

Abstract
Previous work employing five SARS-CoV-2 spike protein receptor-binding domain (RBD) constructs, comprising versions originally developed by Mt. Sinai or the Ragon Institute and later optimized in-house, revealed potential heterogeneity which led to questions regarding variable seropositivity assay performance. Each construct was subjected to N-deglycosylation and subsequent intact mass analysis, revealing significant deviations from predicted theoretical mass for all five proteins. Complementary tandem MS/MS analysis revealed the presence of an additional pyroGlu residue on the N-termini of the two Mt. Sinai RBD constructs, as well as on the N-terminus of the full-length spike protein from which they were derived, thus explaining the observed mass shift and definitively establishing the spike protein N-terminal sequence. Moreover, the observed mass additions for the three Ragon Institute RBD constructs were identified as variable N-terminal cleavage points within the signal peptide sequence employed for recombinant expression. To resolve this issue and minimize heterogeneity for further seropositivity assay development, the best-performing RBD construct was further optimized to exhibit complete homogeneity, as determined by both intact mass and tandem MS/MS analysis. This new RBD construct has been validated for seropositivity assay performance, is available to the greater scientific community, and is recommended for use in future assay development.
Keywords: SARS-CoV-2, COVID-19, spike protein, mass spectrometry, intact mass, bottom-up proteomics
Introduction
The onset of the COVID-19 pandemic spurred extensive research into the properties of the SARS-CoV-2 spike protein due to its antigenic role. The spike protein, one of the four structural proteins, exists as a trimer on the surface of the virus, with each individual monomer heavily glycosylated to avoid detection by the immune system.1,2 The receptor-binding domain (RBD) of the spike protein specifically targets angiotensin-converting enzyme 2 (ACE2) on the surface of host cells for entry.3 Antibodies against spike protein and RBD antigens have been demonstrated to correlate well with effective neutralization of the virus.4−6 In fact, the vaccines approved under the United States Food and Drug Administration’s (FDA’s) Emergency Use Authorization were created to train the immune system against this target, along with multiple serology-based assays intended to facilitate detection of patient antibodies generated in response to prior infection.7−12 For this latter approach, initial studies focused on two recombinant versions of RBD, one created at Mt. Sinai13 and one created at the Ragon Institute of MGH, MIT, and Harvard,14 both of which were made widely available to the scientific community to improve global understanding of COVID-19 infection and subsequent immunity. More recently, improved methods for expression and purification of these recombinant RBD constructs allowed for broader distribution of these resource reagents (Figure S1).11 This innovation directly facilitated the expansion of population seropositivity assays such as a pilot National Institutes of Health collaborative study, which indicated that COVID-19 infection had occurred at a rate approximately 4.6 times that predicted by PCR-based testing.10
During the optimization process for these improved RBD constructs, it was noted that those originating from the Mt. Sinai RBD exhibited lower sensitivity in enzyme-linked immunosorbent assay (ELISA)-based serology assays than those originating from the Ragon RBD.10,11 While the inclusion of a C-terminal streptavidin-binding protein (SBP) sequence within the improved Ragon RBD construct was demonstrated to be responsible for the improved sensitivity of this construct to seropositivity assay antibodies,11 the observation of additional heterogeneity within these recombinant RBD construct populations, specifically altered molecular weight profiles observed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Figure S2), raised further questions about which specific properties might play a role in RBD seropositivity assay performance. Although the glycosylation profiles of both RBD constructs, along with those of the intact spike protein, have been extensively characterized,15−18 less attention has been paid to the role of the eukaryotic signal peptide sequence selected for the recombinant RBD expression process. Each RBD construct bears a distinct eukaryotic signal sequence which is cleaved by signal peptidase in a process more dependent on residue charge than specific amino acid sequence.19,20 However, signal peptide cleavage can be variable when individual protein domains are recombinantly expressed, especially in the case of RBD, with an uncertain N-terminus selected from the genomic sequence of the SARS-CoV-2 spike protein (UniProtKB entry: SPIKE_SARS2).21,22 As both the Mt. Sinai and Ragon Institute RBDs bear distinct N-terminal sequences,13,14 it was hypothesized that this difference may contribute to the heterogeneity observed for the improved versions of these constructs during the optimization process.
In contrast to traditional peptide-based (or “bottom-up”) proteomic methods, analysis of intact proteins by mass spectrometry can reveal the distribution and relative abundance of intact, modified protein forms (“proteoforms”) within a sample population.23 Such “top-down” methods are particularly well-suited for the detection of unexpected protein features, which can then be further characterized by targeted fragmentation or complementary proteomic strategies. Therefore, we subjected five recombinant RBD constructs, comprising the original and improved versions of the Mt. Sinai and Ragon Institute RBD proteins,11,13,14 to intact mass analysis to identify potential heterogeneity. We identified significant deviations from the theoretical mass across all five constructs, which we determined were due to variable N-terminal processing by subsequent proteolytic digest and high-resolution tandem MS/MS. We then verified that the N-terminus of the full-length spike protein was subjected to similar variable processing by tandem MS/MS, isolating the cause of the heterogeneity to the signal peptide sequence. Finally, we optimized a new RBD construct and demonstrated complete homogeneity by combined intact mass and tandem MS/MS analysis. This latest construct is available to the greater scientific community and recommended for use in further COVID-19 assay development.
