

Abstract
Helicobacter pylori infection constitutes one of the major risk factors for the development of gastric diseases including gastric cancer. The activation of nuclear factor‐kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) via classical and alternative pathways is a hallmark of H. pylori infection leading to inflammation in gastric epithelial cells. Tumor necrosis factor receptor‐associated factor (TRAF)‐interacting protein with forkhead‐associated domain (TIFA) was previously suggested to trigger classical NF‐κB activation, but its role in alternative NF‐κB activation remains unexplored. Here, we identify TRAF6 and TRAF2 as binding partners of TIFA, contributing to the formation of TIFAsomes upon H. pylori infection. Importantly, the TIFA/TRAF6 interaction enables binding of TGFβ‐activated kinase 1 (TAK1), leading to the activation of classical NF‐κB signaling, while the TIFA/TRAF2 interaction causes the transient displacement of cellular inhibitor of apoptosis 1 (cIAP1) from TRAF2, and proteasomal degradation of cIAP1, to facilitate the activation of the alternative NF‐κB pathway. Our findings therefore establish a dual function of TIFA in the activation of classical and alternative NF‐κB signaling in H. pylori‐infected gastric epithelial cells.
Keywords: ALPK1, cIAP1, NIK, TRAF
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Helicobacter pylori’s ADP‐heptose binds to the kinase ALPK1 in gastric epithelial cells and directs activation of classical and alternative NF‐κB pathways in a TIFA dependent manner, thereby promoting gastric inflammation.

Introduction
Helicobacter pylori, declared in 1994 as a class I carcinogen, is a Gram‐negative microaerophilic bacterium and one of the etiological risk factors for gastric adenocarcinoma. Infection with H. pylori has a worldwide prevalence of 44% (Zamani et al, 2018). Persistent infection with this non‐invasive pathogen causes chronic gastritis, the progression of which can lead to a plenitude of gastrointestinal and extragastric diseases (Bravo et al, 2018).
Several studies have shown that colonization of gastric epithelial cells by H. pylori leads to the activation of the classical NF‐κB pathway as well as the alternative NF‐κB pathway (Mejías‐Luque et al, 2017; Naumann et al, 2017; Feige et al, 2018). However, there exists still insufficient knowledge about the membrane‐proximal signaling leading to NF‐κB activation by H. pylori. It is well known that the activation of the classical NF‐κB pathway by H. pylori and by several inflammatory cytokines such as TNF and interleukin‐1β (IL‐1β) involves the binding of TAK1 to TRAFs to activate the IκB kinase (IKK) complex leading to the phosphorylation and degradation of IκBα. Thereafter, the released NF‐κB heterodimer composed of RelA and p50 enters the nucleus and regulates expression of target genes (Neumann & Naumann, 2007; Sokolova et al, 2014). In contrast, the alternative NF‐κB pathway is induced by a subset of tumor necrosis factor receptor superfamily members such as the lymphotoxin‐β receptor (LTβR), receptor activator of nuclear factor‐kappa B (RANK), and cluster of differentiation 40 (CD40) (Sun, 2017), and also by H. pylori (Feige et al, 2018) leading to the processing and activation of RelB/p52 heterodimers. The NF‐κB‐inducing kinase (NIK) plays a pivotal role in the activation of the non‐canonical NF‐κB pathway. In the cytosol of resting cells, NIK is bound to a NIK regulatory complex consisting of TRAF3, TRAF2, and cIAP1 or cIAP2 (cIAP1/2) that ensures its constitutive proteasomal degradation (Zarnegar et al, 2008). This process is inhibited upon receptor‐mediated non‐canonical NF‐κB activation, allowing NIK to accumulate and phosphorylate, thereby activating IKKα. The ensuing IKKα‐mediated phosphorylation of p100 is the prerequisite for its proteolytic processing to p52 and the subsequent nuclear translocation of p52‐containing dimers, predominantly the RelB/p52 heterodimers (Xiao et al, 2004).
Resolving the enigma of the unknown effector for NF‐κB activation upon H. pylori infection, the ADP‐glycero‐β‐D‐manno‐heptose (ADP‐heptose), which is an intermediate metabolite in the biosynthesis of lipopolysaccharides of Gram‐negative bacteria, has been recently identified as the key pathogen‐associated molecular pattern in Shigella flexneri, Yersinia pseudotuberculosis, and H. pylori (García‐Weber et al, 2018; Zhou et al, 2018; Pfannkuch et al, 2019). However, little is known about its uptake into the cytosol of cells. The uptake of heptose‐1,7‐bisphosphate (HBP), a precursor of ADP‐heptose, is compromised by dynamin inhibition (Gaudet et al, 2015) and T4SS deficiency (Zimmermann et al, 2017). Exogenously added ADP‐heptose, even without membrane permeabilization, has been shown to trigger NF‐κB activation (Pfannkuch et al, 2019). Interestingly, bacteria without a T3SS, such as enterotoxic Escherichia coli and Burkholderia cenocepacia, are also able to elicit ADP‐heptose‐dependent NF‐κB activation in epithelial cells (Zhou et al, 2018). The ADP‐heptose acts via alpha‐protein kinase 1 (ALPK1) (Zhou et al, 2018). ALPK1 is a serine/threonine kinase belonging to the family of atypical protein kinases, the alpha kinases, which display no homology in their catalytic domains with conventional kinases (Ryazanov et al, 1997). First implicated in the apical protein transport in epithelial cells to maintain polarity (Heine et al, 2005), ALPK1 was identified as a pattern recognition receptor in infections by Gram‐negative bacteria (Milivojevic et al, 2017). The catalytic activity of ALPK1 is required for the self‐oligomerization of TIFA (Milivojevic et al, 2017; Zimmermann et al, 2017). TIFA, a forkhead‐associated (FHA) domain‐containing protein, was first described as a TRAF2‐binding protein for its ability to activate NF‐κB and AP‐1 (Kanamori et al, 2002). Later, it was shown that TIFA undergoes self‐oligomerization and thereby promotes the oligomerization and ubiquitinylation of TRAF6 leading to the activation of the IKK complex (Ea et al, 2004). The FHA domain and the phosphorylation of threonine at position 9 (T9) of TIFA are crucial determinants for the self‐oligomerization of TIFA (Huang et al, 2012).
The molecular basis of the proximal activation of the classical as well as the alternative NF‐κB pathways by H. pylori is still poorly understood. We conducted the present study to define the role of TIFA in the activation of the classical and alternative NF‐κB pathways by H. pylori. We show, on the one hand, the interaction of TIFA with TRAF6 to facilitate TAK1 entry into this complex for the activation of classical NF‐κB upon H. pylori infection. On the other hand, TIFA also binds to TRAF2 of the TRAF3/TRAF2/cIAP1 complex. Our results suggest that this binding event liberates cIAP1 from the complex leading to the destabilization and proteasomal degradation of cIAP1, which is followed by the transient accumulation of NIK, a prerequisite for the activation of the alternative NF‐κB pathway.
Results and Discussion
Recent data have uncovered the lipopolysaccharide metabolite HBP as the activator of ALPK1 – TIFA signaling, transmitting the signal from H. pylori into the host cell leading to the activation of classical NF‐κB signaling (Gall et al, 2017; Zimmermann et al, 2017). Additional studies showed that ADP‐heptose exerts a much stronger capacity to activate ALPK1 (Zhou et al, 2018), which is also observed in H. pylori infection (Pfannkuch et al, 2019). Upon H. pylori infection of AGS gastric epithelial cells, we observed early and transient kinetics of activation of both NF‐κB pathways, whereby the induction of the alternative pathway requires the accumulation of NIK and phosphorylation of p100 (Fig 1A). In contrast, stimulation by 20 ng/ml LTα1β2, a ligand of the LTβR, showed negligible classical NF‐κB activation and delayed activation of alternative NF‐κB signaling (Fig 1A). To investigate whether ALPK1 and TIFA are involved in the activation of the alternative NF‐κB pathway, we used the H. pylori mutant strain (ΔHP0857), where gmhA was mutated leading to the termination of the whole biosynthesis pathway of ADP‐heptose. Upon infection with ΔHP0857, we showed that the activation of the classical NF‐κB pathway is diminished, as seen by the reduced phosphorylation of TAK1, IKKα/β, and IκBα (Fig 1B), in agreement with earlier findings (Stein et al, 2017). Of note, an impact on viability or adherence as described for ΔHP0857 mutants in other H. pylori strains (Yu et al, 2016; Gall et al, 2017; Stein et al, 2017) was not observed for the P1 strain used here. In addition, initiation of the alternative NF‐κB pathway represented by the accumulation of NIK and the phosphorylation of p100 is also strongly attenuated (Fig 1C), hinting that the same trigger is responsible for activating both pathways. Similar effects were observed using NCI‐N87 and HeLa cells (Fig EV1A and B). Furthermore, complementation of ΔHP0857 with amplified gmhA from the P1 strain rescued the ability to induce both NF‐κB pathways (Fig EV1C). We also examined the direct application of ADP‐heptose, which upon binding to ALPK1 triggers its kinase activity toward TIFA (Zhou et al, 2018), to the cells for the induction of both NF‐κB pathways. The accumulation of NIK and the increasing phosphorylation of p100, RelA, and IκBα demonstrated the activation of both NF‐κB pathways by ADP‐heptose in a similar way as by H. pylori infection (Fig 1D). Together, our results indicated that ADP‐heptose is required for the activation of the classical and the alternative NF‐κB pathways upon H. pylori infection.
