

Keywords: cardiovascular disease, glucotoxicity, heart, inflammation, lipotoxicity, obesity, therapy
Abstract
The prevalence of heart failure is on the rise and imposes a major health threat, in part, due to the rapidly increased prevalence of overweight and obesity. To this point, epidemiological, clinical, and experimental evidence supports the existence of a unique disease entity termed “obesity cardiomyopathy,” which develops independent of hypertension, coronary heart disease, and other heart diseases. Our contemporary review evaluates the evidence for this pathological condition, examines putative responsible mechanisms, and discusses therapeutic options for this disorder. Clinical findings have consolidated the presence of left ventricular dysfunction in obesity. Experimental investigations have uncovered pathophysiological changes in myocardial structure and function in genetically predisposed and diet-induced obesity. Indeed, contemporary evidence consolidates a wide array of cellular and molecular mechanisms underlying the etiology of obesity cardiomyopathy including adipose tissue dysfunction, systemic inflammation, metabolic disturbances (insulin resistance, abnormal glucose transport, spillover of free fatty acids, lipotoxicity, and amino acid derangement), altered intracellular especially mitochondrial Ca2+ homeostasis, oxidative stress, autophagy/mitophagy defect, myocardial fibrosis, dampened coronary flow reserve, coronary microvascular disease (microangiopathy), and endothelial impairment. Given the important role of obesity in the increased risk of heart failure, especially that with preserved systolic function and the recent rises in COVID-19-associated cardiovascular mortality, this review should provide compelling evidence for the presence of obesity cardiomyopathy, independent of various comorbid conditions, underlying mechanisms, and offer new insights into potential therapeutic approaches (pharmacological and lifestyle modification) for the clinical management of obesity cardiomyopathy.
1. INTRODUCTION (OVERVIEW OF OBESITY)
1.1. Facts about Prevalence and Health Consequences of Obesity
1.1.1. Epidemiology of obesity.
The World Health Organization (WHO) defines overweight and obesity as a pathological setting with abnormal or excessive fat accumulation. Obesity is generally rooted in a complex interplay between genetic and environmental factors such as culture, socioeconomical status, and lifestyle, leading to an alarming health concern in the 21st century (1–7) (see BOX 1). Body mass index (BMI), estimated by body weight in kilograms divided by the square of height in meters, is the simplest index employed to categorize overweight and obesity in adults. Current guidelines from the US Centers for Disease Control and Prevention and the WHO classify a healthy BMI in the window of 18.5–24.9, whereas a BMI ≥25 kg/m2 is classified as overweight, and a BMI ≥30 kg/m2 is deemed obese, with severe obesity (morbid obesity) listed as a BMI ≥40 kg/m2 (5, 8, 9). Over the past two decades, the worldwide prevalence of overweight and obesity has risen dramatically, mainly driven by socioeconomical and lifestyle changes manifested by lower energy expenditure (physical activity) and increased usage of energy-rich food sources, especially refined carbohydrates (1, 5, 6). Uncorrected obesity unfavorably impacts all aspects of physiological functions, lowers quality of life, and increases the risk of illness and health-care burden worldwide (1, 7, 10). This review will discuss current contemporary knowledge of the causes and underlying mechanisms in adiposity and associated cardiovascular disease (CVD), with an ultimate aim to offer guidance for more effective and targeted antiobesity therapy in particular obesity-induced cardiac anomalies.
1.1.1.1. global epidemiology.
According to the WHO, the overall prevalence of obesity has doubled in the United States and in most of the Westernized countries since 1980 and nearly tripled worldwide between 1975 and 2016 (4, 11, 12). A total of 1.9 billion individuals were classified to be overweight and 650 million adults presented obesity in 2016, representing 39% and 13%, respectively, of world population (3). Specifically, 39% of adults, including 39% of men and 40% of women, met the criteria of overweight and 13% of adults, including 11% of men and 15% of women, reached the threshold of obesity in 2016 (FIGURE 1). The age-justified prevalence of overweight and obesity rose by almost 50% (26.5% in 1980 to 39.0% in 2015) and 80% (7% in 1980 to 12.5% in 2015), respectively (3). In another independent report, the prevalence of obesity jumped from 3.2% to 10.8% in adult men and 6.4% to 14.9% in adult women between 1975 and 2014. In 2014, 0.64% of men and 1.6% of women exhibited morbid obesity (BMI ≥40) (1). More alarmingly, the prevalence of childhood obesity also rose vividly during the last decades with more obese and overweight children growing into overweight and obese adolescents and adults. Based on WHO data, ∼40 million preschool age children under 5 yr were overweight or obese in 2018. More than 340 million children and adolescents between 5 and 19 yr were classified overweight or obese in 2016. Retrospectively, child and adolescent obesity prevalence jumped from 0.7% to 5.6% in boys and 0.9% to 7.8% in girls between 1975 and 2016 (2).
FIGURE 1.

Increase in obesity prevalence over the past 20 yr (1996-2016): According to the World Health Organization’s (WHO) data, 39% of adults aged 18 yr and over (39% of men and 40% of women) were overweight in 2016 worldwide. Overall, about 13% of the world’s adult population (11% of men and 15% of women) were obese in 2016. Reproduced from WHO’s Global Health Observatory (GHO) Data (9) with permission. BMI, body mass index.
1.1.1.2. regional epidemiology.
With the rapidly increased prevalence of obesity worldwide between 1975 and 2016 (2), remarkable regional differences were noted in obesity epidemiology (FIGURE 1). For example, the prevalence of obesity differs dramatically by country, ranging from 3.7% in Japan to 38.2% in the US (13). More than 50% of the global obese population lives in only 10 countries, including the United States, Brazil, China, Egypt, Germany, India, Indonesia, Mexico, Pakistan, and Russia (4). Moreover, dynamics of obesity prevalence exhibits heterogeneity across various countries in the steepness of rise, deceleration, and acceleration of obesity. Although China and India possess abundant obese populations, the prevalence of obesity in these two countries is relatively low due to the population base (∼5.7% and 7.0%, respectively, in 2015) (13). America and Europe still remain the two regions with the highest prevalence of overweight and obesity in 2015 (3). In North America, the occurrence of overweight jumped from 45.3% in 1980 to 64.2% in 2015 and the frequency of obesity rose from 12.9% in 1980 to 28.3% in 2015 (3). The prevalence of obesity was 42.4%, and severe obesity approached 9.2% in the US in 2017–2018. In Europe, the prevalence of overweight changed drastically from 48% in 1980 to 59.6% in 2015 and that of obesity escalated from 14.5% in 1980 to 22.9% in 2015 (3). Along the same line, the numbers of overweight rose from 37.9% in 1980 to 49.6% in 2015 and that of obesity jumped from 11.8% in 1980 to 19.6% in 2015 in the Eastern Mediterranean region. In Africa, the rate of overweight and obesity rose from 18.5% to 34.5% and 6.2% to 12.7%, respectively, between 1980 and 2015. Although with the lowest global ranking, inclinations in overweight and obesity also hiked in the West Pacific region (China, Japan, Philippines, Vietnam, and South Korea) during the past three decades. In particular, the prevalence of overweight more than tripled from 7.8% to 29.9% in China. Likewise, the incidence of overweight escalated from 10.9% in 1980 to 24.3% in 2015, and the rate of obesity rose from 1.7% in 1980 to 6.2% in 2015, in the Southeast Asian region (3). Globally speaking, regions from south Asia, southeast Asia, the Caribbean, and southern Latin America seem to experience the most accelerated increase in BMI value (2).
1.1.2. Disease burden of obesity.
Obesity, when uncorrected, is accompanied with an increased morbidity and mortality of noncommunicable diseases, particularly CVD, musculoskeletal disorders, and certain forms of cancers (breast, ovarian, prostate, liver, kidney, and colon cancers) (5, 6). Obesity results in the onset of a cluster of unfavorable chronic disorders, which commonly trigger profound metabolic pathologies [e.g., hypertension, hyperinsulinemia, dyslipidemia, glucose intolerance, and type 2 diabetes mellitus (T2D)] (14–18). Notably, the World Obesity Federation and the American and Canadian Medical Associations have all affirmed obesity as a chronic developing illness in addition to its role as just a risk factor for other comorbidities (19). In 2015, excess weight underwrote 4.0 million (ranging 2.7–5.3 million) mortalities and 120 million (ranging 84–158 million) disability adjusted life years (DALYs) globally (20, 21). Nearly 39% of deaths and 36% of DALYs associated with high BMI were reported in those with a BMI ≥30 kg/m2. Various obesity-related chronic diseases have been noted for the economic burden in obesity. Among which, CVD accounts for more than two-thirds of mortalities linked with high BMI and 66.3 million DALYs (20). Compared with those with normal weight, individuals who gain substantial weight from young and middle age display a 22% and 49% greater risk of all-cause mortality and CVD mortality, respectively (22).
Obesity increases risks of multiple diseases and poor mental health, all of which might lead to compromised life quality, lower productivity, unemployment, and social hardships and higher healthcare costs (23). For instance, osteoarthritis, a popular aftermath of obesity, is one leading cause of disability and retirement. In the US, the healthcare expense for a single obese individual was estimated to be $1,901 annually in 2014, inferring a total cost of $149.4 billion on obesity nationally. In Europe, overweight- and obesity-induced direct and indirect cost was equivalent to 0.47–0.61% of the gross domestic product (3). According to a systematic review, medical costs of obese people were 32% more compared with lean individuals. Specially, obesity is believed to account for 31.8% of direct or healthcare costs, and 68.1% of indirect costs associated with deficit of productivity and production value (7).
1.2. Etiology of Obesity
1.2.1. Environment: diet consumption and sedentary lifestyle.
The etiology of obesity is multifactorial including genetic, environmental; and behavioral aspects. In general, obesity is usually a result from a prolonged positive energy balance, that is, increased consumption of food consumption in excess of energy expenditure (in the form of heat production) (24). An important dietary determinant of obesity is the increased consumption of sugar in the form of fructose-containing sugars, sucrose, and high-fructose corn syrup, mainly refined carbohydrates used extensively in the modern food industry (25). Evidence from epidemiological studies supports a solid tie between sugar-sweetened beverage usage and BMI (26). However, the perception of obesity is recently switching away from the simplistic notion of energy imbalance, calorie counting, and single isolated nutrients toward overall dietary patterns on the complex physiological determinants of weight regulation (27). In short-term, total calories are most relevant to weight gain regardless of the types of diets. However, in long-term weight control and cardiometabolic health, healthy food-based patterns matter given the synergistic health outcome produced by the combination of foods habitually consumed (27, 28).
Several other lifestyle factors, such as sedentary behavior, circadian alignment, and sleep quality, may interact with diet to influence metabolic risk and disease propensity. Accumulating evidence supports the notion that sedentary behavior is a strong predictor of obesity and detrimental changes in metabolic traits (29, 30). Among various sedentary behaviors, TV watching, computer games, and other electronic entertainments are considered as the main culprits as screen media exposure greatly displaces physical activities and significantly prompts risks of overweight, obesity and T2D in children and adolescent (31–33). While sedentary behaviors prompt the onset of overweight and obesity in spite of “recreational” or “seasonal” physical exercise (34), it is well conceived that lack of proper and regular physical exercise is more strongly associated with obesity prevalence compared with sedentary behaviors themselves (33, 35).
Last but not least, a theory of “fetal programming of adult disease” has evolved linking the increased prevalence of adulthood obesity and metabolic complications with intrauterine and early postnatal environmental stress (36–38). Both under- and overnutrition during intrauterine and early postnatal stages may adversely impact adulthood phenotypes and organ function especially insulin sensitivity, adiposity, myocardial morphology, and function (38–43). Moreover, epigenetic transgenerational inheritance of susceptibility to adiposity or obesity in subsequent generations has been documented with ancestral exposure (maternal or paternal) to environmental toxicants and altered nutrition (44, 45).
1.2.2. Genetics and epigenetics.
Obesity is an anthropometric trait resulting from a complex interplay of genetic and environmental factors. The gene-environment interactions are generally responsible for changes in gene expression and epigenetic modifications leading to excess body fat and obesity (46). Although parental obesity is listed as an important risk factor for childhood and/or adolescent obesity (47), obesity does not usually follow the Mendelian rule of inheritance and genetic factors only explain a small proportion of the development of obesity (1, 48). The obesity trait may be triggered by both single genes (monogenic) and multiple genes (common or multifactorial obesity). In addition to genetic factors, epigenetic contributions (i.e., modifications pre- or posttranslation) also play an essential role in obesogenesis. Loss-of-function mutations in genes including LEP, LEPR, MC4R, PCSK1, ADCY3, hypothalamic proopiomelanocortin (POMC), and SIM1 have all been associated with monogenic obesity (49). Mutations of the leptin gene, noted in rare cases of extremely obese children/mice, underscores a role for leptin-modulated energy balance through melanocortin-dependent/independent mechanisms (50, 51). Leptin normally sends signals of sufficient fuel storage from adipose tissues to the hypothalamus which promotes satiety by production of α-melanocyte-stimulating hormone (α-MSH) and its binding to the MC4 receptor (MC4R) in response to leptin stimulation. Mutations (in the forms of frame shift, missense or deletion) in POMC and MC4R have been reported in severe obesity (52–55). Indeed, mutations in MC4R are deemed the most prevalent mutations in monogenic obesity (52). Up to 5% of individuals with childhood obesity are carriers of heterozygous MC4R mutations (56, 57). Enzymes including prohormone convertase 1 (PCSK1) processes melanocortin peptides (58). Heterozygous mutations in PCSK1 were noted to alter POMC processing and promote obesity associated with glucocorticoid deficiency and hypogonadotropic hypogonadism (59).
The majority of obese subjects is representatives of “common obesity.” Recently, genome-wide association study (GWAS) analysis has paved the way in the elucidation of the complex genetics underlying common obesity. Up-to-date, large-scale GWAS has identified over 800 single nucleotide polymorphisms tightly associated with BMI (60, 61). These GWAS findings revealed various loci associated with obesity related genes in energy and lipid metabolism (FTO, RPTOR, and MAP2K5) (61), insulin response [TCF7L2 and insulin receptor substrate-1 (IRS1)] (61), adipogenesis [CEBPA, PPARG, bone morphogenetic protein (BMP)-2, HOXC/miR196, SPRY1, TBX15, and PEMT] (62), and neurocircuits of appetite and satiety (BDNF, MC4R, and NEGR) (63–65). Although many more loci linked to obesity have been unveiled, merely 5% of the variance of BMI could be explained by genetic factors (60).
Obesogenesis is also regulated by epigenetic changes. Among various forms of epigenetic modifications, DNA cytosine (C) methylation (CpG) may represent the most stable and well-defined epigenetic machinery involved in obesogenesis (66, 67). Saturated fat, refined carbohydrate, and other dietary factors may all strongly affect gene-specific DNA methylation in obesity. For instance, DNA methylation of adipokines leptin and adiponectin is believed to be connected to BMI and LDL cholesterol levels (68). Furthermore, high methylation of POMC is associated with weight regain in those individuals who just achieved weight loss (69). Interestingly, compared with lean subjects, MSH-positive neurons are much more abundant in obese individuals, suggesting a regulatory role for methylation in POMC downstream signaling components (69).
1.2.3. Animal models of obesity.
Animal models of obesity are widely employed in experimental obesity research, which encompass monogenic and polygenic as well as obesogenic diet-induced models (70, 71). Murine obese models usually exhibit hyperphagia and increased energy metabolism and various comorbidities of obesity, including hyperglycemia, insulin resistance, or diabetes-like syndromes (71–74). It is noteworthy that levels of energy expenditure could be mistakenly assessed given that metabolic inactive mass [for example, the “inert” triacylglycerol (TG)] accounts for the majority of increased weight in obesity and major difference in body mass between lean and obese mice (75). Therefore, energy expenditure in murine obesity models should be calculated with special caution, for example, using the animal lean body mass for normalization. Manipulations of genes commonly involved in leptin pathway provoke obesity, including Lepob/Lepob mice or db/db mice and their rat counterparts, the Zucker or Zucker Diabetic Fatty (ZDF) rats. These rodent models present either a loss of leptin (Lepob/Lepob) or a mutation in leptin receptor (Lepdb/Lepdb), imposing severe leptin resistance. Spontaneous mutation that yields Lepob/Lepob mice presents obesity, hyperphagia, hypothermia, and increased energy expenditure capacity. Lepob/Lepob mice also develop elevated circulating glucocorticoid levels and severe insulin resistance associated with hyperglycemia and hyperinsulinemia (76). The phenotype of Lepdb/Lepdb obese mice is reminiscent of that seen in Lepob/Lepob mice (insulin resistance, diabetes mellitus, and hyperglycemia), with marked early onset obesity, albeit with hyperleptinemia in comparison with the Lepob/Lepob mice (71, 77–80). Likewise, obese Zucker (fa/fa or “fatty” rat) and Koletsky rats are leptin receptor deficient. Koletsky rats are characterized by null mutation of leptin receptor, with essentially undetectable leptin receptor mRNA levels, whereas Zucker fatty rats are featured by a genetic processing mistake in leptin receptor (fa/fa mutation), leading to intracellular trapping of the receptor. These murine models of obesity exhibit hyperphagia, increased energy expenditure, compromised glucose tolerance, and growth deficits courtesy of hypothyroidism and low circulating levels of growth hormone (70, 80–83). Despite the fact that monogenic models are practical in the study of specific mechanistic aspects of obesity, they usually cannot truly recapitulate human obesity. Polygenic obesity models are thus more comparable with the polygenic nature of human obesity. Diet-induced rodent obesity models are commonly employed with exposure to high-fat or high-energy diets, prompting an obese phenotype with the extent of weight gain depending on nature and duration of dietary intake. Diet-induced obese rodents usually develop leptin resistance, insulin resistance, and hypertriglyceridemia before the onset of full-blown obesity (71, 84–91). In addition, lesions in specific brain regions, including ventromedial hypothalamus (92) and hypothalamic paraventricular nucleus (93), also result in overt obese phenotype in rodents.
1.3. Obesity and Cardiovascular Disease
1.3.1. Obesity and overall CVD prevalence.
Ample evidence from both clinical and experimental settings supports the role for obesity in the pathogenesis of CVD, including heart failure (HF) (72, 87, 89, 94–106). Not only is obesity closely intertwined with greater prevalence of coexisting risk factors for CVD such as coronary artery disease, hypertension, diabetes mellitus, and obstructive sleep apnea (2, 3), but obesity alone also impacts myocardial structure and pump performance (manifestations of obesity cardiomyopathy) (107). More recently, a new term “cardiometabolic-based chronic disease” (CMBCD) was introduced to boost timely and continued preventive care for cardiometabolic diseases rooted from genetics, environment, and behavior cues. Reported endpoints for CMBCD encompass coronary heart disease, heart failure, and atrial fibrillation (AF), all of which are commonly present in obesity (108). Early evidence for obesity-related CVD includes findings from the Framingham Heart Study that demonstrated an elevated risk of coronary disease, stroke, heart failure, and CVD death in obesity independent of other common risk factors, such as age, gender, smoking, cholesterol, blood pressure, and glucose intolerance (109). Population-based findings also reveal a tight correlation between increased BMI and earlier CVD morbidity (110, 111) or cardiometabolic multimorbidity (112). Moreover, overweight in adolescence is associated with increased CV abnormalities, especially dilated cardiomyopathy (113), and higher all-cause mortality in adulthood (114). Cardiomyopathy, manifested as left ventricular (LV) enlargement and subclinical cardiac dysfunction in the absence of coronary artery disease, is a common cause of HF (107, 115). For instance, elevated BMI in women of childbearing age results in increased risk of cardiomyopathy, particularly dilated and hypertrophic cardiomyopathies (116) (FIGURE 2). With the increasing evidence depicting changes in cardiac structure and function in mildly to moderately obese individuals, it is becoming evident that obesity serves as an independent risk factor for heart failure. This scenario may be expanded to embrace myocardial anomalies in obese subjects that cannot be credited to coronary artery disease, hypertension, diabetes mellitus, or any other confounding etiologies. However, the correlation between BMI and cardiac function gets more complicated with the appearance of “obesity paradox” (117, 118), whereby high BMI appears to confer neutral or even beneficial effects in subgroups who fit into “metabolically healthy obese” category. Better understanding of the mechanisms underlying obesity-related CVD, especially obesity cardiomyopathy, should help to guide clinical decisions according to the comprehensive assessment of obesity. Here we will summarize characteristics of cardiac remodeling, emerging mechanisms, and therapeutic approaches targeting cardiomyopathy in the setting of obesity, with particular emphasis on advances in cardiac metabolic alternations and subcellular abnormalities involved in the development of obesity cardiomyopathy (FIGURE 3).
FIGURE 2.
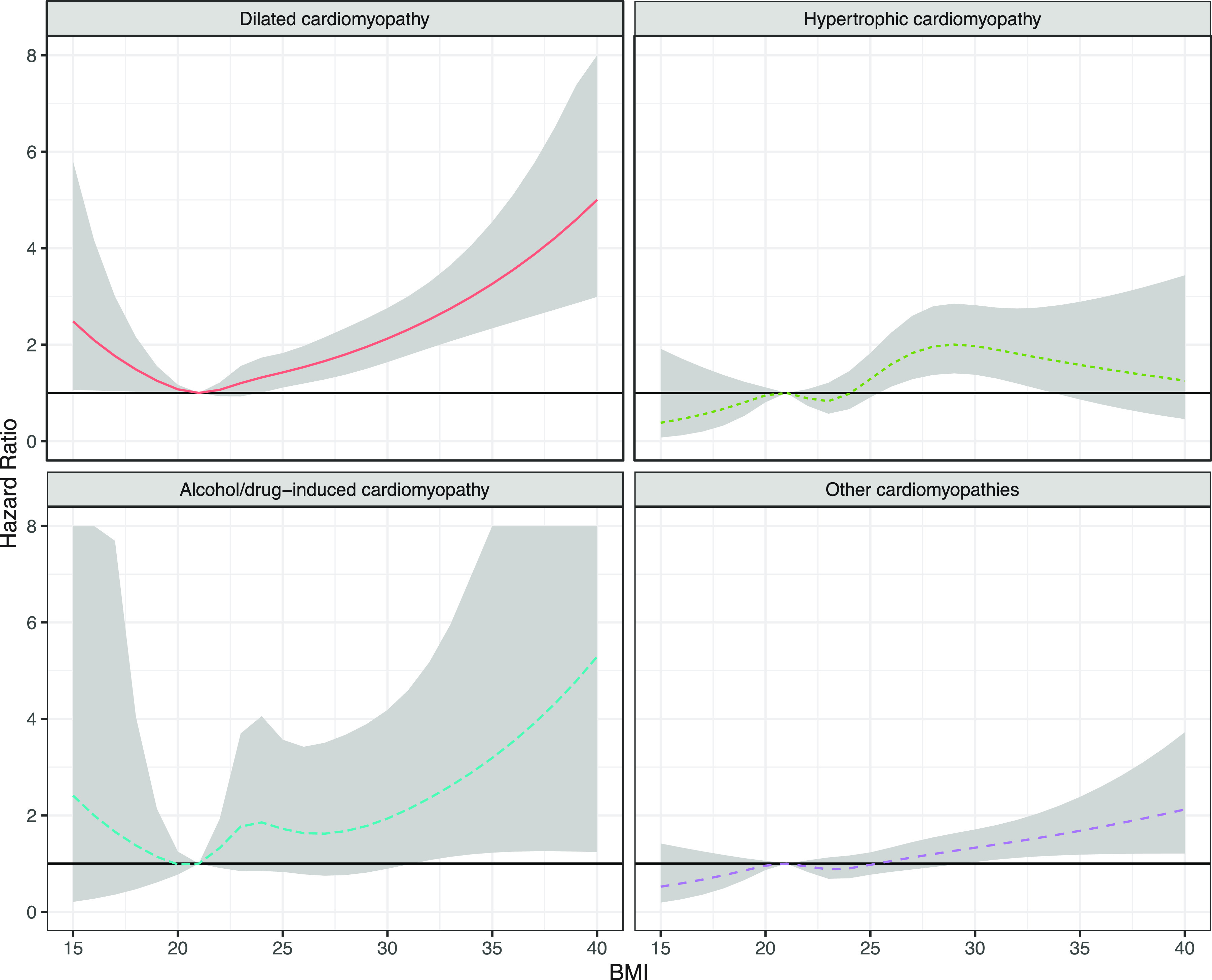
Association between body mass index (BMI) in young women and risk for cardiomyopathies: The model is adjusted for age, year, parity, comorbidities at baseline, smoking, and level of education (n = 1,339,527). Reproduced from Robertson et al. (116) with permission.
FIGURE 3.
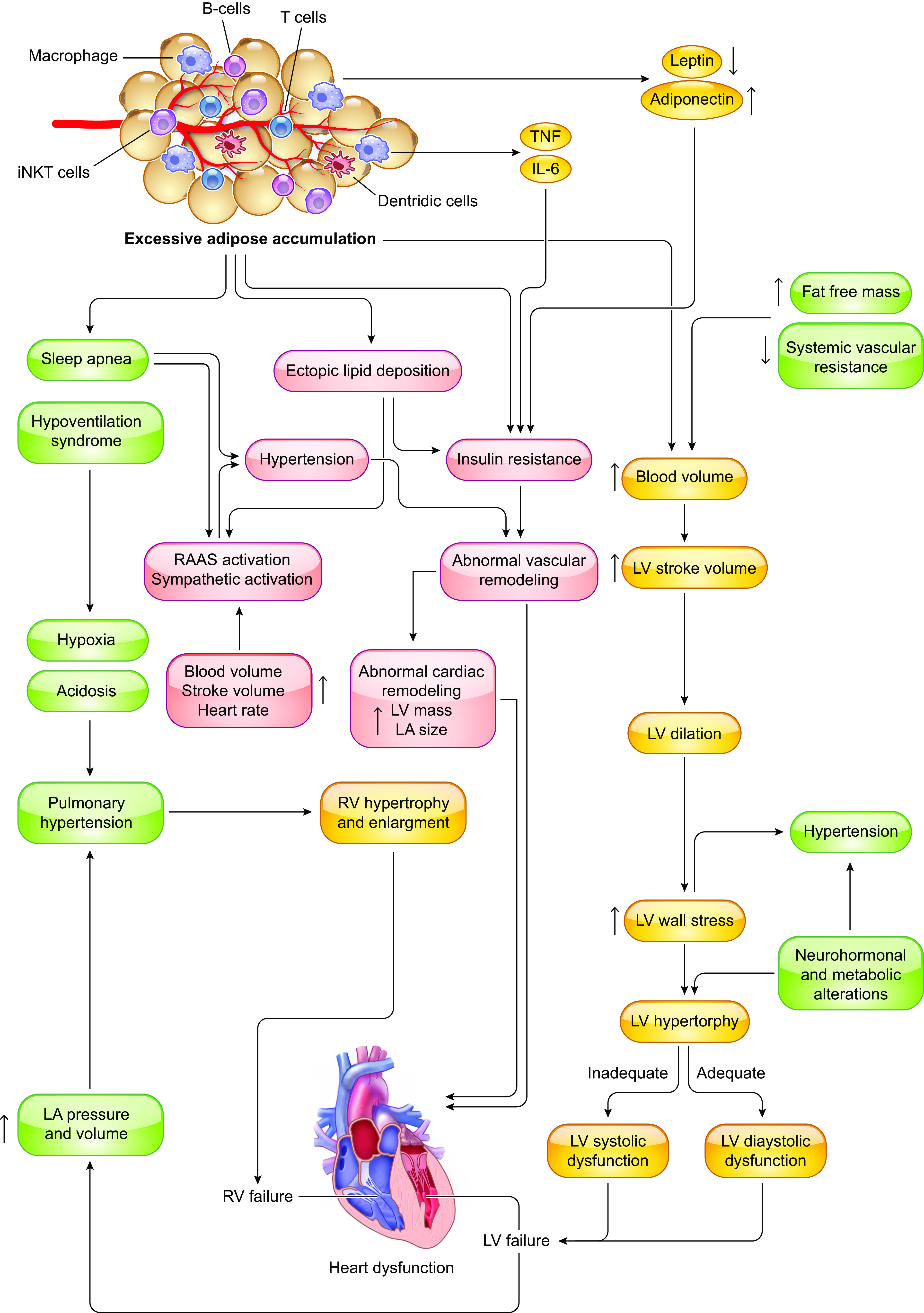
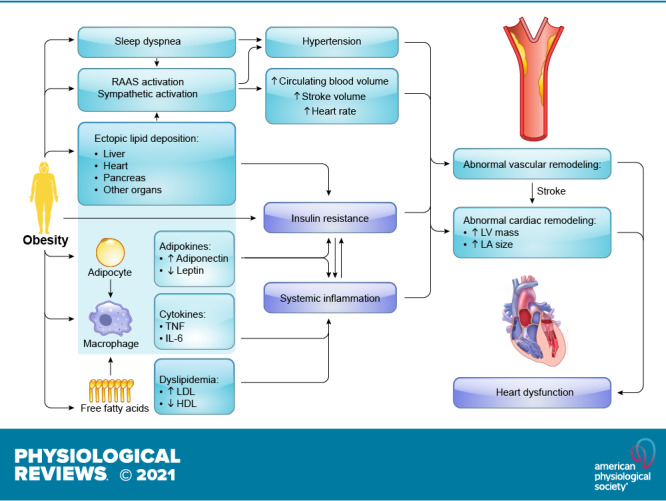
Overall impact of excessive adipose accumulation on cardiac hemodynamic and ventricular function: Under severe obese condition, alterations in ventricular function and abnormalities in cardiac hemodynamic lead to heart failure. Severe obesity induces left ventricular (LV) hypertrophy, which may be eccentric (predominant in normotensive severe obesity) or concentric (predominant in severe obesity and obesity with established systemic hypertension). It is uncertain to what extent metabolic alterations including leptin/insulin resistance, lipid toxicity, and altered renin-angiotensin-aldosterone system (RAAS) may lead to obesity cardiomyopathy. RV, right ventricular; LV, left ventricular; TNF, tumor necrosis factor; IL-6, interleukin-6; iNKT cells, invariant natural killer T cells; LA, left atria.
1.3.2. Impact of obesity on coronavirus disease 2019-associated cardiovascular outcome.
Coronavirus disease 2019 (COVID-19), a pandemic respiratory illness caused by a novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (119), was first detected in late 2019 in Wuhan, China and then spread rapidly across the world. Recent evidence has indicated a close tie between obesity and severity of COVID-19 (120). Clinical data noted an increased risk of COVID-19 severity and mortality in obese patients. In a meta-analysis involving 7,196 patients from 13 independent studies, obesity was associated with an increased risk of critical illness in hospitalized COVID-19 patients (121). In 383 patients with COVID-19, overweight was linked to a 1.84-fold odds, while obesity displayed a 3.40-fold odds of developing severe COVID-19 in comparison with patients with normal weight (122). In another independent study involving 5,279 patients with COVID-19 in the New York City, BMI >40 kg/m2 was the second strongest independent predictor of COVID-19 hospitalization, only after old age (123). Along the same line, the prevalence of obesity was 1.89 times higher in severe COVID-19 patients than the general French population (124). Ample clinical evidence has depicted that CVD and diabetes mellitus are the leading causes of a more severe course of COVID-19 (125). Given the propensity of obesity to prompt CVD including heart failure, hypertension, coronary heart disease, stroke, AF, renal disease, and diabetes mellitus (126–128), being obese itself with high ectopic fat is deemed a unifying risk factor for severe SARS-CoV-2 infection, inflammation, poorly coordinated innate and adaptive responses, inadequate antibody response, cytokine storm, and compromised cardiorespiratory reserve (120, 129). Even in the absence of comorbidities of obesity, the presence of hypertension and metabolic disorders in obesity might result in increased susceptibility to infection via thrombotic events, atherosclerosis, cardiac dysfunction, and impaired immunity (130). Overweight or obese patients exhibit poor endothelial function, cardiorenal and respiratory diseases, which may all negatively impact COVID-19 outcomes (131). On the other side of the coin, reduced physical activity, unhealthy dietary habit, stress, and fear during the COVID-19 pandemic may escalate weight gain and obesity (120). From the molecular biology perspective, SARS-CoV-2 binds to the angiotensin-converting enzyme 2 (ACE2) receptors in the lung and other organs. With overactivated renin-angiotensin-aldosterone system (RAAS) in obesity, the abundant presence of ACE2 in obesity should ease the entrance of SARS-CoV-2 into adipocytes, making adipose tissues an unexpected viral reservoir before the spread of virus to other organs (131). In this context, overweight or obesity should be considered as an independent risk factor for COVID-19 although more scrutiny is warranted to evaluate the clinical management of COVID-19 and associated cardiovascular complications in obesity.
2. CARDIAC REMODELING IN OBESITY
The nomenclature “obesity cardiomyopathy” refers to cardiac morphological, functional, and metabolic abnormalities originating from obesity alone (94, 95). In some cases, the term “metabolic cardiomyopathy” may be used in reference to the broad setting of metabolic disorders including insulin resistance, diabetes mellitus, and obesity (132, 133). In general, long-term obesity is closely associated with cardiac remodeling, characterized as LV hypertrophy, cardiac fibrosis, and diastolic dysfunction that eventually evolves to overt heart failure (FIGURE 3). Fat accumulation, especially disproportionate deposition of metabolically active visceral adipose tissue (VAT) and pericardial fat, drives a higher cardiac output (CO) and workload, and subsequently an enlargement of the LV to meet increased energy requirements (134). Obesity is accompanied by alternations in nutrition, gut microbiota, and neurohumoral activity that compromise cardiac energy metabolism, contractile and relaxation function, and cardiac survival (135, 136) Meanwhile, changes in nutrition status, gut microbiota, and physical exercise are also indispensable for the development of CVD in obesity (137–139). In the absence of lifestyle intervention and targeted drug treatment, obesity impairs cardiac structure and function. It is noteworthy that the cardiac pathological phenotype of obesity varies and can be manifested as either HF with preserved ejection fraction (HFpEF) or reduced ejection fraction (140, 141). In addition to morphological changes, obesity impacts myocardial electrophysiology, resulting in an increased prevalence of AF (142). Obesity-related hemodynamic changes, maladaptive myocardial structural remodeling, and cardiac dysfunction will be briefly summarized (TABLE 1). Regardless of structural or electrophysiological remodeling abnormalities, obesity serves as an independent culprit factor for the occurrence of cardiac hypertrophy and contractile dysfunction (17, 158, 159).
Table 1.
Summary of evidence for cardiomyopathy in obese individuals
| Authors (Year) (Ref. No.) | Patient Information | Presentation of Heart Dysfunction in Obesity |
|---|---|---|
| Dhuper et al. (2011) (143) | 213 obese (BMI = 36.53 ± 0.53) vs. 130 lean subjects | Higher LV mass index, wall thickness, LA index, more aberrant diastolic function by tissue Doppler E/Ea septal, E/Ea lateral, myocardial performance index, Doppler mitral EA ratio, and similar systolic function |
| Shah et al. (2011) (144) | 223 obese and 157 obese diabetic subjects vs. 232 lean subjects | Abnormal cardiac geometry, increased systolic function, and decreased diastolic function |
| Utz et al. (2011) (145) | 65 overweight/obese but otherwise healthy women | Higher myocardial triglyceride levels, reduced left ventricular diastolic, but not systolic function, and increased remodeling index in women with insulin resistance compared with insulin-sensitive women |
| Canepa et al. (2013) (146) | 88 obese vs. 154 nonobese patients | Increased LV mass index, LV posterior wall thickness but not septal wall with increased hypertension; higher LV stroke volume; no changes in LV systolic and diastolic function |
| Olivotto et al. (2013) (147) | 275 adult HCM patients | Increased LV mass and higher risks for developing New York Heart Association (NYHA) functional class III to IV symptoms in obese patients |
| Dahiya et al. (2015) (148) | 35 obese vs. 34 nonobese patients | Lower peak myocardial relaxation velocity, greater filling pressures; Higher LV mass index, left atrial volume index, and LV interventricular septal thickness |
| Rider et al. (2015) (149) | 59 obese vs. 40 normal weight subjects without identifiable CV risk factors | No changes in systolic function; Impaired peak radial and longitudinal diastolic myocardial velocity, prolonged time-to-peak longitudinal diastolic velocity; lower peak longitudinal diastolic strain and time-to-peak longitudinal diastolic strain rate |
| Yagmur et al. (2017) (150) | 40 obese vs. 40 normal weight subjects | No changes in LV diameters and EF; Impaired LV diastolic function (higher transmitral deceleration time, isovolumetric relaxation time, and peak late diastolic tissue Doppler velocity values); Impaired LA reservoir and pump functions |
| Finocchiaro et al. (2018) (151) | 1,033 sudden cardiac death patients (≤35 yr): 212 obese vs. 821 lean subjects | Increased prevalence of left ventricular hypertrophy (12% vs. 2%) and coronary artery disease (12% vs. 3%); less sudden arrhythmic death syndrome (50% vs. 60%) |
| Blomstrand et al. (2018) (152) | 384 patients with T2D, and 184 nondiabetic subjects | Lower LVEF and global longitudinal strain values and increased E/e’ (the ratio between early diastolic mitral flow and annular motion velocities) in obese subjects with or without T2D |
| El Saiedi et al. (2018) (153) | 42 obese vs. 30 healthy children | Higher ratio of transmitral early diastolic filling velocity to septal peak early diastolic myocardial velocity (E/e’) without LVH |
| Balaji et al. (2019) (154) | 504 children with HCM: 140 obese vs. 364 nonobese patients | Increased posterior wall thickness (PWT) but not interventricular septal thickness (IVST) |
| Fumagalli et al. (2019) (155) | 3282 HCM patients: 1,280 preobese and 1,040 obese vs. 962 normal weight subjects | Increased likelihood of NYHA class of III/IV [preobesity, 138 (10.8%); obesity, 215 (20.7%); normal weight, 87 (9.0%)], heart failure (preobesity vs normal weight: HR, 1.192; 95% CI, 0.930–1.1530; obesity vs normal weight: HR, 1.885; 95% CI, 1.485–2.393) and AF |
| Park et al. (2019) (156) | 28,679,891 individuals | Increased incidence of clinical HCM after multivariate adjustment, with a hazard ratio per 1 kg/m2 increase in BMI of 1.063 (95% confidence interval 1.051–1.075) |
| Robertson et al. (2020) (116) | 1,388,571 women (18-45 yr) | Increased risk for cardiomyopathy, particularly for DCM (HR = 4.71 for BMI ≥35 kg/m2 vs. BMI 20-22.5 kg/m2) |
| Litwin et al. (2020) (157) | 11-year follow-up of 254 subjects | Increased BMI was related to increases in LV end-diastolic volume, LV mass, and left atrial volume and decreases in early/late mitral diastolic flow velocity ratio and E-wave deceleration time |
E/Ea, early transmitral flow velocity to early diastolic velocity of the mitral annulus; EA, early to late transmitral flow velocity; HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; LVH, left ventricular hypertrophy; BMI, body mass index; CI, confidence interval; HR, heart rate; AF, atrial fibrillation; T2D, type 2 diabetes.
Although limited data are available, sex difference exists for obesity-associated CVD outcome including a higher mortality risk with increased abdominal fat in female but not male heart failure patients (160). Despite that heart failure and various cardiomyopathies are generally less frequent in women, cardiomyopathies rooted from metabolic derangement seem to be more popular in women than men (161, 162).
Furthermore, most obese individuals suffer from at least one of the comorbidities such as hypertension, sleep apnea, or diabetes mellitus (163, 164). The concurrent presence of coronary heart diseases, hypertension, and the unique obesity cardiomyopathy seems to independently and cooperatively determine anatomic and functional myocardial pathologies in patients with obesity and other comorbidities such as heart failure. Simply launching a defined relationship between obesity and cardiomyopathy is challenging and complicated, since obesity is often present for years before manifestation of any cardiac pathological phenotype. Limited longitudinal data have been accumulated for chronological alterations in cardiac structure and function in obese but otherwise healthy individuals, making it difficult to determine the precise onset timing of “obesity cardiomyopathy” (165).
2.1. Hemodynamics
Although a paradoxical benefit exists in overweight and class I obesity (118, 126, 140, 166–174), it is well documented that obesity produces hemodynamic alterations that generally predispose to unfavorable changes in ventricular structure and function, contributing to the etiology of obesity cardiomyopathy (175) (as summarized in FIGURE 3).
2.1.1. Increased blood volume.
Obese patients have an increased total and central blood volume (140, 176, 177). The increase of blood volume is predominantly the result of elevated renal sodium retention and higher metabolic requirements. The etiology of expanded blood volume and hypertension in obesity, in particular visceral adiposity, involves several pathophysiological processes including activation of sympathetic nervous system and renin-angiotensin-aldosterone system (RAAS), hyperinsulinemia, and natriuretic peptide downregulation, all of which could impair renal capacity to excrete Na, leading to Na retention. Furthermore, overweight/obese individuals generally exhibit a higher level of salt intake, contributing to elevated blood pressure (BP) and blood volume. Obese individuals are also prone to increased salt sensitivity with higher BP hikes with a given amount of salt intake, while genetic variants in salt sensitivity would also affect obesity propensity (178–180).
Obesity commonly causes hypoxemia without concurrent cardiopulmonary anomalies through increased oxygen consumption and decreased lung volumes with normal breathing (181). Along with increased metabolic requirements, increased occurrence of the sleep apnea/hypoventilation syndrome further aggravates hypoxemia in obesity. Hypoxemia promotes erythropoietic activity, as indicated by elevated plasma levels of erythropoietin, the transferrin receptor, and hemoglobin, all of which result in further blood volume elevation (182).
2.1.2. Increased CO.
Similar to arteriovenous shunts in severe liver disease, overweight/obesity, especially abdominal adiposity, is associated with an increased risk of high-output HF (111, 183–186), possibly due to higher filling pressure and increased CO (187, 188). With increased blood volume and stroke volume (SV), CO is generally elevated in overweight/obese subjects with little change in heart rate (189). It should be mentioned that although CO remains relatively high in hypertensive obese patients, it is still lower than that in normotensive obese individuals probably due to increased systemic vascular resistance in hypertension (190).
Abnormal indexes of stroke volume (SV) and CO may be obscured by obesity. Normalization of SV and CO for ideal body surface area (BSA) or height to its age-specific allometric power should provide a more accurate and better estimate for the impact of obesity on LV systolic function (190). Interestingly, fat-free body mass (FFM) is more strongly correlated with SV and CO compared with fat mass and other adiposity variables (191).
Central fat distribution (CFD) is closely correlated with severe abnormalities in body composition and higher CO, independently of FFM/fat mass (FM) in overweight individuals (192). Nonetheless, recent data also suggest that CO may be lower along with higher systemic vascular resistance in central obesity compared with those with peripheral obesity (193, 194).
2.1.3. Increased blood pressure.
Overweight/obesity is a predominant risk factor for the etiology of hypertension (195, 196), although the precise mechanisms coupling hypertension to obesity remain elusive. As aforementioned, Na retention, altered salt sensitivity, overflow of sympathetic nervous system, and activation of RAAS and aldosterone/mineralocorticoid receptor (MR) systems as well as physical compression of kidneys by visceral fat pad may all play an important role in the impaired renal-pressure natriuresis (197, 198). Further evidence has noted impaired tubule-glomerular feedback and renal retention of Na in obese subjects. Elevated BP, dyslipidemia, and hyperglycemia, the three most predominant risk factors for obesity, may be responsible for at least 45%-50% of CVD incidence, particularly coronary heart diseases in obese individuals (14). This is strongly supported by the favorable cardiovascular outcomes in obesity resulting from reductions in BP [telmisartan (199)], glucose [sodium/glucose cotransporter 1 (SGLT1) inhibitor (200)], and lipid [orlistat (201)].
2.1.3.1. arterial stiffness.
