

Abstract
The COVID‐19 pandemic continues to be a severe threat to human health, especially due to current and emerging SARS‐CoV‐2 variants with potential to escape humoral immunity developed after vaccination or infection. The development of broadly neutralizing antibodies that engage evolutionarily conserved epitopes on coronavirus spike proteins represents a promising strategy to improve therapy and prophylaxis against SARS‐CoV‐2 and variants thereof. Herein, a facile multivalent engineering approach is employed to achieve large synergistic improvements in the neutralizing activity of a SARS‐CoV‐2 cross‐reactive nanobody (VHH‐72) initially generated against SARS‐CoV. This synergy is epitope specific and is not observed for a second high‐affinity nanobody against a non‐conserved epitope in the receptor‐binding domain. Importantly, a hexavalent VHH‐72 nanobody retains binding to spike proteins from multiple highly transmissible SARS‐CoV‐2 variants (B.1.1.7 and B.1.351) and potently neutralizes them. Multivalent VHH‐72 nanobodies also display drug‐like biophysical properties, including high stability, high solubility, and low levels of non‐specific binding. The unique neutralizing and biophysical properties of VHH‐72 multivalent nanobodies make them attractive as therapeutics against SARS‐CoV‐2 variants.
Keywords: antibody, antibody fragment, camelid, COVID‐19, polyvalency, polyvalent, protein engineering
This work reports a multivalent engineering strategy to synergistically increase the neutralization potency of nanobodies against SARS‐CoV‐2. An engineered hexavalent nanobody directed against an evolutionarily conserved epitope potently neutralizes SARS‐CoV‐2 and two key variants of concern (B.1.1.7 and B.1.351). This facile approach significantly improves the neutralization breadth and potency of promising nanobodies for potential use in therapeutic applications.
1. Introduction
The COVID‐19 pandemic has claimed millions of lives globally and ravaged human health for more than a year to date.[ 1 ] While SARS‐CoV‐2 vaccines and biologics are vital toward limiting mortality and reducing viral transmission, current clinically‐available options may be insufficient against emergent SARS‐CoV‐2 variants. These variants of concern, including B.1.1.7 (UK), B.1.351 (South African), and P.1 (Brazilian), have been verified in 114 countries including the US, and continue to propagate globally.[ 2 ] Moreover, SARS‐CoV‐2 variants have been linked to evasion of numerous vaccines and targeted therapeutics.[ 3, 4, 5, 6 ] For example, SARS‐CoV‐2 variants in the US have shown resistance to the monoclonal antibody Bamlanivimab (Eli Lilly), which was previously approved for emergency use authorization.[ 3 ] Overall, existing highly transmissible SARS‐CoV‐2 variants and the expected emergence of new ones represent an urgent global health threat for which improved prophylactics and therapeutics are sorely needed.
One promising therapeutic strategy toward addressing the rapidly evolving COVID‐19 pandemic is the development of broadly neutralizing antibodies that engage evolutionarily conserved epitopes on coronavirus spike proteins and retain high affinity and neutralizing activity against SARS‐CoV‐2 variants. Encouragingly, antibodies elicited from exposure to other coronavirus antigens (e.g., SARS‐CoV) have been identified that are cross‐reactive against SARS‐CoV‐2.[ 7, 8, 9, 10 ] However, such antibodies generally possess impaired affinity and neutralizing activity against SARS‐CoV‐2, requiring further optimization before these biomolecules can be considered for therapeutic applications. As a potential solution, multivalency has been employed to enhance the apparent binding affinity and potency of molecular therapeutics for applications against SARS‐CoV‐2[ 11, 12, 13 ] and other viruses,[ 14, 15 ] and is attractive for the optimization of antibodies without requiring in vitro affinity maturation.
Nanobodies, single‐domain fragments of camelid heavy‐chain antibodies, have been developed to target a wide variety of viruses,[ 14, 16, 17, 18, 19 ] frequently with the goal of using them as therapeutic agents. Reformatting nanobodies into multivalent constructs has been proposed to have a number of advantages for such antiviral purposes, including the potential to prevent conformational changes required for the virus to infect host cells.[ 15, 20, 21 ] Recently, there has been a rapid development of many nanobodies targeting SARS‐CoV‐2,[ 8, 12, 13, 22, 23, 24, 25 ] and several have also been tested in multivalent constructs.[ 12, 13, 26 ] However, many previously reported multivalent nanobodies have not been examined for their neutralization activities against SARS‐CoV‐2 variants,[ 8, 12, 13, 22, 23, 24, 25 ] and it is unclear whether they are broadly neutralizing or whether their epitopes are conserved between coronaviruses. Moreover, most previous studies of the impact of multivalency on SARS‐CoV‐2 nanobody affinity and neutralization activity have focused on low valencies (e.g., bivalent and trivalent).[ 8, 12, 13 ] It is likely that higher valencies (e.g., tetravalent and hexavalent) will lead to significant improvements in neutralization activity for nanobodies with modest intrinsic affinity for neutralizing epitopes.
In this work, we evaluated a facile multivalent engineering approach toward enhancing the neutralizing activity of a cross‐reactive nanobody (VHH‐72) previously generated against SARS‐CoV (Figure 1).[ 8 ] We posited that if the modest affinity of VHH‐72 could be significantly improved, this cross‐reactive nanobody may be particularly valuable for neutralizing SARS‐CoV‐2 and its emergent variants. Here we report the engineering and characterization of multivalent nanobodies (as Fc fusion proteins) that display potent and broadly neutralizing activity for wild‐type SARS‐CoV‐2 and the UK (B.1.1.7) and South African (B.1.351) variants, and we show how this performance is superior to that for multivalent nanobodies targeted against non‐conserved epitopes in the receptor‐binding domain (RBD).
Figure 1.
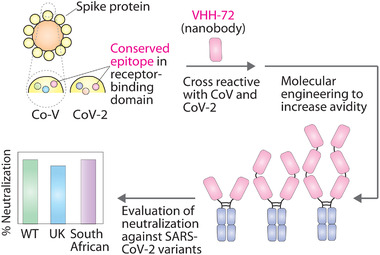
Overview of proposed approach for engineering potent, broadly neutralizing multivalent nanobodies. A nanobody (VHH‐72) targeting an epitope that is conserved between SARS‐CoV and SARS‐CoV‐2 was engineered to increase its neutralization activity toward the SARS‐CoV‐2 virus. The valency of VHH‐72 was systematically increased by incorporating it into bivalent, tetravalent, and hexavalent Fc‐fusion constructs, and their ability to neutralize highly transmissible SARS‐CoV‐2 variants was analyzed.
2. Results
2.1. Multivalent Reformatting Leads to Large Increases in VHH‐72 Neutralization Activity
To evaluate the potential of multivalency to increase the neutralization potency of VHH‐72, we first cloned bivalent, tetravalent and hexavalent constructs as Fc fusion proteins. We also prepared these same constructs for a second higher‐affinity nanobody (KC3.ep3) that recognizes a different epitope than VHH‐72 in the SARS‐CoV‐2 RBD for comparison.[ 27 ] The tetravalent and hexavalent constructs were prepared by connecting the nanobody domains with 15‐residue linkers [(G4S)3]. We found that all the constructs could be readily expressed in HEK293‐6E cells via transient transfection with purification yields of 32–55 mg L−1 for the VHH‐72 constructs and 4–42 mg L−1 for the KC3.ep3 constructs. SDS‐PAGE analysis revealed that the bivalent (≈86–87 kDa), tetravalent (≈113–116 kDa), and hexavalent (≈140–145 kDa) nanobodies demonstrated high purities and expected sizes (Figure 2).
Figure 2.