Figure 1. H. pylori‐induced classical and alternative NF‐κB pathways depend on ADP‐heptose.
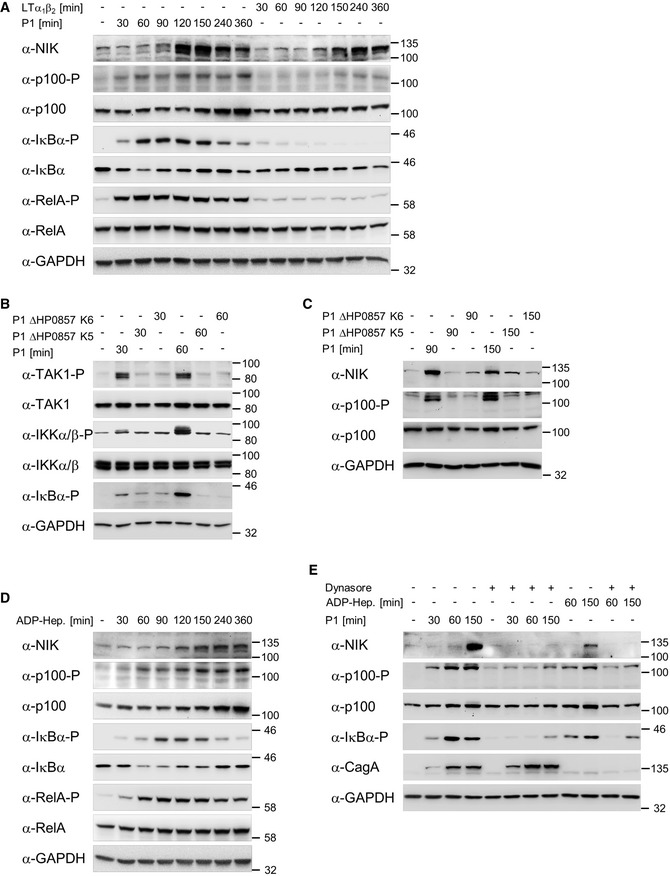
-
AAGS cells were infected with H. pylori or treated with 20 ng/ml LTα1β2 for the indicated times. Total cell lysates were analyzed by immunoblotting using the indicated antibodies.
-
B, CAGS cells were infected with H. pylori P1 wild‐type or isogenic ΔHP0857 (mutated in the gmhA gene) strains. Total cell lysates were analyzed by immunoblotting using the indicated antibodies.
-
DAGS cells were treated with 200 nM synthetic ADP‐heptose for the indicated times. Total cell lysates were analyzed by immunoblotting using the indicated antibodies.
-
EAGS cells were pre‐incubated with 100 μM dynamin GTPase inhibitor Dynasore for 60 min followed by H. pylori infection or addition of ADP‐heptose (200 nM). Total cell lysates were analyzed by immunoblotting using the indicated antibodies. CagA demonstrated a similar infection rate.
Data information: Data shown are representative for at least two independent experiments. GAPDH served as loading control.
Source data are available online for this figure.
Figure EV1. Induction of the classical and alternative NF‐κB pathways by H. pylori depends on ADP‐heptose.
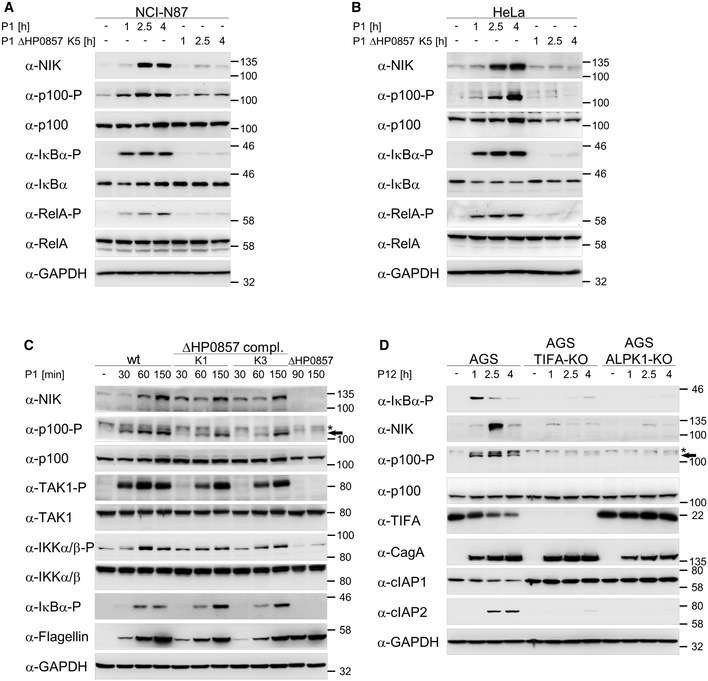
-
A, BInfection of NCI‐N87 and HeLa cells with the H. pylori P1 wild‐type or isogenic ΔHP0857 (mutated in the gmhA gene) strains was performed.
-
CAGS cells were infected with the H. pylori P1 wild‐type strain, isogenic strain mutated in the gmhA gene (ΔHP0857) alone or complemented with P1 gmhA gene. Asterisk denotes an unspecific band and the arrow indicates the band corresponding to phosphorylated p100.
-
DInfection of AGS, TIFA‐KO, and ALPK1‐KO cells with the H. pylori P12 wild‐type strain was performed. Asterisk denotes an unspecific band and the arrow indicates the band corresponding to phosphorylated p100.
Data information: Total cell lysates were analyzed by immunoblotting with the indicated antibodies. Data shown are representative for at least two independent experiments. GAPDH served as loading control.
Source data are available online for this figure.
Heptose‐1,7‐bisphosphate has been shown to enter cells via dynamin‐dependent endocytosis leading to NF‐κB activation (Gaudet et al, 2015). Therefore, we applied the dynamin GTPase inhibitor Dynasore (Macia et al, 2006) to further elucidate the mechanism of ADP‐heptose‐mediated NF‐κB activation during H. pylori infection. In cells pre‐incubated with Dynasore followed by either infection with H. pylori or treatment with exogenous ADP‐heptose, we observed considerably weaker activation of the classical or alternative NF‐κB pathways (Fig 1E). This suggests a possible involvement of dynamin‐dependent processes in the uptake of ADP‐heptose, an interesting concept that warrants further investigations.
To further evaluate the ADP‐heptose‐ALPK1‐TIFA signaling module in conjunction with classical and alternative NF‐κB activation upon H. pylori infection, we generated AGS knockout cells for the ADP‐heptose sensor ALPK1 (ALPK1‐KO) and TIFA (TIFA‐KO) using the CRISPR/Cas9 methodology. All presented TIFA‐KO clones showed an efficient knockout as indicated by the absence of TIFA (Fig 2A). We could not find a functional antibody that detects endogenous ALPK1 in AGS cells; therefore, we used qPCR to assess successful knockout of the ALPK1 transcript in ALPK1‐KO cells (Fig 2B). Any remaining signal probably stems from non‐functional transcripts detected by the TaqMan assay used. The expression of TIFA in ALPK1‐KO clones was preserved (Fig 2A). In ALPK1‐KO and TIFA‐KO cells, the inhibition of both NF‐κB pathways was evident from the reduced phosphorylation of IκBα and p100 and accumulation of NIK upon H. pylori infection (Fig 2A). Of note, although the second ALPK1‐KO clone showed a signal around the size of NIK, no activation of the alternative NF‐κB pathway occurred as seen by the missing phosphorylation of p100 (Fig 2A). The obtained results for the activation of the classical and alternative NF‐κB pathways in wild‐type, as well as ALPK1‐KO and TIFA‐KO AGS cells, were corroborated by infection with another H. pylori wild‐type strain P12 (Fig EV1D). Consequently, we found that TIFA deficiency prevented the translocation of transcription factors RelA and RelB to the nucleus (Fig EV2A). We also observed a significant impediment of the stress‐induced JNK and p38 signaling pathways in TIFA‐KO cells (Fig EV2B). In contrast, TIFA deficiency has no effect on the growth receptor‐mediated MAPK/ERK pathway (Fig EV2B). We further investigated the role of TIFA in NF‐κB activation using additional stimuli. We observed that the classical and alternative NF‐κB pathways, induced by IL‐1β or TNF, and LTα1β2, respectively, remained unaffected in the absence of TIFA (Fig EV2C–E). These data confirm similar results of other studies (Stein et al, 2017; Zimmermann et al, 2017).
