

Abstract
Our knowledge of the coordination of fuel usage in skeletal muscle is incomplete. Whether and how microRNAs are involved in the substrate selection for oxidation is largely unknown. Here we show that mice lacking miR‐183 and miR‐96 have enhanced muscle oxidative phenotype and altered glucose/lipid homeostasis. Moreover, loss of miR‐183 and miR‐96 results in a shift in substrate utilization toward fat relative to carbohydrates in mice. Mechanistically, loss of miR‐183 and miR‐96 suppresses glucose utilization in skeletal muscle by increasing PDHA1 phosphorylation via targeting FoxO1 and PDK4. On the other hand, loss of miR‐183 and miR‐96 promotes fat usage in skeletal muscle by enhancing intramuscular lipolysis via targeting FoxO1 and ATGL. Thus, our study establishes miR‐183 and miR‐96 as master coordinators of fuel selection and metabolic homeostasis owing to their capability of modulating both glucose utilization and fat catabolism. Lastly, we show that loss of miR‐183 and miR‐96 can alleviate obesity and improve glucose metabolism in high‐fat diet‐induced mice, suggesting that miR‐183 and miR‐96 may serve as therapeutic targets for metabolic diseases.
Keywords: fuel metabolism, lipolysis, metabolic flexibility, miR‐183/96, skeletal muscle
Subject Categories: Metabolism, RNA Biology
miR‐183 and miR‐96 are critical for maintaining the oxidative phenotype of skeletal muscle and whole‐body homeostasis. They regulate fuel selection by coordinating glucose usage and fat catabolism via FoxO1, PDK4 and ATGL in skeletal muscle.
Introduction
Skeletal muscle comprises about 55% of the body weight in most mammals, which is critical for both locomotion and metabolic health. As a dynamic tissue, skeletal muscle exhibits a remarkable plasticity upon a variety of environmental challenges, such as contractile activity, external loading, nutrient availability, and humoral factors, by changing the fuel usage of the myofibers to match structural, functional, and metabolic demands (Schiaffino & Reggiani, 2011). The phenomenon of skeletal muscle plasticity in adjusting myocellular fuel choice according to different nutritional circumstances is also known as metabolic flexibility. For instance, when the glucose level is low upon fasting or exercise, the major energy source for skeletal muscle is switched from glucose to other energy sources such as fatty acids (FAs) to preserve glucose level for glucose‐dependent tissues (Kelley, 2005; Goodpaster & Sparks, 2017).
Grown evidence suggests that impaired metabolic flexibility of skeletal muscle likely contributes to the development of certain chronic diseases, including obesity and type 2 diabetes (T2D) (Goodpaster & Sparks, 2017). Decreases in oxidative metabolism are generally considered to be associated with obesity and diabetes. However, it is still not clear whether obesity or T2D is a cause or consequence of the transition from oxidative to glycolytic metabolism in skeletal muscle (Patti et al, 2003; Petersen et al, 2003). It is worth noting that enhancing either oxidative or glycolytic metabolism in skeletal muscle has beneficial metabolic effects in mouse models of obesity and T2D (Izumiya et al, 2008; Gordon et al, 2009; LeBrasseur et al, 2011; Meng et al, 2013). Thus, a better understanding of the molecular basis of fuel metabolism in skeletal muscle will provide valuable clues to enable the development of new agents or interventions to combat metabolic diseases.
It is widely accepted that the substrate‐dependent allosteric mechanism proposed by Randle is not appropriate to explain the insulin resistance in obesity and T2D (Petersen & Shulman, 2018). Moreover, the regulation of oxidative substrate selection is known to be far more complex than originally thought. Besides extensively studied glycolysis and FA oxidation, lipid droplet metabolism has been implicated in metabolic flexibility, since the intracellular fatty acyl‐CoAs used for oxidation can be generated from lipolysis (Bosch et al, 2020). It also has been suggested that the triglyceride (TAG) turnover is not only highly dynamic but also essential for mitochondrial biogenesis and FA oxidation because the lipolytic products can serve as endogenous ligands for metabolic nuclear receptor PPARs (Banke et al, 2010; Haemmerle et al, 2011). However, it remains to be established how the metabolic pathways for glucose and fat utilization in skeletal muscle, including intramuscular lipolysis, are coordinated in oxidative substrate selection.
The substrate utilization in skeletal muscle is tightly controlled at different levels by diverse mechanisms (e.g., allosteric, transcriptional, post‐transcriptional, and posttranslational). MicroRNAs (miRNAs) are small noncoding RNAs that control gene expression at post‐transcriptional level. It has been suggested that miRNAs are highly dynamic in nature and able to influence gene expression more quickly than transcriptional repressors can. Owing to this property, miRNAs are strategically located in loops that confer robustness and precision (Tsang et al, 2007; Inui et al, 2010). Previously, we established miR‐182 as a critical regulator that confers robust and precise controls on glucose utilization in skeletal muscle (Zhang et al, 2016), and however, the roles of miR‐183 and miR‐96, which are expressed as a cluster with miR‐182 and share similar sequences, in fuel metabolism has not been examined, especially with respect to modulating fat utilization.
Here, we show that miR‐183 and miR‐96 double knockout (DKO mice) not only exhibit enhanced muscle oxidative phenotype, altered glucose and lipid homeostasis, as well as altered substrate utilization, but also display resistance to high‐fat diet (HFD)‐induced obesity. Mechanistic studies reveal that miR‐183 and miR‐96 promote glucose utilization by modulating pyruvate dehydrogenase (PDH) which links the glycolytic pathway to the tricarboxylic acid cycle via targeting FoxO1 and PDK4. On the other hand, miR‐183 and miR‐96 inhibit fat utilization by repressing intramuscular TAG lipolysis through targeting FoxO1 and ATGL, which modulates not only the FA supply for mitochondrial oxidation but also the PPARδ‐mediated mitochondrial biogenesis and FA oxidation. Thus, we establish miR‐183 and miR‐96 as master coordinators of fuel selection in skeletal muscle and potential therapeutic targets for metabolic diseases.
Results
Loss of miR‐183 and miR‐96 enhances muscle oxidative phenotype
To define the role of skeletal muscle miR‐183 and miR‐96 in fuel metabolism, we took advantage of mice deficient in both miR‐183 and miR‐96 developed previously, which have normal levels of miR‐182 in skeletal muscle and other tissues, such as liver and white adipose tissue (WAT) (Figs 1A and EV1A) (Xiang et al, 2017). Hematoxylin–eosin (HE) staining results showed that the myofiber nuclei were distributed normally and the number of myofibers was not altered in the skeletal muscles of DKO mice (Fig EV1B). Accordingly, no obvious changes in the mRNA levels of either MyoD or Myf5, two key genes essential for the normal progression of the skeletal muscle developmental program (Rudnicki et al, 1993), were observed in the skeletal muscles of DKO mice (Fig EV1C). These data suggest that miR‐183 and miR‐96 are not required for the development of a normal skeletal muscle system.
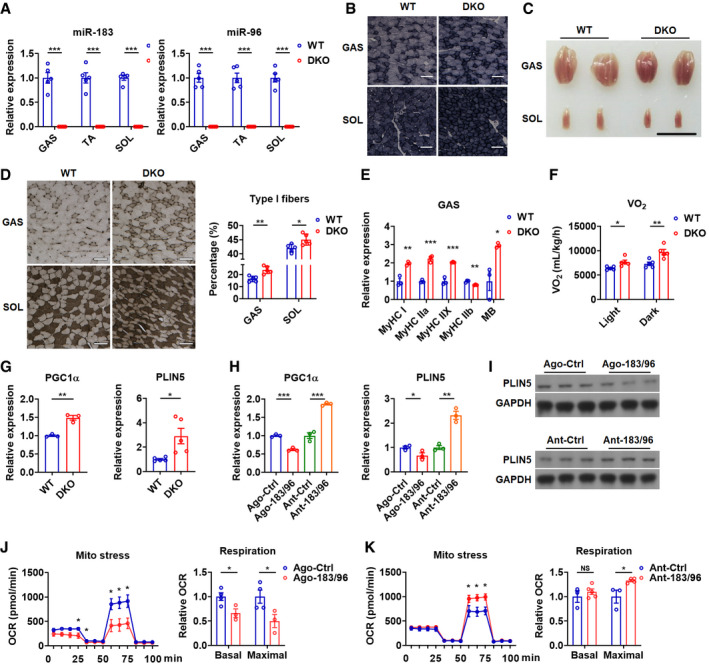
Figure 1. Loss of miR‐183 and miR‐96 enhances muscle oxidative phenotype.
-
ARelative expression of miR‐183 and miR‐96 in GAS, TA and SOL muscles of WT and DKO mice (n = 5).
-
BSDH staining of GAS and SOL muscles in WT and DKO mice (n = 5). Scale bars: 100 μm.
-
CRepresentative photographs of GAS and SOL muscles dissected from WT and DKO mice (n = 5). Scale bar: 1 cm.
-
DATPase staining of GAS and SOL muscles of WT and DKO mice (left). Corresponding percentage of type I fibers were determined (right) (n = 5). Scale bars: 100 μm.
-
ERelative mRNA expression of MHC isoforms in GAS muscles of WT and DKO mice (n = 3).
-
FOxygen consumption rate of WT and DKO mice in light and dark phases (n = 5).
-
GRelative mRNA expression of PGC1α and PLIN5 in GAS muscles of WT and DKO mice (n = 3 for PGC1α analysis; n = 5 for PLIN5 analysis).
-
HRelative mRNA expression of PGC1α and PLIN5 in C2C12 cells transfected with miR‐183 and miR‐96 agomirs (Ago‐183/96) or antagomirs (Ant‐183/96) (n = 3).
-
IWestern blot analysis of PLIN5 in C2C12 cells transfected with Ago‐183/96 or Ant‐183/96.
-
J, KMitochondrial respiration profile of C2C12 cells transfected with Ago‐183/96 (J) or Ant‐183/96 (K). Cellular oxygen consumption rate (OCR) was measured in real time, and corresponding basal and maximal respiration were analyzed by overall OCR (n = 3 or 4 per group).
Data information: Means ± SEM are shown for all panels except for left panels in panel J and K, in which means ± SD are shown. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
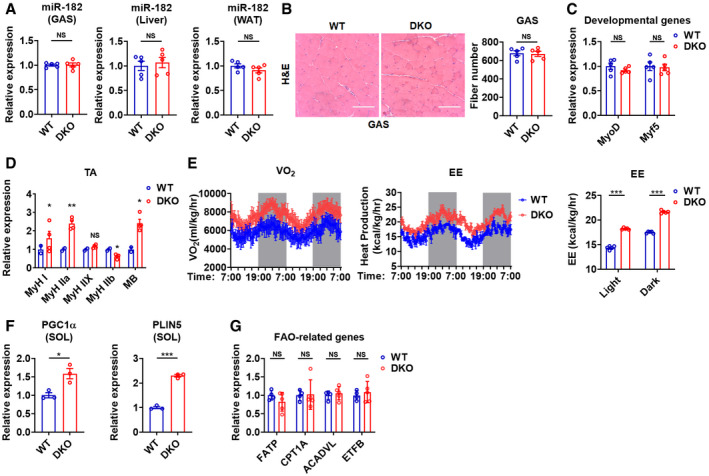
Figure EV1. Loss of miR‐183 and miR‐96 enhances muscle oxidative phenotype, related to Fig 1 .
