

Abstract
On March 10, 2020, the U.S. Food and Drug Administration (FDA) granted accelerated approval to nivolumab in combination with ipilimumab for the treatment of patients with hepatocellular carcinoma (HCC) previously treated with sorafenib. The recommended approved dosage was nivolumab 1 mg/kg i.v. plus ipilimumab 3 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks. The approval was based on data from cohort 4 of CheckMate 040, which randomized patients with advanced unresectable or metastatic HCC previously treated with or who were intolerant to sorafenib to receive one of three different dosing regimens of nivolumab in combination with ipilimumab. Investigator‐assessed overall response rate (ORR) was the primary endpoint, and ORR assessed by blinded independent central review (BICR) was an exploratory endpoint. BICR‐assessed ORR and duration of response (DoR) form the primary basis of the FDA's regulatory decision, and BICR‐assessed ORR was comparable in all three arms at 31%–32% with 95% confidence interval [CI] 18%–47%. The DoR ranged from 17.5 to 22.2 months across the three arms, with overlapping 95% CIs. Adverse events (AEs) were generally consistent with the known AE profiles of nivolumab and ipilimumab, and no new safety events were identified. This article summarizes the FDA review of the data supporting the approval of nivolumab and ipilimumab for the treatment of HCC.
Implications for Practice
Nivolumab and ipilimumab combination therapy is another option for patients with advanced hepatocellular carcinoma who experience radiographic progression during or after sorafenib or sorafenib intolerance. No new toxicities were identified, but, as expected, increased toxicity was observed with the addition of ipilimumab to nivolumab as compared with nivolumab alone, which is also approved for the same indication. Whether to administer nivolumab as a single agent or in combination with ipilimumab is expected to be a joint decision between the oncologist and patient, taking into consideration the potential for a higher likelihood of response and the potentially higher rate of toxicity with the combination.
Keywords: Hepatocellular carcinoma, CheckMate 040, Nivolumab, Ipilimumab
Short abstract
This article summarizes the FDA review of the data supporting the approval of nivolumab and ipilimumab for the treatment of hepatocellular carcinoma.
Introduction
In the U.S., the 5‐year survival rate for hepatocellular carcinoma (HCC) when diagnosed early is 18% but decreases to approximately 2% in patients with metastatic disease [1, 2]. A retrospective cohort study of patients with HCC in the U.S. in the Surveillance, Epidemiology, and End Results (SEER) database between 1973 and 2011 reported that 65.4% were White, 21.0% were Asian/Pacific Islander, 12.2% were Black, and 1.1% were Native Americans/Alaskan Natives [3]. Another retrospective analysis of patients with HCC in the U.S. in the SEER database between 2003 and 2011 found that the incidence of HCC is highest among Asians (18.6 per 100,000 per year), followed by Blacks (15.7 per 100,000 per year), Hispanics (11.8 per 100,000 per year), and non‐Hispanic Whites (7.0 per 100,000 per year) [4]. Until the approval of the combination of atezolizumab and bevacizumab on May 29, 2020, sorafenib and lenvatinib were the only approved drugs for the first‐line treatment of patients with metastatic or unresectable HCC. Currently, regorafenib, cabozantinib, and ramucirumab are the approved therapies for the second‐line treatment of HCC after sorafenib based on studies demonstrating a modest improvement in median survival of 1.2 to 2.8 months over placebo with low overall response rates (ORRs) ranging from 4% to 7% [5, 6, 7]. Under the accelerated approval pathway, the immune checkpoint inhibitors nivolumab and pembrolizumab were approved as single agents based on an ORR of 14.3% (95% confidence interval [CI]: 9– 21) and 17% (95% CI: 11–26), respectively, with a duration of response (DoR) ≥12 months in 56%–59% of responders [8, 9].
Data from clinical trial CA209040 (CheckMate 040) supported the approval of nivolumab and ipilimumab for the treatment of patients with HCC previously treated with sorafenib. The results of this clinical trial have been published [10]. This article summarizes the U.S. Food and Drug Administration (FDA)'s review of the data submitted in the supplemental Biologics Licensing Application and the basis for approval of nivolumab and ipilimumab for this new indication. Table 1 and Table 2 provide the background information for nivolumab and ipilimumab.
Table 1.
