

SUMMARY
Objective:
Osteoarthritis (OA) is a progressive degenerative disease of the articular cartilage caused by an unbalanced activity of proteases, cytokines and other secreted proteins. Since heparan sulfate (HS) determines the activity of many extracellular factors, we investigated its role in OA progression.
Methods:
To analyze the role of the HS level, OA was induced by anterior cruciate ligament transection (ACLT) in transgenic mice carrying a loss-of-function allele of Ext1 in clones of chondrocytes (Col2-rtTA-Cre;Ext1e2fl/e2fl). To study the impact of the HS sulfation pattern, OA was surgically induced in mice with a heterozygous (Ndst1+/−) or chondrocyte-specific (Col2-Cre;Ndst1fl/fl) loss-of-function allele of the sulfotransferase Ndst1. OA progression was evaluated using the OARSI scoring system. To investigate expression and activity of cartilage degrading proteases, femoral head explants of Ndst1+/− mutants were analyzed by qRT-PCR, Western Blot and gelatin zymography.
Results:
All investigated mouse strains showed reduced OA scores (Col2-rtTA-Cre;Ext1e2fl/e2fl: 0.83; 95% HDI 0.72–0.96; Ndst1+/−: 0.83, 95% HDI 0.74–0.9; Col2-Cre;Ndst1fl/fl: 0.87, 95% HDI 0.76–1). Using cartilage explant cultures of Ndst1 animals, we detected higher amounts of aggrecan degradation products in wildtype samples (NITEGE 4.24-fold, 95% HDI 1.05–18.55; VDIPEN 1.54-fold, 95% HDI 1.54–2.34). Accordingly, gelatin zymography revealed lower Mmp2 activity in mutant samples upon RA-treatment (0.77-fold, 95% HDI: 0.60–0.96). As expression of major proteases and their inhibitors was not altered, HS seems to regulate cartilage degeneration by affecting protease activity.
Conclusion:
A decreased HS content or a reduced sulfation level protect against OA progression by regulating protease activity rather than expression.
Keywords: Heparan sulfate, Osteoarthritis, Ndst1, Ext1, Aggrecan degradation, Mmp2, Articular cartilage
Introduction
The mechanical properties of the articular cartilage are determined by a network of collagen fibers providing tensile strength, and glycosaminoglycans (GAGs), predominantly chondroitin sulfate (CS) and hyaluronic acid, giving the tissue its compressive features. This extracellular matrix (ECM) undergoes slow but constant remodeling. During osteoarthritis (OA), the balance between ECM synthesis and degradation shifts towards catabolic processes, including the degradation of collagens and aggrecan by members of the matrix metalloprotease (MMP) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) families1.
The distribution and activity of many secreted proteins like proteases, cytokines and growth factors are regulated by heparan sulfate (HS) carrying proteoglycans (HSPGs)2,3. The synthesis of the HS chain takes place in the Golgi apparatus where the glycosyltransferases EXT1 and EXT2 catalyze the elongation of the polysaccharide chain by adding alternating units of d-glucuronic acid (GlcA) and N-acetyl-d-glucosamine (GlcNAc) to a tetrasaccharide linker attached to serine residues of the core proteins. Simultaneously, the sugar chain is modified by N-deacetylase-N-sulfotransferases (NDST1–4), the d-glucuronyl-C5-epimerase (GLCE), and several 2-O, 3-O and 6-O sulfotransferases. The overlapping expression of these enzymes generates a tissue-specific sulfation pattern, which determines the binding affinities of HS to extracellular proteins and thereby their activity and distribution in the ECM4.
To gain insight into the importance of HS for the regulation of articular cartilage homeostasis, we have investigated the progression of OA in three mouse strains carrying mutations in either Ext1 or Ndst1. While homozygous deletion of Ext1, which results in a complete loss of HS, is embryonically lethal5, mice carrying a chondrocyte-specific, clonal deletion of Ext1 (Col2-rtTA-Cre;Ext1e2fl/e2fl) are viable and fertile6. These mice have been created to mimic the human Multiple Osteochondroma syndrome, which originates in mutations in EXT1 or EXT27,8. Interestingly, while HS-deficient chondrocytes at the border of the growth plate develop into osteochondromas6,9,10, mutant cells in the articular cartilage morphologically resemble hypertrophic chondrocytes (personal observation and11). Since the presence of hypertrophic chondrocytes in the articular cartilage has been described as an early sign of OA12–14, we hypothesized that Col2-rtTA-Cre;Ext1e2fl/e2fl mutants are prone to develop OA.
In contrast to Ext1 deficiency the inactivation of Ndst1 (Ndst1−/−), which determines regions of high sulfation on the HS chain, results in the production of severely undersulfated HS15,16. Homozygous Ndst1−/− mutants die perinatally due to respiratory failure, while heterozygous Ndst1+/− animals are viable and do not show any obvious phenotypic alterations. Nevertheless, mild changes in the HS structure have been described15, which may become important during ageing or under stress.
In this study, we show that Col2-rtTA-Cre;Ext1e2fl/e2fl mice develop reduced OA during ageing and after surgical induction of OA. Similarly, heterozygous and chondrocyte-specific inactivation of Ndst1 lead to less severe OA. Using femoral head explants, we demonstrate that aggrecan and collagen degradation are reduced in the articular cartilage of Ndst1+/− mutants and identify MMP2 as a protease with decreased proteolytic activity.
Methods
Transgenic mice
Transgenic Col2-rtTA-Cre;Ext1e2fl/e2fl [Tg(Col2a1-rtTA,tetO-cre) 22Pjro; Ext1tm1.1Vcs]6, R26R-LacZ [Gt(ROSA)26Sortm1Sor]17; Ndst1fl/fl mice [Ndst1tm1Grob]15, Prx1-Cre [Tg(Prrx1-cre)1Cjt]18 and Col2-Cre [Tg(Col2a1-cre)1Star]19 mice were maintained on a C57Bl/6J genetic background. Up to six females, one breeding pair or single males were kept in air filtered cages (SPF conditions, Green Line, Techniplast) under a 12 h light/dark cycle on regular bedding with nesting material (Abed) as environmental enrichment and food (Sniff) and water ad libidum.
Genotyping was performed by PCR on tail tip DNA (DirectPCR Lysis Reagent; VIAGEN) with primers listed in Table S1. Allele recombination in Col2-rtTA-Cre;Ext1e2fl/e2fl mice was induced by peritoneal injection of 80 mg Doxycycline per kg body weight (kgbw) into lactating dams at postnatal day 8 (P8)6. Mice undergoing surgery received 1 mg/kgbw Doxycycline at 6 weeks.
Surgical induction of OA
OA was induced in the left knee of female mice (8 weeks; approximately 20 gbw) by anterior cruciate ligament transection (ACLT20,21). As a control (sham) the joint capsule of the contralateral leg was incised. Mice were anesthetized with 160 mg Ketamine and 8 mg Xylazine/kgbw. Carprofen (5 mg/kgbw) was provided perioperatively and 3 days post-surgery. Health status was controlled daily. Litters with at least two mutant and control mice were operated in a blinded manner on the same day and caged together. Information about litters and operation groups were included in the statistical evaluation.
Histology
For the histological analysis, knees were dissected, fixed in 4% paraformaldehyde, decalcified in 25% EDTA and embedded in paraffin. 7 μm Sections were stained with Safranin-O. OA was evaluated on frontal sections according to the OARSI-scoring system in a double blinded manner22. Mean scores of the four quadrants of the femoro-tibial joint are displayed. Articular cartilage thickness was measured on frontal sections using ImageJ software23. Immunofluorescence analysis was performed with mouse anti-collagen type X (Hybridoma), rabbit anti-aggrecan (Millipore) and corresponding secondary antibodies (Jackson ImmunoResearch, ThermoFisher)24. β-Galactosidase was detected as described10.