Figure 2. ALPK1 and TIFA are required for induction of the classical NF‐κB pathway in H. pylori infection.
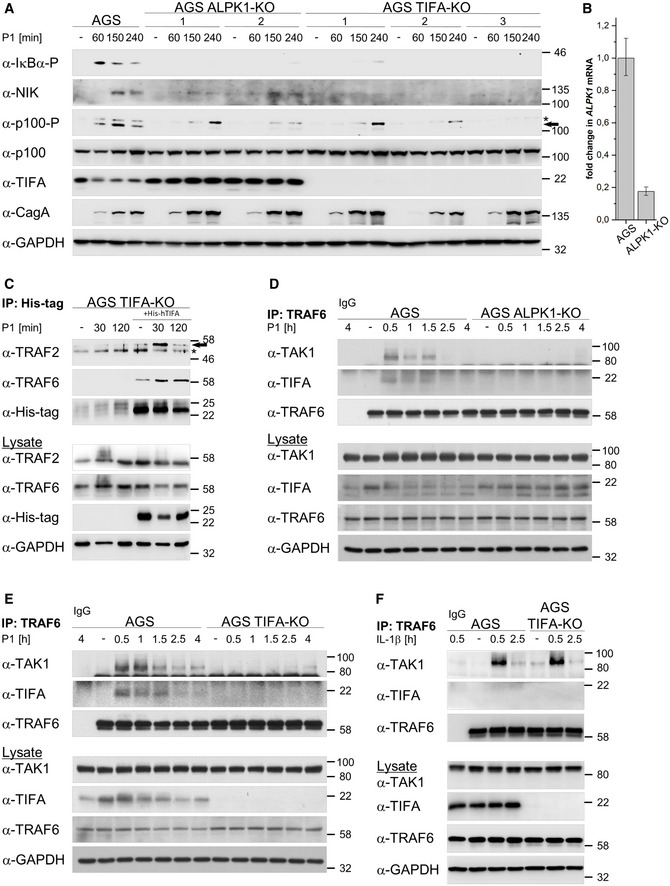
-
AParental AGS, ALPK1‐KO, and TIFA‐KO cells were infected with H. pylori for the times shown. Total cell lysates were analyzed by immunoblotting using the indicated antibodies. CagA demonstrated a similar infection rate of the different cells. Asterisk denotes an unspecific band and the arrow indicates the band corresponding to phosphorylated p100.
-
BTotal RNA was isolated from ALPK1‐KO cells and analyzed using quantitative RT‐PCR for the ALPK1 transcript. Data shown depict the average of triplicate determinations normalized to the RPL13A housekeeping gene. Error bars denote RQ ± RQmin/RQmax.
-
CTIFA‐KO cells were transfected with His‐tagged recombinant TIFA protein 24 h prior to H. pylori infection. Co‐IP with an anti‐His antibody was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies. Asterisk denotes an unspecific band and the arrow indicates the TRAF2 band.
-
D, EAGS and ALPK1‐KO (D) or TIFA‐KO (E) cells were infected with H. pylori for the times shown. Co‐IP with an anti‐TRAF6 antibody or isotype‐matched antibody (IgG) was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies.
-
FAGS and TIFA‐KO cells were treated with 30 ng/ml IL‐1β for the times shown. Co‐IP with an anti‐TRAF6 antibody or isotype‐matched antibody (IgG) was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies.
Data information: Data shown (A, C‐F) are representative for at least two independent experiments. GAPDH served as loading control. Data shown in (B) are from one experiment with three technical repeats.
Source data are available online for this figure.
Figure EV2. TIFA is required for the activation of NF‐κB upon H. pylori infection.
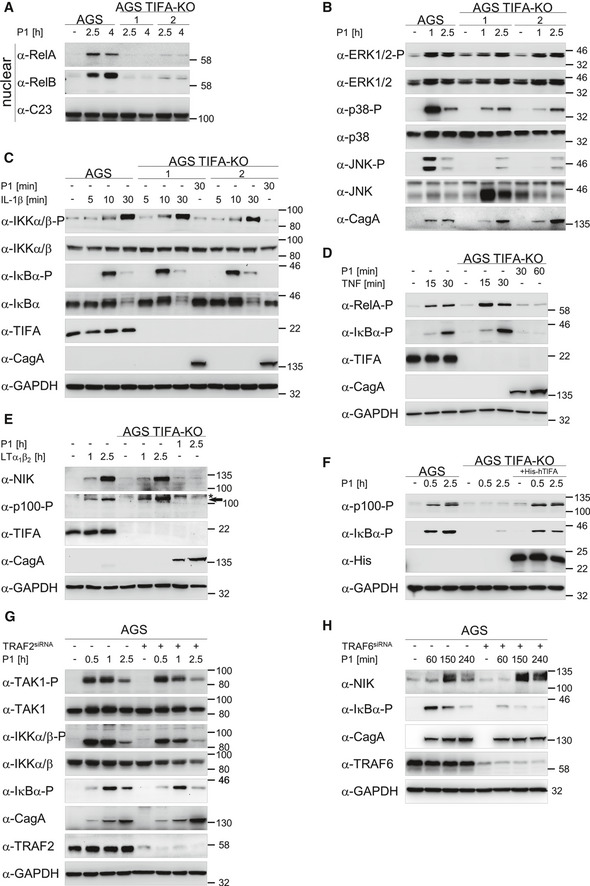
-
AThe nuclear translocation of transcription factors RelA and RelB was analyzed in nuclear fractions of AGS and TIFA‐KO cells. C23 served as loading control.
-
BAnalysis of the activation of ERK1/2, p38, and JNK in AGS and TIFA‐KO cells by immunoblot. CagA demonstrated a similar infection rate of different cells.
-
C–EAnalysis of IL‐1β (C), TNF (D), and LTα1β2 (E) treatment of AGS and TIFA‐KO cells by immunoblotting revealed the specificity of TIFA for H. pylori infection. Cell lysates were analyzed by immunoblotting with the indicated antibodies. Asterisk denotes an unspecific band and the arrow indicates the band corresponding to phosphorylated p100 (E).
-
FTransfection of His‐tagged recombinant TIFA into TIFA‐KO cells rescued the activation of classical and alternative NF‐κB pathways upon H. pylori infection. Cell lysates were analyzed by immunoblotting with the indicated antibodies.
-
GAGS cells were transfected with siRNA against TRAF2 and infected with the H. pylori P1 wild‐type strain. Cell lysates were analyzed by immunoblotting using the indicated antibodies.
-
HAGS cells were transfected with siRNA against TRAF6 and infected with the H. pylori P1 wild‐type strain. Cell lysates were analyzed by immunoblotting using the indicated antibodies.
Data information: Data shown are representative for at least two independent experiments. GAPDH served as loading control.
Source data are available online for this figure.
First identified by its capacity to bind TRAF2 (Kanamori et al, 2002), TIFA was also found to interact with TRAF6 (Ea et al, 2004; Milivojevic et al, 2017). To determine the potential endogenous binding partners of TIFA involved in the activation of both NF‐κB pathways by H. pylori, we transfected TIFA‐KO cells with His‐tagged recombinant TIFA protein, whose ability to rescue the TIFA functionality was shown (Fig EV2F), and performed co‐immunoprecipitation (co‐IP) using an anti‐His antibody. Expectedly, we detected the inducible interaction of TIFA with TRAF6 or TRAF2 (Fig 2C), which was shown to participate in the H. pylori‐mediated activation of the classical or alternative NF‐κB pathways, respectively (Hirata et al, 2006; Sokolova et al, 2014; Feige et al, 2018). To elucidate the potential impact of TRAF2 and TRAF6 on the activation of H. pylori‐induced classical and alternative NF‐κB pathways, respectively, we performed a knockdown of TRAF2 or TRAF6. Knockdown of TRAF2 did not affect the activation of classical NF‐κB signaling (Fig EV2G), whereas the depletion of TRAF6 significantly inhibited the activation of the classical NF‐κB but had no effect on the activation of the alternative NF‐κB pathway (Fig EV2H). This suggests a distinct separation of the classical and alternative NF‐κB pathways upon H. pylori infection on the level of TRAF6 and TRAF2, respectively. Immunoprecipitation of TRAF6 showed a transient interaction with TIFA and TAK1 in AGS cells upon H. pylori infection (Fig 2D and E). Notably, the interaction between TRAF6 and TAK1 seems to depend strictly on the presence of ALPK1 and TIFA as no TAK1 was bound to TRAF6 in ALPK1‐KO and TIFA‐KO cells upon H. pylori infection (Fig 2D and E). In contrast, treatment of these cells with IL‐1β led to TAK1 binding to TRAF6 independent of TIFA interaction with TRAF6, reiterating that the function of TIFA is specific to H. pylori infection (Fig 2F).