-
ARelative expression of miR‐182 mRNA in GAS muscles, liver and inguinal WAT of WT and DKO mice (n = 5).
-
BHE staining of GAS muscles of WT and DKO mice (left). The total fiber number of GAS muscle was determined (right, n = 5).
-
CRelative mRNA expression of MyoD and Myf5 in GAS muscles of WT and DKO mice (n = 5).
-
DRelative expression of MHC isoforms mRNA in TA muscles of WT and DKO mice (n = 2 or 4).
-
EWhole‐body oxygen consumption rate (left) and energy expenditure (middle) of WT and DKO mice in light and dark phases (n = 5). Energy expenditure rate of WT and DKO mice in light and dark phases (n = 5).
-
FRelative mRNA expression of PGC1α and PLIN5 in SOL muscles of WT and DKO mice (n = 3).
-
GRelative mRNA expression of fatty acid oxidation (FAO)‐related genes in GAS muscles of WT and DKO mice (n = 4 for WT; n = 5 for DKO).
Data information: Means ± SEM are shown for all panels. *P < 0.05; **P < 0.01; NS, not significant (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.Source data are available online for this figure.
Interestingly, we found that the succinic dehydrogenase (SDH) staining was increased in either glycolytic, fast (type II) fiber‐enriched gastrocnemius (GAS) or oxidative, slow (type I) fiber‐enriched soleus (SOL) muscles of DKO mice as compared to wild‐type (WT) control mice (Fig 1B), indicating a transition from glycolytic to oxidative metabolism. Since type I fibers are highly oxidative and also known as red fibers, the observation that GAS and SOL muscles of DKO mice appeared redder than that of WT control mice suggests that the increased muscle oxidative phenotype in DKO mice might be attributed to an increase in type I fiber composition (Fig 1C).
To test this possibility, we analyzed the myofiber type composition in DKO mice. As expected, the results of ATPase staining revealed that the percentage of type I fiber was markedly increased in GAS and SOL muscles of DKO mice as compared to WT mice (Fig 1D). Accordingly, a fast‐to‐slow shift in MyHC expression was observed in either GAS or tibialis anterior (TA) muscles of DKO mice (Figs 1E and EV1D). In agreement with the enhanced muscle oxidative phenotype observed in DKO mice, the oxygen consumption and energy expenditure were increased in DKO mice (Figs 1F and EV1E). Consistently, increased mRNA expression of PGC1α and PLIN5, two key genes involved in oxidative metabolism, was observed in GAS and SOL muscles of DKO mice (Figs 1G and EV1F). In contrast, no differences in the mRNA levels of other genes involved in fatty acid transport and oxidation, such as FATP, CPT1A, ACADVL, and ETFB, were observed in GAS muscles of DKO mice (Fig EV1G).
In agreement with these in vivo data, overexpression of miR‐183 and miR‐96 by transfection of pooled agomirs for miR‐183 and miR‐96 (Ago‐183/96) repressed the mRNA and protein expression of PGC1α and PLIN5, while inhibition of miR‐183 and miR‐96 by administration of pooled antagomirs for miR‐183 and miR‐96 (Ant‐183/96) increased the mRNA and protein levels of PGC1α and PLIN5 in C2C12 myocytes (Fig 1H and I). Accordingly, oxygen consumption rate (OCR) analysis revealed that the maximal respiration was decreased by Ago‐183/96 treatment, but increased by Ant‐183/96 treatment in C2C12 myocytes (Fig 1J and K), further supporting the notion that miR‐183 and miR‐96 might play critical roles in regulating muscle oxidative metabolism. Collectively, these in vitro results also suggest that miR‐183 and miR‐96 might regulate the metabolic phenotype of skeletal muscle in a cell autonomous manner.
Loss of miR‐183 and miR‐96 alters glucose and lipid homeostasis
To test whether the glycolytic to more oxidative myofiber type conversion in the skeletal muscle of DKO mice would have any effect on whole‐body metabolic homeostasis, we first examined the blood glucose levels in these mice. No apparent difference in random blood glucose levels was observed between DKO and WT mice (Fig 2A). Interestingly, the fasting blood glucose levels were significantly increased in DKO mice as compared to WT mice (Fig 2B), suggesting that the glucose metabolism is altered in the absence of miR‐183 and miR‐96. Moreover, the glucose tolerance was decreased in DKO mice (Fig 2C), further suggesting that miR‐183 and miR‐96 are required for maintaining normal glucose homeostasis. Accordingly, the insulin levels were increased DKO mice (Fig EV2A), further supporting the notion that DKO mice have altered glucose metabolism. As the pyruvate tolerance and the expression of hepatic gluconeogenic genes were not significantly altered in DKO mice (Fig EV2B and C), we speculated that the gluconeogenic capacity might not be impaired in the liver of DKO mice and the altered glucose metabolism observed might be probably attribute to the abnormality in glucose utilization in the skeletal muscle of DKO mice.
Figure 2. Loss of miR‐183 and miR‐96 alters glucose and lipid homeostasis and fuel metabolism in skeletal muscle.

-
A, BRandom (A) and fasting (B) blood glucose levels of WT and DKO mice (n = 8 for DKO fasted group, n = 10 for all other groups).
-
CGlucose tolerance test (GTT) and area under the curve (AUC) data for GTT in WT and DKO mice (n = 8 for DKO, n = 10 for WT).
-
DHE staining of the iWAT and eWAT of WT and DKO mice. Scale bars: 100 μm.
-
EAnalysis of fat mass of WT and DKO mice by NMR technique (n = 7) (left) and tissue weight of iWAT, eWAT, and iBAT (right) (n = 4).
-
FSerum levels of triglycerides (TAG) and non‐esterified free fatty acids (NEFA) in WT and DKO mice (n = 5).
-
GRespiratory exchange ratio (RER) of WT and DKO mice in light and dark phases (n = 5).
-
HRelative mRNA expression of FoxO1 and PDK4 in GAS muscles of WT and DKO mice (n = 5).
-
IWestern blot analysis of FoxO1, PDK4, p‐PDHA1, and PDHA1 in GAS muscles of WT and DKO mice.
-
JWestern blot analysis of FoxO1, PDK4, p‐PDHA1, and PDHA1 in C2C12 cells transfected with Ago‐183/96 (left) or Ant‐183/96 (right).
-
KRelative mRNA expression of ATGL and HSL in GAS muscles of WT and DKO mice (n = 5).
-
LWestern blot analysis of HSL and ATGL in GAS muscles of WT and DKO mice.
-
MWestern blot analysis of HSL and ATGL in C2C12 cells transfected with Ago‐183/96 (left) or Ant‐183/96 (right).
-
N–QLD area per cell was quantified at the indicated time points to measure LD turnover in C2C12 myocytes transfected with agomiR‐183/96 (N) or antagomir‐183/96 (P) stained with Nile red (n = 5). Representative immunofluorescence images of lipid droplets in C2C12 cells transfected with agomiR‐183/96 (O) or antagomir‐183/96 (Q). Scale bars: 5 μm.
Data information: Means ± SEM are shown for all panels. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
Figure EV2. Loss of miR‐183 and miR‐96 alters glucose and lipid homeostasis and fuel metabolism in skeletal muscle, related to Fig 2 .

-
AThe serum insulin levels of WT and DKO mice were determined (n = 5).
-
BPyruvate tolerance test (PTT) were performed in WT mice and DKO mice (n = 10 for WT; n = 12 for DKO) (left). Area under the curve (AUC) data for PTT tests were calculated (right).
-
CRelative mRNA expression of gluconeogenic genes in the liver of WT and DKO mice (n = 5).
-
DRelative mRNA expression of thermogenic genes in the BAT of WT and DKO mice (n = 5).
-
EFood intake (left) and physical activity (right) of WT and DKO mice (n = 5). Light: light phase; Dark: dark phase.
-
F, GRespiratory exchange ratio (RER) and mean ΔRER of WT and DKO mice in light and dark phases (n = 5).
-
HRelative mRNA expression of lipogenic genes in the liver of WT and DKO mice (n = 5).
-
IHE staining of the liver of WT and DKO mice. Scale bar: 100 μm.
-
JWestern blot analysis of FoxO1, PDK4, p‐PDHA1, and PDHA1 in SOL muscles of WT and DKO mice (left). Western blot analysis of HSL and ATGL in SOL muscles of WT and DKO mice (right).
-
KRelative mRNA expression of glycolytic genes in GAS muscles of WT and DKO mice (n = 5).
-
LLactate, pyruvate, and LDHA mRNA levels were determined in the GAS muscles of WT and DKO mice (n = 5).
-
MLactate, pyruvate, and LDHA mRNA levels were determined in the C2C12 cells treated with Ago‐183/96 or Ant‐183/96 (n = 5).
-
N, OqPCR analysis of relative miR‐183 (N) and miR‐96 (O) levels in different tissues (Ep, epididymal fat; Kid, kidney; Liv, liver; Lun, lung; Mus, skeletal muscle; Pan, pancreas; Spl, spleen) from C57BL/6J mice as indicated (n = 3).
-
P, QRelative mRNA expression (P) and Western analysis (Q) of FoxO1, PDK4, ATGL, HSL, and PLIN5 in the inguinal WAT of WT and DKO mice (n = 5).
-
R, SRelative mRNA expression (R) and Western analysis (S) of FoxO1, PDK4, ATGL, HSL, and PLIN5 in the liver of WT and DKO mice (n = 5).
Data information: Means ± SEM are shown for all panels. *P < 0.05; NS, not significant (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.Source data are available online for this figure.
