

Abstract
INTRODUCTION:
In the arms race between pathogen and host, infecting microbes often escape extracellular defense mechanisms to exploit the nutrient-rich intracellular environment as a replicative niche. In humans, this is countered by the interferon-γ (IFN-γ) response, which confers widespread pathogen resistance in most nucleated cells through the transcriptional induction of hundreds of interferon-stimulated genes (ISGs) encoding putative antimicrobial restriction factors. Remarkably, despite the importance of IFN-γ against all taxonomic classes of intracellular pathogens, many restriction factors elicited by this cytokine remain to be characterized, as do their molecular activities.
RATIONALE:
Identified as the major human macrophage-activating cytokine in 1983, IFN-γ in fact transcriptionally reprograms numerous host cell types to eliminate infection. This includes nonimmune epithelial cell populations, which lack many traditional phagocytic defenses ascribed to IFN-γ stimulation, yet still manage to mount protective cell-autonomous immune responses. To find ISG effectors involved in safeguarding mucosal and barrier tissue types, we conducted a genome-wide CRISPR-Cas9 screen in IFN-γ–activated human epithelial cells for their ability to restrict virulent intracellular pathogens such as Salmonella enterica serovar Typhimurium.
RESULTS:
We identify the ISG apolipoprotein L3 (APOL3) as a potent effector protein capable of killing cytosol-invasive bacteria. The human APOL family is a cluster of six genes that have evolved rapidly under positive selection in simian primates; however, aside from the founding member APOL1, a secreted extracellular protein that forms the trypanolytic factor of human serum, the function of the intracellular APOL family members is unknown. Human cells genetically engineered to lack APOL3 failed to control the replication of multiple cytosol-invasive Gram-negative bacteria after IFN-γ activation. Such findings were validated in primary human intestinal epithelial cells, intestinal myofibroblasts, and venular endothelium—all cellular targets not typically considered part of the immune system. We tracked APOL3 by live microscopy and found that it rapidly relocated to cytosol-exposed bacteria, whereas other APOL family members did not. A combination of superresolution imaging, bioengineered reporters, and cell-free reconstitution revealed that when APOL3 targets pathogens inside IFN-γ–activated cells, it inflicts a lethal insult to the bacterial inner membrane (IM). Here APOL3 synergizes with other ISG-encoded proteins, including guanylate-binding protein 1 (GBP1), that perturb the bacterial O-antigen outer membrane (OM) permeability barrier to allow APOL3 access to the IM underneath. Using a panel of compositionally distinct liposome targets, we found that APOL3 membranolytic activity toward microbial rather than host endomembranes stemmed from an ability to dissolve bacterial polyanionic lipid substrates lacking cholesterol into discoidal lipoprotein complexes; single-particle cryo–electron microscopy found that these complexes resembled apolipoprotein-scaffold “nanodiscs.” Corroborating these findings in live bacteria by native mass spectrometry, we found that APOL3 transitioned from a partially disordered lipid-free state to tightly folded lipoprotein nanodiscs upon extracting lipid from the IM—a process that resulted in rapid death of the bacterium.
CONCLUSION:
Detergents are highly effective antimicrobials used to decontaminate surfaces infected by deadly pathogens. Our results identify APOL3 as an IFN-γ–stimulated host defense protein that has evolved potent detergent-like activity to bestow bactericidal protection in the cytosol of human cells. APOL3 synergizes with other host ISGs in a multipronged attack against the double membrane of Gram-negative bacteria—a formidable barrier that imparts resistance to many classes of antibiotics. This study reveals that antibacterial agents that dismantle this barrier during infection naturally exist inside human cells. That these agents are encoded within the IFN-γ–inducible defense program reinforces the importance of this powerful antimicrobial network for cell-autonomous immunity in humans.
Graphical Abstract

APOL3 kills intracellular bacteria. (A) Negative-stain electron microscopy of recombinant APOL3 (bead) added to Salmonella Typhimurium (periplasm pseudocolored yellow). Destruction of bacterial membrane (blue-bordered inset) is triggered by APOL3 extracting lipid to form lipoproteins (burgundy-bordered inset). (B) Bacterial mutants (ΔwaaL) expressing a truncated O-antigen permit passage of APOL3 through the outer membrane (OM) to the inner membrane (IM); this passage inside cells is facilitated by synergizing ISG-encoded proteins such as GBP1 that co-target cytosol-exposed bacteria.