Nivolumab background information
| Structure | Fully human IgG4 monoclonal antibody. |
|---|---|
| Mechanism of action |
Binds to the PD‐1 cell membrane receptor and blocks the interaction of PD‐1 with its ligands, PD‐L1 and PD‐L2. |
| Pharmacokinetics |
The exposure to nivolumab increases dose proportionally over the dose range of 0.1 to 10 mg/kg administered every 2 weeks. The predicted exposure of nivolumab after a 30‐minute infusion is comparable to that observed with a 60‐minute infusion. Steady‐state concentrations of nivolumab were reached by 12 weeks when administered at 3 mg/kg every 2 weeks, and systemic accumulation was 3.7‐fold. The geometric mean volume of distribution at steady state and coefficient of variation is 6.8 L (27.3%). The geometric mean elimination half‐life is 25 days (77.5%). |
| Prior approvals (before March 10, 2020) |
Melanoma: Patients with unresectable or metastatic melanoma, as a single agent or in combination with ipilimumab. Patients with melanoma with lymph node involvement or metastatic disease who have undergone complete resection, in the adjuvant setting. |
|
Non‐small cell lung cancer: Patients with metastatic NSCLC and progression on or after platinum‐based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA‐approved therapy for these aberrations prior to receiving nivolumab. | |
|
Small cell lung cancer: Patients with metastatic SCLC with progression after platinum‐based chemotherapy and at least one other line of therapy. a | |
|
Renal cell carcinoma: Patients with advanced RCC who have received prior antiangiogenic therapy. Patients with intermediate or poor risk, previously untreated advanced RCC, in combination with ipilimumab. | |
|
Classical Hodgkin lymphoma: Adult patients with cHL that has relapsed or progressed aftera autologous HSCT and brentuximab vedotin, or after three or more lines of systemic therapy that includes autologous HSCT. | |
|
Squamous cell carcinoma of the head and neck: Patients with recurrent or metastatic SCCHN with disease progression on or after a platinum‐based therapy. | |
|
Urothelial carcinoma: Patients with locally advanced or metastatic urothelial carcinoma whoa have disease progression during or following platinum‐containing chemotherapy. Patients with locally advanced or metastatic urothelial carcinoma whoa have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum‐containing chemotherapy. | |
|
Colorectal cancer: Adult and pediatric (12 years and older) patients with MSI‐H or dMMR metastatic CRC that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan, as a single agent or in combination with ipilimumab. a | |
|
Hepatocellular carcinoma: Patients with HCC who have been previously treated with sorafenib. |
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Abbreviations: ALK, anaplastic lymphoma kinase; cHL, classical Hodgkin lymphoma; CRC, colorectal cancer; dMMR, mismatch repair deficient; EGFR, epidermal growth factor receptor; FDA, U.S. Food and Drug Administration; HCC, hepatocellular carcinoma; HSCT, hematopoietic stem cell transplantation; MSI‐H, microsatellite instability high; NSCLC, non‐small cell lung cancer; PD‐1, programmed death‐1; PD‐L1, programmed death‐ligand 1; PD‐L2, programmed death‐ligand 2; RCC, renal cell carcinoma; SCCHN, squamous cell carcinoma of the head and neck; SCLC, small cell lung cancer.
Source: Nivolumab U.S. Package Insert dated September 18, 2019 (Supplement 75).
Table 2.
Ipilimumab background information
| Structure | Recombinant human IgG1 kappa monoclonal antibody. |
|---|---|
| Mechanism of action |
Binds to the CTLA‐4 cell membrane receptor and blocks the interaction of CTLA‐4 with its ligands, CD80 and CD86. |
| Pharmacokinetics |
The pharmacokinetics of ipilimumab is linear in the dose range of 0.3 mg/kg to 10 mg/kg. Following administration of ipilimumab every 3 weeks, the systemic accumulation was 1.5‐fold or less. Steady‐state concentrations of ipilimumab were reached by the third dose; the mean minimum concentration at steady state was 19.4 mcg/mL at 3 mg/kg and 58.1 mcg/mL at 10 mg/kg every 3 weeks. The mean (percent coefficient of variation) terminal half‐life was 15.4 days (34%) and the mean (percent coefficient of variation) CL was 16.8 mL/h (38%). The CL of ipilimumab was unchanged in presence of anti‐ipilimumab antibodies. |
| Prior approvals (before March 10, 2020) |
Treatment of unresectable or metastatic melanoma in adults and pediatric patients (12 years and older). Adjuvant treatment of patients with cutaneous melanoma with pathologic involvement of regional lymph nodes of more than 1 mm who have undergone complete resection, including total lymphadenectomy. Treatment of patients with intermediate or poor risk, previously untreated advanced renal cell carcinoma, in combination with nivolumab. Treatment of adult and pediatric patients 12 years of age and older with MSI‐H or dMMR metastatic colorectal cancer that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan, in combination with nivolumab. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. |
Abbreviations: CL, clearance; CTLA‐4, cytotoxic T‐lymphocyte–associated antigen 4; dMMR, mismatch repair deficient; MSI‐H, microsatellite instability high.
Source: Ipilimumab U.S. Package Insert dated September 20, 2019 (Supplement 104).
CheckMate 040: Clinical Trial Design
CheckMate 040 (CA209040, NCT01658878) is an ongoing, multicenter, multiple parallel cohort, open‐label clinical trial evaluating the efficacy of nivolumab as a single agent and in combination with other agents, including ipilimumab, in patients with histologically confirmed HCC who received prior sorafenib or are sorafenib‐naïve. The primary data supporting the indication are from cohort 4, which employed a two‐stage trial design and randomized 36 patients 1:1:1 (n = 12 each) into one of the following three treatment arms in stage 1: nivolumab 1 mg/kg i.v. plus ipilimumab 3 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks (N1I3 Q3); nivolumab 3 mg/kg i.v. plus ipilimumab 1 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks (N3I1 Q3); or nivolumab 3 mg/kg i.v. every 2 weeks plus ipilimumab 1 mg/kg i.v. every 6 weeks (N3I1 Q6).
Treatment continued until toxicity or clinical or radiographic disease progression. If four or fewer patients in a given treatment arm permanently discontinued trial treatment because of treatment‐related adverse events prior to week 13, then the treatment arm was deemed tolerable and the trial proceeded to stage 2 and enrolled 28 additional patients. The trial was not designed to enable a formal comparison between the three arms.