Expression analysis by qRT-PCR
qRT-PCR was performed on femoral head cDNA (RNeasy Lipid tissue Kit, QIAgen; Maxima First Strand Kit, Fermentas) with the StepOnePlus Real-Time PCR (ThermoFisher Scientific) or CFX96 Touch Real-Time PCR System (BioRad) using EVA Green PCR Master Mix (BioBudget) and primers listed in Table S2.
Aggrecan degradation
Aggrecan degradation was investigated as described25. In brief, femoral heads of 4 weeks old mice were cultured pairwise in medium without and with 10 μM retinoic acid (RA) for 3 days. The GAG concentrations of the culture supernatant, a guanidium hydrochloride (GuHCl) extract of the femoral head and a papain digest of the remaining tissue were determined by dimethylmethylene blue assay to estimate total aggrecan content. Samples from at least two littermates with the same genotype were pooled, dialyzed and freeze-dried. The re-dissolved samples were digested with Chondroitinase ABC (Sigma) and analyzed by Western Blot for VDIPEN and NITIGE neo-epitopes. Signal intensity was quantified against total aggrecan content and background signal of the respective blot.
Western Blot
The following antibodies were used: for MMP and ADAMTS activity: anti-VDIPEN26, and rabbit anti-NITEGE (Polyclonal rabbit α-aggrecan neo (NITEGE) IgG, Thermo Scientific) and the respective IRdye680RD secondary antibody (LI-COR); for MMP2 protein: Rat anti-MMP2 (Chemicon), biotinylated goat anti-rabbit IgG (Vector Laboratories) and IRdye 800CW Streptavidin (LI-COR). Signal detection was performed with Odyssey CLx (LI-COR).
MMP activity
For gelatin zymography recombinant mouse/rat MMP2 (rMMP2, R&D systems) or supernatants of femoral head cultures were separated on 8%SDS PAGE gels containing 0.1% gelatin. Renaturation, digestion and staining were carried out as described27. Degradation bands were quantified with Photoshop CS 4 (Adobe) or ImageStudioLite (LICOR) software.
Statistical analysis
To estimate the effects of genotype, age, and other predictors on measured response quantities such as cartilage thickness, zymogram signals, etc. we described the relations between the responses and predictors as hierarchical generalized linear models (GLMs)28, with the exception of the response OA scores, for which we used a beta regression (see Supplementary Information). Strictly positive response quantities (except OA scores) were log-transformed for this analysis to enable use of Gaussian noise distributions for the transformed response29. OA scores were normalized to range 0–1. Weakly informative prior distributions were assumed for the slopes and intercepts of the GLMs, as proposed by R-package rstanarm, version 2.17.4 (https://CRAN.R-project.org/package=rstanarm; details in Supplementary Information).
Between groups of related measurements, e.g., measurements for animals of the same litter, intercepts were allowed to vary according to a joint noise distribution, constituting a second level of our hierarchical probability models. The posteriors of the probability models were numerically inferred by Bayesian analysis with rstanarm30. The agreement of the models with reality was checked by graphical posterior predictive checks (PPCs), i.e., comparisons of response distributions simulated with the fitted model vs distributions of actual measurements. If deviations in the PPCs were observed, the model was modified, typically by including an interaction, as e.g., in the case of the log cartilage thickness. If several models were considered for the same response, the best model was identified by comparing the competing models with respect to their ability to generalize to unseen data in an approximate leave-one-out cross-validation31.
Detailed information on the statistical outcomes and the number of animals used in each experiment can be found in Tables S3 and S4.
Results
Clonal deletion of Ext1 in chondrocytes protects from OA
To investigate the role of HS in the maintenance of the articular cartilage, we aged Col2-rtTA-Cre;Ext1e2fl/e2fl mice after recombination of the allele at postnatal day 8 (P8). Introduction of a R26R-LacZ reporter allele (Col2-rtTA-Cre;Ext1e2fl/e2fl;R26R-LacZ) identified clusters of mutant, hypertrophic-appearing cells, which are surrounded by wildtype articular cartilage [Fig. 1(A)]. Since hypertrophic chondrocytes in the articular cartilage are regarded as a sign of early OA12–14, we investigated OA development in knee joints of 3, 6, 12 and 18 month old mice using the OARSI scoring system22. No obvious signs of OA were detected in 3 month (3m) old wildtype or Col2-rtTA-Cre;Ext1e2fl/e2fl mice (mean OARSI scores 3m: 0.32 and 0.17, respectively), while at 6m and 12m first signs of cartilage degeneration were observed in both genotypes (OARSI scores 6m: 0.83 and 0.73; 12m: 1.10 and 1.10). At 18m control mice displayed mild to moderate signs of OA (OARSI score: 2.17) [Fig. 1(B)]. Surprisingly, at this stage Col2-rtTA-Cre;Ext1e2fl/e2fl mutants did not develop the expected increased signs of OA, but seemed to develop less OA (OARSI score 0.67). There was a clear positive effect of age on the observed OA scores (increase over time: 0.13 OARSI score units per month; 95% highest density intervals (HDI) 0.08–0.18). Importantly, OA scores of mutant mice increased less than those of their wildtype littermates (mean increase factor: −0.09, 95% HDI −0.16 to −0.03; Tables S3 and S4).
Fig. 1. Clonal deletion of Ext1 in Col2-rtTA-Cre;Ext1e2fl/e2fl mice protects against OA.