Activation of cell-autonomous defense by the immune cytokine interferon-γ (IFN-γ) is critical to the control of life-threatening infections in humans. IFN-γ induces the expression of hundreds of host proteins in all nucleated cells and tissues, yet many of these proteins remain uncharacterized. We screened 19,050 human genes by CRISPR-Cas9 mutagenesis and identified IFN-γ–induced apolipoprotein L3 (APOL3) as a potent bactericidal agent protecting multiple non–immune barrier cell types against infection. Canonical apolipoproteins typically solubilize mammalian lipids for extracellular transport; APOL3 instead targeted cytosol-invasive bacteria to dissolve their anionic membranes into human-bacterial lipoprotein nanodiscs detected by native mass spectrometry and visualized by single-particle cryo–electron microscopy. Thus, humans have harnessed the detergent-like properties of extracellular apolipoproteins to fashion an intracellular lysin, thereby endowing resident nonimmune cells with a mechanism to achieve sterilizing immunity.
Cell-autonomous immunity operates across all three domains of life to defend against infection (1). This ancient form of host defense protects against intracellular pathogens through direct and indirect effector mechanisms (1). In vertebrates, these effector mechanisms can be mobilized by the type II cytokine interferon-γ (IFN-γ), which regulates the transcription of hundreds of IFN-stimulated genes (ISGs) to help combat bacteria, viruses, parasites, and fungi in a wide variety of host cell types (2). Human population genetics and animal models have firmly established the importance of IFN-γ signaling in organismal defense (3, 4), yet few ISGs with direct pathogen-neutralizing activity have been characterized. This is especially true within human mucosal or stromal cell lineages that are historically viewed as separate from the classical immune system. These cell lineages, known more for their role in shaping organ architecture and creating tissue boundaries, nonetheless mount protective responses when appropriately instructed by activating signals such as IFN-γ (5, 6). The mechanisms and protein machineries involved in this nonclassical or “structural” arm of immunity remain poorly understood (6).
Discovery of human APOL3 as an antibacterial ISG
We searched for new antimicrobial ISGs in human epithelial (HeLa CCL2) cells, using the virulent Gram-negative bacteria Salmonella enterica serovar Typhimurium (Stm) as an initial infection model. Here, bulk replication arises from a subpopulation (~10%) of infected cells in which Stm escape their entry vacuole to rapidly proliferate in the cytosol and serve as a reservoir for dissemination (7). Using fluorescence-activated cell sorting (FACS) analysis, we found that IFN-γ specifically controlled this subpopulation, completely preventing the appearance of cells laden with cytosolic hyper-replicating bacteria (HR epithelial cells) without affecting slowly replicating Stm within vacuolar (Lamp1+) compartments (SR epithelial cells) (Fig. 1A). To identify the protective host factors, we performed a genome-wide CRISPR-Cas9 screen and retrieved single guide RNAs (sgRNAs) selectively enriched in IFN-γ–activated HR cells failing to restrict Stm (Fig. 1B). ISGs were simultaneously defined by RNA sequencing (RNA-seq) profiles from IFN-γ–activated versus unactivated Stm-infected HeLa cells. Stringent significance thresholds (P < 0.001; mRNA > 4-fold induced) identified two major hits exclusive to IFN-γ–activated cells (Fig. 1B and table S1): the master IFN transcription factor STAT1 and the primate-specific apolipoprotein L family member APOL3, a gene whose product we found to be robustly and specifically induced by IFN-γ (fig. S1A) but has not previously been linked to bacterial infection.
Fig. 1. Genome-wide identification of human APOL3 as an antibacterial ISG.

(A) FACS of HeLa cells infected with GFP-expressing Salmonella enterica Typhimurium (StmGFP). HR and SR gates are percentages of infected cells. Below, 3D confocal microscopy and calculated bacterial load per cell from each population (mean ± SEM). (B) Genome-wide CRISPR-Cas9 screen schema and gene-level enrichment scores in HR versus SR populations in the presence or absence of IFN-γ. Each gene is plotted against relative (fold) induction of its mRNA in IFN-γ–activated cells determined by RNA-seq. (C) StmGFP growth by FACS at 6 hours (left) and gentamicin protection assays (right) in APOL3-deficient HeLa cells (ΔAPOL3) genetically complemented with APOL3 (bottom row) or empty retroviral control (top two rows). Fold is given as relative to 1-hour starting time point (input). (D) Increase in bacterial load [relative to wild-type (WT) cells] recovered from APOL3- or STAT1-deficient IFN-γ–activated HeLa cells after the indicated time. **P < 0.01, ***P < 0.001 (one-way ANOVA); ns, not significant. (E) Human primary intestinal myofibroblasts treated with APOL3 siRNA or nontargeting scrambled control (siCtrl) (immunoblot, bottom right) and infected with StmmScarlet. Shown are representative final micrographs (10 hours) and quantification of HR Stm (foci 10 to 35 μm) every hour (mean ± SD, n = 3, representative of two independent experiments. In (C) and (D), data are means ± SEM from four independent experiments and FACS plots representative of at least four independent experiments. ϕ denotes a nonspecific band. Scale bar, 75 μm.