Besides glucose metabolism, lipid homeostasis was also investigated in DKO mice. Interestingly, the HE staining results indicate that the size of the inguinal WAT (iWAT) and epididymal WAT (eWAT) in DKO mice was smaller than that in WT mice, respectively (Fig 2D). The body composition analysis by using NMR technique revealed a significant decrease in overall fat mass of in DKO mice (Fig 2E). Consistently, the weight of either iWAT or eWAT was markedly reduced in DKO mice (Fig 2E). No difference in the weight of interscapular brown adipose tissue (iBAT) was noticed between WT and DKO mice (Fig 2E). Accordingly, the mRNA expression of thermogenic genes, UCP1 and PRDM16, was not altered in the BAT of DKO mice, indicating that the BAT‐mediated adaptive thermogenesis might remain unaltered in DKO mice (Fig EV2D). Interestingly, decreased serum TAG and NEFA levels were observed in DKO mice, which might be probably attributed to the reduced adiposity (Fig 2F). As DKO mice had a lean phenotype, while the food intake and physical activity were not significantly altered in DKO mice (Fig EV2E), we speculated that the fat utilization might be increased in DKO mice. In accordance with this hypothesis, results from in vivo indirect calorimetry showed that the respiratory exchange ratio (RER) was decreased in DKO mice in both light and dark phases, suggesting a shift in substrate utilization toward carbohydrates relative to fat (Figs 2G and EV2F). Notably, the differences in RER between the light and dark phases were smaller in DKO mice than those in WT mice, further suggesting that the metabolic flexibility is altered in the absence of miR‐183 and miR‐96 (Fig EV2G). It is worth noting that, although DKO mice had defects in glucose utilization, the increase in glucose levels would not drive hepatic de novo lipogenesis, as evident from unaltered mRNA expression of lipogenic genes in the liver and normal liver histology in DKO mice (Fig EV2H and I).
Loss of miR‐183 and miR‐96 alters the fuel metabolism in skeletal muscle
Since FoxO1‐PDK4‐PDH axis plays an essential role in glucose usage in skeletal muscle (Petersen & Shulman, 2018), we examined this axis in DKO mice. Interestingly, the mRNA and protein levels of either FoxO1 or PDK4 were increased in GAS muscles of DKO mice as compared to WT mice (Fig 2H and I). Accordingly, the phosphorylation of alpha subunit of PDH (PDHA1) was increased in GAS muscles of KO mice, suggesting that the glucose utilization was suppressed in the absence of miR‐183 and miR‐96 (Fig 2H and I). Similar results were observed in SOL muscles of DKO mice as compared to WT mice (Fig EV2J). In agreement with the in vivo data, Ago‐183/96 treatment decreased the protein levels of FoxO1 and PDK4 and the phosphorylation of PDHA1, while administration of Ant‐183/96 increased the FoxO1 and PDK4 protein levels and the PDHA1 phosphorylation in myocytes (Fig 2J). These in vitro results also suggest that miR‐183 and miR‐96 regulate the glucose utilization through FoxO1‐PDK4‐PDH axis in skeletal muscle in a cell autonomous manner. Notably, the mRNA expression of glycolytic genes, HK1, PFK, and PKM was not changed in GAS muscles of DKO mice (Fig EV2K). Given that the FoxO1‐PDK4‐PDH axis was impaired in DKO mice, we speculated that the loss of miR‐183 and miR‐96 might affect glycolytic flux mainly through targeting the PDH, which links the glycolytic pathway to the tricarboxylic acid cycle. In agreement with the above findings, the pyruvate levels were increased in GAS muscles of DKO mice (Fig EV2L). However, the lactate levels in GAS muscles of DKO mice were not increased, which might be attributed to the decreased expression of LDHA, which is responsible for the pyruvate‐to‐lactate flux (Fig EV2L). Consistent results could be obtained in myocytes transfected with Ago‐183/96 or Ant‐183/96 (Fig EV2M).
As PLIN5 plays a role in channeling FAs from lipid droplet toward mitochondria (MacPherson & Peters, 2015), the increased expression of PLIN5 in the skeletal muscle of DKO mice (Fig 1G and I) prompted us to examine whether the intramuscular lipolysis, a critical process that supports FA oxidation by supplying FAs for mitochondrial oxidation and providing ligands to activate PPAR signaling, was also affected. Interestingly, the mRNA and protein levels of both ATGL and HSL, two key TAG lipases, were markedly increased in GAS muscles of DKO mice as compared to WT mice (Fig 2K and L), suggesting that the intramuscular lipolysis was enhanced in the absence of miR‐183 and miR‐96. Increased protein levels of ATGL and HSL were also observed in SOL muscles of DKO mice (Fig EV2J). In agreement with these in vivo data, Ago‐183/96 treatment could decrease the protein levels of ATGL and HSL, while Ant‐183/96 administration was able to elevate the protein levels of these two lipases in myocytes (Fig 2M), indicating that miR‐183 and miR‐96 regulate the expression of ATGL and HSL in skeletal muscle in a cell autonomous manner. In line with these observations, Ago‐183/96 treatment repressed the rate of lipid droplet turnover, while Ant‐183/96 administration accelerated the rate of lipid droplet turnover in muscle cells (Fig 2N–Q). Based on these findings and the above observations (Fig 1), we speculated that miR‐183 and miR‐96 might control fat utilization in skeletal muscle by regulating TAG lipolysis and FA oxidation.
Since miR‐183 and miR‐96 are ubiquitously expressed in a variety of tissues (Fig EV2N and O), we then examined the expression of the aforementioned genes in other metabolic tissues, including WAT and liver, to exclude the possibility that the altered expression of these genes in other tissues due to miR‐183 and miR‐96 deficiency would also have contribution in DKO mice. We found that the expression of FoxO1, PDK4, ATGL, HSL, and PLIN5 was not altered in the WAT of DKO mice (Fig EV2P and Q). Increases in the mRNA expression of FoxO1 and PDK4 were observed in the liver of DKO mice, and however, the protein levels of FoxO1 and PDK4 were not changed (Fig EV2R and S). Both mRNA and protein levels of ATGL, HSL, and PLIN5 were not changed in the liver of DKO mice (Fig EV2R and S). Collectively, although our above data do not exclude the possibility that lacking miR‐183 and miR‐96 in other tissues might also have an effect on metabolic homeostasis, we speculated that the metabolic phenotype observed in DKO mice above might be likely attributed to the deficiency of miR‐183 and miR‐96 in skeletal muscle.
FoxO1 and PDK4 are target genes of miR‐183 and miR‐96 in skeletal muscle
As miR‐182 modulates glucose utilization in skeletal muscle by targeting FoxO1 and PDK4 (Karolina et al, 2011), while miR‐183, miR‐96, and miR‐182 share similar sequences, we then tested whether FoxO1 and PDK 4 also are target genes of miR‐183 and miR‐96. As expected, we identified a putative target site for both miR‐183 and miR‐96 in either FoxO1‐3′UTR or PDK4‐3′UTR, which is conserved across different species (Fig 3A and B). Moderate overexpression of either miR‐183 or miR‐96 by specific agomirs could decrease the mRNA and protein levels of FoxO1 and PDK4, while inhibition of either miR‐183 or miR‐96 by specific antagomirs could increase the mRNA and protein levels of FoxO1 and PDK4 in myocytes (Fig 3C–F). Similar results were obtained when Ago‐183/96 or Ant‐183/96 were used for transfection (Fig EV3, EV4, EV5). Additionally, administration of Ago‐183/96 could inhibit the activity of the reporter containing FoxO1‐3′UTR or PDK4‐3′UTR with the miRNA‐responsive element, while Ant‐183/96 administration could increase the activity of these two reporters in cells, respectively (Fig EV3D). Consistently, overexpression of either miR‐183 or miR‐96 by specific mimics could repress the activity of these two reporters (Fig EV3E and F). Collectively, these results suggest that both FoxO1 and PDK4 are target genes of miR‐183 and miR‐96, which contribute to the regulation of glucose utilization in the skeletal muscle by miR‐183 and miR‐96.
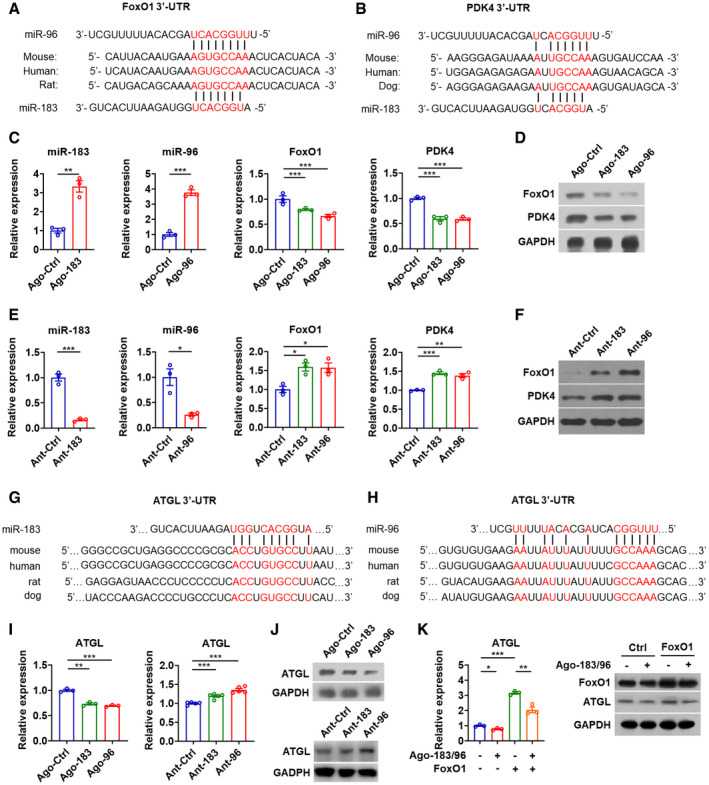
Figure 3. FoxO1, PDK4, and ATGL are target genes of miR‐183 and miR‐96 in skeletal muscle.
-
A, BSequence alignment of miR‐183 and miR‐96 and their target sites in the 3′UTR of FoxO1 (A) or PDK4 (B) from various species.
-
CRelative expression of miR‐183 and miR‐96 in C2C12 myocytes transfected with miR‐183 or miR‐96 agomir (left) (n = 3). Relative mRNA expression of FoxO1 and PDK4 in C2C12 cells treated with Ago‐183 or Ago‐96 (right) (n = 3).
-
DWestern blot analysis of FoxO1 and PDK4 in C2C12 cells treated with Ago‐183 or Ago‐96.
-
ERelative expression of miR‐183 and miR‐96 in C2C12 myocytes transfected with miR‐183 or miR‐96 antagomir (left) (n = 3). Relative mRNA expression of FoxO1 and PDK4 in C2C12 cells treated with Ant‐183 or Ant‐96 (right) (n = 3).
-
FWestern blot analysis of FoxO1 and PDK4 in C2C12 cells treated with Ant‐183 or Ant‐96.
-
G, HSequence alignment of miR‐183 (G) and miR‐96 (H) and their target sites in the 3′UTR of ATGL from various species.
-
I, JRelative mRNA expression (I, left) and Western blot analysis (J, top) of ATGL in C2C12 cells transfected with agomir for miR‐183 (Ago‐183) or miR‐96 (Ago‐96). Relative mRNA expression (I, right) and Western blot analysis (J, bottom) of ATGL in C2C12 cells transfected with antagomir for miR‐183 (Ant‐183) or miR‐96 (Ant‐96). (n = 5 per group for RT–PCR analysis).
