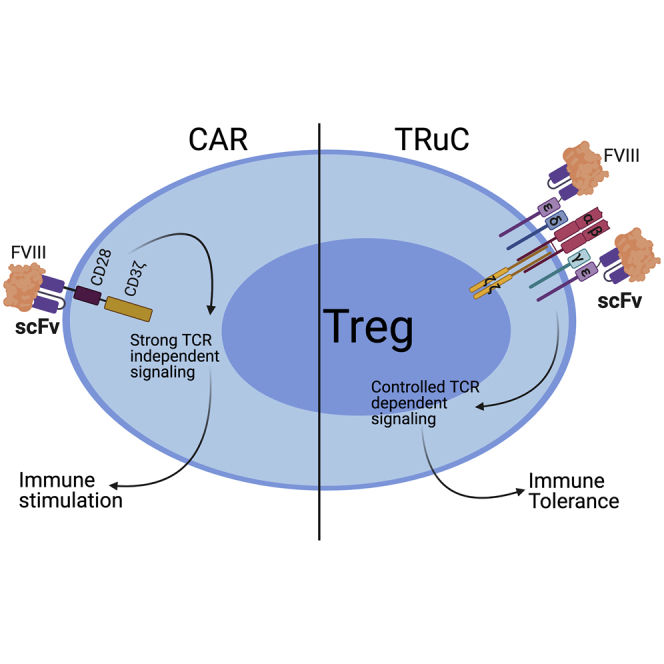
Rana and colleagues compare Treg engineering platforms to suppress anti-drug antibodies using hemophilia as a model. They show that dysregulated signaling using high-affinity CAR-Tregs leads to loss of suppression. In contrast, TCR fusion TRuC-Tregs more faithfully mimic physiological TCR signaling, delivering the desired immune suppression in an antigen-specific manner.
Introduction
FoxP3-expressing regulatory T cells (Tregs) are crucial drivers of central and peripheral tolerance and are therefore an ideal cellular therapeutic tool for antigen-specific tolerance induction. In human clinical trials, single infusions of polyclonal Tregs can successfully prevent or attenuate autoimmune disease1,2 as well as allogeneic hematopoietic cell or solid organ transplant rejection,3,4 thus reducing dependency on immunosuppressive drugs. Tregs can further be redirected for antigen specificity using cutting-edge cellular engineering mechanisms, thereby improving targeted suppression at lower effective doses.5,6
One strategy to engineer specificity in polyclonal Tregs is to express a chimeric antigen receptor (CAR), which is a synthetic molecule that combines extracellular single-chain variable fragments (scFvs) of an antibody with primary T cell receptor (TCR) signaling and costimulatory moieties.7 Alternatively, this strategy can potentially be applied to FoxP3-engineered conventional T cells (Tconvs) under conditions of Treg scarcity.8, 9, 10 CAR expression combines antigen specificity and cell signaling without the requirement for major histocompatibility complex (MHC) class II restriction in a diverse patient group. CAR-engineered Tconvs have been shown to be highly effective at eradicating B cell leukemias that are resistant to standard therapies, whereas studies with CAR Tregs show promise in models of autoimmune disease11,12 and allograft rejection,5,13 with a first-in-man clinical trial soon to be launched for solid organ transplantation (phase I/II STEADFAST trial, TX200; Sangamo Therapeutics). To date, CAR Treg design has been modeled on CAR Tconv constructs for cancer by employing second-generation CD3ζ and costimulatory CD28 or 4-1BB signaling domains.14 Moreover, most CAR molecules have been designed to recognize a membrane-bound surface antigen, with a major gap in understanding the mechanism of action for soluble antigen(s). Additional questions remain, such as the potential impact(s) of inserting a high-affinity scFv on the Treg function, as well as signal strength and kinetics of a synthetic CAR in comparison to its endogenous TCR counterpart.
In this study, we used a high-affinity CAR specific for coagulation factor (F)VIII to suppress inhibitory antibody responses to replacement FVIII therapy in a murine model of hemophilia A (HA). Mutations in the F8 gene can lead to reduced, misfolded, or complete lack of expression of FVIII in the blood, with severity of the condition consistent with the degree of residual clotting activity.15,16 Inhibitory antibodies to exogenously infused FVIII can neutralize the therapeutic protein in up to 30% of severe HA patients, thus interfering with treatment. A growing body of evidence suggests that immunomodulation by Tregs could offer a new treatment strategy in HA.17 We have previously shown that cellular therapy with either ex vivo-expanded polyclonal Tregs or FoxP3-transduced Tconv (i.e., de novo Tregs) enriched for antigen specificity is tolerogenic in a murine model of severe HA.18,19
Here, we sought to understand the effect of high-affinity CAR signaling on Treg stability, cytokine production, in vivo persistence, and suppressive capacity. We analyzed the contribution of proximal and distal CD3ζ immune receptor tyrosine-based activation motifs (ITAMs), as well as CD28 signaling motifs. We explored the alternative TCR fusion construct (TRuC), which was synthesized by fusing the FVIII scFv to the N terminus of the TCRε subunit.20 We confirmed that incorporation of the TRuC construct into the TCR-CD3 complex limited surface receptor density and more faithfully mimicked physiological TCR signaling. In vivo, TRuC Tregs suppressed the development of adaptive immune responses to FVIII. Complexing TCR-based signaling with scFv recognition has not been tested earlier in Tregs and has the potential to engage not just the complete TCR machinery in an MHC-unrestricted manner but can also subject the cell to negative-feedback mechanisms that are rapidly induced by TCR engagement to regulate signal output in response to antigen.21
Results
Generation of a FVIII-directed CAR for engineered specificity
We synthesized a second-generation FVIII CAR construct comprising a high-affinity (10−11 M−1) extracellular human scFv (BO2C11, which was specific for the highly immunogenic C2 domain of FVIII),22 complexed to the transmembrane and intracellular murine CD28 costimulatory and CD3ζ signaling domains (Figure 1A). Tregs from BALB/c Foxp3IRES-GFP mice, which express GFP under control of the mouse Foxp3 promoter, were magnetically enriched and purified by fluorescence-activated cell sorting (FACS; >98% FoxP3GFP+ cells). Following transduction of activated Tregs with the FVIII CAR-pMYs-IRES-mScarlet retroviral vector, mScarlet- and FoxP3GFP+-co-expressing Tregs were FACS sorted for a 2nd time (>98% mScarlet+FoxP3GFP+) and ex vivo expanded for a short period (3−4 days) to obtain ∼2-fold expansion (Figure 1B). This was done in order to minimize phenotypic or functional differences that may arise as a result of prolonged ex vivo culture.
Figure 1.

FVIII CAR Tregs bind and respond to FVIII stimulation in vitro
(A) Schematic of the FVIII CAR construct. The variable light (VL) and heavy (VH) regions of the FVIII-specific scFv, hinge, transmembrane, and intracellular signaling regions of murine CD28 and CD3ζ signaling domains are indicated. ITAM tyrosine motifs in the CD3ζ chain are indicated (Y), as well as tyrosine to phenylalanine point mutations (Y-F) in ITAM1 and ITAM3. (B) Schematic of Treg isolation, anti (α)-CD3/28 microbead activation, and retroviral vector transduction of FVIII CAR and FVIII TRuC Tregs. Representative plots showing pre-and post-sort purity analysis at the Treg isolation step and after the transduction step. (C) Representative density plots of FVIII CAR Tregs (indicated by mScarlet fluorescent reporter protein) to show surface expression of the Myc tag, detected with α-Myc PE, surface binding of 1 IU/mL FVIIIFc detected with α-human Fc conjugated to Alexa Fluor (AF) 647. (D) In vitro upregulation of Treg-associated activation markers CD69, LAP, GITR, FoxP3, CD49b, Ki67, and CD28 by BDD-FVIII- or FVIIIFc-stimulated murine CAR Tregs at 48 h. Controls include unstimulated cells, cells stimulated with an irrelevant protein, FIXFc, or α-Fc antibody. (E) In vitro proliferation of CTV-labeled FVIII CAR Tregs, 72 h post-stimulation with BDD-FVIII, FVIIIFc, FIXFc, or no stimulation. Proliferation assay was carried out in the absence of exogenous IL-2. Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by 1-way ANOVA test with Dunnett’s multiple comparisons test relative to control (Ctrl; unstimulated) for (D).
Detection of the c-Myc epitope tag on transduced Tregs confirmed surface scFv expression. Binding of Fc-conjugated B domain-deleted (BDD)-FVIII (FVIIIFc) was highly sensitive (Figures 1C and S1). Transduced murine FVIII CAR Tregs upregulated Treg-associated activation markers CD69, latency associated peptide (LAP), glucocorticoid-induced TNFR-related protein (GITR), FoxP3, Ki67, and surface CD28 upon stimulation with BDD-FVIII or FVIIIFc for 48 h (Figure 1D). In vitro-stimulated CellTrace Violet (CTV)-labeled CAR Tregs proliferated in an antigen-specific manner (division index 3.20 ± 0.007; Figure 1E), although proliferation was limited in the absence of exogenous interleukin (IL)-2 in the culture media. Lack of activation/proliferation in unstimulated cells or upon stimulation with an irrelevant antigen, FIXFc, suggested that tonic signaling in the absence of cognate ligand did not occur.