Validating our results, two independent CRISPR deletions of APOL3 (ΔAPOL3) rendered IFN-γ–primed cells unable to fully restrict Stm hyperreplication in the cytosol, a deficiency restored by APOL3 cDNA complementation (Fig. 1C and fig. S1B). These defects were not due to impaired bacterial uptake and were only evident after cytokine priming (fig. S1C). Forced expression of APOL3 in unprimed cells did not have a significant effect on Stm replication (fig. S1, D to F), indicating that APOL3 is necessary but not itself sufficient for bacterial control. APOL3 was required for restriction of other cytosol-invasive bacteria [Shigella flexneri, Burkholderia thailandensis, or a hyper–cytosol-invasive Stm mutant (StmΔsifA) (8)] but not vacuole-residing bacteria such as Salmonella Typhi (9) or an injectisome-deficient Stm mutant (StmΔinvA::pR1203) that is unable to initiate vacuolar escape (Fig. 1D). That S. flexneri was less susceptible to APOL3-mediated restriction may hint at the presence of resistance mechanisms for this professional cytosol-dwelling human pathogen. APOL3 likewise operated in primary human intestinal epithelium, intestinal myofibroblasts, and vascular endothelium, where small interfering RNA (siRNA) silencing of IFN-γ–stimulated APOL3 expression led to a significant loss of Stm control (Fig. 1E and fig. S2, A to C). Thus, APOL3 is an IFN-γ–inducible restriction factor that controls cytosol-invasive pathogens in human tissue cells originating outside of the hematopoietic compartment.
APOL3 targets cytosol-invasive bacteria
The human APOL family is a cluster of six genes that have evolved rapidly under positive selection in primates (10). Aside from APOL1, a secreted protein associating with high-density lipoprotein (HDL) to form the trypanolytic factor of human serum (11, 12), protective functions for the remaining five family members that are intracellular and lack a secretion signal are unknown. We examined the entire family and found that most APOL genes were highly induced by IFN-γ in a STAT1-dependent manner (fig. S3A), yet only cells chromosomally deficient in APOL3 failed to restrict Stm (fig. S3B). Using live microscopic imaging to investigate its subcellular location, we found that ectopically expressed APOL3 fused to monomeric NeonGreen fluorescent protein (APOL3mnGFP), but not mnGFP fused to the other family members, rapidly relocated to Stm and proceeded to “coat” bacteria over a ~20- to 45-min period (Fig. 2A, fig. S3C, and movie S1). Such APOL3 coating occurred in unprimed cells, indicating that cytokine activation was needed for APOL3 expression but not its relocation. Even so, once APOL3 relocated to bacteria within IFN-γ–activated cells, it conferred antibacterial activity; identification and mutation of a hydrophobic patch (LAP137QSS) (fig. S3D) that prevented bacterial targeting also abolished restriction (Fig. 2B). Targeting was specific to cytosol-exposed bacterium, because vacuole-confined StmΔinvA::pR1203 failed to recruit APOL3 unless released into the cytosol with the lysosomotropic agent l-leucyl-l-leucine methyl ester (LLOMe), which restored both bacterial targeting and subsequent restriction (Fig. 2C). Moreover, APOL3 targeted cytosolic S. flexneri, B. thailandensis, and Listeria monocytogenes but not compartmentalized S. Typhi (fig. S4A), which is generally in keeping with the susceptibility profiles of these pathogens to APOL3 restriction. Notably, sterile damage triggered by LLOMe was sufficient to mobilize APOL3 to the lumen of ruptured LAMP1+ endolysosomes independent of bacteria (fig. S4B and movie S2). It is therefore likely that initial APOL3 targeting signals are damage-associated molecular patterns (DAMPs) induced by pathogenic bacteria when they rupture their LAMP1+ vacuole to enter the cytosol, akin to the defense protein Galectin-8 (13).