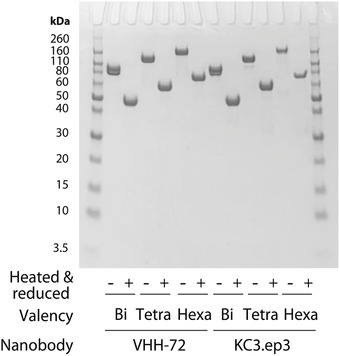
SDS‐PAGE analysis of the multivalent nanobody‐Fc fusion proteins evaluated in this work. Bivalent (Bi), tetravalent (Tetra), and hexavalent (Hexa) nanobodies (produced as Fc fusion proteins) for two nanobodies (VHH‐72 and KC3.ep3) were evaluated via SDS‐PAGE for protein samples either without reduction and heating (−) or with reduction and heating (+).
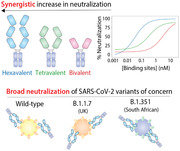
We next evaluated the impact of valency on the ability of the VHH‐72 multivalent constructs to neutralize the wild‐type SARS‐CoV‐2 pseudovirus (Figure 3A). To identify potential synergistic impacts of valency on neutralization activity, we evaluated the neutralization activity as a function of the concentration of the nanobody binding domains instead of the multivalent antibody concentration. For example, this results in 1 nm hexavalent antibody being reported at 6 nm based on the concentration of binding domains. As expected, the bivalent VHH‐72 fusion protein displays modest neutralization activity (IC50 of 3.3±1.8 nm; Figure 3A), which is >170‐fold weaker than observed for the bivalent version of the higher‐affinity nanobody (KC3.ep3, IC50 of 0.019 ± 0.003 nm; Figure 3B). However, increasing the valency of VHH‐72 results in large synergistic increases in neutralization potency, as the tetravalent nanobody construct displays an order‐of‐magnitude improvement (IC50 of 0.34 ± 0.072 nm) and the hexavalent nanobody construct is improved by two orders of magnitude (IC50 of 0.035 ± 0.003 nm; Figure 3A). We observed that the neutralization activity of VHH‐72 could be improved beyond that of a previously reported neutralizing antibody,[ 28 ] S309 (IC50 of 1.9 ± 0.9 nm). Interestingly, the higher affinity nanobody (KC3.ep3) did not display synergistic improvements in neutralization activity (Figure 3B).
Figure 3.
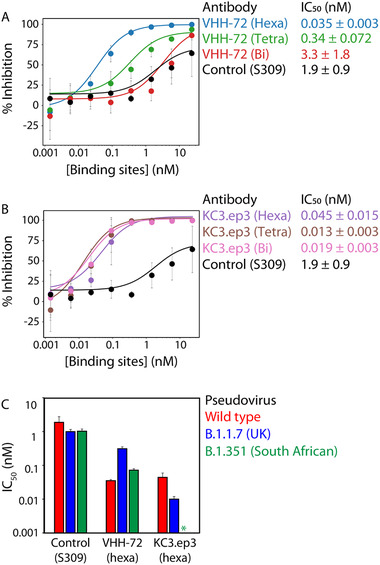
Multivalency increases the broadly neutralizing activity of VHH‐72 in a synergistic manner. Neutralization of wild‐type SARS‐CoV‐2 pseudovirus by multivalent A) VHH‐72 and B) KC3.ep3 nanobodies (as Fc fusion proteins) relative to a control mAb (S309). Nanobody concentrations (x‐axis) are reported on the basis of the number of nanobody domains, not the multivalent antibody concentration. C) The neutralization activity of hexavalent VHH‐72 and KC3.ep3 against B.1.1.7 (UK) and B.1.351 (South African) SARS‐CoV‐2 pseudovirus variants relative a control mAb (S309). In (A) and (B), the data are averages of two or three independent repeats, and the error bars are standard deviations. In (C), the data are averages of four independent repeats, and the error bars are standard deviations.
Given the increasing prevalence of SARS‐CoV‐2 variants containing one or more RBD mutations, we next tested the ability of the hexavalent nanobodies to neutralize pseudoviruses of the B.1.1.7 (UK) and B.1.351 (South African) variants (Figure 3C). Given that VHH‐72 binds to an evolutionarily conserved epitope in the RBD of SARS‐CoV‐2 distinct from the epitope recognized by KC3.ep3, we suspected VHH‐72 may be more broadly neutralizing than KC3.ep3. Indeed, we find hexavalent VHH‐72 potently neutralizes the SARS‐CoV‐2 pseudoviruses of the B.1.1.7 and B.1.351 variants in addition to wild‐type (Figure 3C). Hexavalent VHH‐72 potently neutralized both the B.1.1.7 (IC50 of 0.31 ± 0.044 nm) and B.1.351 (IC50 of 0.072 ± 0.0075 nm) pseudoviruses. In contrast, hexavalent KC3.ep3 retains similar neutralization activity toward the B.1.1.7 pseudovirus (IC50 of 0.010 ± 0.0020 nm) as the wild‐type, but it is unable to neutralize the B.1.351 pseudovirus. For comparison, the neutralization activity of the S309 (control) toward the pseudovirus variants was also examined. S309 neutralized both the B.1.1.7 (IC50 of 0.99 ± 0.17 nm) and the B.1.351 (IC50 of 1.0 ± 0.16 nm) pseudoviruses similarly.
2.2. VHH‐72 Recognizes the RBD in a Manner Distinct from Other Common SARS‐CoV and SARS‐CoV‐2 Antibodies
To examine the unique neutralizing properties of multivalent versions of VHH‐72 and KC3.ep3, we next characterized their binding affinities and epitopes. First, we examined the monovalent affinity of each nanobody for the S1 protein from wild‐type SARS‐CoV‐2 and two variants [UK (B.1.1.7) and South African (B.1.351); Figure 4]. VHH‐72 displayed similar binding affinity for wild‐type, UK, and South African S1 proteins (K D of 29–60 nm), although its affinity for the South African variant was reduced twofold. However, KC3.ep3 maintained high affinity for wild‐type and UK S1 proteins (K D of 3–4 nm) but lost binding to the South African S1 protein. This finding is consistent with the inability of hexavalent KC3.ep3 to neutralize the pseudovirus of the South African variant (Figure 3C). We also initially sought to examine the apparent binding affinity of multivalent VHH‐72 and KC3.ep3 constructs to immobilized SARS‐CoV‐2 RBD in an ELISA‐like format. However, we observed similar binding profiles for i) multivalent constructs of the same nanobody with different valencies (e.g., bivalent vs. hexavalent VHH‐72) and ii) multivalent constructs for different nanobodies with the same valency (e.g., bivalent VHH‐72 vs. bivalent KC3.ep3), which was likely due to strong avidity effects. We have previously reported that it is more difficult to observe differences in apparent affinity for multivalent nanobodies (e.g., two different bivalent nanobodies) than for their monovalent counterparts.[ 27 ]
Figure 4.
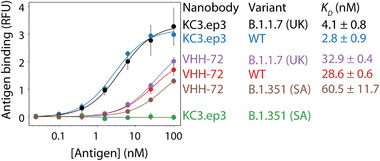
VHH‐72 broadly recognizes the S1 proteins from highly transmissible SARS‐CoV‐2 variants. The monovalent affinities of VHH‐72 and KC3.ep3 were evaluated against the wild‐type, B.1.1.7 (UK) and B.1.351 (South African, SA) SARS‐CoV‐2 S1 proteins for nanobodies displayed on the surface of yeast. The data are averages of two independent repeats, and the error bars are standard deviations.