-
KRelative mRNA expression of ATGL and Western blot analysis of FoxO1 and ATGL in C2C12 cells transfected with Ago‐183/96 and FoxO1 as indicated (n = 3).
Data information: Means ± SEM are shown for all panels. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
Figure EV3. FoxO1 and PDK4 are target genes of miR‐183 and miR‐96 in skeletal muscle, related to Fig 3 .

-
ARelative mRNA expression of FoxO1 (left) and PDK4 (right) in C2C12 cells treated with Ago‐183/96 or Ant‐183/96 (n = 3 for FoxO1 analysis; n = 5 for PDK4 analysis).
-
B, CWestern blot analysis of FoxO1 (B) and PDK4 (C) in C2C12 cells treated with Ago‐183/96 or Ant‐183/96.
-
DThe activity of the reporter containing the 3′UTR of FoxO1 or PDK4 was determined in HEK 293T cells transfected with Ago‐183/96, or Ant‐183/96 as indicated (n = 6 for FoxO1 3′UTR; n = 5 for PDK4 3′UTR).
-
EThe relative expression of miR‐183 and miR‐96 in C2C12 cells transfected with miR‐183 mimics (left) or miR‐96 mimics (right) as indicated (n = 3).
-
FThe activity of the reporter containing the 3′UTR of FoxO1 (left) and PDK4 (right) was determined in HEK 293T cells transfected with mimics for miR‐183 (183 mimics) or miR‐96 (96 mimics) (n = 6 for FoxO1 3′UTR; n = 5 for PDK4 3′UTR).
-
GThe activity of the reporter containing the 3′UTR of ATGL was determined in HEK 293T cells transfected with Ago‐183/96, or Ant‐183/96 as indicated (n = 6).
-
HThe activity of the reporter containing the 3′UTR of ATGL was determined in HEK 293T cells transfected with mimics for miR‐183 (183 mimics) or miR‐96 (96 mimics) (n = 6).
-
IRelative mRNA expression of ATGL in C2C12 cells treated with Ago‐183/96 or Ant‐183/96 (n = 3).
-
JSequence alignment of miR‐182 and its target site in the 3′UTR of ATGL from various species.
-
KRelative mRNA expression of ATGL in C2C12 cells treated with Ago‐182 or Ant‐182 (left) (n = 3). Western blot analysis of ATGL in C2C12 cells treated with Ago‐182 or Ant‐182 (right).
-
LRelative mRNA expression of miR‐182 (left) and primary miR‐183 cluster (right) in C2C12 cells treated with Ant‐183/96 (n = 3).
-
MRelative mRNA expression of miR‐183, miR‐96, miR‐182, FoxO1, PDK4, ATGL, HSL, PGC1α, and PLIN5 in Cas9‐mediated miR‐183 and miR‐96 knockout C2C12 cells (n = 3).
Data information: Means ± SEM are shown for all panels. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.Source data are available online for this figure.
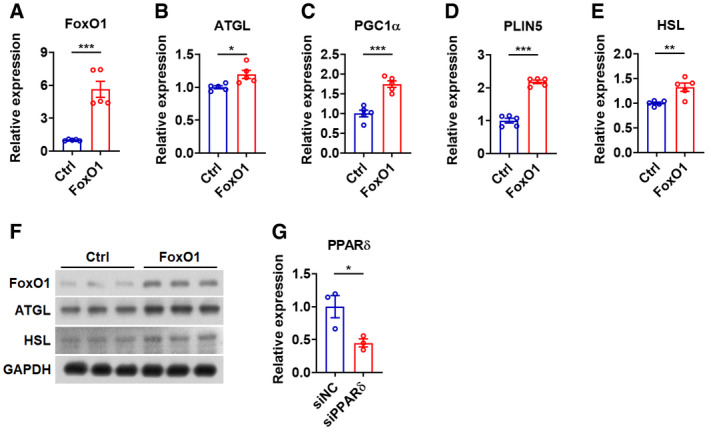
Figure EV4. miR‐183 and miR‐96 modulate fat usage via FoxO1 and ATGL in muscle cells, related to Fig 4 .
-
A–ERelative mRNA expression of FoxO1 (A), ATGL (B), PGC1α (C), PLIN5 (D), and HSL (E) in C2C12 cells transfected with FoxO1 (n = 5).
-
FWestern blot analysis of FoxO1, ATGL, and HSL in C2C12 cells transfected with FoxO1.
-
GRelative mRNA expression of PPARδ in C2C12 cells transfected with siPPARδ (n = 3). siNC: negative control oligos for siRNA.
Data information: Means ± SEM are shown for all panels. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.Source data are available online for this figure.
Figure EV5. miR‐183 and miR‐96 levels in skeletal muscles alter upon HFD feeding and loss of miR‐183 and miR‐96 protects mice from diet‐induced obesity and glucose intolerance, related to Figs 5 and 6 .
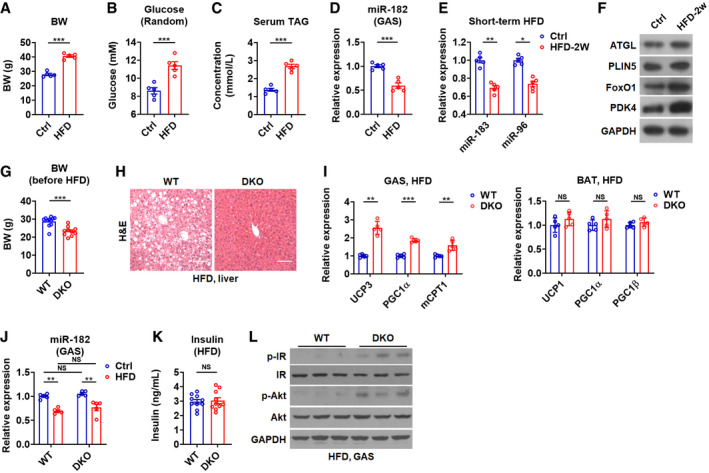
- Body weight of C57BL/6 mice after a HFD feeding for 2 months (n = 5).
- Random glucose levels of C57BL/6 mice after a HFD feeding for 2 months (n = 5).
- Serum TAG levels of C57BL/6 mice after a HFD feeding for 2 months (n = 5).
- Relative mRNA expression of miR‐182 in GAS muscles of C57BL/6 mice after HFD feeding (n = 5).
- Relative mRNA expression of miR‐183 and miR‐96 in the GAS muscles of mice after 2 weeks of HFD feeding (n = 5).
- Western blot analysis of ATGL, PLIN5, FoxO1, and PDK4 in the GAS muscles of mice after 2 weeks of HFD feeding.
- Body weight of C57BL/6 mice before a HFD feeding (n = 10–11).
- H&E staining of the liver of WT and DKO mice after a HFD feeding Scale bar: 100 μm.
- Relative mRNA expression of UCP3, PGC1α, and mCPT1 in GAS muscles of WT and DKO mice after a HFD feeding (n = 5) (left). Relative mRNA expression of UCP1, PGC1α, and PGC1β in the BAT of WT and DKO mice after a HFD feeding (n = 5) (right).
- Relative mRNA expression of miR‐182 in GAS muscles of WT and DKO mice after a HFD feeding (n = 5).
- Serum insulin levels in WT and DKO mice after a HFD feeding (n = 10) were determined.
- Western blot analysis of p‐IR, IR, p‐Akt, and Akt in GAS muscles of WT and DKO mice after a HFD feeding.
Data information: Means ± SEM are shown for all panels. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.Source data are available online for this figure.
ATGL is a direct target gene of miR‐183 and miR‐96 in skeletal muscle
To explore molecular mechanisms underlying the effect of miR‐183 and miR‐96 on the fat utilization in skeletal muscle, we checked the literatures and Targetscan to identify putative miR‐183 and miR‐96 target genes that may be involved in the regulation of TAG lipolysis and FA oxidation. Interestingly, we found that ATGL itself is a predicted miR‐183 and miR‐96 target gene. Putative target sites for miR‐183 and miR‐96 in the ATGL‐3′UTR were identified, which are conserved across different species (Fig 3G and H). Moderate overexpression of either miR‐183 or miR‐96 by using corresponding agomir decreased the mRNA and protein levels of ATGL, while inhibition of either miR‐183 or miR‐96 by using corresponding antagomir increased the mRNA and protein levels of ATGL in myocytes (Fig 3I and J). Accordingly, Ago‐183/96 treatment repressed the luciferase activity of the reporter containing ATGL‐3′UTR with miRNA‐responsive elements in cells, while Ant‐183/96 treatment enhanced the luciferase activity of this reporter (Fig EV3G). In line with these data, overexpression of either miR‐183 or miR‐96 by corresponding mimics significantly repressed the luciferase activity of the reporter containing ATGL‐3′UTR with miRNA‐responsive elements in cells (Fig EV3H). Consistently, Ago‐183/96 treatment decreased the mRNA levels of ATGL, while Ant‐183/96 treatment increased the mRNA expression of ATGL (Fig EV3I). These results indicate that ATGL is a direct target gene of miR‐183 and miR‐96 in muscle cells.
According to the prediction by Targetscan, miR‐182 and miR‐96 share same target site in the ATGL‐3′UTR (Fig EV3J). As expected, miR‐182 could negatively regulate the ATGL mRNA and protein expression, indicating that ATGL is also a target gene of miR‐182 (Fig EV3K). As Ago‐183/96 treatment did not affect the expression of either pri‐miR‐183c or miR‐182 (Fig EV3L), we speculated that miR‐183 and miR‐96 could directly affect the expression of target genes, FoxO1, PDK4, and ATGL, independent of miR‐182. We also employed another approach to inhibit the action of miR‐183 and miR‐96 by using CRISPR/Cas9‐mediated gene knockout in C2C12 myocytes. In agreement with our above findings, without changing the expression of miR‐182, knockout of miR‐183 and miR‐96 could not only elevate the mRNA levels of target genes, FoxO1, PDK4, and ATGL, but also increase the mRNA expression of those genes altered in GAS muscles of DKO mice, such as HSL, PGC1α, and PLIN5 (Fig EV3M).