Fig. 2. Human APOL3 targets and inflicts damage to cytosolic bacteria.

(A) APOL3mnGFP targeting StmRFP by live imaging in HeLa cells (movie S1). Percentage of total Stm targeted by HA-tagged APOL family members (2 hours) is shown at right. (B) StmRFP targeting and replication in IFN-γ–primed ΔAPOL3 cells complemented with the indicated APOL3 variant. (C) Deconvolved wide-field images of APOL3mnGFP targeting vacuole-confined StmRFP (StmΔinvA::pR1203) with or without vacuole release with LLOMe; fold replication is shown at right. (D) Inner membrane (IM) integrity as measured by minDmnGFP aggregation within Stm in HeLa cells expressing APOL3RFP at 2 hours with or without IFN-γ. Quantification reflects aggregation in APOL3-coated versus uncoated bacteria or total bacteria in WT versus ΔAPOL3 cells via Fisher’s exact test. (E) Arabinose-induced GFP in Stm targeted by APOL3FLAG in HeLa cells with or without IFN-γ. Maximal-intensity GFP/mCherry ratios are shown (mean ± SD, n = 50). (F) Immunofluorescence and SIM of APOL3HA and LPS on Stm with or without IFN-γ at selected times. Mid-2D z-planes are shown. Quantification of LPS penetrance (25 bacilli, mean ± SEM, n = 3) and 3D surface rendering are shown below. Blue arrows indicate cryo-immunogold EM staining of APOL3GFP in Stm-infected HeLa cells; OM, outer membrane. Micrographs are representative of at least three independent experiments. Data are means ± SEM [(A), (B), (C)] with significance by one-way ANOVA at 6 hours. ***P < 0.001. Scale bars, 5 μm [(A), (B), (C), and (E)], 2 μm (D), 1 μm (F).
To assess the fate of APOL3-coated bacteria, we engineered reporter strains to diagnose Stm fitness inside human cells. Stm inner membrane (IM) integrity was tracked via minD, a bacterial cell division protein that loses its lateral membrane oscillatory behavior when IM potential is perturbed (14). Loss of both IM localization and oscillation (demarcated by minD aggregates) was significantly elevated in APOL3-coated versus uncoated Stm and reversed in ΔAPOL3 cells treated with IFN-γ (Fig. 2D and movies S3 and S4). Stm lacking the Cpx-driven IM repair pathway (StmΔcpxR:FRT) had exacerbated damage and was more susceptible to APOL3-driven restriction (fig. S5, A to C), underscoring the importance of bacterial membrane repair for resisting APOL3-dependent immunity. In addition, a dual-reporter Stm strain responsive to de novo arabinose-induced GFP expression became completely unresponsive to this stimulus when targeted by APOL3 (Fig. 2E), indicating that these bacteria were compromised in their ability to respond transcriptionally to external cues. These fitness defects were accompanied by a marked transformation of the APOL3 bacterial coat: Superresolution structured illumination microscopy (SIM) revealed that APOL3 breached the lipopolysaccharide (LPS) outer membrane (OM) to penetrate the bacterial cytoplasm in IFN-γ–activated cells by 2.5 hours of infection (Fig. 2F and movie S5). Cryo–immunoelectron microscopy confirmed this penetrance (Fig. 2F), with clearance of APOL3-targeted bacteria within IFN-γ cells occurring shortly thereafter (fig. S5, C and D, and movie S6).
Human APOL3 exhibits bactericidal activity
The above results suggested that APOL3 could be directly exerting antibacterial effects. Critically, these effects were observed exclusively on bacteria within IFN-γ–activated human cells, where, in addition to being susceptible to OM penetration by APOL3, bacteria displayed irregular staining for the LPS O-antigen (fig. S5E), an essential component of the OM permeability barrier. Other ISGs could therefore facilitate direct APOL3 bactericidal activity by increasing OM permeability, which would normally exclude such a large hydrophobic agent. We generated recombinant rAPOL3 (15) to test this possibility directly (fig. S6A). Cytosol-enriched Stm extracted from ΔAPOL3 IFN-γ–activated cells were highly susceptible to direct rAPOL3 killing, whereas Stm from vacuoles, from unprimed cells, or grown in broth were resistant despite equivalent APOL3 binding (Fig. 3A and fig. S6, B and C). Similar results were obtained with Stm extracted from APOL3-silenced, IFN-γ–activated primary human intestinal myofibroblasts (fig. S6D). Live imaging revealed that Stm released from IFN-γ–activated cells had sustained transient perturbations to their cell wall that enabled rAPOL3 to gain initial access to the OM (loss of periplasm ssTorA-GFP) and to permeabilize the IM (uptake of zombie-UV) (Fig. 3B). These events were rapid, beginning 2 to 3 min after rAPOL3 exposure (fig. S6E and movies S7 to S9) and explain why IFN-γ priming is required for APOL3 antibacterial activity inside human cells.