We also evaluated the epitopes and binding mechanisms of the nanobodies via competition analysis using ACE2 and antibodies that recognize distinct classes of RBD epitopes (Figure 5).[ 29 ] For this analysis, soluble biotinylated RBD (5 nm) was first incubated with soluble ACE2, mAbs, or nanobody Fc‐fusion proteins at a range of concentrations (0.05–500 nm). Next, the mixture were incubated with yeast expressing monovalent VHH‐72 on their surface. Finally, the ability of biotinylated RBD to bind to monovalent VHH‐72 in the presence of various concentrations of competing protein/antibody was detected via flow cytometry.
Figure 5.
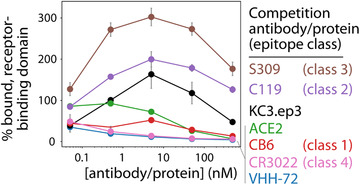
VHH‐72 competes with ACE2 and multiple classes of SARS‐CoV‐2 antibodies for binding to the receptor‐binding domain. The epitope of VHH‐72 was analyzed via competition analysis using SARS‐CoV‐2 RBD. First, ACE2 receptor and distinct classes of antibodies (0.05–500 nm) were incubated with biotinylated RBD (5 nm). Next, the mixtures were incubated with yeast displaying monovalent VHH‐72, and the relative % RBD binding to VHH‐72 was evaluated via flow cytometry. The data are averages of two independent repeats, and the error bars are standard deviations.
As expected, VHH‐72 competes most strongly with itself for RBD binding (Figure 5). We also find that ACE2 competes with VHH‐72 for RBD binding, which is consistent with the neutralization activity of VHH‐72 (Figure 3). VHH‐72 also competes with the class 4 antibody CR3022 (Figure 5), which is consistent with previous findings.[ 8 ] Moreover, we find that VHH‐72 competes with a class 1 antibody (CB6).[ 30 ] This is surprising because VHH‐72 does not compete with KC3.ep3, and we have previously found that KC3.ep3 competes with CB6.[ 27 ] Finally, VHH‐72 does not compete with either class 2 (C199) or 3 (S309) antibodies.[ 28, 29 ] In fact, C119 and especially S309 enhanced VHH‐72 binding to RBD, suggesting that these mAbs may bind in a manner that increases the exposure, or stabilizes the structure, of the VHH‐72 epitope (Figure 5).
2.3. Multivalent VHH‐72 Fusion Proteins Display Drug‐Like Biophysical Properties
For use of affinity proteins in diverse biomedical and therapeutic applications, it is important that they possess favorable biophysical properties, including high stability, high solubility, and low off‐target binding. Therefore, we evaluated the folding stability, percent monomer, and non‐specific binding of the multivalent nanobodies analyzed in this study (Figure 6). The melting temperatures of the VHH‐72 nanobodies were high (68–69 °C) and similar for bivalent, tetravalent, and hexavalent constructs, suggesting that multivalent engineering did not destabilize them (Figure 6A). However, the KC3.ep3 multivalent constructs displayed significant destabilization for the tetravalent and hexavalent nanobodies. We also observed that the VHH‐72 multivalent nanobodies displayed high percentages of monomeric protein (>95%) after a single Protein A purification step (Figure 6B), suggesting that these proteins are particularly soluble and well behaved. We observed lower percentages of monomeric protein for the KC3.ep3 multivalent antibodies (90–95%). Finally, all of the multivalent antibodies displayed low levels of non‐specific binding to soluble membrane proteins relative to a positive control antibody (emibetuzumab) with high levels of non‐specific binding (Figure 6C). Given that the polyspecificity reagent (PSR; soluble membrane proteins) has been previously validated to be a strong indicator of antibody polyreactivity,[ 31, 32, 33 ] these findings demonstrate that the multivalent nanobodies possess extremely low levels of off‐target binding. These results collectively demonstrate that the broadly neutralizing VHH‐72 nanobodies display favorable biophysical properties and are expected to be robust agents for diverse biomedical and therapeutic applications.
Figure 6.
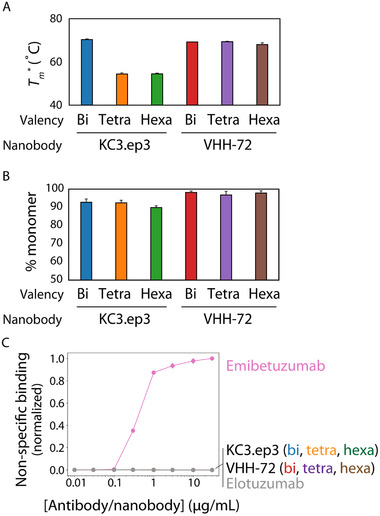
Multivalent nanobodies demonstrate drug‐like biophysical properties. A) Apparent midpoint temperatures of the first unfolding transition (T m *) of nanobody‐Fc fusion proteins. B) % monomer of nanobody‐Fc fusion proteins evaluated using size‐exclusion chromatography. C) Non‐specific binding of nanobody‐Fc fusion proteins. The nanobody‐Fc fusion proteins were immobilized on Protein A‐coated magnetic beads, incubated with biotinylated soluble membrane proteins, and non‐specific interactions were evaluated using flow cytometry. Control mAbs with high (emibetuzumab) and low (elotuzumab) levels of non‐specific interactions were also analyzed for reference. The data are averages of four (A), three (B), and two (C) independent repeats, and the error bars are standard deviations.
3. Discussion
We have demonstrated the use of multivalent nanobody engineering for potently neutralizing SARS‐CoV‐2. Our work builds on previous reports of using multivalency to increase the neutralization activity of nanobodies in vitro[ 12, 14 ] and reduce viral symptoms in vivo.[ 19 ] Interestingly, we find that the effects of increased valency are distinct and dependent upon the individual nanobody. We demonstrate that the neutralization activity of a moderate affinity, cross‐reactive nanobody (VHH‐72) increases substantially when formatted as tetravalent and hexavalent constructs compared to as a bivalent construct. However, we did not observe similar increases in neutralization activity for a higher affinity nanobody that binds to a SARS‐CoV‐2 specific epitope (KC3.ep3), indicating that the increased activity obtained via multivalency is dependent on the epitope‐specific properties and intrinsic affinity of the nanobody. Importantly, we have examined neutralization activity as a function of the concentration of binding domains rather than the concentration of the overall complex. The highest examined concentration is ≈24 nm in terms binding domains. This corresponds to a concentration of ≈12 nm bivalent nanobody, ≈6 nm tetravalent nanobody, and ≈4 nm hexavalent nanobody. Thus, the increase in neutralization activity observed for the higher valency constructs of VHH‐72 results from a synergistic improvement in activity of these constructs rather than simply an increase in the number of binding domains.
An examination of the monovalent affinities of VHH‐72 and KC3.ep3 for the S1 proteins from different SARS‐CoV‐2 variants provides significant insight into the expected neutralization behavior. Monovalent VHH‐72 retains binding to both the B.1.1.7 and B.1.351 variants (Figure 4), and multivalent constructs of this nanobody neutralize both variants (Figure 3C). KC3.ep3 retains binding to the B.1.1.7 variant but loses observable binding to the B.1.351 variant (Figure 4) and, as expected, multivalent constructs of KC3.ep3 show neutralization activity toward the B.1.1.7 variant but not the B.1.351 variant (Figure 3C). Nevertheless, the differences in the monovalent affinities of the VHH‐72 and KC3.ep3 nanobodies for the different variants (Figure 4) are not fully predictive of their relative neutralization activities (Figure 3C), which may be due to the effects of the multivalent molecular architecture on the apparent nanobody affinity (avidity) and neutralization activity.