ITAM mutations in FVIII CAR Tregs increase persistence
In vitro stimulation of FVIII CAR Tconvs with either BDD-FVIII or FVIIIFc resulted in a significant loss of viability (Figure 2A). We hypothesized that the additive signaling effect contributed by all three pairs of CD3ζ ITAMS might be responsible for activation-induced cell death (AICD) in these transduced cells.23, 24, 25 We therefore mutated either the proximal (ITAM1−) or distal (ITAM3−) tyrosine residues in CD3ζ, as indicated in the schematic in Figure 1A. Mutating either ITAM1− or ITAM3− in order to disrupt the extent of CD3ζ signaling significantly prevented AICD in BDD-FVIII-stimulated FVIII CAR Tconv (Figure 2A). On the other hand, mutating both the proximal and distal residues (ITAM1−3−) completely abrogated CAR signaling and had no effect on viability (Figure 2A). Interestingly, FVIII-stimulated wild-type (WT) CAR Tregs did not develop AICD or any associated cytotoxicity (Figure 2B), indicating comparative resistance to apoptosis. Single ITAM mutations did not markedly impede CAR Treg function, as expression of activation markers, CD69 and Ki67, did not differ significantly among FVIII-stimulated WT or ITAM1−- or ITAM3−-mutated CAR Tregs (Figures S2A−S2C). Following adoptive transfer and subsequent BDD-FVIII administration, all three CAR Treg variants had similar division indices in vivo (WT 7.5 ± 0.8, ITAM1− 7.3 ± 0.4, ITAM3− 8.1 ± 2.2; Figure 2C). ITAM1− CAR Tregs exhibited increased persistence in spleens of recipient BALB/c mice with a deletion in exon 16 of the F8 gene (F8e16−/−) as compared to WT CAR Tregs (0.30% ± 0.1% versus 0.12% ± 0.09%/splenic CD4+ T cells, day 3 post-adoptive transfer; Figure 2D). We incorporated the ITAM1− mutation to all further modifications of the FVIII CAR construct.
Figure 2.

ITAM1− mutation on CD3ζ increases CAR Treg persistence in vivo but does not improve suppressive function
(A) BDD-FVIII or FVIIIFc stimulation of WT, ITAM1−, and ITAM3− but not ITAM1−ITAM3− FVIII-transduced CAR Tconvs leads to cell death of activated cells, whereas stimulation with FIXFc does not affect viability in vitro. Viability is normalized to 100% using unstimulated controls. (B) BDD-FVIII or FVIIIFc stimulation of WT, ITAM1−, or ITAM3− FVIII CAR Tregs does not affect viability in vitro. Viability is normalized to 100% using unstimulated control Tregs. (C) Proliferation fit statistics indicating division index of CTV-labeled, adoptively transferred WT, ITAM1−, or ITAM3− FVIII CAR Tregs on day 3. A representative proliferation histogram for ITAM1− FVIII CAR Tregs is indicated. (D) Flow cytometric detection of CTV-labeled, adoptively transferred (1 × 106 cells/mouse) WT, ITAM1−, or ITAM3− FVIII CAR Tregs from splenocytes of recipient animals on days 3 and 8. Recipient mice were intravenously injected with 1.5 IU of BDD-FVIII 1 day following adoptive transfer (n = 4). Frequencies of adoptively transferred FVIII CAR Tregs in splenic CD4+ T cells are indicated. (E) Timeline for investigating in vivo prevention of inhibitor formation by CAR Treg cellular therapy. 1 × 106 freshly isolated tTregs, 5 × 105 WT, ITAM1−, ITAM3− FVIII CAR Tregs, or WT FVIII CAR Tregs expanded in the presence of rapamycin were adoptively transferred into BALB/c F8e16−/− recipient mice (n = 6−10/group). Mice received 4 weekly i.v. injections of 1.5 IU BDD-FVIII before functional inhibitors were quantified by (F) Bethesda assay and (G) α-FVIII IgG1 ELISA. Control mice received only BDD-FVIII injections without cell transfer. Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by 1-way ANOVA test with Tukey’s multiple comparisons (A) or Dunnett’s comparisons (F and G). Multiple unpaired t tests (D).
The suppressive capacity of WT, ITAM1−, and ITAM3− FVIII CAR Tregs was then assessed in vivo. Naive BALB/c F8e16−/− recipient mice were infused with 5 × 105 sorted FVIII CAR Tregs, followed by 4 weekly intravenous (i.v.) injections of 1.5 international units (IU) BDD-FVIII (Figure 2E). To our surprise, adoptively transferred FVIII CAR Tregs were found to be immune stimulatory, escalating the formation of inhibitors in recipient animals (Figure 2F). Mice that received CAR Treg therapy developed high titer inhibitors (55.5 ± 6.4 BU/mL), as compared to controls that only received FVIII injections (7.3 ± 1.0 Bethesda unit [BU]/mL). In contrast, freshly isolated polyclonal thymic Tregs (tTregs; 1 × 106 tTregs) were suppressive (2.4 ± 1.7 BU/mL). αFVIII immunoglobulin G1 (IgG1) levels corroborated these findings (Figure 2G). Ex vivo expansion of CAR Tregs in the presence of rapamycin26 did not restore the suppressive function in FVIII CAR Tregs, although it was able to prevent inhibitor escalation to some extent (14.9 ± 5.4 BU/mL), indicating that signaling pathways downstream of CD3ζ such as mTOR might regulate CAR signaling effects.
Dose-dependent dysregulation of signaling
We evaluated cytokine secretion and transcription factor co-expression by BDD-FVIII-stimulated CAR Tregs. Activated WT CAR Tregs produced high levels of IL-10, IL-4, and interferon (IFN)-γ, comparable to FVIII CAR Tconvs, and low levels of IL-2 and IL-17 at 48 h (Figure 3A). Intracellular staining confirmed a heterogenous cytokine profile in the transduced Treg population, with predominant expression of IFN-γ in the FoxP3+ population (Figures 3B and S3). This heterogenous profile was also observed at the transcription factor level with upregulation of IRF4, T-bet, and GATA3, typically associated with either T helper (Th)1 or Th2 response, as quantified by both flow cytometry and real-time RT-PCR (Figures 3C, 4A, and 4B).
Figure 3.

Altered cytokine, transcription factor expression, signaling, and in vitro suppression of FVIII CAR Tregs
(A) Detection of IL-2, IL-10, IL-4, IL-17, IL-35, and IFN-γ from supernatants of sorted and BDD-FVIII-stimulated FVIII CAR Tconvs or Tregs at 48 h in vitro. Cells were cultured in the absence of exogenous IL-2. (B) Intracellular cytokine staining of cell-sorted FVIII CAR Tregs stimulated with high-dose (5 IU/mL) or low-dose (0.1 IU/mL) BDD-FVIII or left unstimulated (Ctrl) for 36 h in vitro. (C) Flow cytometric analysis for transcription factors: IRF4, T-bet, GATA3, and RORγt in cell-sorted and BDD-FVIII-stimulated FVIII CAR Tregs at 48 h in vitro. Unstimulated (Ctrl) or BDD-FVIII- or FIX-stimulated groups are indicated. (D) Normalized in vitro suppression of CTV-labeled FVIII CAR Tconv proliferation when co-cultured with FVIII CAR Tregs at the indicated Treg:Tconv ratios. Cells were stimulated with high-dose (5 IU/mL) or low-dose (0.1 IU/mL) BDD-FVIII or left unstimulated for 72 h in vitro. Percentage suppression calculated as ([mean proliferation Tconv − mean proliferation Treg + Tconv]/[mean proliferation Tconv]) × 100%. (E) Phospho-flow cytometry for pAKT (T308), pAKT (S473), pERK, and pS6 at indicated times following stimulation with low-dose (0.1 IU/mL) or high-dose (5 IU/mL) BDD-FVIII or 2:1 ratio of α-CD3/28 microbeads:CAR Tregs. Representative histogram plots for the same (filled in histograms represent 0 min stimulation). Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by multiple t tests for (A) and 1-way ANOVA with Tukey’s multiple comparisons for (B) and (C). 2-way ANOVA test with Tukey’s multiple comparisons for (D) and (E).
We sought to understand the basis for this dysregulation by first looking at the effect of antigen dose. We speculate that tTregs might exhibit a high affinity for self-antigen at antigen doses that might be too low for Tconv stimulation.27 Whether high antigen dose can, in turn, destabilize Treg-suppressive function is not known.28 We performed an in vitro suppression assay to determine if FVIII CAR Tregs could suppress the proliferation of FVIII CAR Tconv responders when stimulated with high-dose (5 IU/mL) or low-dose (0.1 IU/mL) BDD-FVIII. Low-dose BDD-FVIII-stimulated FVIII CAR Tregs were able to suppress the proliferation of FVIII CAR Tconvs even at low Treg:Tconv ratios (Figure 3D). This suppressive effect was lost on stimulation with high-dose BDD-FVIII. Non-specific suppression was observed at a high Treg:Tconv ratio, which could be attributed to competition for antigen and IL-2. We then performed phospho-flow analysis of signaling molecules downstream of TCR/CD3ζ, which are amplified by CD28 engagement, such as the PI3K-PDK1-AKT and the MAPK/ERK pathways.29,30 We observed greatly enhanced phosphorylation of AKT (S473) and S6 kinases at 30−60 min, with a rapid transient response time for ERK at 10 min in high-dose, BDD-FVIII-stimulated FVIII CAR Tregs (Figure 3E). In contrast, dampened phosphorylation of AKT (T308), pAKT (S473), pS6, and pERK was seen in transduced Tregs on TCR triggering (anti-CD3/28 microbeads) or low-dose BDD-FVIII CAR stimulation. FVIII CAR Tconvs triggered with CD3/28 microbeads were used as a positive control for pS6, pERK, and pAKT (S473), which confirmed that TCR signaling in Tconvs was much more robust as compared to Tregs (Figure S5A).