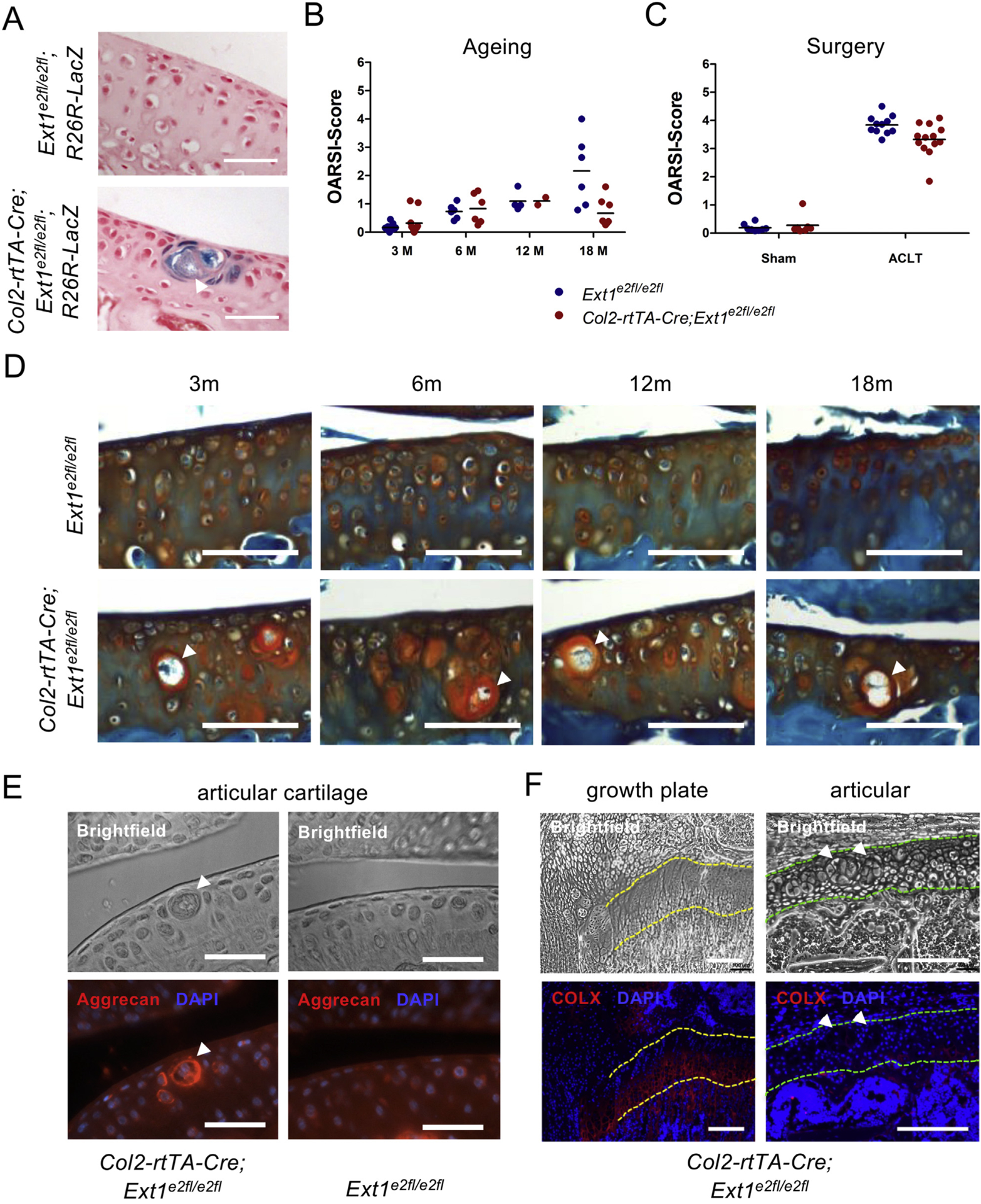
(A) Cre activity in 6 week old Col2-rtTA-Cre;Ext1e2fl/e2fl;R26R-LacZ samples was detected by β-Galactosidase staining of sagittal sections. Recombined cells are clustered and enlarged in size. Doxycycline administration at P8. Scale Bar: 50 μm; counterstaining: Nuclear Fast Red. White arrow head indicates cluster of recombined cells. (B) OARSI scores of Ext1e2fl/e2fl and Col2-rtTA-Cre;Ext1e2fl/e2fl mice, black line indicates mean. OA scores increase over time in both genotypes. At 18 months Col2-rtTA-Cre;Ext1e2fl/e2fl animals display reduced OA scores compared to Ext1e2fl/e2fl littermates (3m: n = 11, 6m: n = 6, 12m: n ≥ 2, 18m: n ≥ 6). Effect sizes: genotype 0.49 (95% HDI −0.27–1.25), age 0.13 (95% HDI 0.08–0.18), genotype and age 0.09 (95% HDI −0.16 to −0.03). Doxycycline administration at P8, analysis at 3, 6, 12 and 18 months. (C) OARSI scores of wildtype and Col2-rtTA-Cre;Ext1e2fl/e2fl mice after surgery. Col2-rtTA-Cre;Ext1e2fl/e2fl mice show reduced OA scores after ACLT (M: ACLT n = 13, sham n = 7; WT: ACLT n = 11, sham n = 8). Effect size: 0.83 (95% HDI 0.72–0.96). Doxycycline administration at 6 weeks, ACLT at 8 weeks, analysis at 12 weeks. (D) Safranin-O staining of frontal sections through knee joints of wildtype and Col2-rtTA-Cre;Ext1e2fl/e2fl mice at 3, 6, 12, and 18 months of age reveals decreased cartilage erosion in Col2-rtTA-Cre;Ext1e2fl/e2fl animals. Clusters of enlarged cells are present at all analyzed timepoints. Doxycycline administration at P8. Scale bar: 100 μm. White arrow heads indicate clusters of recombined cells. (E, F) Immunofluorescence labeling of aggrecan (E) and COLX (F) on sagittal sections of 4 weeks old Col2-rtTA-Cre;Ext1e2fl/e2fl mice. Increased aggrecan levels surround clusters of mutant cells in the articular cartilage. COLX is not expressed in mutant cells of the articular cartilage, while a clear signal is detected in hypertrophic chondrocytes of the growth plate on the same section. Doxycycline administration at P8. Scale Bar E: 50 μm, F: 200 μm. White arrow heads exemplify clusters of recombined cells. Outlines of growth plate (yellow) and articular cartilage (green) are highlighted by dashed lines.
At all analyzed time points, the observed clusters of enlarged, mutant cells in the articular cartilage did not obviously change in morphology, number or size [Fig. 1(D)]. Safranin-O and immunofluorescence staining for aggrecan on sections of P28 mice showed that these clusters were surrounded by a thick, proteoglycan-rich matrix containing increased levels of aggrecan [Fig. 1(E)]. The loss of HS seems thus to alter the ECM composition surrounding the HS-deficient cells. To further characterize the mutant cells, we analyzed the expression of the hypertrophy marker Collagen type X (COLX) by immunofluorescence. Although COLX was clearly present in the growth plate, we could not detect COLX in mutant or wildtype articular chondrocytes of the same section indicating that, although enlarged, the HS-deficient cells are molecularly distinct from hypertrophic chondrocytes of the growth plate [Fig. 1(F)].
To confirm the role of HS in OA pathogenesis, we surgically induced OA in Col2-rtTA-Cre;Ext1e2fl/e2fl mutants by ACLT20,21 at 8 weeks of age after recombining the Ext1 allele at 6 weeks. At 12 weeks, knee joints were dissected and Safranin-O stained frontal sections were scored for OA. As expected, control littermates developed severe signs of OA, while reduced scores were observed in Col2-rtTA-Cre;Ext1e2fl/e2fl mutants (mean OARSI scores: 3.8, and 3.3; median reduction factor of 0.83; 95% HDI 0.72–0.96; Fig. 1(C), Tables S3 and S4). Sham-operated contralateral legs showed no signs of OA pointing against a systemic effect of the operation or a substantial shift in weight bearing (OARSI scores: 0.19 (Ext1e2fl/e2fl) and 0.28 (Col2-rtTA-Cre;Ext1e2fl/e2fl)). In summary, the surgical model confirmed the findings observed in aged mice indicating that the HS level affects the severity of OA.
Ndst1-mutant mice display reduced OA progression
Since the clonal inactivation of Ext1 in Col2-rtTA-Cre;Ext1e2fl/e2fl mice makes it difficult to investigate the overall function of HS during OA pathogenesis, we analyzed OA progression in mice carrying a heterozygous loss-of-function allele of Ndst1. We surgically induced OA in control and Ndst1+/− mice at 8 weeks of age and analyzed the femorotibial joints as described. Sham-operated knees showed no obvious signs of cartilage erosion (mean OARSI scores: 0.11 and 0.17). In contrast, the ACLT-operated joints displayed severe OA including loss of the articular surface lamina and severe erosion of the articular cartilage occasionally reaching the subchondral bone [Fig. 2(A)]. We found reduced OA progression in heterozygous Ndst1+/− mice compared to controls (OARSI scores: 2.9 and 3.6; median reduction factor: 0.82, 95% HDI 0.74–0.90; Fig. 2(B), Tables S3 and S4). To evaluate whether complete loss of Ndst1 might have a similarly protective effect, we investigated mice carrying a chondrocyte-specific deletion of Ndst1 (Col2-Cre;Ndst1fl/fl). In contrast to the strong phenotypes seen in Ndst1−/− animals, Col2-Cre;Ndst1fl/fl mice were viable and fertile, and displayed no obvious morphological alterations of the skeleton. Sham surgery did not trigger OA development in Ndst1fl/fl or Col2-Cre;Ndst1fl/fl animals (OARSI scores: 0.05 and 0.12), while surgical induction led to severe OA (OARSI scores: 3.4 and 3.0; Fig. 2(A) and (B)). As observed for the Ndst1+/− mutants, the genotype had a modest, but clearly negative effect on OA progression in Col2-Cre;Ndst1fl/fl mice (median reduction factor 0.87, 95% HDI 0.76–1.00; Tables S3 and S4).