We used competition analysis to evaluate the epitope of the VHH‐72, which deserves further consideration. This assay determines whether an antibody's epitope overlaps with the ACE2 binding site and the epitopes of antibodies that compete with ACE2. It also examines competition with antibodies that bind to the RBD either only in the “up” conformation or in both the “up” and “down” conformations. Competition with ACE2 indicates an antibody's ability to prevent binding to this receptor and infection of host cells. This can also be tested directly by examining binding of an antibody to RBD that has been preincubated with ACE2. Binding of an antibody to the RBD in the “up” or “down” conformation of the RBD can also provide an indication of the antibody's epitope. The “down” conformation of the RBD represents the state in which RBD, in the context of the spike trimer, is inaccessible for binding to ACE2, while the “up” conformation represents a state that is thought to be less stable in which the RBD can bind to ACE2.[ 34 ] However, the use of the trimeric spike protein in such competition assays is complicated because the RBD can be present in either the “up” or “down” conformation, and further, it has been shown that the conformation of the three individual RBDs can differ within a single trimer.[ 34, 35 ] Instead, competition for RBD binding with different classes of antibodies validated for binding to each conformation, as done in this work, provides a facile method for determining the RBD state in which a nanobody or antibody can bind.
Our measurements detect antibodies that compete with VHH‐72 for binding to the RBD, either due to binding to an overlapping epitope or through steric hindrance. As expected, we observed that VHH‐72 strongly competes with CR3022 (Figure 5) given that they have been reported to possess overlapping epitopes.[ 8 ] It is notable that CR3022, like VHH‐72, was initially identified against SARS‐CoV.[ 36 ] CR3022 binds to a class 4 epitope, indicating that it does not compete for the same epitope as ACE2 and binds only when the RBD is in the “up” conformation.[ 29 ] A crystal structure of VHH‐72 in complex with the SARS‐CoV RBD has previously been reported (PBD: 6WAQ).[ 8 ] Despite binding to an epitope that overlaps with that of CR3022, the angle at which VHH‐72 binds to the RBD differs from that of CR3022, and in this way, VHH‐72 neutralizes SARS‐CoV‐2 through steric competition with the ACE2 receptor.[ 8 ] In agreement with this report, we find that VHH‐72 competes with soluble ACE2 (Figure 5).
Interestingly, we also observed competition between VHH‐72 and a class 1 antibody (CB6; Figure 5). This is notable because class 1 antibodies, including CB6, bind to the RBD only in the “up” conformation. However, the epitopes of CB6 and CR3022 do not overlap[ 30 ] despite the fact that VHH‐72 is competitive with both of them. The observed competition between VHH‐72 and CB6 likely results from steric competition between the two antibodies. Further, we have previously shown that KC3.ep3 also strongly competes for RBD binding with CB6.[ 27 ] However, we did not observe competition between VHH‐72 and KC3.ep3, demonstrating the limitations of strictly defining antibody epitopes using competition studies with class‐specific antibodies.
Conversely, competition was not observed between VHH‐72 and either class 2 (C119) and 3 (S309) antibodies. Class 2 antibodies, such as C119, are defined as those that compete for the ACE2 binding site and bind the RBD in both the “up” and “down” conformations. Class 3 antibodies do not compete with the ACE2 binding site but also bind to the RBD in both the “up” and “down” conformations. Our results indicate that VHH‐72 competes strongly with antibodies that bind the RBD only in the “up” conformation and binds stronger in the presence of antibodies that can bind the RBD in both the “up” and “down” conformations. It has been previously observed that S309 can enhance neutralization activity when combined in cocktails with other antibodies.[ 28 ] Our results indicate that combining VHH‐72 with either class 2 or 3 antibodies can lead to enhanced binding, which has the potential to also increase neutralization activity.
Finally, we have demonstrated that the VHH‐72 multivalent nanobodies have highly favorable, drug‐like biophysical properties. This is particularly interesting in the case of hexavalent VHH‐72 nanobodies that have the most broadly neutralizing activity. It is surprising that this large and complex multivalent nanobody displays such high percentage monomer after Protein A purification. Moreover, it is notable that it is not destabilized in the manner observed for KC3.ep3 due to multivalent engineering. Based on the protein sequences, the theoretical isoelectric points (pIs) of the multivalent nanobody‐Fc constructs for VHH‐72 differ from those for KC3.ep3. As isolated nanobodies and Fc fragments, the theoretical pIs are 4.96 (VHH‐72), 6.51 (KC3.ep3), and ≈6.3–6.5 (Fc with hinge and tags). The VHH‐72 multivalent constructs have pIs of 6.25 (bivalent), 6.07 (tetravalent), and 5.93 (hexavalent). However, the theoretical pIs of KC3.ep3 multivalent constructs are 6.37 (bivalent), 6.5 (tetravalent), and 6.51 (hexavalent). Thus, the greatest differences in solubility between these two nanobodies would be expected for the hexavalent nanobodies. It is also encouraging that the multivalent nanobodies show little non‐specific binding, even at high valencies, which in our experience is a relatively common problem when using multivalent antibodies.
Nanobodies, and antibodies more generally, are currently of great interest for use as therapeutic agents for treatment of SARS‐CoV‐2, and several antibodies and antibody cocktails have been approved for this purpose.[ 37, 38, 39 ] We have demonstrated that engineering multivalent nanobody constructs has the potential to rapidly improve the properties of promising nanobodies to prepare them for use in therapeutic applications. However, there are a few potential barriers to the use of nanobodies as anti‐viral therapeutics, including their smaller size in comparison to mAb drugs and non‐human origin. One potential limitation of nanobodies for therapeutic applications is their short half‐lives due to their small sizes.[ 40, 41, 42 ] Increased valency could contribute to an extension of half‐life due to the larger size of the multivalent nanobodies. Multivalent constructs have previously been explored for extending the half‐life of nanobodies.[ 42, 43 ] Further, the presence of Fc domains is expected to extend the half‐lives of these multivalent constructs[ 44, 45 ] and aid in immune responses.[ 46 ]
Another possible limitation of using nanobodies as therapeutics applications is their potential to produce anti‐drug immune responses. Both the nanobodies examined in this study, VHH‐72 and KC3.ep3, have camelid frameworks, and administration of non‐human antibodies as therapeutics has the potential to elicit immune responses against these molecules. However, camelid antibodies have been reported to share greater sequence similarity with human antibodies than with antibodies from other commonly immunized species, such as mice.[ 47 ] While immunogenicity of nanobodies is difficult to predict and direct analysis is beyond the scope of this study, nanobodies have been successfully humanized and administered as therapeutics. The first therapeutic nanobody, caplacizumab, was approved by the FDA in 2019,[ 48 ] demonstrating the potential of nanobodies in therapeutic applications. Overall, the combination of potent and broad neutralization activities, favorable biophysical properties, and expected long half‐lives of multivalent VHH‐72 Fc fusion proteins makes them attractive candidates for therapeutic applications.
4. Conclusions
The SARS‐CoV‐2 pandemic has presented widespread disruption to human life, and despite recent advances in therapeutic development, the virus as well as newly emerging variants continue to pose a threat to global health. The development of potent and broadly neutralizing agents is urgently needed. Our multivalent VHH‐72 nanobodies demonstrate synergistic improvements in neutralization activity without the need for affinity maturation. This approach, particularly for nanobodies specific for conserved epitopes across different coronaviruses, is expected to be broadly useful for combating viral variants that emerge during the current and future pandemics.