Mutations in CD28 signaling motifs impact the FVIII CAR Treg cytokine profile
Cytokine signaling is responsive to signals emanating from both the TCR and the costimulatory receptor. Since CD28 is known to increase the rate of pCD3ζ, potentiate TCR signaling, and increase effector cytokine production,31 we generated targeted mutations in the CD28 signaling motifs, YMNM or PYAP, known to bind PI3K and LCK, respectively (Figure 4A). CD28-Y170F or CD28-AYAA substitution mutations did not negatively affect the upregulation of activation markers CD69, CD28, Ki67, or CTLA-4 in response to BDD-FVIII stimulation in vitro (Figure 4B). Notably, both the CD28-Y170F and CD28-AYAA mutations significantly reduced production of IFN-γ and IL-4 in BDD-FVIII-stimulated CAR Tregs, although this was also accompanied by diminished IL-10 production (Figure 4C). In vivo, however, CD28-Y170F and CD28-AYAA FVIII CAR Tregs were still unable to suppress inhibitor formation (4.25 ± 2.15, 9.1 ± 1.0, and 2.87 ± 1.58 BU/mL for control, CD28-Y170F, and CD28-AYAA groups, respectively; Figure 4D), although we did not observe the high inhibitor escalation seen in Figures 2F and 2G.
Figure 4.

Targeted mutations in the CAR CD28 signaling domain do not confer suppressive ability
(A) Schematic representation of FVIII CAR constructs with targeted Y170F or AYAA mutations in the CD28 PI3K- and LCK-binding sites, respectively. (B) Y170F- or AYAA-targeted mutations in the CD28 signaling chain of the FVIII CAR construct do not adversely affect upregulation of CD69, CD28, Ki67, or CTLA-4 in transduced Tregs. MFIs for WT FVIII CAR Tregs compared to Y170F or AYAA variants at 48 h in vitro (WT, top panel; CD28-Y170F, middle panel; CD28-AYAA, bottom panel). Cells are either left unstimulated (Ctrl), stimulated with 5 IU/mL BDD-FVIII, or stimulated with an irrelevant protein, FIX, as indicated. (C) Y170F- or AYAA-targeted mutations in the CD28 signaling chain of the FVIII CAR construct downregulate the production of multiple cytokines, including IL-2, IL-10, IL-4, IL-17, and IFN-γ. WT, Y170F, or AYAA variants of FVIII CAR Tregs were stimulated with 5 IU/mL BDD-FVIII or left unstimulated, and cytokines were assayed after 48 h in vitro. (D) 5 × 105 WT-, Y170F-, or AYAA-mutated FVIII CAR Tregs were adoptively transferred into BALB/c F8e16−/− recipient mice (n = 4−10/group). Mice received 4 weekly i.v. injections of 1.5 IU BDD-FVIII before functional inhibitors were assayed by the Bethesda assay. Control mice received only BDD-FVIII injections without cell transfer. Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by 1-way ANOVA test with Dunnett’s multiple comparisons for (B) and (D); 2-way ANOVA with Dunnett’s multiple comparisons for (C).
Constitutive IL-10 expression does not restore CAR Treg function
As a second strategy, we incorporated the murine IL-10 coding sequence downstream of the auto-cleaving P2A peptide sequence in WT, CD28-Y170F, or CD28-AYAA knock-in variants of FVIII CAR (Figure 5A). IL-10 is an important modulator in Tregs that is known to regulate the production of both Th1 and Th2 cytokines.32,33 IL-10 coding CARs constitutively produced IL-10 (1,617−2,260 pg/mL), which increased 1.3- to 2.1-fold on in vitro BDD-FVIII stimulation (Figure 5B). IFN-γ, IL-2, IL-17, IL-4, and tumor necrosis factor (TNF)-α levels were either completely abrogated or significantly diminished in IL-10-overexpressing WT, CD28-Y170F, or CD28-AYAA CAR Tregs (Figure 5B). Notably, IL-10 overexpression did not affect the ability of the CAR Tregs to proliferate in response to BDD-FVIII stimulation in vitro (Figure 5C). However, constitutive overexpression of IL-10 in adoptively transferred FVIII CAR Tregs or combined with targeted mutations in CD28 was unable to tolerize recipient BALB/c F8e16−/− mice (4.5 ± 1.5, 41.3 ± 9.2, 36 ± 12.3, and 29.1 ± 8.17 BU/mL for control, WT-IL-10, CD28-AYAA-IL-10, and CD28-Y170F-IL-10 cohorts, respectively; Figures 5D and 5E).
Figure 5.

IL-10-overexpressing FVIII CAR Tregs are not suppressive
(A) Schematic showing incorporation of the murine IL-10 sequence into WT, Y170F, or AYAA FVIII CAR constructs, separated by a P2A self-cleaving peptide sequence. (B) Effect of constitutive IL-10 expression on production of cytokines: IL-10, IL-4, IL-17, IFN-γ, IL-2, and TNF-α. Cell-sorted WT, WT-IL-10, CD28 AYAA-IL-10, or CD28 Y170F-IL-10 FVIII CAR Tregs were either left unstimulated (Ctrl) or stimulated with BDD-FVIII or FIX. Cell supernatants were estimated for cytokine production at 48 h in vitro. (C) CD28 AYAA-IL-10 or CD28 Y170F-IL-10 mutations do not adversely affect proliferation of activated FVIII CAR Tregs. In vitro proliferation of CTV-labeled WT-IL-10, CD28 AYAA-IL-10, or CD28 Y170F-IL-10 FVIII CAR Tregs, 72 h post-stimulation with low-dose (0.1 IU/mL) or high-dose (5 IU/mL) BDD-FVIII or no stimulation (Ctrl). Representative histogram plots to compare proliferation and graphical representation of division indices are shown. (D) 5 × 105 WT-IL-10, CD28 AYAA-IL-10, or CD28 Y170F-IL-10 FVIII CAR Tregs were adoptively transferred into BALB/c F8e16−/− recipient mice (n = 6−10/group). Mice received 4 weekly i.v. injections of 1.5 IU BDD-FVIII before functional inhibitors were assayed by the Bethesda assay. Control mice received only BDD-FVIII injections without cell transfer. (E) αFVIII IgG1 ELISA for the same. Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001 by 2-way ANOVA with Sidak’s comparisons for (B), 1-way ANOVA test with Tukey’s multiple comparisons for (C), and 1-way ANOVA test with Dunnett’s comparison for (D) and (E).
Integration of TRuC into the TCR-CD3 complex regulates surface expression
A recent report showed that fusing anti-CD19 scFv to the N termini of any of the five TCR subunits results in incorporation of TRuCs into the TCR-CD3 complex.20 This approach significantly improved tumor cell lysis as compared to high-affinity CD19-CAR T cells, which correlated with differences in intracellular signaling events between the two constructs. We fused FVIII scFv to murine CD3ε in order to generate FVIII TRuC Tregs (Figure 6A). FVIII TRuC was surface expressed in transduced Tregs and bound FVIIIFc in vitro (Figure 6B). However, we observed under conditions of comparable reporter protein expression (mScarlet median fluorescence intensity [MFI]) that surface scFv expression, detected by the Fab antibody, was markedly lower for FVIII TRuC as compared to FVIII CAR (p = 0.0002; Figure 6C). This indicates that TRuC surface expression is limited by the number of TCR-CD3 complexes on the transduced Treg. In order to confirm FVIII TRuC integration into the TCR-CD3 complex, we co-transduced the murine T cell hybridoma 5KCα−/β−, which are TCR-α/β deficient, with FVIII TRuC or FVIII CAR and a murine TCR-α/β-expressing construct. We observed that FVIII CAR was surface expressed, bound FVIIIFc, and upregulated the activation marker CD69 independent of TCR-α/β expression (Figure 6D). In contrast, FVIIIFc binding and CD69 upregulation in FVIII TRuC-expressing 5KCα−/β− cells were dependent on co-transduction with TCR-α/β, which confirms that TRuC integrates into the TCR-CD3 complex (Figure 6D). Next, we tested the requirement for both TCR-α/β and CD3 for TRuC incorporation. HEK293 cells were transfected with FVIII CAR or TRuC and either a murine TCR-α/β- or CD3δγεζ-expressing construct or both. Whereas FVIIIFc binding by FVIII CAR was independent of TCR-α/β or the CD3 complex, FVIII TRuC surface expression and FVIIIFc binding depended on co-transfection of both TCR-α/β and CD3 (Figure S6).34,35
Figure 6.

The TCR-CD3 complex regulates TRuC surface expression
(A) Surface organization and schematic representation of the FVIII TRuC construct. The VL and VH regions of the FVIII-specific scFv, linker, extracellular, transmembrane, and intracellular signaling regions of murine CD3ε domains are indicated. (B) Representative density and histogram plot of TRuC-transduced Tregs (indicated by mScarlet reporter protein) to show binding of 1 IU/mL FVIIIFc and detection with α-human IgG Fc conjugated to AF647. (C) Comparison of surface scFv expression between CAR and TRuC Tregs at comparable transduction levels (indicated by mScarlet MFI) by α-human F(ab′)2 binding and detection with α-goat AF647. (D) Surface expression of FVIII TRuC is dependent on incorporation into the TCR-CD3 complex. Single or co-transduction of FVIII CAR or TRuC and TCR-α/β into the TCR-α/β-deficient murine T cell line 5KCα−/β−. Incubation with 1 IU FVIIIFc and detection of frequencies of mScarlet+ cells that bind FVIIIFc by αFc conjugated to AF647 indicates dependence of TRuC but not CAR surface expression and CD69 upregulation on co-transduction with TCR-α/β. Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001 by unpaired t test for (C).