As ATGL is also a direct target gene of FoxO1 (Chakrabarti & Kandror, 2009), to test whether the effect of miR‐183 and miR‐96 on ATGL expression was not solely dependent on FoxO1, FoxO1 expression plasmid lacking 3′UTR and Ago‐183/96 were co‐transfected into the myocytes. In agreement with the notion that ATGL is a direct target gene of miR‐183 and miR‐96, overexpression of FoxO1 transcripts lacking 3′UTR could not totally abolish the repressive effect of miR‐183 and miR‐96 on both mRNA and protein expression of ATGL (Fig 3K), suggesting that miR‐183 and miR‐96 regulate the ATGL expression either directly or indirectly through FoxO1. Collectively, these results also indicate that miR‐183 and miR‐96 might regulate ATGL‐mediated intramuscular lipolysis, thereby controlling the FA supply for mitochondrial oxidation in skeletal muscle.
miR‐183 and miR‐96 modulate fat usage via FoxO1 and ATGL in muscle cells
As intracellular lipolysis might also contribute to the mitochondrial oxidation by generating lipid ligands for PPAR activation and PPARδ is expressed at the highest level in skeletal muscle (Gan et al, 2011; Fritzen et al, 2020), we then tested whether an increase in ATGL expression would upregulate the expression of PPARδ target genes involved in the regulation of mitochondrial biogenesis and FA oxidation, including PGC1α and PLIN5. Consistent with our current knowledge, overexpression of ATGL increased the mRNA levels of PGC1α and PLIN5 in myocytes (Fig 4A). It is worth noting that, since HSL is also regulated by PPARδ (Tanaka et al, 2003), overexpression of ATGL increased the mRNA expression of HSL, which might in turn further enhance the ATGL‐activated lipolysis (Fig 4A). Importantly, knockdown of ATGL by specific siRNA significantly attenuated the effect of Ant‐183/96 treatment on the mRNA expression of PGC1α, PLIN5, and HSL in myocytes, indicating that ATGL might be a primary target gene mediating the effect of miR‐183 and miR‐96 (Fig 4B–D). In addition, overexpression of FoxO1 transcripts lacking 3′UTR not only increased the mRNA expression of PGC1α, PLIN5, and HSL in myocytes (Fig EV4, EV5), but also attenuated the suppressive effect of miR‐183 and miR‐96 overexpression on the mRNA expression of ATGL, PGC1α, PLIN5, and HSL (Fig 4E–H), further supporting the notion that both ATGL and its upstream regulator FoxO1 are direct target genes of miR‐183 and miR‐96. More importantly, knockdown of PPARδ by specific siRNA significantly attenuated the effect of Ant‐183/96 treatment on the mRNA expression of PGC1α, PLIN5, and HSL (Figs 4I and EV4G). Collectively, our above results suggest that miR‐183 and miR‐96 modulate the fat utilization in skeletal muscle by regulating lipolysis via targeting FoxO1 and ATGL, which increases not only the FA supply for mitochondrial oxidation but also the availability of lipid ligands that promote the PPARδ‐mediated mitochondrial biogenesis and FA oxidation.
Figure 4. miR‐183 and miR‐96 modulate fat usage via FoxO1 and ATGL in muscle cells.

-
ARelative mRNA expression of ATGL, PGC1α, PLIN5, and HSL in C2C12 cells transfected with ATGL (n = 5).
-
BRelative mRNA expression and Western blot analysis of ATGL in C2C12 cells transfected with Ant‐183/96 and siRNA of ATGL (siATGL) as indicated (n = 3 per group for RT–PCR analysis).
-
C, DRelative mRNA expression (C) and Western blot analysis (D) of PGC1α, PLIN5, and HSL in C2C12 cells transfected with Ant‐183/96 and siATGL as indicated (n = 3 per group for RT–PCR analysis).
-
E, FRelative mRNA expression of ATGL (E) and Western blot analysis (F) of FoxO1 and ATGL in C2C12 cells transfected with Ago‐183/96 and FoxO1 as indicated (n = 5 per group for RT–PCR analysis).
-
G, HRelative mRNA expression (G) and Western blot analysis (H) of PGC1α, PLIN5, and HSL in C2C12 cells transfected with Ago‐183/96 and FoxO1 as indicated (n = 5 per group for RT–PCR analysis).
-
IRelative mRNA expression of PGC1α, PLIN5, and HSL in C2C12 cells transfected with Ant‐183/96 and siRNA of PPARδ (siPPARδ).
Data information: Means ± SEM are shown for all panels. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control (Student’s t‐test). The loading control was reused in Fig 4F and H. All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
miR‐183 and miR‐96 levels are low in oxidative muscles and downregulated upon HFD feeding
In agreement with the notion that ATGL is a direct target gene of miR‐183 and miR‐96, we found that the expression of miR‐183 and miR‐96 was lower in oxidative SOL muscles than that in glycolytic GAS muscles, while the mRNA and protein levels of ATGL were higher in SOL muscles than that in GAS muscles (Fig 5A–C). Notably, the expression of PPARδ‐regulated genes, HSL and PLIN5, was also higher in SOL muscles than that in GAS muscles (Fig 5B and C). The finding that the expression of miR‐183 and miR‐96 was negatively correlated with the lipolytic and oxidative capacity of myofibers further supports the notion that miR‐183 and miR‐96 serve as negative regulators of the fat utilization in skeletal muscle (Fig 5A–C). Interestingly, similar to what we observed for miR‐182 (Zhanget al, 2016), the expression of miR‐183 and miR‐96 in skeletal muscle was downregulated after 2 months of HFD feeding, which was accompanied with increased expression of ATGL, HSL, and PLIN5 (Figs 5D–F and EV5A–D). These results suggest that the downregulation of miR‐183 and miR‐96 might contribute to the increased levels of ATGL, HSL, and PLIN5 in the skeletal muscle of HFD‐fed mice. Notably, both the downregulation of miR‐183 and miR‐96 and the upregulation of their target genes could be observed in GAS muscles of mice after 2 weeks of HFD feeding (Fig EV5E and F), further supporting the notion that the decrease in miR‐183 and miR‐96 expression might be involved in the upregulation of their target genes.
Figure 5. miR‐183 and miR‐96 levels are low in oxidative muscles and downregulated upon HFD feeding.

- Relative expression of miR‐183 and miR‐96 mRNAs in GAS and SOL muscles of C57 BL/6 mice (n = 3).
- Relative mRNA expression of ATGL, HSL, and PLIN5 in GAS and SOL muscles of C57 BL/6 mice (n = 3).
- Western blot analysis of ATGL, HSL, and PLIN5 in GAS and SOL muscles of C57 BL/6 mice.
- Relative expression of miR‐183 and miR‐96 mRNAs in GAS muscles of C57 BL/6 mice fed with HFD for 2 months (n = 5).
- Relative mRNA expression of ATGL, HSL, and PLIN5 in GAS muscles of C57 BL/6 mice fed with HFD for 2 months (n = 5).
- Western blot analysis of ATGL, HSL, and PLIN5 in GAS muscles of C57 BL/6 mice fed with HFD for 2 months.
- The role of skeletal muscle miR‐183 and miR‐96 in fuel metabolism. The effects of miR‐183 and miR‐96 deficiency (genetic ablation) and downregulation (HFD feeding), and low levels of miR‐183 and miR‐96 (more oxidative myofibers) on gene expression, enzyme, or transcription factor activity, and metabolic pathways involved in fuel usage are shown by up/down arrows or cross in red.
Data information: Means ± SEM are shown for all panels. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
Thus, we establish miR‐183 and miR‐96 as master regulators of fuel metabolism in skeletal muscle. miR‐183 and miR‐96 orchestrate the fuel usage in skeletal muscle through both glucose and fat utilization pathways via their direct targets FoxO1, PDK4, and ATGL (Fig 5G). Based on our above findings, we also speculated that the downregulation of miR‐183 and miR‐96 in skeletal muscle upon HFD feeding would facilitate the fat utilization via enhancing the TAG lipolysis and FA oxidation, thereby serving as a protective mechanism to reduce the lipid overloading at the expense of decreasing the glucose usage (Fig 5G).
Loss of miR‐183 and miR‐96 protects mice from HFD‐induced obesity
Lastly, since our observation in this study suggest that miR‐183 and miR‐96 act as negative regulators of fat utilization and downregulation of miR‐183 and miR‐96 may serve as a protective mechanism to reduce the lipid overloading (Fig 5G), we tested whether loss of miR‐183 and miR‐96 could be protective against HFD‐induced obesity. Interestingly, we found that the DKO mice were resistant to HFD‐induced obesity, as evident from reduced body and fat weight in DKO mice as compared to WT mice after HFD feeding (Fig 6A–C). Importantly, although these DKO mice had smaller body weights than those of WT mice before HFD challenge, the weight gain was much greater in WT mice than that in DKO mice, further suggesting that DKO mice were resistant to HFD feeding (Figs 6B and EV5G). We also found that loss of miR‐183 and miR‐96 could reduce the susceptibility to HFD‐induced hepatic steatosis (Fig EV5H). The levels of serum TAG and NEFA were lower in DKO mice than those in WT mice after HFD treatment (Fig 6D). As expected, the mRNA and protein levels of either FoxO1 or ATGL were increased in GAS muscles of HFD‐fed DKO mice as compared to those in HFD‐fed WT mice (Fig 6E and F). Accordingly, increased mRNA levels of PGC1α, PLIN5, and HSL were observed in GAS muscles of HFD‐fed DKO mice (Fig 6G and H). These results suggest that the increased fat utilization in skeletal muscles due to miR‐183 and miR‐96 deficiency might contribute to the resistance to HFD‐induced obesity. In line with these data and the increased energy expenditure observed for DKO mice (Fig EV1E), the expression of genes involved in energy expenditure was also elevated in GAS muscles of HFD‐fed DKO mice (Fig EV5I). Consistent with the observation in the BAT of DKO mice under normal chow diet condition (Fig EV2D), the expression of genes involved in energy expenditure was not altered in the BAT of HFD‐fed DKO mice (Fig EV5I), suggesting that BAT‐mediated energy expenditure did not participate in the metabolic benefits observed here. Again, the expression of miR‐182 in GAS muscles was similar between DKO and WT mice after HFD feeding, suggesting that miR‐182 in skeletal muscle was not involved (Fig EV5J).
Figure 6. Loss of miR‐183 and miR‐96 protects mice from diet‐induced obesity and glucose intolerance.

-
ARepresentative photographs of WT and DKO mice after HFD feeding for 2 months. Scale bar: 1 cm.
-
B, CBody weight, body weight gain after HFD feeding (B) and tissue weight of iWAT and eWAT (C) of WT and DKO mice fed with HFD for 2 months (n = 10–11).
-
DConcentration of serum triglyceride (TAG) (left) and non‐esterified fatty acid (NEFA) (right) in WT and DKO mice after HFD feeding for 2 months (n = 5).
-
ERelative mRNA expression of FoxO1, PDK4, and ATGL in GAS muscles of WT and DKO mice fed with HFD for 2 months (n = 5).
-
FWestern blot analysis of FoxO1, PDK4, p‐PDHA1, PDHA1, and ATGL in GAS muscles of WT and DKO mice fed with HFD for 2 months.
-
G, HRelative mRNA expression of PGC1α, PLIN5 (G), and HSL (H) in GAS muscles of WT and DKO mice fed with HFD for 2 months (n = 5).
-
IFasting blood glucose levels (left) and random blood glucose levels (right) in WT and DKO mice after HFD feeding (n = 10–11).
-
JGlucose tolerance test (GTT) and area under the curve (AUC) data for GTT in WT and DKO mice after HFD feeding (n = 10–11).