Fig. 2. Ndst1+/− and Col2-Cre;Ndst1fl/fl animals develop less severe OA after ACLT surgery.
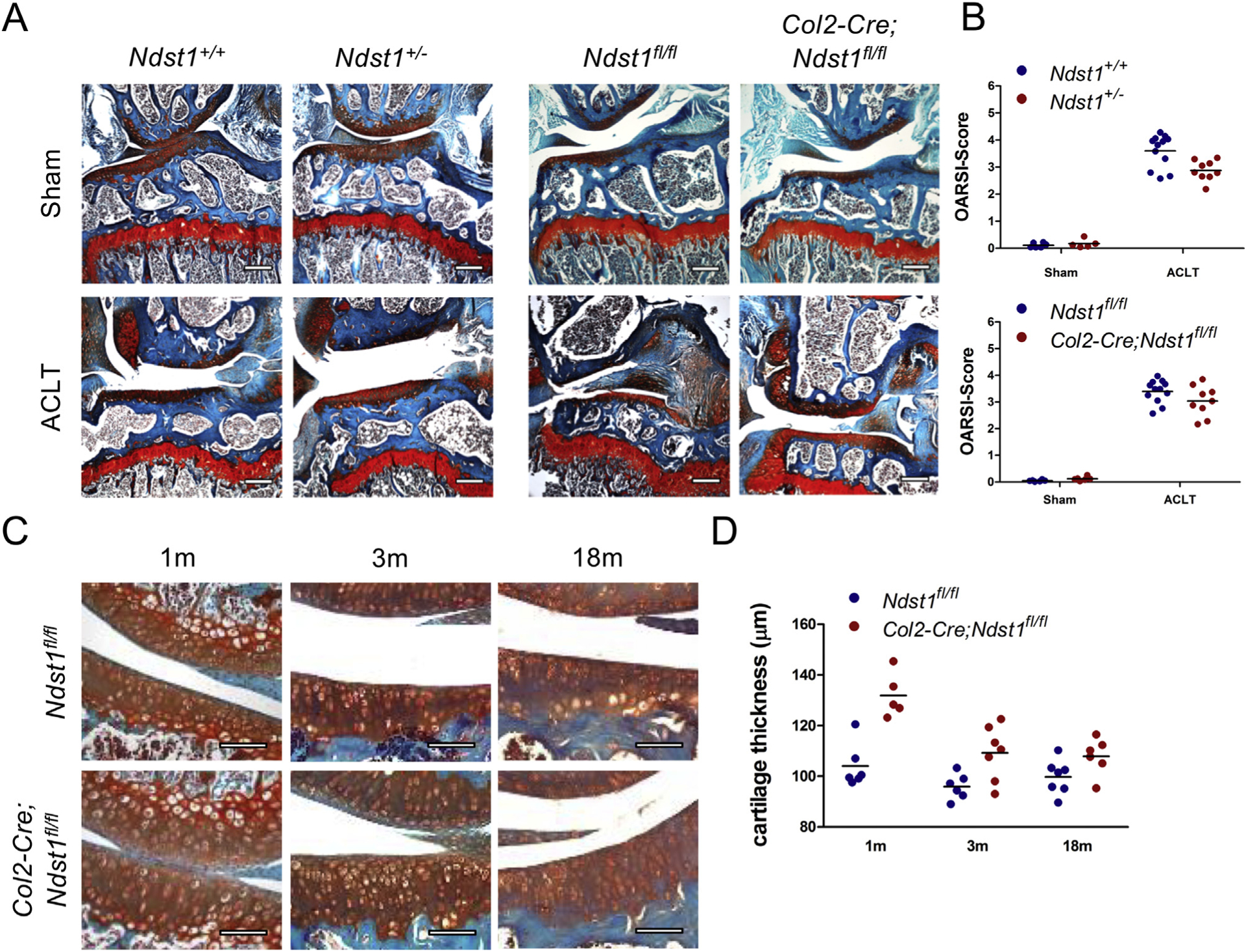
(A) Safranin-O staining of frontal sections of knee joints from Ndst1+/+, Ndst1+/−, Ndst1fl/fl and Col2-Cre;Ndst1fl/fl mice after surgery. Both Ndst1 mutants display reduced signs of OA compared to wildtype littermates. ACLT surgery at 8 weeks, analysis at 12 weeks. Scale Bar: 200 μm. (B) OA scores of individual mice, black lines indicate mean (Ndst1+/+: ACLT n = 12, sham n 6; Ndst1+/−: ACLT n = 9, sham n = 5; Ndst1fl/fl: ACLT n = 12, sham n = 6; Col2-Cre;Ndst1fl/fl: ALCT n = 9, sham n = 6). OA score change factors associated to mutation: Ndst1+/− 0.82 (95% HDI 0.74–0.90), Ndst1fl/fl 0.87 (95% HDI 0.76–1.00). (C) Safranin-O staining of sagittal sections from Ndst1fl/fl and Col2-Cre;Ndst1fl/fl mice at 1, 3, and 18 months of age. Col2-Cre;Ndst1fl/fl mutants display an increased thickness of the articular cartilage. Scale Bar: 100 μm. (D) Quantification of cartilage thickness, black lines indicate mean (1m: n ≥ 5, 3m: n ≥ 6, 18m: n ≥ 6). Thickness change due to mutation: 1.24 (95% HDI 1.15–1.33).
Chondrocyte-specific loss of Ndst1 alters joint morphology
During the histological evaluation of Col2-Cre;Ndst1fl/fl knees we noticed morphological changes of the articular cartilage [Fig. 2(C)]. Analysis at 1, 3 and 18 months of age revealed an increased thickness in Col2-Cre;Ndstfl/fl mutants at all investigated time points (1m: 131 μm and 104 μm; 3m: 109 μm and 95 μm; 18m: 108 μm and 100 μm; mean increase in thickness: 1.24-fold; 95% HDI 1.15–1.33; Fig. 2(D), Tables S3 and S4). An increased articular cartilage thickness might contribute to the reduced OA scores by delaying the complete erosion of the cartilage down to the subchondral bone. We did, however, not detect differences in articular cartilage thickness in heterozygous Ndst1+/− mutants (data not shown), which are also protected against OA. This indicates that additional degenerating mechanisms are regulated by the altered HS structure.
Expression of ECM-degrading proteases is unaltered in Ndst1+/− mice
To test if the expression of main proteases of the articular cartilage is affected by the altered HS structure, we analyzed the expression of Mmp2, Mmp3, Mmp9, Mmp13, Adamts4 and Adamts51,32,33 in Ndst1+/− femoral head cultures treated with RA to induce cartilage degradation34 by qRT-PCR. We did not detect differences in the expression of any of the proteases between Ndst1+/− mutants and wildtype littermates (Fig. 3(A); Tables S3 and S4). On the activity level, MMPs are controlled by members of the tissue inhibitor of metalloproteinases (TIMP) family35. We thus quantified the expression of Timp1, Timp2 and Timp3 in the same RNA samples, but did not detect differences in their expression between Ndst1+/− mutants and controls (Fig. 3(B); Tables S3 and S4).
Fig. 3. The expression levels of major proteases and protease inhibitors are not altered in Ndst1+/− mice.