Methods
Production of RBD Proteins
DNA clones for the production of RBD proteins were generated as previously described in Mehalko et al.11 Final DNA expression constructs for M67 (#166018), M68 (#166019), and M96 (#166020) are available from the Addgene repository (www.addgene.org). Expression and purification were also carried out as previously described.11 Briefly, Expi293F cells were transiently transfected according to the manufacturer’s instructions (ThermoFisher). Enhancers were added at 18–20 h post-transfection and the cultures were set at 32 °C. The supernatants were harvested via centrifugation at 96 h post-transfection. The purification of the constructs was then performed using nickel-charged magnetic beads (GenScript) equilibrated in 1× phosphate-buffered saline (PBS), which were added to harvested supernatants and allowed to mix for 1 h at RT. The beads were then washed with 1× PBS and eluted with 1× PBS with 0.5 M Imidazole. Samples were electrophoresed on a 10–20% Tris–Glycine SDS-PAGE gel and those positive for protein as visualized by Coomassie stain were pooled. Finally, the purified proteins were filtered through a low-protein binding 0.2 μm syringe filter, aliquoted into 1.7 mL microtubes, and flash-frozen in liquid nitrogen.
Intact Mass Analysis of RBD Proteins
Each purified RBD sample was subjected to N-deglycosylation via Rapid PNGase F (New England Biolabs) following the manufacturer’s protocol. Samples of 20 μg were prepared in duplicate, brought to a final volume of 20 μL in an Eppendorf Protein LoBind tube using Optima H2O (Thermo Fisher Scientific), incubated at 50 °C for 5 min, reduced in a final concentration of 20 mM dithiothreitol (DTT) at room temperature (RT) for 5 min, and diluted 1:5 in buffer A (5% Optima acetonitrile (ACN), 95% Optima H2O, 0.2% mass spectrometry grade formic acid (FA); all Thermo Fisher Scientific). N-deglycosylated RBD proteins were then concentrated and desalted via Elut OMIX C4 pipette tip (100 μL volume, Agilent Technologies), using the following scheme: Activation in 100% Optima ACN (5 × 50 μL), equilibration in 0.2% FA in Optima H2O (5 × 50 μL), sample binding (15 × 50 μL), washing in 0.2% FA in Optima H2O (8 × 50 μL), and elution in 80% Optima ACN and 0.2% FA in Optima H2O (10 × 10 μL). Eluted proteins were then diluted 1:5 in buffer A and transferred to autosampler vials.
Intact N-deglycosylated RBD proteins were analyzed by liquid chromatography coupled on-line with mass spectrometry (LC-MS). Reverse-phase separation was performed on a Vanquish Flex chromatographic system (Thermo Fisher Scientific) using a MabPac RP analytical column (4 μm, 1500 Å, 3 × 50 mm, Thermo Fisher Scientific) maintained at 50 °C. Proteins were separated at 0.5 mL/min over an 8 min gradient of buffer B (47.5% Optima ACN, 47.5% Optima isopropanol (IPA, Thermo Fisher Scientific), 5% Optima H2O, and 0.2% FA) from 2 to 100% followed by re-equilibration at 2% buffer B (total run time 10 min). The outlet of the column was routed through a divert valve into a heated electrospray ionization (HESI) source connected to an Exactive Plus EMR mass spectrometer (Thermo Fisher Scientific).
Low-resolution intact mass (MS1) spectra were acquired over a 600–2500 m/z window at 8750 FT resolution (at 200 m/z), averaging 10 microscans, with an AGC target value of 3 × 106, 200 ms maximum inject time, 80% S-lens RF level, 15 V source-induced dissociation, and capillary temperature of 320 °C. The resulting MS1 spectra were manually averaged followed by deconvolution using the ReSpect algorithm in BioPharma Finder 3.2 (Thermo Fisher Scientific). The deconvolution parameters set were a 20 ppm deconvolution mass tolerance, 6–10 minimum adjacent charges (low and high model mass), 0% relative abundance threshold, 2:2 left/right peak shape, peak detection minimum significance measure of 1 standard deviation, 95% peak detection quality measure, peak model width factor of 1, 0.01 intensity threshold scale, and the noise compensation set to true. Neutral average masses (M) were reported with two decimal places. The mass error was calculated as the difference between the reported average mass and the theoretical average mass.
High-resolution intact mass (MS1) spectra were acquired over a 600–2000 m/z window at 140 000 FT resolution (at 200 m/z), averaging four microscans, with an AGC target value of 1 × 106, 200 ms maximum inject time, 80% S-lens RF level, and capillary temperature of 320 °C. The resulting MS1 spectra were manually averaged followed by deconvolution using Xtract in BioPharma Finder 3.2. The deconvolution parameters set were a signal-to-noise threshold of 3, 0% relative abundance threshold, three minimum detected charge states, 80% fit factor, 25% remainder threshold, minimum intensity of 1, and an expected intensity error of 3. Neutral monoisotopic masses (M) were reported with three decimal places. The mass error was calculated as the difference between the reported monoisotopic mass and the theoretical monoisotopic mass.