In resting cells, the alternative NF‐κB pathway is dormant due to the constant proteasomal degradation of NIK mediated by the NIK regulatory complex consisting of TRAF3, TRAF2, and cIAP1/2 (Zarnegar et al, 2008). Thus, we addressed whether the requirement of TIFA for the induction of the H. pylori‐mediated alternative NF‐κB pathway was linked to the regulation of the NIK regulatory complex. We transfected TIFA‐KO cells with His‐tagged recombinant TIFA protein and observed a fast interaction of TRAF2, TRAF3, and cIAP1 with co‐immunoprecipitated His‐tagged recombinant TIFA in H. pylori‐infected cells (Fig 3A). In contrast, none of these proteins was found in the co‐IP with His‐tagged recombinant TIFA in LTα1β2‐treated cells (Fig 3A), suggesting that the association of TIFA with the NIK regulatory complex is specific for H. pylori infection. Moreover, the association of TIFA with the NIK regulatory complex depends on ADP‐heptose as shown by the inhibition of dynamin (Fig 3B). Furthermore, in ALPK1‐KO cells, TIFA was unable to interact with the NIK regulatory complex (Fig 3C). Therefore, our data strongly imply that ADP‐heptose triggers the specific interaction of TIFA with the NIK regulatory complex in H. pylori infection. The immunoprecipitation of TRAF2 after H. pylori infection demonstrated a transient interaction of TIFA with TRAF2 (Fig 3D), similar to the interaction with TRAF6 (Fig 2D and E). Interestingly, in the TRAF2 co‐IP, we noted a decline in the interaction with cIAP1 at early time‐points and an occurrence of cIAP2 in the complex at later time‐points in H. pylori‐infected AGS but not TIFA‐KO cells (Fig 3D). Accordingly, in the lysates, we observed a decline of the cIAP1 protein and an increase in the expression of cIAP2 in H. pylori‐infected AGS cells but not in TIFA‐KO cells (Fig 3D, lower panels). It is important to note that upon H. pylori infection, we observed an early reduction in the level of endogenous cIAP1 that stabilizes at a lower plateau in AGS cells (Fig EV3A). On the contrary, in ALPK1‐KO and TIFA‐KO cells, which are defective in NF‐κB signaling, H. pylori infection had no effect on the level of cIAP1 (Fig EV3A). This phenomenon could explain the observations we made in the TRAF2 co‐IP experiments. Given that no change to the level of cIAP1 was detected in H. pylori‐infected AGS cells treated with the proteasome inhibitor MG‐132 thirty minutes after infection (Fig EV3B), our results suggest that infection by H. pylori elicits the proteasome‐dependent transient turnover of cIAP1, which leads to the accumulation of NIK. The proteasome‐dependent turnover of cIAP1 was corroborated by the detection of K48‐linked ubiquitinylation of cIAP1 upon H. pylori infection (Fig EV3C). Upon H. pylori infection, it is plausible that the binding of TIFA to the NIK regulatory complex contributes to this event due to the displacement of cIAP1 from the complex and its destabilization. The transient interaction of TIFA with the NIK regulatory complex, however, is not directly mediated by cIAP1 because in cIAP1‐depleted cells, we still found the transient, albeit enhanced, binding of TIFA to TRAF2 and TRAF3 (Fig 3E). Interestingly, we also observed an inducible decline of TRAF2 in the lysates of cIAP1‐depleted cells (Fig 3E).
Figure 3. ALPK1‐mediated TIFA oligomerization is required for induction of the alternative NF‐κB pathway in H. pylori infection.
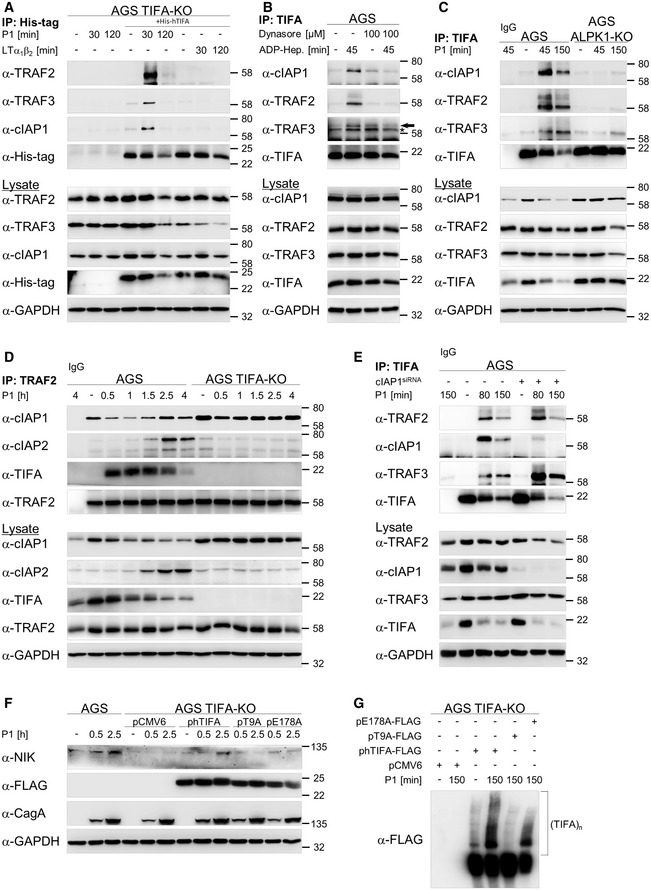
- TIFA‐KO cells were transfected with His‐tagged recombinant TIFA protein 24 h prior to H. pylori infection or treatment with 30 ng/ml LTα1β2. Co‐IP with an anti‐His antibody was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies.
- AGS cells were pre‐incubated with 100 μM dynamin GTPase inhibitor Dynasore for 60 min followed by addition of ADP‐heptose (200 nM). Co‐IP with an anti‐TIFA antibody was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies. Asterisk indicates an unspecific band and the arrow indicates the TRAF3 band.
- AGS and ALPK1‐KO cells were infected with H. pylori for the times shown. Co‐IP with an anti‐TIFA antibody or isotype‐matched antibody (IgG) was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies.
- AGS and TIFA‐KO cells were infected with H. pylori for the times shown. Co‐IP with an anti‐TRAF2 antibody or isotype‐matched antibody (IgG) was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies.
- AGS cells were transfected with siRNA against cIAP1. Co‐IP with an anti‐TIFA antibody or isotype‐matched antibody (IgG) was performed. Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies.
- TIFA‐KO cells were transfected with empty vector plasmid (pCMV6), wild‐type human (phTIFA), phosphorylation‐defective mutant (pT9A), or TRAF6 binding‐defective mutant (pE178A) FLAG‐tagged TIFA plasmids. Total cell lysates were analyzed by immunoblotting using the indicated antibodies.
- TIFA‐KO cells were transfected with empty vector plasmid (pCMV6), wild‐type human (phTIFA), phosphorylation‐defective mutant (pT9A), or TRAF6 binding‐defective mutant (pE178A) FLAG‐tagged TIFA plasmids. Total cell lysates were separated by native gel electrophoresis under non‐reducing conditions.
Data information: Data shown are representative for at least two independent experiments. GAPDH served as loading control.
Source data are available online for this figure.
Figure EV3. Degradation of cIAP1 and synthesis of cIAP2 upon H. pylori infection in AGS, ALPK1‐KO, and TIFA‐KO cells.
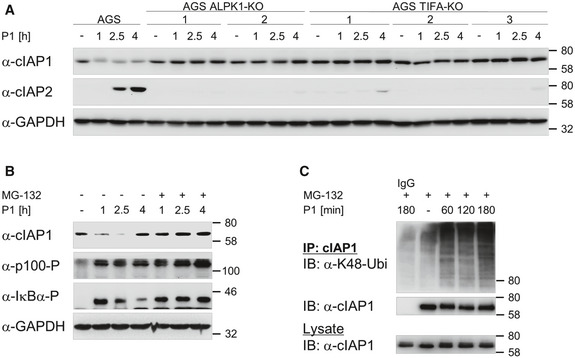
- AGS, ALPK1‐KO, and TIFA‐KO cells were infected with H. pylori. Total cell lysates were analyzed by immunoblotting for the expression of cIAP1 and cIAP2.
- AGS cells were treated with 10 µM MG‐132 thirty minutes after infection with H. pylori. Total cell lysates were analyzed by immunoblotting for the expression of cIAP1 and phosphorylation of p100 and IκBα.