Arterial stiffness, a devastating pathological process featured by progressively loss of distensibility of large arteries, emerges as an independent risk factor exacerbating the development of CVD in obesity. Excess caloric intake prompts the onset and development of vascular stiffness to compromise vascular function via endothelial dysfunction, extracellular matrix remodeling, calcification, and inflammation (202–204). Arterial stiffness is usually monitored clinically using pulse wave velocity (205, 206). Weight gain and metabolic disorders occur before, or concomitant with, arterial stiffening (189, 207–212). This reduction of cushioning capacity imposes multiple consequences on cardiovascular health, including elevated systolic blood pressure, incident hypertension, increased penetration of pulsatile energy into microvasculature of target organs, and a rise in coronary perfusion pressure and myocardial wall stress, all of which prompt LV remodeling, dysfunction, and heart failure (202, 203, 211, 213, 214). Thus arterial stiffness may be evaluated through comprehensive assessment of arterial distensibility in the clinical setting to pinpoint vascular health and predict future cardiovascular risk. Many, although not all, studies have found that weight loss reduces arterial stiffness, indicating the reversible nature of obesity-induced vascular stiffness (212, 215, 216). A better understanding of arterial stiffness in overweight and obesity should assist therapeutic strategies to reduce obesity-associated inordinate CVD risks.
2.1.3.2. peripheral resistance.
Hypertension is evoked by increased CO and/or increased peripheral vascular resistance (PVR) (217). Normotensive obese patients commonly display low PVRs (175, 218). Total PVR generally correlates inversely with BMI while it is positively correlated with waist/hip ratio in stress conditions, indicating a much more determining role for central obesity (as opposed to BMI) in the genesis of elevated PVR (219).
2.1.3.3. blood pressure.
Overweight/obesity possesses a close relationship with high BP (195, 220, 221). Resistant hypertension is more prevalent with obesity (222). Compared with lean subjects, obese subjects often exhibit a higher incidence of nondipping hypertension, based on a 24-h BP obtained using an ambulatory blood pressure monitor (223, 224). Patients displaying a nondipping pattern suffer from more hypertension-induced organ damage such as LV hypertrophy, microalbuminuria, and stroke, leading to poor overall cardiovascular outcomes (224). Interestingly, overweight and obesity tend to exhibit lower central systolic blood pressure as compared with lean patients, especially in women (225).
In hypertensive patients receiving antihypertensive therapy, a higher BMI value is typically linked with much smaller BP reduction-associated benefit in LV remodeling and LV systolic function (226), which contributes to higher CVD mortality in obese hypertensive. Weight loss using antiobesity medications such as orlistat is associated with an overt BP drop in overweight/obese subjects (227). Furthermore, bariatric surgery may also serve as a useful alternative strategy for BP control in obese hypertensive patients (228).
A sexual dimorphism was noted in the hemodynamics pattern in middle-aged overweight or obese hypertensive individuals, in particular with respect to total PVR (195). Although the sex-specific intrinsic BP regulatory mechanisms are absent in nonobese subjects, multiple sex differences exist in terms of regulation of BP and hemodynamics in overweight/obese individuals at the molecular, cellular, and tissue levels. In particular, a number of governing machineries involved in the development of hypertension and CVD, including the sympathetic nervous system, the RAAS, and the immune system, display a sexual dimorphism (229). Moreover, age also plays a key role as nonobese premenopausal women exhibit a much higher degree of cardioprotection than age-matched men (although such benefits diminished with onset of menopause) (229). Furthermore, both sex chromosome complements and sex hormones such as estrogen and testosterone likely participate in gender-related differences in BP and CVD. The advantage in female longevity may limit cell senescence signaling and hypertensive organ damage in nonobese women. Last but not least, certain sex-related difference in lifestyle, such as smoking, drinking, and dietary intake, may also influence BP and CVD between men and women (230). Importantly, female advantage in cardioprotection disappears with obesity, reminiscent to that seen in diabetes mellitus (231–235).
2.1.4. Increased LV wall stress.
One of the main obesity-associated changes in hemodynamics is the rising LV wall stress and tension. In normotensive obesity, elevated central blood volume, stroke volume, and CO are the main driving forces for LV wall stress, which predisposes LV dilatation, and eccentric hypertrophy (FIGURE 3). Eccentric LV hypertrophy (discussed in much more detail in sect. 2.2) is likely a compensatory machinery for elevated LV wall stress and contributes to diastolic dysfunction in obesity. Systolic dysfunction may occur latter as a result of excessive wall stress if LV wall thickening fails to keep pace with chamber dilatation (236). In response to elevated tension development during systole, myocardial fibers thicken (LV hypertrophy) to maintain systolic stress at a normal range. To the contrary, elevation in resting or diastolic wall stress or tension gradually lengthens myocardial fiber, which should improve the work efficiency of ventricular chambers although it may not normalize or compensate diastolic wall stress in obese individuals (237).
2.1.5. Pulmonary hemodynamics.
Uncorrected obesity is a common comorbidity for pulmonary hypertension (PH) and influences the severity of pulmonary arterial hypertension (PAH), commonly acknowledged as primary pulmonary hypertension. A close relationship between obesity and PH is has been shown by echocardiography-based and invasive hemodynamic measurements (127). Apart from epidemiological evidence, several groups have revealed a link between obesity-related metabolic defects and pulmonary vascular remodeling utilizing various genetic and diet-induced models (238, 239). The precise pathogenic cue for obesity-induced PH is complex and undefined. Obesity exerts pathologic impact on systemic circulation through external environment of pulmonary vascular cells. With the increasingly recognized role for perivascular adipose tissue (PVAT) in CVD, adipocyte defect in obesity leads to abrupted release of endocrine and paracrine adipokines (240). Leptin and adiponectin denote two most prevalent adipokines in the pathogenesis of PH in overweight/obese subjects involving endothelial dysfunction (241, 242). Leptin-deficient (ob/ob) mice spontaneously develop PAH and pulmonary vascular remodeling (238). In addition, estrogen and its metabolites are deemed risk factors for the etiology of PAH as the effects of leptin deletion are attenuated by inhibition of endogenous estrogen production (238). It is perceived that obesity-evoked systemic and local inflammatory responses, such as inflammation, oxidative stress, PVAT expansion, and insulin resistance, all contribute to irreversible maladaptive pulmonary vasculature remodeling, thus fostering the onset and progression of PH (243).
A number of secondary factors may also contribute to the etiology of PH in obese individuals including increased LV filling pressure and pulmonary capillary wedge pressure (PCWP) associated with LV failure (244). In fact, elevated pulmonary BP due to left heart disease is perhaps the most common type of PH. Increased LV filling pressure greatly jacks up pulmonary venous pressure and PCWP, resulting in elevated pulmonary artery pressure and right ventricular (RV) end-diastolic and right atrial pressures, the process of which is aggravated by pathological stress such as sleep apnea and obesity hypoventilation (238). Furthermore, hypoxic vasoconstriction and subsequent pulmonary arteriolar remodeling as a result of repetitive nocturnal hypoxemia may prompt right-ventricular hypertrophy and PH (245, 246). Moreover, obesity-associated chronic hyperuricemia, an independent risk factor for PH, reduces local flow within pulmonary vessels, possibly related to lowered nitric oxide production and elevated endothelin, resulting in endothelial dysfunction and ultimately rises in pulmonary artery pressures (247, 248).
2.2. Structural Changes in Obesity
2.2.1. Measurements.
2.2.1.1. echocardiography.
Obesity leads to unfavorable changes in cardiac structure and function. With the use of echocardiography, magnetic resonance imaging (MRI), and radionucleotide, derangement in LV structure and function is noted in obesity, such as concentric remodeling and compromised diastolic and systolic function (249, 250). Moreover, left atrial (LA) enlargement is frequently noted in obesity as well (251). Two-dimensional echocardiography and tissue doppler imaging are employed to assess LV structure, myocardial systolic and diastolic function. This approach revealed much higher LV structure such as posterior and septal wall thickness, LV mass (LVM), the LVM/height and the relative wall thickness (RWT), in obese (including young, otherwise-healthy women) than nonobese individuals (252). However, LVM index (g/m2) remained essentially unchanged in obesity.
2.2.1.2. magnetic resonance imaging.
With the use of cardiac MRI, a modality, which offers significant advantages over two-dimensional (2-D) echocardiography in estimating LV mass and volume, LV mass and end-diastolic volume are shown to be positively associated with obesity severity in men and women (253). Although MRI seems to offer a more accurate and reliable measurement of LVM (254), echocardiography is still more frequently employed in the clinics due to its noninvasive nature and moderate cost
2.2.1.3. positron emission tomography.
Cardiac rubidium 82 (Rb-82) positron emission tomography (PET) has been employed for evaluation of myocardial perfusion imaging in the clinical setting courtesy of its diagnostic accuracy and low risk of radiation exposure. In obese individuals who are usually prone to soft tissue attenuation artifact with poor acoustic echocardiogram ranges, cardiac PET perfusion imaging offers a high prognostic value irrespective of BMI (255).
2.2.2. LV remodeling.
LV remodeling refers to changes in the size, shape, or structure of LV chamber and occurs more frequently in obesity (256). The Bogalusa heart study revealed an underlying role for obesity in LV hypertrophy and remodeling (257, 258). Indeed, ample evidence has suggested a positive correlation between BMI and LV mass (259) (TABLE 1). Obese subjects usually possess a high risk of LV hypertrophy (LVH) defined by increased ventricular mass. The prevalence of LV hypertrophy in obese normotensive subjects was ∼14%, much higher than lean counterparts (5%) (260). The occurrence rate of LV hypertrophy can be up to 78% in morbid obese individuals (261), including adverse changes in LV mass, volume, geometry, and composition, ultimately resulting in impaired LV function and cardiomyopathy (256). Severe obesity and hypertension frequently coexist and impose an additive impact on LV hypertrophy (94, 262).
2.2.2.1. changes in lv mass and volume.
LV remodeling is characterized by overt changes in cardiac chamber diameter, wall thickness, volume, mass, and LV ejection fraction (EF) using imaging techniques (263). Multiple factors may promote rises in LV mass in obese individuals, with a clear positive relationship between LV mass (LVM) and seriousness of obesity. LV diastolic chamber size (diameter or volume) is also used to evaluate cardiac remodeling in obese subjects (256). LV end-diastolic volume (LVEDV) indexed to BSA is commonly employed to measure LV volume. LVEDV has been related to BSA to a power of 1.5 (264).
Physiological LV remodeling, such as exercise-induced elevation in LV end diastolic dimension (LVEDD) and LV mass, normally reverses with the cessation of physiological stimuli (e.g., exercise training). However, obesity-induced pathological remodeling seems to progress continuously and irreversibly, denoting an adverse clinical prognosis. In routine practice and clinical studies, LVM was calculated from LVEDD and interventricular septum and posterior wall thickness using echocardiography and cardiac magnetic resonance, according to the Devereux’s formula.
Although ventricular weight is typically normalized to body weight in experimental research, overweight subjects have lower ventricular mass/body weight ratio, yielding an artificial difference as a result of the increased denominator in obesity (265). Recent observations showed that patients with obesity displayed a higher LV mass when appropriately indexed to height2.7(266). LV mass indexed to height2.7 is more appropriately employed to avoid misleading predictions of CVD risk in obesity. Classical assessment or indexation of LV mass may underestimate or overestimate the degree of hypertrophy in obese adults. Bioelectric impedance analysis (BIA) is used to yield more precise measurements of overweight and obesity than BMI. The Strong Heart Study (SHS) has demonstrated that fat-free body mass (FFM), calculated using BIA, is the main variable determining levels of LV mass in obesity, instead of adipose mass, the waist/hip ratio, height, or height2.7 (267). Changes in LVM display a much tighter association with FFM, suggesting the utility of FFM as a more refined parameter for normalization of LV mass (268).
2.2.2.2. four-tiered classification of lv geometry.
LV hypertrophy (LVH) in obesity is typically categorized into concentric or eccentric based on RWT measured by echocardiography. However, in the traditional classification, eccentric LVH was considered a lower risk profile compared with concentric LVH, mostly as a result of indeterminate hypertrophy (269, 270). Tantiore, based on whether or not LV concentricity (measured by LV mass/LVEDV0.67) and LVEDV are increased, cardiac magnetic resonance is used to refine the classification of LVH into a four geometric patterns as 1) “thick hypertrophy” (increased concentricity without increased EDV); 2) “dilated hypertrophy” (increased EDV without increased concentricity); 3) “thick and dilated hypertrophy” (increased concentricity with increased EDV); and 4) “indeterminate hypertrophy” (increased LVM with neither increased concentricity nor EDV) (271).
Previous research has shown the presence of concentric LV remodeling associated with decreased systolic and diastolic function in young otherwise-healthy obese women (249). A recent follow-up study included 1,699 cases of obesity cardiomyopathy with an incidence of 5.9 per 100,000 observation years. Among these, 481 were classified dilated cardiomyopathy, 246 cases were hypertrophic cardiomyopathy, 61 individuals met the criteria of alcohol/drug-induced cardiomyopathy, and 509 exhibited other forms of cardiomyopathies. Increased BMI significantly escalated the risk of these types of cardiomyopathies especially dilated cardiomyopathy, with a fivefold increase in the risk of cardiomyopathy in those with severe obesity (116) (FIGURE 2).
2.2.2.3. other factors.
Obstructive sleep apnea (OSA) also serves as an independent factor of high LV mass index and abnormal LV geometry in obesity, likely due to increased blood pressure, heart rate, intermittent hypoxia, sympathetic tone, and negative intrathoracic pressure during airway obstruction (272). It seems that degree of sustained hypoxemia, rather than the number of apneic and hypopneic episodes, contributes to the development of LV hypertrophy (261, 273). Obese individuals with OSA displayed increased left atrial volume index, which predisposes them to atrial fibrillation and HF (274). Additionally, the increased presence of hypertension, especially resistant hypertension, in patients with OSA is also tied with LV hypertrophy or increased wall thickness (261).
Insulin resistance, a hallmark of obesity, also participates in the pathogenesis of LV hypertrophy and diastolic dysfunction in obesity. Furthermore, the impact of insulin resistance on LV remodeling and function may be influenced by sex and BMI. In one study, insulin resistance was associated with increased LV mass and LV wall thickness in obese women but not men (275). Indeed, obese women exhibit both eccentric and concentric hypertrophy, whereas obese male counterparts predominantly display concentric hypertrophy. It is well known that concentric hypertrophy serves as a much stronger predictive of CVD mortality compared with eccentric hypertrophy (276). Impaired insulin metabolic signaling plays a predominant role in insulin resistance-elicited LV dysfunction (277). The existence of impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) predisposes the development of LV hypertrophy in normotensive individuals (278).
2.2.3. LA remodeling.
Obesity is often joined with atrial electrostructural remodeling, including alterations in atrial size, conduction, histology (lipodosis), and levels of profibrotic and proinflammatory mediators (279). The accepted methods to evaluate LA size include uniaxial anterior-posterior dimension and LA volume indexed to body surface area or height (280). LA volume is progressively enlarged and corelates well with LV mass (weakly with blood pressure) in obesity (281). However, the reported prevalence of LA enlargement in obese individuals is somewhat variable. This is probably related to the presence of obesity paradox, duration of obesity, the presence of comorbidities, and different methods utilized for body size normalization. For example, the relationship of obesity to LA enlargement is confounded by the existence of hypertension. Both BMI and BP are risk factors for LA enlargement although BP displays a weaker correlation (282). According to a 10-yr longitudinal study, obesity imposes a stronger clinical relevance than hypertension with respect to LA enlargement (283). Of note, atrial remodeling secondary to obesity is characterized by progressive impairment of atrial structure, electrophysiological function and electro-anatomical integrity, leading to a proarrhythmic status (284).
2.2.4. RV hypertrophy.
Obesity serves as an independent risk factor for RV hypertrophy. Obese individuals display a higher RV mass and larger RV end-diastolic volumes (RVEDV) compared with lean counterparts (76). Indeed, RV mass was 6% and 14% greater, respectively, in overweight and obese groups. These authors noted that every 5 kg/m2 rise in BMI was associated with a 1.3 g higher RV mass and 8.65 mL higher RVEDV. A significant correlation was identified between BMI and RV mass or RVEDV (285). Assessment of RV wall thickness using echocardiography in normal, overweight, and obese participants has revealed much higher RV free wall thickness in overweight (37 ± 6 mm) and obese (38 ± 5 mm for BMI of 30–34.9; 49 ± 9 mm for BMI ≥35) participants compared with lean (33 ± 6 mm) participants (286).
Factors indicating in RV hypertrophy include OSA and chronic PAH. One study revealed that high-respiratory disturbance index (RDI) is correlated with higher BMI than low-RDI (190). Simultaneously, a significant difference was noted in RV wall thickness between high RDI and low-RDI groups, the effect of which was independent of hypertension and pulmonary function. These findings convincingly support the notion that obstructive sleep apnea might be a link between obesity and RV hypertrophy (287).
Chronic PAH may serve as another independent factor for RV hypertrophy. RV diastolic stiffness is elevated by PAH and is associated with RV disease severity. Histological analyses revealed elevated cardiomyocyte number and RV fibrosis in PAH patients. In addition, RV diastolic stiffness and passive tension at different sarcomere lengths were overtly increased in PAH patients. At the molecular level, phosphorylation of sarcomeric protein titin, an essential governor of sarcomeric stiffening, was overtly lower in RV tissues from PAH group. In this context, increased RV fibrosis may serve as a compensatory mechanism to combat the elevated RV afterload in obesity (288).
2.2.5. Aortic valve stenosis.
Earlier studies have shown conflicting results with regards to the relationship between BMI and aortic valve stenosis (AVS) (289–291). However, recent large cohort observational studies have depicted an association between increased adiposity and AVS risk (292, 293). A Swedish study including 71,817 individuals found a positive relationship between obesity and risk of AVS. Obesity with a BMI ≥30 had an 80% higher risk of AVS. Abdominal obesity (waist circumference ≥102 cm for men and ≥88 cm for women) was tightly correlated with a 30% higher occurrence rate of AVS (292). Later the Denmark group including 108,304 individuals revealed similar results with risk of AVS and aortic valve replacement much greater those with both high BMI and high waist-hip ratio or waist circumference (293). At this point, the precise mechanism behind AVS risk in obesity is unclear although structural or metabolic changes in the heart may play a role. Obesity leads to higher blood pressure that may impose geometric changes on LV and aortic valve (294) as well as damage to endothelial cells (295). Metabolically, elevated plasma lipids lead to lipid deposits on aortic valve leaflets and provoke valvular interstitial damage (295, 296). At the same time, lipid deposition activates inflammatory responses, resulting in differentiation of valvular interstitial cells into osteoblasts and thus calcification of valve leaflets (297).
2.3. Cardiac Dysfunction in Obesity
2.3.1. LV systolic dysfunction in obesity.
Conventional 2-D or M-mode echocardiography is classified as the most common technique for measurement of systolic function by examining stroke volume, ejection fraction, transmitral velocity and fractional shortening. LV EF is reported to be normal (253) or even supranormal in obesity-induced cardiac remodeling (TABLE 1), which also underpins the high prevalence of HF with preserved ejection fraction (HFpEF) in obese patients (115). However, increased stroke volume (SV) has been observed in overweight and obese patients, in parallel with increasing levels of fat free body mass (FFM) (191) and CFD (192, 298), indicating elevated systolic load in preclinical obesity cardiomyopathy. Fractional shortening (FS) evaluates circumferential LV myocardial contractility by measuring the percentage change in LV diameter during systole. LV midwall fractional shortening is believed to be more suitable than LV endocardial fractional shortening for the assessment of regional systolic function in LV hypertrophy, particularly concentric geometry. Most studies have demonstrated a slight (261) to moderate (299) decrease of LV midwall factional shortening accompanied by LV hypertrophy in obesity, indicating reduced systolic contraction. To this point, gastric bypass surgery was shown to effectively reverse LV remodeling along with improved midwall shortening (300).
Notably, ample observations have substantiated subclinical depressed LV systolic function in obesity cardiomyopathy. Although EF can be normal even in severe obesity, longstanding obesity for more than 20 yr is associated with overtly impaired LV systolic and diastolic function (301). Accurate measurements using more sensitive modalities have revealed subclinical systolic dysfunction associated with obesity, as evidenced by proportionally decreased myocardial tissue velocity and strain index with escalating severities of obesity. Myocardial strain typically refers to load-dependent deformation (shortening, lengthening, or thickening) of the myocardium, which is estimated as the ratio of the distances between two points during expansion and contraction (302). Myocardial strain has shown prognostic significance courtesy of the ability to identify subclinical abnormalities in LV and RV function before onset of overt cardiomyopathy and HF (286, 303). This is evidenced by the biventricular strain abnormities that are already present in obese children (302, 304, 305).
Tissue Doppler imaging (TDI), a new echocardiographic modality that detects low-velocity, high-amplitude myocardial motion, has emerged as one of the most widely applied noninvasive tools for the quantitative assessment of myocardial systolic and diastolic function. The TDI-derived parameters of myocardial velocities, including myocardial systolic velocity (Sm, Sa), early diastolic velocity (Em, Ea), late diastolic myocardial velocity (Am), and LV diastolic pressure (E/Em, E/Ea) are already reduced at the preclinical or early stage of cardiomyopathy in obesity (249, 250) and drop further with increased adiposity (249). Tissue Doppler myocardial strain and strain rate imaging are also employed to decipher subclinical cardiac outcomes of isolated obesity. Subclinical cardiac manifestations shown by decreased LV global longitudinal peak strain rate are correlated with BMI, duration of obesity, and increasing age (303). Patients with increasing degree of obesity show diminished myocardial velocity, and strain index, in light of normal ranges of ejection fraction from conventional 2-D echo measurement. These observations indicate diminished LV systolic function even in patients with mild obesity (250).
However, strain measurements based on TDI are angle dependent with the use of the Doppler device which produces simultaneous opposite deformations in the long and short axes. Whereas speckle tracking echocardiography offers more precise and angle-independent readouts of LV dimensions and strains. Speckle tracking relies solely on tracking of characteristic speckle patterns generated by interference of ultrasound beams with myocardial tissues (306). There is evidence suggesting that subclinical LV systolic dysfunction assessed by speckle-tracking global longitudinal strain is associated with abdominal adiposity but not BMI (307).
Another LV derangement found in obesity using speckle tracking echocardiography or MRI is increased LV torsion and untwisting rate. Whether altered LV rotational function is a compensatory machinery for compromised LV contractility or an outcome of insufficient diastolic filling requires further investigation. Systolic torsion and diastolic untwisting are found elevated in the early phases of diastolic dysfunction and are then normalized or reduced in the advanced stage (308). These emerging techniques should help to define earlier cardiac abnormalities associated with overweight and obesity.
2.3.2. LV diastolic dysfunction in obesity.
Diastolic dysfunction develops either alone or in concert with systolic dysfunction and typically precedes the onset of systolic failure (as shown in multiple clinical studies listed in TABLE 1). The classic diastolic dysfunction, which denotes abnormalities in relaxation or dampened myocardial compliance, or both, is featured by a higher impedance to LV filling, resulting in an inappropriately elevated diastolic pressure (309). A cross-sectional survey of 1,275 individuals aged 60 to 86 yr found that LV diastolic dysfunction is a common situation among the elderly, although EF is preserved in this setting (270). As it is the case with systolic dysfunction, comprehensive studies using TDI and other techniques have been performed to examine diastolic function in obesity. Current consensus recommends the application of early (E) to late (A) diastolic transmitral flow velocity (E/A) as a surrogate for diastolic function and E to early diastolic mitral annular tissue velocity (E/e’) as a more sensitive indicator for LV filling pressures (309). In fact, multivariate analyses demonstrated that BMI is independently associated with higher E, A, and E/e’ and even overweight is linked to diastolic dysfunction, as evidenced by reduced E’ and higher E/e’ in the overweight subjects (310). Obesity in young otherwise-healthy women was found to contribute to concentric LV remodeling and impaired LV relaxation as evidenced by decreased early diastolic myocardial velocity (Em) (249). In addition, hemodynamic data obtained from invasive studies showed that obesity is associated increased LV end-diastolic pressure (298). LV diastolic anomalies appear to be frequent in obese individuals without any clinically evident heart diseases triggered by diabetes mellitus, hypertension, or coronary artery disease.
2.3.3. RV dysfunction in obesity.
Obesity not only leads to RV hypertrophy but also RV dysfunction. Larger RV stroke volume (RVSV) and lower RV ejection fraction (RVEF) are noted in overweight and obese individuals after adjusting for demographics, height, education, and CVD risk factors. Even after adjustment of LV parameters, differences still prevailed. RVSV was increased in overweight and obese individuals. RVEF was mildly but notably lower in overweight and obese individuals (285).
Finally, overtly decreased diastolic and systolic velocities of RV free wall motion were noted in overweight and obese individuals in comparison with lean subjects. Meanwhile, RV systolic and diastolic velocities as well as strain indexes were significantly depressed in overweight and obese individuals in comparison with controls. Alongside with increased BMI, the degree of decline of RV function and strain rate were progressively worsened (286).
2.3.4. Atrial fibrillation.
AF is yet another severe malady commonly seen in obesity. Although the precise etiological nature of obesity-induced atrial arrhythmias remains elusive, LA remodeling manifested by an increased LA dimension may be a key decisive factor. Obesity predisposes to the onset of AF mainly through LA structural and electrophysiological changes, elevated BP, LV hypertrophy, and LV diastolic dysfunction (142, 284). Large ambispective cohort and longitudinal obese ovine findings have revealed that obesity early on in life and progressive weight gain are closely associated with more episodes, prolongation, and greater cumulative duration of AF (311, 312). In a 21-yr cohort study involving 3,248 patients with paroxysmal AF, greater BMI paralleled larger LA volume and predicted incremental progression to permanent AF (313). Based on the electrophysiological findings in LA and pulmonary vein from obese individuals, decreased posterior LA voltage and shortened or unchanged effective refractory period in LA and PVs were found to coincide with elevated LA pressure, LA volume and lower LA strain (314). Infiltration of LA epicardial fat may also serve as a unique substrate for AF in obesity (312). Epidemiological evidence indicates that weight loss greatly lowers AF burden and recurrence incidence following pharmacological treatment in obese patients (163). The Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) study denoted one of the largest multicenter clinical trials for AF, involving 4,060 patients. Although earlier reports noted an association between obesity and higher risk of AF, multivariate analysis from AFFIRM study found an interesting “obesity paradox” for AF outcomes, where overweight and obesity were in fact related to a decreased all-cause mortality (315).
2.3.5. Clinical HF and obesity.
Uncorrected obesity is a major culprit factor for heart failure (HF), independent of other CVD risk factors, including diabetes mellitus, ischemic heart diseases, dyslipidemia and hypertension. In the international cohort study of international Sarcomeric Human Cardiomyopathy Registry (SHARE) with a median follow-up of 6.8 yr, BMI >30 was independently correlated with HF and arrhythmias irrespective of genotype, age and sex (155). Clinical manifestation and pathophysiology are often redundant between obesity and HF, including decreased LV function, cardiac remodeling, neurohormonal activation (sympathetic nervous system and RAAS). Obese patients are commonly characterized by reduced exercise capacity, increased cardiac filling pressure, Na retention, plasma volume expansion, and a normal LV ejection fraction. These individuals exhibit one of the three following phenotypes. First, HF in obesity occurs in association with hypervolemia, including Na retention, plasma volume expansion, and cardiac enlargement, although cardiac index and circulating levels of natriuretic peptides are not significantly elevated. It is believed that RAAS activation and increased aldosterone levels may underscore hypervolemia in obesity (175, 185). In addition, increased leptin levels directly activate both RAAS and sympathetic nervous system (179, 185). Second, elevated natriuretic peptides in obesity lower systemic vascular resistance, leading to LV dilatation, increased RV and LV filling pressures, resulting in high-output HF and glomerular hyperfiltration (188). This is consistent with increased LV end-diastolic pressure in obesity, indicative of diastolic dysfunction (95). LV and possibly RV dilation are likely a result of increased CO in obesity. In subpopulations of obese patients, elevation of natriuretic peptides may contribute to decreased systemic vascular resistance and elevated CO (175, 316). Endogenous natriuretic peptides are normally constrained by neprilysin produced by adipocytes (317). However, the release of natriuretic peptides greatly exceeds the degradative capacity of adipocyte-related neprilysin with continuous stretch of ventricles, resulting in systemic vasodilatation and high output HF (188). In addition, increased natriuretic peptides may result in glomerular hyperfiltration due to low afferent arteriolar resistance in obesity-related high output HF (318). Third, it has been noted that older female obese patients often exhibit HFpEF with modestly elevated ventricular dimensions, frequent AF, increased natriuretic peptides and glomerular malfunction and plasma volume expansion reminiscent of high output HF (183). As the LV dilates wall stress rises thereby prompting secondary eccentric hypertrophy. With proportionated LV hypertrophy in response to ventricular dilation, systolic function remains preserved (due to normalized wall stress), resulting in the onset of HFpEF. If LV hypertrophy is unable to keep pace with dilation (inadequate hypertrophy), LV wall stress overtly rises to promote systolic dysfunction (319, 320). In this context, obesity is deemed a primary factor for the etiology of HFpEF (321, 322). To distinguish among these three phenotypes and optimize therapeutic interventions in obese individuals, several key parameters may be used including exercise intolerance, rises in ventricular filling pressures, and LV ejection fraction. In particular, recognition of HF in obesity is urgently needed to initiate large-scale clinical trials enrolling obese patients with various forms of HF.
2.4. Obesity Paradox
Emerging evidence suggests that overweight or obesity is associated with an improved survival in patients afflicted with CVD (175, 323, 324). In general, HF patients who are underweight display the highest cardiovascular mortality and hospitalization prevalence, as opposed to those who are overweight or obese (324, 325). This is profoundly noted in epidemiological findings with a better prognosis in obese patients with HF, a phenomenon termed “obesity paradox” (167). Not only does obesity paradox exists in HF, intervention cardiac procedure such as percutaneous coronary intervention (PCI) also exhibits a much lower mortality rate in overweight and obese patients in comparison with lean subjects (166).
2.4.1. Potential rationales underlying obesity paradox.
Several scenarios may be considered for obesity paradox. First, the severity of HF may be overestimated in obese patients because of the concurrent comorbidities such as dyspnea (171). Second, nearly all available data concerning “obesity paradox” at this point use BMI as the gold standard to categorize obesity. In this regard, 60,335 participants were followed up for 10 yr comparing BMI with body composition indices as predictors of CVD death. The results yielded a stronger association between BMI and CVD mortality (326). However, compared with other adiposity measures, BMI does not accurately reflect various cardiometabolic risks in obesity since it cannot distinguish fat mass from fat-free mass (327, 328). Lower FFM is pertinent to increased risk of death in the lower BMI range (329, 330). Measurement for abdominal obesity using waist-to-height ratio (WHR) and waist circumference (WC) should better pinpoint fat mass and decipher cardiometabolic risks associated with obesity (331). Independent of BMI, abdominal obesity is associated with impaired LV contractile and diastolic function and associated with higher mortality risk in adults (251, 307, 332). Furthermore, increased pericardial fat is related to the pathogenesis of obesity-related CVDs and displays a stronger correlation with heart structure and function than the more general obesity indicators (333).
2.4.2. Other risk factors of CVD outcomes in obesity.
Obesity is often a product of intertwined nutritional and lifestyle risk factors including smoking (334). A population-based study noted escalating prevalence and mortality of CVD with increasing BMI in diabetic patients following exclusion of smoking, poor metabolic control, and short duration of follow-up (126). The relationship between BMI and CVD incidence is more linear in subgroups without any comorbidities or smoking (335), whereas the increased risk of death in underweight (J-shape association) is apparent in ever smokers in a subgroup analysis of smoking status (336). Sex differences also impact the correlation between obesity and CVD outcome. For instance, increased abdominal fat, assessed by WHR, seems to be tied with a higher mortality risk in female but not male HF patients (160). Cardiorespiratory fitness (CRF) is a measure of how well the lungs and CV system perform during physical activity (168). High CRF has long been considered a predictor of lower CVD risks and better prognosis (168, 337), while low CRF results in greater impairment in LV strain (338) and chronic disability due to CVD (339). The obesity paradox was less pronounced among diabetes patients with high CRF, although mortality risk decreased with increasing BMI in patients with low CRF (173).
Furthermore, emerging evidence has started to shed light on the existence of the heterogeneity in obesity as defined by BMI. The metabolically healthy obese (MHO) refers to an obesity trait in the absence of dyslipidemia, insulin resistance, hypertension, diabetes mellitus, and any of the classical cardiometabolic factors, conferring less prevalence for CVD (340). However, previous studies have provided inconsistent results about the association of MHO and CVD. In fact, a large proportion of MHO may be converted to an unhealthy phenotype over time with an associated higher CVD incidence compared with a normal-weight group (341). Therefore, MHO should not be considered as healthy, as existing clinical approaches can hardly identify a “stably” benign obese subgroup (342).
3. BASIC MECHANISM OF OBESITY CARDIOMYOPATHY
3.1. Adipose Tissue Dysfunction and Inflammation
Obesity is characterized by chronic activation of inflammation, commonly associated with and a contributing factor for insulin resistance and T2D (104, 343, 344). Ample recent evidence has depicted an important role for dysfunctional adipose tissues in inflammation, insulin resistance and cardiac abnormalities. Obesity is a progressive pathological process whereby adipocytes and resident immune cells are activated and subsequently release vast secretory factors. In addition, various immune cells are recruited to adipose tissues, where they are converted into an active inflammatory phenotype to produce and release proinflammatory cytokines (343). Ample evidence has revealed an essential role for alterations in these adipose tissue-released factors, termed adipocytokines, including adiponectin, leptin, resistin, nitric oxide, interleukins, tumor necrosis factor-α (TNF-α), and other inflammatory mediators in the development of CVD via an autocrine, paracrine or endocrine fashion (FIGURE 4). Other local and systemic influences, such as insulin resistance (IR), renin-angiotensin-aldosterone system (RAAS) activation, lipotoxicity, and interstitial fibrosis, may act in synergy with adipocytokines in the onset and development of CVD.
FIGURE 4.
Adipose tissue dysfunction and inflammation in obesity that directly and indirectly aggravates cardiomyopathy: In lean state, adipocytes secrete various endocrine factors that maintain metabolic homeostasis. In response to chronic energy excess, infiltration of proinflammatory immune cells and hypertrophic adipose expansion along with a lack of adipogenesis can be observed in adipose tissues, which is accompanied by altered secretion of adipose tissue hormones, cytokines, metabolites, and exosomal miRNAs. Overall, the changes in hypertrophic adipose tissue contribute to insulin resistance, impaired glucose, and lipid metabolism and low-grade systemic inflammation and exert local effects that exacerbate cardiomyopathy in obesity. IL-10, -13, -4, interleukin 10, 13, 4; SFRP5, secreted frizzled-related protein 5; TNFα, tumor necrosis factor-α; IL-6, -1β, interleukin 6, 1β; IFN-γ, interferon γ; MCP1, monocyte chemoattractant protein 1; CXCL5, C-X-C motif chemokine ligand 5; FFAs, free fatty acids; SCFAs, short-chain fatty acids; FAHFAs, fatty acid esters of hydroxy fatty acids; 12,13-DHOME, 12,13-dihydroxy-(9Z)-octadecenoic acid; BCAAs, branched-chain amino acids. Created with BioRender.com.
3.1.1. Ectopic adipose tissue in obesity cardiomyopathy.
There exist two main kinds of adipose tissues, white adipose tissue (WAT) and brown adipose tissue (BAT), among which WAT is predominant in mammals while BAT resides in several regions such as interscapular and supraclavicular areas (345). Adipose tissues can also be divided into different anatomical depots, VAT, and subcutaneous adipose tissue (SAT) (346). Adipose tissue dysfunction, especially WAT dysfunction, plays an imperative role in the pathogenesis of various metabolic diseases and CVDs (133).
3.1.1.1. pathological features of adipose tissue in obesity.
The expansion of adipose tissue includes hyperplasia/adipogenesis (new adipocytes differentiated from precursor cells) and hypertrophy (the increase in adipocyte size) (347).
Interestingly, pathological expansion of adipose tissue in obesity is accompanied with a decline of hyperplasia and adipogenesis (347, 348). Subcutaneous adipose tissue (SAT) serves as the largest reservoir for lipid storage that prevent excess lipids accumulating in peripheral organs such as liver, heart, and muscles in physiological conditions. The limited expansion and dysfunction of SAT due to impaired adipogenesis lead to a hypertrophic expansion of adipose cells and increased fibrosis in adipose tissue (348–350). Hypertrophic adipose depots present different biochemical properties, including elevated lipolysis, increased secretion of inflammatory cytokines, and reduced secretion of anti-inflammatory adipokines (351, 352). Lipotoxicity caused by excessive hypertrophic fat accumulation leads to insulin resistance and functional deficits in both adipose tissue and other organs including liver, heart, muscle, and pancreas (164, 344, 353, 354).
Insulin resistance is regarded as a springboard linking adipose tissue accumulation in obesity to CVD. Although inflammation of adipose tissues is required for physiological expansion of adipose tissues in healthy individuals, evidence from both clinical and experimental studies strongly suggests an important relationship between systemic inflammation caused by maladaptive adipose tissues and insulin resistance in obesity (355).
3.1.1.2. ectopic fat deposition.
As SAT fails to store excess fat, lipids accumulate in visceral adipose tissues and other tissues, which would normally contain only small fractions of fat, including liver, skeletal muscle, heart, and pancreas (356). Certain ectopic fat depots, such as vascular adipose tissue (357), intrahepatic fat, and intramuscular fat, predominantly generate systemic effects (358), while adipose tissue surrounding heart [epicardial adipose tissue (EAT) and paracardial adipose tissue (PAT)] and most of the blood vessels (PVAT) are pertinent to adverse local CV effects (357, 359).
Adipose tissues surrounding the heart are classified into two layers: epicardial adipose tissue (EAT) and paracardial adipose tissue (PAT) based on anatomic location. EAT is the fat depot between myocardium and pericardial visceral layer and is anatomically and functionally contiguous with myocardium (360). Paracardial fat is located outside of the visceral pericardium. Perivascular adipose tissue (PVAT) refers to adipose tissue surrounding blood vessels, which plays a key role in the maintenance of vascular function. PVAT normally possesses antiatherogenic functions by secreting biologically active factors (361, 362). However, PVAT becomes dysfunctional in obesity with an increased level of proinflammatory adipokine secretion, which induces oxidative stress in vessels and leads to endothelial dysfunction, impaired vasodilation, and stiffening of vessels. All of these events may lead to vascular dysfunction and CVD (361, 363).
3.1.2. Local effects of EAT.
EAT is a marker of visceral adiposity and cardiac lipotoxicity and sets the stage for cardiac dysfunction in adiposity by promoting inflammation and metabolic disorders in the heart (364, 365). Epicardial fat directly exerts local effects on cardiac structure and function, including elevated LV mass, deranged RV geometry, and impaired relaxation (366, 367). Of note, therapeutic interventions targeting adipokines greatly reduce the risk of heart failure, while drugs promoting accumulation of epicardial adipocytes or inflammation may exacerbate CVD, including AF, coronary artery disease, and HF (365, 368).
Both mechanical and biomolecular factors may underlie the impact of EAT on heart structure and function (367). First, increased EAT puts additional mechanical stress on both ventricles and increases cardiac workload, leading to LV hypertrophy. In addition, increased EAT also exaggerates atrial enlargement and ventricular diastolic dysfunction owing to physical obstruction of cardiac filling. Second, infiltration of adipocytes from EAT to the myocardium facilitates aberrant cardiac metabolism by promoting excessive myocardial FFA usage and development of lipotoxicity. Third, in obesity, EAT is enriched with a transcriptome associated with genes governing inflammation, and endothelial dysfunction (364, 369). EAT serves as a local transducer of systemic inflammation to the myocardium and a source of local secretion of proinflammatory adipocytokines that promotes myocardial disarray, cardiac fibrosis, and stiffness in uncorrected obesity. As the metabolic profile shifts in obesity and EAT abnormally expands, both resident and infiltrated immune cells generate a proinflammatory microenvironment with cardiomyocytes, which influents cardiac metabolism and contractility (344, 360). In addition, increases in EAT was shown to elicit local electromechanical responses in atrial tissues though released cytokines, such as TNF and IL-6, as well as FFAs.
3.1.3. Systemic effects of adipose tissue.
Adipose tissue is considered an endocrine organ and an “ancestral immune organ” (110, 370, 371). Generally speaking, adipokines are hormones, metabolites, exosomal microRNAs, cytokines and chemokines secreted by adipose tissue and sent to the targets in other organs, such as adiponectin, leptin, resistin, FFA, transforming growth factor-β (TGF-β), interleukins, fibroblast growth factor 21 (FGF21), bone morphogenetic protein (BMP)-4, BMP-7, and many others (370, 372). Obesity is characterized by a decrease in anti-inflammatory adipokines and an increase in proinflammatory ones. This shift impacts on multiple functions such as appetite, energy balance, endothelial function, and immunity, promoting the development of chronic, systemic inflammation, and insulin resistance, processes that are thought central to the obesity-associated CVD (344, 373).
3.1.3.1. dysregulation of adipokines.
3.1.3.1.1. Decreased anti-inflammatory adipokines.
Adiponectin is one of the principal adipokines with insulin-sensitizing and anti-inflammatory properties. Plasma levels of adiponectin are declined in obesity (374–377). In a study of 933 middle‐aged subjects, low plasma adiponectin levels were independently correlated with increased LV hypertrophy (378). Adiponectin evokes cardioprotection against pathological cardiac remodeling and ischemia injuries partially through reducing myocardial oxidative stress, suppressing inflammation, and improving energy supply (379). Although clinical studies are limited, these cardioprotective effects are potentially translational.
Secretion of cardiac FGF21 is increased in response to obesity to evoke cardioprotection (380), while FGF21 deficiency predisposes the susceptibility of obesity-related cardiomyopathy in mice (381). Interleukin 10 (IL-10) is generally considered as an anti-inflammatory cytokine. As IL-10 levels are declined in obesity, systemic IL-10 administration was found to markedly ameliorate LA remodeling and vulnerability to AF in diet-induced obesity (382). Interleukin 33 (IL-33), another anti-inflammatory adipokine from the IL-1 family, participates in type 2-like immune responses (383, 384). Levels of IL-33 were suppressed in obesity and were correlated with natriuretic peptides in hearts (385). Secreted frizzled-related protein 5 (SFRP5) is another anti-inflammatory adipokine secreted from WAT and suppresses inflammation in WAT (386, 387). FAM19A5, a novel adipokine, inhibits neointimal formation through sphingosine-1-phosphate receptor 2-G12/13-RhoA signaling (388, 389).
3.1.3.1.2. Increased proinflammatory adipokines.
Dysfunctional adipose tissue in obesity releases an abundance of proinflammation factors, including leptin, resistin, chemokine, and cytokines such as transforming growth factor-α (TNF-α), interleukin-6 (IL-6), IL-1, and monocyte chemoattractant protein-1 (MCP-1) (390).
Leptin, a 16-kDa peptide hormone product of the ob gene, is one of the proinflammatory adipokines secreted by adipocytes (391). The level of leptin is elevated in obesity to coincide with cardiac hypertrophy through binding of leptin to the short form leptin receptor in rat hearts (392). The adiponectin/leptin ratio is a marker for adipose tissue status, exhibiting an inverse correlation with low-grade chronic inflammation (393). Leptin deficiency mitigates diet-induced and preestablished obesity through polarizing macrophages to anti-inflammatory phenotypes, while hyperleptinemia is frequently accompanied by insulin resistance, T2D, and increased prevalence of CVD (394).
Resistin is an adipokine secreted by macrophages within adipose tissues in obesity. Increased levels of resistin in obesity mediate LV hypertrophy (252) and systolic dysfunction (395) possibly through fostering contractile dysfunction (396), inflammation, and endothelial activation (397). However, the precise role of resistin remains unclear in obesity as experimental evidence either failed to confirm the association between resistin and obesity (398) or favored dissociation between insulin resistance and elevated resistin levels in obesity (334).
Levels of TGF-β1 were overtly increased in LV from obese compared with lean rabbits, possibly contributing to cardiac collagen deposition (399). IL-6 deficiency promotes insulin resistance and cardiac lipid accumulation, interstitial fibrosis, and inflammation in high-fat diet-induced obesity (400). C-X-C motif chemokine ligand-14 (CXCL14) is an adipokine secreted in BAT, which mediates the communication between brown fat and macrophage in response to thermogenic activation (401). Osteopontin (OPN) is another proinflammatory cytokine abruptly elevated in adipose tissue from obese individuals. OPN functions as a key mediator in promoting macrophage proliferation for local adipose tissue in obesity (402, 403). Endocannabinoids, another proinflammatory adipokine, have an important role in regulating energy intake, storage, and consumption (404). Leukotriene B4 (LTB4) facilitates the development of insulin resistance in a macrophage-dependent manner in obesity (405).