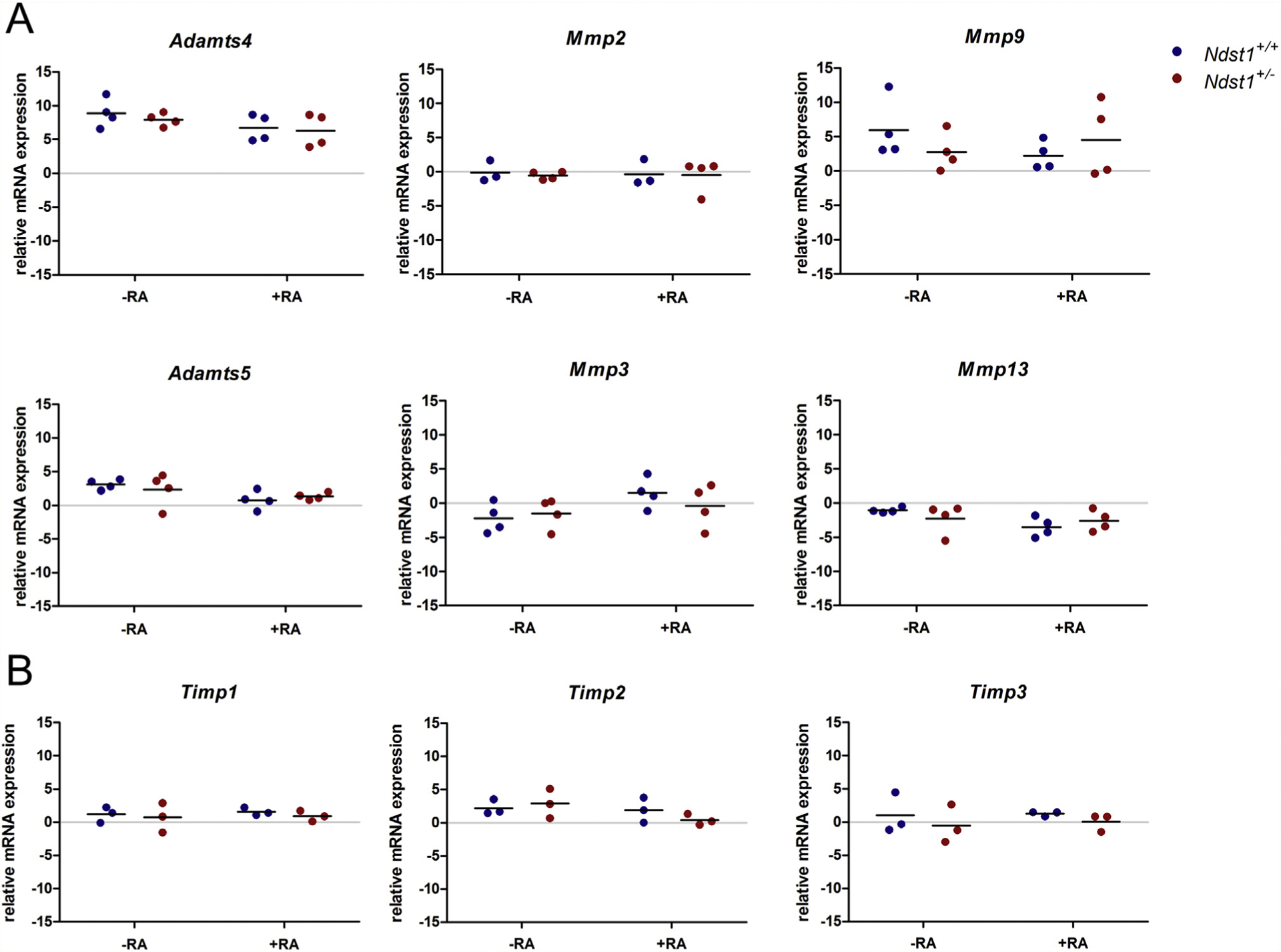
qRT-PCR analysis of protease and protease inhibitor expression in femoral heads cultured for 3 days in the presence or absence of RA. Scatter plots display ΔCT values relative to B2M, black lines indicate means (mRNA pools from n ≥ 3 litters; no statistical difference; Tables S3 and S4).
Aggrecan degradation is decreased in Ndst1+/− cartilage explants
Since HS regulates the activity of many extracellular proteins, we next asked if protease function is altered by changes in the HS structure. The degradation of aggrecan by metalloproteases of the ADAMTS and MMP families results in the generation of the neo-epitopes VDIPEN36 and NITEGE37, respectively. To investigate aggrecan degradation, we cultured femoral heads of 4 weeks old Ndst1+/+ and Ndst1+/− mice and treated them with RA34. The presence of each neo-epitope was analyzed in the culture medium and in GuHCl extracts of the explant tissue by Western Blot25. In the supernatant of untreated femur heads, VDIPEN and NITEGE neo-epitopes were only detected in few samples. Higher levels of both epitopes were present in GuHCl extracts of these tissues, but the signal intensity could not always be quantified [Fig. 4(A) and (B)]. Nevertheless, the obtained data hint at decreased levels of NITEGE neo-epitopes in Ndst1+/− samples. After RA treatment, both neo-epitopes could reproducibly be detected in the culture medium and GuHCl extracts. In accordance with the elevated OA scores higher levels of aggrecan degradation products were found in the supernatants (VDIPEN: 1.43-fold; NITEGE: 2.13-fold) and tissue extracts of wildtype mice (VDIPEN: 1.52-fold; NITEGE: 2.50-fold) [Fig. 4(A) and (B)]. Taken together, there was a clear effect of the genotype on the detected VDIPEN and NITEGE signals after RA treatment (wildtype-associated mean increases VDIPEN: 1.54; 95% HDI 1.00–2.34; NITEGE: 4.24; 95% HDI 1.05–18.6; Tables S3 and S4).
Fig. 4. Protease activity is reduced in Ndst1+/− mutants.