Bottom-Up Mass Spectrometry Analysis of RBD Proteins
Aliquots of full-length spike and each purified RBD sample were subjected to N-deglycosylation via Rapid PNGase F following the manufacturer’s protocol. Samples of 20 μg were brought to a final volume of 20 μL in an Eppendorf Protein LoBind tube using Optima H2O. Full-length spike underwent denaturation in the supplied buffer at 80 °C for 2 min, cooling at room temperature for 10 min, and incubation with PNGase F at 50 °C for 30 min. The purified RBDs were incubated with PNGase F at 50 °C for 10 min. N-deglycosylated proteins were then precipitated with ice-cold acetone at −20 °C for 1 h. Proteins were pelleted by centrifugation at max speed for 5 min and acetone was decanted. Pellets were washed with 1 mL ice-cold acetone, pelleted by centrifugation, and acetone was decanted. The pellets were left to air-dry for 20 min. The pellets were then resuspended in 20 μL of 8 M deionized urea in Optima H2O. DTT was added to a final concentration of ∼9 mM to reduce disulfide bonds for 1 h. IAA was added to a final concentration of ∼36 mM to alkylate free cysteines for 45 min in the dark. DTT was added to a final concentration of ∼38 mM to quench the alkylation reaction. The reduced and alkylated proteins were then diluted with 135 μL of 100 mM ammonium bicarbonate to dilute the urea concentration to below 2 M. Proteins were digested overnight (∼17 h) at 37 °C with 500 ng of sequencing grade modified trypsin (Promega). The digestion was quenched with 3 μL of concentrated FA. Digested proteins were brought to a final concentration of 0.2% trifluoroacetic acid (TFA; Thermo Fisher Scientific) and 5% ACN using 20% Optima ACN and 2% TFA in Optima H2O for desalting using Pierce C18 spin columns (Thermo Fisher Scientific) following the manufacturer’s protocol. Eluted proteins were then evaporated to dryness using a SpeedVac, resuspended in 48 μL of buffer A, and transferred to autosampler vials.
Trypsin digested, N-deglycosylated full-length spike, and RBD proteins were analyzed by liquid chromatography coupled on-line with mass spectrometry (LC-MS/MS). Reverse-phase separation was performed on an UltiMate 3000 chromatographic system (Thermo Fisher Scientific) using an Acclaim PepMap 100 C18 HPLC trap column (5 μm, 100 Å, 0.1 × 20 mm, Thermo Fisher Scientific) and Acclaim PepMap 100 C18 HPLC analytical column (3 μm, 100 Å, 0.075 × 500 mm, Thermo Fisher Scientific) maintained at 60 °C. Peptides were first loaded onto the trap column at 3 μL/min at 2% buffer B (95% Optima ACN, 5% Optima H2O, and 0.2% FA) over 10 min from the loading pump. The divert valve then switched to separate the peptides at 300 nL/min from the NC pump over a 90 min gradient of buffer B from 2 to 25%, ramp to 90% B in 1 min, column wash at 90% B for 3 min, ramp to 2% B in 2 min, and equilibration at 2% B for 15 min (total run time 120 min). Peptides were ionized by a Nanospray Flex ion source (Thermo Fisher Scientific) with a stainless steel emitter coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific).
Each digested sample was injected three times for analysis by different fragmentation methods. All MS instrument methods obtained intact mass (MS1) spectra at 120 000 FT resolution (at 200 m/z), an AGC target of 4 × 105, 50 ms maximum inject time, 30% S-lens RF level, and capillary temperature of 275 °C. All fragmentation scans (MS2) were taken at 30 000 FT resolution (at 200 m/z). The instrument was operated in top speed mode with a 3 s cycle time, and precursor ions were placed on an exclusion list after one scan for 60 s for all injections. The first injection isolated precursors with 2–8 charges by the quadrupole with a width of 2 m/z for higher-energy collisional dissociation (HCD) at 28% normalized collision energy (NCE). A product ion trigger was used to trigger an electron transfer dissociation (ETD) event with supplemental activation by HCD (EThcD) and a collision-induced dissociation (CID) event if one of the following masses was observed within the top-20 product ions: 204.0867 m/z (HexNAc), 138.0545 m/z (HexNAcFragment), 366.1396 m/z (HexNAcHex), 145.0495 m/z (HexFragmentA), 127.039 m/z (HexFragmentB), 292.103 m/z (NeuAc), and 274.092 m/z (NeuAc-H2O). EThcD scans used a quadrupole isolation window of 3 m/z, instrument calibrated reaction parameters with supplemental activation by HCD at 25% NCE, an AGC target of 1 × 105, and 120 ms maximum injection time. CID scans used a quadrupole isolation window of 2 m/z, an AGC target of 5 × 104, 30% fixed collision energy for 10 ms at an activation q of 0.25, and 60 ms maximum injection time. The second injection isolated precursors with 2–24 charges by the quadrupole with a width of 1.6 m/z for ETD events using the instrument calibrated reaction parameters, an AGC target of 5 × 104, and 54 ms maximum injection time. The third injection isolated precursors with 2–24 charges by the quadrupole with a width of 1.6 m/z for EThcD events using the instrument calibrated reaction parameters, supplemental activation of 15% NCE, an AGC target of 5 × 104, and 54 ms maximum injection time.