3.1.4. Activation of immune cells.
As discussed above, adipocytes can become inflamed and secrete a variety of inflammatory adipokines. However, with the identification of macrophages in adipose tissue (AT), activation of immune cells is known to promote the release of the majority of inflammatory molecules in obese animals and humans (FIGURE 4). Adipose tissue macrophages (ATMs) possess a vital role in this process and can be divided into two main phenotypes, including M1, the classically activated macrophages, and M2, the alternatively activated macrophages (406).
3.1.4.1. m1/m2 paradigm in obesity.
Generally speaking, M2 macrophages produce anti-inflammatory adipokines and help maintain AT homeostasis in the lean state by playing an important role in phagocytosing necrotic or apoptotic myocytes, tissue remodeling, and cardiac fibrosis after injury (407, 408). On the other hand, the number of AT M1 macrophages rises in obesity and correlates with AT inflammation and insulin resistance as mediated by increasing proinflammatory cytokines (350, 406, 409). Macrophages ranges from under 10% in AT in lean mice and humans, the levels of which may exceed 40%–50% through local proliferation (410) and infiltration in obese humans and leptin-deficient obese rodents (411).
EAT can be a major source of cardiac M1 macrophages, thereby contributing to cardiac anomaly- associated obesity. In addition, resident macrophages exist in the healthy heart in low numbers, where they typically assume an M2 phenotype but can be inflamed during obesity (412). Cardiac macrophages expand in humans and mice with diastolic dysfunction owing to monocyte recruitment and hematopoiesis in bone marrow and spleen (413). The systemic and cardiac inflammation associated with obesity and pathological cardiac remodeling is largely mediated by M1 macrophages (412).
Although accumulation and activation of immune cells has been extensively reported in obesity, the signals that trigger and magnify these inflammatory changes remain to be deciphered. A number of scenarios are speculated for obesity-linked inflammation including hypoxia, adipocyte and cardiomyocyte death, gut microbiota, and alternation of circulating metabolic substrates, which help to program metabolic, inflammatory, and functional traits of AT immune cells (344, 414–422).
3.1.4.2. role of macrophages in obesity-induced cardiomyocyte hypertrophy and dysfunction.
Macrophages mediate LV inflammation and remodeling through several perceived mechanisms. First, macrophages promote pathological hypertrophy and impair systolic and diastolic function in the heart through proinflammatory cytokines (TNF-α, IL-1β, IL-6, IL-12, and IL-23), which in turn stimulate mitogen-activated protein kinase (MAPK) and NF-κB signaling and inhibit the Akt-mechanistic target of rapamycin kinase (mTOR) cascade in neighboring cardiomyocytes (412, 423). Second, macrophages secrete matrix metalloproteinases (MMPs) to degrade extracellular matrix (ECM) and phagocytose necrotic cardiomyocytes. Macrophages promote myocardial stiffness and indirectly compromise myocardial relaxation through activating fibroblasts and promoting collagen deposition as well as secreting ECM proteins (412, 413). Third, M1 macrophages also exacerbate systemic complications of cardiometabolic syndrome, including insulin resistance and hypertension through a cascade of cytokines (412, 414, 424, 425). Last but not least, the lipid-buffering capacity and lipolysis become defective in obese adipose tissue macrophages (ATMs), leading to excessive lipid release into the bloodstream and subsequently development of cardiac lipotoxicity (409, 426).
Taken together from the aforementioned findings, macrophage is a promising target to prevent and mitigate obesity-induced cardiomyopathy. Ezetimibe, a potent cholesterol absorption inhibitor, mitigated interstitial fibrosis and coronary arterial thickening in the heart through ameliorating cardiac macrophage infiltration in db/db mice (427). Mineralocorticoid receptor (MR) inhibition using a low-dose spironolactone (LSp) specifically increased M2 macrophage and attenuated inflammation in the heart, leading to restored diastolic function and lessened fibrosis in the heart (428).
3.1.4.3. other immune cells in obesity.
The phenotypic switch of AT macrophages may be more complex than the classical M1–M2 paradigm. Multiple phenotypes along the M1/M2 spectrum exist, each with potentially distinct functions, phenotypes, and markers (429, 430). Fatty acids (FAs) are believed to function as a main drive for metabolic activation of ATMs in obese adipose tissue. Alterations in lipid metabolism may be predominantly responsible for inflammatory activation in metabolically activated macrophages (431). Evidence from human subcutaneous adipose tissue (SAT) revealed that fat mass enlargement is associated with accumulation of CD206+/CD16- macrophages, which exert a pronounced proangiogenic response on AT-derived endothelial and progenitor cells, and various cardiovascular pathologies (432).
In addition to macrophage, several other Immune cells are highlighted as important factors in inflammatory responses that may impact global energy metabolism. For instance, natural killer (NK) cells, mast cells, and innate lymphoid cells (ILCs) as well as the cytokines derived from them are necessary to foster polarization of proinflammatory macrophages and obesity-associated insulin resistance and metabolic complications (433–435), whereas, invariant NK T cells, conventional dendritic cells, and the associated Cd40 signaling pathway are poised to counteract obesity-induced inflammation and promote the immunometabolic status in heart (436–438).
3.1.5. Insulin resistance and inflammation cross talk in obesity.
While insulin resistance seems to precede and contribute to AT inflammation (439), most studies support that inflammation, in addition to caloric imbalance, may impose a culprit role in the pathogenesis of insulin resistance (344, 418, 424, 440).
3.1.5.1. the ikk/nf-κb pathway.
It has been suggested that activation of IKKβ/NF-κB signaling cascade fosters insulin resistance through IKK-mediated serine phosphorylation of insulin receptor substrate 1 (IRS-1) or insulin receptor (IR) (441), which results in compromised tyrosine phosphorylation of IR1-1 and postreceptor signaling molecules such as glucose transporter type 4 (GLUT4), resulting in insulin resistance (440, 442).
Surprisingly, although adipose-specific TANK binding kinase 1 (TBK1) deficiency attenuates high-fat diet-induced obesity, it promotes activation of nuclear factor-κB (NF-κB), with upregulation of MCP-1 production from adipocytes and macrophage infiltration into adipose tissue, associated with worsened insulin resistance in high-fat diet-fed mice (441). To this point, Amlexanox, an inhibitor of noncanonical IκB kinases IKKε and TBK1, overtly lowered Hemoglobin A1c and improves insulin sensitivity in a subgroup of obese patients (443).
3.1.5.2. the nlrp3 cascade.
The canonical NF-κB pathway is initiated by the activation of the pattern recognition receptors (PRRs). The Nod-like receptor (NLR) family of PRRs represented by NLRP3 inflammasome serves as innate immune sensors and participates in the recognition of “danger signals,” resulting in caspase-1 activation and release of interleukin-1β (IL-1β)/IL-18. A rise in body weight in patients, sheep, and mice has been shown to be accompanied with NLRP3-inflammasome activation in the heart and other organs (444, 445). Antiobesity measures such as caloric restriction and exercise were shown to lower levels of adipose NLRP3, suppress inflammation, and improve insulin sensitivity in obesity. In response to lipotoxicity mainly manifested as intracellular ceramide rise, NLRP3 inflammasome promotes caspase-1 cleavage in macrophages and adipose tissue. This is supported by ablation of obesity-associated inflammasome activation in liver and fat depots along with improved insulin signaling with NLRP3 knockout (446). In the heart, NLRP3 ablation resisted pacing-induced AF in the face of high-fat intake, denoting a key role for NLRP3 inflammasome in obesity-induced atrial arrhythmogenesis (444). In another independent study, although deficiency of NLRP3 and the adaptor protein apoptosis-associated speck-like protein containing CARD domain (ASC or Pycard) retarded obesity-induced systemic inflammation, LV concentric remodeling and diastolic dysfunction without affecting cardiac hypertrophic response to high-fat diet-induced obesity. In addition, deficiency in NLRP3 and ASC was found to protect against obesity-induced metabolic derangement including compromised insulin signaling and steatosis in hearts and livers (445).
3.1.5.3. toll-like receptors: tlr2 and tlr4.
Toll-like receptors (TLRs), another major family of PRRs, plays a critical role in early innate immunity by recognizing the pathogen-associated molecular patterns as well as endogenous damage-associated molecular patterns (DAMPs). Intake of high-energy Western diet (high refined carbohydrates and saturated fat) has been shown to elicit adaptive pancreatic β-cell proliferation to offset peripheral insulin resistance. Interestingly, both TLR2 and TLR4 may suppress high-fat diet-induced replication of β-cells. When both receptors are removed, replication of β-cells but not α-cells is activated resulting in expanded β-islets and hyperinsulinemia in diet-induced obesity (447).
3.1.5.4. c-jun-nh2-terminal kinase and mapk.
C-Jun NH2-terminal kinase (JNK) and other mitogen-activated protein kinases (MAPKs), such as p38 MAPK, are activated in response to stress stimuli including saturated FAs, IL-1β, TNFα, and endoplasmic reticulum (ER) stress, to promote insulin resistance through serine and threonine phosphorylation of IRS, as well as dampened downstream postreceptor signaling (344, 440). In particular, immunophenotyping examination indicated a vital role for JNK activation in proinflammatory macrophage polarization (448).
3.2. Metabolic Disturbances
3.2.1. Insulin resistance.
The term “insulin resistance” (IR) commonly makes reference to a decline in metabolic response to insulin in target cells, or at the whole-organism level, demand of higher insulin levels to reduce blood glucose (449). Insulin sensitivity denotes the capacity of insulin to bind insulin receptor and evokes intrinsic tyrosine kinase activity of the receptor, leading to phosphorylation of IRS1/2 and activation of phosphatidylinositol-3 kinase (PI3K) and MAPK (450). IRS-1/IRS-2-mediated activation of PI3K mediates metabolic actions of insulin (i.e., increasing glucose uptake), whereas MAPK pathway (Grb2/Sos/Ras/ERK) mainly governs growth and remodeling responses in the heart. In addition, insulin also participates in the regulation of fat metabolism such as suppression of fatty acid oxidation (FAO) (449, 450). A number of postreceptor components, including serine/threonine protein kinase Akt, protein tyrosine phosphatase 1B (PTP1B), and mammalian target of rapamycin (mTOR), have been reported to mediate insulin-induced glucose metabolism (451–453).
Loss of insulin sensitivity or IR represents a cardinal trait of obesity and T2D and contributes to onset and development of heart diseases. The heart is an important insulin responsive organ and can become insulin resistant in obesity (454, 455). Given the key role of insulin as the predominant anabolic hormone in growth, development, glucose, protein, and lipid metabolism, insulin resistance is known to directly provoke adverse cardiovascular sequelae in obesity. Of note, metabolic milieu in the setting of insulin resistance is featured by elevated circulating levels of glucose, free fatty acids, and triglycerides, as well as dysregulated substrate supply from the periphery to the heart, along with elevated fatty acid oxidation, decreased glucose uptake and oxidation, and altered gene expression in cardiomyocytes (456). Compromised insulin-stimulated glucose uptake and dampened postinsulin receptor signaling have been considered a hallmark in the hearts from obese individuals, to various degrees pending on severity and duration of adiposity.
3.2.1.1. evidence for insulin resistance in obesity.
Ample evidence has depicted the presence of insulin resistance preceding the onset of LV remodeling and contractile dysfunction in obesity, favoring a vital role for insulin resistance in the pathogenesis of obesity cardiomyopathy. Not surprisingly, insulin resistance cardiomyopathy shares many commonalities with obesity cardiomyopathy.
Mounting findings have denoted a cardinal role for systemic insulin resistance in the etiology of cardiac dysfunction in patients with obesity. This theory is convincingly supported by the presence of insulin resistance in obesity whereas all adiposity measures (e.g., BMI, waist circumference, skinfold thicknesses, and bioimpedance) are positively correlated with the insulin sensitivity marker homeostatic model assessment for insulin resistance (HOMA-IR) (275, 457). In CARDIA (Coronary Artery Risk Development in Young Adults) study involving 3,179 patients, changes in HOMA-IR were monitored in nondiabetic populations based on severity of insulin resistance (low IR, moderate IR, and high IR). It was noted that severe form of IR in early adulthood was closely associated with accentuated LV wall thickness and worse longitudinal systolic strain, as well as early diastolic strain rate at middle age, depending on the severity of obesity (458). In addition to insulin resistance, glucose intolerance represents another key denominator for LV dysfunction. Examination of LV parameters and glucose tolerance in 2,623 Framingham Study individuals (in the absence of myocardial infarction and heart failure) revealed a tight correlation between glucose intolerance and LV mass/wall thickness, with a more pronounced effect in women than men. Adjustment for BMI considerably weakened the correlation between LV/LA parameters and HOMA-IR, with nonsignificant finding in the normal glucose tolerance group (275). Indeed, patients with both impaired IFG and IGT displayed greater LV mass, LV mass index, and lower Doppler early peak rapid filling velocity to peak atrial filling velocity ratio compared with those individuals with IFG alone, exhibiting a 10-fold higher susceptibility of preclinical LV hypertrophy. LV mass index was associated with WC, C-reactive protein, and 2-h oral glucose tolerance test (278).
Cardiac insulin resistance, on the other hand, dampens cardiac metabolism and function in obesity. A decline in insulin-stimulated cardiac glucose metabolism was observed much sooner in response to obesity in comparison with that in skeletal muscles, adipose tissues, and liver (roughly 1.5 wk vs. 3 wk after the initiation of high-fat feeding) (454). This suggests that obesity-induced cardiac dysfunction may be attributable to local insulin resistance and changes in cardiac metabolism rather than the global systemic insulin resistance (454). The same study also reported reduction in Akt-mediated insulin signaling and GLUT4 levels in cardiac insulin resistance (454). However, a later study offered contrary finding where increased cardiac glucose uptake; enhanced mitochondrial oxidation of palmitoyl carnitine, glutamate, and succinate; and greater basal insulin signaling were present in high-fat diet-fed mice compared with those of chow-fed mice, despite the presence of systemic insulin resistance (459). Given the high metabolic dynamics in the heart, different pathways may be involved in the response to hyperinsulinemia in the heart. In fact, it was shown that IGF-1 receptor-mediated Akt activation promoted cardiac hypertrophy in ob/ob mice and mTOR was responsible for the suppression of autophagy flux through alternative upstream pathways such as ERK signaling but not Akt (451, 460).
3.2.1.2. metabolic mediators of insulin resistance.
A number of scenarios have been speculated for insulin resistance in obesity, with positive energy balance as a result of high caloric intake and low physical activity being the most important factor. However, caloric imbalance cannot fully address the multifaceted metabolic traits in insulin resistance. Accumulating evidence has suggested a rather important role for altered inter-organ communication using various metabolites as messengers in insulin resistance. Of note, targeted and untargeted metabolomics have significantly enriched our knowledge of the role of lipids, amino acids and glucose in promoting insulin resistance (461).
3.2.1.2.1. Lipids.
Hypertrophic adipocytes become resilient to the antilipolytic action of insulin and impose a reduced ability to lipid storage, leading to accumulation of fat in, inter alia, muscle and liver cells, (461). More than five decades ago, Randle and colleagues (462) reported that lipids can provoke insulin resistance in diaphragm and hearts. Later studies confirmed that ectopic deposition of fatty acids and lipid metabolites, such as long-chain acyl-CoA esters, diacylglycerol (DAG), triacylglycerol (TG), and ceramide in muscles is closely related to the onset and development of insulin resistance (463–465). Schulman and colleagues (466) also revealed similar mechanism for diacylglycerol-induced insulin resistance in the liver through PKC-ε activation and decreased IRS-2 tyrosine phosphorylation. More recently, beneficial properties of certain lipid categories on insulin signaling have emerged, such as gut microbiota-generated short-chain fatty acids (acetate, propionate, and butyrate) (467), dietary unsaturated fatty acids (468), fatty acid esters of hydroxyl fatty acids (469), and phospholipids.
3.2.1.2.2. Amino acids.
The circulating levels of amino acids (AAs) are tightly correlated with insulin resistance in obese humans (461). In particular, branched-chain AAs (BCAAs), representing ∼20% of protein intake, aromatic AAs, and certain AA metabolites have garnered attention as to their role in insulin resistance. BCAAs offer various physiological and metabolic benefits including stimulation of pancreatic insulin secretion, adipogenesis, milk production, and immune function. Elevated serum BCAA levels were reported in obesity and insulin resistance decades ago (470). Following its initial perceived pathophysiological role as a reliable marker for obesity and T2D (471, 472), more studies have surfaced implicating a vital role for dysregulated BCAAs in the etiology of other chronic diseases such as cancers (473) and CVD (474), possibly mediated by the development of T2D. BCAAs are believed to evoke hyperactivation of mTOR in muscles, leading to impaired insulin signaling (471). Furthermore, high levels of circulating BCAAs may exacerbate cardiac insulin resistance and myocardial contractile dysfunction through fostering mitochondria dysfunction and mTOR upregulation (474, 475).
3.2.1.2.3. Glucose.
Current nutrition regimen consists of excessive carbohydrates from digestible polysaccharides to refined sugars which impose unfavorable health effects in human, a phenomenon commonly being referred to as “carbotoxicity” (476). Among various contributing factors for carbotoxicity, alteration in GLUT4, the insulin-responsive glucose transporter, serves as the major contributor for dampened glucose uptake in muscle and adipose tissues in obesity. Although loss of GLUT4 does not necessarily induce obesity, it drastically elevated serum glucose and insulin, decreased tissue glucose uptake, and imposed hypertension, reminiscent of noninsulin-dependent diabetes mellitus in human (477).
Other than GLUT4, sodium/glucose cotransporter 1 (SGLT1) functions as the main Na-dependent glucose cotransporter in heart (478). Cardiac SGLT1 (the main cardiac isoform) was shown to evoke an important role in acute IRI likely by way of facilitated glucose uptake, particularly in insulin resistance where GLUT4 regulation is compromised, although SGLT1 inhibition reduces obesity, incident diabetes, HF, and death (479, 480). In addition, SGLT6 [sodium-myoinositol cotransporter-1 (STIM1)] senses hyperglycemia and triggers the production of reactive oxygen species (ROS) in heart (481). SGLT2 is an emerging therapeutic target in the treatment of type 2 diabetes. However, SGLT2 is not expressed in the heart (478). Recent evidence suggested that SGLT2 inhibitors promoted energy expenditure, attenuated inflammation (polarizing M2 macrophages in WAT and liver) and insulin resistance in obesity, which may offer indirect cardiovascular protection (482).
In addition, mitsugumin 53 (MG53) is myokine/cardiokine secreted from hearts and skeletal muscle following high glucose or high insulin challenge to bind with the extracellular domain of the insulin receptor, thus allosterically inhibiting insulin signaling. Hyperglycemia is believed to be associated with elevated circulating MG53 in humans and rodents with diabetes mellitus (483).
3.2.2. Alternations of cardiac metabolism.
Alteration in fuel metabolism plays vital roles in both ATP-producing and non-ATP-producing energy homeostasis and pathogenesis of heart dysfunction in obesity. Cardiomyocytes switch main energy supply from carbohydrates to fatty acids during perinatal period, in concert with elevated mitochondrial oxidative phosphorylation and FAO (423). The heart maintains profound metabolic flexibility under stress such as nutrient excess, leading to changes in cardiac energetics and contractile function (135). For example, elevated LV wall stress in obesity evokes rises in myocardial oxygen consumption. The rise in substrate supply in obese hearts triggers an increase in FAO in conjunction with suppressed glucose oxidation. Obesity promotes a switch in gene expression favoring FAO over glucose oxidation, which is initially adaptive although further dampens insulin sensitivity and metabolic flexibility over time, resulting in impaired cardiac efficiency and cardiac contractile anomalies (FIGURE 5). Regardless of precipitating factors, sustained metabolic derangements in obesity promote oxidative stress, inflammation, insulin resistance, lipotoxicity, and energy deprivation, all of which promote progression of HF (135).
FIGURE 5.
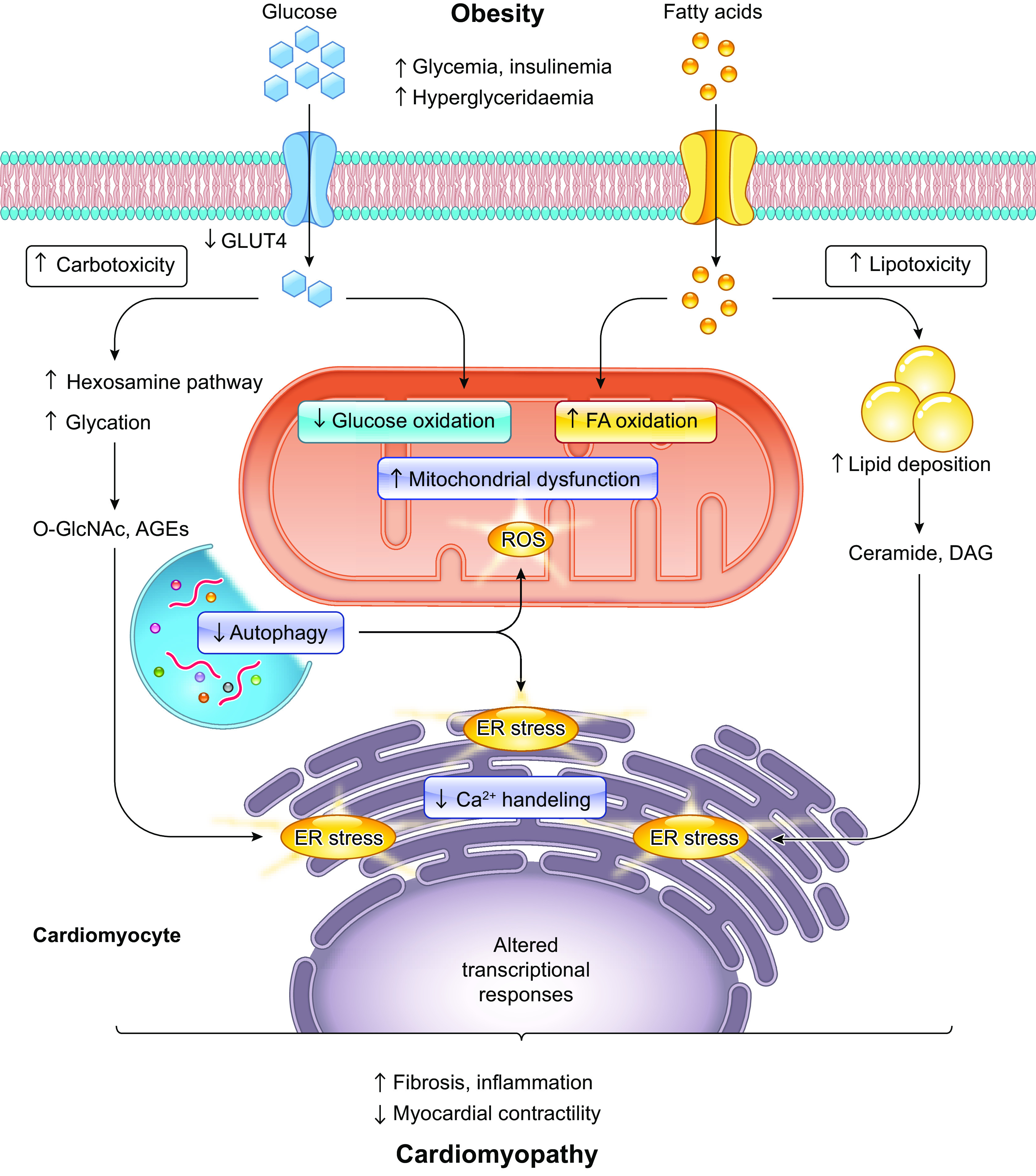
Metabolic stress and organelle dysfunction in obesity cardiomyopathy: Obesity leads to decreased myocardial glucose uptake and oxidation, increased fatty acid oxidation (FAO), and altered cardiomyocyte gene expression. Increased triglyceride accumulation and their products, such as ceramides and DAG, cause majority of lipotoxicity in hearts. Different metabolic pathways such as hexosamine and advanced glycation end-product (AGE) pathways have been identified as pro-oxidative processes and are usually elevated in uncorrected obesity. Autophagy activity in the heart declines with obesity, and its insufficiency is involved in the accumulation of reactive oxygen species (ROS) and the development of endoplasmic reticulum (ER) stress, leading to obesity-related cardiometabolic diseases. GLUT4, glucose transporter type 4; DAG, diacylglycerol; O-GlcNAc, β-linked N-acetylglucosamine; AGEs; advanced glycation end-products; FA, fatty acid; ROS, reactive oxygen species. Created with BioRender.com.
3.2.2.1. lipids.
The fine interplay of uptake, metabolism and oxidation of fatty acids (FAs) is necessary to maintain ATP and lipid homeostasis and membrane biosynthesis in the heart. At the energy level, obesity cardiac dysfunction is characterized by elevated myocardial oxygen consumption, dampened cardiac efficiency, and overwhelmed oxidative stress, denoting a likely role for increased FAO in cardiac dysfunction. A multivariate, stepwise regression evaluation concluded that high BMI value was the sole independent determinant of higher myocardial oxygen consumption and lower LV efficiency (484). It is perceived that insulin resistance, evaluated using glucose area under the curve, functions as the independent predictor of elevated myocardial fatty acid uptake, utilization, and oxidation (484).
High FAO seems to be responsible for reduced oxygen efficiency and accumulation of fatty acid, derivatives that further compromises cardiac efficiency by uncoupling mitochondria (485). However, later studies suggested that the heart exhibits impaired capacity of FAO in the face of excess lipid supply in obesity, a notion that is supported by the beneficial effect of FAO stimulation on cardiac pathology. Given that malonyl CoA generation via acetyl CoA carboxylase 2 (ACC2) suppresses the entrance of long chain fatty acids into mitochondria, cardiac-specific knockout of ACC2 maintained FAO and, to our surprise, attenuated obesity-induced cardiac dysfunction (486). Shao and associates (487) suggested that elevated cardiac FAO protected against cardiomyopathy in chronically obese mice, in part, by preservation of mitochondrial function through parkin-mediated mitophagy. In addition, reduced cardiac efficiency noted in obesity should be attributed to mechanisms other than increased FAO, for instance, mitochondrial uncoupling and ROS (488).
An important hallmark of altered cardiac metabolism in obesity and other metabolic anomalies is the excess of substrate supply compared with demand for ATP synthesis. Even with elevated FAO, obese hearts are still filled with excess lipid accumulation prompting lipotoxic cardiomyopathy. Cardiac lipotoxicity denotes accumulation of excess fatty acids and associated triglyceride in parenchymal cardiomyocytes resulting cell death and cardiac anomalies (106, 353, 489, 490). Ultrastructural examination using electron microscopy has unveiled that obesity is associated with enlarged sarcoplasmic reticulum, disordered alignment of myofilaments, abnormal morphology of mitochondria, and numerous lipid droplets between myofibrils in cardiac tissues (491). Long-chain nonesterified fatty acids and their products, such as ceramides and diacylglycerols (DAGs), are mainly responsible for lipotoxic sequalae (492–494). Intramyocardial lipid overload was found to corelate with contractile dysfunction and alterations in gene expression in obese Zucker Diabetic Fatty (ZDF) rats, reminiscent of human failing hearts with lipid overload (495). It is conceived that presence of obesity-induced DAG accumulation in the heart signified the development of cardiac insulin resistance through PKCα-dependent inhibition of Akt, p70s6k activation, and IRS-1 Ser332/336 phosphorylation (496). Not surprisingly, pharmacological and genetic strategies targeting lipotoxicity yield salutary actions on the heart and overall metabolic health (381, 497, 498). For example, troglitazone therapy lowers obesity-induced myocardial TG and ceramide and prevents apoptosis and loss of cardiac function in obese ZDF rats (499).
Intramyocardial lipid deposition is often accompanied with upregulated nuclear receptors, peroxisome proliferator-activated receptor-α (PPARα)-regulated genes, myosin heavy chain-β, and proinflammatory cytokines including TNF-α. PPARα has long been known to govern fatty acid metabolism in the heart and mediate the development of lipotoxicity (490). PPARβ and PPARδ, on the other hand, participate in the homeostasis of both fatty acid and glucose metabolism. Given that PPARα generally governs genes involved in fatty acid metabolism, including uptake, storage and oxidation in the heart, emerging studies have tried to clarify how PPARα provokes the imbalance of lipid metabolism. It was recently demonstrated that fatty acids (FAs) upregulate glycogen synthase kinase-3α (GSK-3α) and thus phosphorylate PPARα at Ser280 in its ligand-binding domain, resulting in elevated transcription of a subset of PPARα targets and its biased activation, that is, only activation of its transcriptional activity of FA uptake and storage but not oxidation (500). The activator protein 1 members JunD and miR-494-3p may also play a role in lipid accumulation as levels of JunD and miR-494-3p were found dysregulated to correlate with myocardial TG content and echocardiographic indexes of LV dysfunction in obese human hearts (501).
3.2.2.2. glucose.
In the setting of obesity, glucose uptake and oxidation are suppressed (135). Obesity-induced change in insulin signaling directly impedes insulin-stimulated GLUT4 translocation and glucose uptake. Restoring glucose oxidation is believed to improve obesity-induced cardiac injury. PPARα activation could increase glucose oxidation in the heart and alleviate contractile dysfunction in obesity (502). However, profound rises in intracellular glucose and sustained stimulation of glucose uptake and oxidation remodeled the cardiac metabolic network and dampened metabolic flexibility. In this context of high-glucose stimulus, high-fat diet-induced FAO is obtuse, leading to overwhelmed oxidative stress and cardiac dysfunction (503).
As a result of insulin resistance and elevated FAO, obesity is associated with dampened glucose uptake and utilization for ATP synthesis, albeit with possibly elevated accessary glucose metabolism flux (135). For example, nutrient excess in obesity may promote hexosamine biosynthetic pathway resulting in protein posttranslational modification by O-GlcNAc transferase (OGT)-mediated O-linked β-N-acetylglucosamine (O-GlcNAc) moieties. Overexpression of adipose OGT suppresses lipolysis and exacerbates diet-induced obesity (504). AMPK, on the other hand, inhibits O-GlcNAcylation through regulation of glutamine:fructose-6-phosphate aminotransferase (GFAT) phosphorylation to alleviate O-GlcNAcylation of target proteins such as troponin T. Blockade of O-GlcNAcylation using inhibitors of the GFAT mitigates cardiomyocyte hypertrophy (505).
3.2.2.3. other substrates.
In pathological cardiac hypertrophy and HF, the ATP-generating modality is altered to promote energy production from glycolysis, anaplerosis, lactate, BCAAs, and ketone bodies (506). However, glycolysis and utilization of lactate, BCAAs, and ketone bodies cannot adequately compensate for loss of glucose oxidation in obesity, thus prompting energy deficit and onset of HF (423). Chronic loss of insulin sensitivity imposes blunted BCAA oxidation in the liver and adipose tissues, resulting in increased workload of BCAA oxidation in muscles (507). In addition, BCAA oxidation is vital for cardiac function. Suppression of the essential step for BCAA oxidation namely branched-chain α-keto acid dehydrogenase (BCKDH) (with BCATm being the rate-limiting enzyme for BCAA oxidation) reduces cardiac systolic function in mice whereas 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid, a pharmacological inhibitor of BCKDH kinase, to lower plasma BCAAs, enhances cardiac BCAA degradation and thus preserves heart contractility (474).
3.2.3. Mitochondrial dysfunction.
Mitochondrial dysfunction, including the inability to generate ATP, disturbed mitochondrial dynamics, insufficient mitophagy, and accumulation of reactive oxygen species (ROS), plays a rather unique role in the onset of cardiomyopathy in obesity (105, 508–511). First, evidence was obtained that the inability of the mitochondria in obese heart to appropriately utilize glucose and the consequent switch from glucose to FAO results in metabolic inflexibility (485) (FIGURE 5). As the reliance on FAO requires a greater oxygen consumption and free fat acids (FFAs) instinctively causes mitochondrial uncoupling, these changes pose a significant threat to cardiovascular health through reduced ATP generation, decreased cardiac efficiency, and as a consequence, defects in contractile function (512, 513).
Over the past decades, many advances have been achieved toward understanding mitochondrial biogenesis, dynamics, quality control and their involvement in the progression of obesity-related cardiomyocyte dysfunction. Mitochondrial proliferation was increased in db/db hearts (514). Morphological transition from mitochondria network to fragmented mitochondria was noted in obese cardiomyocytes (515). Palmitate challenge in neonatal rat cardiomyocytes initially turned on mitochondrial respiration, along with increased mitochondrial polarization and ATP production, while sustained incubation of palmitate (>8 h) evoked ROS production and mitochondrial fission (516). Lipid overload-induced cardiomyocyte apoptosis and cardiac dysfunction were likely due to altered posttranslational modifications of the mitochondrial fission and fusion proteins, including increased ubiquitination of A-kinase anchor protein 121 (AKAP121), dynamin-related protein 1 (Drp-1), and proteolytic processing of optic atrophy 1 (OPA1) (515, 516).
Dysfunctional mitochondria are removed by a specialized form of autophagy, referred to as mitophagy. Imbalanced mitochondrial biogenesis and mitophagy also occurs in the development of metabolic cardiomyopathy. Mitophagy is induced by high-fat-diet consumption, while deficiency in mitophagy aggravates high-fat intake-induced cardiomyopathy (517, 518). Mitochondria and endoplasmic reticulum (ER) are interconnected organelles, numerous proteins were proposed to tether the two organelles together at specific sites, referred to as mitochondria-associated ER membranes (MAMs) (519). Interestingly, despite that disruption of MAMs instigates aberrant Ca2+ signaling and cardiac anomalies, a recent study suggested that high-glucose-induced FUNDC1-mediated MAMs formation and mitochondrial calcium overload in the cardiomyocytes, leading to functional cardiac abnormalities (519, 520).
3.3. Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) is most essential for Ca2+ storage, lipid biosynthesis, and protein sorting and processing. A wide variety of gene products pass through the ER lumen, governing both physiological and pathophysiological processes (521, 522). Cellular perturbations in obesity interrupt ER homeostasis, resulting in the buildup of unfolded/misfolded proteins and pronounced ER stress (523, 524). To sustain ER homeostasis, cells utilize protein quality-control systems through unfolded protein response (UPR), endoplasmic reticulum associated degradation (ERAD), and autophagy (525) (FIGURE 6). The ER-resident proteins are predominantly removed by ERAD for proteasomal degradation inside ER, while protein aggregates within the ER lumen may be destroyed through autophagy degradation (524, 525). When misfolded proteins accumulate in ER, UPR can be triggered by activation of three main UPR sensors, inositol-requiring enzyme 1α, protein kinase R-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6, on ER membrane, to cope with unfolded and misfolded proteins (521). When the UPR is unable to handle unfolded and/or misfolded proteins in the ER, the ER-initiated apoptotic signaling is turned on (523).
FIGURE 6.
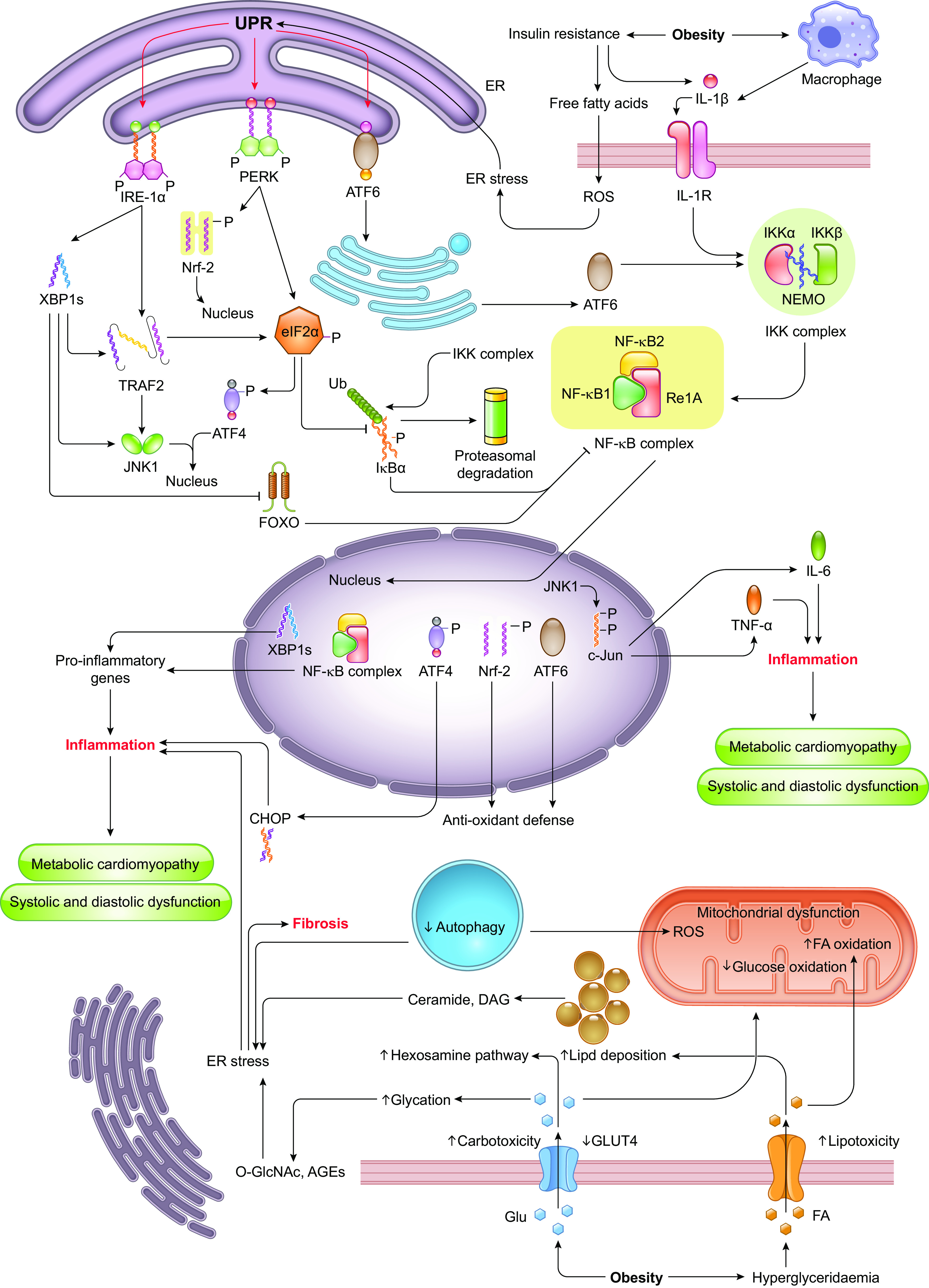
Obesity-mediated endoplasmic reticulum (ER) stress underlies cardiometabolic disorders: Obesity induces certain conditions such as hyperglyceridemia, glycemia, insulin resistance, and macrophage activation, all of which, in turn switches on several pathways culminating in ER stress, inflammation, and ultimately cardiometabolic disorders. Insulin resistance induces ER stress, which activates three branches of unfolded protein response (UPR) including ATF6, PERK, and IRE-1α. IRE-1α/TRAF2/JNK1/c-Jun pathway induces inflammation via TNF-α and IL-6, while PERK/eIF2α/IκBα pathway and ATF6 trigger NF-κB complex-mediated inflammation. Macrophage also produces IL-1β, which activates IKK and NF-κB complexes leading to inflammation. Hyperglyceridemia and glycemia induce carbotoxicity and lipotoxicity, which instigates metabolic alterations resulting in ER stress and inflammation. IRE-1α, inositol-requiring protein-1; PERK, protein kinase RNA-like ER kinase; ATF6, activating transcription factor-6; IKK, I-κb-kinase; NF-κB, nuclear factorκB; IκBα, I-κb-α; Ub, ubiquitin; eIF2α, eukaryotic initiation factor 2α; Nrf-2, nuclear factor-E2-related factor; XBP1s, X-box-binding-protein-1 spliced; TRAF2, TNF receptor-associated factor 2; JNK1, C-Jun NH2-terminal kinase 1; ATF4, activating transcription factor-4; FOXO, forkhead box O; c-Jun, a transcription factor; CHOP, C/EBP homologous protein; FA, fatty acid; Glu, glucose; GLUT4, glucose transporter type 4; DAG, diacylglycerol; O-GlcNAc, β-linked N-acetylglucosamine; AGEs, advanced glycation end-products.
SR/ER and ER stress in obesity are also closely related to obesity in the heart (524). Levels of PERK were suppressed in the liver and heart, following 16 and 8 wk, respectively, following high-fat diet intake (526). High-fat diet intake upregulated levels of GRP94 and CHOP in left ventricles (LV) from 5-mo-old Lee-Sung pigs (527). Palmitate increased ER stress in H9C2 cells, manifested as levels of p-PERK, p-eIF2α, and transmission electron microscopy examination (528). Furthermore, palmitate-triggered ER stress induced adiponectin resistance through AMPK phosphorylation and reduced APPL1 levels (528). Chronic ER chaperone tauroursodeoxycholic acid treatment rescued against increased systolic blood pressure, glucose intolerance, cardiac hypertrophy, and cardiac contractile dysfunction in obese mice through reconciliation of obesity-associated drop in sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) function, rises in serine phosphorylation of IRS, total and phosphorylated cJun, as well as ER stress markers Bip, peIF2α, and pPERK (77).
3.4. Calcium Handling
Ample vital mitochondrial function including ATP production and mitochondrial metabolism are heavily controlled by Ca2+ signal, an essential intracellular second messenger that reaches the mitochondrial intermembrane space and then mitochondrial matrix, to participate in the regulation of proteins, enzymes and transporters (e.g., AMPK, pyruvate dehydrogenase kinase 4, pyruvate dehydrogenase phosphorylation, and GLUT4) required for ATP synthesis and mitochondrial metabolism (529). Intracellular and mitochondrial Ca2+ is tightly regulated in a narrow range to preserve the overall cellular Ca2+ homeostasis and cardiomyocyte contractility and defective Ca2+ handling (both intracellular and mitochondrial) is supposed to play a major role in obesity-related cardiac dysfunctions noted as prolonged diastolic relaxation, decreased fractional shortening, and compromised ejection fraction (530–533). Besides, extracardiac findings have suggested other potential mechanisms underlying calcium overload and energy metabolism. For instance, elevated intracellular Ca2+ in hepatocytes during obesity inhibits insulin-stimulated Akt phosphorylation and its key downstream signaling molecules by inhibiting membrane localization of pleckstrin homology domains (534). Obesity-induced increase in PDK4 activity augments MAM formation to promote Ca2+ transfer from SR/ER to mitochondria and suppresses insulin signaling in skeletal muscle (535).
3.4.1. Calcium handling in sarcoplasmic reticulum.
During the process of excitation-contraction coupling in cardiomyocytes, activation of voltage-gated L-type Ca2+ channels evokes Ca2+ releases from sarcoplasmic reticulum (SR) via ryanodine receptors (RyRs), a process commonly known as Ca2+-induced Ca2+ release. Cardiac relaxation is driven by Ca2+ reuptakes via SR Ca2+-ATPase (SERCA) and Ca2+ extrusion from sarcolemma Na+ and Ca2+ exchanger. Dephosphorylated phospholamban at Thr17 and Ser16 inhibits SERCA2a and suppresses Ca2+ uptake (536). Animal studies have reported prolonged Ca2+ clearance including reduced intracellular Ca2+ amplitude and decay in obese hearts, coinciding with prolonged relaxation (537–540). Increased SR Ca2+ leak via RyR2 plays a critical role in the progression of HF and cardiac arrhythmia. Suppressed SERCA2a activity is known to contribute to impaired relaxation and Ca2+ handling in ob/ob mice and sucrose-fed rats (538). However, reduced SERCA2a activity may result from SERCA oxidation in ob/ob mice, while SERCA2a dysfunction was associated with inhibited phosphorylation of phospholamban at Ser16 and Thr17 in sucrose-fed rats. More evidence has indicated unaltered expression of SERCA2a and phospholamban (or phosphorylation) in obese hearts, indicating possible involvement of posttranslational modification of SERCA2a (539). Results from studies in animal models with HF suggested that inhibition of SERCA2a activity is associated with the sulfonylation at Cys674 and nitration at Tyr294/295 (541). Downregulation of SIRT1, a class III histone deacetylase, increased SERCA2a acetylation at Lysine 492, leading to reduced SERCA2a activity and cardiac defects (542, 543). Moreover, myocardial dysfunction was correlated with impaired L-type Ca2+ channel activity in the absence of altered SERCA2a function and L-type Ca2+ channel protein levels in high-fat-fed rats (544). Among various mechanisms proposed for altered Ca2+ regulation in obesity, stromal interaction molecule 1 (STIM1) is believed to regulate cardiac Ca2+ signaling by sensing reduced SR Ca2+ level and interact with plasma membrane Orai channels to induce Ca2+ influx, leading to prolonged action potential duration and cardiac hypertrophy (545). SMIT1 overexpression exacerbated glucotoxicity and sensitized cardiomyocytes to hyperglycemia (481). Nonetheless, further study is needed to discern if this mechanism prevails in obesity cardiomyopathy.