Data information: Means ± SEM are shown for all panels. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.001 versus control (Student’s t‐test). All experiments were performed at least three times, and representative data are shown.
Source data are available online for this figure.
Notably, although the mRNA and protein levels of FoxO1 and PDK4 and the phosphorylation of PDHA1 were all higher in GAS muscles of HFD‐fed DKO mice than those in HFD‐fed WT mice (Fig 6E and F), the fasting glucose levels in HFD‐fed DKO mice were decreased rather than increased as compared to HFD‐fed WT mice (Fig 6I). Additionally, the random glucose levels were also reduced in HFD‐fed DKO mice as compared to HFD‐fed WT mice (Fig 6I). Moreover, an increase in glucose tolerance was observed in HFD‐fed DKO mice as compared to HFD‐fed WT mice (Fig 6J). Consistently, after HFD feeding, the increase in insulin levels in DKO mice could no longer be observed (Figs EV2A and 5K). Moreover, we found that the insulin signaling was increased in GAS muscles of DKO mice after HFD feeding (Fig EV5L). Based on these results, we speculated that, upon HFD challenge, WT mice gained more weight, which led to the impaired glucose metabolism, in contrast, DKO mice were resistant to HFD‐induced obesity probably due to the increased fat usage and energy expenditure in skeletal muscle, which might ameliorate the deleterious effects of HFD feeding. Our study also indicates that miR‐183 and miR‐96 may serve as therapeutic targets for metabolic diseases.
Discussion
Metabolic flexibility is the capacity for the organism to adapt fuel oxidation to fuel availability, which is important for maintaining metabolic homeostasis. Impaired metabolic flexibility has been implicated in the development of metabolic diseases (Corpeleijn et al, 2009; Aucouturier et al, 2011; Goodpaster & Sparks, 2017; Boschet al, 2020). In skeletal muscle, energy is obtained mainly from glucose and FAs through glycolysis and β‐oxidation, respectively. Skeletal muscle is highly susceptible to changes in glucose and FA availability and is able to adjust the fuel usage to match metabolic demands according to environmental circumstances (Kelley & Mandarino, 2000; Overmyer et al, 2015; Petersen & Shulman, 2018). Thus, a better understanding of the molecular mechanism underlying the regulation of intramuscular glucose and lipid metabolism will not only provide new insight into the fuel selection in skeletal muscle but also open new avenues for the pharmacological treatment of metabolic diseases. In this study, we identified miR‐183 and miR‐96 as critical regulators of both glucose and fat utilization in skeletal muscle, which may serve as therapeutic targets.
It is generally accepted that decreased β‐oxidation in skeletal muscle is associated with metabolic diseases, including obesity and diabetes (Koh et al, 2019; Sanchez‐Gonzalez et al, 2020). However, the relationship between the metabolic disorders and the shift in substrate utilization in skeletal muscle remains not fully understood. In this study, we show that the expression of miR‐183 and miR‐96 is downregulated in the skeletal muscle of mice upon HFD feeding. Since inhibition of miR‐183 and miR‐96 is able to reprogram the fuel metabolism by accelerating the fat utilization and suppressing the glucose usage, we speculate that the downregulation of miR‐183 and miR‐96 upon HFD feeding might serve as a protective mechanism to reduce the lipid overloading at the expense of impairing normal glucose homeostasis. Thus, our study also suggests that the downregulation of miR‐183 and miR‐96 after HFD challenge might be involved in the development of fasting hyperglycemia.
Beside liberation from intramyocellular TAG stores, FA could be obtained from plasma sources as albumin‐bound FA or through hydrolysis of circulating lipoproteins. The imported FAs enter the oxidative process and TAG synthesis, depending on the metabolic status of the cells. In resting condition, FAs are used for TAG synthesis in lipid droplet, which localized adjacent to mitochondria, as the first destination instead of undergoing oxidation in the mitochondria. Serving as an energy pool, lipid droplet is essential for intracellular lipid storage, turnover, trafficking, and protection against lipotoxicity. Consistent with the notion that lipolytic release of lipid ligands from lipid droplets is required for the activation of target genes essential for maintaining normal mitochondrial oxidation and respiration, growing evidence suggests that ATGL (Zimmermann et al, 2004; Haemmerle et al, 2006) has regulatory effect on mitochondrial biogenesis and oxidative capacity (Haemmerleet al, 2011; Meex et al, 2015). Here, we found that miR‐183 and miR‐96 modulate intramuscular lipolysis by targeting ATGL and its upstream regulator FoxO1, thereby controlling the energy availability and substrate utilization. Furthermore, we show that PPARδ mediates the effect of miR‐183 and miR‐96 on the expression of genes involved in mitochondrial biogenesis and FA oxidation. Thus, we uncovered previously undescribed mechanisms not only underlying the regulation of fat utilization but also linking the intramyocellular lipolysis to PPARδ‐mediated transcription and FA oxidation.
Recent studies suggest that PLIN5 has an important role in promoting lipolysis and FA oxidation (Wolins et al, 2006; Wang et al, 2011; Bosma et al, 2013). PLIN5 is able to participate in the formation of lipid droplet‐mitochondria contacts and facilitate the direct transfer of FAs released during lipolysis from lipid droplets to mitochondria in skeletal muscle (Wolinset al, 2006; Wanget al, 2011; Bosma et al, 2012; Bosmaet al, 2013; MacPherson & Peters, 2015; Sanders et al, 2015). Using two super‐resolution imaging approaches (Gemmink et al, 2018), it has been shown that PLINs localize to lipid droplet at distinct sites, with abundance of PLIN5 at lipid droplet‐mitochondria tethering sites, supporting the notion that PLIN5 is involved in facilitating liberating fatty acids from the lipid droplet for mitochondrial fat oxidation rather than creating a lipolytic barrier (Pollak et al, 2013). In addition, PLIN5 can either act as a direct target gene of PPARδ or serve as an activator of the PGC1α to increase the transcription of genes involved in mitochondrial biogenesis, electron transport chain complexes, and FA oxidation (Bindesboll et al, 2013; Wang et al, 2019; Najt et al, 2020). To be noted, during fasting, PLIN5 has been suggested to decrease lipotoxicity by promoting interaction of lipid droplets with mitochondria. In this study, we found that the enhanced intramuscular lipolysis due to miR‐183 and miR‐96 deficiency was accompanied with an increase in PLIN5 expression, which in turn increases the lipolysis and oxidation. These findings also indicate that miR‐183 and miR‐96 might also function to promote the interaction between lipid droplet and mitochondria and decrease the lipotoxicity in skeletal muscle.
As miR‐183 and miR‐96 are expressed with comparable expression levels in skeletal muscle, liver, and WAT (Fig EV2N and O), the expression of ATGL, HSL, and PLIN5 in the liver and WAT of DKO mice was determined. We found that the mRNA and protein levels of both ATGL, HSL, and PLIN5 were not altered in either the liver or WAT of DKO mice (Fig EV2, EV3, EV4, EV5), suggesting that the lipolysis and subsequent oxidation might not be altered in these two tissues of DKO mice. Although an increase in the mRNA levels of FoxO1 and PDK4 was observed in the liver of DKO mice, we could not detect any changes in protein levels of FoxO1 and PDK4 in both liver and WAT of DKO mice (Fig EV2, EV3, EV4, EV5). Whether there were compensatory mechanisms in the liver and WAT obscuring the detection of expression changes of the aforementioned genes after the loss of miR‐183 and miR‐96 requires further investigation. Although we could not exclude the possibility that lacking miR‐183 and miR‐96 in other tissues might also have effects on the metabolic homeostasis, our in vivo and in vitro data suggest that the metabolic phenotype observed in DKO mice might be likely attributed to the deficiency of miR‐183 and miR‐96 in skeletal muscle and the skeletal muscle but not the liver and WAT might be the major site where miR‐183 and miR‐96 regulate the fuel usage and systematic metabolism.
Since miR‐183, miR‐96, and miR‐182 are expressed from the same cluster and share same targets mentioned in this study, to test whether the metabolic phenotype in DKO mice and the in vitro findings observed were attributed to the alteration of miR‐182 levels, we checked the expression of miR‐182 in the absence of miR‐183 and miR‐96 or after Ant‐183/96 treatment. We found that the miR‐182 levels in the GAS muscle, liver, and WAT were all not altered in DKO mice (Fig EV1A). qPCR analysis revealed that Ant‐183/96 treatment did not affect the levels of pri‐miRNA of miR‐183/96/182 (Fig EV3L), which is in agreement with the notions that antagomirs are exclusively localized in the cytosol and pri‐miRNAs are located in the nucleus (Krutzfeldt et al, 2007; Stenvang et al, 2012). In line with the fact that the seed sequences for miR‐183, miR‐96, and miR‐182 are not exactly the same, and differences could be found in other positions of antagomirs, we found that Ant‐183/96 transfection could not affect the levels of miR‐182 in C2C12 cells (Fig EV3L), suggesting that Ant‐183/96 display high sequence specificity (Robertson et al, 2010; Stenvanget al, 2012). Additionally, CRISPR/Cas9‐mediated gene knockout of miR‐183 and miR‐96 could elevate the mRNA levels of FoxO1, PDK4, ATGL, as well as HSL, PGC1α and PLIN5 without changing the miR‐182 levels in C2C12 myocytes (Fig EV3M). Collectively, based on these data, we speculate that the altered metabolic phenotype and expression of target genes observed in current study were not due to the changes of miR‐182 levels. It is also worth noting that, after HFD feeding, the GAS miR‐182 levels were decreased in DKO mice to the same extent as in WT mice (Fig EV5J), also indicating that the miR‐182 in GAS muscles might not contribute to the metabolic phenotype observed in DKO mice upon HFD challenge.
Taken together, our study establishes miR‐183 and miR‐96 as master regulators of fuel usage by coordinating both glucose and lipid metabolism in skeletal muscle (Fig 5G). Mechanistically, miR‐183 and miR‐96 regulate glucose usage by controlling PDH activity via targeting FoxO1 and PDK4 and modulate fat usage by affecting intracellular lipolysis and oxidative capacity via targeting FoxO1 and ATGL (Fig 5G). Our findings suggest that a shift in fuel usage from glucose toward fat in skeletal muscle due to the loss of miR‐183 and miR‐96 would alter the glucose homeostasis in mice fed with normal chow, while enhancing the fat catabolism in skeletal muscle by miR‐183 and miR‐96 deficiency might have beneficial effects upon HFD challenge.