TRuC Tregs exhibit controlled signaling
BDD-FVIII stimulation of FVIII TRuC Tregs in vitro led to upregulation of CD69, Ki67, CD28, and FoxP3 and a 5-fold increase in CTLA-4 expression (Figure 7A). In contrast to FVIII CAR Tregs, signaling molecules pAKT (S473), pERK, and pS6 were considerably dampened in TRuC Tregs, similar to levels observed on TCR triggering with anti-CD3/28 microbeads (Figure 7B). This was also confirmed by western blot for pERK and pS6 (Figure 7C). FVIII TRuC Tconvs triggered with CD3/28 microbeads were used as a positive control for pS6, pERK, and pAKT (S473), which confirmed that Tconv signaling was much more robust as compared to Tregs (Figure S5B).
Figure 7.

TRuC Tregs exhibit controlled signaling in vitro
(A) Upregulation of Treg-associated activation markers CD69, Ki67, CD28, CTLA-4, and FoxP3 by BDD-FVIII-stimulated murine TRuC Tregs at 48 h in vitro. Controls include unstimulated cells or stimulation with an irrelevant protein, FIX. (B) Comparison of pAKT (S473), pERK, or pS6 at indicated times following stimulation with low-dose (0.1 IU/mL) or high-dose (5 IU/mL) BDD-FVIII or TCR triggering with α-CD3/28 microbeads by flow cytometry for WT CAR (solid line) or TRuC (dotted line) Tregs. (C) Comparison of western blot analysis for pERK and pS6 at indicated times following stimulation with high-dose (5 IU/mL, blue line) or low-dose (0.1 IU/mL, pink line) BDD-FVIII. Densitometric analysis for pERK and pS6 for WT CAR Treg (solid line) or TRuC Treg (dotted line) is indicated. (D) Detection of IL-2, IL-10, IL-4, IL-17, and IFN-γ cytokines from BDD-FVIII-stimulated TRuC and CAR Treg cell supernatants at 48 h in vitro. (E) Normalized in vitro suppression of CTV-labeled FVIII TRuC Tconv proliferation when co-cultured with FVIII TRuC Tregs at the indicated Treg:Tconv ratios. Cells were stimulated with high-dose (5 IU/mL) or low-dose (0.1 IU/mL) BDD-FVIII or left unstimulated for 72 h in vitro. Percentage suppression calculated as ([mean proliferation Tconv − mean proliferation Treg + Tconv]/[mean proliferation Tconv]) × 100%. Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001 by 1-way ANOVA with Dunnett’s comparisons for (A), multiple unpaired t tests for (B), 2-way ANOVA test with Tukey’s multiple comparison for (C) and (D), and 2-way ANOVA with Sidak’s multiple comparisons for (E).
Consistent with dampened signaling, BDD-FVIII-stimulated TRuC Tregs secreted significantly lower levels of cytokines IL-2, IL-4, IL-17, IL-10, and IFN-γ as compared to WT CAR Tregs (Figure 7D). This was also confirmed by intracellular cytokine staining (Figure S7A), which showed low but significantly elevated frequencies of IL-10 in BDD-FVIII-stimulated TRuC Tregs (Figure S7B) and insignificant IFN-γ expression. Similar to FVIII CAR Tregs, FVIII TRuC Tregs were able to suppress the in vitro proliferation of TRuC Tconv responders when stimulated with low-dose (0.1 IU/mL) BDD-FVIII (Figure 7E). This suppressive effect was lost on stimulation with high-dose BDD-FVIII. We also tested to see if engineered Tregs induce cytolysis by evaluating granzyme B and CD107a upregulation. Neither CAR nor TRuC Treg upregulated cytolytic markers in response to BDD-FVIII or CD3/28 stimulation, whereas CAR and TRuC Tconvs exhibited a substantial increase in both granzyme B and CD107a expression on CD3/28 stimulation and a less-significant increase on BDD-FVIII stimulation (Figures S8A and S8B).
TRuC Tregs are suppressive in vivo but have limited persistence
We investigated whether controlled signaling by TRuC Tregs was sufficient to maintain a suppressive phenotype in vitro and in vivo. In vivo, naive BALB/c F8e16−/− recipient mice were infused with 5 × 105-sorted TRuC Tregs or polyclonal tTregs (5 × 105 or 1 × 106) followed by 8 weekly i.v. injections of 1.5 IU BDD-FVIII (Figure 8A). FVIII TRuC Tregs were more effective at suppressing inhibitor formation as compared to polyclonal tTregs (Figures 8B and 8C). 7 out of 8 animals in the TRuC Treg group did not develop detectable inhibitors (average [avg] BU/mL 0.23 ± 0.23) at 4 weeks, whereas 100% of mice in the control group developed high-titer inhibitors >5 BU/mL (avg BU/mL 36.48 ± 9.66, p = 0.02). At 8 weeks, mice in the FVIII TRuC Treg group had a mean inhibitor titer of 15.4 ± 10.4 BU/mL as compared to the control group (avg BU/mL 151.4 ± 48.6, p = 0.004; Figure 8B). Anti-FVIII IgG1 titers were also significantly lower in the FVIII TRuC Treg group (5,238 ± 3,862 ng/mL) as compared to the polyclonal tTreg groups at 8 weeks (5 × 105 tTreg group: 28,429 ± 3,862 ng/mL, p = 0.004, 1 × 106 tTreg group: 2,1821 ± 8,020 ng/mL, p = 0.04), suggesting a more sustained tolerogenic effect for FVIII TRuC Tregs (Figure 8C). Although adoptively transferred Tregs persisted only transiently (7−14 days), we observed that BDD-FVIII stimulation was required for increased persistence in vivo. Repeated BDD-FVIII injections led to 1.75-fold (p = 0.0001) and 1.17-fold (p = 0.036) higher numbers of CAR Tregs and TRuC Tregs, respectively, on day 3 (Figure 8D).
Figure 8.

TRuC Tregs maintain a suppressive phenotype in vivo
(A) Timeline for assessing in vivo prevention of inhibitor formation by TRuC Treg cellular therapy. 5 × 105 TRuC Treg and 5 × 105 or 1 × 106 freshly isolated tTregs were adoptively transferred into BALB/c F8e16−/− recipient mice (n = 5−8/group). Mice received 8 weekly i.v. injections of 1.5 IU BDD-FVIII, and plasma samples were analyzed after the 4th and 8th injection for (B) functional inhibitors by Bethesda assay and (C) α-FVIII IgG1 ELISA. Control mice received only BDD-FVIII injections without Treg transfer. (D) In vivo persistence of 1 × 106 adoptively transferred FVIII CAR or TRuC Tregs following 3×/week BDD-FVIII (+FVIII) or mock (−FVIII) injections (n = 3/group). Total number of adoptively transferred (mScarlet+ FoxP3GFP+), engineered Tregs per 1 × 106 splenic CD4+ T cells is indicated on days 3, 7, and 14 post-adoptive transfer. (E) Effect of repeated in vitro BDD-FVIII stimulations on the phenotype of FVIII CAR or TRuC Tregs. Expression of PD1, CD69, LAP, CTLA-4, and FoxP3 and frequencies of FoxP3+ cells were estimated at each time point following 4 daily stimulations with BDD-FVIII. Controls were either mock stimulated or stimulated with an irrelevant antigen, FIX. Solid lines represent CAR Tregs, and dotted lines represent TRuC Tregs. (F) In vivo Treg stability of 1 × 106 FVIII CAR or TRuC Tregs assessed pre- and post-adoptive transfer in BALB/c F8e16−/− recipient mice (n = 6/group). One-half of the recipient animals received 2 daily BDD-FVIII injections. Representative density plots and bar graph of mScarlet and FoxP3GFP+ co-expression following BDD-FVIII (+FVIII) or mock (−FVIII) injection are indicated on day 3. (F) Data points are averages ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001 by 2-way ANOVA with Sidak’s multiple comparisons for (B) and (C) and 2-way ANOVA with Tukey’s multiple comparisons for (D)−(F).
BDD-FVIII-stimulated CAR and TRuC Tregs maintain a Treg phenotype
We next asked whether BDD-FVIII stimulation would affect CAR and TRuC Treg stability. To address this, we performed repeated (daily) stimulations of CAR Treg and TRuC Treg with BDD-FVIII in vitro and evaluated their phenotype after each stimulation. FoxP3 frequencies were unchanged in both BDD-FVIII-stimulated CAR and TRuC Tregs (Figure 8E). PD1, CD69, LAP, CD69, and FoxP3 MFIs were significantly elevated in BDD-FVIII-stimulated CAR and TRuC Tregs, as compared to unstimulated controls or FIX-stimulated controls (Figure 8E).
Finally, we analyzed the phenotype of sorted mScarlet+FoxP3GFP+ cells prior to and post-adoptive transfer into BALB/c F8e16−/− mice. Pre-adoptive transfer, mScarlet+FoxP3GFP+ cells were 92%–95% FoxP3+ with a good correlation between FoxP3 and GFP (Figure 8F). Cohorts of recipient mice were either left untreated or injected with BDD-FVIII every 2 days, starting 1 day post-adoptive transfer. On day 3 post adoptive transfer, splenic CD4+ T cells were magnetically enriched, and frequencies of mScarlet+FoxP3GFP+ cells were evaluated. mScarlet+FoxP3GFP+ CAR Treg and TRuC Treg both retained GFP expression, which correlated with FoxP3 expression (CAR Treg 93.18% ± 0.36%, TRuC Treg 92.75% ± 0.3% mScarlet+FoxP3GFP+ cells). Treg phenotype was not affected by BDD-FVIII stimulation in vivo (CAR Treg 93.71% ± 0.7%, TRuC Treg 90.54% ± 0.3% mScarlet+FoxP3GFP+ cells) (Figure 8F). Overall, these results demonstrate that CAR- and TRuC-mediated signaling preserves the regulatory phenotype of engineered Tregs.