Cardiac systolic dysfunction in various obese animals is mainly characterized by slightly decreases in the amplitude and decay rate of cardiomyocyte shortening, which may attribute to the decreased amplitude of Ca2+ transients (104, 537, 538, 546, 547). It was observed that diminished cardiac function in the fat-fed dogs was associated with the reduced RyR2 activity, which is not due to changes of protein expression but the increased phosphorylation of RyR2 (548). However, the results from the obese Zucker rats exhibited cardiomyocyte dysfunction, which was related to the decreased expression of L-type Ca2+ channel and eventually contributed to the prolonged duration of AP (549). Additionally, it has been suggested that the posttranslational modifications, including S-glutathionylation, S-nitrosylation, and disulfide oxidation could regulate the activity and function of RyR2 in HF (550). Further research needs to establish whether these PTM of RyR2 participate in the cardiac dysfunction of obesity.
3.4.2. Ca2+ overload and mitochondria.
Mitochondrial Ca2+ homeostasis is regulated by various organelles. The ER can release Ca2+ into the mitochondria quickly through Ca2+ channels in the MAM area to maintain Ca2+ homeostasis of mitochondria in physiological settings (551). There are a variety of Ca2+-regulated transporters in the MAM region, including inositol 1,4,5-trisphosphate receptor (IP3R) and ryanodine receptors (RyRs) located on the ER and voltage-dependent anion channel 1 (VDAC1) and mitochondrial Ca2+ uniporter (MCU) located on the mitochondrial membrane (551). One of the most important Ca2+ transport complexes between ER and mitochondria is formed by VDAC1 and IP3R, which are bridged together by Grp75 (551). Both increased MAM formation or the MAM Ca2+ channel hyperactivation would result in mitochondria uptake of excessive Ca2+ in stress condition, prompting mitochondrial Ca2+ overload, mitochondrial permeability transition pore (mPTP) opening, mitochondrial dysfunction, and ultimately cell death (551).
Appropriate Ca2+ handling is essential to mitochondria, where Ca2+ uptake supports oxidative phosphorylation (OXPHOS) and ATP production to limit excessive ROS production during cardiac contraction. ROS production is significantly increased in cardiomyocytes from obese animals (552, 553), and these rises correlate with prolonged relaxation in the heart (546, 554). Data from our group revealed that loss of mitochondrial autophagy receptor FUNDC1 accentuated high-fat diet-induced cardiac remodeling, functional and mitochondrial anomalies likely through rises in type 3 IP3R and mitochondrial Ca2+ overload (555). Recent evidence revealed a role for diastolic SR Ca2+ leak via RyR2 and type 2 IP3R (IP3R2) in mitochondrial Ca2+ overload, ROS production and cardiac dysfunction (537, 556). Furthermore, electrically evoked Ca2+ transients were smaller and slower, associated with the increased IP3 level in ob/ob cardiomyocytes (557). In addition, it was suggested that ROS production was associated with increased carbonyl oxidation of SERCA2a and abnormal relaxation in ob/ob mice (538). Moreover, overexpression of mitochondrial Na+/Ca2+ exchanger (NCLX), the primary mitochondrial Ca2+ extrusion machinery in cardiomyocytes, plays a protective effect in the clearance of mitochondrial Ca2+ overload, alleviating cardiomyocyte necrosis and HF (558), although its role in obesity cardiomyopathy remains unclear.
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a multifunctional serine/threonine kinase to mediate various physiological responses upon β-adrenergic activation (559). CaMKII is upregulated and mediates Ca2+ handling in various cardiac diseases including obesity-related cardiomyopathy through phosphorylating and regulating RyR, IP3R2, and phospholamban (559). Activation of CaMKII is suspected to provoke mitochondrial dysfunction, oxidative stress, ER stress, inflammation, and cell apoptosis in palmitate-induced cell hypertrophy and fibrosis in H9C2 cells (560). Sustained CaMKII activation interrupts intracellular Ca2+ homeostasis. For instance, CaMKII phosphorylates RyR to affect its open probability and promotes arrhythmogenic spontaneous SR Ca2+ release in heart diseases (559). In addition, CaMKII may facilitate mitochondrial Ca2+ entry through upregulation of MCU conductance, resulting in mitochondrial Ca2+ overload, mPTP opening and dissipation of mitochondrial inner membrane potential (561). In this context, CaMKII links ER stress and mitochondrial dysfunction through Ca2+ transfer between ER and mitochondria.
3.5. Autophagy
Autophagy is an evolutionarily conserved process to engulf cytoplasmic cargos and organelles through autophagosomes, fusion with lysosomes to form autophagolysosomes for production of ATP and macromolecules. Autophagy serves as a double-edged sword with physiological levels of autophagy being cytoprotective whereas unchecked or excessive autophagy being self- destructive for cannibalistic cell death - a form of nonapoptotic cell death or “type II programmed cell death.” Three types of autophagy are identified modality of autophagosome delivery into lysosomes: microautophagy, macroautophagy and chaperone-mediated autophagy (133, 562, 563).
Autophagy levels alter in response to variations in nutrient status such as fat/caloric intake and may unfavorably affect local (such as in a metabolically active organ) or global metabolism to promote metabolic derangement. Both elevated and suppressed autophagy have been noted in metabolic disorders including obesity (547, 564–567), due to an interplay among genetic factors, environment and energy imbalance. Three major risks factors in obesity, dyslipidemia, hypertension and insulin resistance/hyperglycemia are believed to be responsible for disturbed autophagy response in obesity. Meanwhile, deranged autophagy, in particular loss of autophagy, fosters metabolic derangement, insulin resistance and obesogenesis (133). In addition, considering the essential role for autophagy in the regulation of cardiac homeostasis, one should not neglect the direct contribution of autophagy derangement in cardiac proteotoxic pathology (aka, cardiac proteinopathy), and development of cardiomyopathies (568–575). Ample evidence has depicted that activation of autophagy through autophagy inducers or the master regulator of lysosomal function transcription factor EB (TFEB) effectively rescues cardiac protein quality control, cardiac remodeling and contractile function under various pathological settings (568, 570, 571, 576).
3.5.1. Cardiac autophagy in obesity.
Autophagy defects are closely associated with adiposity. For example, mice with global or tissue-specific (e.g., liver and pancreas) ablation of autophagy associated proteins including Atg7, Becn2, Bif, LAMP2, and Tfeb present obesogenic phenotypes or are predisposed to diet-induced or genetic adiposity (133). Autophagy also governs pathological sequalae of obesity, mainly through buildup of autophagy substrates including protein aggregates, lipid droplets, and damaged mitochondria (563, 577).
While the molecular mechanisms of autophagy have been extensively examined in obesity, how obesity-related autophagy changes in cardiomyocytes remains unclarified. Although research has shown that autophagosomes are normally formed in obesity, autophagy flux is disrupted because of defects in lysosome and impaired fusion of autophagosomes and lysosomes to preclude autophagic degradation (578, 579). Lack of an additional accumulation of LC3-II and p62 in chloroquine-treated high-fat-fed hearts favored the notion that elevated autophagosomes may be secondary to compromised autophagosome turnover (565). Impaired autophagy flux contributes to the development of cardiac dysfunction caused by obesity (580, 581), while the underlying mechanisms remain unclear. For instance, both genetic and diet-induced models of obesity noted a drastic loss in cardiac autophagy in concordant with downregulated unc-51 like kinase-1 (ULK1) (582). Of note, cardiomyocyte-specific knockout of Ulk1 disrupts autophagy and mimics the increase in cardiac lipoprotein lipase (LPL) levels occurred in obesity models. In the heart, LPL prompts myocardial fatty acid buildup via intravascular triglyceride (TG) hydrolysis. Concurrent ablation of LPL and ULK1 mitigated high-fat intake-evoked aberration in triglyceride (TG), diacylglycerol, and cardiac function, indicating a protective role of ULK1-mediated autophagy in obesity-induced cardiomyopathy through the regulation of lipid metabolism (582).
It was noted that although autophagic flux is transiently activated by high-fat diet consumption, peaking at 6 wk, it is ultimately attenuated (518, 580, 581). However, mitophagy, evaluated with Mito-Keima, continues to increase even after 2-mo high-fat-diet feeding (518). Deletion of Parkin partially impaired mitophagy, increased lipid accumulation, and exacerbated cardiac dysfunction during high-fat-diet feeding (518). These findings denote an essential role of mitophagy in cardiac protection against high-fat uptake.
3.5.2. Lipophagy in the heart during obesity.
Lipid storage can be evaluated using lipophagy, a specific subset of selective autophagy that targets lipid droplets and catabolizes their components into FFAs and glycerol (583). Altered cardiac lipophagy has been solidified in murine obese models, denoting a role for lipophagy in high myocardial lipid accumulation (584). Transmission electron microscopy can be employed to monitor the size and quantity of lipid droplets, along with lipid droplet-associated double-membrane structures, corresponding to autophagosomes (583). Consistent with the changes of macroautophagy in the heart during obesity, data from both immunoblotting and electron microscopy revealed more lipid droplets and autophagosomes with fewer autolysosomes in the hearts of high-fat diet-fed mice, denoting inhibition of autophagosome degradation (584). Among various possible scenarios, compromised ability of FGF21 to promote autophagy/lipophagy was shown to exacerbate lipid accumulation and structural derangements in obese murine hearts (381).
3.5.3. Autophagy stimulation and cardioprotective effects.
Obesity-related cardiac anomalies are associated with the disruptions of signaling pathways involved in cardiomyocyte autophagy, while activation of autophagy/mitophagy retards cardiac dysfunction in obesity (487, 547, 580). Injection of Tat-Beclin1 effectively inhibits lipid accumulation and protects against cardiac dysfunction by restoring autophagy or mitophagy (518, 582). SGLT2 inhibitors induce a transcriptional paradigm that mimics nutrient, and oxygen deprivation and activates autophagy, thereby mitigating cardiomyocyte dysfunction (585). Overexpression of ALDH2 increased Beclin‐1, Atg7, and AMPK as well as decreased mTOR have been shown to lead to autophagy induction and alleviated palmitic acid-induced cardiac dysfunction via autophagy regulation in a SUV39H-Sirt1-PGC-1α deacetylation-dependent manner (586).
4. THERAPEUTIC IMPLICATIONS OF OBESITY CARDIOMYOPATHY
4.1. Management of Obesity
Long-term pharmacotherapy of obesity is structured on reduction of energy intake, facilitation of satiety, lowering sensation of hunger, and caloric absorption, along with lifestyle modification intervention (587). In severe or morbid obesity, intragastric balloon insertion, shock therapies, and bariatric surgery can be applied (587–589). Nonetheless, effective antiobesity therapy has been greatly hindered by the lack of a better understanding of the precise interplay between genetic and nongenetic factors (e.g., nutrition and environmental cues) in the etiology of obesity and obesity complications.
4.1.1. Cornerstone: weight loss.
Despite the debatable benefits of obesity paradox (590), constellation of findings suggested weight loss, in particular by way of cardiorespiratory fitness and regular exercise, plays an indispensable role in the prevention and management of CVD in obese individuals (591). Obesity-related cardiac remodeling and dysfunction is prevented or even be reversed with purposeful weight loss (591, 592). For example, a switch from high-fat to low-fat diet in obese mice with heart failure drastically lowered the body weight gain, retarded cardiac remodeling, as well as improved cardiac insulin sensitivity and diastolic function (593), denoting a beneficial effect of weight loss. This is echoed by the ample beneficial responses on cardiac geometry and function in overweight and obese individuals with bariatric surgery or lesser levels of weight loss using caloric restriction (CR) (TABLE 2). Furthermore, a retrospective study found that weigh loss between annual physical examinations displayed beneficial effects in general populations (616). Nonetheless, coexisting chronic diseases and the duration and severity of overweight and physical limitations should be considered to assess the health risks and benefits of treatment options. Efficient strategies with evidence-based support to improve health and quality of life mainly include lifestyle intervention, pharmacotherapy, and bariatric surgery (591).
Table 2.
Effects of weight loss on cardiac function and morphology in human subjects
| Authors (Year) (Ref. No.) | Sample Size | Intervention and Follow-Ip | Baseline BMI | BMI after Weight Loss | Cardiac Function after Weight Loss | Cardiac Structure after Weight Loss |
|---|---|---|---|---|---|---|
| Naylor et al. (2008) (594) | 23 | Resistance training, 8 wk | 32.5 ± 1.9 | 30.2 ± 2.6 | Improved early diastolic myocardial velocities | No change in LV geometry |
| Amaro-Gahete et al. (2021) (595) | 12 | Physical exercise training, 12 wk | 32.1 ± 3.6 | 31.4 ± 2.7 | Increased LV end diastolic diameter | Not assessed |
| Serrano-Ferrer et al. (2014) (596) | 39 | Lifestyle intervention (diet and exercise), 3 mo | 31.9 ± 3.5 | 28.8 ± 3.2 | Improved RV global longitudinal strain and early diastolic strain rate; No change in TDI indices | Not assessed |
| Haufe et al. (2012) (597) | 170 | Hypocaloric diets, 6 mo | 32.9 ± 4.4 | 30.4 ± 4.3 (reduced carbohydrate), 30.6 ± 3.9 (reduced fat) | No change in LV function | Lower LV mass |
| Viljanen et al. (2009) (598) | 34 | Hypocaloric diet, 6 wk | 33.7 ± 0.7 | 29.9 ± 0.7 | Improved cardiac output, decreased blood pressure | Lower LV mass |
| Karimian et al. (2017) (599) | 32 | Hypocaloric diet, 6 wk | 40.3 ± 6.6 | 33.2 ± 6.1 | Reduced blood pressure and partially normalized diastolic dysfunction | Not assessed |
| Varli et al. (2010) (600) | 13 | Hypocaloric diet plus orlistat, 6 mo | 39.9 ± 4.3 | 36.1 ± 4.6 | Improved LV diastolic function | Lower LV mass |
| Gulsin et al. (2020) (601) | 87 | Hypocaloric diet or aerobic training, 12 wk | 36.6 ± 5.5 | 34.5 (routine care), 33.0 (exercise), 30.3 (low-energy diet) | No change in LV diastolic function | Less concentric LV remodeling and aortic stiffness |
| Andersson et al. (2016) (602) | 68 | Dietary intervention, 2 yr | 32.6 ± 0.6 | 31.0 (Nordic nutrition recommend diet), 29.4 (Paleolithic-type diet) | Improved cardiac function | Lower LV mass |
| de las Fuentes et al. (2009) (603) | 60 | Dietary intervention, 2 yr | 37 ± 3 | 4.1 ± 8.8 kg loss in weight | No change in LV diastolic and systolic function | Lower LV mass |
| Leung et al. (2016) (604) | 8 | Bariatric surgery, 9 mo | 44 ± 9 | 35 ± 6 | Improved LV global longitudinal strain and LV EF | Not assessed |
| Giudici et al. (2020) (215) | 26 | Bariatric surgery, 8 mo | 47.9 ± 7.1 | 33.4 ± 6.9 | Reduced carotid arterial stiffness and improved LV diastolic function | No change in LV geometry or mass |
| Kaier et al. (2014) (605) | 52 | Bariatric surgery, 6 mo | 42.4 ± 4.6 | 31.5 ± 2.6 | Improved RV and LV global strain, EF | Lower LV mass |
| Garza et al. (2010) (606) | 57 | Bariatric surgery, 3.6 yr | 49 ± 9 | 35 ± 8 | No change in LV function, RV function or EF | Reduced cardiac remodeling |
| Ikonomidis et al. (2007) (607) | 60 | Bariatric surgery, 3 yr | 48.7 ± 7.8 | 23 ± 1 | Improved LV diastolic and aortic function | Reduced LV hypertrophy |
| Hsuan et al. (2010) (608) | 66 | Bariatric surgery, 3 mo | 43.3 ± 6.3 | 34.1 ± 5.6 | Improved peak systolic mitral annular velocity and diastolic indices | Lower LV size, relative wall thickness, LV mass index; no change in chamber size |
| Algahim et al. (2010) (609) | 15 | Bariatric surgery, 2 yr | 46.7 ± 1.7 | 32.4 | Not assessed | Decreased LV mass |
| Owen et al. (2011) (300) | 423 | Bariatric surgery, 2 yr | 47.9 ± 7.0 | 32.2 ± 7.8 | Improved LV and RV function | Decreased cardiac remodeling |
| Jhaveri et al. (2009) (610) | 17 | Bariatric surgery, 17 mo | 44.1 ± 4.2 | 29.9 ± 4.7 | No change in LVEDV, RVEDV, or EF | Decreased LV and RV mass |
| Lin et al. (2011) (611) | 30 | Bariatric surgery, 16 mo; Diet, 8 mo | 39 ± 6 | 36 ± 7 (diet), 29 ± 5 (bariatric surgery) | Improved LV diastolic function | Lower LV mass |
| Valezi and Machado (2011) (612) | 43 | Bariatric surgery, 1 yr | 41.8 ± 4.4 | 28.4 ± 3.8 | Increased EF, improved diastolic function | Lower LV mass, interventricular septum, posterior wall thickness |
| Luaces et al. (2012) (613) | 41 | Bariatric surgery, 1 yr | 47.41 | 30.43 | Decreased early mitral velocity, increased mitral inflow E/A ratio | Decreased cardiac remodeling |
| Rider et al. (2009) (614) | 30 | Bariatric surgery or diet, 1 yr | 39.7 ± 7.6 | 32.2 ± 5.3 | Improved LV diastolic function | Decreased LV and RV mass |
| Alpert et al. (2015) (615) | 67 | After bariatric surgery | 46.3 ± 5.2 | 34.5 ± 5.7 | Reduced QT interval | Decreased LV mass normalized to height |
E/a ratio, early-to-atrial wave ratio; EF, ejection fraction; LV, left ventricle; LVEDV/RVEDV, left/right ventricular end-diastolic volume; RV, right ventricle; TDI, Tissue doppler imaging.
4.1.2. Lifestyle intervention.
Lifestyle interventions are geared to modify dietary habits and physical activity. Given their low cost, great convenience, improvement in the quality of life and the minimal risk of complications, lifestyle interventions are often recommended as the first option for obesity management. Of 130 severely obese participants randomized, one-year intensive lifestyle intervention resulted in a reduction of ∼10 kg in weight and favorable changes in CVD risk factors (617). Weight regain is a common situation whenever a lifestyle intervention program is failing to produce additional weight loss and patients tend to be reluctant to maintain the long-term behavioral modification. Interestingly, a 20-yr follow-up study found that a better adherence to healthy dietary patterns weakens the genetic association with weight gain (618). Individuals at high genetic risk for obesity were proposed to receive greater benefits from improved diet quality (618). Physical inactivity is closely associated with chronic subclinical myocardial damage, while physical activity is suggested to attenuate obesity-associated cardiac remodeling (619). The salutatory impact of habitual physical activity is partially obtained from its effects on traditional cardiovascular risk factors. At the cellular level, regular physical activity contributes to maintain vascular homeostasis and to reduce systemic and cardiac inflammation (620, 621).
Although excessive CR has generated some debatable or even detrimental outcomes on cardiac geometry and contractile function (622–624), emerging studies have highlighted the potential benefits of CR on obesity-associated heart diseases (622). Thirty-four obese although otherwise healthy subjects consumed a very low-calorie diet for 6 wk. Consumption of very low-calorie diet decreased weight, myocardial fatty acid uptake, triglyceride content, mass and cardiac work. Although global insulin sensitivity was improved one-third, with insulin-stimulated myocardial glucose uptake remained unchanged (598). The INFINITE study involving 180 older (65–79 yr) obese men and women examined the effects of combined aerobic exercise with CR. The investigators noted that combination of aerobic exercise with even moderate CR (−250 kcal/day) seems to be more efficacious for cardiorespiratory fitness, fatigue and disability, and glucose control compared with exercise alone (suggested to be as effective as higher-dose CR −250 kcal/day) (625). Therefore, implementing CR with an aerobic exercise program would greatly potentiate the benefits on the heart.
4.1.3. Pharmacotherapy.
4.1.3.1. food and drug administration-approved medications for weight loss.
Drug therapy in combination with lifestyle modification, produce beneficial effects on weight control and various obesity complications, particularly CVDs (589, 626). Development and approval of antiobesity drugs, however, have been challenging due to the adverse effects (cardiac arrhythmia, cataracts and neurotoxicity) of earlier weight loss medications such as 2,4,-dinitrophernol and fenfluramine. The food and drug administration (FDA) criteria for approval of antiobesity drugs have been stringent ever since the release of standard guideline in mid-1990s. A drug must elicit a significant placebo-adjusted weight loss of >5% in 1 yr or over 35% patients should reach >5% weight loss, in addition to overt improvement of metabolic biomarkers including blood pressure, blood lipid and glucose levels (627). Up-to-date, five drugs have been approved by FDA (three monotherapies: orlistat, lorcaserin, liraglutide and two combined therapies: phentermine/topiramate, naltrexone/bupropion) for obesity. The European Medicines Agency (EMA) has approved only three drug therapies (orlistat, bupropion/naltrexone, and liraglutide). All these medications yield a placebo-adjusted weight loss of >5% over a year. Among which, phentermine-topiramate and liraglutide display the highest efficacy of >5% weight loss whereas liraglutide and naltrexone-bupropion exhibit the lowest rate of discontinuation of medication due to appearance of adverse events (628–631).
Orlistat, a selective pancreatic lipase inhibitor to alleviate intestinal digestion and absorption of fat, is approved by both FDA and EMA to induce weight loss in conjunction with caloric restriction. Orlistat is also indicated to reduce the risk of T2D and dyslipidemia, independent of its weight loss efficacy (627). Orlistat needs to be taken with meals and is indicated for patients with a BMI >30 or >28 when other risk factors (e.g., hypertension, diabetes, and hyperlipidemia) are present. Pooled data from randomized clinical trials suggested beneficial effects of orlistat in glucose tolerance and T2D development in obese individuals with impaired fasting glucose.
Lorcaserin, a hypothalamic 5-HT2C receptor agonist that acts on anorexigenic POMC neurons in the hypothalamus, is an appetite suppressant approved by the FDA in 2012 as an adjunct to caloric restriction and physical exercise for weight management in patients with BMI >30 or >27 with comorbidities (e.g., hypertension, diabetes, dyslipidemia). Lorcaserin is selective for 5-HT2C and may reduce risks associated with earlier medications of this group (such as hallucination, PAH and cardiac valvular insufficiency), Lorcaserin generates an annual weight loss of ∼3.2–3.6 kg in addition to improved metabolic indices including blood pressure and blood lipids.
Liraglutide is a glucagon-like peptide-1 (GLP-1) agonist of an incretin-derived hormone which helps to control glucose homeostasis, and food intake, Liraglutide was initially developed for treatment of T2D given its incretin property. Liraglutide not only reduces glucose levels and promotes satiety but also retards gastric emptying and lowers bodyweight in a dose-dependent manner (628). It is the only injectable antiobesity medication that promotes weight loss by fostering satiety via hypothalamic stimulation and retarding gastric emptying. It is approved by FDA and EMA for patients with BMI >30 or >27 with obesity-related comorbidities.
Phentermine/topiramate is classified as a sympathomimetic that stimulates noradrenaline release and inhibits appetite, while topiramate is an anticonvulsant that maximizes body weight loss associated with phentermine use although the mechanism behind appetite suppression remains at large. It is considered an effective weight loss agent with reported 6.6–8.6 kg weight loss over a period of 12 mo.
Naltrexone/bupropion is an opiate antagonist, whereas bupropion is a weak dopamine and noradrenaline reuptake inhibitor. Monotherapy of these medications has been used for management of addiction to nicotine and alcohol. Not surprisingly, combination therapy was approved for the FDA and EMA to elicit central nervous system reward effects on food intake and satiety though antagonistic feedback inhibition. for obesity treatment. Naltrexone/bupropion promotes satiety via stimulation of POMC-mediated release of melanocyte-stimulating hormone (MSH) to lower food intake and facilitate energy expenditure. Naltrexone/bupropion is indicated for BMI >30 or >27 in association with at least one of the obesity-related comorbidities.
4.1.3.2. insulin secretagogues.
In addition to the FDA-approved antiobesity medications, a number of drugs commonly used in metabolic diseases such as T2D may also offer benefit in clinical obesity management. Dipeptidyl peptidase 4 (DPP-4) inhibitors are used through suppression of degradation of incretin hormones to maintain plasma glucagon-like peptide (GLP-1) and gastric inhibitory polypeptide (GIP) levels, resulting in facilitation of insulin secretion in pancreatic β cells and inhibition of glucagon release in pancreatic α cells as well as diverse GLP-1 biological response through GLP-1 receptor ubiquitously expressed in various tissues (632). More evidence has suggested that inhibition of DPP-4 using linagliptin improved obesity-associated insulin resistance and inflammation via intervening the M1/M2 macrophage phenotypical switch (633–636). Earlier evidence noted little adverse effects in body weight, ischemic incidence, heart rate and blood pressure in patients using DPP-4 inhibitors. Recent findings favor beneficial clinical outcomes of DPP-4 (637, 638). For example, risk of all-cause mortality may be drastically lowered when insulin is administered with DPP-4 inhibitors compared with insulin plus non-DPP-4 inhibitors (639).
4.1.3.3. potential therapeutic targets.
4.1.3.3.1. Autophagy.
Both clinical and experimental findings support an essential role for autophagy in maintaining metabolic homeostasis in obesity (640). To this end, autophagy-targeting compounds have been applied in clinical or preclinical settings. It has been well documented that induction of autophagy is mainly derived from nutrient sensing signaling of mTOR, AMPK, and the insulin-IGF1 cascade (87, 133, 453). Hyperactivation of mTOR provokes metabolic derangement by suppressed autophagy (641). Therefore, nutrient sensing through mTOR is cardinal for metabolic regulation and serves as a likely target for intervention of autophagy in metabolic derangement and lysosomal dysfunction (642). For example, the transcriptional factor TFEB mediates mTOR phosphorylation to govern autophagy flux and sustained transcriptional regulation of autophagy (643). Many drugs with proven benefits in the therapeutics of obesity and T2D involve autophagy regulatory mechanism, including metformin, thiazolidinedione pioglitazone, and DPP-4 inhibitors, as summarized in our recent review (133). Nonetheless, it is noteworthy that these metabolic regulatory medications may produce off-target effects thus it is premature to credit autophagy as their only and main mechanism for metabolic regulation in obesity.
4.1.3.3.2. Adiponectin.
As an anti-inflammatory cytokine, chronic overexpression of adiponectin provokes abrupt rises in subcutaneous fat, and retards diet-induced insulin resistance (644). It was revealed that classical antidiabetic agents including PPARγ agonists (e.g., thiazolidinediones) are capable of stimulating circulating levels of adiponectin. More evidence has revealed beneficial effect of adiponectin in hepatomegaly, liver steatosis, and liver injury in obesity. Although the precise mechanism of action behind adiponectin-elicited metabolic benefit remains elusive, the ability of adiponectin to promote carnitine palmitoyltransferase I and hepatic FAO may play a role, with both enzymes or processes heavily involved in fatty acid synthesis (645).
4.1.3.3.3. Antiinflammatory agents.
Obesity is commonly associated with a chronic low-grade inflammation. Not surprisingly, anti-inflammatory drugs are potentially effective in obesity, with low risks of adverse effects. Salsalate, a prodrug of salicylate to inhibit IKKβ/NF-κB, improves glycemic control in patients with T2D (646). Along the same line, inhibition of proinflammatory cytokines also shows promises in metabolic regulation. For example, IL-1 receptor antagonists (anakinra) have been shown to benefit glycemia and β-cell function (647). TNF-α blockade with etanercept, on the other hand, failed to display major response in insulin sensitivity although improvement on circulating inflammatory cytokines and glucose were noted (648).
4.1.3.3.4. Aldosterone antagonists: eplerenone.
As mentioned earlier, Na retention is a central component of obesity-related HFpEF, where diuretics serve as a logical therapeutic regimen (322). Obese patients are usually responsive to diuretics albeit with worsened renal function following natriuresis. Given the overproduction of aldosterone in obesity due to hyperactivation of RAAS and adipokine leptin, Na retention in obesity may be better addressed using aldosterone antagonists. Moreover, adipocytes also synthesize aldosterone directly, and elevated neprilysin in obesity diminishes natriuretic peptide-induced suppression on aldosterone secretion. In addition to Na retention, hyperaldosteronism also promotes epicardial adipose tissue inflammation and therefore onset of microvascular rarefaction and fibrosis in myocardium. In this case, perivisceral fat transforms into a proinflammatory phenotype in a mineralocorticoid receptor-dependent manner (649). Recent work also implicates increased aldosterone and coronary artery MR activation in promotion of Western diet-induced cardiomyocyte stiffness and diastolic dysfunction (650).
4.1.3.3.5. PPARγ agonists thiazolidinediones.
Drugs targeting on insulin secretion (secretagogues such as sulfonylureas and meglitinides) or insulin sensitivity [insulin sensitizers such as thiazolidinediones (TZDs) and metformin] are the mainstream therapeutic options for T2D. In particular, TZDs remain a first-line therapeutic option for T2D, courtesy of the antihyperglycemic properties elicited by PPARγ, which regulate essential genes involved in glucose and lipid metabolism.
Given that obesity is the single most independent risk factor for T2D, many pharmacotherapies employed in the management of T2D received some favorable indications in obesity including the FDA-approved antidiabetic drugs metformin, insulin therapy, PPARγ agonists sodium–glucose cotransporter 2 (SGLT2) inhibitors, sulfonylureas, meglitinides, dipeptidyl peptidase 4 (DPP-4), and inhibitors (589, 629, 651–653). Moreover, novel targets recognized in the therapy of T2D such as free fatty acid receptor 1 (also known as G protein-coupled receptor 40), glucokinase, and protein tyrosine phosphatase 1B have not been fully validated in the clinical management of obesity and obesity complications. Given the complexity and the multifactorial nature of obesity, a multitarget approach is worth of exploration (654).
4.1.3.3.6. Epigenetically based pharmacotherapy.
Epigenetics has seen a drastic development over the last years although it remains challenging to apply effective epigenetically based pharmacotherapy for obesity probably due to incomplete picture of epigenome regulation by metabolic stress (655). This is particular helpful for early life deleterious epigenetic programming (656). Epigenetic therapies inhibiting DNA Methyltransferase DNA methyltransferases (DNMTs) or histone deacetylases (HDACs) have shown some promises. Three HDAC inhibitors, as well as two DNMT inhibitors are FDA-approved for cancer therapy (657). Several HDAC and DNMT inhibitors as well as candidates targeting ncRNAs are currently under preclinical studies. Despite the benefit for Na restriction and nutrient supplement to control DNA methylation, employment of such measures to control lipid and glucose levels in obesity is still debatable. One obvious disadvantage is the likely unspecific and profound epigenetic deregulation nature of these drugs. Generally speaking, epigenetic-based therapy offers a potential way to prevent and mitigate chronic diseases through altering or correcting epigenetic abnormalities (658). Higher levels of LDL particles and lower levels of HDL particles were noted in coronary heart disease in morbid obese postmenopausal women (659). Circulating proinflammatory biomarkers offer potential utility in predicting the risk of coronary artery disease through its positive correlation with pericoronary fat (660). However, a number of drawbacks still hinder the clinical applications of these traditional biomarkers. Given the lack of efficient biomarkers for early diagnosis of cardiac anomalies and the emerging role of epigenetic traits in obesity, “BMI”-associated epigenetic biomarkers have gained some popularities in the early diagnosis and management of obesity-induced CVD. Genomic imprinting is a process of epigenetic modification which allows the gene to be expressed in a parent-of-origin specific manner. In this vein, investigators identified a nonclassical imprinted gene dysregulation in the Trim28 haploinsufficiency-induced obesity (661). This notion of alternate epigenetic trajectories in obesity points to a new direction for pharmacological drug development targeting the potential “on-and-off” switch for obesity phenotype manifestation.
4.1.4. Bariatric surgery.
In comparison to lifestyle interventions and pharmacological agents, bariatric surgery has long been established as a more effective and durable intervention for weight-loss and remission of cardiovascular risks (662, 663). Three major types of bariatric surgery are widely conceived (664). Laparoscopic adjustable gastric banding involves placing an inflatable silicone band around the upper stomach, without anatomical gut changes, to reversibly restrict the transit of ingested food. Roux-en-Y gastric bypass restricts food intake by reconstructing the alimentary canal, such that food bypasses majority of the gastrointestinal tract. The lately developed vertical-sleeve gastrectomy involves excision of more than half of the stomach to accelerate the gastric emptying. The use of the latter two procedures has escalated due to larger weight-loss (∼25% and 30%, respectively) and less reoperation rates than gastric banding (662, 665).
With the well-perceived cardiovascular risks in obesity, proven clinical benefits are solidified for bariatric surgery, especially after Roux-en-Y gastric bypass and vertical-sleeve gastrectomy (666). At a 2-yr follow up, gastric bypass surgery reversed LV remodeling and preserved LV and RV function, along with weight-loss (300). Compared with gastric banding, Roux-en-Y gastric bypass restricts reduced more abdominal visceral fat and ameliorated LV remodeling and aortic stiffness caused by obesity (667). Furthermore, bariatric surgery leads to remission of obesity-related insulin resistance, hypertension, and other metabolic complications (665, 668). TABLE 2 summarizes proven benefits of bariatric surgery on overweight- and obesity-induced cardiac remodeling and contractile dysfunction including reduced LV mass, wall thickness, as well as systolic and diastolic functional indices. Nonetheless, it is noteworthy that a cadre of clinical observations failed to note discernable changes in certain aspects (such as systolic or diastolic function) in obese individuals despite clear weight loss, indicating possible disparate response of weight loss on reversibility of cardiac remodeling and contractile dysfunction in obese individuals. Among the pleiotropic effects of bariatric surgery, complicated mechanisms that contribute to the prolonged cardiometabolic improvements following bariatric surgery include stable weight-loss, sustained satiety, subsequent changes in eating habits and gastrointestinal hormone activity, whereas further investigation is required (669).
As with all procedures, potential risks must be weighed against its benefits before proceeding with bariatric surgery. Limitations remain, including the high cost initially, potential risks for weight regain, surgical revisions, short- and long-term complications, and the lifelong requirement for nutritional supplementation (665). Furthermore, pharmacological intervention is still a viable approach to most cardiovascular anomalies. Collectively, Roux-en-Y gastric bypass and vertical-sleeve gastrectomy are feasible alternatives to the treatment of severe and/or refractory obesity, taking into account the potential risks and high cost.
5. SUMMARY AND CONCLUSION
Here we provided a contemporary review that iterates the evidence for the existence of an entity termed obesity cardiomyopathy and putative mechanisms from the perspectives of genetics and epigenetics for its pathogenesis, as well as therapeutic options/implications for this disorder. Clinical findings have underscored the presence of ventricular dysfunction in obesity independent of hypertension, coronary heart disease and other conventional causes of CVD. Experimental evidence has also confirmed pathophysiological changes in myocardial structure and function in genetically predisposed and diet-induced obesity. Contemporary understanding of the mechanisms underlying obesity cardiomyopathy include metabolic disturbances (insulin resistance, abnormal glucose transport, increased FAs, lipotoxicity, and amino acid derangement), changes in intracellular Ca2+ homeostasis, oxidative stress, autophagy dysregulation, myocardial fibrosis, cardiac autonomic neuropathy (denervation or adrenergic and renin-angiotensin aldosterone overflow), inflammation, small coronary vessel disease (microangiopathy), impaired coronary flow reserve, and coronary artery endothelial dysfunction. In addition, epigenetic modifications also participate in the etiology of obesity cardiomyopathy. Ample evidence has been engaged toward the management of obesity cardiomyopathy, although effective and targeted medications and procedures are still lacking. Nonpharmacological approaches such as lifestyle modification (e.g., exercise and diet control) may also benefit heart health in obesity (670).
GRANTS
Work carried out in the authors’ laboratories has been supported in part by National Institutes of Health, American Diabetes Association (7-13-BS-142-BR), National Key R&D Program of China (2017YFA0506000), University of Wyoming Faculty Grant-in-Aid, Natural Science Foundation of China (81770261, 82000351, and 81521001), Science and Technology Innovation Project of the Chinese Academy of Medical Sciences (Health and Longevity Pilot Special Project 2019-RC-HL-021), Shanghai Municipal Science and Technology Committee of Shanghai outstanding academic leaders plan no. 20XD1420900, and the Training Program of Excellent Academic Leaders of Shanghai Health Mission (2018BR25).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.R. and S.W. prepared figures; J.R., N.N.W., and J.R.S. drafted manuscript; J.R., N.N.W., S.W., J.R.S. and Y.Z. edited and revised manuscript; J.R., N.N.W., S.W., J.R.S. and Y.Z. approved final version of manuscript.
ACKNOWLEDGEMENTS
We wholeheartedly appreciate all members in Jun Ren and Yingmei Zhang’s research group for valuable contributions and discussion to this project. We sincerely apologize to those authors whose important work cannot be included due to space limit.
REFERENCES
- 1.Bluher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol 15: 288–298, 2019. doi: 10.1038/s41574-019-0176-8. [DOI] [PubMed] [Google Scholar]
- 2.Abarca-Gómez L, Abdeen ZA, Hamid ZA, Abu-Rmeileh NM, Acosta-Cazares B, Acuin C, et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 390: 2627–2642, 2017. doi: 10.1016/S0140-6736(17)32129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism 92: 6–10, 2019. doi: 10.1016/j.metabol.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384: 766–781, 2014. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA 309: 71–82, 2013. doi: 10.1001/jama.2012.113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdelaal M, Le Roux CW, Docherty NG. Morbidity and mortality associated with obesity. Ann Transl Med 5: 161, 2017. doi: 10.21037/atm.2017.03.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tremmel M, Gerdtham UG, Nilsson PM, Saha S. Economic burden of obesity: a systematic literature review. Int J Environ Res Public Health 14: 435, 2017. doi: 10.3390/ijerph14040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dietz WH. The response of the US Centers for Disease Control and Prevention to the obesity epidemic. Annu Rev Public Health 36: 575–596, 2015. doi: 10.1146/annurev-publhealth-031914-122415. [DOI] [PubMed] [Google Scholar]
- 9.World Health Organtization (WHO). Prevalence of Obesity Among Adults, BMI >= 30 (age-standardized estimate) (%). https://www.who.int/data/gho/data/indicators/indicator-details/GHO/prevalence-of-obesity-among-adults-bmi-=-30-(age-standardized-estimate)-(-). [2021].
- 10.Chopra S, Malhotra A, Ranjan P, Vikram NK, Singh N. Lifestyle-related advice in the management of obesity: a step-wise approach. J Educ Health Promot 9: 239, 2020. doi: 10.4103/jehp.jehp_216_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keating C, Backholer K, Peeters A. Prevalence of overweight and obesity in children and adults. Lancet 384: 2107–2108, 2014. doi: 10.1016/s0140-6736(14)62367-9. [DOI] [PubMed] [Google Scholar]
- 12.Morgen CS, Sorensen TI. Obesity: global trends in the prevalence of overweight and obesity. Nat Rev Endocrinol 10: 513–514, 2014. doi: 10.1038/nrendo.2014.124. [DOI] [PubMed] [Google Scholar]
- 13.Organisation for Economic Co-operation and Development (OECD). Obesity Update. https://www.oecd.org/els/health-systems/Obesity-Update-2017.pdf. [2017].