Key inclusion criteria were Eastern Cooperative Oncology Group performance status 0–1, ineligibility for surgical or locoregional treatment of HCC, and radiographic progression during or after sorafenib or protocol‐defined sorafenib intolerance. Patients with hepatitis B (HBV) or hepatitis C (HCV) infections, or uninfected patients, were eligible to enroll. Patients were ineligible on the basis of the following key exclusion criteria: active coinfection with HBV and HCV or HBV and hepatitis D, or known fibrolamellar, sarcomatoid, or mixed cholangiocarcinoma HCC.
The primary efficacy endpoints for cohort 4 were investigator‐assessed ORR and DoR, based on RECIST version 1.1 [11]. Key secondary endpoints included progression‐free survival (PFS) and overall survival (OS). ORR by blinded independent central review (BICR) assessment based on RECIST 1.1 was an exploratory endpoint.
In addition to data from cohort 4, the application included data from cohorts 1 and 2 of CheckMate 040 to provide support for the contribution of treatment effect of ipilimumab to the effect observed with the combination of nivolumab and ipilimumab in the indicated population. The data included from cohorts 1 and 2 evaluated single‐agent nivolumab in patients with HCC who received prior sorafenib.
Results
Efficacy
Demographic and baseline patient and disease characteristics are summarized in Table 3. Cohort 4 enrolled a total of 148 patients (i.e., the N1I3 Q3, N3I1 Q3, and N3I1 Q6 arms enrolled 50, 49, and 49 patients, respectively). Two randomized patients were not treated (one each in the N1I3 Q3 and N3I1 Q6 arms) because they no longer met the trial eligibility criteria. The minimum follow‐up duration across all three arms was approximately 28.2 months. The leading cause of trial treatment discontinuation in the N1I3 Q3, N3I1 Q3, and N3I1 Q6 arms was disease progression (50%, 69%, and 67%, respectively), followed by treatment‐emergent adverse events (TEAEs) (22%, 6%, and 2%, respectively).
Table 3.
Demographics and baseline disease characteristics of randomized subjects
| Demographics and characteristics | N1I3 Q3 (n = 50), n (%) | N3I1 Q3 (n = 49), n (%) | N3I1 Q6 (n = 49), n (%) |
|---|---|---|---|
| Sex, male | 43 (86) | 37 (76) | 40 (82) |
| Age, median (min, max), years | 60.5 (18–80) | 65.0 (34–83) | 58.0 (32–79) |
| Race | |||
| White | 12 (24) | 20 (41) | 15 (31) |
| Black or African American | 1 (2) | 1 (2) | 3 (6) |
| Asian | 37 (74) | 27 (55) | 30 (61) |
| Region | |||
| U.S./Canada | 9 (18) | 11 (22) | 9 (18) |
| Europe | 7 (14) | 11 (22) | 12 (25) |
| Asia | 34 (68) | 27 (55) | 28 (57) |
| BCLC stage C | 43 (86) | 45 (92) | 46 (94) |
| Child‐Pugh Scorea | |||
| 5 | 41 (82) | 38 (78) | 32 (65) |
| 6 | 9 (18) | 9 (18) | 15 (31) |
| Extrahepatic spread and/or vascular invasion | |||
| Vascular invasion present | 18 (36) | 13 (27) | 19 (39) |
| Extrahepatic spread present | 40 (80) | 40 (82) | 42 (86) |
| Extrahepatic spread or vascular invasion present | 43 (86) | 44 (90) | 45 (92) |
| AFP ≥400 μg/L | 25 (50) | 18 (37) | 22 (45) |
| ECOG PS | |||
| 0 | 31 (62) | 25 (51) | 25 (51) |
| 1 | 19 (38) | 24 (49) | 24 (49) |
| HCC etiology | |||
| Active HCV | 7 (14) | 15 (31) | 13 (27) |
| Active HBVb | 29 (58) | 21 (43) | 26 (53) |
| Alcoholic liver disease | 8 (16) | 8 (16) | 6 (12) |
| Prior nonsystemic therapies for HCC | |||
| Nonsurgical local therapies | 29 (58) | 33 (67) | 29 (59) |
| Surgery | 36 (72) | 36 (73) | 28 (57) |
| Sorafenib status at enrollment | |||
| Intolerant | 3 (6) | 1 (2) | 2 (4) |
| Progressor | 46 (92) | 47 (96) | 46 (94) |
| Years from initial diagnosis to randomization | |||
| <1 | 12 (24) | 5 (10) | 11 (22) |
| ≥1–2 | 12 (24) | 20 (41) | 14 (29) |
| ≥2 | 26 (52) | 24 (49) | 24 (49) |
The inclusion criteria required the Child‐Pugh score (CPS) A5 or A6, but two patients each with higher CPS were enrolled into the N3I1 Q3 and N3I1 Q6 arms.
Patients with active hepatitis B must have an HBV DNA viral load <100 IU/mL at screening and must be on antiviral therapy per regional standard of care guidelines prior to initiation of trial therapy.
Abbreviations: AFP, alpha‐feto protein; BCLC, Barcelona Clinic Liver Cancer; ECOG PS, Eastern Cooperative Oncology Group performance status; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; N1I3 Q3, nivolumab 1 mg/kg i.v. plus ipilimumab 3 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; N3I1 Q3, nivolumab 3 mg/kg i.v. plus ipilimumab 1 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; N3I1 Q6, nivolumab 3 mg/kg i.v. every 2 weeks plus ipilimumab 1 mg/kg i.v. every 6 weeks.