All files were searched against their respective sequence-specific database (single entry) and a common contaminants database (obtained from the Max Planck Institute of Biochemistry, Martinsried; 245 entries; download date: Sep 26, 2019) in Proteome Discoverer version 2.4.1.15 (Thermo Fisher Scientific) using SEQUEST and MS Amanda nodes. All cysteines were fixed with carbamidomethyl. For analyses of full-length spike proteins, variable modifications included N-terminus acetyl, N-Terminus Met-loss, N-terminus Met-loss + acetyl, Asn/Gln/Arg deamination, Met oxidation, peptide N-terminal Gln to pryoGlu, Asn Hex(5)HexNAc(2), Asn Hex(7)HexNAc(2), Ser/Thr Hex(1)HexNAc(1)NeuAc(2), and Ser/Thr Hex(2)HexNAc(2)NeuAc(2). Full-length spike protein searches allowed for three missed trypsin cleavages, four maximum modifications per peptide, and a maximum peptide mass of 10 kDa. For analyses of the RBD domains, variable modifications included N-terminus acetyl, N-Terminus Met-loss, N-terminus Met-loss + acetyl, Asn/Gln/Arg deamination, Met oxidation, peptide N-terminal Gln to pryoGlu, Asn/Ser/Thr HexNAc(1), Asn/Ser/Thr Hex(1)HexNAc(1), Ser/Thr Hex(1)HexNAc(1)NeuAc(2), and Ser/Thr Hex(2)HexNAc(2)NeuAc(2). RBD protein searches allowed for four missed trypsin cleavages, four maximum modifications per peptide, and a maximum peptide mass of 15 kDa. All searches had a precursor mass tolerance of 10 ppm for SEQUEST and 5 ppm for MS Amanda. Fragment mass tolerances were set to 0.02 Da. Manual analysis was performed on all protein N-terminal peptides for sequence validation, with resulting graphical fragment maps generated in ProSight Lite.24 The observed peptide neutral monoisotopic mass was manually calculated using the following equation
| 1 |
where M was the neutral monoisotopic mass, p was the observed monoisotopic mass for the peptide at charge state z, and 1.00727 Da was the mass of H+. The resulting value was rounded to four decimal places.
Raw files for both intact mass and tandem MS/MS analyses, along with BioPharma Finder reports, Proteome Discoverer search result files, protein sequences used to create custom RBD or full-length spike protein search databases, and converted peak list files (.mzXML), are available for download from the MassIVE repository with identifier MSV000087585.
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA was carried out as previously reported.10 Briefly, 200 ng of the purified RBD proteins were antigens against a monoclonal antibody specific to the SARS-CoV-2 RBD domain (mAb 109). A serial 3-fold dilution scheme from a 2.5 μg/mL stock of the monoclonal antibody was used. Measurements were taken in triplicate.
Results and Discussion
Intact RBD Mass Measurements
Five RBD constructs that have been previously published13,14 and optimized for high yield11 were analyzed by intact mass spectrometry to investigate the potential causes of observed heterogeneity (Figure S2). The Mt. Sinai construct consists of an N-terminal signal sequence comprising the first 14 residues of the SARS-CoV-2 spike protein, residues 319–541 of the RBD domain, and a C-terminal His6 tag resulting in a theoretical average mass of 25921.25 Da (Figure S3). The Ragon Institute construct consists of an N-terminal tissue plasminogen activator (TPA) signal sequence, residues 319–529 of the RBD domain, and a C-terminal combined HRV3C protease cleavage site, His8, and streptavidin-binding peptide tag, resulting in a theoretical average mass of 31185.80 Da (Figure S4). The remaining three RBD constructs (M67: optimized Mt. Sinai construct; M68: optimized Ragon Institute construct; M69: optimized Ragon Institute construct with only a His8 C-terminal tag (Figure S5)) were gene-optimized by ATUM, cloned into improved expression vectors, and proteins were purified at the Frederick National Laboratory for Cancer Research (FNLCR).11 The purified proteins were first treated with PNGase F to remove all N-glycosylations, thereby reducing the complexity and heterogeneity of the samples. The intact N-deglycosylated RBD proteins were then subjected to intact LC-MS analysis on an Exactive Plus EMR mass spectrometer to visualize the proteoforms present within each sample (Figures S6 and S7; Tables S1−S5). Example spectra from the optimized Mt. Sinai (M67) and Ragon Institute (M68) RBD constructs are shown in Figure 1. The most abundant species for all five proteins corresponded to the major O-glycans of Hex(1)HexNAc(1)NeuAc(2) [+947.84 Da] and Hex(2)HexNAc(2)NeuAc(2) [+1313.18 Da] as previously reported.15 However, each protein showed different discrepancies in observed masses when compared to the reported theoretical mass. The M67 construct had an unknown mass addition of 111 Da after accounting for the O-glycosylation. The M68 construct produced the predicted mass of RBD at about 50% fractional abundance after accounting for the O-glycosylation (Table S4). However, abundant unexplained mass shifts of 87 and 245 Da from each O-glycan peak were observed. Each unknown mass shift did not correspond to a different combination of glycosylations or a known post-translational modification. The original published version of each construct was also observed to bear these mass shifts (Figure S6A,C).
Figure 1.

Intact Mass Analysis of COVID RBD. (A) Optimized Mt. Sinai construct (M67) and (B) optimized Ragon construct (M68) following PNGase F treatment. The insets correspond to the ReSpect deconvolution of the respective MS1 scans, with significant species labeled. Both constructs had abundant O-glycans of Hex(1)HexNAc(1)NeuAc(2) and Hex(2)HexNAc(2)NeuAc(2). The observed O-glycan masses in the M67 construct had an additional unknown mass of 111 Da. The M68 construct had an unknown mass shift of 87 Da from each abundant O-glycan and a mass shift of 158 Da from the first unknown shift, for a total of 245 Da from the unmodified protein.