5. Experimental Section
Nanobody Expression and Purification
Multivalent nanobodies were cloned into human IgG1 Fc‐fusion vectors. Tetravalent and hexavalent constructs contain three repeats of a G4S linker connecting each nanobody domain. Nanobodies were expressed in HEK293‐6E cells (National Research Council of Canada) as previously described.[ 49, 50 ] Briefly, cells were cultured at 37 °C with 5% CO2 and agitation at 250 rpm in 50 mL mini bioreactors (Fisher Scientific, NC0664085). Cells were transfected with 15 µg plasmid and 45 µg polyethylenimine after reaching a density of 1.5–2 million cells mL−1. Cells were fed with 20% w/v Yeastolate (BD Sciences, 292804) 24–48 h after transfection. Six days after transfection, cells were centrifuged at 3500–4000 ×g for 40 min. Following centrifugation, the supernatant was transferred to a new tube and incubated with Protein A agarose beads (Thermo Fisher Scientific, 20333) overnight at 4 °C with mild agitation. Protein A beads were collected in vacuum filter columns (Fisher, PI89898) and washed with 1×PBS. Nanobodies were eluted from Protein A beads in 0.1 m glycine at pH 3 and immediately buffer exchanged into 1×PBS using Zeba desalting columns (Fisher, PI89892). Nanobodies were sterile filtered through a 0.22 µm filter (Millex, SLGV004SL), aliquoted, and frozen at −80 °C until use. Nanobody concentration was determined measuring absorbance at 280 nm, and purity was examined by SDS‐PAGE (Invitrogen, WG1203BOX).
Construction and Preparation of SARS‐CoV‐2 Pseudovirus Variants
Plasmids encoding the expression of mutant spike proteins B.1.1.7 (UK) and B.1.351 (South African) were generated via modification of the following plasmid: pCMV3 SARS‐CoV2 S Untagged Delta 19AA C‐term plasmid encoding the SARS‐CoV‐2 spike (S) protein with a 19‐amino acid deletion at the C‐terminus. First, to generate inserts, gBlocks Gene Fragments (Integrated DNA Technologies) encoding the UK variant of the SARS‐CoV‐2 spike protein (HV69‐70del, Y144del, N501Y, A570D, D614G, P681H, T716I, S982A, and D1118H) and the South African variant of the SARS‐CoV‐2 spike protein (L18F, D80A, D215G, 242‐244del, R246I, K417N, E484K, N501Y, D614G, and A701V) with a 19‐amino acid deletion at the C‐terminus were amplified via PCR using Phusion polymerase (Thermo Fisher, F‐530) following the manufacturer's protocol. To prepare digested vector, the gene encoding wild‐type spike protein with a 19‐amino acid deletion at the C‐terminus was excised from pCMV3 SARS‐CoV2 S Untagged Delta 19AA C‐term plasmid via restriction digestion with Kpn1 and XbaI (New England Biolabs), followed by treatment with calf intestinal alkaline phosphatase (New England Biolabs, M0525). Inserts and vectors were assembled via Hifi DNA assembly kit (New England Biolabs, E2621) following the manufacturer's protocol, and sequences were confirmed via Sanger sequencing.
Pseudovirus Neutralization Assay
Pseudovirus preparations and neutralization assays were informed by a previous report.[ 51 ] For pseudovirus preparation, Lenti‐X 293T cells (Takara, 632180) were first seeded at 5 × 105 cells per well in 6‐well plates in RPMI media with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) and cultured at 37 °C with 5% CO2. Upon reaching target density (50–70% confluent), cells were transfected with third generation lentivirus plasmids: HDM‐Hgpm2 plasmid (BEI number NR‐52517) encoding HIV Gag‐Pol under CMV promoter, HDM‐tat1b plasmid (BEI, NR‐52518) encoding HIV Tat under CMV promoter, pRC‐CMV‐Rev1b plasmid (BEI number NR‐52519) encoding HIV Rev, pHAGE‐CMV‐Luc2‐IRES‐ZsGreen‐W (BEI number NR‐52516) lentiviral transfer plasmid encoding co‐expression of luciferase and ZsGreen, and pCMV3 SARS‐CoV2 S Untagged Delta 19AA C‐term plasmid encoding the SARS‐CoV‐2 spike (S) protein with a 19‐amino acid deletion at the C‐terminus. For the transfection, lipofectamine 2000 was used following the manufacturer's protocol along with the respective plasmid masses (mg) per well: 0.22, 0.22, 0.22, 1.00, and 0.34. Then, 24 h post‐transfection, cell media was removed, discarded, and switched to fresh RPMI with 10% FBS and 1% P/S. Next, 72 h post‐transfection, cell supernatant was collected and processed through a 0.45 µm polyethersulfone membrane filter. Cell supernatant was then incubated overnight at 4 °C overnight with Lenti‐X Concentrator (Takara, 631232) solution following the manufacturer's protocol. The next day, the mixture was centrifuged at 1500 ×g for 45 min, supernatant was removed, and the virus particles were resuspended in Opti‐MEM (100 µL per well of virus harvest), aliquoted, and frozen at −80 °C.
For pseudovirus neutralization assays, 293T‐ACE2 cells (BEI, NR‐52511) were seeded at 10 000 cells per well in white bottom 96‐well plates (Corning, 3917) in DMEM (10% FBS and 1% P/S) and cultured at 37 °C and 5% CO2. Then, 24 h post‐seeding, fourfold serial dilutions of various treatments were prepared. Briefly, antibody, nanobody, or controls were combined with an equal volume of pseudovirus at 350 tissue culture infectious units (as determined by flow cytometry[ 27 ]) per well and incubated for 1 h at 37 °C. Following the incubation, mixtures containing pseudovirus and treatment or control were added to 293T‐ACE2 cells along with polybrene (final concentration of 5 µg mL−1). Then, 48 h post‐infection, the 96‐well plates were equilibrated to room temperature for 10 min, and 100 µL of media was removed from each well. Then, luciferase substrate (Promega ONE‐Glo, E6110) was prepared following the manufacturer's protocol, and 100 µL was added to each well. The plates were incubated at room temperature for 10 min, and bioluminescence was measured using a Molecular Devices SpectraMax microplate reader (500 ms integration per well).
Nanobody Specificity Analysis for SARS‐CoV‐2 Variants
Yeast cells (5 × 104 per sample) expressing nanobodies (VHH‐72 or KC3.ep3) were washed twice with PBSB (PBS + 1 g L−1 BSA) and incubated with wild‐type (Acro, S1N‐C82E8), UK (Acro, S1N‐C52Hr) or South African (Acro, S1N‐C52Hm) variants of S1 protein in 1% milk at room temperature for 3 h with mild agitation. Post antigen incubation, the cells were washed once by centrifuging and re‐suspending in ice‐cold PBSB and then incubated with mouse anti‐Myc antibody (Cell Signaling, #2276S) at 1:1000 dilution and chicken anti‐His antibody (Invitrogen, PA1‐9531) at 1:1000 dilution on ice for 20 min. Following primary antibody incubation, the cells were washed once with ice‐cold PBSB and then incubated with goat anti‐mouse AlexaFluor 488 (Invitrogen, A11001) and donkey anti‐chicken AlexaFluor 647 (Jackson Immunoresearch, 703‐606‐155) on ice for 4 min. After the secondary incubation, the cells were washed once with ice‐cold PBSB and analyzed by flow cytometry.