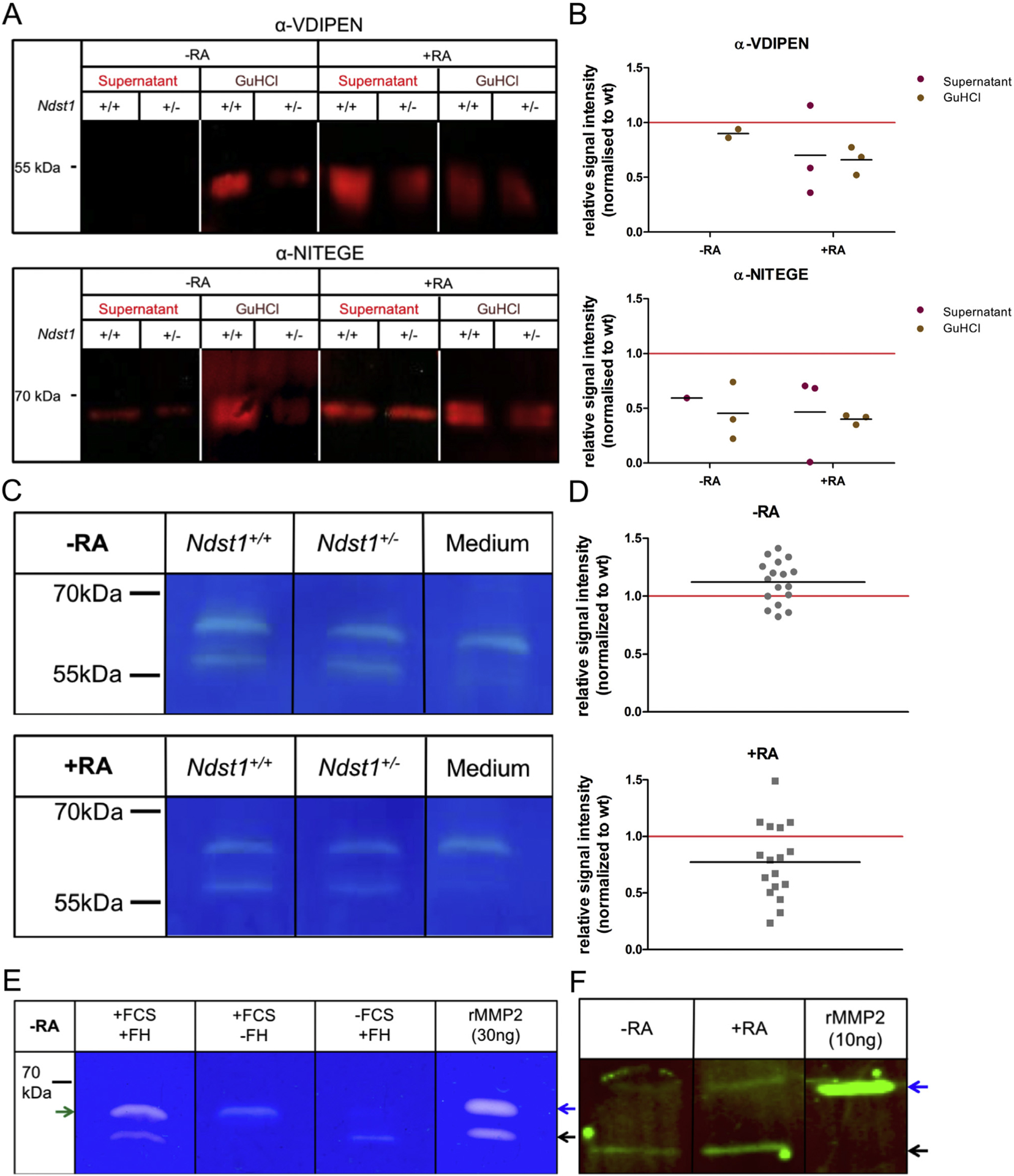
(A) Western Blot analysis of the aggrecan neo-epitopes VDIPEN and NITEGE in supernatants and GuHCl extracts of femoral head explants cultured in the presence or absence of RA. Signal intensities are decreased in RA-treated Ndst1+/− samples. (B) Scatter plots represent relative values of individual litters (littermates pooled by genotype; n ≥ 3 litters, normalized to wildtype (wt)). Wildtype-associated factors of mean increase of digestion signal: NITEGE 4.24 (95% HDI 1.05–18.6), VDIPEN 1.54 (95% HDI 1.00–2.34). (C) Gelatin zymography of supernatants of femoral head explants cultivated with or without RA. A protease of ~65 kDa is contained in the culture medium. At ~58 kDa a femoral head-specific protease is present, which is less active in Ndst1+/− samples upon RA treatment. (D) Scatter plots represent quantified signal intensities from individual femoral heads normalized to wildtype (wt), black lines indicate means (n = 17 mice, from 6 litters). Effect size without RA treatment: 1.09 (95% HDI 0.90–1.34), effect size upon RA treatment: 0.77 (95% HDI 0.60–0.96). (E) Gelatin zymography of supernatants of femoral head cultures compared to recombinant mouse/rat MMP2 protein (rMMP2). Black arrow: active MMP2; blue arrow: proMMP2; green arrow: unspecific protease from FCS. (F) Western Blot analysis of MMP2 in the supernatant of femoral head cultures with and without RA treatment. Blue arrow: proMMP2; black arrow: active MMP2 (n ≥ 3).
MMP2 activity is controlled by extracellular proteoglycans
As aggrecan degradation is reduced in Ndst1+/− mice, we wondered whether the digestion of collagens by MMPs is also affected. Analysis of MMP activity in the supernatant of femoral head cultures by gelatin zymography38,39 revealed two prominent degradation bands [Fig. 4(C)]. The larger protease of around 65 kDa was also present in the fetal calf serum (FCS)-containing culture medium, while the smaller enzyme of around 58 kDa was specifically detected in the femoral head explants. Zymography using the supernatant of serum-free explant cultures confirmed that the larger degradation signal was caused by the FCS-containing medium, while the ~58 kDa protease was specific for the cartilage explants [Fig. 4(E)]. Without RA, no difference in protease activity could be detected under either culture condition between Ndst1+/− and control samples (combined median effect factor 1.09, 95% HDI 0.90–1.34; Fig. 4(D), Tables S3 and S4). Importantly, after RA treatment the activity of the ~58 kDa protease was reduced in Ndst1+/− samples (combined median factor: 0.77, 95% HDI 0.60–0.96; Fig. 4(D), Tables S3 and S4). Additionally, we found a second pair of degradation bands of ~100 kDa under serum-free conditions. The smaller degradation signal was hardly detectable in untreated cultures, but was clearly induced by RA treatment. Again, Ndst1+/− mutants showed a decreased activity of this protease (mean reduction factor: 0.69, 95% HDI: 0.44–1.14; Fig. S1, Tables S3 and S4).
Considering the sizes of proteases typically found in cartilage, the two enzymes likely represent activated MMP2 (58 kDa) and MMP9 (82 kDa)40,41. The presence of MMP2 in the zymogram was confirmed by comparison of the activity bands from supernatants of femur head cultures with recombinant mouse/rat MMP2 (rMMP2; Fig. 4(E)). Furthermore, we detected MMP2 in the supernatant of femoral head explants by Western Blot analysis [Fig. 4(F)].
Discussion
In this study, we investigated the role of HS during OA pathogenesis and found that reduced HS levels and sulfation attenuated OA development. We demonstrate that the altered HS composition in Ndst1+/− mutants leads to a reduced degradation of aggrecan and collagens by metalloproteases and identify MMP2 as one protease with altered activity.
Similar to our results, mice lacking the HS-carrier Syndecan 4 (Sdc4) show reduced OA progression after surgical OA induction. Accordingly, increased expression of Sdc4 has been found in OA samples of human patients and rats upon forced exercise, and administration of a Sdc4-specific antibody decreased OA progression in a surgical OA model42. Other studies found reduced OA progression in mice lacking the HS-carrying exon three of Perlecan (Hspg2Δ3−/Δ3−;43) and lower numbers of osteophytes in mice lacking Perlecan in all joint tissues except for cartilage44. These results strongly support our hypothesis that reduced HS levels inhibit the progression of OA.
While inactivation of Sdc4, Perlecan or Ext1 decreases the HS level, inactivation of Ndst1 leads to severely reduced N-, 6-O and 2-O-sulfation of the HS chain15. The lower OA scores detected in both Ndst1 mutants indicate that not only the HS level but also the sulfation pattern affect the integrity of the articular cartilage. This finding is substantiated by analyses of mice lacking the secreted endosulfatases Sulf1 or Sulf2, which remove 6-O sulfate groups from HS chains. Both mutants developed more severe OA upon ageing and after surgery45. Furthermore, injection of recombinant Sulf1 into knee joints of wildtype mice reduced surgically induced OA progression46, further supporting a protective effect of decreased HS sulfation.
Interestingly, a recent comprehensive study analyzing the expression of 13 core proteins and 25 enzymes of the HS biosynthesis pathway in healthy and osteoarthritic human cartilage revealed that 45% of the investigated genes, including EXT1 and NDST1, showed an increased expression in OA tissues47. Furthermore, the degree of 6-O sulfation was increased in the OA samples, supporting a role of the HS structure as a critical regulator of OA progression also in humans.
As outlined above, HS regulates the distribution and activity of many secreted proteins. While an effect on cytokine and growth factor signaling cannot be ruled out, our results strongly support a reduced activity of ADAMTS and MMP proteases in Ndst1+/− mutants and identify MMP2 as one enzyme with reduced activity. Previous studies revealed that the expression of Mmp13 and Adamts5 is increased in the OA cartilage of Sulf1- and Sulf2-deficient mice45. In contrast, we could not detect altered expression levels of major cartilage-degrading metalloproteases or their inhibitors in Ndst1+/− mutants, strongly pointing to a post-transcriptional regulation of protease activity.