Efficacy results for the randomized population (i.e., intent‐to‐treat [ITT] population) are summarized in Table 4 and Figure 1. BICR‐assessed ORR per RECIST 1.1 was similar in all three treatment arms in the ITT analysis (N1I3 Q3 [32%; 95% CI: 20–47], N3I1 Q3 [31%; 95% CI: 18–45], and N3I1 Q6 [31%; 95% CI: 18–45]). The median BICR‐assessed DoR in the ITT analysis for the N3I1 Q3 arm was 22.2 months (95% CI: 4.4, not available [NA]) compared with 17.5 months [95% CI: 8.3, NA] and 16.6 months [95% CI: 4.3, NA] in the N1I3 Q3 and N3I1 Q6 arms, respectively. Despite these differences, the range of DoR and the proportion of patients with DoR ≥12 and DoR ≥24 months were similar in the three arms. Median OS and BICR‐assessed PFS were longer in the N1I3 Q3 arm compared with the other two arms in the ITT analysis, but the 95% CIs for the three arms overlapped. BICR‐assessed ORR per RECIST 1.1 for the N1I3 Q3 arm for the per protocol population (n = 49) was similar to that of the ITT population (n = 50) (33% [95% CI: 20–48] and 32% [95% CI: 20–47], respectively). Tumor expression of programmed death‐ligand 1 (PD‐L1) did not appear to affect the ORR, but definitive conclusions regarding the impact of this biomarker on response are precluded by the small number of patients in the subgroup and the exploratory nature of the analysis.
Table 4.
Efficacy results per blinded independent central review assessment
| Efficacy results | N1I3 Q3 (n = 50) | N3I1 Q3 (n = 49) | N3I1 Q6 (n = 49) |
|---|---|---|---|
| Overall response ratea , b | |||
| n | 16 | 15 | 15 |
| % (95% CI) | 32 (20–47) | 31 (18–45) | 31 (18–45) |
| CR | 4 (8) | 3 (6) | 0 |
| PR | 12 (24) | 12 (25) | 15 (31) |
| Overall response rate by PD‐L1 status, n/N (%)c | |||
| PD‐L1 ≥1 | 3/10 (30) | 3/10 (30) | 4/8 (50) |
| PD‐L1 <1 | 12/39 (31) | 12/38 (32) | 11/40 (28) |
| Duration of response, monthsb | |||
| Median (95% CI)d | 17.5 (8.3, NA) | 22.2 (4.4, NA) | 16.6 (4.3, NA) |
| Range | 4.6, 30.5+ | 4.2, 29.9+ | 4.1+, 32.0+ |
| DoR ≥12 months, n (%) | 9 (56) | 10 (67) | 8 (53) |
| DoR ≥24 months, n (%) | 5 (31) | 4 (27) | 5 (33) |
| Progression‐free survival, median (95% CI), monthsb | 3.9 (2.6–8.3) | 1.6 (1.4–6.9) | 2.6 (1.3–4.5) |
| OS, median (95% CI), monthsd | 22.8 (9.4, NA) | 12.5 (7.6–16.4) | 12.8 (7.4–33.0) |
Database lock: March 22, 2019; minimum follow‐up: 28.2 months.
Overall response rate (ORR) is sum of CR and PR; confidence interval based on the Clopper and Pearson method.
ORR, DoR, and PFS as assessed by blinded independent central review based on RECIST version 1.1.
PD‐L1 expression was defined as the percent of tumor cells with membrane staining in a minimum of 100 evaluable tumor cells based on the Dako PD‐L1 immunohistochemistry assay.
Kaplan‐Meier estimate.
Abbreviations: CI, confidence interval; CR, complete response; DoR, duration of response; NA, not available; N1I3 Q3, nivolumab 1 mg/kg i.v. plus ipilimumab 3 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; N3I1 Q3, nivolumab 3 mg/kg i.v. plus ipilimumab 1 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; N3I1 Q6, nivolumab 3 mg/kg i.v. every 2 weeks plus ipilimumab 1 mg/kg i.v. every 6 weeks; OS, overall survival; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival; PR, partial response.
Figure 1.