Nanobody Epitope Analysis
Soluble (5 nm) biotinylated SARS‐CoV‐2 RBD (ACRO Biosystems, SPD‐C82E9) was incubated for 2 h at room temperature with mild agitation with varying concentrations (500, 50, 5, 0.5, and 0.05 nm) of soluble ACE2 (RayBiotech, 230‐30165), nanobody‐Fc fusion protein, or mAb. After 2 h, 1 × 105 yeast cells displaying VHH‐72 were added to each sample. Cells were incubated with biotinylated RBD and ACE2 receptor or blocking antibody in 1% milk with 1:1000 dilution mouse anti‐Myc antibody (Cell Signaling, #2276) for 3 h at room temperature with mild agitation. Following incubation, cells were washed once with ice‐cold PBSB. Cells were incubated with 1:1000 dilution of streptavidin AlexaFluor 647 (Invitrogen, S21374) and 1:200 dilution of goat anti‐mouse AlexaFluor 488 (Invitrogen, A11001) on ice for 4 min. Cells were washed once with ice‐cold PBSB. Cells were resuspended in ice‐cold PBSB and analyzed by flow cytometry. The RBD binding signal for cells displaying VHH‐72 was quantified and plotted as a function of receptor or blocking antibody concentration.
Nanobody Polyspecificity Analysis
PSR was prepared as previously reported.[ 31 ] Briefly, CHO cells (109, Gibco, A29133) were centrifuged and washed sequentially with PBSB and Buffer B (50 mm HEPES, 0.15 m NaCl, 2 mm CaCl2, 5 mm KCl, 5 mm MgCl2, 10% Glycerol, pH 7.2) through centrifugation and resuspension. Following the washes, cell pellets were resuspended in Buffer B (5 mL) supplemented with protease inhibitor (Sigma Aldrich, 4693159001). Resuspended cells were homogenized with a Dounce Tissue Grinder for 90 s (three cycles of 30 s). The cell suspension was then sonicated for 90 s (three cycles of 30 s). The prepared cell suspension was then centrifuged at 40 000 ×g for 1 h. Following centrifugation, the supernatant was discarded.
The pellet (enriched cell membrane fraction) was resuspended in Buffer B with a Dounce homogenizer. Concentration of protein in the resuspended solution was then determined using an assay kit (BioRad, 5000116) compatible with the presence of detergents. The enriched membrane fraction was diluted to a concentration of 1 mg mL−1 in solubilization buffer (pH 7.2) containing 50 mm HEPES, 0.15 m NaCl, 2 mm CaCl2, 5 mm KCl, 5 mm MgCl2, 1% n‐dodecyl‐b‐D‐maltopyranoside (Sigma Aldrich, D4641), and protease inhibitor (Sigma Aldrich, 11873580001). The solution was then rotated end‐over‐end overnight at 4 °C. The soluble membrane protein fraction was then centrifuged at 40 000 ×g for 1 h and the supernatant was collected. The final concentration of supernatant was measured again and diluted to a final concentration of 1 mg mL−1.
Sulfo‐NHS‐LC‐biotin (Thermo Fisher Scientific, PI21335) was dissolved at a concentration of ≈11.5 mg mL−1 in distilled water. Stock solutions of Sulfo‐NHS‐LC‐biotin (150 mL) and PSR reagent (4.5 mL at 0.8–0.9 mg mL−1) were end‐over‐end mixed at room temperature for 45 min. The reaction was quenched with hydroxylamine (10 mL, 1.5 m at pH 7.2). Biotinylated PSR was then aliquoted and stored frozen at −80 °C until use.
Protein A‐coated magnetic beads (Invitrogen, 88846) were washed three times with PBSB and incubated with antibodies at various concentrations in 96‐well plates (VWR, 650261) overnight at 4 °C. Next, the 96‐well plates were centrifuged at 2500 ×g for 4 min, and the beads were resuspended in PBSB to wash. Washing procedure was repeated twice. The beads were resuspended in a 10× dilution of biotinylated PSR, and the 96‐well plate was incubated on ice for 20 min. Beads were washed once with ice‐cold PBSB. Beads were then labeled with secondary antibodies through an incubation with 1:1000 dilution of streptavidin AlexaFluor 647 (Invitrogen, S32357) and 1:1000 dilution of goat anti‐human Fc F(ab’)2 AlexaFluor 488 (Invitrogen, H10120). For secondary incubation, the 96‐well plate was placed on ice for 4 min. Beads were then washed with ice‐cold PBSB once, resuspended in ice‐cold PBSB, and analyzed via flow cytometry. Two control antibodies, namely elotuzumab (low non‐specific binding control) and emibetuzumab (high non‐specific binding control), were analyzed to normalize results from all replicates to a range between 0 and 1. These controls were composed of the variable regions of elotuzumab and emibetuzumab grafted onto a common IgG1 framework. The control antibodies were two‐step purified [Protein A followed by size‐exclusion chromatography (SEC)] prior to analysis of polyspecificity.
Analytical Size‐Exclusion Chromatography
Multivalent nanobody Fc‐fusion purity after Protein A purification was evaluated using SEC with a Shimadzu Prominence HPLC System comprised of a LC‐20AT pump, SIL‐20AC autosampler, and FRC10A fraction collector. Multivalent nanobodies in PBS (pH 7.4) were diluted to 0.1 mg mL−1 for analytical SEC. 100 µL of protein sample was then injected into the column (Superdex 200 Increase 10/300 GL column; GE, 28990944). Samples were analyzed at 0.75 mL min−1 using a PBS running buffer supplemented with 200 mm arginine (pH 7.4). Absorbance at 280 nm was monitored. The percentage of protein monomer was evaluated by analyzing the area under the peak between the exclusion and solvent elution times (8 to 22 min).
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
J.M.Z., J.S.S., and A.A.D. contributed equally to this work. J.M.Z., A.A.D., J.S.S., and P.M.T. designed the research, J.M.Z. cloned and produced multivalent nanobodies (with help from A.A.D. and J.S.S.), T.M.L., M.C., and M.G.M.B. planned and constructed the wild‐type reagents for the pseudovirus assay, J.S.S. constructed the pseudovirus variants and performed pseudovirus experiments, J.M.Z., A.A.D., and E.K.M. performed biophysical characterization of nanobodies, and J.M.Z., A.A.D., J.S.S., M.D.S., and P.M.T. analyzed the data. J.M.Z., J.S.S., A.A.D., and P.M.T. wrote the paper with input from the co‐authors.
Acknowledgements
The authors thank Michel Nussenzweig, Pamela Bjorkman, Christopher Barnes, and Anna Gazumyan for providing class 2 and 3 SARS‐CoV‐2 antibodies. Moreover, the authors thank Adam Lauring for providing reagents and advice related to the pseudovirus neutralization assay. The authors also thank members of the Tessier lab for their helpful suggestions. This work was supported by the National Institutes of Health (RF1AG059723 and R35GM136300 to P.M.T., F32 GM137513 to J.S.S., and R01 AI 51588 01 to M.C.), National Science Foundation (CBET 1159943, 1605266, and 1813963 to P.M.T. and Graduate Research Fellowship to M.D.S.), Biointerfaces Institute (to P.M.T.), the Albert M. Mattocks Chair (to P.M.T.), MICHR Education PTSP 2020 (U069943 to M.G.M.B.), COVID‐19: CVC Impact Research Ignitor Grant Award (to M.C.), University of Michigan MICHR Accelerating Synergy Award (to M.C.), and University of Michigan Institutional Funds (to T.M.L.).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.WHO Coronavirus (COVID‐19) Dashboard With Vaccination Data. https://covid19.who.int/ (accessed: April 2021).