To test if the activity of MMP2 is directly regulated by HS, we investigated its physical interaction with HS isolated from E16.5 mouse embryos by affinity chromatography but could not detect evidence for binding (data not shown). This is in contrast to a recent study based on activity assays and molecular modeling, which proposed the formation of MMP2-TIMP3-HS ternary complexes48. However, these experiments were based on the addition of HS to in vitro formed complexes and the subsequent analysis of MMP activity, while the interaction of HS and MMP2 was not directly addressed.
Our experiments show that not only the activity of MMP2 but also MMP9 and not yet identified members of the ADAMTS family are downregulated in Ndst1+/− mutants. The regulation of multiple proteases strongly points to a superordinate mechanism. This might include the HS-dependent regulation of activating proteases like MMP14, which activates MMP249, or MMP2, which in turn activates MMP950, or inhibitors of the TIMP family, which regulate different proteases51.
Besides reduced enzyme activity the degradation process is likely also affected by an altered structure or an increased thickness of the articular cartilage as seen in Col2-Cre;Ndst1fl/fl mutants. Although an increased thickness was not detected in Ndst1+/− mutants, subtle alterations might contribute to the decreased OARSI scores.
One surprising result of our experiments was that the enlarged, HS-deficient cells in the articular cartilage of Col2-rtTA-Cre;Ext1e2fl/e2fl mice did not advance OA progression. Analysis of COLX expression demonstrated that the mutant cells do not resemble hypertrophic chondrocytes of the growth plate. Morphological hypertrophy therefore does not necessarily reflect a distinct differentiation status. The Ext1-deficient cells are surrounded by a dense, Safranin-O-positive ECM, which contains increased amounts of perlecan11 and aggrecan. An increase in ECM production has also been observed after treatment of chondrocytes with heparanase or the HS antagonist Surfen52–54. Furthermore, in a parallel study we demonstrate that mice with a severely decreased HS-content (Ext1gt/gt) or an altered sulfation pattern (Hs2st1−/−) show increased levels of CS, mostly bound to aggrecan55. The upregulation of CS-carrying proteoglycans and other ECM components might thus be a common compensatory reaction of chondrocytes to an altered HS structure and might contribute to an improved quality of the articular cartilage.
While our data identify HS as a regulator of articular cartilage homeostasis, the results should be interpreted considering the following limitations: First, while the combined study of several mouse lines strongly supports a protective effect of reduced HS level or modification, the investigated mouse mutants, especially the Col2-Cre;Ndst1fl/fl mutants, show only a modest decrease in OA. Second, we investigated female mice and used the contralateral knee for the sham operation. Due to the higher susceptibility of males for OA and the potentially shift in weight bearing to the sham-operated leg this might lead to an underestimation of the effect of HS on OA severity. Third, the investigation of aggrecan degradation is based on limited sample numbers and the exact mechanism by which protease activity is reduced has still to be deciphered. Fourth, OA was investigated in genetic mouse models likely leading to a developmentally altered articular cartilage quality. The consequences of modifying HS function during OA progression have therefore to be clarified in future studies. Nevertheless, together with the recently published human data47, our results support a role of the HS structure in determining the quality of the articular cartilage at multiple levels including cartilage thickness, ECM composition and the activity of proteases, and identify HS as a promising target for future therapies.
Supplementary Material
Acknowledgement
We thank the members of the animal facility for excellent mouse husbandry.
Role of the funding source
The project was funded by the DFG grants Vo620/10 and Vo620/14 to AV and FOR2722 to BB.
Footnotes
Animal experiments
All animal experiments were carried out according to the institutional guidelines of the University of Duisburg-Essen and approved by the animal welfare officer. Mouse husbandry was approved by the city of Essen (Az: 32-2-11-80-71/348) in accordance with § 11 (1) 1a of the “Tierschutzgesetz”. Work with transgenic animals was approved by the “Bezirksregierung Düsseldorf” (Az: 53.02.01-D-1.55/12, Anlagen-Nr. 1,464) in accordance with § eight Abs. four Satz two of the “Gentechnikgesetz”.
The induction of Cre-recombination by doxycycline administration was approved by the “Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV) Nordrhein-Westfalen” (Az: 84–02.05.20.12.156), as well as the surgical induction of OA (Az: 84–02.04.2011A280).
Conflict of interest
The authors declare no conflicts of interest.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.joca.2020.04.002.
References
- 1.Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta 2012;1824:133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007;446:1030–7. [DOI] [PubMed] [Google Scholar]
- 3.Yan D, Lin X. Shaping morphogen gradients by proteoglycans. Cold Spring Harbor Perspectives in Biology 2009;1. a002493–a002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin X, Wei G, Shi Z, Dryer L, Esko JD, Wells DE, et al. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev Biol 2000;224:299–311. [DOI] [PubMed] [Google Scholar]
- 6.Jones KB, Piombo V, Searby C, Kurriger G, Yang B, Grabellus F, et al. A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A 2010;107:2054–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn J, Lüdecke H-J, Lindow S, Horton WA, Lee B, Wagner MJ, et al. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat Genet 1995;11: 137–43. [DOI] [PubMed] [Google Scholar]
- 8.Stickens D, Zak BM, Rougier N, Esko JD, Werb Z. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 2005;132:5055–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumoto Y, Matsumoto K, Irie F, Fukushi J, Stallcup WB, Yamaguchi Y. Conditional ablation of the heparan sulfate-synthesizing enzyme Ext1 leads to dysregulation of bone morphogenic protein signaling and severe skeletal defects. J Biol Chem 2010;285:19227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piombo V, Jochmann K, Hoffmann D, Wuelling M, Vortkamp A. Signaling systems affecting the severity of multiple osteochondromas. Bone 2018;111:71–81. [DOI] [PubMed] [Google Scholar]
- 11.Sgariglia F, Candela ME, Huegel J, Jacenko O, Koyama E, Yamaguchi Y, et al. Epiphyseal abnormalities, trabecular bone loss and articular chondrocyte hypertrophy develop in the long bones of postnatal Ext1-deficient mice. Bone 2013;57: 220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vignon E, Arlot M, Hartmann D, Moyen B, Ville G. Hypertrophic repair of articular cartilage in experimental osteoarthrosis. Ann Rheum Dis 1983;42:82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von der Mark K, Kirsch T, Nerlich A, Kuss A, Weseloh G, Glückert K, et al. Type X collagen synthesis in human osteoarthritic cartilage. Arthritis Rheum 1992;35:806–11. [DOI] [PubMed] [Google Scholar]
- 14.Pfander D, Swoboda B, Kirsch T. Expression of early and late differentiation markers (proliferating cell nuclear antigen, syndecan-3, annexin VI, and alkaline phosphatase) by human osteoarthritic chondrocytes. Am J Pathol 2001;159:1777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development 2005;132:3777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pallerla SR, Pan Y, Zhang X, Esko JD, Grobe K. Heparan sulfate Ndst1 gene function variably regulates multiple signaling pathways during mouse development. Dev Dynam 2007;236: 556–63. [DOI] [PubMed] [Google Scholar]