- AGS cells were treated with 10 µM MG‐132 thirty minutes after infection with H. pylori. Cells were harvested in lysis buffer containing 1% SDS. Lysates were diluted 1:10 in lysis buffer without SDS and subjected to IP with antibody against cIAP1 or an isotype‐matched antibody (IgG). Eluates and total cell lysates were analyzed by immunoblotting using the indicated antibodies.
Data information: Data shown are representative for at least two independent experiments. GAPDH served as loading control.
Source data are available online for this figure.
Significance of the phosphorylation‐defective TIFA mutant (T9A) as well as the mutant defective in TRAF6 binding (E178A) for the activation of classical NF‐κB signaling upon bacterial infection has been shown elsewhere (Milivojevic et al, 2017). We observed the rescue of the alternative NF‐κB pathway when TIFA‐KO cells were complemented with wild‐type TIFA but not the TIFA mutant T9A (Fig 3F). In contrast, we witnessed a weak but nevertheless present restoration of the alternative NF‐κB pathway by the mutant E178A (Fig 3F). This could be explained by the notion that self‐oligomerization of TIFA, a prerequisite for NF‐κB activation, is preserved in the E178A mutant but absent in the T9A mutant (Fig 3G), confirming the results of Gaudet et al (2015).
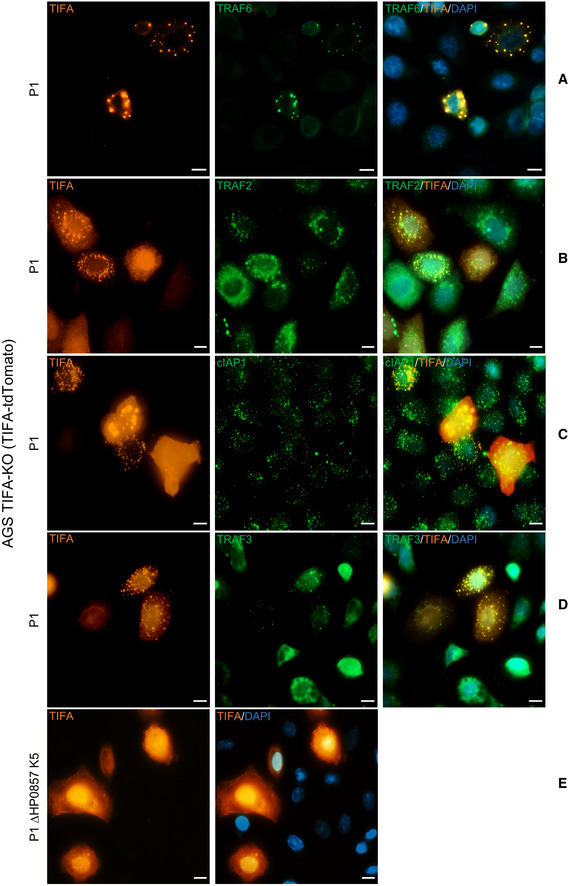
To corroborate the findings on the interaction of TIFA with different effector molecules of the classical and alternative NF‐κB pathways, we transiently transfected TIFA‐KO cells with TIFA‐tdTomato, which enabled us to visualize the formation of the TIFAsomes upon H. pylori infection (Fig 4), a phenomenon first described by Zimmermann et al (2017). Subsequent immunofluorescence staining of TRAF6, TRAF2, cIAP1, or TRAF3 (Fig 4A–D) revealed a partial co‐localization with the TIFAsomes (Fig 4), which was analyzed using Costes’ automatic threshold (Bolte & Cordelières, 2006) (Figs EV4B and EV5). The formation of TIFAsomes was absent in ΔHP0857‐infected (Fig 4E) and uninfected TIFA‐KO cells (Fig EV4A) transiently transfected with TIFA‐tdTomato.
Figure 4. Co‐localization of proteins involved in the activation of classical and alternative NF‐κB pathways with the TIFAsomes in H. pylori infection.
-
A–DIn TIFA‐KO cells transiently expressing TIFA‐tdTomato, co‐localization of TRAF6 (A), TRAF2 (B), cIAP1 (C), and TRAF3 (D) with TIFA‐tdTomato, which formed TIFAsomes upon infection with H. pylori P1 wild‐type strain, were detected by immunofluorescence staining. The nuclei were counterstained with DAPI. Scale bar = 10 µm.
-
ETIFA‐KO cells transiently expressing TIFA‐tdTomato were infected with H. pylori P1 strain mutated in the gmhA gene (ΔHP0857). Scale bar = 10 µm.
Data information: Data shown are representative for at least two independent experiments.
Figure EV4. Co‐localization of TRAF6 with TIFAsomes formed upon H. pylori infection.
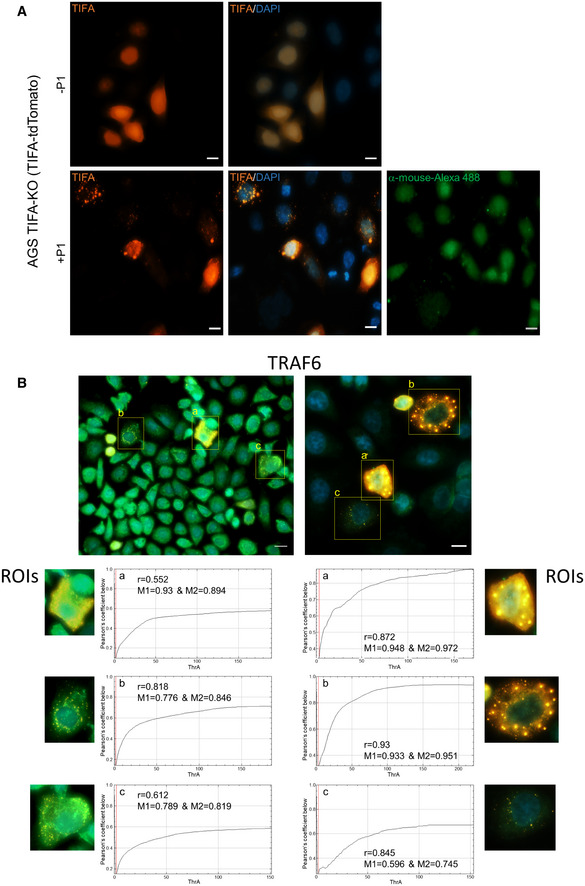
- TIFA‐KO cells transiently expressing TIFA‐tdTomato were left uninfected or infected with H. pylori for 60 min. TIFAsomes were detected by epifluorescence microscopy. Secondary antibody control with no primary antibody added was also shown. The nuclei were counterstained with DAPI. Scale bar = 10 µm.
- TIFA‐KO cells were transiently transfected with TIFA‐tdTomato and infected with H. pylori for 60 min, followed by immunofluorescence detection of TRAF6. Specific regions of interest (ROIs) in the micrographs were analyzed for co‐localization between TRAF6 and TIFAsomes. The Fiji software with the plug‐in JACoP using Costes’ automatic threshold was used. Presented are the values for Pearson’s coefficient (r) and Manders’s coefficient (M1&M2). Scale bar = 20 µm (left) and 10 µm (right).
Figure EV5. Co‐localization of TRAF3, TRAF2, or cIAP1 with TIFAsomes formed upon H. pylori infection.
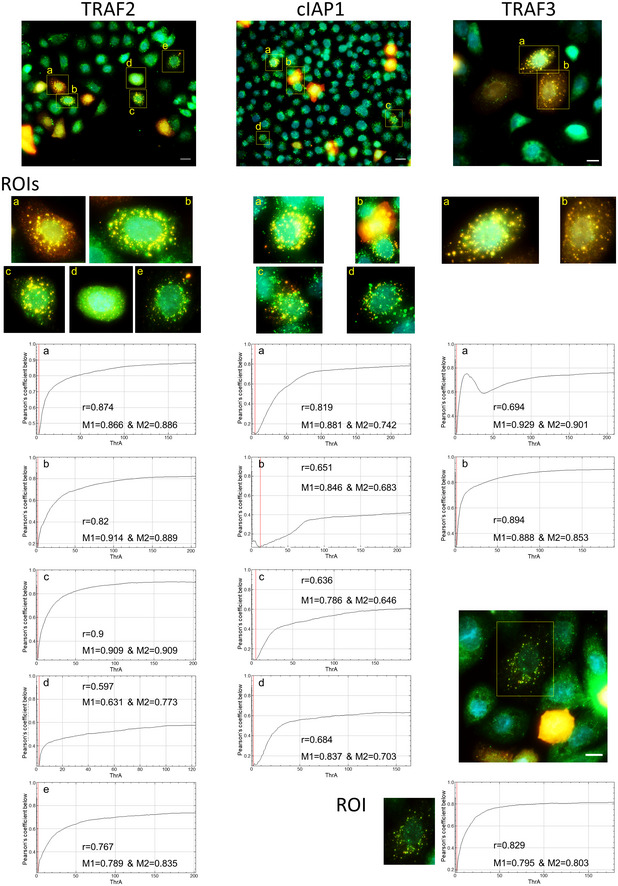
TIFA‐KO cells were transiently transfected with TIFA‐tdTomato and infected with H. pylori for 60 min, followed by immunofluorescence detection of TRAF2, cIAP1, and TRAF3. Specific regions of interest (ROIs) in the micrographs were analyzed for co‐localization of TRAF2, cIAP1, or TRAF3 with TIFAsomes. The Fiji software with the plug‐in JACoP using Costes’ automatic threshold was used. Presented are the values for Pearson’s coefficient (r) and Manders’s coefficient (M1&M2). Scale bar = 20 µm (TRAF2, cIAP1) and 10 µm (TRAF3).