N-Terminus Confirmation by Bottom-Up Mass Spectrometry
The deviations from theoretical intact mass exhibited by the five RBD proteins were further explored by tryptic digestion of PNGase F-treated samples and subsequent analysis by tandem LC-MS/MS on an Orbitrap Fusion Lumos mass spectrometer. While initial searches against the respective predicted protein sequence provided complete coverage across each protein, the N-terminal peptide (Figure 2A) was identified at relatively low abundance in the M67 sample with respect to the remainder of the protein, leading to the hypothesis that the observed mass shifts may be located at the N-termini of these RBD constructs. A manual search of the M67 and original Mt. Sinai RBD data successfully localized the unknown mass shift to the N-terminal peptide, demonstrating that the additional 111 Da was due to a Gln residue N-terminal to the expected RBD tryptic peptide and modified to a pyroGlu (Figure 2B). This residue originated from the signal sequence of the full-length spike protein (1MFVFLVLLPLVSSQ14), suggesting that the preferred cleavage point is at Ser13, as predicted with 72% probability by the SignalP cleavage site prediction algorithm.25 Generally, secreted proteins in eukaryotes contain an N-terminal signal sequence that is recognized by the ER.19,20,26,27 This sequence is broken into three parts: charged N-terminal domain, hydrophobic core, and hydrophilic C-terminal domain.20,28 The hydrophobic core is essential for the signal peptide to insert in the ER membrane while the C-terminal domain is recognized by signal peptidase I for cleavage and release from the membrane.20,28 Positions −1 and −3 from the cleavage site, which are primarily occupied by small amino acids, are two factors influencing where the peptidase will cleave.27,29 Together, these results indicate that Gln14 from the full-length spike protein signal sequence is not compatible in the −1 position for cleavage by signal peptidase I and that Ser13 is a better candidate in the −1 position as predicted, leaving the Gln residue with the mature protein as observed.
Figure 2.

Determining the N-terminus of RBD Protein by EThcD. (A) Predicted N-terminal peptide as found in M68, M69, and original Ragon Institute constructs. (B) N-terminal peptide found in M67 and original Mt. Sinai constructs containing an additional Gln residue. This Gln residue was modified to a pyroGlu residue, as noted by the yellow box. (C) N-terminal peptide found in M68, M69, and original Ragon Institute constructs containing an additional Ser residue. (D) N-terminal peptide found in M68, M69, and original Ragon Institute constructs containing additional Ser-Ala-Ser residues. Carbamidomethylated Cys is noted in blue, and deamidated Asn residues are noted by gray boxes. Observed c/z ions are noted by red flags between residues, while observed b/y ions are noted by blue flags. All peptides were found to be O-glycosylated with Hex(1)HexNAc(1)NeuAc(2) on the first Thr residue (T5 of RBD; T323 of full-length spike) as noted by the yellow box with glycan pictogram. The complementary ion pairs resulting from the backbone cleavage of T5/E6 and E6/S7 confirm the glycosylation localization.
The N-terminus of a previously published recombinant full-length spike protein30 was also investigated to determine whether the cleavage of the signal peptide sequence observed in the Mt. Sinai and M67 RBD constructs also occurred in this construct. SignalP predicts that the most probable cleavage point is Cys15 at about 54%, while the probability of cleaving at Ser13, similarly to the RBD, is about 30%. N-deglycosylated full-length spike protein was digested by trypsin and analyzed by LC-MS/MS in a similar manner to the RBD proteins. Manual analysis revealed that the N-terminus of full-length spike protein contained an N-terminal pyroGlu residue similar to those found in the Mt. Sinai and M67 RBD constructs (Figure 3). Therefore, it is likely that the spike protein signal peptide sequence will also consistently be cleaved after Ser13, marking the pyroGlu at position 14 as the start of the mature spike protein sequence.
Figure 3.
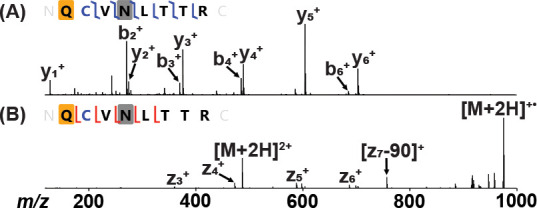
Confirming the N-terminal tryptic peptide of the full-length spike protein. (A) HCD fragmentation of the doubly charged peptide. The yellow box corresponds to a pyroGlu modification, the blue C corresponds to carbamidomethylated Cys, and the gray box corresponds to a deamidated Asn. (B) ETD fragmentation of the doubly charged peptide. The 90 Da loss on the observed z7 ion corresponds to the radical loss of the alkylated cysteine side chain (•S–CH2–CO–NH2).32
Since the mass shift in the M67 construct was localized to the N-terminus from the signal sequence, it was hypothesized that the unknown mass shifts of 87 and 245 Da in the M68, M69, and original Ragon Institute RBD samples may also be due to additional residues from variable cleavage of their respective signal sequences. The Ragon Institute RBD construct includes a TPA signal sequence31 prior to the RBD sequence (1MDAMKRGLCCVLLLCGAVFVSPSAS25), with probable cleavage points predicted by SignalP at Ser25 (34%), Ala24 (14%), and Pro22 (11%). A manual search of the data confirmed that the unknown mass shifts were also due to additional residues from the TPA signal sequence. The 87 Da shift was due to an additional Ser residue at the N-terminus (Figure 2C), while the addition of 245 Da was due to additional Ser-Ala-Ser residues at the N-terminus (Figure 2D). This indicated that the predicted cleavage at Ser25 occurred about 50% of the time, with additional cleavages occurring upstream (Tables S3–S5), suggesting that the charged Arg residue in the +1 position may variably influence cleavage by signal peptidase I.