- 14.Van Gaal LF, Maggioni AP. Overweight, obesity, and outcomes: fat mass and beyond. Lancet 383: 935–936, 2014. doi: 10.1016/S0140-6736(13)62076-0. [DOI] [PubMed] [Google Scholar]
- 15.Elia L, Condorelli G. RNA (Epi)genetics in cardiovascular diseases. J Mol Cell Cardiol 89: 11–16, 2015. doi: 10.1016/j.yjmcc.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 16.Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol 26: 968–976, 2006. doi: 10.1161/01.atv.0000216787.85457.f3. [DOI] [PubMed] [Google Scholar]
- 17.Cavalera M, Wang J, Frangogiannis NG. Obesity, metabolic dysfunction, and cardiac fibrosis: pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl Res 164: 323–335, 2014. doi: 10.1016/j.trsl.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5.24 million UK adults. Lancet 384: 755–765, 2014. doi: 10.1016/S0140-6736(14)60892-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bray GA, Kim KK, Wilding JP, World Obesity Foundation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev 18: 715–723., 2017. doi: 10.1111/obr.12551. [DOI] [PubMed] [Google Scholar]
- 20.Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med 377: 13–27, 2017. doi: 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aune D, Sen A, Prasad M, Norat T, Janszky I, Tonstad S, Romundstad P, Vatten LJ. BMI and all cause mortality: systematic review and non-linear dose-response meta-analysis of 230 cohort studies with 3.74 million deaths among 30.3 million participants. BMJ 353: i2156, 2016. doi: 10.1136/bmj.i2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C, Ye Y, Zhang Y, Pan XF, Pan A. Weight change across adulthood in relation to all cause and cause specific mortality: prospective cohort study. BMJ 367: l5584, 2019. doi: 10.1136/bmj.l5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadde KM, Martin CK, Berthoud HR, Heymsfield SB. Obesity: pathophysiology and management. J Am Coll Cardiol 71: 69–84, 2018. doi: 10.1016/j.jacc.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall KD, Sacks G, Chandramohan D, Chow CC, Wang YC, Gortmaker SL, Swinburn BA. Quantification of the effect of energy imbalance on bodyweight. Lancet 378: 826–837, 2011. doi: 10.1016/S0140-6736(11)60812-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanhope KL. Sugar consumption, metabolic disease and obesity: the state of the controversy. Crit Rev Clin Lab Sci 53: 52–67, 2016. doi: 10.3109/10408363.2015.1084990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Millar L, Rowland B, Nichols M, Swinburn B, Bennett C, Skouteris H, Allender S. Relationship between raised BMI and sugar sweetened beverage and high fat food consumption among children. Obesity 22: E96–103, 2014. doi: 10.1002/oby.20665. [DOI] [PubMed] [Google Scholar]
- 27.Fung TT, Rimm EB, Spiegelman D, Rifai N, Tofler GH, Willett WC, Hu FB. Association between dietary patterns and plasma biomarkers of obesity and cardiovascular disease risk. Am J Clin Nutr 73: 61–67, 2001. doi: 10.1093/ajcn/73.1.61. [DOI] [PubMed] [Google Scholar]
- 28.McNaughton SA, Ball K, Mishra GD, Crawford DA. Dietary patterns of adolescents and risk of obesity and hypertension. J Nutr 138: 364–370, 2008. doi: 10.1093/jn/138.2.364. [DOI] [PubMed] [Google Scholar]
- 29.Liou YM, Liou TH, Chang LC. Obesity among adolescents: sedentary leisure time and sleeping as determinants. J Adv Nurs 66: 1246–1256, 2010. doi: 10.1111/j.1365-2648.2010.05293.x. [DOI] [PubMed] [Google Scholar]
- 30.Keith SW, Redden DT, Katzmarzyk PT, Boggiano MM, Hanlon EC, Benca RM, Ruden D, Pietrobelli A, Barger JL, Fontaine KR, Wang C, Aronne LJ, Wright SM, Baskin M, Dhurandhar NV, Lijoi MC, Grilo CM, DeLuca M, Westfall AO, Allison DB. Putative contributors to the secular increase in obesity: exploring the roads less traveled. Int J Obes (Lond) 30: 1585–1594, 2006. doi: 10.1038/sj.ijo.0803326. [DOI] [PubMed] [Google Scholar]
- 31.Hu FB, Li TY, Colditz GA, Willett WC, Manson JE. Television watching and other sedentary behaviors in relation to risk of obesity and type 2 diabetes mellitus in women. JAMA 289: 1785–1791, 2003. doi: 10.1001/jama.289.14.1785. [DOI] [PubMed] [Google Scholar]
- 32.Chahal H, Fung C, Kuhle S, Veugelers PJ. Availability and night-time use of electronic entertainment and communication devices are associated with short sleep duration and obesity among Canadian children. Pediatr Obes 8: 42–51, 2013. doi: 10.1111/j.2047-6310.2012.00085.x. [DOI] [PubMed] [Google Scholar]
- 33.Katzmarzyk PT, Barreira TV, Broyles ST, Champagne CM, Chaput JP, Fogelholm M, Hu G, Johnson WD, Kuriyan R, Kurpad A, Lambert EV, Maher C, Maia J, Matsudo V, Olds T, Onywera V, Sarmiento OL, Standage M, Tremblay MS, Tudor-Locke C, Zhao P, Church TS. Physical activity, sedentary time, and obesity in an international sample of children. Med Sci Sports Exerc 47: 2062–2069, 2015. doi: 10.1249/MSS.0000000000000649. [DOI] [PubMed] [Google Scholar]
- 34.Healy GN, Wijndaele K, Dunstan DW, Shaw JE, Salmon J, Zimmet PZ, Owen N. Objectively measured sedentary time, physical activity, and metabolic risk: the Australian Diabetes, Obesity and Lifestyle Study (AusDiab). Diabetes Care 31: 369–371, 2008. doi: 10.2337/dc07-1795. [DOI] [PubMed] [Google Scholar]
- 35.Maher CA, Mire E, Harrington DM, Staiano AE, Katzmarzyk PT. The independent and combined associations of physical activity and sedentary behavior with obesity in adults: NHANES 2003-06. Obesity 21: E730–737, 2013. doi: 10.1002/oby.20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gali Ramamoorthy T, Begum G, Harno E, White A. Developmental programming of hypothalamic neuronal circuits: impact on energy balance control. Front Neurosci 9: 126, 2015. doi: 10.3389/fnins.2015.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thornburg KL. The programming of cardiovascular disease. J Dev Origins Health Dis 6: 366–376, 2015. doi: 10.1017/S2040174415001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong M, Zheng Q, Ford SP, Nathanielsz PW, Ren J. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J Mol Cell Cardiol 55: 111–116, 2013. doi: 10.1016/j.yjmcc.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q, Zhu C, Sun M, Maimaiti R, Ford SP, Nathanielsz PW, Ren J, Guo W. Maternal obesity impairs fetal cardiomyocyte contractile function in sheep. FASEB J 33: 2587–2598, 2019. doi: 10.1096/fj.201800988R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kandadi MR, Hua Y, Zhu M, Turdi S, Nathanielsz PW, Ford SP, Nair S, Ren J. Influence of gestational overfeeding on myocardial proinflammatory mediators in fetal sheep heart. J Nutr Biochem 24: 1982–1990, 2013. doi: 10.1016/j.jnutbio.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Ge W, Hu N, George LA, Ford SP, Nathanielsz PW, Wang XM, Ren J. Maternal nutrient restriction predisposes ventricular remodeling in adult sheep offspring. J Nutr Biochem 24: 1258–1265, 2013. doi: 10.1016/j.jnutbio.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Dong F, Ford SP, Fang CX, Nijland MJ, Nathanielsz PW, Ren J. Maternal nutrient restriction during early to mid gestation up-regulates cardiac insulin-like growth factor (IGF) receptors associated with enlarged ventricular size in fetal sheep. Growth Horm IGF Res 15: 291–299, 2005. doi: 10.1016/j.ghir.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 43.Chen LW, Aubert AM, Shivappa N, Bernard JY, Mensink-Bout SM, Geraghty AA, Mehegan J, Suderman M, Polanska K, Hanke W, Jankowska A, Relton CL, Crozier SR, Harvey NC, Cooper C, Hanson M, Godfrey KM, Gaillard R, Duijts L, Heude B, Hebert JR, McAuliffe FM, Kelleher CC, Phillips CM. Maternal dietary quality, inflammatory potential and childhood adiposity: an individual participant data pooled analysis of seven European cohorts in the ALPHABET consortium. BMC Med 19: 33, 2021. doi: 10.1186/s12916-021-01908-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King SE, Skinner MK. Epigenetic transgenerational inheritance of obesity susceptibility. Trends Endocrinol Metab 31: 478–494, 2020. doi: 10.1016/j.tem.2020.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong Y, Xue Y, Li X, Zhang Z, Zhou W, Marcolongo P, Benedetti A, Mao S, Han L, Ding G, Sun Z. Inter- and transgenerational effects of paternal exposure to inorganic arsenic. Adv Sci 8: 2002715, 2021. doi: 10.1002/advs.202002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sargent J. Obesity: obesity turns the hepatic epigenetic clock forward. Nat Rev Endocrinol 11: 2, 2015. doi: 10.1038/nrendo.2014.198, 10.1038/nrendo.2014.194. 25365923, 25350063 [DOI] [PubMed] [Google Scholar]
- 47.Beyerlein A, Kusian D, Ziegler AG, Schaffrath-Rosario A, von Kries R. Classification tree analyses reveal limited potential for early targeted prevention against childhood overweight. Obesity 22: 512–517, 2014. doi: 10.1002/oby.20628. [DOI] [PubMed] [Google Scholar]
- 48.Osorio JO. Looking at the epigenetic link between obesity and its consequences–the promise of EWAS. Nat Rev Endocrinol 10: 249, 2014. doi: 10.1038/nrendo.2014.42. [DOI] [PubMed] [Google Scholar]
- 49.Saeed S, Bonnefond A, Tamanini F, Mirza MU, Manzoor J, Janjua QM, Din SM, Gaitan J, Milochau A, Durand E, Vaillant E, Haseeb A, De Graeve F, Rabearivelo I, Sand O, Queniat G, Boutry R, Schott DA, Ayesha H, Ali M, Khan WI, Butt TA, Rinne T, Stumpel C, Abderrahmani A, Lang J, Arslan M, Froguel P. Loss-of-function mutations in ADCY3 cause monogenic severe obesity. Nat Genet 50: 175–179, 2018. doi: 10.1038/s41588-017-0023-6. [DOI] [PubMed] [Google Scholar]
- 50.Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci 15: 367–378, 2014. 10.1038/nrg3687, doi: 10.1038/nrn3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong M, Ren J. What fans the fire: insights into mechanisms of leptin in metabolic syndrome-associated heart diseases. Curr Pharm Design 20: 652–658, 2014. doi: 10.2174/138161282004140213160930. [DOI] [PubMed] [Google Scholar]
- 52.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 348: 1085–1095, 2003. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 53.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 19: 155–157, 1998. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 54.Nordang GB, Busk OL, Tveten K, Hanevik HI, Fell AK, Hjelmesaeth J, Holla OL, Hertel JK. Next-generation sequencing of the monogenic obesity genes LEP, LEPR, MC4R, PCSK1 and POMC in a Norwegian cohort of patients with morbid obesity and normal weight controls. Mol Genet Metab 121: 51–56, 2017. doi: 10.1016/j.ymgme.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 55.Farooqi IS, Drop S, Clements A, Keogh JM, Biernacka J, Lowenbein S, Challis BG, O’Rahilly S. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes 55: 2549–2553, 2006. doi: 10.2337/db06-0214. [DOI] [PubMed] [Google Scholar]
- 56.Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, O'Rahilly S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest 106: 271–279, 2000. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 106: 253–262, 2000. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pritchard LE, Turnbull AV, White A. Pro-opiomelanocortin processing in the hypothalamus: impact on melanocortin signalling and obesity. J Endocrinol 172: 411–421, 2002. doi: 10.1677/joe.0.1720411. [DOI] [PubMed] [Google Scholar]
- 59.Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O’Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet 16: 303–306, 1997. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 60.Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, Iwata N, Ikegawa S, Hirata M, Matsuda K, Iwasaki M, Yamaji T, Sawada N, Hachiya T, Tanno K, Shimizu A, Hozawa A, Minegishi N, Tsugane S, Yamamoto M, Kubo M, Kamatani Y. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat Genet 49: 1458–1467, 2017. doi: 10.1038/ng.3951. [DOI] [PubMed] [Google Scholar]
- 61.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518: 197–206, 2015. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Magi R, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518: 187–196, 2015. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boender AJ, van Rozen AJ, Adan RA. Nutritional state affects the expression of the obesity-associated genes Etv5, Faim2, Fto, and Negr1. Obesity (Silver Spring) 20: 2420–2425, 2012. doi: 10.1038/oby.2012.128. [DOI] [PubMed] [Google Scholar]
- 64.Horstmann A, Kovacs P, Kabisch S, Boettcher Y, Schloegl H, Tonjes A, Stumvoll M, Pleger B, Villringer A. Common genetic variation near MC4R has a sex-specific impact on human brain structure and eating behavior. PLoS One 8: e74362, 2013. doi: 10.1371/journal.pone.0074362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ho EV, Klenotich SJ, McMurray MS, Dulawa SC. Activity-based anorexia alters the expression of bdnf transcripts in the mesocorticolimbic reward circuit. PLoS One 11: e0166756, 2016. doi: 10.1371/journal.pone.0166756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022, 2011. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther doi: 10.1016/j.pharmthera.2016.03.005. 161: 52–66, 2016. [DOI] [PubMed] [Google Scholar]
- 68.Houde AA, Legare C, Biron S, Lescelleur O, Biertho L, Marceau S, Tchernof A, Vohl MC, Hivert MF, Bouchard L. Leptin and adiponectin DNA methylation levels in adipose tissues and blood cells are associated with BMI, waist girth and LDL-cholesterol levels in severely obese men and women. BMC Med Genet 16: 29, 2015. doi: 10.1186/s12881-015-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crujeiras AB, Campion J, Diaz-Lagares A, Milagro FI, Goyenechea E, Abete I, Casanueva FF, Martinez JA. Association of weight regain with specific methylation levels in the NPY and POMC promoters in leukocytes of obese men: a translational study. Regul Pept 186: 1–6, 2013. doi: 10.1016/j.regpep.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 70.Wallis N, Raffan E. The genetic basis of obesity and related metabolic diseases in humans and companion animals. Genes 11: 1378, 2020. doi: 10.3390/genes11111378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schurmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Muller TD, Tschop MH. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol 14: 140–162, 2018. doi: 10.1038/nrendo.2017.161. [DOI] [PubMed] [Google Scholar]
- 72.Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta 1832: 1136–1148, 2013. doi: 10.1016/j.bbadis.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Relling DP, Esberg LB, Fang CX, Johnson WT, Murphy EJ, Carlson EC, Saari JT, Ren J. High-fat diet-induced juvenile obesity leads to cardiomyocyte dysfunction and upregulation of Foxo3a transcription factor independent of lipotoxicity and apoptosis. J Hypertens 24: 549–561, 2006. doi: 10.1097/01.hjh.0000203846.34314.94. [DOI] [PubMed] [Google Scholar]
- 74.Ren J, Sowers JR, Walsh MF, Brown RA. Reduced contractile response to insulin and IGF-I in ventricular myocytes from genetically obese Zucker rats. Am J Physiol Heart Circ Physiol 279: H1708–H1714, 2000. doi: 10.1152/ajpheart.2000.279.4.H1708. [DOI] [PubMed] [Google Scholar]
- 75.Butler AA, Kozak LP. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes 59: 323–329, 2010. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 14: 141–148, 1978. doi: 10.1007/BF00429772. [DOI] [PubMed] [Google Scholar]
- 77.Ceylan-Isik AF, Sreejayan N, Ren J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J Mol Cell Cardiol 50 : 107–116, 2011.doi: 10.1016/j.yjmcc.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 78.Zhu X, Jiang S, Hu N, Luo F, Dong H, Kang YM, Jones KR, Zou Y, Xiong L, Ren J. Tumour necrosis factor-alpha inhibition with lenalidomide alleviates tissue oxidative injury and apoptosis in ob/ob obese mice. Clin Exp Pharmacol Physiol 41: 489–501, 2014. doi: 10.1111/1440-1681.12240. [DOI] [PubMed] [Google Scholar]
- 79.Dong F, Ren J. Fidarestat improves cardiomyocyte contractile function in db/db diabetic obese mice through a histone deacetylase Sir2-dependent mechanism. J Hypertens 25: 2138–2147, 2007. doi: 10.1097/HJH.0b013e32828626d1. [DOI] [PubMed] [Google Scholar]
- 80.Ren J, Dong F, Cai GJ, Zhao P, Nunn JM, Wold LE, Pei J. Interaction between age and obesity on cardiomyocyte contractile function: role of leptin and stress signaling. PLoS One 5: e10085, 2010. doi: 10.1371/journal.pone.0010085. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Mul JD, van Boxtel R, Bergen DJ, Brans MA, Brakkee JH, Toonen PW, Garner KM, Adan RA, Cuppen E. Melanocortin receptor 4 deficiency affects body weight regulation, grooming behavior, and substrate preference in the rat. Obesity (Silver Spring) 20: 612–621, 2012. doi: 10.1038/oby.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ren J, Ma H. Impaired cardiac function in leptin-deficient mice. Curr Hypertens Rep 10: 448–453, 2008. doi: 10.1007/s11906-008-0084-0. [DOI] [PubMed] [Google Scholar]
- 83.Ren J, Walsh MF, Jefferson L, Natavio M, Ilg KJ, Sowers JR, Brown RA. Basal and ethanol-induced cardiac contractile response in lean and obese Zucker rat hearts. J Biomed Sci 7: 390–400, 2000. doi: 10.1007/BF02255814. [DOI] [PubMed] [Google Scholar]
- 84.Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol Regul Comp Integr Physiol 273: R725–R730, 1997. doi: 10.1152/ajpregu.1997.273.2.R725. [DOI] [PubMed] [Google Scholar]
- 85.Hintz KK, Aberle NS, Ren J. Insulin resistance induces hyperleptinemia, cardiac contractile dysfunction but not cardiac leptin resistance in ventricular myocytes. Int J Obes Relat Metab Disord 27: 1196–1203, 2003. doi: 10.1038/sj.ijo.0802389. [DOI] [PubMed] [Google Scholar]
- 86.de Bem AF, Krolow R, Farias HR, de Rezende VL, Gelain DP, Moreira JC, Duarte J, de Oliveira J. Animal models of metabolic disorders in the study of neurodegenerative diseases: an overview. Front Neurosci 14: 604150, 2021. doi: 10.3389/fnins.2020.604150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Turdi S, Kandadi MR, Zhao J, Huff AF, Du M, Ren J. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol 50: 712–722, 2011. doi: 10.1016/j.yjmcc.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ceylan AF, Wang S, Kandadi MR, Chen J, Hua Y, Pei Z, Nair S, Ren J. Cardiomyocyte-specific knockout of endothelin receptor a attenuates obesity cardiomyopathy. Biochim Biophys Acta Mol Basis Dis 1864: 3339–3352, 2018. doi: 10.1016/j.bbadis.2018.07.020. [DOI] [PubMed] [Google Scholar]
- 89.Ceylan-Isik AF, Kandadi MR, Xu X, Hua Y, Chicco AJ, Ren J, Nair S. Apelin administration ameliorates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol 63: 4–13, 2013.doi: 10.1016/j.yjmcc.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 90.Ren J, Zhu BH, Relling DP, Esberg LB, Ceylan-Isik AF. High-fat diet-induced obesity leads to resistance to leptin-induced cardiomyocyte contractile response. Obesity (Silver Spring) 16: 2417–2423, 2008. doi: 10.1038/oby.2008.381. [DOI] [PubMed] [Google Scholar]
- 91.Hua Y, Zhang Y, Dolence J, Shi GP, Ren J, Nair S. Cathepsin K knockout mitigates high-fat diet-induced cardiac hypertrophy and contractile dysfunction. Diabetes 62: 498–509, 2013. doi: 10.2337/db12-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Penicaud L, Larue-Achagiotis C, Le Magnen J. Endocrine basis for weight gain after fasting or VMH lesion in rats. Am J Physiol Endocrinol Metab 245: E246–E252, 1983. doi: 10.1152/ajpendo.1983.245.3.E246. [DOI] [PubMed] [Google Scholar]
- 93.Tokunaga K, Matsuzawa Y, Fujioka S, Kobatake T, Keno Y, Odaka H, Matsuo T, Tarui S. PVN-lesioned obese rats maintain ambulatory activity and its circadian rhythm. Brain Res Bull 26: 393–396, 1991. doi: 10.1016/0361-9230(91)90012-9. [DOI] [PubMed] [Google Scholar]
- 94.Aurigemma GP, de Simone G, Fitzgibbons TP. Cardiac remodeling in obesity. Circ Cardiovasc Imaging 6: 142–152, 2013. doi: 10.1161/CIRCIMAGING.111.964627. [DOI] [PubMed] [Google Scholar]
- 95.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev 88: 389–419, 2008. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ortega FB, Lavie CJ, Blair SN. Obesity and cardiovascular disease. Circ Res 118: 1752–1770, 2016. doi: 10.1161/CIRCRESAHA.115.306883. [DOI] [PubMed] [Google Scholar]
- 97.Gupta PP, Fonarow GC, Horwich TB. Obesity and the obesity paradox in heart failure. Can J Cardiol 31: 195–202, 2015. [DOI] [PubMed] [Google Scholar]
- 98.Li L, Hua Y, Dong M, Li Q, Smith DT, Yuan M, Jones KR, Ren J. Short-term lenalidomide (Revlimid) administration ameliorates cardiomyocyte contractile dysfunction in ob/ob obese mice. Obesity (Silver Spring) 20: 2174–2185, 2012. doi: 10.1038/oby.2012.106. [DOI] [PubMed] [Google Scholar]
- 99.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med 347: 305–313, 2002. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 100.Kenchaiah S, Sesso HD, Gaziano JM. Body mass index and vigorous physical activity and the risk of heart failure among men. Circulation 119: 44–52, 2009. doi: 10.1161/CIRCULATIONAHA.108.807289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ren J, Zhang Y. Emerging potential of therapeutic targeting of autophagy and protein quality control in the management of cardiometabolic diseases. Biochim Biophys Acta 1852: 185–187, 2015. doi: 10.1016/j.bbadis.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 102.Zhang Y, Yuan M, Bradley KM, Dong F, Anversa P, Ren J. Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: role of insulin signaling and mitochondrial function. Hypertension 59: 680–693, 2012.doi: 10.1161/HYPERTENSIONAHA.111.181867. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103.Turdi S, Hu N, Ren J. Tauroursodeoxycholic acid mitigates high fat diet-induced cardiomyocyte contractile and intracellular Ca2+ anomalies. PLoS One 8: e63615, 2013. doi: 10.1371/journal.pone.0063615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cao L, Qin X, Peterson MR, Haller SE, Wilson KA, Hu N, Lin X, Nair S, Ren J, He G. CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J Mol Cell Cardiol 92: 185–195, 2016. doi: 10.1016/j.yjmcc.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Ren J. Frontiers in research: obesity and health. Introduction. Clin Exp Pharmacol Physiol 39: 158–160, 2012. doi: 10.1111/j.1440-1681.2012.05671.x. [DOI] [PubMed] [Google Scholar]
- 106.Zhang Y, Sowers JR, Ren J. Pathophysiological insights into cardiovascular health in metabolic syndrome. Exp Diabetes Res 2012: 320534, 2012. doi: 10.1155/2012/320534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wong C, Marwick TH. Obesity cardiomyopathy: diagnosis and therapeutic implications. Nat Clin Pract Cardiovasc Med 4: 480–490, 2007. doi: 10.1038/ncpcardio0964. [DOI] [PubMed] [Google Scholar]
- 108.Mechanick JI, Farkouh ME, Newman JD, Garvey WT. Cardiometabolic-based chronic disease, adiposity and dysglycemia drivers: JACC state-of-the-art review. J Am Coll Cardiol 75: 525–538, 2020. doi: 10.1016/j.jacc.2019.11.044, 10.1016/S0735-1097(20)31152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hubert HB, Feinleib M, McNamara PM, Castelli WP. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation 67: 968–977, 1983. doi: 10.1161/01.CIR.67.5.968. [DOI] [PubMed] [Google Scholar]
- 110.Khan SS, Ning H, Wilkins JT, Allen N, Carnethon M, Berry JD, Sweis RN, Lloyd-Jones DM. Association of body mass index with lifetime risk of cardiovascular disease and compression of morbidity. JAMA Cardiol 3: 280–287, 2018. doi: 10.1001/jamacardio.2018.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aune D, Sen A, Norat T, Janszky I, Romundstad P, Tonstad S, Vatten LJ. Body mass index, abdominal fatness, and heart failure incidence and mortality: a systematic review and dose-response meta-analysis of prospective studies. Circulation 133: 639–649, 2016. doi: 10.1161/CIRCULATIONAHA.115.016801. [DOI] [PubMed] [Google Scholar]
- 112.Kivimäki M, Kuosma E, Ferrie JE, Luukkonen R, Nyberg ST, Alfredsson L, Batty GD, Brunner EJ, Fransson E, Goldberg M, Knutsson A, Koskenvuo M, Nordin M, Oksanen T, Pentti J, Rugulies R, Shipley MJ, Singh-Manoux A, Steptoe A, Suominen SB, Theorell T, Vahtera J, Virtanen M, Westerholm P, Westerlund H, Zins M, Hamer M, Bell JA, Tabak AG, Jokela M. Overweight, obesity, and risk of cardiometabolic multimorbidity: pooled analysis of individual-level data for 120,813 adults from 16 cohort studies from the USA and Europe. Lancet Public Health 2: e277–e285, 2017. doi: 10.1016/S2468-2667(17)30074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Robertson J, Schaufelberger M, Lindgren M, Adiels M, Schiöler L, Torén K, McMurray J, Sattar N, Åberg M, Rosengren A. Higher body mass index in adolescence predicts cardiomyopathy risk in midlife. Circulation 140: 117–125, 2019. doi: 10.1161/CIRCULATIONAHA.118.039132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Twig G, Yaniv G, Levine H, Leiba A, Goldberger N, Derazne E, Ben-Ami Shor D, Tzur D, Afek A, Shamiss A, Haklai Z, Kark JD. Body-mass index in 2.3 million adolescents and cardiovascular death in adulthood. N Engl J Med 374: 2430–2440, 2016. doi: 10.1056/NEJMoa1503840. [DOI] [PubMed] [Google Scholar]
- 115.Kosmala W, Sanders P, Marwick TH. Subclinical myocardial impairment in metabolic diseases. JACC Cardiovasc Imaging 10: 692–703, 2017. doi: 10.1016/j.jcmg.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 116.Robertson J, Lindgren M, Schaufelberger M, Adiels M, Björck L, Lundberg CE, Sattar N, Rosengren A, Åberg M. Body mass index in young women and risk of cardiomyopathy: a long-term follow-up study in Sweden. Circulation 141: 520–529, 2020. doi: 10.1161/CIRCULATIONAHA.119.044056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang S, Ren J. Obesity paradox in aging: from prevalence to pathophysiology. Prog Cardiovasc Dis 61: 182–189, 2018. doi: 10.1016/j.pcad.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 118.Elagizi A, Kachur S, Lavie CJ, Carbone S, Pandey A, Ortega FB, Milani RV. An overview and update on obesity and the obesity paradox in cardiovascular diseases. Prog Cardiovasc Dis 61: 142–150, 2018. doi: 10.1016/j.pcad.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 119.Wu L, O'Kane AM, Peng H, Bi Y, Motriuk-Smith D, Ren J. SARS-CoV-2 and cardiovascular complications: From molecular mechanisms to pharmaceutical management. Biochem Pharmacol 178: 114114, 2020. doi: 10.1016/j.bcp.2020.114114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aghili SM, Ebrahimpur M, Arjmand B, Shadman Z, Pejman Sani M, Qorbani M, Larijani B, Payab M. Obesity in COVID-19 era, implications for mechanisms, comorbidities, and prognosis: a review and meta-analysis. Int J Obes (Lond) 45: 998–1016, 2021. doi: 10.1038/s41366-021-00776-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sharma A, Garg A, Rout A, Lavie CJ. Association of obesity with more critical illness in COVID-19. Mayo Clin Proc 95: 2040–2042, 2020. doi: 10.1016/j.mayocp.2020.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cai Q, Chen F, Wang T, Luo F, Liu X, Wu Q, He Q, Wang Z, Liu Y, Liu L, Chen J, Xu L. Obesity and COVID-19 severity in a designated hospital in Shenzhen. Diabetes Care 43: 1392–1398, 2020. doi: 10.2337/dc20-0576. [DOI] [PubMed] [Google Scholar]
- 123.Petrilli CM, Jones SA, Yang J, Rajagopalan H, O’Donnell L, Chernyak Y, Tobin KA, Cerfolio RJ, Francois F, Horwitz LI. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 369: 1966, 2020. doi: 10.1016/j.mayocp.2020.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Caussy C, Pattou F, Wallet F, Simon C, Chalopin S, Telliam C, Mathieu D, Subtil F, Frobert E, Alligier M, Delaunay D, Vanhems P, Laville M, Jourdain M, Disse E, COVID Outcomes HCL Consortium and Lille COVID–Obesity Study Group. Prevalence of obesity among adult inpatients with COVID-19 in France. Lancet Diabetes Endocrinol 8: 562–564, 2020. doi: 10.1016/S2213-8587(20)30160-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323: 1239–1242, 2020. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 126.Edqvist J, Rawshani A, Adiels M, Björck L, Lind M, Svensson AM, Gudbjörnsdottir S, Sattar N, Rosengren A. BMI, mortality, and cardiovascular outcomes in type 1 diabetes: findings against an obesity paradox. Diabetes Care 42: 1297–1304, 2019. doi: 10.2337/dc18-1446. [DOI] [PubMed] [Google Scholar]
- 127.Frank RC, Min J, Abdelghany M, Paniagua S, Bhattacharya R, Bhambhani V, Pomerantsev E, Ho JE. Obesity is associated with pulmonary hypertension and modifies outcomes. J Am Heart Assoc 9: e014195, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hingorani AD, Finan C, Schmidt AF. Obesity causes cardiovascular diseases: adding to the weight of evidence. Eur Heart J 41: 227–230, 2020. doi: 10.1093/eurheartj/ehz569. [DOI] [PubMed] [Google Scholar]
- 129.Sattar N, McInnes IB, McMurray JJ. Obesity is a risk factor for severe COVID-19 infection: multiple potential mechanisms. Circulation 142: 4–6, 2020. doi: 10.1161/CIRCULATIONAHA.120.047659. [DOI] [PubMed] [Google Scholar]
- 130.Stefan N, Birkenfeld AL, Schulze MB, Ludwig DS. Obesity and impaired metabolic health in patients with COVID-19. Nat Rev Endocrinol 16: 341–342, 2020. doi: 10.1038/s41574-020-0364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sanchis-Gomar F, Lavie CJ, Mehra MR, Henry BM, Lippi G. Obesity and outcomes in COVID-19: when an epidemic and pandemic collide. Mayo Clin Proc 95: 1445–1453, 2020. doi: 10.1016/j.mayocp.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res 113: 389–398, 2017. doi: 10.1093/cvr/cvx012. [DOI] [PubMed] [Google Scholar]
- 133.Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol 14: 356–376, 2018. doi: 10.1038/s41574-018-0009-1. [DOI] [PubMed] [Google Scholar]
- 134.Turer AT, Hill JA, Elmquist JK, Scherer PE. Adipose tissue biology and cardiomyopathy: translational implications. Circ Res 111: 1565–1577, 2012. doi: 10.1161/CIRCRESAHA.111.262493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kolwicz SC, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113: 603–616, 2013. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gibb AA, Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res 123: 107–128, 2018. doi: 10.1161/CIRCRESAHA.118.312017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hardy OT, Czech MP, Corvera S. What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes 19: 81–87, 2012. doi: 10.1097/MED.0b013e3283514e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lavallard VJ, Meijer AJ, Codogno P, Gual P. Autophagy, signaling and obesity. Pharmacol Res 66: 513–525, 2012. doi: 10.1016/j.phrs.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 139.Kotsis V, Nilsson P, Grassi G, Mancia G, Redon J, Luft F, Schmieder R, Engeli S, Stabouli S, Antza C, Pall D, Schlaich M, Jordan J, WG on Obesity, Diabetes, the High Risk Patient, European Society of Hypertension. New developments in the pathogenesis of obesity-induced hypertension. J Hypertens 33: 1499–1508, 2015. doi: 10.1097/HJH.0000000000000645. [DOI] [PubMed] [Google Scholar]
- 140.Lavie CJ, Alpert MA, Arena R, Mehra MR, Milani RV, Ventura HO. Impact of obesity and the obesity paradox on prevalence and prognosis in heart failure. JACC Heart Fail 1: 93–102, 2013. doi: 10.1016/j.jchf.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 141.Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 18: 400–423, 2021. doi: 10.1038/s41569-020-00480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lavie CJ, Pandey A, Lau DH, Alpert MA, Sanders P. Obesity and atrial fibrillation prevalence, pathogenesis, and prognosis: effects of weight loss and exercise. J Am Coll Cardiol 70: 2022–2035, 2017. doi: 10.1016/j.jacc.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 143.Dhuper S, Abdullah RA, Weichbrod L, Mahdi E, Cohen HW. Association of obesity and hypertension with left ventricular geometry and function in children and adolescents. Obesity (Silver Spring, MD) 19: 128–133, 2011. doi: 10.1038/oby.2010.134. [DOI] [PubMed] [Google Scholar]
- 144.Shah AS, Khoury PR, Dolan LM, Ippisch HM, Urbina EM, Daniels SR, Kimball TR. The effects of obesity and type 2 diabetes mellitus on cardiac structure and function in adolescents and young adults. Diabetologia 54: 722–730, 2011. doi: 10.1007/s00125-010-1974-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Utz W, Engeli S, Haufe S, Kast P, Hermsdorf M, Wiesner S, Pofahl M, Traber J, Luft FC, Boschmann M, Schulz-Menger J, Jordan J. Myocardial steatosis, cardiac remodelling and fitness in insulin-sensitive and insulin-resistant obese women. Heart 97: 1585–1589, 2011. doi: 10.1136/hrt.2011.224451. [DOI] [PubMed] [Google Scholar]
- 146.Canepa M, Sorensen LL, Pozios I, Dimaano VL, Luo HC, Pinheiro AC, Strait JB, Brunelli C, Abraham MR, Ferrucci L, Abraham TP. Comparison of clinical presentation, left ventricular morphology, hemodynamics, and exercise tolerance in obese versus nonobese patients with hypertrophic cardiomyopathy. Am J Cardiol 112: 1182–1189, 2013. doi: 10.1016/j.amjcard.2013.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Olivotto I, Maron BJ, Tomberli B, Appelbaum E, Salton C, Haas TS, Gibson CM, Nistri S, Servettini E, Chan RH, Udelson JE, Lesser JR, Cecchi F, Manning WJ, Maron MS. Obesity and its association to phenotype and clinical course in hypertrophic cardiomyopathy. J Am Coll Cardiol 62: 449–457, 2013. doi: 10.1016/j.jacc.2013.03.062. [DOI] [PubMed] [Google Scholar]
- 148.Dahiya R, Shultz SP, Dahiya A, Fu J, Flatley C, Duncan D, Cardinal J, Kostner KM, Byrne NM, Hills AP, Harris M, Conwell LS, Leong GM. Relation of reduced preclinical left ventricular diastolic function and cardiac remodeling in overweight youth to insulin resistance and inflammation. Am J Cardiol 115: 1222–1228, 2015. doi: 10.1016/j.amjcard.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 149.Rider OJ, Ajufo E, Ali MK, Petersen SE, Nethononda R, Francis JM, Neubauer S. Myocardial tissue phase mapping reveals impaired myocardial tissue velocities in obesity. Int J Cardiovasc Imaging 31: 339–347, 2015. doi: 10.1007/s10554-014-0548-z. [DOI] [PubMed] [Google Scholar]
- 150.Yagmur J, Cansel M, Kurtoglu E, Hidayet S, Acıkgoz N, Ermis N, Ozyalin F. Assessment of left atrial volume and function by real time three-dimensional echocardiography in obese patients. Echocardiography 34: 210–216, 2017. doi: 10.1111/echo.13417. [DOI] [PubMed] [Google Scholar]
- 151.Finocchiaro G, Papadakis M, Dhutia H, Cole D, Behr ER, Tome M, Sharma S, Sheppard MN. Obesity and sudden cardiac death in the young: clinical and pathological insights from a large national registry. Eur J Prev Cardiolog 25: 395–401, 2018. doi: 10.1177/2047487317751291. [DOI] [PubMed] [Google Scholar]
- 152.Blomstrand P, Sjöblom P, Nilsson M, Wijkman M, Engvall M, Länne T, Nyström FH, Östgren CJ, Engvall J. Overweight and obesity impair left ventricular systolic function as measured by left ventricular ejection fraction and global longitudinal strain. Cardiovasc Diabetol 28: 113, 2018. doi: 10.1186/s12933-018-0756-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.El Saiedi SA, Mira MF, Sharaf SA, Al Musaddar MM, El Kaffas RM, AbdelMassih AF, Barsoum IH. Left ventricular diastolic dysfunction without left ventricular hypertrophy in obese children and adolescents: a Tissue Doppler Imaging and Cardiac Troponin I Study. Cardiol Young 28: 76–84, 2018. doi: 10.1017/S1047951117001627. [DOI] [PubMed] [Google Scholar]
- 154.Balaji S, DiLorenzo MP, Fish FA, Etheridge SP, Aziz PF, Russell MW, et al. Impact of obesity on left ventricular thickness in children with hypertrophic cardiomyopathy. Pediatr Cardiol 40: 1253–1257, 2019. doi: 10.1007/s00246-019-02145-9. [DOI] [PubMed] [Google Scholar]
- 155.Fumagalli C, Maurizi N, Day SM, Ashley EA, Michels M, Colan SD, Jacoby D, Marchionni N, Vincent-Tompkins J, Ho CY, Olivotto I, for the SHARE Investigators. Association of obesity with adverse long-term outcomes in hypertrophic cardiomyopathy. JAMA Cardiol 5: 65, 2020. doi: 10.1001/jamacardio.2019.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Park JB, Kim DH, Lee H, Hwang IC, Yoon YE, Park HE, Choi SY, Kim YJ, Cho GY, Han K, Ommen SR, Kim HK. Obesity and metabolic health status are determinants for the clinical expression of hypertrophic cardiomyopathy. Eur J Prev Cardiol 27: 2849–1857, 2020. doi: 10.1177/2047487319889714. [DOI] [PubMed] [Google Scholar]
- 157.Litwin SE, Adams TD, Davidson LE, McKinlay R, Simper SC, Ranson L, Hunt SC. Longitudinal changes in cardiac structure and function in severe obesity: 11-year follow-up in the Utah Obesity Study. J Am Heart Assoc 9: e014542, 2020. doi: 10.1161/jaha.119.014542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bonow RO, Eckel RH. Diet, obesity, and cardiovascular risk. N Engl J Med 348: 2057–2058, 2003. doi: 10.1056/NEJMp030053. [DOI] [PubMed] [Google Scholar]
- 159.Burgio E, Lopomo A, Migliore L. Obesity and diabetes: from genetics to epigenetics. Mol Biol Rep 42: 799–818, 2015. doi: 10.1007/s11033-014-3751-z. [DOI] [PubMed] [Google Scholar]
- 160.Streng KW, Voors AA, Hillege HL, Anker SD, Cleland JG, Dickstein K, Filippatos G, Metra M, Ng LL, Ponikowski P, Samani NJ, van Veldhuisen DJ, Zwinderman AH, Zannad F, Damman K, van der Meer P, Lang CC. Waist-to-hip ratio and mortality in heart failure. Eur J Heart Fail 20: 1269–1277, 2018. doi: 10.1002/ejhf.1244. [DOI] [PubMed] [Google Scholar]
- 161.Norhammar A, Schenck-Gustafsson K. Type 2 diabetes and cardiovascular disease in women. Diabetologia 56: 1–9, 2013. doi: 10.1007/s00125-012-2694-y, 10.1007/s00125-013-3012-z. 23949610, [DOI] [PubMed] [Google Scholar]
- 162.Murphy E, Amanakis G, Fillmore N, Parks RJ, Sun J. Sex differences in metabolic cardiomyopathy. Cardiovasc Res 113: 370–377, 2017. doi: 10.1093/cvr/cvx008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nalliah CJ, Sanders P, Kottkamp H, Kalman JM. The role of obesity in atrial fibrillation. Eur Heart J 37: 1565–1572, 2016. doi: 10.1093/eurheartj/ehv486. [DOI] [PubMed] [Google Scholar]
- 164.Yazici D, Sezer H. Insulin resistance, obesity and lipotoxicity. Adv Exp Med Biol 960: 277–304, 2017. doi: 10.1007/978-3-319-48382-5_12. [DOI] [PubMed] [Google Scholar]
- 165.Owan T, Litwin SE. Is there a cardiomyopathy of obesity? Curr Heart Fail Rep 4: 221–228, 2007. doi: 10.1007/s11897-007-0016-3. [DOI] [PubMed] [Google Scholar]
- 166.Bundhun PK, Li N, Chen MH. Does an obesity paradox really exist after cardiovascular intervention?: a systematic review and meta-analysis of randomized controlled trials and observational studies. Medicine 94: e1910, 2015. doi: 10.1097/md.0000000000001910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lavie CJ, McAuley PA, Church TS, Milani RV, Blair SN. Obesity and cardiovascular diseases: implications regarding fitness, fatness, and severity in the obesity paradox. J Am Coll Cardiol 63: 1345–1354, 2014. doi: 10.1016/j.jacc.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 168.Oktay AA, Lavie CJ, Kokkinos PF, Parto P, Pandey A, Ventura HO. The interaction of cardiorespiratory fitness with obesity and the obesity paradox in cardiovascular disease. Prog Cardiovasc Dis 60: 30–44, 2017. doi: 10.1016/j.pcad.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 169.Pozzo J, Fournier P, Lairez O, Vervueren PL, Delmas C, Elbaz M, Carrie D, Galinier M, Roncalli J. Obesity paradox: origin and best way to assess severity in patients with systolic HF. Obesity 23: 2008–2015, 2002. doi: 10.1002/oby.21216. [DOI] [PubMed] [Google Scholar]
- 170.Shah RV, Abbasi SA, Heydari B, Rickers C, Jacobs DR, Wang L, Kwong RY, Bluemke DA, Lima JA, Jerosch-Herold M. Insulin resistance, subclinical left ventricular remodeling, and the obesity paradox: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol 61: 1698–1706, 2013. doi: 10.1016/j.jacc.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stokes A, Preston SH. Smoking and reverse causation create an obesity paradox in cardiovascular disease. Obesity 23: 2485–2490 2015. doi: 10.1002/oby.21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Strandberg TE, Strandberg AY, Salomaa VV, Pitkälä KH, Tilvis RS, Sirola J, Miettinen TA. Explaining the obesity paradox: cardiovascular risk, weight change, and mortality during long-term follow-up in men. Eur Heart J 30: 1720–1727, 2009. doi: 10.1093/eurheartj/ehp162. [DOI] [PubMed] [Google Scholar]
- 173.Whelton SP, McAuley PA, Dardari Z, Orimoloye OA, Brawner CA, Ehrman JK, Keteyian SJ, Al-Mallah M, Blaha MJ. Association of BMI, fitness, and mortality in patients with diabetes: evaluating the obesity paradox in the Henry Ford Exercise Testing Project (FIT Project) Cohort. Diabetes Care 43: 677–682, 2020. doi: 10.2337/dc19-1673. [DOI] [PubMed] [Google Scholar]
- 174.Zamora E, Lupón J, Enjuanes C, Pascual-Figal D, de Antonio M, Domingo M, Comín-Colet J, Vila J, Peñafiel J, Farré N, Alonso N, Santesmases J, Troya M, Bayés-Genís A. No benefit from the obesity paradox for diabetic patients with heart failure. Eur J Heart Fail 18: 851–858, 2016. doi: 10.1002/ejhf.576. [DOI] [PubMed] [Google Scholar]
- 175.Alpert MA, Lavie CJ, Agrawal H, Kumar A, Kumar SA. Cardiac effects of obesity: pathophysiologic, clinical, and prognostic consequences-a review. J Cardiopulm Rehabil Prev 36: 1–11, 2016. doi: 10.1097/HCR.0000000000000147. [DOI] [PubMed] [Google Scholar]
- 176.Alpert MA, Lavie CJ, Agrawal H, Aggarwal KB, Kumar SA. Obesity and heart failure: epidemiology, pathophysiology, clinical manifestations, and management. Transl Res 164: 345–356, 2014. doi: 10.1016/j.trsl.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 177.Lavie CJ, Ventura HO, Milani RV, Arena R. Critical impact of fitness in the prevention and treatment of heart failure. Am Heart J 169: 194–196, 2015. doi: 10.1016/j.ahj.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 178.Rust P, Ekmekcioglu C. Impact of salt intake on the pathogenesis and treatment of hypertension. Adv Exp Med Biol 956: 61–84, 2017. doi: 10.1016/j.ahj.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 179.Kawarazaki W, Fujita T. The role of aldosterone in obesity-related hypertension. Am J Hypertens 29: 415–423, 2016. doi: 10.1093/ajh/hpw003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hattori T, Murase T, Takatsu M, Nagasawa K, Matsuura N, Watanabe S, Murohara T, Nagata K. Dietary salt restriction improves cardiac and adipose tissue pathology independently of obesity in a rat model of metabolic syndrome. J Am Heart Assoc 3: e001312, 2014. doi: 10.1161/jaha.114.001312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Littleton SW, Tulaimat A. The effects of obesity on lung volumes and oxygenation. Respir Med 124: 15–20, 2017. doi: 10.1016/j.rmed.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 182.Schmidt W. Effects of intermittent exposure to high altitude on blood volume and erythropoietic activity. High Alt Med Biol 3: 167–176, 2002. doi: 10.1089/15270290260131902. [DOI] [PubMed] [Google Scholar]