The formation of oligomeric TIFA‐TRAF6 complexes is required for propagating classical NF‐κB activation (Milivojevic et al, 2017). Our results confirm this notion and extend the knowledge that TAK1 binding to TRAF6 upon H. pylori infection depends on the presence of TIFA. At the same time, our data also show that TRAF3, TRAF2, and cIAP1 could be involved in the formation of TIFAsomes. Therefore, different TIFAsome assemblies exist for the activation of the classical and alternative NF‐κB pathways by H. pylori. This dual activation of both NF‐κB pathways leads to complex gene regulation that could trigger a variety of cellular events including not only the induction of inflammatory cytokines by the classical NF‐κB pathway, but also other physiological changes leading to gastric pathologies due to the alternative NF‐κB pathway (Merga et al, 2016).
In conclusion, our study has demonstrated that TIFA is involved not only in classical but also alternative NF‐κB signaling. This dual function of TIFA seems to be essential only for H. pylori‐ but not for ligand‐mediated activation of classical and alternative NF‐κB pathways.
Materials and Methods
Cell culture and bacteria
AGS (ATCC, CRL‐1739), NCI‐N87 (ATCC, CRL‐5822), HeLa (ATCC, CCL‐2), and AGS ALPK1‐KO or TIFA‐KO cells were maintained in RPMI 1640 medium (Gibco®/Life Technologies) supplemented with 10% fetal calf serum (FCS) at 37 °C in an incubator with humidified 5% CO2 atmosphere. One day before infection with H. pylori, the growth medium was changed to RPMI 1640 supplemented with 0.2% FCS. For immunofluorescence staining, AGS TIFA‐KO cells were seeded into µ‐slide 8‐well chambered coverslip (ibidi, #80826) at 25000 cells per well.
Helicobacter pylori strain P1 was grown on GC agar plates supplemented with 10% horse serum (Gibco®/Life Technologies), 5 μg/ml trimethoprim, 1 μg/ml nystatin, and 10 μg/ml vancomycin under microaerophilic conditions at 37°C for 48 h before infection. The antibiotics were obtained from Sigma‐Aldrich. For the strain P1 ΔHP0857, deficient in the gene gmhA, the GC agar plates were additionally supplemented with 10 µg/ml chloramphenicol. For infection, the H. pylori culture was prepared in phosphate‐buffered saline (PBS) and eukaryotic cells were infected at a multiplicity of infection (MOI) of 100.
To terminate the production of ADP‐heptose by H. pylori, we generated a knockout mutant in one of the involved biosynthesis genes (phosphoheptose isomerase gmhA, HP0857). For this purpose, we amplified gmhA together with the two flanking genes HP0856 and HP0858 using primers 972F (5’‐ATGGAATTTTATAAAAAACAAACTTTA) and 972R (5’‐ATGAAAAAAATCTTAGTCATAGGC) giving rise to a polymerase chain reaction (PCR) product of 3,051 bp. This PCR fragment was subsequently cloned into the pGEM‐T plasmid (Promega). The gmhA gene was then opened by PCR using primers 973F (5’‐AACGATGTTTTGATAGGGATTTC) and 973R (5’‐TTTTACCCCCAACGCTTCCAC) followed by ligation of the chloramphenicol resistance gene cassette (cat R, about 1 kb EcoRV/SmaII fragment) obtained from plasmid pTnMax1 (Backert et al, 2000), which was fused to an rpsL cassette, resulting in streptomycin‐sensitive (rpsL encodes dominant streptomycin sensitivity) and chloramphenicol‐resistant transformants as described (Tegtmeyer et al, 2020). This construct was then used to mutagenize H. pylori by natural transformation and double cross‐over recombination according to standard procedures (Moese et al, 2001). Correct insertion of the cat R‐rpsL cassette into gmhA on the H. pylori genome was confirmed by PCR using the above‐mentioned primers. Chloramphenicol‐resistant transformants were selected by five days of cultivation on GC agar plates containing 10% horse serum, 1 μg/ml nystatin, 5 μg/ml trimethoprim, 10 μg/ml vancomycin, 10 µg/ml chloramphenicol, and 10 µg/ml streptomycin. For genetic complementation of gmhA, the above‐described 3,051 bp PCR product containing the wild‐type gmhA gene was used to replace the cat R‐rpsL in the mutant by natural transformation (Barrozo et al, 2013). Contra‐selection was then applied to screen for streptomycin‐resistant H. pylori. Correct integration of the wild‐type gmhA gene was again confirmed by PCR.
The following compounds were used at the indicated final concentration: Dynasore (Merck Millipore, #324410‐10MG) 100 µM; MG‐132 (Selleckchem, #S2619) 10 µM; and ADP‐heptose (InvivoGen, #tlrl‐adph) 200 nM.
Generation of knockout AGS cell lines using CRISPR/Cas9
The knockout of TIFA and ALPK1 in AGS cells was performed using reagents from IDT as follows: Alt‐R® CRISPR‐Cas9 crRNA Hs. Cas9.TIFA.1.AC: AATCTGACCATGCACCTGTA TGG; Alt‐R® CRISPR‐Cas9 crRNA Hs. Cas9.ALPK1.1.AC: CGAGTTAAGGACCCTGATCC AGG; Alt‐R® CRISPR‐Cas9 tracrRNA; ATTO™ 550 (#1075927); and Alt‐R® S.p. HiFi Cas9 Nuclease V3 (#1081060), and LipofectamineTM CRISPRMAXTM transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, the crRNA and tracrRNA were mixed equimolar at 1 µM in nuclease‐free duplex buffer (IDT), heated at 95 °C for 5 min, and cooled to RT to produce gRNA. The gRNA (1 µM, 1.5 µl) was combined with Cas9 (1 µM, 1.5 µl) and Cas9 PLUSTM reagent (CRISPRMAX kit, 0.6 µl) in 21.4 µl Opti‐MEM medium (Gibco®/Life Technologies, #31985070) to form the ribonucleoprotein (RNP) complex and incubated for 5 min at RT. The RNP (total volume 25 µl) was combined with CRISPRMAX transfection reagent 1.2 µl in 23.8 µl Opti‐MEM and incubated for another 20 min at RT. The transfection mixture was added to a well in a 96‐well culture plate followed by the addition of 40,000 AGS cells. On the next day, the cells were trypsinized and used for clonal selection by the limiting dilution method.
Reverse transcription and quantitative PCR
Total RNA was isolated from cells using the NucleoSpin® RNA Plus kit (Macherey‐Nagel). The total RNA was reverse transcribed into cDNA using the RT2 HT First Strand kit (Qiagen) on the C1000 Thermal cycler (Bio‐Rad). For the analysis of the ALPK1 transcript, the TaqMan® assay Hs01567926_m1 was used (Thermo Fisher Scientific). The quantitative PCR was performed using the StepOnePlus™ Real‐Time PCR System (Applied Biosystems, Thermo Fisher Scientific). The Comparative CT Method (ΔΔC T) was used to quantify relative changes in the target mRNA.
Transfection of siRNA, protein, and plasmids
On the day before siRNA transfection, cells were seeded at a density of 1 x 106 per 60‐mm culture dish. Transfection was performed using siLentFect™ (Bio‐Rad, #1703362) according to the manufacturer’s protocol. Cell culture medium was changed to Opti‐MEM prior to transfection. The siRNA against TRAF2 (Santa Cruz Biotechnology, sc‐29509) or TRAF6 (Santa Cruz Biotechnology, sc‐36717) was used at a final concentration of 20 nM. A scrambled siRNA was used as negative control (Dharmacon, #D‐001810‐10). The medium was changed to RPMI 1640 medium containing 10% FCS six hours after transfection. Protein transfection of His‐tagged recombinant TIFA protein was performed as follows: 5 µg recombinant human TIFA protein (Novus Biologicals, NBP1‐99103‐50 µg) was combined with 10 µl Cas9 PLUS™ reagent in 250 µl Opti‐MEM and incubated at RT for 5 min. Six microliters of CRISPRMAX transfection reagent was diluted in 250 µl Opti‐MEM and combined with the diluted Cas9 PLUSTM reagent/recombinant TIFA solution, followed by incubation at RT for 20 min before adding dropwise to the cells in the dishes.