Refinement of RBD N-Terminus
After variable peptide sequence cleavage had been identified within the original Ragon Institute, M68, and M69 RBD constructs, further construct optimization was performed with the goal of eliminating N-terminal heterogeneity while maintaining high protein yield and high ELISA sensitivity.11 To do this, the starting position for the RBD protein was altered while retaining the TPA signal sequence. intact mass analysis of each N-deglycosylated construct was then performed to visualize the impact of each change in starting position on resulting RBD protein heterogeneity. The construct that produced an apparently homogeneous RBD population, referred to as M96 (Figure S8), had a starting position of Phe318 (Figure 4A; Table S6). The homogeneity of this new construct was then evaluated by tryptic digestion and subsequent LC-MS/MS. The digestion results confirmed that the only species present in this sample was the correct sequence of RBD (Figure 4B). Therefore, this suggests that the insertion of a large, aromatic group helps solidify the recognized positions by signal peptidase I, thus facilitating the generation of a consistent RBD protein population.
Figure 4.

(A) Intact Mass Analysis of COVID RBD following PNGase F treatment of the M96 construct, which modified the Ragon RBD sequence to include F318 of the spike protein. The inset corresponds to the ReSpect deconvolution result. The major O-glycans of Hex(1)HexNAc(1)NeuAc(2) [+947.3430 Da] and Hex(2)HexNAc(2)NeuAc(2) [+1312.4552 Da] were observed in this construct as well as a fully unmodified species. (B) Confirmation of the N-terminus by EThcD. Carbamidomethylated Cys is noted in blue, and deamidated Asn residues are noted by gray boxes. Observed c/z ions are noted by red flags between residues, while observed b/y ions are noted by blue flags. The peptide was found to be O-glycosylated with Hex(1)HexNAc(1)NeuAc(2) on the first Thr residue (T6 of RBD; T323 of full-length spike) as noted by the yellow box with glycan pictogram. The complementary ion pairs resulting from the backbone cleavage of T6/E7 and E7/S8 confirm the glycosylation localization.
The M96 RBD construct was subsequently validated for COVID-19 seropositivity performance by ELISA in parallel with the previously optimized M67, M68, and M69 RBD constructs (Figure S9). The resulting data indicate that M96 performs near-identically to M68, meaning that variable N-terminal processing was not likely responsible for previously observed variable RBD seropositivity assay performance11 and that seropositivity data previously obtained using M68 were not negatively impacted by the presence of multiple proteoforms. Nevertheless, the newly optimized and fully homogeneous M96 RBD construct is recommended for use in further downstream assay development to minimize the potential for uncertainty provided by use of a variably heterogeneous reagent panel.
Conclusions
Accurate recombinant standard proteins are necessary to ensure that proper conclusions can be drawn from experiments and serological assays. Moreover, analyzing proteins by intact mass spectrometry can reveal new details unavailable from or difficult to obtain by other proteomic methods, allowing increased confidence in the quality of key recombinant standard proteins used for ongoing COVID-19 assay development. Sources of N-terminal sequence heterogeneity were identified and verified within five previously published SARS-CoV-2 spike protein RBD constructs, along with the full-length spike protein construct. A new RBD construct, M96, was then created and verified to exhibit a homogeneous N-terminus consistent with the predicted protein sequence. This new construct is available to the greater scientific community and recommended for future use in the development of COVID-19 assays to ensure consistency and validity of results.
Acknowledgments
The authors thank Dr. Florian Krammer (Icahn School of Medicine, Mt. Sinai; through BEI resources) and Dr. Aaron Schmidt (Ragon Institute of MGH, MIT and Harvard) for graciously providing their original RBD DNA constructs. We would like to thank the NIAID Vaccine Research Center for providing the construct to create the monoclonal antibody against the SARS-CoV-2 RBD domain.
Glossary
Abbreviations Used
- RBD
receptor-binding domain
- ACE2
angiotensin-converting enzyme 2
- FDA
United States Food and Drug Administration
- SBP
streptavidin-binding protein
- LC-MS
liquid chromatography coupled on-line with mass spectrometry
- LC-MS
liquid chromatography coupled on-line with tandem mass spectrometry
- TPA
tissue plasminogen activator
- FNCLR
Frederick National Laboratory for Cancer Research
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00349.
Structural features of the constructs analyzed (Figure S1); SDS-PAGE image of all constructs (Figure S2); the Predicted Mt. Sinai RBD protein sequence (Figure S3); the predicted Ragon RBD protein sequence (Figure S4); the predicted M69 RBD protein sequence (Figure S5); ReSpect deconvolution of initial RBD constructs (Figure S6); intact mass resolution comparison of M68 (Figure S7); predicted M96 RBD protein sequence (Figure S8); comparative ELISA sensitivity of the four RBD constructs (Figure S9); intact mass analysis of the original Mt. Sinai construct (Table S1); intact mass analysis of M67 (Table S2); intact mass analysis of the original Ragon construct (Table S3); mass analysis of M68 (Table S4); intact mass analysis of M69 (Table S5); intact mass analysis of M98 (Table S6) (PDF)
Manually annotated spectra of all MS2 scans shown (PDF)
Author Contributions
M.D., J.M., K.S., V.W., and Z.P. cloned, expressed, and purified the RBD protein constructs. R.D., M.D., and C.D. prepared samples for and performed mass spectrometry experiments. R.D., M.D., D.E., and C.D. analyzed the resulting intact mass and tandem MS/MS data. R.D., D.E., and C.D. conceptualized and wrote the paper.
This project has been funded with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
The authors declare no competing financial interest.