- 183.Obokata M, Reddy YN, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 136: 6–19, 2017. doi: 10.1161/CIRCULATIONAHA.116.026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Olivier A, Pitt B, Girerd N, Lamiral Z, Machu JL, McMurray JJ, Swedberg K, van Veldhuisen DJ, Collier TJ, Pocock SJ, Rossignol P, Zannad F, Pizard A. Effect of eplerenone in patients with heart failure and reduced ejection fraction: potential effect modification by abdominal obesity. Insight from the EMPHASIS-HF trial. Eur J Heart Fail 19: 1186–1197, 2017. doi: 10.1002/ejhf.792. [DOI] [PubMed] [Google Scholar]
- 185.Packer M. Leptin-aldosterone-neprilysin axis: identification of its distinctive role in the pathogenesis of the three phenotypes of heart failure in people with obesity. Circulation 137: 1614–1631, 2018. doi: 10.1161/CIRCULATIONAHA.117.032474. [DOI] [PubMed] [Google Scholar]
- 186.Rosengren A, Aberg M, Robertson J, Waern M, Schaufelberger M, Kuhn G, Aberg D, Schioler L, Toren K. Body weight in adolescence and long-term risk of early heart failure in adulthood among men in Sweden. Eur Heart J 38: 1926–1933, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cerit L, Kemal H, Gunsel A, Duygu H. High-output heart failure: with or without obesity. J Am Coll Cardiol 69: 112–113, 2017. [P doi: 10.1016/j.jacc.2016.08.080, 10.1016/S0735-1097(17)33501-5. [DOI] [PubMed] [Google Scholar]
- 188.Reddy YN, Melenovsky V, Redfield MM, Nishimura RA, Borlaug BA. High-output heart failure: a 15-year experience. J Am Coll Cardiol 68: 473–482, 2016. doi: 10.1016/j.jacc.2016.05.043. [DOI] [PubMed] [Google Scholar]
- 189.Wójtowicz J, Łempicka A, Łuczyński W, Szczepański W, Zomerfeld A, Semeran K, Bossowski A. Central aortic pressure, arterial stiffness and echocardiographic parameters of children with overweight/obesity and arterial hypertension. Adv Clin Exp Med 26: 1399–1404, 2017. doi: 10.17219/acem/65485. [DOI] [PubMed] [Google Scholar]
- 190.Palmieri V, de Simone G, Arnett DK, Bella JN, Kitzman DW, Oberman A, Hopkins PN, Province MA, Devereux RB. Relation of various degrees of body mass index in patients with systemic hypertension to left ventricular mass, cardiac output, and peripheral resistance (The Hypertension Genetic Epidemiology Network Study). Am J Cardiol 88: 1163–1168, 2001. doi: 10.1016/S0002-9149(01)02054-9. doi:. [DOI] [PubMed] [Google Scholar]
- 191.Collis T, Devereux RB, Roman MJ, de Simone G, Yeh J, Howard BV, Fabsitz RR, Welty TK. Relations of stroke volume and cardiac output to body composition: the strong heart study. Circulation 103: 820–825, 2001. doi: 10.1161/01.CIR.103.6.820. [DOI] [PubMed] [Google Scholar]
- 192.de Simone G, Devereux RB, Kizer JR, Chinali M, Bella JN, Oberman A, Kitzman DW, Hopkins PN, Rao DC, Arnett DK. Body composition and fat distribution influence systemic hemodynamics in the absence of obesity: the HyperGEN Study. Am J Clin Nutr 81: 757–761, 2005. doi: 10.1093/ajcn/81.4.757. [DOI] [PubMed] [Google Scholar]
- 193.Farag NH, Matthews SC, Brzezinski E, Nelesen RA, Mills PJ. Relationship between central obesity and cardiovascular hemodynamic indices in postmenopausal women. Fertil Steril 81: 465–467, 2004. doi: 10.1016/j.fertnstert.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 194.Neeland IJ, Gupta S, Ayers CR, Turer AT, Rame JE, Das SR, Berry JD, Khera A, McGuire DK, Vega GL, Grundy SM, de Lemos JA, Drazner MH. Relation of regional fat distribution to left ventricular structure and function. Circ Cardiovasc Imaging 6: 800–807, 2013. doi: 10.1161/CIRCIMAGING.113.000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Syme C, Shin J, Richer L, Gaudet D, Paus T, Pausova Z. Sex differences in blood pressure hemodynamics in middle-aged adults with overweight and obesity. Hypertension 74: 407–412, 2019. doi: 10.1161/HYPERTENSIONAHA.119.13058. [DOI] [PubMed] [Google Scholar]
- 196.Sainju NK, Shah RK, Joshi SK. Screening for hypertension and obesity in rural population of Nepal. Kathmandu Univ Med J (KUMJ) 16: 4–7, 2018. [PubMed] [Google Scholar]
- 197.DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol 10: 364–376, 2014. doi: 10.1038/nrendo.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Grassi G, Mark A, Esler M. The sympathetic nervous system alterations in human hypertension. Circ Res 116: 976–990, 2015. doi: 10.1161/CIRCRESAHA.116.303604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.He H, Yang D, Ma L, Luo Z, Ma S, Feng X, Cao T, Yan Z, Liu D, Tepel M, Zhu Z. Telmisartan prevents weight gain and obesity through activation of peroxisome proliferator-activated receptor-delta-dependent pathways. Hypertension 55: 869–879, 2010. doi: 10.1161/HYPERTENSIONAHA.109.143958. doi:. [DOI] [PubMed] [Google Scholar]
- 200.Lin B, Koibuchi N, Hasegawa Y, Sueta D, Toyama K, Uekawa K, Ma M, Nakagawa T, Kusaka H, Kim-Mitsuyama S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol 13: 148, 2014. doi: 10.1186/s12933-014-0148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lindgärde F. The effect of orlistat on body weight and coronary heart disease risk profile in obese patients: the Swedish Multimorbidity Study. J Intern Med 248: 245–254, 2000. doi: 10.1046/j.1365-2796.2000.00720.x. [DOI] [PubMed] [Google Scholar]
- 202.Chirinos JA, Segers P, Hughes T, Townsend R. Large-artery stiffness in health and disease: JACC state-of-the-art review. J Am Coll Cardiol 74: 1237–1263, 2019. doi: 10.1016/j.jacc.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Weber T, Chirinos JA. Pulsatile arterial haemodynamics in heart failure. Eur Heart J 39: 3847–3854, 2018. doi: 10.1093/eurheartj/ehy346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wildman RP, Mackey RH, Bostom A, Thompson T, Sutton-Tyrrell K. Measures of obesity are associated with vascular stiffness in young and older adults. Hypertension 42: 468–473, 2003. doi: 10.1161/01.HYP.0000090360.78539.CD. [DOI] [PubMed] [Google Scholar]
- 205.Hudson LD, Kinra S, Wong IC, Viner RM. Arterial stiffening, insulin resistance and acanthosis nigricans in a community sample of adolescents with obesity. Int J Obes (Lond) 41: 1454–1456, 2017. doi: 10.1038/ijo.2017.105. [DOI] [PubMed] [Google Scholar]
- 206.Putarek K, Banfic L, Pasalic M, Krnic N, Spehar Uroic A, Rojnic Putarek N. Arterial stiffness as a measure of cardiovascular risk in obese adolescents and adolescents with diabetes type 1. J Pediatr Endocrinol Metab 31: 1315–1323, 2018. doi: 10.1515/jpem-2018-0137. [DOI] [PubMed] [Google Scholar]
- 207.Czippelova B, Turianikova Z, Krohova J, Wiszt R, Lazarova Z, Pozorciakova K, Ciljakova M, Javorka M. Arterial stiffness and endothelial function in young obese patients–vascular resistance matters. J Atheroscler Thromb 26: 1015–1025, 2019. doi: 10.5551/jat.47530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fernandes-Silva MM, Shah AM, Claggett B, Cheng S, Tanaka H, Silvestre OM, Nadruz W, Borlaug BA, Solomon SD. Adiposity, body composition and ventricular-arterial stiffness in the elderly: the Atherosclerosis Risk in Communities Study. Eur J Heart Fail 20: 1191–1201, 2018. doi: 10.1002/ejhf.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Orr JS, Gentile CL, Davy BM, Davy KP. Large artery stiffening with weight gain in humans: role of visceral fat accumulation. Hypertension 51: 1519–1524, 2008. doi: 10.1161/HYPERTENSIONAHA.108.112946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Strasser B, Arvandi M, Pasha EP, Haley AP, Stanforth P, Tanaka H. Abdominal obesity is associated with arterial stiffness in middle-aged adults. Nutr Metab Cardiovasc Dis 25: 495–502, 2015. doi: 10.1016/j.numecd.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 211.Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King CA, Lob HE, Santhanam L, Mitchell G, Cohen RA, Seta F. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension 62: 1105–1110, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wildman RP, Farhat GN, Patel AS, Mackey RH, Brockwell S, Thompson T, Sutton-Tyrrell K. Weight change is associated with change in arterial stiffness among healthy young adults. Hypertension 45: 187–192, 2005. doi: 10.1161/01.HYP.0000152200.10578.5d. [DOI] [PubMed] [Google Scholar]
- 213.Aggoun Y, Farpour-Lambert NJ, Marchand LM, Golay E, Maggio AB, Beghetti M. Impaired endothelial and smooth muscle functions and arterial stiffness appear before puberty in obese children and are associated with elevated ambulatory blood pressure. Eur Heart J 29: 792–799, 2008. doi: 10.1093/eurheartj/ehm633. [DOI] [PubMed] [Google Scholar]
- 214.Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA 308: 875–881, 2012. doi: 10.1001/2012.jama.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Giudici A, Palombo C, Kozakova M, Morizzo C, Losso L, Nannipieri M, Berta R, Hughes AD, Cruickshank JK, Khir AW. Weight loss after bariatric surgery significantly improves carotid and cardiac function in apparently healthy people with morbid obesity. Obes Surg 30: 3776–3783, 2020. doi: 10.1007/s11695-020-04686-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jain S, Gurubhagavatula I, Townsend R, Kuna ST, Teff K, Wadden TA, Chittams J, Hanlon AL, Maislin G, Saif H, Broderick P, Ahmad Z, Pack AI, Chirinos JA. Effect of CPAP, weight loss, or CPAP plus weight loss on central hemodynamics and arterial stiffness. Hypertension 70: 1283–1290, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Saxena T, Ali AO, Saxena M. Pathophysiology of essential hypertension: an update. Expert Rev Cardiovasc Ther 16: 879–887, 2018. doi: 10.1080/14779072.2018.1540301. [DOI] [PubMed] [Google Scholar]
- 218.Weismann D, Wiedmann S, Bala M, Frantz S, Fassnacht M. [Obesity and heart failure] Internist (Berl) 56: 121–126, 2015. doi: 10.1007/s00108-014-3535-5. [DOI] [PubMed] [Google Scholar]
- 219.Jern S, Bergbrant A, Bjorntorp P, Hansson L. Relation of central hemodynamics to obesity and body fat distribution. Hypertension 19: 520–527, 1992. doi: 10.1161/01.HYP.19.6.520. [DOI] [PubMed] [Google Scholar]
- 220.Fazliana M, Liyana AZ, Omar A, Ambak R, Mohamad NN, Shamsudin UK, Salleh NA, Aris T. Effects of weight loss intervention on body composition and blood pressure among overweight and obese women: findings from the MyBFF@home study. BMC Womens Health 18: 93, 2018. doi: 10.1186/s12905-018-0592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dong Y, Ma J, Song Y, Ma Y, Dong B, Zou Z, Prochaska JJ. Secular trends in blood pressure and overweight and obesity in Chinese boys and girls aged 7 to 17 years from 1995 to 2014. Hypertension 72: 298–305, 2018. doi: 10.1161/HYPERTENSIONAHA.118.11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Oliveras A, de la Sierra A. Resistant hypertension: patient characteristics, risk factors, co-morbidities and outcomes. J Hum Hypertens 28: 213–217, 2014. doi: 10.1038/jhh.2013.77. [DOI] [PubMed] [Google Scholar]
- 223.Macumber IR, Weiss NS, Halbach SM, Hanevold CD, Flynn JT. The association of pediatric obesity with nocturnal non-dipping on 24-hour ambulatory blood pressure monitoring. Am J Hypertens 29: 647–652, 2016. doi: 10.1093/ajh/hpv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kotsis V, Stabouli S, Bouldin M, Low A, Toumanidis S, Zakopoulos N. Impact of obesity on 24-hour ambulatory blood pressure and hypertension. Hypertension 45: 602–607, 2005. doi: 10.1161/01.HYP.0000158261.86674.8e. [DOI] [PubMed] [Google Scholar]
- 225.Pichler G, Martinez F, Vicente A, Solaz E, Calaforra O, Lurbe E, Redon J. Influence of obesity in central blood pressure. J Hypertens 33: 308–313, 2015. doi: 10.1097/HJH.0000000000000393. [DOI] [PubMed] [Google Scholar]
- 226.Gerdts E, de Simone G, Lund BP, Okin PM, Wachtell K, Boman K, Nieminen MS, Dahlof B, Devereux RB. Impact of overweight and obesity on cardiac benefit of antihypertensive treatment. Nutr Metab Cardiovasc Dis 23: 122–129, 2013. doi: 10.1016/j.numecd.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 227.Sahebkar A, Simental-Mendia LE, Kovanen PT, Pedone C, Simental-Mendia M, Cicero AF. Effects of orlistat on blood pressure: a systematic review and meta-analysis of 27 randomized controlled clinical trials. J Am Soc Hypertens 12: 80–96, 2018. doi: 10.1016/j.jash.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 228.Thereaux J, Lesuffleur T, Czernichow S, Basdevant A, Msika S, Nocca D, Millat B, Fagot-Campagna A. Multicentre cohort study of antihypertensive and lipid-lowering therapy cessation after bariatric surgery. Br J Surg 106: 286–295, 2019. doi: 10.1002/bjs.10999. [DOI] [PubMed] [Google Scholar]
- 229.Wu J, Dai F, Li C, Zou Y. Gender differences in cardiac hypertrophy. J Cardiovasc Transl Res 13: 73–84, 2020. doi: 10.1007/s12265-019-09907-z. [DOI] [PubMed] [Google Scholar]
- 230.Gerdts E, Regitz-Zagrosek V. Sex differences in cardiometabolic disorders. Nat Med 25: 1657–1666, 2019. doi: 10.1038/s41591-019-0643-8. [DOI] [PubMed] [Google Scholar]
- 231.Colafella KM, Denton KM. Sex-specific differences in hypertension and associated cardiovascular disease. Nat Rev Nephrol 14: 185–201, 2018. doi: 10.1038/nrneph.2017.189, 10.1038/nrrheum.2018.18. [DOI] [PubMed] [Google Scholar]
- 232.Faulkner JL, Belin DC. Sex differences in mechanisms of hypertension associated with obesity. Hypertension 71: 15–21, 2018. doi: 10.1161/HYPERTENSIONAHA.117.09980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fu Q. Sex differences in sympathetic activity in obesity and its related hypertension. Ann N Y Acad Sci 1454: 31–41, 2019. doi: 10.1111/nyas.14095. [DOI] [PubMed] [Google Scholar]
- 234.Karlsson T, Rask-Andersen M, Pan G, Hoglund J, Wadelius C, Ek WE, Johansson A. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med 25: 1390–1395, 2019. doi: 10.1038/s41591-019-0563-7. [DOI] [PubMed] [Google Scholar]
- 235.Ren J, Ceylan-Isik AF. Diabetic cardiomyopathy: do women differ from men? Endocrine 25: 73–83, 2004. doi: 10.1385/ENDO:25:2:073. [DOI] [PubMed] [Google Scholar]
- 236.Barzizza F. [Obesity and the heart] Minerva Gastroenterol Dietol 47: 229–234, 2001. [PubMed] [Google Scholar]
- 237.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest 56: 56–64, 1975. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Mair KM, Harvey KY, Henry AD, Hillyard DZ, Nilsen M, MacLean MR. Obesity alters oestrogen metabolism and contributes to pulmonary arterial hypertension. Eur Respir J 53: 1801524, 2019. doi: 10.1183/13993003.01524-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Khaing P, Pandit P, Awsare B, Summer R. Pulmonary circulation in obesity, diabetes, and metabolic syndrome. Compr Physiol 10: 297–316, 2019. doi: 10.1002/cphy.c190018. [DOI] [PubMed] [Google Scholar]
- 240.de Carvalho MH, Colaco AL, Fortes ZB. [Cytokines, endothelial dysfunction, and insulin resistance] Arq Bras Endocrinol Metabol 50: 304–312, 2006. doi: 10.1590/S0004-27302006000200016. [DOI] [PubMed] [Google Scholar]
- 241.Huertas A, Tu L, Thuillet R, Le Hiress M, Phan C, Ricard N, Nadaud S, Fadel E, Humbert M, Guignabert C. Leptin signalling system as a target for pulmonary arterial hypertension therapy. Eur Respir J 45: 1066–1080, 2015. doi: 10.1183/09031936.00193014. [DOI] [PubMed] [Google Scholar]
- 242.Tonelli AR, Aytekin M, Feldstein AE, Dweik RA. Leptin levels predict survival in pulmonary arterial hypertension. Pulm Circ 2: 214–219, 2012. doi: 10.4103/2045-8932.97607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Perrotta F, Nigro E, Mollica M, Costigliola A, D'Agnano V, Bianco A, Guerra G. . Pulmonary hypertension and obesity: focus on adiponectin. Int J Mol Sci 20: 912, 2019. doi: 10.3390/ijms20040912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Friedman SE, Andrus BW. Obesity and pulmonary hypertension: a review of pathophysiologic mechanisms. J Obes 2012: 505274, 2012. doi: 10.1155/2012/505274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Senaratna CV, Perret JL, Lodge CJ, Lowe AJ, Campbell BE, Matheson MC, Hamilton GS, Dharmage SC. Prevalence of obstructive sleep apnea in the general population: a systematic review. Sleep Med Rev 34: 70–81, 2017. doi: 10.1016/j.smrv.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 246.Warricker F, Islam Z, Shah BN. Lesson of the month 1: obesity hypoventilation (Pickwickian) syndrome: a reversible cause of severe pulmonary hypertension. Clin Med 17: 578–581, 2017. doi: 10.7861/clinmedicine.17-6-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Nagaya N, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Nakanishi N, Yamagishi M, Kunieda T, Miyatake K. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med 160: 487–492, 1999. doi: 10.1164/ajrccm.160.2.9812078. [DOI] [PubMed] [Google Scholar]
- 248.Zharikov SI, Swenson ER, Lanaspa M, Block ER, Patel JM, Johnson RJ. Could uric acid be a modifiable risk factor in subjects with pulmonary hypertension? Med Hypotheses 74: 1069–1074, 2010. doi: 10.1016/j.mehy.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Peterson LR, Waggoner AD, Schechtman KB, Meyer T, Gropler RJ, Barzilai B, Dávila-Román VG. Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. J Am Coll Cardiol 43: 1399–1404, 2004. doi: 10.1016/j.jacc.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 250.Wong CY, O’Moore-Sullivan T, Leano R, Byrne N, Beller E, Marwick TH. Alterations of left ventricular myocardial characteristics associated with obesity. Circulation 110: 3081–3087, 2004. doi: 10.1161/01.CIR.0000147184.13872.0F. [DOI] [PubMed] [Google Scholar]
- 251.Tugcu A, Russo C, Jin Z, Homma S, Nakanishi K, Elkind MS, Rundek T, Sacco RL, Di Tullio MR. Association of body size metrics with left atrial phasic volumes and reservoir function in the elderly. Eur Heart J Cardiovasc Imaging 19: 1157–1164, 2018. doi: 10.1093/ehjci/jex236. [DOI] [PubMed] [Google Scholar]
- 252.Norman G, Norton GR, Libhaber CD, Michel F, Majane OH, Millen AM, Sareli P, Woodiwiss AJ. Independent associations between resistin and left ventricular mass and myocardial dysfunction in a community sample with prevalent obesity. Int J Cardiol 196: 81–87, 2015. doi: 10.1016/j.ijcard.2015.05.184. [DOI] [PubMed] [Google Scholar]
- 253.Turkbey EB, McClelland RL, Kronmal RA, Burke GL, Bild DE, Tracy RP, Arai AE, Lima JA, Bluemke DA. The impact of obesity on the left ventricle: the Multi-Ethnic Study of Atherosclerosis (MESA). JACC Cardiovasc Imaging 3: 266–274, 2010. doi: 10.1016/j.jcmg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Oktay AA, Lavie CJ, Milani RV, Ventura HO, Gilliland YE, Shah S, Cash ME. Current perspectives on left ventricular geometry in systemic hypertension. Prog Cardiovasc Dis 59: 235–246, 2016. doi: 10.1016/j.pcad.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 255.Chow BJ, Dorbala S, Di Carli MF, Merhige ME, Williams BA, Veledar E, Min JK, Pencina MJ, Yam Y, Chen L, Anand SP, Ruddy TD, Berman DS, Shaw LJ, Beanlands RS. Prognostic value of PET myocardial perfusion imaging in obese patients. JACC Cardiovasc Imaging 7: 278–287, 2014. doi: 10.1016/j.jcmg.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 256.Alpert MA, Karthikeyan K, Abdullah O, Ghadban R. Obesity and cardiac remodeling in adults: mechanisms and clinical implications. Prog Cardiovasc Dis 61: 114–123, 2018. doi: 10.1016/j.pcad.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 257.Lai CC, Sun D, Cen R, Wang J, Li S, Fernandez-Alonso C, Chen W, Srinivasan SR, Berenson GS. Impact of long-term burden of excessive adiposity and elevated blood pressure from childhood on adulthood left ventricular remodeling patterns: the Bogalusa Heart Study. J Am Coll Cardiol 64: 1580–1587, 2014. doi: 10.1016/j.jacc.2014.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhang H, Zhang T, Li S, Guo Y, Shen W, Fernandez C, Harville E, Bazzano LA, Urbina EM, He J, Chen W. Long-term excessive body weight and adult left ventricular hypertrophy are linked through later-life body size and blood pressure: The Bogalusa Heart Study. Circ Res 120: 1614–1621, 2017. doi: 10.1161/CIRCRESAHA.116.310421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sivanandam S, Sinaiko AR, Jacobs DR Jr, Steffen L, Moran A, Steinberger J. Relation of increase in adiposity to increase in left ventricular mass from childhood to young adulthood. Am J Cardiol 98: 411–415, 2006. doi: 10.1016/j.amjcard.2006.02.044. [DOI] [PubMed] [Google Scholar]
- 260.de Simone G, Devereux RB, Roman MJ, Alderman MH, Laragh JH. Relation of obesity and gender to left ventricular hypertrophy in normotensive and hypertensive adults. Hypertension 23: 600–606, 1994. doi: 10.1161/01.HYP.23.5.600. [DOI] [PubMed] [Google Scholar]
- 261.Avelar E, Cloward TV, Walker JM, Farney RJ, Strong M, Pendleton RC, Segerson N, Adams TD, Gress RE, Hunt SC, Litwin SE. Left ventricular hypertrophy in severe obesity: interactions among blood pressure, nocturnal hypoxemia, and body mass. Hypertension 49: 34–39, 2007. doi: 10.1161/01.HYP.0000251711.92482.14. [DOI] [PubMed] [Google Scholar]
- 262.Lorber R, Gidding SS, Daviglus ML, Colangelo LA, Liu K, Gardin JM. Influence of systolic blood pressure and body mass index on left ventricular structure in healthy African-American and white young adults: the CARDIA study. J Am Coll Cardiol 41: 955–960, 2003. doi: 10.1016/S0735-1097(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 263.Aimo A, Gaggin HK, Barison A, Emdin M, Januzzi JL Jr.. Imaging, biomarker, and clinical predictors of cardiac remodeling in heart failure with reduced ejection fraction. JACC Heart Fail 7: 782–794, 2019. doi: 10.1016/j.jchf.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 264.Gutgesell HP, Rembold CM. Growth of the human heart relative to body surface area. Am J Cardiol 65: 662–668, 1990. doi: 10.1016/0002-9149(90)91048-B. [DOI] [PubMed] [Google Scholar]
- 265.Cuspidi C, Meani S, Negri F, Giudici V, Valerio C, Sala C, Zanchetti A, Mancia G. Indexation of left ventricular mass to body surface area and height to allometric power of 2.7: is the difference limited to obese hypertensives? J Hum Hypertens 23: 728–734, 2009. doi: 10.1038/jhh.2009.16. [DOI] [PubMed] [Google Scholar]
- 266.Kulkarni A, Gulesserian T, Lorenzo J, Haroonian Y, Ngyuyen M, Lo Y, Wang D, Hsu D, Kaskel F, Mahgerefteh J. Left ventricular remodelling and vascular adaptive changes in adolescents with obesity. Pediatr Obes 13: 541–549, 2018. doi: 10.1111/ijpo.12278. [DOI] [PubMed] [Google Scholar]
- 267.Bella JN, Devereux RB, Roman MJ, O’Grady MJ, Welty TK, Lee ET, Fabsitz RR, Howard BV. Relations of left ventricular mass to fat-free and adipose body mass: the strong heart study. The Strong Heart Study Investigators. Circulation 98: 2538–2544, 1998. doi: 10.1161/01.CIR.98.23.2538. [DOI] [PubMed] [Google Scholar]
- 268.D’Ascenzi F, Pelliccia A, Cameli M, Lisi M, Natali BM, Focardi M, Giorgi A, D’Urbano G, Causarano A, Bonifazi M, Mondillo S. Dynamic changes in left ventricular mass and in fat-free mass in top-level athletes during the competitive season. Eur J Prev Cardiolog 22: 127–134, 2015. doi: 10.1177/2047487313505820. [DOI] [PubMed] [Google Scholar]
- 269.Abbott A. Project set to map marks on genome. Nature 463: 596–597, 2010. doi: 10.1038/463596b, 10.1038/463596a. , 20130621 [DOI] [PubMed] [Google Scholar]
- 270.Abhayaratna WP, Marwick TH, Smith WT, Becker NG. Characteristics of left ventricular diastolic dysfunction in the community: an echocardiographic survey. Heart 92: 1259–1264, 2006. doi: 10.1136/hrt.2005.080150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Khouri MG, Peshock RM, Ayers CR, de Lemos JA, Drazner MH. A 4-tiered classification of left ventricular hypertrophy based on left ventricular geometry: the Dallas heart study. Circ Cardiovasc Imaging 3: 164–171, 2010. doi: 10.1161/CIRCIMAGING.109.883652. [DOI] [PubMed] [Google Scholar]
- 272.Thomas JJ, Ren J. Obstructive sleep apnoea and cardiovascular complications: perception versus knowledge. Clin Exp Pharmacol Physiol 39: 995–1003, 2012. doi: 10.1111/1440-1681.12024. [DOI] [PubMed] [Google Scholar]
- 273.Cioffi G, Russo TE, Stefenelli C, Selmi A, Furlanello F, Cramariuc D, Gerdts E, de Simone G. Severe obstructive sleep apnea elicits concentric left ventricular geometry. J Hypertens 28: 1074–1082, 2010. doi: 10.1097/HJH.0b013e328336c90a. [DOI] [PubMed] [Google Scholar]
- 274.Otto ME, Belohlavek M, Romero-Corral A, Gami AS, Gilman G, Svatikova A, Amin RS, Lopez-Jimenez F, Khandheria BK, Somers VK. Comparison or cardiac structural and functional changes in obese otherwise healthy adults with versus without obstructive sleep apnea. Am J Cardiol 99: 1298–1302, 2007. doi: 10.1016/j.amjcard.2006.12.052. [DOI] [PubMed] [Google Scholar]
- 275.Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, Nesto RW, Wilson PW, Vasan RS. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation 107: 448–454, 2003. doi: 10.1161/01.CIR.0000045671.62860.98. [DOI] [PubMed] [Google Scholar]
- 276.Rider OJ, Lewandowski A, Nethononda R, Petersen SE, Francis JM, Pitcher A, Holloway CJ, Dass S, Banerjee R, Byrne JP, Leeson P, Neubauer S. Gender-specific differences in left ventricular remodelling in obesity: insights from cardiovascular magnetic resonance imaging. Eur Heart J 34: 292–299, 2013. doi: 10.1093/eurheartj/ehs341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Riehle C, Abel ED. Insulin signaling and heart failure. Circ Res 118: 1151–1169, 2016. doi: 10.1161/CIRCRESAHA.116.306206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Capaldo B, Di BP, Iaccarino M, Roman MJ, Lee ET, Devereux RB, Riccardi G, Howard BV, de Simone G. Cardiovascular characteristics in subjects with increasing levels of abnormal glucose regulation: the Strong Heart Study. Diabetes Care 36: 992–997, 2013. doi: 10.2337/dc12-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Abed HS, Samuel CS, Lau DH, Kelly DJ, Royce SG, Alasady M, Mahajan R, Kuklik P, Zhang Y, Brooks AG, Nelson AJ, Worthley SG, Abhayaratna WP, Kalman JM, Wittert GA, Sanders P. Obesity results in progressive atrial structural and electrical remodeling: implications for atrial fibrillation. Heart Rhythm 10: 90–100, 2013. doi: 10.1016/j.hrthm.2012.08.043. [DOI] [PubMed] [Google Scholar]
- 280.Pritchett AM, Jacobsen SJ, Mahoney DW, Rodeheffer RJ, Bailey KR, Redfield MM. Left atrial volume as an index of left atrial size: a population-based study. J Am Coll Cardiol 41: 1036–1043, 2003. doi: 10.1016/S0735-1097(02)02981-9. [DOI] [PubMed] [Google Scholar]
- 281.Sasson Z, Rasooly Y, Gupta R, Rasooly I. Left atrial enlargement in healthy obese: prevalence and relation to left ventricular mass and diastolic function. Can J Cardiol 12: 257–263, 1996. [PubMed] [Google Scholar]
- 282.McManus DD, Xanthakis V, Sullivan LM, Zachariah J, Aragam J, Larson MG, Benjamin EJ, Vasan RS. Longitudinal tracking of left atrial diameter over the adult life course: clinical correlates in the community. Circulation 121: 667–674, 2010. doi: 10.1161/CIRCULATIONAHA.109.885806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Stritzke J, Markus MR, Duderstadt S, Lieb W, Luchner A, Döring A, Keil U, Hense HW, Schunkert H. The aging process of the heart: obesity is the main risk factor for left atrial enlargement during aging the MONICA/KORA (monitoring of trends and determinations in cardiovascular disease/cooperative research in the region of Augsburg) study. J Am Coll Cardiol 54: 1982–1989. 2009. doi: 10.1016/j.jacc.2009.07.034. [DOI] [PubMed] [Google Scholar]
- 284.Magnani JW, Hylek EM, Apovian CM. Obesity begets atrial fibrillation: a contemporary summary. Circulation 128: 401–405, 2013. doi: 10.1161/CIRCULATIONAHA.113.001840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Chahal H, McClelland RL, Tandri H, Jain A, Turkbey EB, Hundley WG, Barr RG, Kizer J, Lima JA, Bluemke DA, Kawut SM. Obesity and right ventricular structure and function: the MESA-Right Ventricle Study. Chest 141: 388–395, 2012. doi: 10.1378/chest.11-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wong CY, O’Moore-Sullivan T, Leano R, Hukins C, Jenkins C, Marwick TH. Association of subclinical right ventricular dysfunction with obesity. J Am Coll Cardiol 47: 611–616, 2006. doi: 10.1016/j.jacc.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 287.Guidry UC, Mendes LA, Evans JC, Levy D, O'Connor GT, Larson MG, Gottlieb DJ, Benjamin EJ. Echocardiographic features of the right heart in sleep-disordered breathing–The Framingham Heart Study. Am J Respir Crit Care Med 164: 933–938, 2001. doi: 10.1164/ajrccm.164.6.2001092. [DOI] [PubMed] [Google Scholar]
- 288.Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmüller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 128: 2016–2025, 2013. doi: 10.1161/CIRCULATIONAHA.113.001873. [DOI] [PubMed] [Google Scholar]
- 289.Lindroos M, Kupari M, Valvanne J, Strandberg T, Heikkilä J, Tilvis R. Factors associated with calcific aortic valve degeneration in the elderly. Eur Heart J 15: 865–870, 1994. doi: 10.1093/oxfordjournals.eurheartj.a060602. [DOI] [PubMed] [Google Scholar]
- 290.Messika-Zeitoun D, Bielak LF, Peyser PA, Sheedy PF, Turner ST, Nkomo VT, Breen JF, Maalouf J, Scott C, Tajik AJ, Enriquez-Sarano M. Aortic valve calcification: determinants and progression in the population. Arterioscl Thrombo Vasc Biol 27: 642–648, 2007. doi: 10.1161/01.ATV.0000255952.47980.c2. [DOI] [PubMed] [Google Scholar]
- 291.Dumesnil JG, Pibarot P. The obesity paradox in aortic stenosis: to be or not to be. J Am Coll Cardiol 62: 1691–1693, 2013. doi: 10.1016/j.jacc.2013.05.047. [DOI] [PubMed] [Google Scholar]
- 292.Larsson SC, Wolk A, Håkansson N, Bäck M. Overall and abdominal obesity and incident aortic valve stenosis: two prospective cohort studies. Eur Heart J 38: 2192–2197, 2017. doi: 10.1093/eurheartj/ehx140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Kaltoft M, Langsted A, Nordestgaard BG. Obesity as a causal risk factor for aortic valve stenosis. J Am Coll Cardiol 75: 163–176, 2020. doi: 10.1016/S0735-1097(20)30790-7, 10.1016/j.jacc.2019.10.050. [DOI] [PubMed] [Google Scholar]
- 294.Otto CM, Prendergast B. Aortic-valve stenosis–from patients at risk to severe valve obstruction. N Engl J Med 371: 744–756, 2014. doi: 10.1056/NEJMra1313875. [DOI] [PubMed] [Google Scholar]
- 295.Pawade TA, Newby DE, Dweck MR. Calcification in aortic stenosis: the skeleton key. J Am Coll Cardiol 66: 561–577, 2015. doi: 10.1016/j.jacc.2015.05.066. [DOI] [PubMed] [Google Scholar]
- 296.Varbo A, Benn M, Smith GD, Timpson NJ, Tybjaerg-Hansen A, Nordestgaard BG. Remnant cholesterol, low-density lipoprotein cholesterol, and blood pressure as mediators from obesity to ischemic heart disease. Circ Res 116: 665–673, 2015. doi: 10.1161/CIRCRESAHA.116.304846. [DOI] [PubMed] [Google Scholar]
- 297.Smith JG, Luk K, Schulz CA, Engert JC, Do R, Hindy G, Rukh G, Dufresne L, Almgren P, Owens DS, Harris TB, Peloso GM, Kerr KF, Wong Q, Smith AV, Budoff MJ, Rotter JI, Cupples LA, Rich S, Kathiresan S, Orho-Melander M, Gudnason V, O'Donnell CJ, Post WS, Thanassoulis G, Cohorts for Heart and Aging Research in Genetic Epidemiology (CHARGE) Extracoronary Calcium Working Group. Association of low-density lipoprotein cholesterol-related genetic variants with aortic valve calcium and incident aortic stenosis. JAMA 312: 1764–1771, 2014. doi: 10.1001/jama.2014.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Powell BD, Redfield MM, Bybee KA, Freeman WK, Rihal CS. Association of obesity with left ventricular remodeling and diastolic dysfunction in patients without coronary artery disease. Am J Cardiol 98: 116–120, 2006. doi: 10.1016/j.amjcard.2006.01.063. [DOI] [PubMed] [Google Scholar]
- 299.Ballo P, Zacà V, Giacomin E, Galderisi M, Mondillo S. Impact of obesity on left ventricular systolic function in hypertensive subjects with normal ejection fraction. Int J Cardiol 141: 316–320, 2010. doi: 10.1016/j.ijcard.2008.11.129. [DOI] [PubMed] [Google Scholar]
- 300.Owan T, Avelar E, Morley K, Jiji R, Hall N, Krezowski J, Gallagher J, Williams Z, Preece K, Gundersen N, Strong MB, Pendleton RC, Segerson N, Cloward TV, Walker JM, Farney RJ, Gress RE, Adams TD, Hunt SC, Litwin SE. Favorable changes in cardiac geometry and function following gastric bypass surgery: 2-year follow-up in the Utah obesity study. J Am Coll Cardiol 57: 732–739, 2011. doi: 10.1016/j.jacc.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Kishi S, Armstrong AC, Gidding SS, Colangelo LA, Venkatesh BA, Jacobs DR, Carr JJ, Terry JG, Liu K, Goff DC, Lima JA. Association of obesity in early adulthood and middle age with incipient left ventricular dysfunction and structural remodeling: the CARDIA study (Coronary Artery Risk Development in Young Adults). JACC Heart Fail 2: 500–508, 2014. doi: 10.1016/j.jchf.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Amzulescu MS, De Craene M, Langet H, Pasquet A, Vancraeynest D, Pouleur AC, Vanoverschelde JL, Gerber BL. Myocardial strain imaging: review of general principles, validation, and sources of discrepancies. Eur Heart J Cardiovasc Imaging 20: 605–619, 2019. doi: 10.1093/ehjci/jez041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Orhan AL, Uslu N, Dayi SU, Nurkalem Z, Uzun F, Erer HB, Hasdemir H, Emre A, Karakus G, Soran O, Gorcsan J, Eren M. Effects of isolated obesity on left and right ventricular function: a tissue doppler and strain rate imaging study. Echocardiography 27: 236–243, 2010. doi: 10.1111/j.1540-8175.2009.01024.x. [DOI] [PubMed] [Google Scholar]
- 304.Labombarda F, Zangl E, Dugue AE, Bougle D, Pellissier A, Ribault V, Maragnes P, Milliez P, Saloux E. Alterations of left ventricular myocardial strain in obese children. Eur Heart J Cardiovasc Imaging 14: 668–676, 2013. doi: 10.1093/ehjci/jes238. [DOI] [PubMed] [Google Scholar]
- 305.Di SG, Pacileo G, Del GE, Natale F, Limongelli G, Verrengia M, Rea A, Fratta F, Castaldi B, D'Andrea A, Calabrò P, Miele T, Coppola F, Russo MG, Caso P, Perrone L, Calabrò R. Abnormal myocardial deformation properties in obese, non-hypertensive children: an ambulatory blood pressure monitoring, standard echocardiographic, and strain rate imaging study. European Heart J 27: 2689–2695, 2006. doi: 10.1093/ehjci/jes238. doi:. [DOI] [PubMed] [Google Scholar]
- 306.Amundsen BH, Helle-Valle T, Edvardsen T, Torp H, Crosby J, Lyseggen E, Stoylen A, Ihlen H, Lima JA, Smiseth OA, Slordahl SA. Noninvasive myocardial strain measurement by speckle tracking echocardiography - Validation against sonomicrometry and tagged magnetic resonance imaging. J Am Coll Cardiol 47: 789–793, 2006. doi: 10.1016/j.jacc.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 307.Russo C, Sera F, Jin Z, Palmieri V, Homma S, Rundek T, Elkind MS, Sacco RL, Di TM. Abdominal adiposity, general obesity, and subclinical systolic dysfunction in the elderly: a population-based cohort study. Eur J Heart Fail 18: 537–544, 2016. doi: 10.1002/ejhf.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Park SJ, Miyazaki C, Bruce CJ, Ommen S, Miller FA, Oh JK. Left ventricular torsion by two-dimensional speckle tracking echocardiography in patients with diastolic dysfunction and normal ejection fraction. J Am Soc Echocardiogr 21: 1129–1137, 2008. doi: 10.1016/j.echo.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 309.Mitter SS, Shah SJ, Thomas JD. A test in context: E/A and E/e' to assess diastolic dysfunction and LV filling pressure. J Am Coll Cardiol 69: 1451–1464, 2017. doi: 10.1016/j.jacc.2016.12.037, 10.1016/S0735-1097(17)34840-4. [DOI] [PubMed] [Google Scholar]
- 310.Russo C, Jin Z, Homma S, Rundek T, Elkind MS, Sacco RL, Di Tullio MR. Effect of obesity and overweight on left ventricular diastolic function: a community-based study in an elderly cohort. J Am Coll Cardiol 57: 1368–1374, 2011. doi: 10.1016/j.jacc.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Feng T, Vegard M, Strand LB, Laugsand LE, Mørkedal B, Aune D, Vatten L, Ellekjær H, Loennechen JP, Mukamal K, Janszky I. Weight and weight change and risk of atrial fibrillation: the HUNT study. Eur Heart J 40: 2859–2866, 2019. doi: 10.1093/eurheartj/ehz390. [DOI] [PubMed] [Google Scholar]
- 312.Mahajan R, Lau DH, Brooks AG, Shipp NJ, Manavis J, Wood JP, Finnie JW, Samuel CS, Royce SG, Twomey DJ, Thanigaimani S, Kalman JM, Sanders P. Electrophysiological, electroanatomical, and structural remodeling of the atria as consequences of sustained obesity. J Am Coll Cardiol 66: 1–11, 2015. doi: 10.1016/j.jacc.2015.04.058. [DOI] [PubMed] [Google Scholar]
- 313.Tsang TS, Barnes ME, Miyasaka Y, Cha SS, Bailey KR, Verzosa GC, Seward JB, Gersh BJ. Obesity as a risk factor for the progression of paroxysmal to permanent atrial fibrillation: a longitudinal cohort study of 21 years. Eur Heart J 29: 2227–2233, 2008. doi: 10.1093/eurheartj/ehn324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Munger TM, Dong YX, Masaki M, Oh JK, Mankad SV, Borlaug BA, Asirvatham SJ, Shen WK, Lee HC, Bielinski SJ, Hodge DO, Herges RM, Buescher TL, Wu JH, Ma CS, Zhang YH, Chen PS, Packer DL, Cha YM. Electrophysiological and hemodynamic characteristics associated with obesity in patients with atrial fibrillation. J Am Coll Cardiol 60: 851–860, 2012. doi: 10.1016/j.jacc.2012.03.042. [DOI] [PubMed] [Google Scholar]
- 315.Badheka AO, Rathod A, Kizilbash MA, Garg N, Mohamad T, Afonso L, Jacob S. Influence of obesity on outcomes in atrial fibrillation: yet another obesity paradox. Am J Med 123: 646–651, 2010. doi: 10.1016/j.amjmed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 316.Santos C, Marques da Silva P. Hemodynamic patterns in obesity associated hypertension. BMC Obes 5: 13, 2018. doi: 10.1186/s40608-018-0190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Standeven KF, Hess K, Carter AM, Rice GI, Cordell PA, Balmforth AJ, Lu B, Scott DJ, Turner AJ, Hooper NM, Grant PJ. Neprilysin, obesity and the metabolic syndrome. Int J Obes (Lond) 35: 1031–1040, 2011. doi: 10.1038/ijo.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Liao MT, Wu XM, Chang CC, Liao CW, Chen YH, Lu CC, Lin YT, Chang YY, Hung CS, Lin LC, Lai CL, Lin LY, Wu VC, Ho YL, Wu KD, Lin YH, Group TS, TAIPAI Study Group. The association between glomerular hyperfiltration and left ventricular structure and function in patients with primary aldosteronism. Int J Med Sci 12: 369–377, 2015. doi: 10.7150/ijms.10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Alpert MA. Obesity cardiomyopathy: pathophysiology and evolution of the clinical syndrome. Am J Med Sci 321: 225–236, 2001. doi: 10.1097/00000441-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 320.Xu X, Hua Y, Nair S, Bucala R, Ren J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension 63: 490–499, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Galinier M, Pathak A, Roncalli J, Massabuau P. [Obesity and cardiac failure]. Arch Mal Coeur Vaiss 98: 39–45, 2005. , 15714862 [PubMed] [Google Scholar]
- 322.Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT, Granzier H, Hummel SL, Kass DA, Redfield MM, Sam F, Wang TJ, Desvigne-Nickens P, Adhikari BB. Research priorities for heart failure with preserved ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 141: 1001–1026, 2020. doi: 10.1161/CIRCULATIONAHA.119.041886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Kim SH, Despres JP, Koh KK. Obesity and cardiovascular disease: friend or foe? Eur Heart J 37: 3560–3568B, 2016. doi: 10.1093/eurheartj/ehv509. [DOI] [PubMed] [Google Scholar]
- 324.Sharma A, Lavie CJ, Borer JS, Vallakati A, Goel S, Lopez-Jimenez F, Arbab-Zadeh A, Mukherjee D, Lazar JM. Meta-analysis of the relation of body mass index to all-cause and cardiovascular mortality and hospitalization in patients with chronic heart failure. Am J Cardiol 115: 1428–1434, 2015. doi: 10.1016/j.amjcard.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 325.Doehner W, Gerstein HC, Ried J, Jung H, Asbrand C, Hess S, Anker SD. Obesity and weight loss are inversely related to mortality and cardiovascular outcome in prediabetes and type 2 diabetes: data from the ORIGIN trial. Eur Heart J 41: 2668–2677, 2020. doi: 10.1093/eurheartj/ehaa293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Ortega FB, Sui X, Lavie CJ, Blair SN. Body mass index, the most widely used but also widely criticized index: would a criterion standard measure of total body fat be a better predictor of cardiovascular disease mortality? Mayo Clinic Proceed 91: 443–455, 2016. doi: 10.1016/j.mayocp.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Ross R, Neeland IJ, Yamashita S, Shai I, Seidell J, Magni P, Santos RD, Arsenault B, Cuevas A, Hu FB, Griffin BA, Zambon A, Barter P, Fruchart JC, Eckel RH, Matsuzawa Y, Després JP. Waist circumference as a vital sign in clinical practice: a Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat Rev Endocrinol 16: 177–189, 2020. doi: 10.1038/s41574-019-0310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Hudda MT, Fewtrell MS, Haroun D, Lum S, Williams JE, Wells JC, Riley RD, Owen CG, Cook DG, Rudnicka AR, Whincup PH, Nightingale CM. Development and validation of a prediction model for fat mass in children and adolescents: meta-analysis using individual participant data. BMJ 366: l4293, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Larsson SC, Bäck M, Rees JM, Mason AM, Burgess S. Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: a Mendelian randomization study. Eur Heart J 41: 221–226, 2020. doi: 10.1093/eurheartj/ehz388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Lee DH, Keum N, Hu FB, Orav EJ, Rimm EB, Willett WC, Giovannucci EL. Predicted lean body mass, fat mass, and all cause and cause specific mortality in men: prospective US cohort study. BMJ 362: k2575, 2018. doi: 10.1136/bmj.k2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Ashwell M, Gunn P, Gibson S. Waist-to-height ratio is a better screening tool than waist circumference and BMI for adult cardiometabolic risk factors: systematic review and meta-analysis. Obes Rev 13: 275–286, 2012. doi: 10.1111/j.1467-789X.2011.00952.x. [DOI] [PubMed] [Google Scholar]