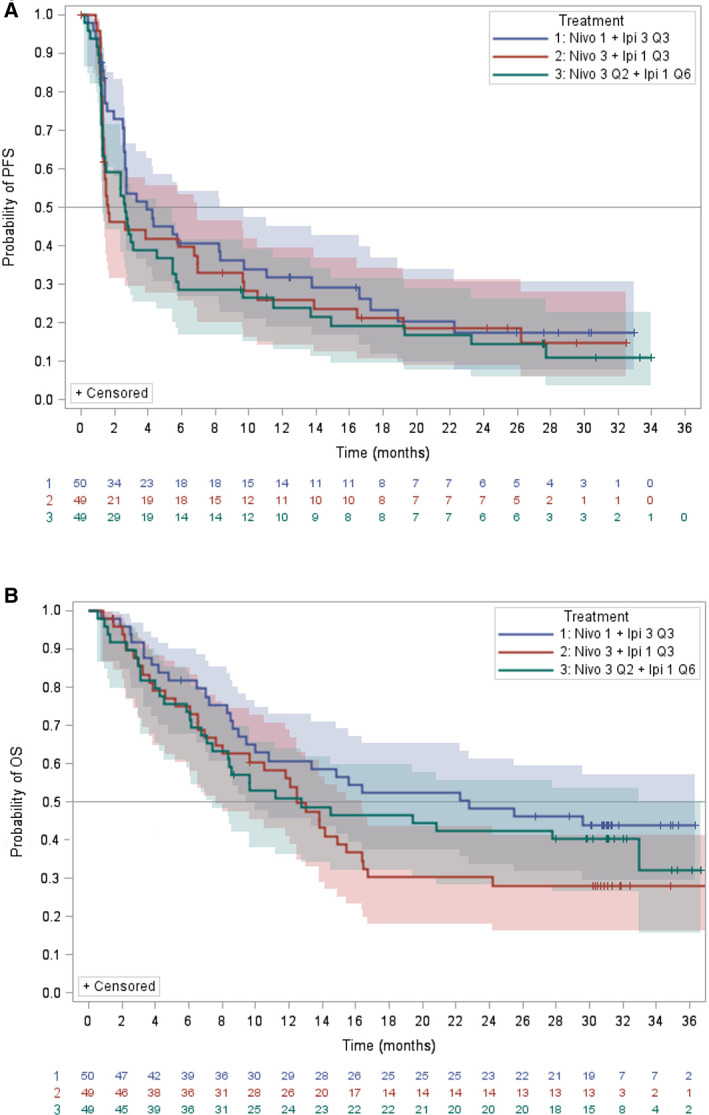
Kaplan‐Meier curves of progression‐free survival and overall survival by treatment arm. Abbreviations: Nivo 1 + Ipi 3 Q3, nivolumab 1 mg/kg i.v. plus ipilimumab 3 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; Nivo 3 + Ipi 1 Q3, nivolumab 3 mg/kg i.v. plus ipilimumab 1 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; Nivo 3 Q2 + Ipi 1 Q6, nivolumab 3 mg/kg i.v. every 2 weeks plus ipilimumab 1 mg/kg i.v. every 6 weeks; OS, overall survival; PFS, progression‐free survival.
Safety
The primary safety population consisted of 146 patients who received at least one dose of trial drug (49, 49, and 48 patients in the N1I3 Q3, N3I1 Q3, and N3I1 Q6 arms, respectively). The observed TEAEs were generally consistent with the known safety profiles of nivolumab and ipilimumab and the underlying disease. No new safety signals for the combination were identified based on review of this application.
Grade 3–4 TEAEs were reported in 76%, 65%, and 69% of patients in the N1I3 Q3, N3I1 Q3, and N3I1 Q6 arms, respectively, with the most frequently reported TEAEs of any grade being the following:
N1I3 Q3 arm: pruritus (53%), diarrhea (39%), decreased appetite (35%), and cough (35%)
N3I1 Q3 arm: pruritus (47%), increased AST (31%), and abdominal pain (29%)
N3I1 Q6 arm: pyrexia (33%), pruritus (31%), diarrhea (29%), and decreased appetite (29%)
Adverse events (AEs) leading to drug discontinuation occurred in 14 (29%), 5 (10%), and 7 (15%) of patients in the N1I3 Q3, N3I1 Q3, and N3I1 Q6 arms, respectively. Of these events, the most frequently reported were diarrhea (N1I3 Q3 arm) and increased AST (N3I1 Q3 arm) with none reported in more than one patient in the N3I1 Q6 arm.
Regarding immune‐mediated AEs (imAEs) captured through 100 days after the last dose of trial drugs, rash and hypothyroidism/thyroiditis were the most frequently reported any‐grade imAEs within each arm. Rash was reported in 35%, 29%, and 17% patients in the N1I3 Q3, N3I1 Q3, and N3I1 Q6 arms, respectively. Hypothyroidism/thyroiditis was reported in 22%, 14%, and 17% in the N1I3 Q3, N3I1 Q3, and N3I1 Q6 arms, respectively. For the N1I3 Q3 and N3I1 Q3 arms, hepatitis was the third most reported imAE (20% and 12%, respectively), whereas for the N3I1 Q6 arm, hepatitis tied with adrenal insufficiency and hyperthyroidism for the third most reported imAE (6% each).
Table 5 provides a summary of the AEs occurring in patients in the N1I3 Q3 arm. Additionally, 14 patients (9.6%) experienced virologic breakthrough in cohort 4 (compared with six in cohorts 1 and 2), of which six were in the N1I3 Q3 arm. HBV virologic breakthrough was defined as at least 1 log increase in HBV DNA for those patients with detectable HBV DNA at baseline. HCV virologic breakthrough was defined as a 1 log increase in HCV RNA from baseline.
Table 5.