Fig. 3. Human APOL3 has direct bactericidal activity.

(A), Viability of Stm extracted from ΔAPOL3 cells with or without IFN-γ and exposed to rAPOL3 (3 hours). (B) Live imaging of cytosol-extracted Stm:ssTorA-GFP treated with 568-labeled rAPOL3 (5 μM) and membrane-impermeable Zombie-UV dye with quantitation (n = 100). (C) Bacterial viability after pulsing with the indicated agent followed by rAPOL3 exposure for 3 hours. (D) Median lethal dose (LD50) at 3 hours after in vivo or ex vivo sensitization of Stm. (E) rAPOL3 domain analysis (10 μM) at 3 hours after incubation with EDTA-pulsed Stm; Hydrophobicity and amphipathicity (μH) plot above. AH, amphipathic helix; TM, transmembrane domain. (F) Sensitivity of Stm and E. coli LPS truncation mutants to rAPOL3 (10 μM) in potassium gluconate (KGl). (G) EM micrographs of E. coliΔhldE exposed to His6-rAPOL3 for 5 min and detected with 5-nm Ni2+-gold beads. Data are means ± SEM from three to five independent experiments [(A), (B), (C), (E), and (F)] or are representative of three independent experiments [(D) and (G)]. *** P < 0.001. Scale bar in (B), 2 μm.
Stm sensitization in situ could be pheno-copied in cell-free settings. Preexposure of Stm to sublethal concentrations of five different OM-destabilizing agents facilitated direct rAPOL3-induced killing in each case (Fig. 3C). S. flexneri, B. thailandensis, Escherichia coli, and L. monocytogenes were likewise sensitized to rAPOL3 by small amounts of EDTA or lysozyme (Fig. 3C). Relative to the bona fide antimicrobial peptides human β-defensin–2 (hBD-2) and mouse RegIIIβ (16, 17), rAPOL3 was more active on an equimolar basis by a factor of 5 to 16, confirming its status as a powerful antibacterial lysin (Fig. 3D). Putative transmembrane domains and amphipathic helical (AH) repeats identified in silico were required for APOL3 bactericidal activity (Fig. 3E and fig. S7). In some cases, N-terminal (rAPOL379–176) and C-terminal (rAPOL3179–333) fragments harboring these motifs were more toxic than the full-length protein, possibly because they are small enough to penetrate bacterial cell walls without prior damage, as seen for L. monocytogenes (which lacks an OM) and DH5α E. coli (possessing only a short O-antigen) (fig. S8, A and B). However, these smaller fragments were missing motifs for intracellular trafficking and they could not fully target bacterial pathogens inside human cells, resulting in loss of killing activity. Only full-length APOL3 could restore such activity in situ (fig. S8, C to F).
A panel of Stm and E. coli mutants with progressive O-antigen truncations mimicking OM damage underscored its potency. Here, all strains lacking a complete polymerized O-antigen were directly killed by rAPOL3 in cytosolic salt concentrations (Fig. 3F). This suggests that potentiating agents inside host cells need only to perturb the outer O-antigen barrier to facilitate APOL3 killing because lipid A and core sugars are vulnerable to its attack. Notably, the trypanolytic human APOL1 ion-channel protein (12) failed to kill truncated Stm mutants under these conditions (fig. S9A), revealing mechanistic differences with APOL3. Indeed, immuno–electron microscopy revealed that rAPOL3 localized to large pores spanning both the OM and IM before complete cell wall disintegration and blebbing ensued at higher dosage (Fig. 3G and fig. S9B). Biophysical measurements supported EM analysis: Bacterial membrane depolarization coupled with loss of fluidity, IM integrity, and cellular ATP in addition to cytosolic leakage all preceded bacteriolysis and were dependent on OM permeabilization (fig. S9, C to H). Thus, rAPOL3 exhibits potent membranolytic activity upon weakening of the OM permeability barrier, as seen within IFN-γ–activated human cells.