- 332.Kosmala W, Jedrzejuk D, Derzhko R, Przewlocka-Kosmala M, Mysiak A, Bednarek-Tupikowska G. Left ventricular function impairment in patients with normal-weight obesity: contribution of abdominal fat deposition, profibrotic state, reduced insulin sensitivity, and proinflammatory activation. Circ Cardiovasc Imaging 5: 349–356, 2012. doi: 10.1161/CIRCIMAGING.111.969956. [DOI] [PubMed] [Google Scholar]
- 333.Fox CS, Gona P, Hoffmann U, Porter SA, Salton CJ, Massaro JM, Levy D, Larson MG, D'Agostino RB, O'Donnell CJ, Manning WJ. Pericardial fat, intrathoracic fat, and measures of left ventricular structure and function: the Framingham Heart Study. Circulation 119: 1586–1591, 2009. doi: 10.1161/CIRCULATIONAHA.108.828970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Li M, Fisette A, Zhao XY, Deng JY, Mi J, Cianflone K. Serum resistin correlates with central obesity but weakly with insulin resistance in Chinese children and adolescents. Int J Obes (Lond) 33: 424–439, 2009. doi: 10.1038/ijo.2009.44. [DOI] [PubMed] [Google Scholar]
- 335.Iliodromiti S, Celis-Morales CA, Lyall DM, Anderson J, Gray SR, Mackay DF, Nelson SM, Welsh P, Pell JP, Gill JM, Sattar N. The impact of confounding on the associations of different adiposity measures with the incidence of cardiovascular disease: a cohort study of 296,535 adults of white European descent. Eur Heart J 39: 1514–1520, 2018. doi: 10.1093/eurheartj/ehy057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Sun YQ, Burgess S, Staley JR, Wood AM, Bell S, Kaptoge SK, Guo Q, Bolton TR, Mason AM, Butterworth AS, Di AE, Vie GÅ, Bjørngaard JH, Kinge JM, Chen Y, Mai XM. Body mass index and all cause mortality in HUNT and UK Biobank studies: linear and non-linear mendelian randomisation analyses. BMJ 364: l1042, 2019. doi: 10.1136/bmj.l1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Shah RV, Murthy VL, Colangelo LA, Reis J, Venkatesh BA, Sharma R, Abbasi SA, Goff DC, Carr JJ, Rana JS, Terry JG, Bouchard C, Sarzynski MA, Eisman A, Neilan T, Das S, Jerosch-Herold M, Lewis CE, Carnethon M, Lewis GD, Lima JA. Association of fitness in young adulthood with survival and cardiovascular risk: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. JAMA Intern Med 176: 87–95, 2016. doi: 10.1001/jamainternmed.2015.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Pandey A, Park B, Martens S, Ayers C, Neeland IJ, Haykowsky MJ, Nelson MD, Sarma S, Berry JD. Relationship of cardiorespiratory fitness and adiposity with left ventricular strain in middle-age adults (from the Dallas Heart Study). Am J Cardiol 120: 1405–1409, 2017. doi: 10.1016/j.amjcard.2017.07.031. [DOI] [PubMed] [Google Scholar]
- 339.Henriksson H, Henriksson P, Tynelius P, Ekstedt M, Berglind D, Labayen I, Ruiz JR, Lavie CJ, Ortega FB. Cardiorespiratory fitness, muscular strength, and obesity in adolescence and later chronic disability due to cardiovascular disease: a cohort study of 1 million men. Eur Heart J 41: 1503–1510, 2020. doi: 10.1093/eurheartj/ehz774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Vecchie A, Dallegri F, Carbone F, Bonaventura A, Liberale L, Portincasa P, Fruhbeck G, Montecucco F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur J Intern Med 48: 6–17, 2018. doi: 10.1016/j.ejim.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 341.Eckel N, Li Y, Kuxhaus O, Stefan N, Hu FB, Schulze MB. Transition from metabolic healthy to unhealthy phenotypes and association with cardiovascular disease risk across BMI categories in 90 257 women (the Nurses' Health Study): 30 year follow-up from a prospective cohort study. Lancet Diabetes Endocrinol 6: 714–724, 2018. doi: 10.1016/S2213-8587(18)30137-2. [DOI] [PubMed] [Google Scholar]
- 342.Eckel N, Meidtner K, Kalle-Uhlmann T, Stefan N, Schulze MB. Metabolically healthy obesity and cardiovascular events: a systematic review and meta-analysis. Eur J Prev Cardiol 23: 956–966, 2016. doi: 10.1177/2047487315623884. [DOI] [PubMed] [Google Scholar]
- 343.Ray I, Mahata SK, De RK. Obesity: an immunometabolic perspective. Front Endocrinol (Lausanne) 7: 157, 2016. doi: 10.3389/fendo.2016.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res 126: 1549–1564, 2020. doi: 10.1161/CIRCRESAHA.119.315896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Zwick RK, Guerrero-Juarez CF, Horsley V, Plikus MV. Anatomical, physiological, and functional diversity of adipose tissue. Cell Metab 27: 68–83, 2018. doi: 10.1016/j.cmet.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Oikonomou EK, Antoniades C. The role of adipose tissue in cardiovascular health and disease. Nat Rev Cardiol 16: 83–99, 2019. doi: 10.1038/s41569-018-0097-6. [DOI] [PubMed] [Google Scholar]
- 347.Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol 20: 242–258, 2019. doi: 10.1038/s41580-018-0093-z. [DOI] [PubMed] [Google Scholar]
- 348.Hammarstedt A, Gogg S, Hedjazifar S, Nerstedt A, Smith U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol Rev 98: 1911–1941, 2018. doi: 10.1152/physrev.00034.2017. [DOI] [PubMed] [Google Scholar]
- 349.Jialal I, Devaraj S. Subcutaneous adipose tissue biology in metabolic syndrome. Horm Mol Biol Clin Investig 33: 20170074, 2018. doi: 10.1515/hmbci-2017-0074. [DOI] [PubMed] [Google Scholar]
- 350.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117: 175–184, 2007. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Fruhbeck G, Mendez-Gimenez L, Fernandez-Formoso JA, Fernandez S, Rodriguez A. Regulation of adipocyte lipolysis. Nutr Res Rev 27: 63–93, 2014. doi: 10.1017/S095442241400002X. [DOI] [PubMed] [Google Scholar]
- 352.Song Z, Xiaoli AM, Yang F. Regulation and metabolic significance of de novo lipogenesis in adipose tissues. Nutrients 10: 1383, 2018. doi: 10.3390/nu10101383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Zhang Y, Ren J. Role of cardiac steatosis and lipotoxicity in obesity cardiomyopathy. Hypertension 57: 148–150, 2011. doi: 10.1161/HYPERTENSIONAHA.110.164178. [DOI] [PubMed] [Google Scholar]
- 354.Turdi S, Ge W, Hu N, Bradley KM, Wang X, Ren J. Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. J Mol Cell Cardiol 55: 117–129, 2013. doi: 10.1016/j.yjmcc.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 355.Bluher M. Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance? Clin Sci (Lond) 130: 1603–1614, 2016. doi: 10.1042/CS20160005. [DOI] [PubMed] [Google Scholar]
- 356.Neeland IJ, Poirier P, Després JP. Cardiovascular and metabolic heterogeneity of obesity: clinical challenges and implications for management. Circulation 137: 1391–1406, 2018. doi: 10.1161/CIRCULATIONAHA.117.029617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Gonzalez N, Moreno-Villegas Z, Gonzalez-Bris A, Egido J, Lorenzo O. Regulation of visceral and epicardial adipose tissue for preventing cardiovascular injuries associated to obesity and diabetes. Cardiovasc Diabetol 16: 44, 2017. doi: 10.1186/s12933-017-0528-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Piche ME, Tchernof A, Despres JP. Obesity phenotypes, diabetes, and cardiovascular diseases. Circ Res 126: 1477–1500, 2020. doi: 10.1161/CIRCRESAHA.120.316101. [DOI] [PubMed] [Google Scholar]
- 359.Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, Vasan RS, Murabito JM, Meigs JB, Cupples LA, D'Agostino RB, O’Donnell CJ. Abdominal visceral and subcutaneous adipose tissue compartments–sssociation with metabolic risk factors in the Framingham Heart Study. Circulation 116: 39–48, 2007. doi: 10.1161/CIRCULATIONAHA.106.675355. [DOI] [PubMed] [Google Scholar]
- 360.Rafeh R, Viveiros A, Oudit GY, El-Yazbi AF. Targeting perivascular and epicardial adipose tissue inflammation: therapeutic opportunities for cardiovascular disease. Clin Sci (Lond) 134: 827–851, 2020. doi: 10.1042/CS20190227. [DOI] [PubMed] [Google Scholar]
- 361.Qi XY, Qu SL, Xiong WH, Rom O, Chang L, Jiang ZS. Perivascular adipose tissue (PVAT) in atherosclerosis: a double-edged sword. Cardiovasc Diabetol 17: 134, 2018. doi: 10.1186/s12933-018-0777-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Scheja L, Heeren J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat Rev Endocrinol 15: 507–524, 2019. doi: 10.1038/s41574-019-0230-6. [DOI] [PubMed] [Google Scholar]
- 363.Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol 174: 3496–3513, 2017. doi: 10.1111/bph.13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J Am Coll Cardiol 71: 2360–2372, 2018. doi: 10.1016/j.jacc.2018.03.509. [DOI] [PubMed] [Google Scholar]
- 365.Le Jemtel TH, Samson R, Ayinapudi K, Singh T, Oparil S. Epicardial adipose tissue and cardiovascular disease. Curr Hypertens Rep 21: 36, 2019. doi: 10.1007/s11906-019-0939-6. [DOI] [PubMed] [Google Scholar]
- 366.Iacobellis G, Ribaudo MC, Zappaterreno A, Iannucci CV, Leonetti F. Relation between epicardial adipose tissue and left ventricular mass. Am J Cardiol 94: 1084–1087, 2004. doi: 10.1016/j.amjcard.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 367.Iacobellis G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat Rev Endocrinol 11: 363–371, 2015. doi: 10.1038/nrendo.2015.58. [DOI] [PubMed] [Google Scholar]
- 368.Wong CX, Sun MT, Odutayo A, Emdin CA, Mahajan R, Lau DH, Pathak RK, Wong DT, Selvanayagam JB, Sanders P, Clarke R. Associations of epicardial, abdominal, and overall adiposity with atrial fibrillation. Circ Arrhythm Electrophysiol 9: e004378, 2016. doi: 10.1161/circep.116.004378. [DOI] [PubMed] [Google Scholar]
- 369.Voelter-Mahlknecht S. Epigenetic associations in relation to cardiovascular prevention and therapeutics. Clin Epigenetics 8: 4, 2016. doi: 10.1186/s13148-016-0170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Schiano C, Vietri MT, Grimaldi V, Picascia A, Pascale MR, Napoli C. Epigenetic-related therapeutic challenges in cardiovascular disease. Trends Pharmacol Sci 36: 226–235, 2015. doi: 10.1016/j.tips.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 371.Grant RW, Dixit VD. Adipose tissue as an immunological organ. Obesity (Silver Spring, MD) 23: 512–518, 2015. doi: 10.1002/oby.21003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Luo YX, Tang X, An XZ, Xie XM, Chen XF, Zhao X, Hao DL, Chen HZ, Liu DP. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J 38: 1389–1398, 2017. doi: 10.1093/eurheartj/ehw138. [DOI] [PubMed] [Google Scholar]
- 373.Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res 118: 1786–1807, 2016. doi: 10.1161/CIRCRESAHA.115.306885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci 18: 1321, 2017. doi: 10.3390/ijms18061321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Fang H, Judd RL. Adiponectin regulation and function. Compr Physiol 8: 1031–1063, 2018. [DOI] [PubMed] [Google Scholar]
- 376.Ghantous CM, Azrak Z, Hanache S, Abou-Kheir W, Zeidan A. Differential role of leptin and adiponectin in cardiovascular system. Intl J Endocrinol 2015: 1–13, 2015. doi: 10.1155/2015/534320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med 10: 1384–1389, 2004. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Pääkkö T, Ukkola O, Ikäheimo M, Kesäniemi YA. Plasma adiponectin levels are associated with left ventricular hypertrophy in a random sample of middle-aged subjects. Ann Med 42: 131–137, 2010. doi: 10.3109/07853890903449827. [DOI] [PubMed] [Google Scholar]
- 379.Essick EE, Ouchi N, Wilson RM, Ohashi K, Ghobrial J, Shibata R, Pimentel DR, Sam F. Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling. Am J Physiol Heart Circ Physiol 301: H984–H993, 2011. doi: 10.1152/ajpheart.00428.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Patel V, Adya R, Chen J, Ramanjaneya M, Bari MF, Bhudia SK, Hillhouse EW, Tan BK, Randeva HS. Novel insights into the cardio-protective effects of FGF21 in lean and obese rat hearts. PLoS One 9: e87102, 2014. doi: 10.1371/journal.pone.0087102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Ruperez C, Lerin C, Ferrer-Curriu G, Cairo M, Mas-Stachurska A, Sitges M, Villarroya J, Giralt M, Villarroya F, Planavila A. Autophagic control of cardiac steatosis through FGF21 in obesity-associated cardiomyopathy. Int J Cardiol 260: 163–170, 2018. doi: 10.1016/j.ijcard.2018.02.109. [DOI] [PubMed] [Google Scholar]
- 382.Kondo H, Abe I, Gotoh K, Fukui A, Takanari H, Ishii Y, Ikebe Y, Kira S, Oniki T, Saito S, Aoki K, Tanino T, Mitarai K, Kawano K, Miyoshi M, Fujinami M, Yoshimura S, Ayabe R, Okada N, Nagano Y, Akioka H, Shinohara T, Akiyoshi K, Masaki T, Teshima Y, Yufu K, Nakagawa M, Takahashi N. Interleukin 10 treatment ameliorates high-fat diet-induced inflammatory atrial remodeling and fibrillation. Circ Arrhythm Electrophysiol 11: e006040, 2018. doi: 10.1161/circep.117.006040. [DOI] [PubMed] [Google Scholar]
- 383.de Oliveira MF, Talvani A, Rocha-Vieira E. IL-33 in obesity: where do we go from here? Inflamm Res 68: 185–194, 2019. doi: 10.1007/s00011-019-01214-2. [DOI] [PubMed] [Google Scholar]
- 384.Mohamed BA, Hartmann N, Tirilomis P, Sekeres K, Li W, Neef S, Richter C, Zeisberg EM, Kattner L, Didié M, Guan K, Schmitto JD, Lehnart SE, Luther S, Voigt N, Seidler T, Sossalla S, Hasenfuss G, Toischer K. Sarcoplasmic reticulum calcium leak contributes to arrhythmia but not to heart failure progression. Sci Transl Med 10: eaan0724, 2018. doi: 10.1126/scitranslmed.aan0724. [DOI] [PubMed] [Google Scholar]
- 385.Vianello E, Dozio E, Tacchini L, Frati L, Corsi Romanelli MM. ST2/IL-33 signaling in cardiac fibrosis. Int J Biochem Cell Biol 116: 105619, 2019. doi: 10.1016/j.biocel.2019.105619. [DOI] [PubMed] [Google Scholar]
- 386.Shen HH, Yang CY, Kung CW, Chen SY, Wu HM, Cheng PY, Lam KK, Lee YM. Raloxifene inhibits adipose tissue inflammation and adipogenesis through Wnt regulation in ovariectomized rats and 3 T3-L1 cells. J Biomed Sci 26: 62, 2019. doi: 10.1186/s12929-019-0556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Teliewubai J, Ji H, Lu Y, Bai B, Yu S, Chi C, Xu Y, Zhang Y. SFRP5 serves a beneficial role in arterial aging by inhibiting the proliferation, migration and inflammation of smooth muscle cells. Mol Med Rep 18: 4682–4690, 2018. doi: 10.3892/mmr.2018.9467. [DOI] [PubMed] [Google Scholar]
- 388.Wang Y, Chen D, Zhang Y, Wang P, Zheng C, Zhang S, Yu B, Zhang L, Zhao G, Ma B, Cai Z, Xie N, Huang S, Liu Z, Mo X, Guan Y, Wang X, Fu Y, Ma D, Wang Y, Kong W. Novel adipokine, FAM19A5, inhibits neointima formation after injury through sphingosine-1-phosphate receptor 2. Circulation 138: 48–63, 2018. doi: 10.1161/CIRCULATIONAHA.117.032398. [DOI] [PubMed] [Google Scholar]
- 389.Webster AL, Yan MS, Marsden PA. Epigenetics and cardiovascular disease. Can J Cardiol 29: 46–57, 2013. doi: 10.1016/j.cjca.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 390.Engin A. The pathogenesis of obesity-associated adipose tissue inflammation. Adv Exp Med Biol 960: 221–245, 2017. doi: 10.1007/978-3-319-48382-5_9. [DOI] [PubMed] [Google Scholar]
- 391.Ren J. Leptin and hyperleptinemia–from friend to foe for cardiovascular function. J Endocrinol 181: 1–10, 2004. doi: 10.1677/joe.0.1810001. [DOI] [PubMed] [Google Scholar]
- 392.Rajapurohitam V, Gan XT, Kirshenbaum LA, Karmazyn M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ Res 93: 277–279, 2003. doi: 10.1161/01.RES.0000089255.37804.72. [DOI] [PubMed] [Google Scholar]
- 393.Fruhbeck G, Catalan V, Rodriguez A, Gomez-Ambrosi J. Adiponectin-leptin ratio: a promising index to estimate adipose tissue dysfunction. Relation with obesity-associated cardiometabolic risk. Adipocyte 7: 57–62, 2018. doi: 10.1080/21623945.2017.1402151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Zhou Y, Yu X, Chen H, Sjöberg S, Roux J, Zhang L, Ivoulsou AH, Bensaid F, Liu CL, Liu J, Tordjman J, Clement K, Lee CH, Hotamisligil GS, Libby P, Shi GP. Leptin deficiency shifts mast cells toward anti-inflammatory actions and protects mice from obesity and diabetes by polarizing M2 macrophages. Cell Metab 22: 1045–1058, 2015. doi: 10.1016/j.cmet.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.McManus DD, Lyass A, Ingelsson E, Massaro JM, Meigs JB, Aragam J, Benjamin EJ, Vasan RS. Relations of circulating resistin and adiponectin and cardiac structure and function: the Framingham Offspring Study. Obesity (Silver Spring) 20: 1882–1886, 2012. doi: 10.1038/oby.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Kim M, Oh JK, Sakata S, Liang I, Park W, Hajjar RJ, Lebeche D. Role of resistin in cardiac contractility and hypertrophy. J Mol Cell Cardiol 45: 270–280, 2008. doi: 10.1016/j.yjmcc.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Maggio AB, Wacker J, Montecucco F, Galan K, Pelli G, Mach F, Beghetti M, Farpour-Lambert NJ. Serum resistin and inflammatory and endothelial activation markers in obese adolescents. J Pediatr 161: 1022–1027, 2012. doi: 10.1016/j.jpeds.2012.05.063. [DOI] [PubMed] [Google Scholar]
- 398.Lee JH, Chan JL, Yiannakouris N, Kontogianni M, Estrada E, Seip R, Orlova C, Mantzoros CS. Circulating resistin levels are not associated with obesity or insulin resistance in humans and are not regulated by fasting or leptin administration: cross-sectional and interventional studies in normal, insulin-resistant, and diabetic subjects. J Clin Endocrinol Metab 88: 4848–4856, 2003. doi: 10.1210/jc.2003-030519. [DOI] [PubMed] [Google Scholar]
- 399.Carroll JF, Tyagi SC. Extracellular matrix remodeling in the heart of the homocysteinemic obese rabbit. Am J Hypertens 18: 692–698, 2005. doi: 10.1016/j.amjhyper.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 400.Chen F, Chen D, Zhao X, Yang S, Li Z, Sanchis D, Jin L, Qiang X, Wang K, Xu Y, Zhang Y, Ye J. Interleukin-6 deficiency facilitates myocardial dysfunction during high fat diet-induced obesity by promoting lipotoxicity and inflammation. Biochim Biophys Acta Mol Basis Dis 1863: 3128–3141, 2017. doi: 10.1016/j.bbadis.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 401.Cereijo R, Gavalda-Navarro A, Cairo M, Quesada-Lopez T, Villarroya J, Moron-Ros S, Sanchez-Infantes D, Peyrou M, Iglesias R, Mampel T, Turatsinze JV, Eizirik DL, Giralt M, Villarroya F. CXCL14, a brown adipokine that mediates brown-fat-to-macrophage communication in thermogenic adaptation. Cell Metab 28: 750–763.e6-+, 2018. doi: 10.1016/j.cmet.2018.07.015. [DOI] [PubMed] [Google Scholar]
- 402.Sawaki D, Czibik G, Pini M, Ternacle J, Suffee N, Mercedes R, Marcelin G, Surenaud M, Marcos E, Gual P, Clément K, Hue S, Adnot S, Hatem SN, Tsuchimochi I, Yoshimitsu T, Hénégar C, Derumeaux G. Visceral adipose tissue drives cardiac aging through modulation of fibroblast senescence by osteopontin production. Circulation 138: 809–822, 2018. doi: 10.1161/CIRCULATIONAHA.117.031358. [DOI] [PubMed] [Google Scholar]
- 403.Tardelli M, Zeyda K, Moreno-Viedma V, Wanko B, Grun NG, Staffler G, Zeyda M, Stulnig TM. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol Metab 5: 1131–1137, 2016. doi: 10.1016/j.molmet.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Hillard CJ. Circulating endocannabinoids: from whence do they come and where are they going? Neuropsychopharmacology 43: 155–172, 2018. doi: 10.1038/npp.2017.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, Oh DY, Li P, Osborn O, Olefsky JM. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest 127: 1019–1030, 2017. doi: 10.1172/JCI90350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Chylikova J, Dvorackova J, Tauber Z, Kamarad V. M1/M2 macrophage polarization in human obese adipose tissue. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 162: 79–82, 2018. doi: 10.5507/bp.2018.015. [DOI] [PubMed] [Google Scholar]
- 407.Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab 18: 355–367, 2013. doi: 10.1016/j.cmet.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737, 2011. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Castoldi A, de Souza CN, Camara NO, Moraes-Vieira PM. The macrophage switch in obesity development. Front Immunol 6: 637, 2015. doi: 10.3389/fimmu.2015.00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, Shen Y, Czech MP, Aouadi M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab 19: 162–171, 2014. doi: 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Mouton AJ, Li X, Hall ME, Hall JE. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ Res 126: 789–806, 2020. doi: 10.1161/CIRCRESAHA.119.312321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F, Nahrendorf M. Cardiac macrophages promote diastolic dysfunction. J Exp Med 215: 423–440, 2018. doi: 10.1084/jem.20171274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Boutens L, Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia 59: 879–894, 2016. doi: 10.1007/s00125-016-3904-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Snodgrass RG, Boß M, Zezina E, Weigert A, Dehne N, Fleming I, Brüne B, Namgaladze D. Hypoxia potentiates palmitate-induced pro-inflammatory activation of primary human macrophages. J Biol Chem 291: 413–424, 2016. doi: 10.1074/jbc.M115.686709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Tang WH, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res 120: 1183–1196, 2017. doi: 10.1161/CIRCRESAHA.117.309715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, Shao M, Zhao F, He S, Yang L, Zhang M, Nan F, Li J, Liu J, Liu J, Jia W, Qiu Y, Song B, Han JD, Rui L, Duan SZ, Liu Y. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol 18: 519–529, 2017. doi: 10.1038/ni.3709. [DOI] [PubMed] [Google Scholar]
- 418.Khodabandehloo H, Gorgani-Firuzjaee S, Panahi G, Meshkani R. Molecular and cellular mechanisms linking inflammation to insulin resistance and β-cell dysfunction. Transl Res 167: 228–256, 2016. doi: 10.1016/j.trsl.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 419.Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M, Gordon JI. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A 105: 16767–16772, 2008. doi: 10.1073/pnas.0808567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461: 1282–1286, 2009. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med 8: 12, 2016. doi: 10.1186/s13073-016-0265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Castaner O, Goday A, Park YM, Lee SH, Magkos F, Shiow S, Schroder H. The gut microbiome profile in obesity: a systematic review. Int J Endocrinol 2018: 4095789, 2018. doi: 10.1155/2018/4095789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 15: 387–407, 2018. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 424.Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest 127: 1–4, 2017. doi: 10.1172/JCI92035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Ying W, Fu W, Lee YS, Olefsky JM. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat Rev Endocrinol 16: 81–90, 2020. doi: 10.1038/s41574-019-0286-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18: 816–830, 2013. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, Yasuda O, Ogawa H, Kim-Mitsuyama S. Ezetimibe ameliorates cardiovascular complications and hepatic steatosis in obese and type 2 diabetic db/db mice. J Pharmacol Exp Ther 335: 70–75, 2010. doi: 10.1124/jpet.110.170373. [DOI] [PubMed] [Google Scholar]
- 428.Bender SB, DeMarco VG, Padilla J, Jenkins NT, Habibi J, Garro M, Pulakat L, Aroor AR, Jaffe IZ, Sowers JR. Mineralocorticoid receptor antagonism treats obesity-associated cardiac diastolic dysfunction. Hypertension 65: 1082–1088, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, Nguyen HC, Chegireddy K, Kim J, Habertheuer A, Vallabhajosyula P, Kambayashi T, Won KJ, Lazar MA. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A 115: E5096–E5105, 2018. doi: 10.1073/pnas.1802611115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, Keren-Shaul H, David E, Zmora N, Eldar SM, Lubezky N, Shibolet O, Hill DA, Lazar MA, Colonna M, Ginhoux F, Shapiro H, Elinav E, Amit I. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178: 686–698.e14, 2019. doi: 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20: 614–625, 2014. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Bourlier V, Zakaroff-Girard A, Miranville A, De Barros S, Maumus M, Sengenes C, Galitzky J, Lafontan M, Karpe F, Frayn KN, Bouloumié A. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation 117: 806–815, 2008. doi: 10.1161/CIRCULATIONAHA.107.724096. [DOI] [PubMed] [Google Scholar]
- 433.O’Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, Sun JC. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity 45: 428–441, 2016. doi: 10.1016/j.immuni.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Štimac D, Wunderlich FT, Brüning JC, Mandelboim O, Polić B. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol 16: 376–385, 2015. doi: 10.1038/ni.3120. [DOI] [PubMed] [Google Scholar]
- 435.Lee BC, Kim MS, Pae M, Yamamoto Y, Eberlé D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, You J, Mosher W, Brady HJ, Shoelson SE, Lee J. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab 23: 685–698, 2016. doi: 10.1016/j.cmet.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Huh JY, Park J, Kim JI, Park YJ, Lee YK, Kim JB. Deletion of CD1d in adipocytes aggravates adipose tissue inflammation and insulin resistance in obesity. Diabetes 66: 835–847, 2017. doi: 10.2337/db16-1122. [DOI] [PubMed] [Google Scholar]
- 437.Macdougall CE, Wood EG, Loschko J, Scagliotti V, Cassidy FC, Robinson ME, Feldhahn N, Castellano L, Voisin MB, Marelli-Berg F, Gaston-Massuet C, Charalambous M, Longhi MP. Visceral adipose tissue immune homeostasis is regulated by the crosstalk between adipocytes and dendritic cell subsets. Cell Metab 27: 588–601.e4, 2018. doi: 10.1016/j.cmet.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Wolf D, Jehle F, Michel NA, Bukosza EN, Rivera J, Chen YC, et al. Coinhibitory suppression of T cell activation by CD40 protects against obesity and adipose tissue inflammation in mice. Circulation 129: 2414–2425, 2014. doi: 10.1161/CIRCULATIONAHA.113.008055. [DOI] [PubMed] [Google Scholar]
- 439.Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, Clement N, Moes S, Colombi M, Meier JA, Swierczynska MM, Jeno P, Beglinger C, Peterli R, Hall MN. Insulin resistance causes inflammation in adipose tissue. J Clin Invest 128: 1538–1550, 2018. doi: 10.1172/JCI96139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest 127: 43–54, 2017. doi: 10.1172/JCI88880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, Skorobogatko Y, Saltiel AR. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell 172: 731–743, 2018. doi: 10.1016/j.cell.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest 129: 3990–4000, 2019. doi: 10.1172/JCI129187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Oral EA, Reilly SM, Gomez AV, Meral R, Butz L, Ajluni N, Chenevert TL, Korytnaya E, Neidert AH, Hench C, Rus D, Horowitz JF, Poirier B, Zhao P, Lehmann K, Jain M, Yu R, Liddle C, Ahmadian M, Downes M, Evans RM, Saltiel AR. Inhibition of IKK 3 and TBK1 improves glucose control in a subset of patients with type 2 diabetes. Cell Metab 26: 157–170.e7, 2017. doi: 10.1016/j.cmet.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Scott L, Jr., Fender AC, Saljic A, Li L, Chen X, Wang X, Linz D, Lang J, Hohl M, Twomey D, Pham TT, Diaz-Lankenau R, Chelu MG, Kamler M, Entman ML, Taffet GE, Sanders P, Dobrev D, Li N. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc Res, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Sokolova M, Sjaastad I, Louwe MC, Alfsnes K, Aronsen JM, Zhang L, Haugstad SB, Bendiksen BA, Ogaard J, Bliksoen M, Lien E, Berge RK, Aukrust P, Ranheim T, Yndestad A. NLRP3 inflammasome promotes myocardial remodeling during diet-induced obesity. Front Immunol 10: 1621, 2019. doi: 10.3389/fimmu.2019.01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17: 179–188, 2011. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Ji Y, Sun S, Shrestha N, Darragh LB, Shirakawa J, Xing Y, He Y, Carboneau BA, Kim H, An D, Ma M, Oberholzer J, Soleimanpour SA, Gannon M, Liu C, Naji A, Kulkarni RN, Wang Y, Kersten S, Qi L. Toll-like receptors TLR2 and TLR4 block the replication of pancreatic β cells in diet-induced obesity. Nat Immunol 20: 677–686, 2019. doi: 10.1038/s41590-019-0396-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339: 218–222, 2013. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med 23: 804–814, 2017. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 6: a009191, 2014. doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Ren J, Samson WK, Sowers JR. Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol 31: 2049–2061, 1999. doi: 10.1006/jmcc.1999.1036. [DOI] [PubMed] [Google Scholar]
- 452.Panzhinskiy E, Ren J, Nair S. Pharmacological inhibition of protein tyrosine phosphatase 1B: a promising strategy for the treatment of obesity and type 2 diabetes mellitus. Curr Med Chem 20: 2609–2625, 2013. doi: 10.2174/0929867311320210001. [DOI] [PubMed] [Google Scholar]
- 453.Ren J, Anversa P. The insulin-like growth factor I system: physiological and pathophysiological implication in cardiovascular diseases associated with metabolic syndrome. Biochem Pharmacol 93: 409–417, 2015. doi: 10.1016/j.bcp.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 454.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A, Russell KS, Kim JK. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes 54: 3530–3540, 2005. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 455.Deng JY, Huang JP, Lu LS, Hung LM. Impairment of cardiac insulin signaling and myocardial contractile performance in high-cholesterol/fructose-fed rats. Am J Physiol Heart Circ Physiol 293: H978–H987, 2007. doi: 10.1152/ajpheart.01002.2006. [DOI] [PubMed] [Google Scholar]
- 456.Abel ED, O'Shea KM, Ramasamy R. Insulin resistance: metabolic mechanisms and consequences in the heart. Arterioscler Thromb Vasc Biol 32: 2068–2076, 2012. doi: 10.1161/ATVBAHA.111.241984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Nightingale CM, Rudnicka AR, Owen CG, Wells JC, Sattar N, Cook DG, Whincup PH. Influence of adiposity on insulin resistance and glycemia markers among U.K. Children of South Asian, black African-Caribbean, and white European origin: child heart and health study in England. Diabetes Care 36: 1712–1719, 2013. doi: 10.2337/dc12-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Kishi S, Gidding SS, Reis JP, Colangelo LA, Venkatesh BA, Armstrong AC, Isogawa A, Lewis CE, Wu C, Jacobs DR, Liu K, Lima JA. Association of insulin resistance and glycemic metabolic abnormalities with lv structure and function in middle age: The CARDIA Study. JACC Cardiovasc Imaging 10: 105–114, 2017. doi: 10.1016/j.jcmg.2016.02.033. [DOI] [PubMed] [Google Scholar]
- 459.Gupte AA, Minze LJ, Reyes M, Ren Y, Wang X, Brunner G, Ghosn M, Cordero-Reyes AM, Ding K, Pratico D, Morrisett J, Shi ZZ, Hamilton DJ, Lyon CJ, Hsueh WA. High-fat feeding-induced hyperinsulinemia increases cardiac glucose uptake and mitochondrial function despite peripheral insulin resistance. Endocrinology 154: 2650–2662, 2013. doi: 10.1210/en.2012-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Pires KM, Buffolo M, Schaaf C, David Symons J, Cox J, Abel ED, Selzman CH, Boudina S. Activation of IGF-1 receptors and Akt signaling by systemic hyperinsulinemia contributes to cardiac hypertrophy but does not regulate cardiac autophagy in obese diabetic mice. J Mol Cell Cardiol 113: 39–50, 2017. doi: 10.1016/j.yjmcc.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Gancheva S, Jelenik T, Álvarez-Hernández E, Roden M. Interorgan metabolic crosstalk in human insulin resistance. Physiol Rev 98: 1371–1415, 2018. [DOI] [PubMed] [Google Scholar]
- 462.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281: 785–789, 1963. doi: 10.1016/S0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 463.Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB, Storlien LH. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46: 983–988, 1997. doi: 10.2337/diab.46.6.983, 10.2337/diabetes.46.6.983. [DOI] [PubMed] [Google Scholar]
- 464.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 51: 2005–2011, 2002. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 465.Furler SM, Poynten AM, Kriketos AD, Lowy AJ, Ellis BA, Maclean EL, Courtenay BG, Kraegen EW, Campbell LV, Chisholm DJ. Independent influences of central fat and skeletal muscle lipids on insulin sensitivity. Obes Res 9: 535–543, 2001. doi: 10.1038/oby.2001.70. [DOI] [PubMed] [Google Scholar]
- 466.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279: 32345–32353, 2004. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 467.Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol 11: 577–591, 2015. doi: 10.1038/nrendo.2015.128. [DOI] [PubMed] [Google Scholar]
- 468.Oliveira V, Marinho R, Vitorino D, Santos GA, Moraes JC, Dragano N, Sartori-Cintra A, Pereira L, Catharino RR, da Silva AS, Ropelle ER, Pauli JR, De Souza CT, Velloso LA, Cintra DE. Diets containing alpha-linolenic (omega 3) or Oleic (omega 9) fatty acids rescues obese mice from insulin resistance. Endocrinology 156: 4033–4046, 2015. doi: 10.1210/en.2014-1880. [DOI] [PubMed] [Google Scholar]
- 469.Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, Dhaneshwar AS, Hammarstedt A, Smith U, McGraw TE, Saghatelian A, Kahn BB. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 159: 318–332, 2014. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Felig P, Marliss E, Cahill GF. Jr.,Plasma amino acid levels and insulin secretion in obesity. N Engl J Med 281: 811–816, 1969. doi: 10.1056/NEJM196910092811503. [DOI] [PubMed] [Google Scholar]
- 471.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS, Jr., Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326, 2009. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, Schmidt AF, Imamura F, Stewart ID, Perry JR, Marney L, Koulman A, Karoly ED, Forouhi NG, Sjögren RJ, Näslund E, Zierath JR, Krook A, Savage DB, Griffin JL, Chaturvedi N, Hingorani AD, Khaw KT, Barroso I, McCarthy MI, O'Rahilly S, Wareham NJ, Langenberg C. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: a Mendelian randomisation analysis. PLoS Med 13: e1002179, 2016. doi: 10.1371/journal.pmed.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 20: 1193–1198, 2014. doi: 10.1038/nm.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 133: 2038–2049, 2016. doi: 10.1161/CIRCULATIONAHA.115.020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Wang W, Zhang F, Xia Y, Zhao S, Yan W, Wang H, Lee Y, Li C, Zhang L, Lian K, Gao E, Cheng H, Tao L. Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol 311: H1160–H1169, 2016. doi: 10.1152/ajpheart.00114.2016. [DOI] [PubMed] [Google Scholar]
- 476.Kroemer G, López-Otín C, Madeo F, de Cabo R. Carbotoxicity-noxious effects of carbohydrates. Cell 175: 605–614, 2018. doi: 10.1016/j.cell.2018.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, Houseknecht K, Katz EB, Charron MJ. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med 3: 1096–1101, 1997. doi: 10.1038/nm1097-1096. [DOI] [PubMed] [Google Scholar]
- 478.Bertrand L, Auquier J, Renguet E, Angé M, Cumps J, Horman S, Beauloye C. Glucose transporters in cardiovascular system in health and disease. Pflugers Arch 472: 1385–1399, 2020. doi: 10.1007/s00424-020-02444-8. [DOI] [PubMed] [Google Scholar]
- 479.Seidelmann SB, Feofanova E, Yu B, Franceschini N, Claggett B, Kuokkanen M, Puolijoki H, Ebeling T, Perola M, Salomaa V, Shah A, Coresh J, Selvin E, MacRae CA, Cheng S, Boerwinkle E, Solomon SD. Genetic variants in SGLT1, glucose tolerance, and cardiometabolic risk. J Am Coll Cardiol 72: 1763–1773, 2018. doi: 10.1016/j.jacc.2018.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Yoshii A, Nagoshi T, Kashiwagi Y, Kimura H, Tanaka Y, Oi Y, Ito K, Yoshino T, Tanaka TD, Yoshimura M. Cardiac ischemia-reperfusion injury under insulin-resistant conditions: SGLT1 but not SGLT2 plays a compensatory protective role in diet-induced obesity. Cardiovascular diabetology 18: 85, 2019. doi: 10.1186/s12933-019-0889-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Van Steenbergen A, Balteau M, Ginion A, Ferté L, Battault S, Ravenstein CM, Balligand JL, Daskalopoulos EP, Gilon P, Despa F, Despa S, Vanoverschelde JL, Horman S, Koepsell H, Berry G, Hue L, Bertrand L, Beauloye C. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci Rep 7: 41166, 2017. doi: 10.1038/srep41166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, Mayoux E, Kaneko S, Ota T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 20: 137–149, 2017. doi: 10.1016/j.ebiom.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Wu HK, Zhang Y, Cao CM, Hu X, Fang M, Yao Y, Jin L, Chen G, Jiang P, Zhang S, Song R, Peng W, Liu F, Guo J, Tang L, He Y, Shan D, Huang J, Zhou Z, Wang DW, Lv F, Xiao RP. Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation 139: 901–914, 2019. doi: 10.1161/CIRCULATIONAHA.118.037216. [DOI] [PubMed] [Google Scholar]
- 484.Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T, Gropler RJ. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 109: 2191–2196, 2004. doi: 10.1161/01.CIR.0000127959.28627.F8. [DOI] [PubMed] [Google Scholar]
- 485.Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, Sutton E, Jamil AA, Parassol N, Clarke K. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol 106: 447–457, 2011. doi: 10.1007/s00395-011-0156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Kolwicz SC Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res 111: 728–738, 2012. doi: 10.1161/CIRCRESAHA.112.268128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Shao D, Kolwicz SC, Wang P, Roe ND, Villet O, Nishi K, Hsu YA, Flint GV, Caudal A, Wang W, Regnier M, Tian R. Increasing fatty acid oxidation prevents high fat diet induced cardiomyopathy through regulating parkin mediated mitophagy. Circulation 142: 983–997, 2020. doi: 10.1161/CIRCULATIONAHA.119.043319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258, 2010. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 489.Rayner JJ, Banerjee R, Holloway CJ, Lewis AJ, Peterzan MA, Francis JM, Neubauer S, Rider OJ. The relative contribution of metabolic and structural abnormalities to diastolic dysfunction in obesity. Int J Obes (Lond) 42: 441–447, 2018. doi: 10.1038/ijo.2017.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A 100: 1226–1231, 2003. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Leopoldo AS, Sugizaki MM, Lima-Leopoldo AP, do Nascimento AF, Luvizotto Rde A, de Campos DH, Okoshi K, Dal Pai-Silva M, Padovani CR, Cicogna AC. Cardiac remodeling in a rat model of diet-induced obesity. Can J Cardiol 26: 423–429, 2010. doi: 10.1016/S0828-282X(10)70440-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Ren J, Relling DP. Leptin-induced suppression of cardiomyocyte contraction is amplified by ceramide. Peptides 27: 1415–1419, 2006. doi: 10.1016/j.peptides.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 493.Colligan PB, Relling DP, Ren J. Ceramide attenuates high glucose-induced cardiac contractile abnormalities in cultured adult rat ventricular myocytes. Cell Mol Biol (Noisy-le-Grand) 48: OL251–257, 2002. [PubMed] [Google Scholar]
- 494.Relling DP, Hintz KK, Ren J. Acute exposure of ceramide enhances cardiac contractile function in isolated ventricular myocytes. Br J Pharmacol 140: 1163–1168, 2003. doi: 10.1038/sj.bjp.0705510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18: 1692–1700, 2004. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 496.Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res 89: 148–156, 2011. doi: 10.1093/cvr/cvq266. [DOI] [PubMed] [Google Scholar]
- 497.Marín-Royo G, Gallardo I, Martínez-Martínez E, Gutiérrez B, Jurado-López R, López-Andrés N, Gutiérrez-Tenorio J, Rial E, Bartolomé MA, Nieto ML, Cachofeiro V. Inhibition of galectin-3 ameliorates the consequences of cardiac lipotoxicity in a rat model of diet-induced obesity. Dis Model Mech 11: dmm032086, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Mori J, Patel VB, Abo Alrob O, Basu R, Altamimi T, Desaulniers J, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail 7: 327–339, 2014. doi: 10.1161/CIRCHEARTFAILURE.113.000672. [DOI] [PubMed] [Google Scholar]
- 499.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 97: 1784–1789, 2000. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Nakamura M, Liu T, Husain S, Zhai P, Warren JS, Hsu CP, Matsuda T, Phiel CJ, Cox JE, Tian B, Li H, Sadoshima J. Glycogen synthase kinase-3α promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metab 29: 1119–1134, 2019. doi: 10.1016/j.cmet.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Costantino S, Akhmedov A, Melina G, Mohammed SA, Othman A, Ambrosini S, Wijnen WJ, Sada L, Ciavarella GM, Liberale L, Tanner FC, Matter CM, Hornemann T, Volpe M, Mechta-Grigoriou F, Camici GG, Sinatra R, Lüscher TF, Paneni F. Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur Heart J 40: 997–1008, 2019. doi: 10.1093/eurheartj/ehy903. [DOI] [PubMed] [Google Scholar]
- 502.Golfman LS, Wilson CR, Sharma S, Burgmaier M, Young ME, Guthrie PH, Van Arsdall M, Adrogue JV, Brown KK, Taegtmeyer H. Activation of PPARgamma enhances myocardial glucose oxidation and improves contractile function in isolated working hearts of ZDF rats. Am J Physiol Endocrinol Metab 289: E328–E336, 2005. doi: 10.1152/ajpendo.00055.2005. [DOI] [PubMed] [Google Scholar]