- 2.Davies N. G., Abbott S., Barnard R. C., Jarvis C. I., Kucharski A. J., Munday J. D., Pearson C. A. B., Russell T. W., Tully D. C., Washburne A. D., Wenseleers T., Gimma A., Waites W., Wong K. L. M., van Zandvoort K., Silverman J. D., Diaz‐Ordaz K., Keogh R., Eggo R. M., Funk S., Jit M., Atkins K. E., Edmunds W. J., Science 2021, 372, eabg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann M., Arora P., Groß R., Seidel A., Hörnich B. F., Hahn A. S., Krüger N., Graichen L., Hofmann‐Winkler H., Kempf A., Winkler M. S., Schulz S., Jäck H.‐M., Jahrsdörfer B., Schrezenmeier H., Müller M., Kleger A., Münch J., Pöhlmann S., Cell 2021, 184, 2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Planas D., Bruel T., Grzelak L., Guivel‐Benhassine F., Staropoli I., Porrot F., Planchais C., Buchrieser J., Rajah M. M., Bishop E., Albert M., Donati F., Prot M., Behillil S., Enouf V., Maquart M., Smati‐Lafarge M., Varon E., Schortgen F., Yahyaoui L., Gonzalez M., De Sèze J., Péré H., Veyer D., Sève A., Simon‐Lorière E., Fafi‐Kremer S., Stefic K., Mouquet H., Hocqueloux L., et al., Nat. Med. 2021, 27, 917. [DOI] [PubMed] [Google Scholar]
- 5.Wang P., Nair M. S., Liu L., Iketani S., Luo Y., Guo Y., Wang M., Yu J., Zhang B., Kwong P. D., Graham B. S., Mascola J. R., Chang J. Y., Yin M. T., Sobieszczyk M., Kyratsous C. A., Shapiro L., Sheng Z., Huang Y., Ho D. D., Nature 2021, 593, 130. [DOI] [PubMed] [Google Scholar]
- 6.Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J. C. C., Muecksch F., Rutkowska M., Hoffmann H. H., Michailidis E., Gaebler C., Agudelo M., Cho A., Wang Z., Gazumyan A., Cipolla M., Luchsinger L., Hillyer C. D., Caskey M., Robbiani D. F., Rice C. M., Nussenzweig M. C., Hatziioannou T., Bieniasz P. D., eLife 2020, 9, e61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wec A. Z., Wrapp D., Herbert A. S., Maurer D. P., Haslwanter D., Sakharkar M., Jangra R. K., Dieterle M. E., Lilov A., Huang D., Tse L. V., Johnson N. V., Hsieh C. L., Wang N., Nett J. H., Champney E., Burnina I., Brown M., Lin S., Sinclair M., Johnson C., Pudi S., Bortz R., Wirchnianski A. S., Laudermilch E., Florez C., Fels J. M., O'Brien C. M., Graham B. S., Nemazee D., et al., Science 2020, 369, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wrapp D., De Vlieger D., Corbett K. S., Torres G. M., Wang N., Van Breedam W., Roose K., van Schie L., Hoffmann M., Pöhlmann S., Graham B. S., Callewaert N., Schepens B., Saelens X., McLellan J. S., Cell 2020, 181, 1004.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan M., Wu N. C., Zhu X., Lee C. C. D., So R. T. Y., Lv H., Mok C. K. P., Wilson I. A., Science 2020, 368, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian X., Li C., Huang A., Xia S., Lu S., Shi Z., Lu L., Jiang S., Yang Z., Wu Y., Ying T., Emerging Microbes Infect. 2020, 9, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koenig P. A., Das H., Liu H., Kümmerer B. M., Gohr F. N., Jenster L. M., Schiffelers L. D. J., Tesfamariam Y. M., Uchima M., Wuerth J. D., Gatterdam K., Ruetalo N., Christensen M. H., Fandrey C. I., Normann S., Tödtmann J. M. P., Pritzl S., Hanke L., Boos J., Yuan M., Zhu X., Schmid‐Burgk J. L., Kato H., Schindler M., Wilson I. A., Geyer M., Ludwig K. U., Hällberg B. M., Wu N. C., Schmidt F. I., Science 2021, 371, eabe6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoof M., Faust B., Saunders R. A., Sangwan S., Rezelj V., Hoppe N., Boone M., Billesbølle C. B., Puchades C., Azumaya C. M., Kratochvil H. T., Zimanyi M., Deshpande I., Liang J., Dickinson S., Nguyen H. C., Chio C. M., Merz G. E., Thompson M. C., Diwanji D., Schaefer K., Anand A. A., Dobzinski N., Zha B. S., Simoneau C. R., Leon K., White1 K. M., Chio U. S., Gupta M., Jin M., et al., Science 2020, 370, 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiang Y., Nambulli S., Xiao Z., Liu H., Sang Z., Duprex W. P., Schneidman‐Duhovny D., Zhang C., Shi Y., Science 2020, 370, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Detalle L., Stohr T., Palomo C., Piedra P. A., Mas V., Millar A., Power U. F., Stortelers C., Allosery K., Melero J. A., Delpa E., Antimicrob. Agents Chemother. 2016, 60, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hultberg A., Temperton N. J., Rosseels V., Koenders M., Gonzalez‐Pajuelo M., Schepens B., Ibañez L. I., Vanlandschoot P., Schillemans J., Saunders M., Weiss R. A., Saelens X., Melero J. A., Verrips C. T., Van Gucht S., de Haard H. J., PLoS One 2011, 6, e17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laursen N. S., Friesen R. H. E., Zhu X., Jongeneelen M., Blokland S., Vermond J., Van Eijgen A., Tang C., Van Diepen H., Obmolova G., Van Der Neut Kolfschoten M., Zuijdgeest D., Straetemans R., Hoffman R. M. B., Nieusma T., Pallesen J., Turner H. L., Bernard S. M., Ward A. B., Luo J., Poon L. L. M., Tretiakova A. P., Wilson J. M., Limberis M. P., Vogels R., Brandenburg B., Kolkman J. A., Wilson I. A., Science 2018, 362, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ibañez L. I., De Filette M., Hultberg A., Verrips T., Temperton N., Weiss R. A., Vandevelde W., Schepens B., Vanlandschoot P., Saelens X., J. Infect. Dis. 2011, 203, 1063. [DOI] [PubMed] [Google Scholar]
- 18.Cardoso F. M., Ibanez L. I., Van den Hoecke S., De Baets S., Smet A., Roose K., Schepens B., Descamps F. J., Fiers W., Muyldermans S., Depicker A., Saelens X., J. Virol. 2014, 88, 8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pant N., Hultberg A., Zhao Y., Svensson L., Pan‐Hammarström Q., Johansen K., Pouwels P. H., Ruggeri F. M., Hermans P., Frenken L., Borén T., Marcotte H., Hammarström L., J. Infect. Dis. 2006, 194, 1580. [DOI] [PubMed] [Google Scholar]
- 20.Schotte L., Strauss M., Thys B., Halewyck H., Filman D. J., Bostina M., Hogle J. M., Rombaut B., J. Virol. 2014, 88, 4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss M., Schotte L., Thys B., Filman D. J., Hogle J. M., J. Virol. 2016, 90, 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanke L., Vidakovics Perez L., Sheward D. J., Das H., Schulte T., Moliner‐Morro A., Corcoran M., Achour A., Karlsson Hedestam G. B., Hällberg B. M., Murrell B., McInerney G. M., Nat. Commun. 2020, 11, 4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi X., Liu X., Wang C., Zhang X., Li X., Hou J., Ren L., Jin Q., Wang J., Yang W., Nat. Commun. 2020, 11, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huo J., Bas A. L., Ruza R. R., Duyvesteyn H. M. E., Mikolajek H., Malinauskas T., Tan T. K., Rijal P., Dumoux M., Ward P. N., Ren J., Zhou D., Harrison P. J., Weckener M., Clare D. K., Vogirala V. K., Radecke J., Moynié L., Zhao Y., Gilbert‐jaramillo J., Knight M. L., Tree J. A., Buttigieg K. R., Coombes N., Elmore M. J., Carroll M. W., Carrique L., Shah P. N. M., James W., Townsend A. R., et al., Nat. Struct. Mol. Biol. 2020, 27, 846. [DOI] [PubMed] [Google Scholar]