Discussion
Dysregulation of Treg signaling contributes to pathogenesis of many autoimmune conditions and highlights safety concerns in clinical Treg therapy.36 Cell-intrinsic molecular events responsible for this altered Treg function are not completely understood. Here, we redirected Treg specificity to clotting FVIII using two different approaches: expression of CAR or TRuC. We observed that CAR signaling can overstimulate signaling molecules and lead to a pro-inflammatory profile in transduced Tregs. We sought to understand the basis for this dysregulated profile by focusing on antigen or receptor dose/density and cytokine profile.
The magnitude of CAR signaling is dependent on scFv affinity, receptor density, antigen dose, choice of co-stimulatory molecule, and cytokine signals, among other factors. It is known that CAR scFv can bind to antigen with up to 1,000-fold higher affinity than TCRs, although the effect of affinity on signaling and functionality is not well studied.37 It can be agreed that CAR signaling in both Tconv and Treg is more rapid and intense compared to TCR stimulation.38,39 This can result in over-activation and apoptosis of CAR Tconvs affecting in vivo persistence.40,41 Over-activation and potentially fatal cytokine release syndrome are important safety concerns following administration of CAR T cell therapies for cancer.42 Approaches for tapering the magnitude of CAR signaling, such as introducing targeted mutations in CD3ζ ITAMs, have been carried out previously, with some success,43, 44, 45 although this strategy has not been tested for CAR Tregs. A decrease in the number of ITAM pairs from three to two or even one in the CAR construct can increase selectivity and prevent off-target effects by increasing the activation threshold.45,46 Our studies show that although Tregs are inherently more resistant to AICD, introducing the ITAM1− mutation in CD3ζ (CD247) improved CAR Treg persistence in vivo. Interestingly, tTregs are reported to preferentially express the alternatively spliced θ isoform of CD247, which naturally lacks the ITAM3 domain, rather than the CD3ζ isoform.47
The TCR repertoire in Tregs is mostly distinct from that of Tconvs, with an increased tendency toward self-specificity.48 Signals elicited by the TCR are greatly dampened in Tregs.49 This regulation is evident in both primary signal initiation and co-stimulatory signal potentiation, such that several signaling molecules like CD3ζ, SLP76, MAPK/ERK, AKT, or S6 and calcium flux are attenuated in Tregs.49 In particular, Tregs are shown to be defective in pAKT at S473, thus displaying reduced pAKT substrates.50 We confirmed a similar pattern of dampened signaling of these pathways upon TCR triggering of CAR-transduced Tregs, which was distinct from CAR Tconv signaling. Conversely, CAR stimulation of these same cells resulted in increased phosphorylation of many of these signaling mediators, including strong pAKT S473, affirming that CAR signaling differs from endogenous Treg signaling at multiple signaling nodes.51 We do not yet know the effect that unrestricted CD28z signaling would have on inhibitory signaling motifs such as the inhibitory immunoreceptor tyrosine-based inhibitory motif (ITIM) or the immunoreceptor tyrosine-based switch motif (ITSM) commonly overexpressed in Tregs such as CTLA-4, PD1, and T cell Ig and ITIM domain (TIGIT).52,53 These motifs are responsible for the inhibition of TCR function and are thought to be critical for Treg-immunosuppressive function. For this study, we did not test the 4-1BB co-receptor as its incorporation into the CAR construct has previously been shown to be detrimental to Treg function.54
It has been reported that Tregs can augment proliferation of T cells under strong stimulatory conditions.27 We were able to confirm this, as mice that were infused with FVIII CAR Tregs developed high inhibitor titers. Mutating either the PI3K or LCK binding motifs in the CD28 signaling domain partially controlled the exacerbation of inhibitors in recipient mice. However, this was insufficient to confer suppressive activity, as recipient mice still developed inhibitors in response to BDD-FVIII injections.
CAR Treg stimulation in vitro was accompanied by significant production of IFN-γ, TNF-α, IL-10, and IL-4. Altered cytokine production by CAR Tregs has also been reported in murine models of graft-versus-host disease (GvHD),54,55 where IFN-γ production by CAR Tregs as well as target cell lysis in a granzyme B-dependent manner was observed.56 Given that IL-10 has been shown to block antigen-specific T cell cytokines such as PI3K/AKT-induced IFN-γ by inhibiting the CD28 signaling pathway,57,58 we demonstrated that constitutive expression of murine IL-10 in FVIII CAR Tregs was able to completely abrogate IFN-γ production. However, IL-10-overexpressing FVIII CAR Tregs were unable to suppress the development of inhibitors in recipient mice, and combining IL-10 overexpression with CD28-YMNM or PYAP mutations did not contribute to suppression. Since IL-10 is also reported to promote the germinal center response and IgG class switching,59, 60, 61 additional studies are needed to determine the effect of IL-10 dose, constitutive versus inducible expression, and localized versus systemic IL-10 delivery for optimizing tolerance to FVIII.
An important consideration for CAR Tregs specific to soluble antigens like FVIII is whether contact-dependent mechanisms are essential for suppression, either via direct contact with antigen-bound B cell or dendritic cell or by modulation of antigen-presenting cell (APC) function via co-stimulatory molecule binding and/or trogocytosis.62 A recent report demonstrated transient suppressive activity of human CAR Tregs specific to the A2 domain of FVIII in a murine HA model, although the use of a xenogeneic system made it difficult to fully determine the extent of suppression.63 It is not known whether the affinity of the A2 CAR used in that study was significantly lower to that of the BO2C11 antibody used here, which has a very high affinity of 10−11 M−1.22,64 One notable difference between the two studies is that in vitro suppression was enhanced by the presence of autologous peripheral blood mononuclear cells (PBMCs), indicating a requirement for contact-mediated suppression, most likely with APC. We believe that these two independent studies are not contradictory but rather raise important questions about the role of scFv affinity and requirement for contact-dependent mechanisms of suppression.
We tested an alternative approach to engineer antigen-specific Tregs by tethering FVIII scFv to the CD3ε subunit of the TCR-CD3 complex, which can overcome the limitations of destabilizing effects mediated by rapid and strong CAR signaling. We and others20 observed TRuC to be expressed on the cell surface as a component of the TCR-CD3 complex. In fact, incorporation of TRuC into the TCR-CD3 complex regulated receptor density on the transduced cell surface, likely contributing to modulation of signaling. It is also possible that the TRuC-TCR-CD3 complex is subject to internalization and re-expression following single or repeated exposure to antigen,65 which further protects the transduced cell from chronic activation or exhaustion. Further studies are required to confirm this. A related study targeting a CAR to the TRAC locus was shown to avert tonic signaling in a mouse model of acute lymphoblastic leukemia by a mechanism of CAR internalization and post-stimulation replenishment of cell-surface CAR expression.66 TRuCs employ the entire TCR complex to signal, whereas CARs utilize only the CD3ζ moiety of the complex with limited signaling capacity and lack intrinsic autoregulation, although recent studies indicate that CARs can interact with endogenous TCR molecules.21,67,68 FVIII TRuC Tregs were phenotypically stable and did not express cytolytic markers. Functionally, FVIII TRuC Tregs were immunosuppressive and prevented the formation of inhibitors to FVIII.
In conclusion, we provide evidence that differences in signaling strength and kinetics via CAR and TRuC receptors can strongly affect Treg function. The incorporation of TCR-like signaling to engineered receptors has recently shown promise in multiple solid and liquid tumor models due to more controlled signaling and lower cytokine release.20,69,70 Our data show that the TRuC platform can potentially improve the suppressive capacity of engineered Tregs in anti-drug antibody formation. The design of the next generation of antigen-specific Tregs will most likely emphasize finetuning activation in order to improve effectiveness and persistence, whereas reducing exhaustion and anergy.45,66
Materials and methods
Mice
BALB/c Foxp3IRES-GFP (Foxp3GFP) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). HA BALB/c F8e16−/− mice were originally provided by Dr. David Lillicrap (Queens University, Ontario, Canada). Animals were housed under specific pathogen-free conditions at Indiana University (Indianapolis, IN, USA) and treated under Institutional Animal Care and Use Committee-approved protocols. Both male and female mice were used as Treg donors for in vitro studies. Male mice were used for studies involving adoptive transfer or inhibitor formation.
CAR and TRuC constructs
The FVIII scFv was derived from an Epstein-Barr virus (EBV)-transformed B cell clone obtained from a HA patient (originally developed by Jacquemin and colleagues22, kindly provided to us by Dr. David Scott, Uniformed Services University, Bethesda, MD, USA). This B cell clone (BO2C11) produces IgG4 directed against amino acid residues 2,125−2,332 of human FVIII, which corresponds to the carboxyl-terminus of C1 (residues 2,125−2,172) and the C2 (residues 2,173−2,332) light-chain domains.22 The scFv was constructed from the variable heavy (VH) and light (VL) sequences (Creative Biolabs, Shirley, NJ, USA) and fused to second-generation murine 28z CAR signaling sequences (kind gift from Dr. Angelica Loskog, Uppsala University, Sweden). Hinge regions from either murine CD28 or CD8 were incorporated with no observed differences in signaling. A Myc tag was cloned into the original construct (GenScript, Piscataway, NJ, USA). Single amino-acid mutations in ITAMs 1, 3, or 1 + 3 of CD3ζ or in the CD28 signaling domains were introduced by site-directed mutagenesis (GenScript, Piscataway, NJ, USA). Murine IL-10 was cloned downstream of the CAR sequence, separated by a P2A cleavage sequence (GenScript, Piscataway, NJ, USA). FVIII-specific TRuC was generated by complexing the BO2C11 FVIII scFv sequence to the N terminus of murine CD3ε by a flexible linker (G4S)X3 (GenScript, Piscataway, NJ, USA).