Notes
Colleagues within the top-down proteomics field are strongly urged to consider what precautions can be taken to protect their ion optics before pursuing these deceptively accessible low-MW protein targets. During attempted analyses of N-deglycosylated, C4-desalted RBD constructs by targeted top-down LC-MS/MS on an Orbitrap Fusion Lumos (data not shown), the source ion optics sustained damage sufficiently severe to require replacement of the ion funnel and MP00 to restore the instrument to working order (albeit with permanent residual ion optics charging). Careful consideration of such experiments is therefore recommended to avoid similar instrument damage (although it should be noted that the ion optics of the Exactive Plus EMR, which contain an S-lens sustained no ill effects from these analyses).
Supplementary Material
References
- Huang Y.; Yang C.; Xu X. F.; Xu W.; Liu S. W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant O. C.; Montgomery D.; Ito K.; Woods R. J. Analysis of the SARS-CoV-2 spike protein glycan shield reveals implications for immune recognition. Sci. Rep. 2020, 10, 14991 10.1038/s41598-020-71748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Park Y. J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 281–292. 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R.; Shan C.; Duan X.; Chen Z.; Liu P.; Song J.; Song T.; Bi X.; Han C.; Wu L.; Gao G.; Hu X.; Zhang Y.; Tong Z.; Huang W.; Liu W. J.; Wu G.; Zhang B.; Wang L.; Qi J.; Feng H.; Wang F. S.; Wang Q.; Gao G. F.; Yuan Z.; Yan J. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- Suthar M. S.; Zimmerman M. G.; Kauffman R. C.; Mantus G.; Linderman S. L.; Hudson W. H.; Vanderheiden A.; Nyhoff L.; Davis C. W.; Adekunle O.; Affer M.; Sherman M.; Reynolds S.; Verkerke H. P.; Alter D. N.; Guarner J.; Bryksin J.; Horwath M. C.; Arthur C. M.; Saakadze N.; Smith G. H.; Edupuganti S.; Scherer E. M.; Hellmeister K.; Cheng A.; Morales J. A.; Neish A. S.; Stowell S. R.; Frank F.; Ortlund E.; Anderson E. J.; Menachery V. D.; Rouphael N.; Mehta A. K.; Stephens D. S.; Ahmed R.; Roback J. D.; Wrammert J. Rapid Generation of Neutralizing Antibody Responses in COVID-19 Patients. Cell Rep. Med. 2020, 1, 100040 10.1016/j.xcrm.2020.100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Wang P.; Nair M. S.; Yu J.; Rapp M.; Wang Q.; Luo Y.; Chan J. F.; Sahi V.; Figueroa A.; Guo X. V.; Cerutti G.; Bimela J.; Gorman J.; Zhou T.; Chen Z.; Yuen K. Y.; Kwong P. D.; Sodroski J. G.; Yin M. T.; Sheng Z.; Huang Y.; Shapiro L.; Ho D. D. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Polack F. P.; Thomas S. J.; Kitchin N.; Absalon J.; Gurtman A.; Lockhart S.; Perez J. L.; Perez Marc G.; Moreira E. D.; Zerbini C.; Bailey R.; Swanson K. A.; Roychoudhury S.; Koury K.; Li P.; Kalina W. V.; Cooper D.; Frenck R. W. Jr.; Hammitt L. L.; Tureci O.; Nell H.; Schaefer A.; Unal S.; Tresnan D. B.; Mather S.; Dormitzer P. R.; Sahin U.; Jansen K. U.; Gruber W. C. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden L. R.; El Sahly H. M.; Essink B.; Kotloff K.; Frey S.; Novak R.; Diemert D.; Spector S. A.; Rouphael N.; Creech C. B.; McGettigan J.; Khetan S.; Segall N.; Solis J.; Brosz A.; Fierro C.; Schwartz H.; Neuzil K.; Corey L.; Gilbert P.; Janes H.; Follmann D.; Marovich M.; Mascola J.; Polakowski L.; Ledgerwood J.; Graham B. S.; Bennett H.; Pajon R.; Knightly C.; Leav B.; Deng W.; Zhou H.; Han S.; Ivarsson M.; Miller J.; Zaks T. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoff J.; Le Gars M.; Shukarev G.; Heerwegh D.; Truyers C.; de Groot A. M.; Stoop J.; Tete S.; Van Damme W.; Leroux-Roels I.; Berghmans P. J.; Kimmel M.; Van Damme P.; de Hoon J.; Smith W.; Stephenson K. E.; De Rosa S. C.; Cohen K. W.; McElrath M. J.; Cormier E.; Scheper G.; Barouch D. H.; Hendriks J.; Struyf F.; Douoguih M.; Van Hoof J.; Schuitemaker H. Interim Results of a Phase 1-2a Trial of Ad26.COV2.S Covid-19 Vaccine. N. Engl. J. Med. 2021, 384, 1824–1835. 10.1056/NEJMoa2034201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp-Thomas C.; Kalish H.; Drew M.; Hunsberger S.; Snead K.; Fay M. P.; Mehalko J.; Shunmugavel A.; Wall V.; Frank P.; Denson J. P.; Hong M.; Gulten G.; Messing S.; Hicks J.; Michael S.; Gillette W.; Hall M. D.; Memoli M. J.; Esposito D.; Sadtler K. Standardization of ELISA protocols for serosurveys of the SARS-CoV-2 pandemic using clinical and at-home blood sampling. Nat. Commun. 2021, 12, 113 10.1038/s41467-020-20383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehalko J.; Drew M.; Snead K.; Denson J. P.; Wall V.; Taylor T.; Sadtler K.; Messing S.; Gillette W.; Esposito D. Improved production of SARS-CoV-2 spike receptor-binding domain (RBD) for serology assays. Protein Expression Purif. 2021, 179, 105802 10.1016/j.pep.2020.105802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F.; Simon V. Serology assays to manage COVID-19. Science 2020, 368, 1060–1061. 10.1126/science.abc1227. [DOI] [PubMed] [Google Scholar]