- 25.Custódio T. F., Das H., Sheward D. J., Hanke L., Pazicky S., Pieprzyk J., Sorgenfrei M., Schroer M. A., Gruzinov A. Y., Jeffries C. M., Graewert M. A., Svergun D. I., Dobrev N., Remans K., Seeger M. A., Mcinerney G. M., Murrell B., Hällberg B. M., Löw C., Nat. Commun. 2020, 11, 5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moliner‐Morro A., Sheward D. J., Karl V., Vidakovics L. P., Murrell B., McInerney G. M., Hanke L., Biomolecules 2020, 10, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zupancic J. M., Desai A. A., Schardt J. S., Pornnoppadol G., Makowski E. K., Smith M. D., Kennedy A. A., Garcia de Mattos Barbosa M., Cascalho M., Lanigan T. M., Tai A. W., Tessier P. M., Cell Chem. Biol. 2021, 28, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinto D., Park Y. J., Beltramello M., Walls A. C., Tortorici M. A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., Peter A., Guarino B., Spreafico R., Cameroni E., Case J. B., Chen R. E., Havenar‐Daughton C., Snell G., Telenti A., Virgin H. W., Lanzavecchia A., Diamond M. S., Fink K., Veesler D., Corti D., Nature 2020, 583, 290. [DOI] [PubMed] [Google Scholar]
- 29.Barnes C. O., Jette C. A., Abernathy M. E., Dam K. M. A., Esswein S. R., Gristick H. B., Malyutin A. G., Sharaf N. G., Huey‐Tubman K. E., Lee Y. E., Robbiani D. F., Nussenzweig M. C., West A. P., Bjorkman P. J., Nature 2020, 588, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi R., Shan C., Duan X., Chen Z., Liu P., Song J., Song T., Bi X., Han C., Wu L., Gao G., Hu X., Zhang Y., Tong Z., Huang W., Liu W. J., Wu G., Zhang B., Wang L., Qi J., Feng H., Wang F. S., Wang Q., Gao G. F., Yuan Z., Yan J., Nature 2020, 584, 120. [DOI] [PubMed] [Google Scholar]
- 31.Xu Y., Roach W., Sun T., Jain T., Prinz B., Yu T., Torrey J., Thomas J., Bobrowicz P., Vasquez M., Wittrup K. D., Krauland E., Protein Eng., Des. Sel. 2013, 26, 663. [DOI] [PubMed] [Google Scholar]
- 32.Jain T., Sun T., Durand S., Hall A., Houston N. R., Nett J. H., Sharkey B., Bobrowicz B., Caffry I., Yu Y., Cao Y., Lynaugh H., Brown M., Baruah H., Gray L. T., Krauland E. M., Xu Y., Vásquez M., Wittrup K. D., Proc. Natl. Acad. Sci. USA 2017, 114, 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hötzel I., Theil F. P., Bernstein L. J., Prabhu S., Deng R., Quintana L., Lutman J., Sibia R., Chan P., Bumbaca D., Fielder P., Carter P. J., Kelley R. F., mAbs 2012, 4, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C.‐L., Abiona O., Graham B. S., Mclellan J. S., Science 2020, 367, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walls A. C., Park Y. J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Cell 2020, 181, 281.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ter Meulen J., van den Brink E. N., Poon L. L. M., Marissen W. E., Leung C. S. W., Cox F., Cheung C. Y., Bakker A. Q., Bogaards J. A., van Deventer E., Preiser W., Doerr H. W., Chow V. T., de Kruif J., Peiris J. S. M., Goudsmit J., PLoS Med. 2006, 3, e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baum A., Fulton B. O., Wloga E., Copin R., Pascal K. E., Russo V., Giordano S., Lanza K., Negron N., Ni M., Wei Y., Atwal G. S., Murphy A. J., Stahl N., Yancopoulos G. D., Kyratsous C. A., Science 2020, 369, 1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baum A., Ajithdoss D., Copin R., Zhou A., Lanza K., Negron N., Ni M., Wei Y., Mohammadi K., Musser B., Atwal G. S., Oyejide A., Goez‐Gazi Y., Dutton J., Clemmons E., Staples H. M., Bartley C., Klaffke B., Alfson K., Gazi M., Gonzalez O., Dick E., Carrion R., Pessaint L., Porto M., Cook A., Brown R., Ali V., Greenhouse J., Taylor T., et al., Science 2020, 370, 1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen P., Nirula A., Heller B., Gottlieb R. L., Boscia J., Morris J., Huhn G., Cardona J., Mocherla B., Stosor V., Shawa I., Adams A. C., Van Naarden J., Custer K. L., Shen L., Durante M., Oakley G., Schade A. E., Sabo J., Patel D. R., Klekotka P., Skovronsky D. M., N. Engl. J. Med. 2020, 384, 229.33113295 [Google Scholar]
- 40.Cortez‐Retamozo V., Lauwereys M., Hassanzadeh Gh G., Gobert M., Conrath K., Muyldermans S., De Baetselier P., Revets H., Int. J. Cancer 2002, 98, 456. [DOI] [PubMed] [Google Scholar]
- 41.Van Roy M., Ververken C., Beirnaert E., Hoefman S., Kolkman J., Vierboom M., Breedveld E., ’t Hart B., Poelmans S., Bontinck L., Hemeryck A., Jacobs S., Baumeister J., Ulrichts H., Arthritis Res. Ther. 2015, 17, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iqbal U., Trojahn U., Albaghdadi H., Zhang J., O'Connor‐Mccourt M., Stanimirovic D., Tomanek B., Sutherland G., Abulrob A., Br. J. Pharmacol. 2010, 160, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sparkes A., De Baetselier P., Brys L., Cabrito I., Sterckx Y. G. J., Schoonooghe S., Muyldermans S., Raes G., Bucala R., Vanlandschoot P., Van Ginderachte J. A., Stijlemans B., FASEB J. 2018, 32, 3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Junghans R. P., Anderson C. L., Proc. Natl. Acad. Sci. USA 1996, 93, 5512.8643606 [Google Scholar]
- 45.Andersen J. T., Sandlie I., Drug Metab. Pharmacokinet. 2009, 24, 318. [DOI] [PubMed] [Google Scholar]
- 46.Sinclair N. R. S., Chan P. L., in Morphological and Functional Aspects of Immunity, Springer, Boston, MA: 1971, p. 609. [Google Scholar]
- 47.Rossotti M. A., Bélanger K., Henry K. A., Tanha J., in Immunogenicity and Humanization of Single‐Domain Antibodies, Blackwell Publishing Ltd, Hoboken, NJ: 2021. [DOI] [PubMed] [Google Scholar]
- 48.Jovčevska I., Muyldermans S., in The Therapeutic Potential of Nanobodies, Springer International Publishing, New York: 2019. [Google Scholar]
- 49.Zhang J., Liu X., Bell A., To R., Baral T. N., Azizi A., Li J., Cass B., Durocher Y., Protein Expression Purif. 2009, 65, 77. [DOI] [PubMed] [Google Scholar]
- 50.Desai A. A., Smith M. D., Zhang Y., Makowski E. K., Gerson J. E., Ionescu E., Starr C. G., Zupancic J. M., Moore S. J., Sutter A. B., Ivanova M. I., Murphy G. G., Paulson H. L., Tessier P. M., J. Biol. Chem. 2021, 296, 100508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crawford K. H. D., Eguia R., Dingens A. S., Loes A. N., Malone K. D., Wolf C. R., Chu H. Y., Tortorici M. A., Veesler D., Murphy M., Pettie D., King N. P., Balazs A. B., Bloom J. D., Viruses 2020, 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.