Retroviral transduction
FVIII CAR and TRuC sequences were inserted into the pMYs-IRES-mScarlet retroviral backbone. Transfer plasmids were transfected into the PlatE ecotropic retroviral packaging cell line (Cell Biolabs, San Diego, CA, USA) using either Viafect (Promega, Madison, WI, USA) or polyethylenimine (PEI) transfection reagents, and supernatants were collected after 48 h. CD4+CD25− Tconv or CD4+CD25+ Tregs from BALB/c Foxp3GFP mice were magnetically enriched using a mouse CD4+CD25+ Treg isolation kit (Miltenyi Biotec, Auburn, CA, USA), further purified by cell sorting (FACSAria II or FACSAria SORP, BD Biosciences) and pre-activated for 48 h with a 1:1 bead-to-cell ratio using anti-CD3/28 mouse microbeads (Dynabeads; Invitrogen). High purity was ensured by a “four-way purity” sort followed by post-sort flow analysis (99.5% ± 0.3%). Tregs were cultured in Biotarget serum-free media (Biological Industries, Cromwell, CT, USA) supplemented with 5% fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA, USA), 10,000 IU/mL penicillin, 10 mg/mL streptomycin, 1× GlutaMAX-1, 1 mM sodium pyruvate, 10 mM HEPES, 1× nonessential amino acids, and 10 μM 2-mercaptoethanol. Clinical-grade recombinant hIL-2 (Proleukin/aldesleukin; Prometheus Therapeutics and Diagnostics, San Diego, CA, USA) was added at a final concentration of 1,000 IU/mL. Cells were transduced by spinoculation with retrovirus containing supernatants at 1,200 × g for 90 min in non-tissue culture-treated 6-well plates coated with 20 μg/mL retronectin (Takara Bio, Middleton, WI, USA). Transduced cells were further purified by sorting for FoxP3GFP+mScarlet+ cells and ex vivo expanded for 3−4 days in the presence of anti-CD3/28 microbeads at a 1:1 bead-to-cell ratio. 100 nM rapamycin (LC laboratories, Woburn, MA, USA) was added under some conditions. Cells were rested for 4−6 h prior to functional in vitro or in vivo experiments (Figure 1B).
Flow cytometry
1 × 106 FVIII CAR- or TRuC-transduced Tconvs or Tregs were plated in 12-well plates in Biotarget medium with 5% FBS without IL-2, before stimulation with 0.1, 1, or 5 IU/mL of recombinant human BDD-FVIII (Xyntha; Pfizer, New York, NY, USA), FVIIIFc (Bioverativ, Cambridge, MA, USA), FIXFc (Sanofi Genzyme, Cambridge, MA, USA), and anti-human Fc (5 mg/mL; BioLegend, San Diego, CA, USA) or anti-CD3/28 microbeads (1:1 bead-to-cell ratio). At 24−96 h, cells were first Fc blocked with anti-CD16/32 and then stained using antibodies against CD4 (BV421), GITR (BV510), and RORγt (BV421) from BD Biosciences (San Jose, CA, USA); CD69 (eFluor450), FoxP3 (eFluor660), GATA3 (PE/Cy7), and granzyme B (PerCP-eFluor 710) from eBioscience (San Diego, CA, USA); CD4 (BV421), CTLA-4 (BV421), PD1 (BV605), CD28 (PerCP/Cy5.5), LAP (PE), Ki67 (PE/Cy7), CD69 (PE/Cy7), CD49b (APC/Cy7), GFP (A488), IRF4 (PE), T-bet (BV605), anti-human IgG Fc (purified and AF647 conjugated), IL-10 (BV421), IL-4 (BV711), IL-17 (AF647), IFN-γ (AF700), and CD107a (BV711) from BioLegend (San Diego, CA, USA); and Myc (PE) from R&D Systems (Minneapolis, MN, USA). CD107a staining was performed as described.71 Goat anti-human F(ab′)2 antibody (Invitrogen) and anti-goat AF647 (Jackson ImmunoResearch, West Grove, PA, USA) were used for scFv surface detection. To analyze transcription factor expression, cells were first fixed with 2% paraformaldehyde and permeabilized using the Foxp3/Transcription Factor Staining Buffer (eBioscience, San Diego, CA, USA). Data were analyzed using FCS Express v7 (DeNovo Software, Los Angeles, CA, USA).
Proliferation
For an in vitro proliferation assay, cells were labeled with 3−5 μM CTV (Invitrogen, Carlsbad, CA, USA) prior to stimulation with BDD-FVIII or an irrelevant antigen (FIX; Benefix; Pfizer, New York, NY, USA) and incubated for 72 h at 37°C. CTV dilution in proliferating relative to unstimulated Tregs was quantified via proliferation analysis in FCS Express v.7. For in vivo proliferation and persistence, WT, ITAM1−, ITAM3− FVIII CAR Tregs, or TRuC Tregs were purified by FACS and labeled with 3−5 μM CTV. 1 × 106 Tregs were adoptively transferred into recipient BALB/c F8e16−/− mice (n = 4/group), and 1 day later, mice were i.v. injected with 1.5 IU BDD-FVIII or left untreated. Mice were euthanized on days 3, 7/8, and 14 following adoptive transfer. Spleen CD4+ T cells were magnetically enriched, and CTV+ FoxP3GFP+ mScarlet+ cells were quantified on a BD LSR Fortessa.
In vitro suppression
For in vitro suppression, WT FVIII CAR or FVIII TRuC Tregs were purified by sorting and incubated with 3−5 μM CTV-labeled FVIII CAR Tconv or FVIII TRuC Tconv responder cells, respectively, at varying ratios of Treg:Tconv, whereas keeping Tconv numbers constant. Cells were stimulated with either high-dose (5 IU/mL) or low-dose (0.1 IU/mL) BDD-FVIII and acquired on a BD LSR Fortessa after 72 h at 37°C. Dilution of CTV in proliferating CAR Tconv or TRuC Tconvs was quantified relative to unstimulated cells using proliferation analysis in FCS Express v.7.
Cytokine detection
For intracellular cytokine staining, FVIII CAR- or TRuC-transduced cells were plated in 12-well plates in Biotarget medium with 5% FBS without IL-2 before stimulation with 5 IU/mL of BDD-FVIII, FIX, or anti-CD3/28 microbeads (1:1 bead-to-cell ratio). Following 20−32 h of stimulation, Brefeldin A (3 μg/mL; eBioscience, San Diego, CA, USA) was added for an additional 4 h. Cells were fixed and permeabilized with Cyto-Fast Fix/Perm Buffer (BioLegend), and intracellular cytokine staining was performed for flow cytometry analysis. Additionally, supernatants were collected from stimulated cells at 48 h, and levels of IL-2, IL-4, IL-10, IL-35, IL-17, IL-21, and IFN-γ were quantified by the DuoSet ELISA kits according to the manufacturer’s recommendations (R&D Systems, Minneapolis, MN, USA).
Phospho flow
1 × 106 FVIII CAR or TRuC Tconv or Tregs/well were plated in a 12-well plate in Biotarget medium without FBS or IL-2. Cells were stimulated with BDD-FVIII or anti-CD3/28 microbeads for 0, 10, 30, and 60 min, following which, cells were immediately fixed with 2% paraformaldehyde. Fixed cells were permeabilized with 90% methanol for 30 min, followed by staining for pERK (APC), pS6 (PacBlue), pAKT (S473, PE, APC), and pAKT (T308, APC; Cell Signaling Technology, Danvers, MA, USA), and analyzed by flow cytometry on a BD LSR Fortessa.
Western blot
CAR or TRuC Tregs were stimulated and fixed as described in phospho flow. Fixed cells were lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors (Cell Signaling Technology, Danvers, MA, USA). PAGE-separated lysates were transferred to polyvinylidene fluoride (PVDF) membranes (Transblot Turbo; Bio-Rad, Hercules, CA, USA). Membranes were probed for pS6, pERK, and β-actin (Cell Signaling Technology, Danvers, MA, USA), signal detected on ChemiDoc MP (Bio-Rad, Hercules, CA, USA), and quantified using ImageJ software.
Inhibitor establishment and analysis of plasma samples
BALB/c F8e16−/− HA mice (n = 5−10) received weekly i.v. administrations of 1.5 IU BDD-FVIII. Mice received 5 × 105 FVIII CAR or TRuC Tregs 1 day prior to starting BDD-FVIII injections (Figures 2E and 7B). At 1- and 2-month time points post-adoptive transfer, ∼200 μL blood was collected from the retroorbital plexus using non-treated capillary tubes into 3.8% sodium citrate, and plasma was analyzed for inhibitor formation by the Bethesda assay (measured on a Diagnostica Stago STart Hemostasis Analyzer; Stago, Parsippany, NJ, USA) and anti-FVIII IgG1 ELISA as previously described.72,73 1 BU is defined as the reciprocal of the dilution of test plasma at which 50% of FVIII activity is inhibited.
Statistical analysis
Data shown are mean ± SEM. Statistical significance was determined using the Student’s t test, 1-way or 2-way ANOVA, and multiple comparisons were made using Dunnett’s, Tukey’s, Sidak’s, or Kruskal-Wallis post-tests as indicated, using GraphPad Prism 8 software (GraphPad, La Jolla, CA, USA). Values at p < 0.05 were deemed significant and indicated as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Frequencies of mice that developed inhibitors were compared using Fisher’s exact test.