APOL3 bactericidal activity is facilitated by GBP1
We next considered the identity of host ISGs that weaken the OM permeability barrier of cytosolic bacteria for APOL3 killing. Galectin-8, p62/SQSTM1, and guanylate-binding protein 1 (GBP1) are defense proteins that target cytosol-invasive bacteria in human cells. Galectin-8 and p62/SQSTM1 restrict bacteria through xenophagy (13, 18), whereas GBP1 belongs to a family of IFN-γ–inducible guanosine triphosphatases (GTPases) that establish signaling platforms for cell-autonomous immunity and inflammasome activation (19–21). In human epithelium, GBP1 assists the LPS-responsive caspase-4 inflammasome to initiate pyroptotic cell death (22, 23). APOL3+ bacteria harbored all of these cytosolic defense markers and their proximal adaptors, yet they localized to different microdomains on bacilli and targeting was mutually independent, as shown by genetic ablation of 16 different signaling nodes or components, suggestive of parallel defense pathways (fig. S10, A to C).
Notably, co-targeting was highest for GBP1, which, in addition to recruiting caspase-4, has also been reported to disrupt the O-antigen barrier upon bacterial coating (24). We therefore considered that GBP1 may aid APOL3 killing by facilitating its penetration through the OM. In support of this model, genetic removal of GBP1 in ΔAPOL3 cells (ΔAPOL3/GBP1 double knockouts) rendered cytosol-extracted Stm less vulnerable to killing by exogenous rAPOL3 (Fig. 4A). In our in vitro system, treatment of wild-type Stm with recombinant GBP1 (rGBP1) purified from human cells, which coated bacteria in a GTP-dependent manner (Fig. 4B), was sufficient to sensitize wild-type Stm to killing by rAPOL3 (Fig. 4C). This synergy stemmed from the ability of rGBP1 to increase bacterial OM permeability [measured by uptake of the fluorescent dye NPN (N-phenyl-1-naphthylamine)] and facilitate APOL3 disruption of the IM (Sytox uptake), resulting in a loss of cellular ATP (Fig. 4D). Cellular reconstitution corroborated these findings. Forced expression of both APOL3 and GBP1 transgenes in tandem (fig. S10D) was sufficient to confer antibacterial protection even in unprimed HeLa cells, partly mimicking the actions of IFN-γ (Fig. 4E). This agrees with SIM imaging of the bacterial surface inside IFN-γ–activated cells where penetrating APOL3 foci were localized at regions of low LPS O-antigen intensity; such regions were reduced in HeLa cells doubly deleted for GBP1 and GBP2 (Fig. 4F). Thus, human GBP1 is one host factor that can sensitize bacteria to APOL3 killing, although other factors likely exist.
Fig. 4. Human GBP1 potentiates APOL3 bactericidal activity.

(A) Viability of cytosolic Stm extracted from the indicated HeLa cell genotype (+IFN-γ) and exposed to rAPOL3. (B to D) Stm treated with 5 μM recombinant human GBP1RFP (rGBP1) (1 hour) with or without GTP and imaged by confocal microscopy (B), washed, treated with 5 μM rAPOL3 for 1 hour, and then analyzed by colony counting (C) or ATP (bubble size) in conjunction with both OM and IM permeability by NPN or Sytox uptake, respectively (D). Bubbles represent five independent experiments in technical duplicate. (E) Fold replication of Stm in unprimed HeLa cells expressing doxycycline-inducible (TRE, Tet response element) APOL3, GBP1, or both in tandem separated by the self-cleavable P2A peptide. (F) IFN-γ–activated HeLa cells expressing APOL3FLAG infected with Stm for 2 hours were analyzed by SIM (mid-2D single-plane imaging) after immunostaining for FLAG, GBP1, and Stm LPS (O-antigen). Arrows indicate penetrating APOL3 foci and quantification (n = 50). (G and H) IFN-γ–activated HeLa cells infected with StmGFP were analyzed by live microscopy for HR Stm (foci 10 to 35 μm) and cell death (Sytox+) (G) or whole-cell lysates probed by immunoblot after 3 hours (H). Representative images and immunoblots from one of three independent experiments and quantification of total events per well (% total cells) are shown. Data are means ± SEM from three to five independent experiments. Micrographs in [(B) and (F)] are representative of three independent experiments. Statistics indicate significance by one-way ANOVA [(C) and (G)], two-way ANOVA (E), unpaired t test (F), or nonlinear regression (A). *P < 0.05, ***P < 0.001. Scale bars, 10 μm (B), 1 μm (F), 80 μm (G).