Adverse reactions occurring in ≥10% of patients receiving nivolumab in combination with ipilimumab
| Adverse reaction | N1I3 Q3 arm (n = 49) | |
|---|---|---|
| All grades (%) | Grades 3–4 (%) | |
| Skin and subcutaneous tissue | ||
| Rash | 53 | 8 |
| Pruritus | 53 | 4 |
| Musculoskeletal and connective tissue | ||
| Musculoskeletal pain | 41 | 2 |
| Arthralgia | 10 | 0 |
| Gastrointestinal | ||
| Diarrhea | 39 | 4 |
| Abdominal pain | 22 | 6 |
| Nausea | 20 | 0 |
| Ascites | 14 | 6 |
| Constipation | 14 | 0 |
| Dry mouth | 12 | 0 |
| Dyspepsia | 12 | 2 |
| Vomiting | 12 | 2 |
| Stomatitis | 10 | 0 |
| Abdominal distension | 8 | 0 |
| Respiratory, thoracic, and mediastinal | ||
| Cough | 37 | 0 |
| Dyspnea | 14 | 0 |
| Pneumonitis | 10 | 2 |
| Metabolism and nutrition | ||
| Decreased appetite | 35 | 2 |
| General | ||
| Fatigue | 27 | 2 |
| Pyrexia | 27 | 0 |
| Malaise | 18 | 2 |
| Edema | 16 | 2 |
| Influenza‐like illness | 14 | 0 |
| Chills | 10 | 0 |
| Nervous system | ||
| Headache | 22 | 0 |
| Dizziness | 20 | 0 |
| Endocrine | ||
| Hypothyroidism | 20 | 0 |
| Adrenal insufficiency | 18 | 4 |
| Investigations | ||
| Weight decreased | 20 | 0 |
| Psychiatric | ||
| Insomnia | 18 | 0 |
| Blood and lymphatic system | ||
| Anemia | 10 | 4 |
| Infections | ||
| Influenza | 10 | 2 |
| Upper respiratory tract infection | 6 | 0 |
| Vascular | ||
| Hypotension | 10 | 0 |
Discussion
Cohort 4 of CheckMate 040 demonstrated a clinically meaningful ORR and DoR in patients with HCC previously treated with sorafenib who received nivolumab in combination with ipilimumab. One key limitation of the trial was the lack of a prespecified statistical plan to compare outcomes across the three dosage arms, precluding a conclusive determination regarding whether the N1I3 Q3 regimen requested for approval by Bristol‐Myers Squibb was the optimal dosage for second‐line treatment of HCC. Although the median OS observed in the N1I3 Q3 arm (22.8 months) was numerically higher compared with the median OS observed in the other two arms (approximately 12 months), the lack of stratification factors for randomization in cohort 4 led to imbalances in baseline covariates across the three arms (Table 3), which may have contributed to the differential median OS across the three arms. Furthermore, the small sample size of the subgroups limits further interpretation of these observed differences. From a safety perspective, the variable rates of AEs observed across the arms could be due to imbalances in covariates across arms because of the aforementioned lack of stratification, the higher dose of ipilimumab leading to more AEs, or chance. Overall, there was no discernible regimen with a more favorable benefit‐risk assessment. Ultimately, the FDA approved the N1I3 Q3 regimen after concluding that this dosage regimen is safe and effective for the proposed indication (Table 6).
Table 6.
Overall benefit‐risk assessment
| Dimension | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
| Analysis of condition |
The overall estimated 5‐year survival rate for patients with HCC is 18.4%; 5‐year survival rates are 33% in those diagnosed with localized disease, 11% in those with regional (nodal) involvement, and 2.4% for those with metastatic disease at diagnosis. |
Advanced HCC is a serious disease with a poor prognosis. |
| Current treatment options |
Five drugs (regorafenib, cabozantinib, ramucirumab [for those with AFP ≥400 ng/mL], nivolumab, and pembrolizumab) are approved for the treatment of unresectable or metastatic Child‐Pugh A HCC, following treatment with sorafenib in the first‐line setting. Of these, regorafenib, cabozantinib, and ramucirumab are the only ones with regular approval and demonstrate a modest survival advantage of 1.2 to 2.8 months over placebo and have low ORRs at 4%–7%. Nivolumab and pembrolizumab were approved under the accelerated approval pathway, and these agents had an ORR of 14%–17% with a durable response lasting ≥12 months in 55%–56% of responders. |
The efficacy of the treatment options for patients who progressed on first‐line treatment for advanced HCC is limited, and more second‐line treatment options are needed for patients with advanced HCC. |
| Benefit |
The N1I3 Q3 dosage regimen demonstrated an ORR of 33% (95% CI: 20–48) with 56% of responses durable for ≥12 months and 31% of responses durable for ≥24 months. Supportive data was provided by the N3I1 Q3 and N3I1 Q6 dosage regimens, which demonstrated similar ORRs and durability of responses. |
The magnitude and durability of the ORR observed in patients with advanced HCC who received second‐line treatment of nivolumab in combination with ipilimumab far exceeded the ORR of available therapy (cabozantinib and regorafenib), was clinically meaningful, and represented a significant increase over the response rates that supported the accelerated approval of nivolumab and pembrolizumab as single agents. |
| Risk and risk management |
The observed safety profile was generally consistent with the known safety profile of combination nivolumab and ipilimumab. The most common adverse reactions (occurring in at least 20% of patients) were rash, pruritus, musculoskeletal pain, diarrhea, cough, decreased appetite, fatigue, pyrexia, headache, abdominal pain, nausea, dizziness, hypothyroidism, and decreased weight. The most common severe adverse reactions (occurring in at least 3% of patients) were rash, abdominal pain, ascites, pruritis, and anemia. |
The toxicity profile of nivolumab in combination with ipilimumab was acceptable when assessed in the context of the life‐threatening nature of unresectable HCC that progressed following first‐line treatment with sorafenib. No new significant safety concerned were identified. Significant and serious adverse reactions were primarily immune‐mediated and were adequately addressed by product labeling for nivolumab. |
Abbreviations: AFP, alpha‐feto protein; CI, confidence interval; HCC, hepatocellular carcinoma; N1I3 Q3, nivolumab 1 mg/kg i.v. plus ipilimumab 3 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; ORR, overall response rate; N3I1 Q3, nivolumab 3 mg/kg i.v. plus ipilimumab 1 mg/kg i.v. every 3 weeks for four cycles, followed by nivolumab 240 mg i.v. every 2 weeks; N3I1 Q6, nivolumab 3 mg/kg i.v. every 2 weeks plus ipilimumab 1 mg/kg i.v. every 6 weeks.