Acknowledgments
We would like to thank Dr. Amanda Posgai for critical editing of the manuscript. We would like to acknowledge the Flow Cytometry Core Facility at Indiana University Simon Cancer Center for cell-sorting services and the Center for Immunology and Transplantation at the University of Florida. Murine CD3 WTdelta-F2A-gamma-T2A-epsilon-P2A-zeta pMIA II (Addgene plasmid #52093) and murine TCR OTI-2A.pMIG II (Addgene plasmid #52111) were a gift from Dr. Dario Vignali. We would like to thank Dr. Maki Nakayama for the kind gift of the 5KCα−/β− murine T cell line (NIDDK grant P30DK116073). M.B. is in receipt of a career development award from the National Hemophilia Foundation (NHF). T.M.B. is supported by grants from the National Institutes of Health (P01 AI42288 and R01 DK106191) and The Leona M. and Harry B. Helmsley Charitable Trust.
Author contributions
J.R., S.R.P.K., D.J.P., R.S., M.M.-M., and M.B. performed experiments. J.R., S.R.P.K., D.J.P., T.M.B., and M.B. designed experiments and analyzed and interpreted data. T.M.B. and M.B. wrote the manuscript and supervised the study.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.04.034.
Supplemental information
References
- 1.Bluestone J.A., Buckner J.H., Fitch M., Gitelman S.E., Gupta S., Hellerstein M.K., Herold K.C., Lares A., Lee M.R., Li K. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015;7:315ra189. doi: 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dall’Era M., Pauli M.L., Remedios K., Taravati K., Sandova P.M., Putnam A.L., Lares A., Haemel A., Tang Q., Hellerstein M., Autoimmunity Centers of Excellence Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019;71:431–440. doi: 10.1002/art.40737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawitzki B., Harden P.N., Reinke P., Moreau A., Hutchinson J.A., Game D.S., Tang Q., Guinan E.C., Battaglia M., Burlingham W.J. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. 2020;395:1627–1639. doi: 10.1016/S0140-6736(20)30167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Ianni M., Falzetti F., Carotti A., Terenzi A., Castellino F., Bonifacio E., Del Papa B., Zei T., Ostini R.I., Cecchini D. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 5.Noyan F., Zimmermann K., Hardtke-Wolenski M., Knoefel A., Schulde E., Geffers R., Hust M., Huehn J., Galla M., Morgan M. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am. J. Transplant. 2017;17:917–930. doi: 10.1111/ajt.14175. [DOI] [PubMed] [Google Scholar]
- 6.Bluestone J.A., Tang Q. Treg cells-the next frontier of cell therapy. Science. 2018;362:154–155. doi: 10.1126/science.aau2688. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava S., Riddell S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018;200:459–468. doi: 10.4049/jimmunol.1701155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honaker Y., Hubbard N., Xiang Y., Fisher L., Hagin D., Sommer K., Song Y., Yang S.J., Lopez C., Tappen T. Gene editing to induce FOXP3 expression in human CD4+ T cells leads to a stable regulatory phenotype and function. Sci. Transl. Med. 2020;12:eaay6422. doi: 10.1126/scitranslmed.aay6422. [DOI] [PubMed] [Google Scholar]
- 9.Allan S.E., Alstad A.N., Merindol N., Crellin N.K., Amendola M., Bacchetta R., Naldini L., Roncarolo M.G., Soudeyns H., Levings M.K. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol. Ther. 2008;16:194–202. doi: 10.1038/sj.mt.6300341. [DOI] [PubMed] [Google Scholar]
- 10.Fu R.Y., Chen A.C., Lyle M.J., Chen C.Y., Liu C.L., Miao C.H. CD4+ T cells engineered with FVIII-CAR and murine Foxp3 suppress anti-factor VIII immune responses in hemophilia a mice. Cell. Immunol. 2020;358:104216. doi: 10.1016/j.cellimm.2020.104216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blat D., Zigmond E., Alteber Z., Waks T., Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. 2014;22:1018–1028. doi: 10.1038/mt.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fransson M., Piras E., Burman J., Nilsson B., Essand M., Lu B., Harris R.A., Magnusson P.U., Brittebo E., Loskog A.S. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation. 2012;9:112. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacDonald K.G., Hoeppli R.E., Huang Q., Gillies J., Luciani D.S., Orban P.C., Broady R., Levings M.K. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest. 2016;126:1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson N.A.J., Rosado-Sánchez I., Novakovsky G.E., Fung V.C.W., Huang Q., McIver E., Sun G., Gillies J., Speck M., Orban P.C. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci. Transl. Med. 2020;12:eaaz3866. doi: 10.1126/scitranslmed.aaz3866. [DOI] [PubMed] [Google Scholar]
- 15.Bagnall R.D., Waseem N., Green P.M., Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99:168–174. doi: 10.1182/blood.v99.1.168. [DOI] [PubMed] [Google Scholar]
- 16.Jacquemin M., De Maeyer M., D’Oiron R., Lavend’Homme R., Peerlinck K., Saint-Remy J.M. Molecular mechanisms of mild and moderate hemophilia A. J. Thromb. Haemost. 2003;1:456–463. doi: 10.1046/j.1538-7836.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 17.Cao O., Loduca P.A., Herzog R.W. Role of regulatory T cells in tolerance to coagulation factors. J. Thromb. Haemost. 2009;7(Suppl 1):88–91. doi: 10.1111/j.1538-7836.2009.03417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarkar D., Biswas M., Liao G., Seay H.R., Perrin G.Q., Markusic D.M., Hoffman B.E., Brusko T.M., Terhorst C., Herzog R.W. Ex Vivo Expanded Autologous Polyclonal Regulatory T Cells Suppress Inhibitor Formation in Hemophilia. Mol. Ther. Methods Clin. Dev. 2014;1:14030. doi: 10.1038/mtm.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herzog R.W., Kuteyeva V., Saboungi R., Terhorst C., Biswas M. Reprogrammed CD4+ T Cells That Express FoxP3+ Control Inhibitory Antibody Formation in Hemophilia A Mice. Front. Immunol. 2019;10:274. doi: 10.3389/fimmu.2019.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baeuerle P.A., Ding J., Patel E., Thorausch N., Horton H., Gierut J., Scarfo I., Choudhary R., Kiner O., Krishnamurthy J. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019;10:2087. doi: 10.1038/s41467-019-10097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acuto O., Di Bartolo V., Michel F. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat. Rev. Immunol. 2008;8:699–712. doi: 10.1038/nri2397. [DOI] [PubMed] [Google Scholar]
- 22.Jacquemin M.G., Desqueper B.G., Benhida A., Vander Elst L., Hoylaerts M.F., Bakkus M., Thielemans K., Arnout J., Peerlinck K., Gilles J.G. Mechanism and kinetics of factor VIII inactivation: study with an IgG4 monoclonal antibody derived from a hemophilia A patient with inhibitor. Blood. 1998;92:496–506. [PubMed] [Google Scholar]
- 23.Thill P.A., Weiss A., Chakraborty A.K. Phosphorylation of a Tyrosine Residue on Zap70 by Lck and Its Subsequent Binding via an SH2 Domain May Be a Key Gatekeeper of T Cell Receptor Signaling In Vivo. Mol. Cell. Biol. 2016;36:2396–2402. doi: 10.1128/MCB.00165-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love P.E., Hayes S.M. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb. Perspect. Biol. 2010;2:a002485. doi: 10.1101/cshperspect.a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ardouin L., Boyer C., Gillet A., Trucy J., Bernard A.M., Nunes J., Delon J., Trautmann A., He H.T., Malissen B., Malissen M. Crippling of CD3-zeta ITAMs does not impair T cell receptor signaling. Immunity. 1999;10:409–420. doi: 10.1016/s1074-7613(00)80041-2. [DOI] [PubMed] [Google Scholar]
- 26.Battaglia M., Stabilini A., Roncarolo M.G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi T., Kuniyasu Y., Toda M., Sakaguchi N., Itoh M., Iwata M., Shimizu J., Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 28.Turner M.S., Kane L.P., Morel P.A. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J. Immunol. 2009;183:4895–4903. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kane L.P., Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol. Rev. 2003;192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- 30.Carey K.D., Dillon T.J., Schmitt J.M., Baird A.M., Holdorf A.D., Straus D.B., Shaw A.S., Stork P.J. CD28 and the tyrosine kinase lck stimulate mitogen-activated protein kinase activity in T cells via inhibition of the small G protein Rap1. Mol. Cell. Biol. 2000;20:8409–8419. doi: 10.1128/mcb.20.22.8409-8419.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esensten J.H., Helou Y.A., Chopra G., Weiss A., Bluestone J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity. 2016;44:973–988. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu P., Santner-Nanan B., Hu M., Skarratt K., Lee C.H., Stormon M., Wong M., Fuller S.J., Nanan R. IL-10 Potentiates Differentiation of Human Induced Regulatory T Cells via STAT3 and Foxo1. J. Immunol. 2015;195:3665–3674. doi: 10.4049/jimmunol.1402898. [DOI] [PubMed] [Google Scholar]
- 33.Ito S., Ansari P., Sakatsume M., Dickensheets H., Vazquez N., Donnelly R.P., Larner A.C., Finbloom D.S. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood. 1999;93:1456–1463. [PubMed] [Google Scholar]
- 34.Kappes D.J., Tonegawa S. Surface expression of alternative forms of the TCR/CD3 complex. Proc. Natl. Acad. Sci. USA. 1991;88:10619–10623. doi: 10.1073/pnas.88.23.10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ley S.C., Tan K.N., Kubo R., Sy M.S., Terhorst C. Surface expression of CD3 in the absence of T cell receptor (TcR): evidence for sorting of partial TcR/CD3 complexes in a post-endoplasmic reticulum compartment. Eur. J. Immunol. 1989;19:2309–2317. doi: 10.1002/eji.1830191220. [DOI] [PubMed] [Google Scholar]