We next examined the importance of this co-operative behavior for host defense in ΔAPOL3/GBP1 cells. Synergistic defects in IFN-γ–dependent bacterial restriction were observed in the double knockout (Fig. 4G). This was independent of the noncanonical inflammasome because individual deletion of APOL3, unlike GBP1, did not reduce Stm-triggered cell death, caspase-4 cleavage, or downstream interleukin (IL)–18 processing in IFN-γ–activated cells (Fig. 4, G and H). Thus, both genes operate in distinct host defense pathways that intersect on individual bacteria; in the process of activating the noncanonical inflammasome, GBP1 contributes to the OM damage that renders Stm vulnerable to direct killing by APOL3.
APOL3 dissolves bacterial membranes into nanodisc-like lipoproteins
How does APOL3 discriminate and permeabilize bacterial membranes? We prepared liposomes mimicking bacterial [80:20 phosphatidylethanolamine (PE):phosphatidylglycerol (PG)] or mammalian [60:10:30 phosphatidylcholine (PC):phosphatidylserine (PS):cholesterol] membrane composition and found the former to be >10 times as sensitive to rAPOL3 permeabilization (Fig. 5A). A panel of compositionally distinct liposome targets revealed that this selectivity arose from a preference for acidic phospholipids naturally rich in bacterial membranes that promoted cationic APOL3 binding and permeabilization, coupled with an aversion to the eukaryote-restricted lipid cholesterol, which inhibited lysis (fig. S11, A to C). It also fitted the pH and salt dependency of bacterial killing (fig. S11D). Liposome permeation, like permeabilization of bacteria, required APOL3 TM regions and amphipathic helices and liberated luminal reporter molecules as a function of protein concentration irrespective of their size (0.158 to 40 kDa) or net charge (0 to 3+) (fig. S11, E to G). Thus, rAPOL3 does not impose defined gating properties on the type of molecule released.
Fig. 5. APOL3 dissolves anionic membranes into lipoprotein nanodiscs.
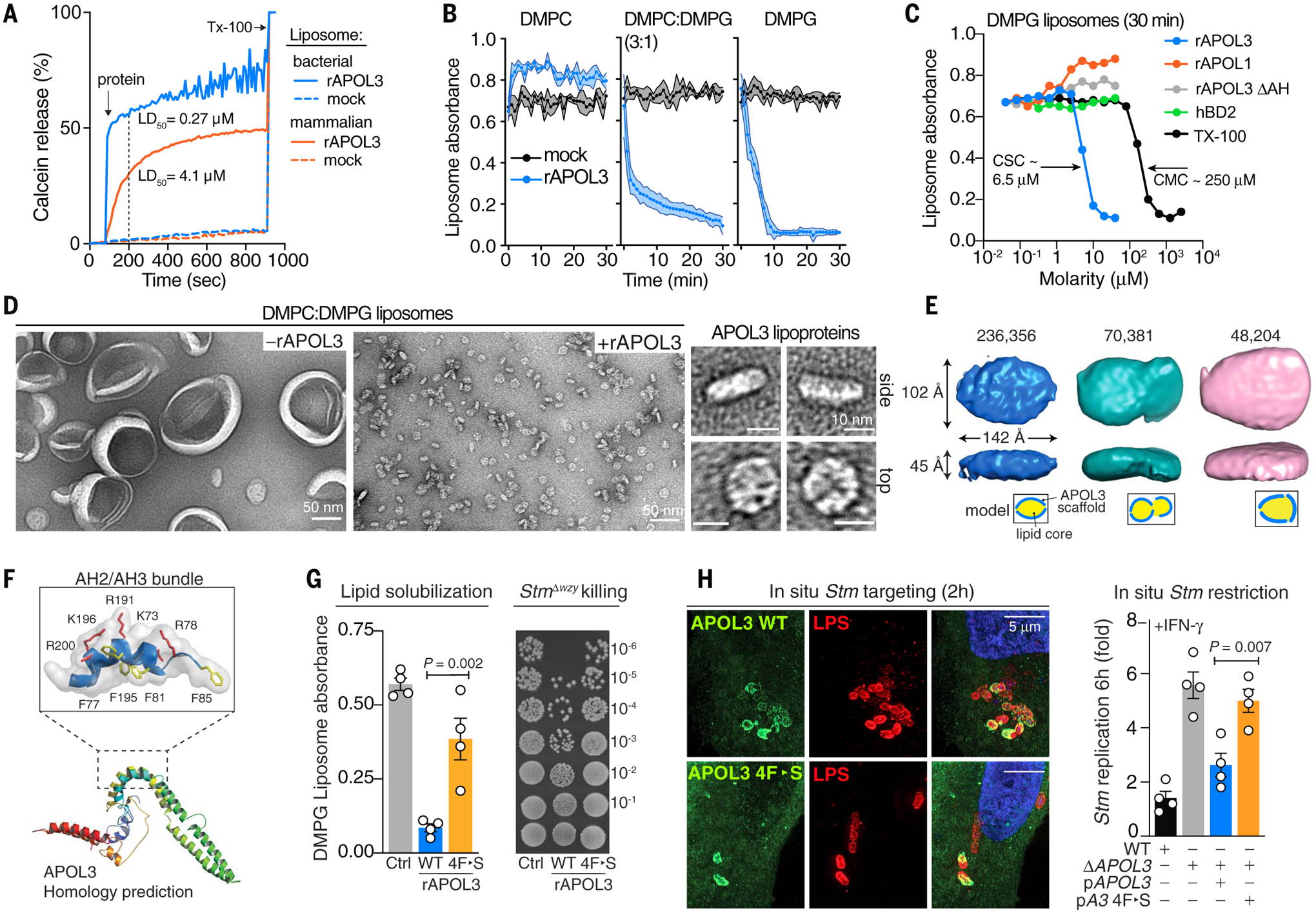
(A) Calcein leakage from “bacterial” (80:20 DOPE:DOPG) or “mammalian” (60:10:30 DOPC/DOPS/cholesterol) liposomes (500 μM lipid) exposed to rAPOL3 (500 nM). Vertical dashed line indicates dosage yielding 50% dye release (LD50) in 200 s. (B and C) Turbidity of liposomes treated with rAPOL3 or indicated reagent over time (B) or after 30 min (C). (D) Negative-stain EM of liposomes before and after addition of rAPOL3 for 30 min as in (B). (E) Single-particle cryo-EM reconstruction of APOL3 lipoprotein nanodiscs. Isosurface