Another important FDA review consideration was whether the application provided sufficient information to establish the contribution of effect of the components of the trial regimen. Cohort 4 of CheckMate 040 was not designed for direct comparison of the effectiveness of nivolumab as a single agent and in combination with ipilimumab. As of the March 20, 2018, database lock for cohorts 1 and 2, the BICR‐assessed ORR according to RECIST 1.1 for nivolumab as a single agent was 14% (95% CI: 9–21). Notably, there was no period of temporal overlap in enrollment across cohorts 1 and 2, and cohort 4; thus, physician bias in the decision to enroll patients into the single‐agent arm (cohorts 1 and 2) versus a combination therapy arm (cohort 4) is unlikely. In an exploratory analysis, the 95% CI for the pooled ORR for the three arms in cohort 4 investigating the combination of nivolumab and ipilimumab (24%–39%) excludes the upper bound of the 95% CI of the estimated ORR for cohorts 1 and 2, which indirectly supports the contribution of ipilimumab to the treatment effect observed with the combination. In addition, the odds ratio using a propensity scoring analysis of cohorts 1 and 2, and cohort 4 with all three treatment arms pooled was similar to the unadjusted odds ratio, 2.5 (95% CI: 1.3–4.6) and 2.7 (95% CI: 1.4–5.2), respectively, providing additional support that nivolumab in combination with ipilimumab results in an ORR that is higher than the ORR achieved with single‐agent nivolumab. Taking into consideration the observed incremental increase in ORR with durability of response with the addition of ipilimumab to nivolumab in patients with HCC in cohort 4 (compared with cohort 1 and 2), along with the demonstrated incremental efficacy (PFS and OS) observed with the combination of ipilimumab and nivolumab in patients with melanoma compared with ipilimumab alone, the FDA considered it is highly unlikely that ipilimumab alone could be driving the observed treatment effect of the combination [12].
To verify clinical benefit with nivolumab in combination with ipilimumab treatment of HCC, an ongoing randomized control trial is investigating nivolumab in combination with ipilimumab (at the N1I3 Q3 dosage regimen) for first‐line treatment of patients with advanced HCC compared with sorafenib or lenvatinib, with the primary endpoint of OS (CheckMate 9DW; NCT04039607). The key secondary endpoints are ORR as assessed by BICR based on RECIST 1.1, DoR, and cancer‐related symptom burden. Single‐agent nivolumab has accelerated approval for the treatment of HCC previously treated with sorafenib based on the data from cohorts 1 and 2 of CheckMate 040, but its confirmatory trial, CheckMate 459, did not meets its primary endpoint of improved OS with nivolumab compared with sorafenib for first‐line treatment of patients with unresectable HCC (hazard ratio, 0.85; 95% CI: 0.72–1.02; p = .075) [13]. Since the approval of nivolumab plus ipilimumab in March 2020, the FDA subsequently approved atezolizumab in combination with bevacizumab in May 2020 for the first‐line treatment of HCC, representing a change in treatment landscape, importantly moving immune checkpoint inhibitors to the first‐line treatment setting for advanced/metastatic disease [14]. Thus, it will be important to determine if there is a role for programmed death‐1 (PD‐1) or PD‐L1 antibodies in combination with ipilimumab for the treatment of patients who received prior treatment with a PD[L]‐1 inhibitor in combination with a vascular endothelial growth factor inhibitor and to evaluate the optimal sequencing of approved therapies.
Two other important limitations of CheckMate 040 arose because the U.S. population represented only 14.9% of the overall trial population: etiology of HCC and enrollment of U.S. minority patient populations, including Black or African American and Hispanic patients. The most common etiology for HCC in this trial was HBV (50.7%), which reflects the regional representation of the trial population that primarily enrolled in Asia (60.1%); only 19.6% enrolled from sites across North America. Although chronic HBV is associated with 50% of all HCC cases worldwide, HBV is less common in the U.S., and 30% of HCC cases in the U.S. have markers of HCV infection [15, 16]. In addition, several studies conducted in Western countries have found that 30%–40% of HCC cases do not have associated HBV or HCV infection and are typically associated with alcohol use, steatohepatitis, or metabolic syndrome [13]. In CheckMate 040, only six patients (4%), two Asian patients and four White patients, were considered to have risk factors for nonalcoholic fatty liver disease or hepatosteatosis, and even among these patients, 50% (three patients) had viral hepatitis infection. Although responses were observed irrespective of HCC etiology, a residual source of uncertainty in this trial is that the distribution of etiologies for HCC in this trial population was not reflective of the overall HCC population in the U.S. Another important limitation is that only 5 of 148 patients (3.4%) were Black or African American and none of the patients were Hispanic; these findings are consistent with trials supporting other approvals for HCC. Given that the reported 5‐year survival in patients with HCC is lowest in Black and Hispanic patients in the U.S. (21.3% [95% CI: 19.5–23.1] and 23.8% [95% CI: 22.3–25.4], respectively) compared with non‐Hispanic White and Asian patients in the U.S. (25.0% [95% CI: 24.1–25.9]; and 26.1% [95% CI: 24.5–27.6], respectively), increased efforts are needed to enroll a more representative patient population in future clinical trials [4].