Materials and Methods
Animal studies
All procedures that involved animal handing were approved by the Institutional Review Board of Institute for Nutrition and Health Science (SINH), Chinese Academy of Sciences (CAS) (2016‐AN‐1, SIBS‐2019‐YH‐1). All in vivo experiments described in this study were in accordance with institutional guidelines for the care and use of animals. miR‐183 and miR‐96 DKO mice were generated as described previously (Xianget al, 2017). All mice of C57BL/6J background were housed at a temperature of 23 ± 3°C and a humidity of 35 ± 5% under a 12 h light/dark cycle in a specific pathogen‐free animal facility. For high‐fat diet, WT or DKO mice were fed 60 kcal% HFD for 2 months or 2 weeks.
Measurement of metabolic profile
Glucose levels were measured under different feeding condition as indicated using a glucose meter (Abbott). Food intake was evaluated by weighting out the grams of food every 12 h (7 AM‐7 PM day 7 PM‐7 AM night). Minispec TD‐NMR Analysers were used to evaluate living body composition. To measure energy expenditure, mice were placed in metabolic cages (Columbus Instruments) to assess their O2 consumption, CO2 production, energy expenditure, and physical activity. The respiratory exchange ratio (RER) and energy metabolism were determined using LabMaster software. The levels of triglyceride and NEFA in mice serum were measured using commercially available kits LabAssayTM Triglyceride and LabAssayTM NEFA (ACS‐ACOD method) (Wako, Japan). Lactate and pyruvate were determined using commercially available kits (Nanjing Jiancheng Bio‐company).
Tolerance tests
Glucose tolerance and pyruvate tolerance test were carried out on animal that had been fasted overnight for 16 h. After determination of fasted glucose levels, each animal received an intraperitoneal (i.p.) injection of 1 or 2 g/kg body weight of glucose or pyruvate as indicated in the figures. Blood glucose levels were detected from tail vein after 15, 30, 60, 90, and 120 min.
Morphological studies
Mice muscle tissue was frozen and fixed in isopentane. Muscle tissue section and cover slides were treated with 0.1% Triton X‐100 in PBS for 10 min. Muscle fiber type was determined by the ATPase staining for pre‐incubation at either pH 10.6 or 4.2 as previously described (Zhang et al, 2014). To determine the percentage of myofibers, more than 500 myofibers were counted for gastrocnemius muscles and more than 200 myofibers were counted for soleus muscles of each mouse.
Histology
H&E staining of mice muscle, liver, and fat tissues was carried out after the tissues were fixed in methanol‐free 4% paraformaldehyde for 24–36 h. For H&E staining, the paraffin‐embedded specimens were sectioned at 5 mm and then stained according to standard protocols. Staining images were taken using a light microscope (Olympus).
Plasmid and RNA oligonucleotide
For construction of the luciferase reporter plasmid, the 3′UTR of FoxO1, ATGL, and PDK4 were amplified from mouse cDNA by PCR, (the primer sequences are provided in Table EV1), and inserted into pRL‐TK vector. GMR‐miR™ microRNA double‐stranded mimics for miR‐183 (UAU GGC ACU GGU AGA AUU CACU), miR‐96 (UUU GGC ACU AGC ACA UUU UUG CU), inhibitor for miR‐183 (AGU GAA UUC UAC CAG UGC CAUA), miR‐96 (AGC AAA AAU GUG CUA GUG CCA AA) and siRNA oligos for FoxO1 (GGA CAA CAA CAG UAA AUUU), ATGL (GGU AUU CUU CAG CUC AUA AAG UG), PPARδ (GGU UCU UCU UCU GGA UCU UGC AG) were obtained from Genepharma. miR‐183 (AGU GAA UUC UAC CAG UGC CAUA), miR‐96 (AGC AAA AAU GUG CUA GUG CCA AA), miR‐182 (CGG UGU GAG UUC UAC CAU UGC CAA A) miR‐Down™ antagomir and miR‐Up™ agomir of miR‐183 (UAU GGC ACU GGU AGA AUU CACU), miR‐96 (UUU GGC ACU AGC ACA UUU UUG CU), miR‐182 (UUU GGC AAU GGU AGA ACU CAC ACCG) were purchased from GenePharma. Nonsense sequence were used as agomir control (UCA CAA CCU CCU AGA AAG AGU AGA) and antagomir control (UCU ACU CUU UCU AGG AGG UUG UGA).
RNA isolation and real‐time RT–PCR
Total RNA was isolated by using TRIzol reagent (Invitrogen) according to the manufacturer’s instruction. Total RNA was reverse transcribed by using PrimeScript RT Reagent Kit (TaKaRa). The microRNA was transcribed in a stem‐loop manner according to the protocols. Real‐time RT–PCR was performed on an ABI 7900 Real‐time PCR System (Applied Biosystems). The primer sequence is provided in Table EV2.
Protein preparation and immunoblotting
Cells were lysed directly with 1× SDS loading buffer (EpiZyme), sonicated and denatured at 95°C for 10 min. Mouse tissue samples were prepared according to a previously published method (Li et al, 2018). Briefly, mouse tissues were homogenized in RIPA lysis buffer (50 mM Tris–HCl, pH7.5, 150 mM NaCl, 1.0 mM EDTA, 0.1% SDS, 1% sodium deoxycholate and 1% Triton X‐100). The buffer was supplemented with Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail (Roche) just prior to use. The protein concentration was determined using a Pierce Coomassie (Bradford) assay kit (Thermo Fisher). Protein lysates were dissolving in 5× SDS loading buffer (EpiZyme) and denatured at 95°C for 10 min. Total protein were fractionated by 10% SDS–PAGE gels (EpiZyme) and transferred to a PVDF membrane (Millipore) for immunoblotting. Immunoblotting was performed with antibodies indicated and detected by ECL system as described previously (31). Antibodies against FoxO1 (Cell Signaling, 2880), PDK4 (Abcam, ab38242), phos‐PDHA1‐S293 (Millipore, ABS204), PDHA1 (Protein tech, 18068‐1‐AP), p‐Akt (Cell Signaling, 4060), Akt (Cell Signaling, 9272), p‐IR (Cell Signaling, 3023), IR (Cell Signaling, 3025), HSP90 (Cell Signaling, 4877), GAPDH (Kang Chen Bio‐tech, LK2003), HSL (Cell Signaling, 4107), ATGL (Cell Signaling, 2138S), PLIN5 (Proteintech, 26951‐1‐AP), and PGC1α (Santa Cruz, sc‐13067) were used for Western blot analysis.
Cell culture, transfection, and luciferase assay
Mouse C2C12 myoblasts and HEK 293T (ATCC) cell were maintained in a humidified incubator at 37°C and 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco). When C2C12 myoblasts grown up to 90% confluent, myoblasts were transferred to differentiation medium consisting of DMEM with 2% horse serum and differentiated for 48 h. Transfection was performed using Lipofectamine 2000 (Invitrogen) according to the standard protocol, using 2 pmol/ml oligos for moderate overexpression, or 1 µg/ml plasmids or 20 pmol/ml oligos for regular cell transfection. Luciferase assays were performed as described before modification (Zhang et al, 2014). For FoxO1, ATGL, and PDK4 3′UTR‐Renilla luciferase reporter assay, pRL‐TK‐FoxO1 or ATGL or PDK4 3′UTR reporter construct was co‐transfected into 293T cells together with firefly luciferase plasmid pGL3 and miR‐183 or miR‐96 mimics, or Ctrl RNA (GenePharma) or microRNA’s inhibitors.
Cells were harvested 48 h after transfection. Luciferase activities were measured using the Dual Luciferase Reporter Assay System (Promega) following the manufacturer’s protocol on a luminometer (Berthold Technologies).
Construction of the miR‐183 and miR‐96 knockout cell line
In brief, site‐specific sgRNAs were predicted following a web tool Crispor (http://crispor.tefor.net/). To minimize off‐target effects, only sgRNAs with a score higher than 5 were selected. To construct the AAV9‐sgRNA transfer plasmids, the AAV: ITR‐U6‐sgRNA (backbone)‐pCBh‐Cre‐WPRE‐hGHpA‐ITR (Addgene, 60229) was used as donor plasmid. Coding sequencing for DsRed was PCR‐amplified and cloned into the donor plasmid through replacing the sequence encoding Cre using Age I and EcoR I sites. Annealed oligonucleotides were constructed into a Cas9‐EGFP expressing vector (pX458, Addgene) using Bbs I site and AAV9‐sgRNA vector (AAV: ITR‐U6‐sgRNA(backbone)‐CMV‐DsRed‐WPRE‐hGHpA‐ITR) using Sap I site. SgRNA‐pX458 and AAV9‐vector plasmids were transiently transfected into C2C12 using Lipofectamine 3000. GFP‐ and DsRed‐positive cells were sorted out using FACS 48 h after transfection and cultured for another two days. To screen out suitable monoclonal cells, plate the polyclonal cells from the selection step at a density of 10 cells/ml in a 96‐well tissue culture plate adding 100 µl per well (i.e., 1 cell per well) and harvest them after 7 days. Genomic DNAs were extracted by QuickExtract (Epicentre) solution and amplified by using designed primer to determine the editing efficiency.
Measurement of oxygen consumption in myocytes
Oxygen consumption and glycolysis were measured using a Seahorse Bioscience XF24 instrument. C2C12 myocytes were culture in 20 wells of XF24 microplate. Mito stress test kit was obtained from Seahorse bioscience. The oxygen consumption rate (OCR) is determined following the manufacturer’s instructions. Furthermore, the basal respiration and maximal respiration were calculated as described (Nicholls et al, 2010).
LD turnover
LD turnover experiment was executed as previous reported (Olzmann et al, 2013). Briefly, the basal LDs contained in C2C12 myocytes were consumed through the treatment of 10 μM triacsin C for16 h. After washing with culture medium, cells were treated with 200 μM oleate for 3 h to induce the endogenic generation of LDs. Finally, cells were cultured with10 μM triacsin C for an additional 3, 6, or 12 h as indicated to evaluate the levels of LD turnover. Respectively, C2C12 cells were transfected with Ago‐183/96 or Ant‐183/96 after incubated with oleate.
Statistical analyses
Sample size was determined based on power analysis and standard practice. Randomization and blinding strategy were used whenever possible. Results are expressed as mean ± SEM. of at least three independent experiments. Statistical significance was assessed by Student’s t‐test. Differences were considered statistically significant at P < 0.05. The software used are Excel and GraphPad Prism 8.0 (GraphPad, San Diego, CA). NIH ImageJ was employed for densitometry analysis of Western blotting.
Author contributions
HW, HY, and YaL designed the experiments and analyzed the data; HW and YaL carried out most of the experiments; MM, YuL, JL, CS, SL, YM, YY, ZT, SS, JY, and YW provided the technical assistance; JJ, LW, and Z‐BJ contributed to the discussion and supervised the project; HW, HY, and YaL wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by MOST of China (2016YFA0500102, 2016YFC1304905); National NSFC (31871195, 31525012, 31900841, 32071166); manned space program from Technology and Engineering Center for Space Utilization, Chinese Academy of Sciences (CAS) (No. 356); CAS projects (Interdisciplinary Innovation Team, ZDBSSSW‐DQC‐02, ZDRW‐ZS‐2017‐1, KFJ‐STS‐ZDTP‐084), CAS Key Laboratory of Nutrition, Metabolism Food Safety (KLNMFS2019‐01); Youth Innovation Promotion Association, CAS (2021261); the Fundamental Research Funds for the Central Universities (JUSRP221001); the Young Elite Scientists Sponsorship Program by CAST; and the NHC Key Laboratory of Food Safety Risk Assessment (2020K02).