Plasmids for rescue experiments were prepared using the QuikChange II XL site‐directed mutagenesis kit from Agilent Technologies (#200521‐5). The original plasmid containing wild‐type human TIFA C‐terminal tagged with Myc‐FLAG (Origene, #RC204357) was mutated with the following mutagenesis primer pairs: s_hTIFA_T9A 5'‐AGACAAGTTACTGTCTCTTCGGCGTCAGCATCTTCAAAACTGG‐3' and as_hTIFA_T9A 5'‐CCAGTTTTGAAGATGCTGACGCCGAAGAGACAGTAACTTGTCT‐3'; s_hTIFA_E178A 5'‐GTTGACTCATTTTCATCCATGGCTGTCGGAGAACTGCTTTGG‐3'; and as_hTIFA_E178A 5'‐CCAAAGCAGTTCTCCGACAGCCATGGATGAAAATGAGTCAAC‐3'. Plasmid transfection was performed using METAFECTENE® PRO transfection reagent (Biontex, #T040‐1.0) according to the manufacturer’s protocol. Three microgram plasmid was transfected per 60‐mm dish using 2 µl transfection reagent per 1 µg plasmid, both diluted in PBS without calcium and magnesium. Cells seeded in the µ‐slides 8‐well chambered coverslips (ibiTreat) were transfected using the same procedure with 0.5 µg of the TIFA‐tdTomato plasmid (Zimmermann et al, 2017) per well. Five hours after transfection, the medium was changed to fresh RPMI 1640 medium containing 10% FCS.
Preparation of whole cell lysates, subcellular fractionation, immunoprecipitation, and immunoblotting
Cells were lysed in whole cell lysis buffer (50 mM Tris/HCl pH7.5, 150 mM NaCl, 5 mM EDTA, 10 mM K2HPO4, 10% glycerol, 1% Triton X‐100, 0.5% NP‐40) supplemented with 1 mM sodium vanadate, 1 mM sodium molybdate, 20 mM sodium fluoride, 10 mM sodium pyrophosphate, 1 mM AEBSF, 20 mM 2‐phosphoglycerate, and protease inhibitor mix (cOmplete™, Mini, EDTA‐free, Roche). Subcellular fractionation was performed as described previously (Studencka‐Turski et al, 2018) to obtain cytosolic and soluble nuclear fractions. Protein concentration was estimated using the Pierce™ BCA protein assay kit (Thermo Fisher Scientific).
For co‐immunoprecipitation, the whole cell lysate was rotated with the respective antibody (1 μg antibody/1 mg protein) overnight at 4°C. The IgG control was an isotype‐matched IgG preparation from non‐immune serum (Sigma‐Aldrich). Pre‐washed Pierce™ protein A/G magnetic beads (Thermo Fisher Scientific, #88802) were added and rotated for 1 h at 4°C. The beads were washed four times with lysis buffer and once with PBS containing 150 mM NaCl and eluted in 2× Laemmli sample buffer. For samples used for IP under denaturing conditions, cells were scraped in lysis buffer containing 1% SDS and heated at 95°C for 15 min followed by centrifugation at 15,700 g for 15 min. Before IP, cell lysates were diluted 1:10 in lysis buffer without SDS.
For SDS–PAGE and immunoblotting, samples were heated at 95°C for 5 min and separated in Tris‐Glycine gels, transferred onto PVDF membranes (Millipore), and blocked for 1 h at RT using 5% skim milk in TBS containing 0.1% Tween 20 (TBS‐T). The primary antibodies were incubated overnight in either 5% BSA or 5% skim milk in TBS‐T at 4 °C. The membranes were washed thrice in TBS‐T. The appropriate HRP‐conjugated secondary antibody (Jackson ImmunoResearch Laboratories) was added at a dilution of 1:10,000 in 5% skim milk in TBS‐T. After 1‐h incubation at RT, the membranes were washed thrice in TBS‐T. The membranes were developed using a chemiluminescent substrate (Millipore, #WBKLS0500). The band pattern was visualized using the ChemoCam Imager (Intas). For native PAGE, Laemmli sample buffer with 0.02% Brilliant Blue G (Sigma‐Aldrich, #B0770) instead of bromophenol blue and without SDS was added to the samples. The samples were not heated prior to protein separation by native PAGE as described (Preissler et al, 2015).
The following primary antibodies were used: GAPDH (Millipore, #MAB374); CagA (Austral Biologicals, #HPM‐5001‐5); Flagellin (Acris, AM00865PU‐N); C23 (sc‐13057), A20 (sc‐166692), RelA (sc‐8008), RelB (sc‐226), cIAP1 (sc‐271419, for denaturing IP), TRAF2 (sc‐136999), and His‐probe (sc‐804) were purchased from Santa Cruz Biotechnology; phospho‐IκBα (#9246), IκBα (#4812), phospho‐RelA (#3031), phospho‐p38 (#4511), p38 (#9212), phospho‐SAPK/JNK (#4671), phospho‐p44/42 (Erk1/2) (#4377), phospho‐p100 (#4810), phospho‐TAK1 (#4508), NIK (#4994), TIFA (#61358), TAK1 (#5206), TRAF6 (#8028), SAPK/JNK (#9252), cIAP1 (#7065), cIAP2 (#3130), p44/42 MAPK (Erk1/2) (#4696), and p100/p52 (#4882) were purchased from Cell Signaling Technology. Anti‐TRAF3 antibody (#700121) was from Invitrogen. Anti‐FLAG antibody (#F3165) was from Sigma‐Aldrich.
Immunofluorescence staining and epifluorescence microscopy
Cells grown and transfected in µ‐slide 8‐well chambered coverslips (ibiTreat) were gently washed with PBS before fixation with 4% PFA for 10 min at RT. The PFA was removed and cells were washed thrice with PBS. The cells were blocked and permeabilized by incubation in blocking solution (10% fetal calf serum and 0.25% Triton X‐100 in TBS) for 1 h at RT on an orbital shaker. The cells were incubated with the primary antibody (TRAF6 (sc‐8409), cIAP1 (sc‐271419), TRAF2 (sc‐136999), or TRAF3 (sc‐6933) from Santa Cruz Biotechnology) diluted in 1/10 of blocking solution for 1 h at RT, followed by the secondary antibody (anti‐mouse Alexa488, Thermo Fisher Scientific) in 1% BSA in TBS‐T for 1 h at RT. The primary antibody was omitted as a secondary antibody control. The cells were counterstained with DAPI (Sigma‐Aldrich, #D9542) to visualize the nuclei. Images were acquired on an Axio Observer 7 equipped with the Colibri 5 RGB‐UV, a camera Axiocam 305 color, and a 40× objective (Zeiss Plan‐Apochromat, NA 1.4, Oil a = 0.13 mm) using the software ZEN 3.0 pro (Carl Zeiss microscopy). The filter sets used were as follows: DAPI, 96 HE BFP; Alexa488, 38 HE GFP; and DsRed, 43 HE DsRed. Z‐stacks were acquired, followed by maximum projection. For co‐localization analysis, the Fiji software with the plug‐in JACoP using Costes’ automatic threshold was used (Bolte & Cordelières, 2006). The analysis included only the ROIs depicted in the images (Figs EV4B and EV5). Presented are the values for Pearson’s coefficient (r) and Manders’s coefficient (M1&M2) (Manders et al, 1992).
Author contributions
GM and MN conceived the study; GM and MCCL designed experiments; GM, MCCL, and OS performed experiments; GM, MCCL, and MN analyzed and interpreted data; GM, MCCL, and MN wrote the manuscript. SB and TFM provided reagents, expertise, and feedback.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We thank S. Reissinger for technical assistance. The work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project‐ID 97850925—SFB 854 (A04) to M.N. Open Access funding enabled and organized by Projekt DEAL.
EMBO reports (2021) 22: e52878.
Data availability
No data were deposited in a public database. Inquiries for reagents used in this study should be addressed to M. Naumann (naumann@med.ovgu.de).