- 36.Eisenstein E.M., Williams C.B. The T(reg)/Th17 cell balance: a new paradigm for autoimmunity. Pediatr. Res. 2009;65:26R–31R. doi: 10.1203/PDR.0b013e31819e76c7. [DOI] [PubMed] [Google Scholar]
- 37.Oren R., Hod-Marco M., Haus-Cohen M., Thomas S., Blat D., Duvshani N., Denkberg G., Elbaz Y., Benchetrit F., Eshhar Z. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J. Immunol. 2014;193:5733–5743. doi: 10.4049/jimmunol.1301769. [DOI] [PubMed] [Google Scholar]
- 38.Salter A.I., Ivey R.G., Kennedy J.J., Voillet V., Rajan A., Alderman E.J., Voytovich U.J., Lin C., Sommermeyer D., Liu L. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018;11:eaat6753. doi: 10.1126/scisignal.aat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davenport A.J., Cross R.S., Watson K.A., Liao Y., Shi W., Prince H.M., Beavis P.A., Trapani J.A., Kershaw M.H., Ritchie D.S. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA. 2018;115:E2068–E2076. doi: 10.1073/pnas.1716266115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Künkele A., Johnson A.J., Rolczynski L.S., Chang C.A., Hoglund V., Kelly-Spratt K.S., Jensen M.C. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol. Res. 2015;3:368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 41.Gargett T., Yu W., Dotti G., Yvon E.S., Christo S.N., Hayball J.D., Lewis I.D., Brenner M.K., Brown M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016;24:1135–1149. doi: 10.1038/mt.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fitzgerald J.C., Weiss S.L., Maude S.L., Barrett D.M., Lacey S.F., Melenhorst J.J., Shaw P., Berg R.A., June C.H., Porter D.L. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017;45:e124–e131. doi: 10.1097/CCM.0000000000002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y., Wang Q.J., Yang S., Kochenderfer J.N., Zheng Z., Zhong X., Sadelain M., Eshhar Z., Rosenberg S.A., Morgan R.A. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 2009;183:5563–5574. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kochenderfer J.N., Yu Z., Frasheri D., Restifo N.P., Rosenberg S.A. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–3886. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feucht J., Sun J., Eyquem J., Ho Y.J., Zhao Z., Leibold J., Dobrin A., Cabriolu A., Hamieh M., Sadelain M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019;25:82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.James J.R. Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci. Signal. 2018;11:eaan1088. doi: 10.1126/scisignal.aan1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawse W.F., Boggess W.C., Morel P.A. TCR Signal Strength Regulates Akt Substrate Specificity To Induce Alternate Murine Th and T Regulatory Cell Differentiation Programs. J. Immunol. 2017;199:589–597. doi: 10.4049/jimmunol.1700369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stritesky G.L., Jameson S.C., Hogquist K.A. Selection of self-reactive T cells in the thymus. Annu. Rev. Immunol. 2012;30:95–114. doi: 10.1146/annurev-immunol-020711-075035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan D., Farache J., Mingueneau M., Mathis D., Benoist C. Imbalanced signal transduction in regulatory T cells expressing the transcription factor FoxP3. Proc. Natl. Acad. Sci. USA. 2015;112:14942–14947. doi: 10.1073/pnas.1520393112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crellin N.K., Garcia R.V., Levings M.K. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood. 2007;109:2014–2022. doi: 10.1182/blood-2006-07-035279. [DOI] [PubMed] [Google Scholar]
- 51.Molinero L.L., Miller M.L., Evaristo C., Alegre M.L. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-κB-dependent manner. J. Immunol. 2011;186:4609–4617. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurtulus S., Sakuishi K., Ngiow S.F., Joller N., Tan D.J., Teng M.W., Smyth M.J., Kuchroo V.K., Anderson A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Invest. 2015;125:4053–4062. doi: 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Gianchecchi E., Fierabracci A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018;9:2374. doi: 10.3389/fimmu.2018.02374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boroughs A.C., Larson R.C., Choi B.D., Bouffard A.A., Riley L.S., Schiferle E., Kulkarni A.S., Cetrulo C.L., Ting D., Blazar B.R. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight. 2019;5:e126194. doi: 10.1172/jci.insight.126194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boardman D.A., Philippeos C., Fruhwirth G.O., Ibrahim M.A., Hannen R.F., Cooper D., Marelli-Berg F.M., Watt F.M., Lechler R.I., Maher J. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am. J. Transplant. 2017;17:931–943. doi: 10.1111/ajt.14185. [DOI] [PubMed] [Google Scholar]
- 56.Cao X., Cai S.F., Fehniger T.A., Song J., Collins L.I., Piwnica-Worms D.R., Ley T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–646. doi: 10.1016/j.immuni.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 57.Joss A., Akdis M., Faith A., Blaser K., Akdis C.A. IL-10 directly acts on T cells by specifically altering the CD28 co-stimulation pathway. Eur. J. Immunol. 2000;30:1683–1690. doi: 10.1002/1521-4141(200006)30:6<1683::AID-IMMU1683>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 58.Kitz A., de Marcken M., Gautron A.S., Mitrovic M., Hafler D.A., Dominguez-Villar M. AKT isoforms modulate Th1-like Treg generation and function in human autoimmune disease. EMBO Rep. 2016;17:1169–1183. doi: 10.15252/embr.201541905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laidlaw B.J., Lu Y., Amezquita R.A., Weinstein J.S., Vander Heiden J.A., Gupta N.T., Kleinstein S.H., Kaech S.M., Craft J. Interleukin-10 from CD4+ follicular regulatory T cells promotes the germinal center response. Sci. Immunol. 2017;2:eaan4767. doi: 10.1126/sciimmunol.aan4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saxena A., Khosraviani S., Noel S., Mohan D., Donner T., Hamad A.R. Interleukin-10 paradox: A potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine. 2015;74:27–34. doi: 10.1016/j.cyto.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guthmiller J.J., Graham A.C., Zander R.A., Pope R.L., Butler N.S. Cutting Edge: IL-10 Is Essential for the Generation of Germinal Center B Cell Responses and Anti-Plasmodium Humoral Immunity. J. Immunol. 2017;198:617–622. doi: 10.4049/jimmunol.1601762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akkaya B., Oya Y., Akkaya M., Al Souz J., Holstein A.H., Kamenyeva O., Kabat J., Matsumura R., Dorward D.W., Glass D.D., Shevach E.M. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat. Immunol. 2019;20:218–231. doi: 10.1038/s41590-018-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoon J., Schmidt A., Zhang A.H., Königs C., Kim Y.C., Scott D.W. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. 2017;129:238–245. doi: 10.1182/blood-2016-07-727834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delignat S., Russick J., Gangadharan B., Rayes J., Ing M., Voorberg J., Kaveri S.V., Lacroix-Desmazes S. Prevention of the anti-factor VIII memory B-cell response by inhibition of Bruton tyrosine kinase in experimental hemophilia A. Haematologica. 2019;104:1046–1054. doi: 10.3324/haematol.2018.200279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu H., Rhodes M., Wiest D.L., Vignali D.A. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. 2000;13:665–675. doi: 10.1016/s1074-7613(00)00066-2. [DOI] [PubMed] [Google Scholar]
- 66.Eyquem J., Mansilla-Soto J., Giavridis T., van der Stegen S.J., Hamieh M., Cunanan K.M., Odak A., Gönen M., Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schamel W.W., Alarcon B., Höfer T., Minguet S. The Allostery Model of TCR Regulation. J. Immunol. 2017;198:47–52. doi: 10.4049/jimmunol.1601661. [DOI] [PubMed] [Google Scholar]
- 68.Ramello M.C., Benzaïd I., Kuenzi B.M., Lienlaf-Moreno M., Kandell W.M., Santiago D.N., Pabón-Saldaña M., Darville L., Fang B., Rix U. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019;12:eaap9777. doi: 10.1126/scisignal.aap9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y., Liu G., Wang J., Zheng Z.Y., Jia L., Rui W., Huang D., Zhou Z.X., Zhou L., Wu X. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci. Transl. Med. 2021;13:eabb5191. doi: 10.1126/scitranslmed.abb5191. [DOI] [PubMed] [Google Scholar]
- 70.Xu Y., Yang Z., Horan L.H., Zhang P., Liu L., Zimdahl B., Green S., Lu J., Morales J.F., Barrett D.M. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018;4:62. doi: 10.1038/s41421-018-0066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Betts M.R., Brenchley J.M., Price D.A., De Rosa S.C., Douek D.C., Roederer M., Koup R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 72.Cao O., Hoffman B.E., Moghimi B., Nayak S., Cooper M., Zhou S., Ertl H.C., High K.A., Herzog R.W. Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia B. Mol. Ther. 2009;17:1733–1742. doi: 10.1038/mt.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Biswas M., Rogers G.L., Sherman A., Byrne B.J., Markusic D.M., Jiang H., Herzog R.W. Combination therapy for inhibitor reversal in haemophilia A using monoclonal anti-CD20 and rapamycin. Thromb. Haemost. 2017;117:33–43. doi: 10.1160/TH16-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.