- 503.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 119: 2818–2828, 2009. doi: 10.1161/CIRCULATIONAHA.108.832915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Yang Y, Fu M, Li MD, Zhang K, Zhang B, Wang S, Liu Y, Ni W, Ong Q, Mi J, Yang X. O-GlcNAc transferase inhibits visceral fat lipolysis and promotes diet-induced obesity. Nat Commun 11: 181, 2020. doi: 10.1038/s41467-019-13914-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Gélinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois-Deruy E, Lauzier B, Gauthier C, Olson AK, Bouchard B, Des Rosiers C, Viollet B, Sakamoto K, Balligand JL, Vanoverschelde JL, Beauloye C, Horman S, Bertrand L. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun 9: 374, 2018. doi: 10.1038/s41467-017-02795-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP. The failing heart relies on ketone bodies as a fuel. Circulation 133: 698–705, 2016. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, Li X, Zhan L, White E, Anthony TG, Rabinowitz JD, Arany Z. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab 29: 417–429, 2019. doi: 10.1016/j.cmet.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Wu NN, Zhang Y, Ren J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med Cell Longev 2019: 9825061, 2019. doi: 10.1155/2019/9825061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med 88: 993–1001, 2010. doi: 10.1007/s00109-010-0663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Wang D, Yin Y, Wang S, Zhao T, Gong F, Zhao Y, Wang B, Huang Y, Cheng Z, Zhu G, Wang Z, Wang Y, Ren J, Liang G, Li X, Huang Z. FGF1(DeltaHBS) prevents diabetic cardiomyopathy by maintaining mitochondrial homeostasis and reducing oxidative stress via AMPK/Nur77 suppression. Signal Transduct Target Ther 6: 133, 2021. doi: 10.1038/s41392-021-00542-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Wold LE, Ceylan-Isik AF, Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol Sin 26: 908–917, 2005. doi: 10.1111/j.1745-7254.2005.00146.x. [DOI] [PubMed] [Google Scholar]
- 512.Niemann B, Rohrbach S, Miller MR, Newby DE, Fuster V, Kovacic JC. Oxidative stress and cardiovascular risk: obesity, diabetes, smoking, and pollution: part 3 of a 3-part series. J Am Coll Cardiol 70: 230–251, 2017. doi: 10.1016/j.jacc.2017.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Rayner JJ, Peterzan MA, Watson WD, Clarke WT, Neubauer S, Rodgers CT, Rider OJ. Myocardial energetics in obesity: enhanced ATP delivery through creatine kinase with blunted stress response. Circulation 141: 1152–1163, 2020. doi: 10.1161/CIRCULATIONAHA.119.042770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466, 2007. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 515.Hu Q, Zhang H, Gutiérrez Cortés N, Wu D, Wang P, Zhang J, Mattison JA, Smith E, Bettcher LF, Wang M, Lakatta EG, Sheu SS, Wang W. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res 126: 456–470, 2020. doi: 10.1161/CIRCRESAHA.119.315252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res 122: 58–73, 2018. doi: 10.1161/CIRCRESAHA.117.311307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Wu H, Wang Y, Li W, Chen H, Du L, Liu D, Wang X, Xu T, Liu L, Chen Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 15: 1882–1898, 2019. doi: 10.1080/15548627.2019.1596482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 124: 1360–1371, 2019. doi: 10.1161/circresaha.118.314607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, Huang K, Xie Z, Zou MH. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136: 2248–2266, 2017. doi: 10.1161/CIRCULATIONAHA.117.030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, Mao X, Huang K, Xie Z, Zou MH. Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation 139: 1913–1936, 2019. doi: 10.1161/CIRCULATIONAHA.118.033552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Pagliassotti MJ, Kim PY, Estrada AL, Stewart CM, Gentile CL. Endoplasmic reticulum stress in obesity and obesity-related disorders: an expanded view. Metabolism 65: 1238–1246, 2016. doi: 10.1016/j.metabol.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol 18: 499–521, 2021. doi: 10.1038/s41569-021-00511-w. [DOI] [PubMed] [Google Scholar]
- 523.Cominacini L, Mozzini C, Garbin U, Pasini A, Stranieri C, Solani E, Vallerio P, Tinelli IA, Pasini AF. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Rad Biol Med 88: 233–242, 2015. doi: 10.1016/j.freeradbiomed.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 524.Ajoolabady A, Wang S, Kroemer G, Klionsky DJ, Uversky VN, Sowers JR, Aslkhodapasandhokmabad H, Bi Y, Ge J, Ren J. ER stress in cardiometabolic diseases: from molecular mechanisms to therapeutics. Endocrine Rev: bnab006, 2021. doi: 10.1210/endrev/bnab006. [DOI] [PubMed] [Google Scholar]
- 525.Hwang J, Qi L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci 43: 593–605, 2018. doi: 10.1016/j.tibs.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Hsu HC, Liu CH, Tsai YC, Li SJ, Chen CY, Chu CH, Chen MF. Time-dependent cellular response in the liver and heart in a dietary-induced obese mouse model: the potential role of ER stress and autophagy. Eur J Nutr 55: 2031–2043, 2016. doi: 10.1007/s00394-015-1017-8. [DOI] [PubMed] [Google Scholar]
- 527.Li SJ, Liu CH, Chu HP, Mersmann HJ, Ding ST, Chu CH, Wang CY, Chen CY. The high-fat diet induces myocardial fibrosis in the metabolically healthy obese minipigs-The role of ER stress and oxidative stress. Clin Nutr 36: 760–767, 2017. doi: 10.1016/j.clnu.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 528.Park M, Sabetski A, Kwan Chan Y, Turdi S, Sweeney G. Palmitate induces ER stress and autophagy in H9c2 cells: implications for apoptosis and adiponectin resistance. J Cell Physiol 230: 630–639, 2015. doi: 10.1002/jcp.24781. [DOI] [PubMed] [Google Scholar]
- 529.Rossi A, Pizzo P, Filadi R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim Biophys Acta Mol Cell Res 1866: 1068–1078, 2019. doi: 10.1016/j.bbamcr.2018.10.016. [DOI] [PubMed] [Google Scholar]
- 530.Pascual M, Pascual DA, Soria F, Vicente T, Hernandez AM, Tebar FJ, Valdes M. Effects of isolated obesity on systolic and diastolic left ventricular function. Heart 89: 1152–1156, 2003. doi: 10.1136/heart.89.10.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Yaylali YT, Fidan-Yaylali G, Can B, Senol H, Kilinc M, Yurtdas M. Predictive power of different obesity measures for the presence of diastolic dysfunction. Turk Kardiyol Dern Ars 46: 651–658, 2018. doi: 10.5543/tkda.2018.10.5543/tkda.2018.12844. [DOI] [PubMed] [Google Scholar]
- 532.Hamilton S, Terentyev D. Proarrhythmic remodeling of calcium homeostasis in cardiac disease; implications for diabetes and obesity. Front Physiol 9: 1517, 2018. doi: 10.3389/fphys.2018.01517, 10.3389/fpls.2018.01517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Xu H, Cui S, Zhang Y, Ren J. Mitochondrial Ca2+ regulation in the etiology of heart failure: Physiological and pathophysiological implications. Acta Pharmacol Sin 41: 1301–1309, 2020. doi: 10.1038/s41401-020-0476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Kang JK, Kim OH, Hur J, Yu SH, Lamichhane S, Lee JW, Ojha U, Hong JH, Lee CS, Cha JY, Lee YJ, Im SS, Park YJ, Choi CS, Lee DH, Lee IK, Oh BC. Increased intracellular Ca(2+) concentrations prevent membrane localization of PH domains through the formation of Ca(2+)-phosphoinositides. Proc Natl Acad Sci U S A 114: 11926–11931, 2017. doi: 10.1073/pnas.1706489114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Thoudam T, Ha CM, Leem J, Chanda D, Park JS, Kim HJ, Jeon JH, Choi YK, Liangpunsakul S, Huh YH, Kwon TH, Park KG, Harris RA, Park KS, Rhee HW, Lee IK. PDK4 augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 68: 571–586, 2019. doi: 10.2337/db18-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.James P, Inui M, Tada M, Chiesi M, Carafoli E. Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature 342: 90–92, 1989. doi: 10.1038/342090a0. [DOI] [PubMed] [Google Scholar]
- 537.Dong F, Fang CX, Yang X, Zhang X, Lopez FL, Ren J. Cardiac overexpression of catalase rescues cardiac contractile dysfunction induced by insulin resistance: role of oxidative stress, protein carbonyl formation and insulin sensitivity. Diabetologia 49: 1421–1433, 2006. doi: 10.1007/s00125-006-0230-7. [DOI] [PubMed] [Google Scholar]
- 538.Li SY, Yang X, Ceylan-Isik AF, Du M, Sreejayan N, Ren J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia 49: 1434–1446, 2006. doi: 10.1007/s00125-006-0229-0. [DOI] [PubMed] [Google Scholar]
- 539.Wold LE, Dutta K, Mason MM, Ren J, Cala SE, Schwanke ML, Davidoff AJ. Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J Mol Cell Cardiol 39: 297–307, 2005. doi: 10.1016/j.yjmcc.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 540.Miklos Z, Kemecsei P, Biro T, Marincsak R, Toth BI, Op den Buijs J, Benis E, Drozgyik A, Ivanics T. Early cardiac dysfunction is rescued by upregulation of SERCA2a pump activity in a rat model of metabolic syndrome. Acta Physiol (Oxf) 205: 381–393, 2012. doi: 10.1111/j.1748-1716.2012.02420.x. [DOI] [PubMed] [Google Scholar]
- 541.Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ Res 107: 228–232, 2010. doi: 10.1161/CIRCRESAHA.110.217570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, Chepurko V, Yang DK, Kwak TH, Eom SH, Park ZY, Yoo YJ, Kim DH, Kook H, Sunagawa Y, Morimoto T, Hasegawa K, Sadoshima J, Vangheluwe P, Hajjar RJ, Park WJ, Kho C. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca(2+)-ATPase in heart failure. Circ Res 124: e63–e80, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Troupes CD, Wallner M, Borghetti G, Zhang C, Mohsin S, von Lewinski D, Berretta RM, Kubo H, Chen X, Soboloff J, Houser S. Role of STIM1 (stromal interaction molecule 1) in hypertrophy-related contractile dysfunction. Circ Res 121: 125–136, 2017. doi: 10.1161/CIRCRESAHA.117.311094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Leopoldo AS, Lima-Leopoldo AP, Sugizaki MM, do Nascimento AF, de Campos DH, Luvizotto RA, Castardeli E, Alves CA, Brum PC, Cicogna AC. Involvement of L-type calcium channel and SERCA2a in myocardial dysfunction induced by obesity. J Cell Physiol 226: 2934–2942, 2011. doi: 10.1002/jcp.22643. [DOI] [PubMed] [Google Scholar]
- 545.Bartoli F, Bailey MA, Rode B, Mateo P, Antigny F, Bedouet K, Gerbaud P, Gosain R, Plante J, Norman K, Gomez S, Lefebvre F, Rucker-Martin C, Ainscough JF, Kearney MT, Bruns AF, Shi J, Appleby HL, Young RS, Shawer HM, Debant M, Gomez AM, Beech DJ, Foster R, Benitah JP, Sabourin J. Orai1 channel inhibition preserves left ventricular systolic function and normal Ca(2+) handling after pressure overload. Circulation 141: 199–216, 2020. doi: 10.1161/CIRCULATIONAHA.118.038891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Balderas-Villalobos J, Molina-Munoz T, Mailloux-Salinas P, Bravo G, Carvajal K, Gomez-Viquez NL. Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome. Am J Physiol Heart Circ Physiol 305: H1344–H1353, 2013. doi: 10.1152/ajpheart.00211.2013. [DOI] [PubMed] [Google Scholar]
- 547.Hu N, Zhang Y. TLR4 knockout attenuated high fat diet-induced cardiac dysfunction via NF-kappaB/JNK-dependent activation of autophagy. Biochim Biophys Acta Mol Basis Dis 1863: 2001–2011, 2017. doi: 10.1016/j.bbadis.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 548.Dincer UD, Araiza A, Knudson JD, Shao CH, Bidasee KR, Tune JD. Dysfunction of cardiac ryanodine receptors in the metabolic syndrome. J Mol Cell Cardiol 41: 108–114, 2006. doi: 10.1016/j.yjmcc.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 549.Lin YC, Huang J, Kan H, Castranova V, Frisbee JC, Yu HG. Defective calcium inactivation causes long QT in obese insulin-resistant rat. Am J Physiol Heart Circ Physiol 302: H1013–H1022, 2012. doi: 10.1152/ajpheart.00837.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 103: 1466–1472, 2008. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Arruda AP, Hotamisligil GS. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab 22: 381–397, 2015. doi: 10.1016/j.cmet.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Carvajal K, Balderas-Villalobos J, Bello-Sanchez MD, Phillips-Farfan B, Molina-Munoz T, Aldana-Quintero H, Gomez-Viquez NL. Ca(2+) mishandling and cardiac dysfunction in obesity and insulin resistance: role of oxidative stress. Cell Calcium 56: 408–415, 2014. doi: 10.1016/j.ceca.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 553.Littlejohns B, Pasdois P, Duggan S, Bond AR, Heesom K, Jackson CL, Angelini GD, Halestrap AP, Suleiman MS. Hearts from mice fed a non-obesogenic high-fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS One 9: e100579, 2014. doi: 10.1371/journal.pone.0100579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71: 310–321, 2006. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 555.Ren J, Sun M, Zhou H, Ajoolabady A, Zhou Y, Tao J, Sowers JR, Zhang Y. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv 6: eabc8561, 2020. doi: 10.1126/sciadv.abc8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 112: 11389–11394, 2015. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Fauconnier J, Lanner JT, Zhang SJ, Tavi P, Bruton JD, Katz A, Westerblad H. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes 54: 2375–2381, 2005. doi: 10.2337/diabetes.54.8.2375. [DOI] [PubMed] [Google Scholar]
- 558.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545: 93–97, 2017. doi: 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol 127: 246–259, 2019. doi: 10.1016/j.yjmcc.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 560.Zhong P, Quan D, Peng J, Xiong X, Liu Y, Kong B, Huang H. Role of CaMKII in free fatty acid/hyperlipidemia-induced cardiac remodeling both in vitro and in vivo. J Mol Cell Cardiol 109: 1–16, 2017. doi: 10.1016/j.yjmcc.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 561.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature 491: 269–273, 2012. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Zhang Y, Whaley-Connell AT, Sowers JR, Ren J. Autophagy as an emerging target in cardiorenal metabolic disease: from pathophysiology to management. Pharmacol Therap 191: 1–22, 2018. doi: 10.1016/j.pharmthera.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Ren J, Zhang Y. Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol Sci 39: 1064–1076, 2018. doi: 10.1016/j.tips.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Ignacio-Souza LM, Bombassaro B, Pascoal LB, Portovedo MA, Razolli DS, Coope A, Victorio SC, de Moura RF, Nascimento LF, Arruda AP, Anhe GF, Milanski M, Velloso LA. Defective regulation of the ubiquitin/proteasome system in the hypothalamus of obese male mice. Endocrinology 155: 2831–2844, 2014. doi: 10.1210/en.2014-1090. [DOI] [PubMed] [Google Scholar]
- 565.Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE, Abel ED. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res 56: 546–561, 2015. doi: 10.1194/jlr.M055152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Juarez-Rojas JG, Reyes-Soffer G, Conlon D, Ginsberg HN. Autophagy and cardiometabolic risk factors. Rev Endo Metab Dis 15: 307–315, 2014. doi: 10.1007/s11154-014-9295-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem 286: 32324–32332, 2011. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123: 5284–5297, 2013. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res 109: 296–308, 2011. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Pan B, Zhang H, Cui T, Wang X. TFEB activation protects against cardiac proteotoxicity via increasing autophagic flux. J Mol Cell Cardiol 113: 51–62, 2017. doi: 10.1016/j.yjmcc.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Ma X, Mani K, Liu H, Kovacs A, Murphy JT, Foroughi L, French BA, Weinheimer CJ, Kraja A, Benjamin IJ, Hill JA, Javaheri A, Diwan A. Transcription factor eb activation rescues advanced alphab-crystallin mutation-induced cardiomyopathy by normalizing desmin localization. J Am Heart Assoc 8: e010866, 2019. doi: 10.1161/jaha.118.010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Wang S, Ren J. Refining the role of autophagy in hypertrophic cardiomyopathy. Int J Cell Sci Mol Biol 42: 555637, 2018. [Google Scholar]
- 573.Zhang Y, Ren J. Autophagy in ALDH2-elicited cardioprotection against ischemic heart disease: slayer or savior? Autophagy 6: 1212–1213, 2010. doi: 10.4161/auto.6.8.13652. [DOI] [PubMed] [Google Scholar]
- 574.Roe ND, Xu X, Kandadi MR, Hu N, Pang J, Weiser-Evans MC, Ren J. Targeted deletion of PTEN in cardiomyocytes renders cardiac contractile dysfunction through interruption of Pink1-AMPK signaling and autophagy. Biochim Biophys Acta 1852: 290–298, 2015. doi: 10.1016/j.bbadis.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Wang S, Ren J. Role of autophagy and regulatory mechanisms in alcoholic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis 1864: 2003–2009, 2018. doi: 10.1016/j.bbadis.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 576.Xu X, Roe ND, Weiser-Evans MC, Ren J. Inhibition of mammalian target of rapamycin with rapamycin reverses hypertrophic cardiomyopathy in mice with cardiomyocyte-specific knockout of PTEN. Hypertension 63: 729–739, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02526. [DOI] [PubMed] [Google Scholar]
- 577.Namkoong S, Cho CS, Semple I, Lee JH. Autophagy dysregulation and obesity-associated pathologies. Mol Cells 41: 3–10, 2018. doi: 10.14348/molcells.2018.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Castaneda D, Gabani M, Choi SK, Nguyen QM, Chen C, Mapara A, Kassan A, Gonzalez AA, Ait-Aissa K, Kassan M. Targeting autophagy in obesity-associated heart disease. Obesity 27: 1050–1058, 2019. doi: 10.14348/molcells.2018.2213. [DOI] [PubMed] [Google Scholar]
- 579.Trivedi PC, Bartlett JJ, Perez LJ, Brunt KR, Legare JF, Hassan A, Kienesberger PC, Pulinilkunnil T. Glucolipotoxicity diminishes cardiomyocyte TFEB and inhibits lysosomal autophagy during obesity and diabetes. Biochim Biophys Acta 1861: 1893–1910, 2016. doi: 10.1016/j.bbalip.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 580.Xu X, Hua Y, Nair S, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol 5: 61–63, 2013. doi: 10.1093/jmcb/mjs055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Zhang Y, Xu X, Ren J. MTOR overactivation and interrupted autophagy flux in obese hearts: a dicey assembly? Autophagy 9: 939–941, 2013. doi: 10.4161/auto.24398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.An M, Ryu DR, Won Park J, Ha Choi J, Park EM, Eun Lee K, Woo M, Kim M. ULK1 prevents cardiac dysfunction in obesity through autophagy-meditated regulation of lipid metabolism. Cardiovasc Res 113: 1137–1147, 2017. doi: 10.1093/cvr/cvx064. [DOI] [PubMed] [Google Scholar]
- 583.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 458: 1131–1135, 2009. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Che Y, Wang ZP, Yuan Y, Zhang N, Jin YG, Wan CX, Tang QZ. Role of autophagy in a model of obesity: A longterm high fat diet induces cardiac dysfunction. Mol Med Rep 18: 3251–3261, 2018. doi: 10.3892/mmr.2018.9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Packer M. Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium-glucose cotransporter 2 inhibitors. Eur J Heart Fail 22: 618–628, 2020. doi: 10.1002/ejhf.1732. [DOI] [PubMed] [Google Scholar]
- 586.Wang S, Wang C, Turdi S, Richmond KL, Zhang Y, Ren J. ALDH2 protects against high fat diet-induced obesity cardiomyopathy and defective autophagy: role of CaM kinase II, histone H3K9 methyltransferase SUV39H, Sirt1, and PGC-1α deacetylation. Int J Obesity 42: 1073–1087, 2018. doi: 10.1038/s41366-018-0030-4. [DOI] [PubMed] [Google Scholar]
- 587.Wharton S. Current perspectives on long-term pharmacotherapy for obesity. Can J Diabetes 40: 184–191, 2015. doi: 10.1016/j.jcjd.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 588.Cordero P, Li J, Oben JA. Epigenetics of obesity: beyond the genome sequence. Curr Opin Clin Nutr Metab Care 18: 361–366, 2015. doi: 10.1097/MCO.0000000000000179. [DOI] [PubMed] [Google Scholar]
- 589.Nair RP, Ren J. Pharmacotherapy of obesity–benefit, bias and hyperbole. CMC 16: 1888–1897, 2009. doi: 10.2174/092986709788186110. [DOI] [PubMed] [Google Scholar]
- 590.Horwich TB, Fonarow GC, Clark AL. Obesity and the obesity paradox in heart failure. Prog Cardiovasc Dis 61: 151–156, 2018. doi: 10.1016/j.pcad.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 591.Bray GA, Fruhbeck G, Ryan DH, Wilding JP. Management of obesity. Lancet 387: 1947–1956, 2016. doi: 10.1016/S0140-6736(16)00271-3. [DOI] [PubMed] [Google Scholar]
- 592.Lavie CJ, Arena R, Alpert MA, Milani RV, Ventura HO. Management of cardiovascular diseases in patients with obesity. Nat Rev Cardiol 15: 45–56, 2018. doi: 10.1038/nrcardio.2017.108. [DOI] [PubMed] [Google Scholar]
- 593.Sankaralingam S, Abo Alrob O, Zhang L, Jaswal JS, Wagg CS, Fukushima A, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Lowering body weight in obese mice with diastolic heart failure improves cardiac insulin sensitivity and function: implications for the obesity paradox. Diabetes 64: 1643–1657, 2015. doi: 10.2337/db14-1050. [DOI] [PubMed] [Google Scholar]
- 594.Naylor LH, Watts K, Sharpe JA, Jones TW, Davis EA, Thompson A, George K, Ramsay JM, O'Driscoll G, Green DJ. Resistance training and diastolic myocardial tissue velocities in obese children. Med Sci Sports Exerc 40: 2027–2032, 2008. doi: 10.1249/MSS.0b013e318182a9e0. [DOI] [PubMed] [Google Scholar]
- 595.Amaro-Gahete FJ, Ponce-Gonzalez JG, Corral-Perez J, Velazquez-Diaz D, Lavie CJ, Jimenez-Pavon D. Effect of a 12-week concurrent training intervention on cardiometabolic health in obese men: a pilot study. Front Physiol 12: 630831, 2021. doi: 10.3389/fphys.2021.630831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Serrano-Ferrer J, Walther G, Crendal E, Vinet A, Dutheil F, Naughton G, Lesourd B, Chapier R, Courteix D, Obert P. P. Right ventricle free wall mechanics in metabolic syndrome without type-2 diabetes: effects of a 3-month lifestyle intervention program. Cardiovasc Diabetol 13: 116, 2014. doi: 10.1186/s12933-014-0116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Haufe S, Utz W, Engeli S, Kast P, Bohnke J, Pofahl M, Traber J, Haas V, Hermsdorf M, Mahler A, Busjahn A, Wiesner S, Otto C, Mehling H, Luft FC, Boschmann M, Schulz-Menger J, Jordan J. Left ventricular mass and function with reduced-fat or reduced-carbohydrate hypocaloric diets in overweight and obese subjects. Hypertension 59: 70–75, 2012. doi: 10.1161/HYPERTENSIONAHA.111.178616. [DOI] [PubMed] [Google Scholar]
- 598.Viljanen AP, Karmi A, Borra R, Pärkkä JP, Lepomäki V, Parkkola R, Lautamäki R, Järvisalo M, Taittonen M, Rönnemaa T, Iozzo P, Knuuti J, Nuutila P, Raitakari OT. Effect of caloric restriction on myocardial fatty acid uptake, left ventricular mass, and cardiac work in obese adults. Am J Cardiol 103: 1721–1726, 2009. doi: 10.1016/j.amjcard.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 599.Karimian S, Stein J, Bauer B, Teupe C. Improvement of impaired diastolic left ventricular function after diet-induced weight reduction in severe obesity. DMSO 10: 19–25, 2017. doi: 10.2147/DMSO.S124541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Varli M, Turhan S, Aras S, Atli T, Erdogan G. Effects of weight loss on ventricular systolic and diastolic functions and left ventricular mass assessed by tissue doppler imaging in obese geriatric women: preliminary report. Aging Clin Exp Res 22: 206–211, 2010. doi: 10.1007/BF03324798. [DOI] [PubMed] [Google Scholar]
- 601.Gulsin GS, Swarbrick DJ, Athithan L, Brady EM, Henson J, Baldry E, Argyridou S, Jaicim NB, Squire G, Walters Y, Marsh AM, McAdam J, Parke KS, Biglands JD, Yates T, Khunti K, Davies MJ, McCann GP. Effects of low-energy diet or exercise on cardiovascular function in working-age adults with type 2 diabetes: a prospective, randomized, open-label, blinded end point trial. Diabetes Care 43: 1300–1310, 2020. doi: 10.2337/dc20-0129. [DOI] [PubMed] [Google Scholar]
- 602.Andersson J, Mellberg C, Otten J, Ryberg M, Rinnstrom D, Larsson C, Lindahl B, Hauksson J, Johansson B, Olsson T. Left ventricular remodelling changes without concomitant loss of myocardial fat after long-term dietary intervention. Int J Cardiol 216: 92–96, 2016. doi: 10.1016/j.ijcard.2016.04.050. [DOI] [PubMed] [Google Scholar]
- 603.de las Fuentes L, Waggoner AD, Mohammed BS, Stein RI, Miller BV, 3rd, Foster GD, Wyatt HR, Klein S, Davila-Roman VG. Effect of moderate diet-induced weight loss and weight regain on cardiovascular structure and function. J Am Coll Cardiol 54: 2376–2381, 2009. doi: 10.1016/j.jacc.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Leung M, Xie M, Durmush E, Leung DY, Wong VW. Weight loss with sleeve gastrectomy in obese type 2 diabetes mellitus: impact on cardiac function. Obes Surg 26: 321–326, 2016. doi: 10.1007/s11695-015-1748-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Kaier TE, Morgan D, Grapsa J, Demir OM, Paschou SA, Sundar S, Hakky S, Purkayastha S, Connolly S, Fox KF, Ahmed A, Cousins J, Nihoyannopoulos P. Ventricular remodelling post-bariatric surgery: is the type of surgery relevant? A prospective study with 3D speckle tracking. Eur Heart J Cardiovasc Imaging 15: 1256–1262, 2014. doi: 10.1093/ehjci/jeu116. [DOI] [PubMed] [Google Scholar]
- 606.Garza CA, Pellikka PA, Somers VK, Sarr MG, Collazo-Clavell ML, Korenfeld Y, Lopez-Jimenez F. Structural and functional changes in left and right ventricles after major weight loss following bariatric surgery for morbid obesity. Am J Cardiol 105: 550–556, 2010. doi: 10.1016/j.amjcard.2009.09.057. [DOI] [PubMed] [Google Scholar]
- 607.Ikonomidis I, Mazarakis A, Papadopoulos C, Patsouras N, Kalfarentzos F, Lekakis J, Kremastinos DT, Alexopoulos D. Weight loss after bariatric surgery improves aortic elastic properties and left ventricular function in individuals with morbid obesity: a 3-year follow-up study. J Hypertens 25: 439–447, 2007. doi: 10.1097/HJH.0b013e3280115bfb. [DOI] [PubMed] [Google Scholar]
- 608.Hsuan CF, Huang CK, Lin JW, Lin LC, Lee TL, Tai CM, Yin WH, Tseng WK, Hsu KL, Wu CC. The effect of surgical weight reduction on left ventricular structure and function in severe obesity. Obesity 18: 1188–1193, 2010. doi: 10.1038/oby.2010.42. [DOI] [PubMed] [Google Scholar]
- 609.Algahim MF, Lux TR, Leichman JG, Boyer AF, Miller CC, Laing ST, Wilson EB, Scarborough T, Yu S, Snyder B, Wolin-Riklin C, Kyle UG, Taegtmeyer H. Progressive regression of left ventricular hypertrophy two years after bariatric surgery. Am J Med 123: 549–555, 2010. doi: 10.1016/j.amjmed.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Jhaveri RR, Pond KK, Hauser TH, Kissinger KV, Goepfert L, Schneider B, Jones DB, Manning WJ. Cardiac remodeling after substantial weight loss: a prospective cardiac magnetic resonance study after bariatric surgery. Surg Obes Relat Dis 5: 648–652, 2009. doi: 10.1016/j.soard.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 611.Lin CH, Kurup S, Herrero P, Schechtman KB, Eagon JC, Klein S, Dávila-Román VG, Stein RI, Dorn GW 2nd, Gropler RJ, Waggoner AD, Peterson LR. Myocardial oxygen consumption change predicts left ventricular relaxation improvement in obese humans after weight loss. Obesity 19: 1804–1812, 2011. doi: 10.1038/oby.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Valezi AC, Machado VH. Morphofunctional evaluation of the heart of obese patients before and after bariatric surgery. Obes Surg 21: 1693–1697, 2011. doi: 10.1007/s11695-011-0431-0. [DOI] [PubMed] [Google Scholar]
- 613.Luaces M, Cachofeiro V, Garcia-Munoz-Najar A, Medina M, Gonzalez N, Cancer E, Rodriguez-Robles A, Canovas G, Antequera P. . Anatomical and functional alterations of the heart in morbid obesity. Changes after bariatric surgery. Rev Esp Cardiol (Engl Ed) 65: 14–21, 2012. doi: 10.1016/j.recesp.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 614.Rider OJ, Francis JM, Ali MK, Petersen SE, Robinson M, Robson MD, Byrne JP, Clarke K, Neubauer S. Beneficial cardiovascular effects of bariatric surgical and dietary weight loss in obesity. J Am Coll Cardiol 54: 718–726, 2009. doi: 10.1016/j.jacc.2009.02.086. [DOI] [PubMed] [Google Scholar]
- 615.Alpert MA, Nusair MB, Mukerji R, Omran J, Mehra A, Ardhanari S, Kumar SA, Terry BE. Effect of weight loss on ventricular repolarization in normotensive severely obese patients with and without heart failure. Am J Med Sci 349: 17–23, 2015. doi: 10.1097/MAJ.0000000000000342. [DOI] [PubMed] [Google Scholar]
- 616.Lee SC, Daimon M, Di TM, Homma S, Hasegawa T, Chiou SH, Nakao T, Hirokawa M, Mizuno Y, Yatomi Y, Yamazaki T, Komuro I. Beneficial effect of body weight control on left ventricular diastolic function in the general population: an analysis of longitudinal data from a health check-up clinic. Eur Heart J Cardiovasc Imaging 19: 136–142, 2018. doi: 10.1093/ehjci/jex219. [DOI] [PubMed] [Google Scholar]
- 617.Goodpaster BH, Delany JP, Otto AD, Kuller L, Vockley J, South-Paul JE, Thomas SB, Brown J, McTigue K, Hames KC, Lang W, Jakicic JM. Effects of diet and physical activity interventions on weight loss and cardiometabolic risk factors in severely obese adults: a randomized trial. JAMA 304: 1795–1802, 2010. doi: 10.1001/jama.2010.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Wang T, Heianza Y, Sun D, Huang T, Ma W, Rimm EB, Manson JE, Hu FB, Willett WC, Qi L. Improving adherence to healthy dietary patterns, genetic risk, and long term weight gain: gene-diet interaction analysis in two prospective cohort studies. BMJ 360: j5644, 2018. doi: 10.1136/bmj.j5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Florido R, Ndumele CE, Kwak L, Pang Y, Matsushita K, Schrack JA, Lazo M, Nambi V, Blumenthal RS, Folsom AR, Coresh J, Ballantyne CM, Selvin E. Physical activity, obesity, and subclinical myocardial damage. JACC Heart Fail 5: 377–384, 2017. doi: 10.1016/j.jchf.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Lobelo F, Rohm Young D, Sallis R, Garber MD, Billinger SA, Duperly J, Hutber A, Pate RR, Thomas RJ, Widlansky ME, McConnell MV, Joy EA. Routine assessment and promotion of physical activity in healthcare settings: a scientific statement from the American Heart Association. Circulation 137: e495–e522, 2018. doi: 10.1161/cir.0000000000000559. [DOI] [PubMed] [Google Scholar]
- 621.Wu NN, Tian H, Chen P, Wang D, Ren J, Zhang Y. Physical exercise and selective autophagy: benefit and risk on cardiovascular health . Cells 8: 1436, 2019. doi: 10.3390/cells8111436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Ayinapudi K, Samson R, Le Jemtel TH, Marrouche NF, Oparil S. Weight reduction for obesity-induced heart failure with preserved ejection fraction. Curr Hypertens Rep 22: 47, 2020. doi: 10.1007/s11906-020-01074-w. [DOI] [PubMed] [Google Scholar]
- 623.Han X, Ren J. Caloric restriction and heart function: is there a sensible link? Acta Pharmacol Sin 31: 1111–1117, 2010. doi: 10.1038/aps.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Han X, Turdi S, Hu N, Guo R, Zhang Y, Ren J. Influence of long-term caloric restriction on myocardial and cardiomyocyte contractile function and autophagy in mice. J Nutr Biochem 23: 1592–1599, 2012. doi: 10.1016/j.jnutbio.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Nicklas BJ, Brinkley TE, Houston DK, Lyles MF, Hugenschmidt CE, Beavers KM, Leng X. Effects of caloric restriction on cardiorespiratory fitness, fatigue, and disability responses to aerobic exercise in older adults with obesity: a randomized controlled Trial. J Gerontol A Biol Sci Med Sci 74: 1084–1090, 2019. doi: 10.1093/gerona/gly159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Khera R, Murad MH, Chandar AK, Dulai PS, Wang Z, Prokop LJ, Loomba R, Camilleri M, Singh S. Association of pharmacological treatments for obesity with weight loss and adverse events a systematic review and meta-analysis. JAMA 315: 2424–2434, 2016. doi: 10.1001/jama.2016.7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Srivastava G, Apovian CM. Current pharmacotherapy for obesity. Nat Rev Endocrinol 14: 12–24, 2018. doi: 10.1038/nrendo.2017.122. [DOI] [PubMed] [Google Scholar]
- 628.Bessesen DH, Van Gaal LF. Progress and challenges in anti-obesity pharmacotherapy. Lancet Diabetes Endocrinol 6: 237–248, 2018. doi: 10.1016/S2213-8587(17)30236-X. [DOI] [PubMed] [Google Scholar]
- 629.Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov 15: 639–660, 2016. doi: 10.1038/nrd.2016.75. [DOI] [PubMed] [Google Scholar]
- 630.Narayanaswami V, Dwoskin LP. Obesity: Current and potential pharmacotherapeutics and targets. Pharmacol Ther 170: 116–147, 2017. doi: 10.1016/j.pharmthera.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Valsamakis G, Konstantakou P, Mastorakos G. New targets for drug treatment of obesity. Annu Rev Pharmacol Toxicol 57: 585–605, 2017. doi: 10.1146/annurev-pharmtox-010716-104735. [DOI] [PubMed] [Google Scholar]
- 632.Bae EJ. DPP-4 inhibitors in diabetic complications: role of DPP-4 beyond glucose control. Arch Pharm Res 39: 1114–1128, 2016. doi: 10.1007/s12272-016-0813-x. [DOI] [PubMed] [Google Scholar]
- 633.Zhuge F, Ni Y, Nagashimada M, Nagata N, Xu L, Mukaida N, Kaneko S, Ota T. DPP-4 inhibition by linagliptin attenuates obesity-related inflammation and insulin resistance by regulating M1/M2 macrophage polarization. Diabetes 65: 2966–2979, 2016. doi: 10.2337/db16-0317. [DOI] [PubMed] [Google Scholar]
- 634.Rohrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Front Immunol 6: 1–20, 2015. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Drucker DJ. The cardiovascular biology of glucagon-like peptide-1. Cell Metab 24: 15–30, 2016. doi: 10.1016/j.cmet.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 636.Zhong JX, Maiseyeu A, Davis SN, Rajagopalan S. DPP4 in cardiometabolic disease recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ Res 116: 1491–1504, 2015. doi: 10.1161/CIRCRESAHA.116.305665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I, SAVOR-TIMI 53 Steering Committee and Investigators . Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 369: 1317–1326, 2013. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 638.Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S, Lachin JM, McGuire DK, Pencina MJ, Standl E, Stein PP, Suryawanshi S, Van de Werf F, Peterson ED, Holman RR, TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 373: 232–242, 2015. doi: 10.1056/NEJMoa1501352. [DOI] [PubMed] [Google Scholar]
- 639.Yen FS, Chiang JH, Hwu CM, Yen YH, Lin BJ, Wei JC, Hsu CC. All-cause mortality of insulin plus dipeptidyl peptidase-4 inhibitors in persons with type 2 diabetes. BMC Endocr Disord 19: 3, 2019. doi: 10.1186/s12902-018-0330-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Maixner N, Bechor S, Vershinin Z, Pecht T, Goldstein N, Haim Y, Rudich A. Transcriptional dysregulation of adipose tissue autophagy in obesity. Physiology (Bethesda) 31: 270–282, 2016. doi: 10.1152/physiol.00048.2015. [DOI] [PubMed] [Google Scholar]
- 641.Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M, Ruegg MA. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab 17: 731–744, 2013. doi: 10.1016/j.cmet.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 642.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296, 2013. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8: 903–914, 2012. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621–2637, 2007. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 112: 91–100, 2003. doi: 10.1172/JCI200317797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Wu Y, Potempa LA, El Kebir D, Filep JG. C-reactive protein and inflammation: conformational changes affect function. Biol Chem 396: 1181–1197, 2015. doi: 10.1515/hsz-2015-0149. [DOI] [PubMed] [Google Scholar]
- 647.Burghardt PR, Kemmerer ES, Buck BJ, Osetek AJ, Yan C, Koch LG, Britton SL, Evans SJ. Dietary n-3:n-6 fatty acid ratios differentially influence hormonal signature in a rodent model of metabolic syndrome relative to healthy controls. Nutr Metab (Lond) 7: 53, 2010. doi: 10.1186/1743-7075-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Wan JB, Huang LL, Rong R, Tan R, Wang J, Kang JX. Endogenously decreasing tissue n-6/n-3 fatty acid ratio reduces atherosclerotic lesions in apolipoprotein E-deficient mice by inhibiting systemic and vascular inflammation. Arterioscler Thromb Vasc Biol 30: 2487–2494, 2010. doi: 10.1161/ATVBAHA.110.210054. [DOI] [PubMed] [Google Scholar]
- 649.Packer M, Kitzman DW. Obesity-related heart failure with a preserved ejection fraction: the mechanistic rationale for combining inhibitors of aldosterone, neprilysin, and sodium-glucose cotransporter-2. JACC Heart Fail 6: 633–639, 2018. doi: 10.1016/j.jchf.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 650.Sowers JR, Habibi J, Jia G, Bostick B, Manrique-Acevedo C, Lastra G, Yang Y, Chen D, Sun Z, Domeier TL, Durante W, Whaley-Connell AT, Hill MA, Jaisser F, DeMarco VG, Aroor AR. Endothelial sodium channel activation promotes cardiac stiffness and diastolic dysfunction in Western diet fed female mice. Metabolism 109: 154223, 2020. doi: 10.1016/j.metabol.2020.154223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Ren DY, Zhang Y. Cardiovascular benefit of SGLT2 inhibitors in the therapeutics of diabetes mellitus: a close look beyond the horizon. Curr Drug Targets 19: 1051–1057, 2018. doi: 10.2174/1389450119666180531102227. [DOI] [PubMed] [Google Scholar]
- 652.Ceylan-Isik AF, Fliethman RM, Wold LE, Ren J. Herbal and traditional Chinese medicine for the treatment of cardiovascular complications in diabetes mellitus. Curr Diabetes Rev 4: 320–328, 2008. doi: 10.2174/157339908786241142. [DOI] [PubMed] [Google Scholar]
- 653.Ren J, Dominguez LJ, Sowers JR, Davidoff AJ. Metformin but not glyburide prevents high glucose-induced abnormalities in relaxation and intracellular Ca2+ transients in adult rat ventricular myocytes. Diabetes 48: 2059–2065, 1999. doi: 10.2337/diabetes.48.10.2059. [DOI] [PubMed] [Google Scholar]
- 654.Ammazzalorso A, Maccallini C, Amoia P, Amoroso R. Multitarget PPARgamma agonists as innovative modulators of the metabolic syndrome. Eur J Med Chem 173: 261–273, 2019. doi: 10.1016/j.ejmech.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 655.Reddy MA, Natarajan R. Recent developments in epigenetics of acute and chronic kidney diseases. Kidney Int 88: 250–261, 2015. ] doi: 10.1038/ki.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Labonte B, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I, Bureau A, Mechawar N, Szyf M, Meaney MJ, Turecki G. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry 69: 722–731, 2012. doi: 10.1001/archgenpsychiatry.2011.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Hamm CA, Costa FF. Epigenomes as therapeutic targets. Pharmacol Ther 151: 72–86, 2015. doi: 10.1016/j.pharmthera.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 658.Vaiserman A. Epidemiologic evidence for association between adverse environmental exposures in early life and epigenetic variation: a potential link to disease susceptibility? Clin Epigenetics 7: 96, 2015. doi: 10.1186/s13148-015-0130-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Mackey RH, McTigue KM, Chang YF, Barinas-Mitchell E, Evans RW, Tinker LF, Lewis CE, Manson JE, Stefanick ML, Howard BV, Phillips LS, Liu S, Kulick D, Kuller LH. Lipoprotein particles and size, total and high molecular weight adiponectin, and leptin in relation to incident coronary heart disease among severely obese postmenopausal women: The Women's Health Initiative Observational Study. BBA clinical 3: 243–250, 2015. doi: 10.1016/j.bbacli.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Maurovich-Horvat P, Kallianos K, Engel LC, Szymonifka J, Schlett CL, Koenig W, Hoffmann U, Truong QA. Relationship of thoracic fat depots with coronary atherosclerosis and circulating inflammatory biomarkers. Obesity 23: 1178–1184, 2015. doi: 10.1002/oby.21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Dalgaard K, Landgraf K, Heyne S, Lempradl A, Longinotto J, Gossens K, Ruf M, Orthofer M, Strogantsev R, Selvaraj M, Lu TT, Casas E, Teperino R, Surani MA, Zvetkova I, Rimmington D, Tung YC, Lam B, Larder R, Yeo GS, O'Rahilly S, Vavouri T, Whitelaw E, Penninger JM, Jenuwein T, Cheung CL, Ferguson-Smith AC, Coll AP, Körner A, Pospisilik JA. Trim28 haploinsufficiency triggers bi-stable epigenetic obesity. Cell 164: 353–364, 2016. doi: 10.1016/j.cell.2015.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Al-Najim W, Docherty NG, Le Roux CW. Food intake and eating behavior after bariatric surgery. Physiol Rev 98: 1113–1141, 2018. doi: 10.1152/physrev.00021.2017. [DOI] [PubMed] [Google Scholar]
- 663.Samson WK. Food intake, eating behavior, and indices of glucose homeostasis following bariatric surgery. Am J Physiol Regul Integr Comp Physiol 315: R668, 2018. doi: 10.1152/ajpregu.00207.2018. [DOI] [PubMed] [Google Scholar]
- 664.Beamish AJ, Olbers T, Kelly AS, Inge TH. Cardiovascular effects of bariatric surgery. Nat Rev Cardiol 13: 730–743, 2016. doi: 10.1038/nrcardio.2016.162. [DOI] [PubMed] [Google Scholar]
- 665.Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. N Engl J Med 376: 254–266, 2017. doi: 10.1056/NEJMra1514009. [DOI] [PubMed] [Google Scholar]
- 666.Cuspidi C, Rescaldani M, Tadic M, Sala C, Grassi G. Effects of bariatric surgery on cardiac structure and function: a systematic review and meta-analysis. Am J Hypertens 27: 146–156, 2014. doi: 10.1093/ajh/hpt215. [DOI] [PubMed] [Google Scholar]
- 667.Rayner JJ, Banerjee R, Francis JM, Shah R, Murthy VL, Byrne J, Neubauer S, Rider OJ. Normalization of visceral fat and complete reversal of cardiovascular remodeling accompany gastric bypass, not banding. J Am Coll Cardiol 66: 2569–2570, 2015. doi: 10.1016/j.jacc.2015.09.071. [DOI] [PubMed] [Google Scholar]
- 668.Schiavon CA, Bersch-Ferreira AC, Santucci EV, Oliveira JD, Torreglosa CR, Bueno PT, Frayha JC, Santos RN, Damiani LP, Noujaim PM, Halpern H, Monteiro FL, Cohen RV, Uchoa CH, de Souza MG, Amodeo C, Bortolotto L, Ikeoka D, Drager LF, Cavalcanti AB, Berwanger O. Effects of bariatric surgery in obese patients with hypertension: The GATEWAY Randomized Trial (Gastric Bypass to Treat Obese Patients With Steady Hypertension). Circulation 137: 1132–1142, 2018. doi: 10.1161/CIRCULATIONAHA.117.032130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Boido A, Ceriani V, Cetta F, Lombardi F, Pontiroli AE. Bariatric surgery and prevention of cardiovascular events and mortality in morbid obesity: mechanisms of action and choice of surgery. Nutr Metab Cardiovasc Dis 25: 437–443, 2015. doi: 10.1016/j.numecd.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 670.Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M. Transcriptional and Epigenetic Regulation of Autophagy in Aging. Autophagy 11: 867–880, 2015. doi: 10.1080/15548627.2015.1034410. [DOI] [PMC free article] [PubMed] [Google Scholar]