EMBO reports (2021) 22: e52247.
Contributor Information
Hao Ying, Email: yinghao@sibs.ac.cn.
Yan Li, Email: liyan0520@jiangnan.edu.cn.
Data availability
No data were deposited in a public database.
References
- Aucouturier J, Duche P, Timmons BW (2011) Metabolic flexibility and obesity in children and youth. Obes Rev 12: e44–53 [DOI] [PubMed] [Google Scholar]
- Banke NH, Wende AR, Leone TC, O'Donnell JM, Abel ED, Kelly DP, Lewandowski ED (2010) Preferential oxidation of triacylglyceride‐derived fatty acids in heart is augmented by the nuclear receptor PPAR alpha. Circ Res 107: 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindesboll C, Berg O, Arntsen B, Nebb HI, Dalen KT (2013) Fatty acids regulate perilipin5 in muscle by activating PPAR delta. J Lipid Res 54: 1949–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Parton RG, Pol A (2020) Lipid droplets, bioenergetic fluxes, and metabolic flexibility. Semin Cell Dev Biol 108: 33–46 [DOI] [PubMed] [Google Scholar]
- Bosma M, Minnaard R, Sparks LM, Schaart G, Losen M, Baets MH, Duimel H, Kersten S, Bickel PE, Schrauwen Pet al (2012) The lipid droplet coat protein perilipin 5 also localizes to muscle mitochondria. Histochem Cell Biol 137: 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma M, Sparks LM, Hooiveld GJ, Jorgensen JA, Houten SM, Schrauwen P, Kersten S, Hesselink MK (2013) Overexpression of PLIN5 in skeletal muscle promotes oxidative gene expression and intramyocellular lipid content without compromising insulin sensitivity. Biochim Biophys Acta 1831: 844–852 [DOI] [PubMed] [Google Scholar]
- Chakrabarti P, Kandror KV (2009) FoxO1 controls insulin‐dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem 284: 13296–13300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpeleijn E, Saris WH, Blaak EE (2009) Metabolic flexibility in the development of insulin resistance and type 2 diabetes: effects of lifestyle. Obes Rev 10: 178–193 [DOI] [PubMed] [Google Scholar]
- Fritzen AM, Lundsgaard AM, Kiens B (2020) Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat Rev Endocrinol 16: 683–696 [DOI] [PubMed] [Google Scholar]
- Gan Z, Burkart‐Hartman EM, Han DH, Finck B, Leone TC, Smith EY, Ayala JE, Holloszy J, Kelly DP (2011) The nuclear receptor PPARbeta/delta programs muscle glucose metabolism in cooperation with AMPK and MEF2. Genes Dev 25: 2619–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmink A, Daemen S, Kuijpers HJH, Schaart G, Duimel H, Lopez‐Iglesias C, van Zandvoort M, Knoops K, Hesselink MKC (2018) Super‐resolution microscopy localizes perilipin 5 at lipid droplet‐mitochondria interaction sites and at lipid droplets juxtaposing to perilipin 2. Biochim Biophys Acta Mol Cell Biol Lipids 1863: 1423–1432 [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Sparks LM (2017) Metabolic flexibility in health and disease. Cell Metab 25: 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BA, Benson AC, Bird SR, Fraser SF (2009) Resistance training improves metabolic health in type 2 diabetes: a systematic review. Diabetes Res Clin Pract 83: 157–175 [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder Set al (2006) Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312: 734–737 [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler Ket al (2011) ATGL‐mediated fat catabolism regulates cardiac mitochondrial function via PPAR‐alpha and PGC‐1. Nat Med 17: 1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui M, Martello G, Piccolo S (2010) MicroRNA control of signal transduction. Nat Rev Mol Cell Biol 11: 252–263 [DOI] [PubMed] [Google Scholar]
- Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, Walsh K (2008) Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab 7: 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolina DS, Armugam A, Tavintharan S, Wong MT, Lim SC, Sum CF, Jeyaseelan K (2011) MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS One 6: e22839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE (2005) Skeletal muscle fat oxidation: timing and flexibility are everything. J Clin Invest 115: 1699–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, Mandarino LJ (2000) Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49: 677–683 [DOI] [PubMed] [Google Scholar]
- Koh J‐H, Johnson ML, Dasari S, LeBrasseur NK, Vuckovic I, Henderson GC, Cooper SA, Manjunatha S, Ruegsegger GN, Shulman GIet al (2019) TFAM enhances fat oxidation and attenuates high‐fat diet‐induced insulin resistance in skeletal muscle. Diabetes 68: 1552–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M (2007) Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 35: 2885–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Walsh K, Arany Z (2011) Metabolic benefits of resistance training and fast glycolytic skeletal muscle. Am J Physiol Endocrinol Metab 300: E3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jiang J, Liu W, Wang H, Zhao L, Liu S, Li P, Zhang S, Sun C, Wu Yet al (2018) microRNA‐378 promotes autophagy and inhibits apoptosis in skeletal muscle. Proc Natl Acad Sci USA 115: E10849–E10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson RE, Peters SJ (2015) Piecing together the puzzle of perilipin proteins and skeletal muscle lipolysis. Appl Physiol Nutr Metab 40: 641–651 [DOI] [PubMed] [Google Scholar]
- Meex RC, Hoy AJ, Mason RM, Martin SD, McGee SL, Bruce CR, Watt MJ (2015) ATGL‐mediated triglyceride turnover and the regulation of mitochondrial capacity in skeletal muscle. Am J Physiol Endocrinol Metab 308: E960–970 [DOI] [PubMed] [Google Scholar]
- Meng ZX, Li S, Wang L, Ko HJ, Lee Y, Jung DY, Okutsu M, Yan Z, Kim JK, Lin JD (2013) Baf60c drives glycolytic metabolism in the muscle and improves systemic glucose homeostasis through Deptor‐mediated Akt activation. Nat Med 19: 640–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najt CP, Khan SA, Heden TD, Witthuhn BA, Perez M, Heier JL, Mead LE, Franklin MP, Karanja KK, Graham MJet al (2020) Lipid droplet‐derived monounsaturated fatty acids traffic via PLIN5 to allosterically activate SIRT1. Mol Cell 77: 810–824.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Darley‐Usmar VM, Wu M, Jensen PB, Rogers GW, Ferrick DA (2010) Bioenergetic profile experiment using C2C12 myoblast cells. J Vis Exp 46: 2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Richter CM, Kopito RR (2013) Spatial regulation of UBXD8 and p97/VCP controls ATGL‐mediated lipid droplet turnover. Proc Natl Acad Sci USA 110: 1345–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmyer K, Evans C, Qi N, Minogue C, Carson J, Chermside‐Scabbo C, Koch L, Britton S, Pagliarini D, Coon Jet al (2015) Maximal oxidative capacity during exercise is associated with skeletal muscle fuel selection and dynamic changes in mitochondrial protein acetylation. Cell Metab 21: 468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone Ret al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100: 8466–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300: 1140–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Shulman GI (2018) Mechanisms of insulin action and insulin resistance. Physiol Rev 98: 2133–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak NM, Schweiger M, Jaeger D, Kolb D, Kumari M, Schreiber R, Kolleritsch S, Markolin P, Grabner GF, Heier Cet al (2013) Cardiac‐specific overexpression of perilipin 5 provokes severe cardiac steatosis via the formation of a lipolytic barrier. J Lipid Res 54: 1092–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson B, Dalby AB, Karpilow J, Khvorova A, Leake D, Vermeulen A (2010) Specificity and functionality of microRNA inhibitors. Silence 1: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R (1993) MyoD or Myf‐5 is required for the formation of skeletal muscle. Cell 75: 1351–1359 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Gonzalez C, Nuevo‐Tapioles C, Herrero Martin JC, Pereira MP, Serrano Sanz S, Ramirez de Molina A, Cuezva JM, Formentini L (2020) Dysfunctional oxidative phosphorylation shunts branched‐chain amino acid catabolism onto lipogenesis in skeletal muscle. EMBO J 39: e103812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders M, Madoux F, Mladenovic L, Zhang H, Ye X, Angrish M, Mottillo E, Caruso J, Halvorsen G, Roush Wet al (2015) Endogenous and synthetic ABHD5 ligands regulate ABHD5‐perilipin interactions and lipolysis in fat and muscle. Cell Metab 22: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Reggiani C (2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91: 1447–1531 [DOI] [PubMed] [Google Scholar]
- Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S (2012) Inhibition of microRNA function by antimiR oligonucleotides. Silence 3: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana Ket al (2003) Activation of peroxisome proliferator‐activated receptor delta induces fatty acid beta‐oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA 100: 15924–15929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J, Zhu J, van Oudenaarden A (2007) MicroRNA‐mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell 26: 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Yuan Y, Wu J, Zhao Y, Gao X, Chen Y, Sun C, Xiao L, Zheng P, Hu Pet al (2019) Plin5 deficiency exacerbates pressure overload‐induced cardiac hypertrophy and heart failure by enhancing myocardial fatty acid oxidation and oxidative stress. Free Radic Biol Med 141: 372–382 [DOI] [PubMed] [Google Scholar]
- Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet‐associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52: 2159–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC, Varma V, Yao‐Borengasser A, Rasouli N, Kern PAet al (2006) OXPAT/PAT‐1 is a PPAR‐induced lipid droplet protein that promotes fatty acid utilization. Diabetes 55: 3418–3428 [DOI] [PubMed] [Google Scholar]
- Xiang L, Chen XJ, Wu KC, Zhang CJ, Zhou GH, Lv JN, Sun LF, Cheng FF, Cai XB, Jin ZB (2017) miR‐183/96 plays a pivotal regulatory role in mouse photoreceptor maturation and maintenance. Proc Natl Acad Sci USA 114: 6376–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Li Y, Yao X, Wang H, Zhao L, Jiang H, Yao X, Zhang S, Ye C, Liu Wet al (2016) miR‐182 regulates metabolic homeostasis by modulating glucose utilization in muscle. Cell Rep 16: 757–768 [DOI] [PubMed] [Google Scholar]
- Zhang D, Wang X, Li Y, Zhao L, Lu M, Yao X, Xia H, Wang Y‐C, Liu M‐F, Jiang Jet al (2014) Thyroid hormone regulates muscle fiber type conversion via miR‐133a1. J Cell Biol 207: 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner‐Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter Aet al (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306: 1383–1386 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
No data were deposited in a public database.