- 17.Soriano P Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 1999;21:70–1. [DOI] [PubMed] [Google Scholar]
- 18.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 2002;33:77–80. [DOI] [PubMed] [Google Scholar]
- 19.Terpstra L, Prud’homme J, Arabian A, Takeda S, Karsenty G, Dedhar S, et al. Reduced chondrocyte proliferation and chon-drodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol 2003;162:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pond MJ, Nuki G. Experimentally-induced osteoarthritis in the dog. Ann Rheum Dis 1973:387–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamekura S, Hoshi K, Shimoaka T, Chung U, Chikuda H, Yamada T, et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis Cartilage 2005;13:632–41. [DOI] [PubMed] [Google Scholar]
- 22.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010;18(Suppl 3):S17–23. [DOI] [PubMed] [Google Scholar]
- 23.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holzer T, Probst K, Etich J, Auler M, Georgieva VS, Bluhm B, et al. Respiratory chain inactivation links cartilage-mediated growth retardation to mitochondrial diseases. J Cell Biol 2019;218:1853–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanton H, Golub SB, Rogerson FM, Last K, Little CB, Fosang AJ. Investigating ADAMTS-mediated aggrecanolysis in mouse cartilage. Nat Protoc 2011;6:388. [DOI] [PubMed] [Google Scholar]
- 26.Fosang AJ, Last K, Maciewicz RA. Aggrecan is degraded by matrix metalloproteinases in human arthritis. Evidence that matrix metalloproteinase and aggrecanase activities can be independent. J Clin Invest 1996;98:2292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woessner JF Jr. Quantification of matrix metalloproteases in tissue samples. Methods Enzymol 1995;248:510–28. [DOI] [PubMed] [Google Scholar]
- 28.Gelman AC, Stern JB, Dunson HS, Vehtari DB, Rubin DBA. Bayesian Data Analysis. 3rd edn. Edition.Taylor & Francis; 2013. [Google Scholar]
- 29.Koch AL. Logarithm in biology .1. Mechanisms generating log-normal distribution exactly. J Theor Biol 1966;12:276–&. [DOI] [PubMed] [Google Scholar]
- 30.Muth C, Oravecz Z, Gabry J. User-friendly Bayesian regression modeling: a tutorial with rstanarm and shinystan. Quant Methods Psychol 2018;14:99–119. [Google Scholar]
- 31.Vehtari A, Gelman A, Gabry J. Practical Bayesian model evaluation using leave-one-out cross-validation and WAIC (vol 27, pg 1413, 2017). Stat Comput 2017;27. 1433–1433. [Google Scholar]
- 32.Martel-Pelletier J, Welsch DJ, Pelletier JP. Metalloproteases and inhibitors in arthritic diseases. Best Pract Res Clin Rheumatol 2001;15:805–29. [DOI] [PubMed] [Google Scholar]
- 33.Zeng GQ, Chen AB, Li W, Song JH, Gao CY. High MMP-1, MMP-2, and MMP-9 protein levels in osteoarthritis. Genet Mol Res 2015;14:14811–22. [DOI] [PubMed] [Google Scholar]
- 34.Little CB, Meeker CT, Fosang AJ. Degradative mechanisms in mouse articular cartilage: use of in vitro models to analyse proteolysis and loss of aggrecan and link protein. Trans Orthop Res Soc 2003;28:699. [Google Scholar]
- 35.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta 2010;1803:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singer II, Kawka DW, Bayne EK, Donatelli SA, Weidner JR, Williams HR, et al. VDIPEN, a metalloproteinase-generated neoepitope, is induced and immunolocalized in articular cartilage during inflammatory arthritis. J Clin Invest 1995;95: 2178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lark MW, Gordy JT, Weidner JR, Ayala J, Kimura JH, Williams HR, et al. Cell-mediated catabolism of aggrecan. J Biol Chem 1995;270:2550–6. [DOI] [PubMed] [Google Scholar]
- 38.Makowski GS, Ramsby ML. Calibrating gelatin zymograms with human gelatinase standards. Anal Biochem 1995;236: 353–6. [DOI] [PubMed] [Google Scholar]
- 39.Troeberg L, Nagase H. Zymography of metalloproteinases. Curr Protocols Protein Sci 2003;33. 21.15.21–21.15.12. [DOI] [PubMed] [Google Scholar]
- 40.Ogata Y, Enghild J, Nagase H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem 1992;267:3581–4. [PubMed] [Google Scholar]
- 41.Van Hul M, Lijnen HR. A functional role of gelatinase A in the development of nutritionally induced obesity in mice. J Thromb Haemostasis 2008;6:1198–206. [DOI] [PubMed] [Google Scholar]
- 42.Echtermeyer F, Bertrand J, Dreier R, Meinecke I, Neugebauer K, Fuerst M, et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med 2009;15: 1072–6. [DOI] [PubMed] [Google Scholar]
- 43.Shu CC, Jackson MT, Smith MM, Smith SM, Penm S, Lord MS, et al. Ablation of perlecan domain 1 heparan sulfate reduces progressive cartilage degradation, synovitis, and osteophyte size in a preclinical model of posttraumatic osteoarthritis. Arthritis Rheum 2016;68:868–79. [DOI] [PubMed] [Google Scholar]
- 44.Kaneko H, Ishijima M, Futami I, Tomikawa-Ichikawa N, Kosaki K, Sadatsuki R, et al. Synovial perlecan is required for osteophyte formation in knee osteoarthritis. Matrix Biol 2013;32:178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otsuki S, Hanson SR, Miyaki S, Grogan SP, Kinoshita M, Asahara H, et al. Extracellular sulfatases support cartilage homeostasis by regulating BMP and FGF signaling pathways. Proc Natl Acad Sci U S A 2010;107:10202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otsuki S, Murakami T, Okamoto Y, Hoshiyama Y, Oda S, Neo M. Suppression of cartilage degeneration by intra-articular injection of heparan sulfate 6-O endosulfatase in a mouse osteoarthritis model. Histol Histopathol 2017;32:725–33. [DOI] [PubMed] [Google Scholar]
- 47.Chanalaris A, Clarke H, Guimond SE, Vincent TL, Turnbull JE, Troeberg L. Heparan sulfate proteoglycan synthesis is dysregulated in human osteoarthritic cartilage. Am J Pathol 2019;189:632–47. [DOI] [PubMed] [Google Scholar]
- 48.Ruiz-Gomez G, Vogel S, Moller S, Pisabarro MT, Hempel U. Glycosaminoglycans influence enzyme activity of MMP2 and MMP2/TIMP3 complex formation - insights at cellular and molecular level. Sci Rep 2019;9:4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994;370:61–5. [DOI] [PubMed] [Google Scholar]
- 50.Fridman R, Toth M, Pena D, Mobashery S. Activation of progelatinase B (MMP-9) by gelatinase A (MMP-2). Canc Res 1995;55:2548–55. [PubMed] [Google Scholar]
- 51.Yamamoto K, Murphy G, Troeberg L. Extracellular regulation of metalloproteinases. Matrix Biol 2015;44–46:255–63. [DOI] [PubMed] [Google Scholar]
- 52.Schuksz M, Fuster MM, Brown JR, Crawford BE, Ditto DP, Lawrence R, et al. Surfen, a small molecule antagonist of heparan sulfate. Proc Natl Acad Sci U S A 2008;105:13075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huegel J, Mundy C, Sgariglia F, Nygren P, Billings PC, Yamaguchi Y, et al. Perichondrium phenotype and border function are regulated by Ext1 and heparan sulfate in developing long bones: a mechanism likely deranged in Hereditary Multiple Exostoses. Dev Biol 2013;377:100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huegel J, Enomoto-Iwamoto M, Sgariglia F, Koyama E, Pacifici M. Heparanase stimulates chondrogenesis and is up-regulated in human ectopic cartilage: a mechanism possibly involved in hereditary multiple exostoses. Am J Pathol 2015;185:1676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bachvarova V, Dierker T, Esko J, Hoffmann D, Kjellen L, Vortkamp A. Chondrocytes respond to an altered heparan sulfate composition with distinct changes of heparan sulfate structure and increased levels of chondroitin sulfate. Matrix Biol 2020, 10.1016/j.matbio.2020.03.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.