- Stadlbauer D.; Amanat F.; Chromikova V.; Jiang K.; Strohmeier S.; Arunkumar G. A.; Tan J.; Bhavsar D.; Capuano C.; Kirkpatrick E.; Meade P.; Brito R. N.; Teo C.; McMahon M.; Simon V.; Krammer F. SARS-CoV-2 Seroconversion in Humans: A Detailed Protocol for a Serological Assay, Antigen Production, and Test Setup. Curr. Protoc. Microbiol. 2020, 57, e100 10.1002/cpmc.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman M.; Gilboa T.; Ogata A. F.; Maley A. M.; Cohen L.; Busch E. L.; Lazarovits R.; Mao C. P.; Cai Y.; Zhang J.; Feldman J. E.; Hauser B. M.; Caradonna T. M.; Chen B.; Schmidt A. G.; Alter G.; Charles R. C.; Ryan E. T.; Walt D. R. Ultrasensitive high-resolution profiling of early seroconversion in patients with COVID-19. Nat. Biomed. Eng. 2020, 4, 1180–1187. 10.1038/s41551-020-00611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A.; Supekar N. T.; Gleinich A. S.; Azadi P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. 10.1093/glycob/cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Allen J. D.; Wrapp D.; McLellan J. S.; Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Baudys J.; Bundy J. L.; Solano M.; Keppel T.; Barr J. R. Comprehensive Analysis of the Glycan Complement of SARS-CoV-2 Spike Proteins Using Signature Ions-Triggered Electron-Transfer/Higher-Energy Collisional Dissociation (EThcD) Mass Spectrometry. Anal. Chem. 2020, 92, 14730–14739. 10.1021/acs.analchem.0c03301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda M.; Morrison L.; Goldman R. N- and O-Glycosylation of the SARS-CoV-2 Spike Protein. Anal. Chem. 2021, 93, 2003–2009. 10.1021/acs.analchem.0c03173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo K. H.; Ranganathan S. Flanking signal and mature peptide residues influence signal peptide cleavage. BMC Bioinf. 2008, 9, S15 10.1186/1471-2105-9-S12-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martoglio B.; Dobberstein B. Signal sequences: more than just greasy peptides. Trends Cell Biol. 1998, 8, 410–415. 10.1016/S0962-8924(98)01360-9. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K. Y.; Wang Q.; Zhou H.; Yan J.; Qi J. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904 e9. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Zhu Y.; Liu M.; Lan Q.; Xu W.; Wu Y.; Ying T.; Liu S.; Shi Z.; Jiang S.; Lu L. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. M.; Kelleher N. L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. 10.1126/science.aat1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellers R. T.; Greer J. B.; Early B. P.; Yu X.; LeDuc R. D.; Kelleher N. L.; Thomas P. M. ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 2015, 15, 1235–1238. 10.1002/pmic.201400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro Armenteros J. J.; Tsirigos K. D.; Sonderby C. K.; Petersen T. N.; Winther O.; Brunak S.; von Heijne G.; Nielsen H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- Nyathi Y.; Wilkinson B. M.; Pool M. R. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Biophys. Acta, Mol. Cell Res. 2013, 1833, 2392–2402. 10.1016/j.bbamcr.2013.02.021. [DOI] [PubMed] [Google Scholar]
- Heijne G. V.Structural and Thermodynamic Aspects of the Transfer of Proteins into and across Membranes. In Current Topics in Membranes and Transport; Bronner F., Ed.; Academic Press, 1985;Vol. 24, pp 151–179. [Google Scholar]
- von Heijne G. The signal peptide. J. Membr. Biol. 1990, 115, 195–201. 10.1007/BF01868635. [DOI] [PubMed] [Google Scholar]
- Auclair S. M.; Bhanu M. K.; Kendall D. A. Signal peptidase I: cleaving the way to mature proteins. Protein Sci. 2012, 21, 13–25. 10.1002/pro.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D.; Mehalko J.; Drew M.; Snead K.; Wall V.; Taylor T.; Frank P.; Denson J. P.; Hong M.; Gulten G.; Sadtler K.; Messing S.; Gillette W. Optimizing high-yield production of SARS-CoV-2 soluble spike trimers for serology assays. Protein Expression Purif. 2020, 174, 105686 10.1016/j.pep.2020.105686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Y.; Song W. T.; Li Y.; Chen W. J.; Yang D.; Zhong G. C.; Zhou H. Z.; Ren C. Y.; Yu H. T.; Ling H. Improved expression of secretory and trimeric proteins in mammalian cells via the introduction of a new trimer motif and a mutant of the tPA signal sequence. Appl. Microbiol. Biotechnol. 2011, 91, 731–740. 10.1007/s00253-011-3297-0. [DOI] [PubMed] [Google Scholar]
- Chalkley R. J.; Brinkworth C. S.; Burlingame A. L. Side-chain fragmentation of alkylated cysteine residues in electron capture dissociation mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1271–1274. 10.1016/j.jasms.2006.05.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.