Conclusion
The FDA's primary assessment of the safety and effectiveness of nivolumab in combination with ipilimumab was based on the single‐arm data from cohort 4 of CheckMate 040. The limitations of the trial design precluded a determination of which regimen was optimal. However, the FDA considered the ORR of 32% with a DoR of 17.5 months evidence that the combination of nivolumab and ipilimumab provided an advantage over available therapy (with ORR ranging from 4% to 7%) and is reasonably likely to predict clinical benefit in the indicated population, as required for accelerated approval. No new toxicities were identified, but, as expected, increased toxicity was observed with the addition of ipilimumab to nivolumab as compared with nivolumab alone, which is also approved for this indication. When determining the optimal treatment strategy for a given patient, it is recommended that physicians and patients evaluate the risk‐benefit relationship for use of nivolumab and nivolumab in combination with ipilimumab, taking into account the clinical condition and preferences of individual patients.
Author Contributions
Conception/design: May Tun Saung, Lorraine Pelosof, Sandra Casak, Martha Donoghue, Steven Lemery, Mengdie Yuan, Lisa Rodriguez, Peter Schotland, Meredith Chuk, Gina Davis, Kirsten B. Goldberg, Marc R. Theoret, Richard Pazdur, Lola Fashoyin‐Aje
Provision of study material or patients: May Tun Saung, Lorraine Pelosof, Sandra Casak, Martha Donoghue, Steven Lemery, Mengdie Yuan, Lisa Rodriguez, Peter Schotland, Meredith Chuk, Gina Davis, Kirsten B. Goldberg, Marc R. Theoret, Richard Pazdur, Lola Fashoyin‐Aje
Collection and/or assembly of data: May Tun Saung, Lorraine Pelosof, Sandra Casak, Martha Donoghue, Steven Lemery, Mengdie Yuan, Lisa Rodriguez, Peter Schotland, Meredith Chuk, Gina Davis, Kirsten B. Goldberg, Marc R. Theoret, Richard Pazdur, Lola Fashoyin‐Aje
Data analysis and interpretation: May Tun Saung, Lorraine Pelosof, Sandra Casak, Martha Donoghue, Steven Lemery, Mengdie Yuan, Lisa Rodriguez, Peter Schotland, Meredith Chuk, Gina Davis, Kirsten B. Goldberg, Marc R. Theoret, Richard Pazdur, Lola Fashoyin‐Aje
Manuscript writing: May Tun Saung, Lorraine Pelosof, Sandra Casak, Martha Donoghue, Steven Lemery, Mengdie Yuan, Lisa Rodriguez, Peter Schotland, Meredith Chuk, Gina Davis, Kirsten B. Goldberg, Marc R. Theoret, Richard Pazdur, Lola Fashoyin‐Aje
Final approval of manuscript: May Tun Saung, Lorraine Pelosof, Sandra Casak, Martha Donoghue, Steven Lemery, Mengdie Yuan, Lisa Rodriguez, Peter Schotland, Meredith Chuk, Gina Davis, Kirsten B. Goldberg, Marc R. Theoret, Richard Pazdur, Lola Fashoyin‐Aje
Disclosures
The authors indicated no financial relationships.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1.Jemal A, Ward EM, Johnson CJ et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J Natl Cancer Inst 2017;109:djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Njei B, Rotman Y, Ditah I et al. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015;61:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ha J, Yan M, Aguilar M et al. Race/ethnicity‐specific disparities in cancer incidence, burden of disease, and overall survival among patients with hepatocellular carcinoma in the United States. Cancer 2016;122:2512–2523. [DOI] [PubMed] [Google Scholar]
- 5.Bruix J, Qin S, Merle P et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017;389:56–66. [DOI] [PubMed] [Google Scholar]
- 6.Abou‐Alfa GK, Meyer T, Cheng AL et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med 2018;379:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu AX, Kang YK, Yen CJ et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α‐fetoprotein concentrations (REACH‐2): A randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2019;20:282–296. [DOI] [PubMed] [Google Scholar]
- 8.El‐Khoueiry AB, Sangro B, Yau T et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu AX, Finn RS, Edeline J et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE‐224): A non‐randomised, open‐label phase 2 trial. Lancet Oncol 2018;19:940–952. [DOI] [PubMed] [Google Scholar]
- 10.Yau T, Kang YK, Kim TY et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The CheckMate 040 randomized clinical trial. JAMA Oncol 2020;6:e204564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisenhauer EA, Therasse P, Bogaerts J et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 12.Squibb Bristol‐Myers. Opdivo (nivolumab) [package insert]. Princeton, NJ: Bristol‐Myers Squibb. Revised November 2020. U.S. Food and Drug Administration Web site. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125554s081lbl.pdf. Accessed December 20, 2020.
- 13.Yau T, Park JW, Finn RS et al. CheckMate 459: A randomized, multi‐center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first‐line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann Oncol 2019;30(suppl 5):v874–v875. [Google Scholar]
- 14.Casak SJ, Donoghue M, Fashoyin‐Aje L et al. FDA approval summary: Atezolizumab plus bevacizumab for the treatment of patients with advanced unresectable or metastatic hepatocellular carcinoma. Clin Cancer Res 2021;27:1836–141. [DOI] [PubMed] [Google Scholar]
- 15.El‐Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118–1127. [DOI] [PubMed] [Google Scholar]
- 16.El‐Serag HB, Rudolph KL. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557–2576. [DOI] [PubMed] [Google Scholar]