References
- Backert S, Ziska E, Brinkmann V, Zimny‐Arndt U, Fauconnier A, Jungblut PR, Naumann M, Meyer TF (2000) Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol 2: 155–164 [DOI] [PubMed] [Google Scholar]
- Barrozo RM, Cooke CL, Hansen LM, Lam AM, Gaddy JA, Johnson EM, Cariaga TA, Suarez G, Peek RM, Cover TLet al (2013) Functional plasticity in the type IV secretion system of Helicobacter pylori . PLoS Pathog 9: e1003189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232 [DOI] [PubMed] [Google Scholar]
- Bravo D, Hoare A, Soto C, Valenzuela MA, Quest AF (2018) Helicobacter pylori in human health and disease: mechanisms for local gastric and systemic effects. World J Gastroenterol 24: 3071–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea C‐K, Sun L, Inoue J‐I, Chen ZJ (2004) TIFA activates IκB kinase (IKK) by promoting oligomerization and ubiquitination of TRAF6. Proc Natl Acad Sci USA 101: 15318–15323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige MH, Vieth M, Sokolova O, Täger C, Naumann M (2018) Helicobacter pylori induces direct activation of the lymphotoxin beta receptor and non‐canonical nuclear factor‐kappa B signaling. Biochim Biophys Acta 1865: 545–550 [DOI] [PubMed] [Google Scholar]
- Gall A, Gaudet RG, Gray‐Owen SD, Salama NR (2017) TIFA signaling in gastric epithelial cells initiates the cag Type 4 secretion system‐dependent innate immune response to Helicobacter pylori infection. MBio 8: e01168–e11117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Weber D, Dangeard A‐S, Cornil J, Thai L, Rytter H, Zamyatina A, Mulard LA, Arrieumerlou C (2018) ADP‐heptose is a newly identified pathogen‐associated molecular pattern of Shigella flexneri. EMBO Rep 19: e46943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet RG, Sintsova A, Buckwalter CM, Leung N, Cochrane A, Li J, Cox AD, Moffat J, Gray‐Owen SD (2015) Cytosolic detection of the bacterial metabolite HBP activates TIFA‐dependent innate immunity. Science 348: 1251–1255 [DOI] [PubMed] [Google Scholar]
- Heine M, Cramm‐Behrens CI, Ansari A, Chu H‐P, Ryazanov AG, Naim HY, Jacob R (2005) α‐Kinase 1, a new component in apical protein transport. J Biol Chem 280: 25637–25643 [DOI] [PubMed] [Google Scholar]
- Hirata Y, Ohmae T, Shibata W, Maeda S, Ogura K, Yoshida H, Kawabe T, Omata M (2006) MyD88 and TNF receptor‐associated factor 6 are critical signal transducers in Helicobacter pylori infected human epithelial cells. J Immunol 176: 3796–3803 [DOI] [PubMed] [Google Scholar]
- Huang C‐C, Weng J‐H, Wei T‐Y, Wu P‐Y, Hsu P‐H, Chen Y‐H, Wang S‐C, Qin D, Hung C‐C, Chen S‐Tet al (2012) Intermolecular binding between TIFA‐FHA and TIFA‐pT mediates Tumor Necrosis Factor Alpha stimulation and NF‐κB activation. Mol Cell Biol 32: 2664–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori M, Suzuki H, Saito R, Muramatsu M, Hayashizaki Y (2002) T2BP, a novel TRAF2 binding protein, can activate NF‐κB and AP‐1 without TNF stimulation. Biochem Biophys Res Commun 290: 1108–1113 [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T (2006) Dynasore, a cell‐permeable inhibitor of Dynamin. Dev Cell 10: 839–850 [DOI] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA (1992) Dynamics of three‐dimensional replication patterns during the S‐phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103: 857–862 [DOI] [PubMed] [Google Scholar]
- Mejías‐Luque R, Zöller J, Anderl F, Loew‐Gil E, Vieth M, Adler T, Engler DB, Urban S, Browning JL, Müller Aet al (2017) Lymphotoxin β receptor signalling executes Helicobacter pylori‐driven gastric inflammation in a T4SS‐dependent manner. Gut 66: 1369–1381 [DOI] [PubMed] [Google Scholar]
- Merga YJ, O'Hara A, Burkitt MD, Duckworth CA, Probert CS, Campbell BJ, Pritchard DM (2016) Importance of the alternative NF‐κB activation pathway in inflammation‐associated gastrointestinal carcinogenesis. Am J Physiol Gastrointest Liver Physiol 310: G1081–G1090 [DOI] [PubMed] [Google Scholar]
- Milivojevic M, Dangeard A‐S, Kasper CA, Tschon T, Emmenlauer M, Pique C, Schnupf P, Guignot J, Arrieumerlou C (2017) ALPK1 controls TIFA/TRAF6‐dependent innate immunity against heptose‐1,7‐bisphosphate of gram‐negative bacteria. PLoS Pathog 13: e1006224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moese S, Selbach M, Zimny‐Arndt U, Jungblut PR, Meyer TF, Backert S (2001) Identification of a tyrosine‐phosphorylated 35 kDa carboxy‐terminal fragment (p35CagA) of the Helicobacter pylori CagA protein in phagocytic cells: processing or breakage? Proteomics 1: 618–629 [DOI] [PubMed] [Google Scholar]
- Naumann M, Sokolova O, Tegtmeyer N, Backert S (2017) Helicobacter pylori: A paradigm pathogen for subverting host cell signal transmission. Trends Microbiol 25: 316–328 [DOI] [PubMed] [Google Scholar]
- Neumann M, Naumann M (2007) Beyond IκBs: alternative regulation of NF‐κB activity. FASEB J 21: 2642–2654 [DOI] [PubMed] [Google Scholar]
- Pfannkuch L, Hurwitz R, Trauisen J, Sigulla J, Poeschke M, Matzner L, Kosma P, Schmid M, Meyer TF (2019) ADP heptose, a novel pathogen‐associated molecular pattern identified in Helicobacter pylori . FASEB J 33: 9087–9099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, Chambers JE, Crespillo‐Casado A, Avezov E, Miranda E, Perez J, Hendershot LM, Harding HP, Ron D (2015) Physiological modulation of BiP activity by trans‐protomer engagement of the interdomain linker. eLife 4: e08961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, Wiedmann M, Erdjument‐Bromage H, Tempst P, Parmer TG, Prostko CRet al (1997) Identification of a new class of protein kinases represented by eukaryotic elongation factor‐2 kinase. Proc Natl Acad Sci USA 94: 4884–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova O, Maubach G, Naumann M (2014) MEKK3 and TAK1 synergize to activate IKK complex in Helicobacter pylori infection. Biochim Biophys Acta 1843: 715–724 [DOI] [PubMed] [Google Scholar]
- Stein SC, Faber E, Bats SH, Murillo T, Speidel Y, Coombs N, Josenhans C (2017) Helicobacter pylori modulates host cell responses by CagT4SS‐dependent translocation of an intermediate metabolite of LPS inner core heptose biosynthesis. PLoS Pathog 13: e1006514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studencka‐Turski M, Maubach G, Feige MH, Naumann M (2018) Constitutive activation of nuclear factor kappa B‐inducing kinase counteracts apoptosis in cells with rearranged mixed lineage leukemia gene. Leukemia 32: 2498–2501 [DOI] [PubMed] [Google Scholar]
- Sun S‐C (2017) The non‐canonical NF‐κB pathway in immunity and inflammation. Nat Rev Immunol 17: 545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegtmeyer N, Neddermann M, Lind J, Pachathundikandi SK, Sharafutdinov I, Gutiérrez‐Escobar AJ, Brönstrup M, Tegge W, Hong M, Rohde Met al (2020) Toll‐like receptor 5 activation by the CagY repeat domains of Helicobacter pylori . Cell Rep 32: 108159 [DOI] [PubMed] [Google Scholar]
- Xiao G, Fong A, Sun S‐C (2004) Induction of p100 Processing by NF‐κB‐inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα‐mediated phosphorylation. J Biol Chem 279: 30099–30105 [DOI] [PubMed] [Google Scholar]
- Yu C‐K, Wang C‐J, Chew Y, Wang P‐C, Yin H‐S, Kao M‐C (2016) Functional characterization of Helicobacter pylori 26695 sedoheptulose 7‐phosphate isomerase encoded by hp0857 and its association with lipopolysaccharide biosynthesis and adhesion. Biochem Biophys Res Commun 477: 794–800 [DOI] [PubMed] [Google Scholar]
- Zamani M, Ebrahimtabar F, Zamani V, Miller WH, Alizadeh‐Navaei R, Shokri‐Shirvani J, Derakhshan MH (2018) Systematic review with meta‐analysis: the worldwide prevalence of Helicobacter pylori infection. Aliment Pharmacol Ther 47: 868–876 [DOI] [PubMed] [Google Scholar]
- Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh W‐C, Mak TWet al (2008) Noncanonical NF‐κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 9: 1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, She Y, Dong Na, Li P, He H, Borio A, Wu Q, Lu S, Ding X, Cao Yet al (2018) Alpha‐kinase 1 is a cytosolic innate immune receptor for bacterial ADP‐heptose. Nature 561: 122–126 [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Pfannkuch L, Al‐Zeer MA, Bartfeld S, Koch M, Liu J, Rechner C, Soerensen M, Sokolova O, Zamyatina Aet al (2017) ALPK1‐ and TIFA‐dependent innate immune response triggered by the Helicobacter pylori Type IV secretion system. Cell Rep 20: 2384–2395 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Data Availability Statement
No data were deposited in a public database. Inquiries for reagents used in this study should be addressed to M. Naumann (naumann@med.ovgu.de).