representation of top three particle classes (number of particles) is shown, with space-constrained model below. Thickness is equivalent to a single DMPC or DMPG bilayer (45 Å). (F) Phyre2 structural homology prediction. Inset indicates arrangement of amphipathic helices (AH) 2 and 3, with four Phe (F) residues on the interior hydrophobic face highlighted in yellow and exterior-facing acidic residues Arg (R) and Lys (K) highlighted in red. (G) Liposome turbidity and viability of StmΔwzy treated with wild-type or mutant rAPOL3. The four Phe residues depicted in (F) were mutated to Ser (S). (H) Complementation of ΔAPOL3 HeLa cells with the indicated APOL3HA genotype evaluated for Stm targeting (left) and IFN-γ–dependent restriction (right). Data are means ± SEM from three or four independent experiments [(B), (G), and (H)] or are representative of three or more independent experiments [(A), (C), and (D)]. Statistics indicate significance by one-way ANOVA.
Instead, we found that APOL3 dissolved anionic liposomes into discoidal lipoprotein complexes that we tracked by real-time optical absorbance (Fig. 5, B and C) and visualized directly by negative-stain EM (Fig. 5D). Subsequent single-particle cryo–electron microscopy (cryo-EM) captured these discoidal complexes in their native state; three modular classes arose from 431,789 particles sampled, but all were ~45 Å in height, indicating a single lipid bilayer bounded by different arrangements of APOL3 (Fig. 5E and fig. S12). This bilayer configuration is reminiscent of apolipoprotein-scaffold nanodiscs and nascent HDL particles (25). Indeed, liposome dissolution by rAPOL3 was accelerated by lipid packing defects induced by temperature shift (fig. S11H) in a manner that resembles other bona fide apolipoproteins such as APOA-1 (26).
The critical solubilizing concentration (CSC) for rAPOL3 was lower than that of a conventional detergent (such as Triton X-100) by a factor of ~40; this represents potent detergent activity and is mechanistically distinct from the antimicrobial activities of hBD-2 and APOL1, which did not trigger liposome clarification (Fig. 5C). We used sub-CSC rAPOL3 concentrations together with unsaturated lipid to slow the detergent-like activities of rAPOL3 sufficiently to capture lipid extraction and blebbing by live confocal imaging of giant unilamellar vesicles (GUVs) and negative-stain EM of liposomes (fig. S13, A and B, and movies S10 and S11). These effects were completely lost with rAPOL3ΔAH that could not undergo lipid-triggered α-helical conversion for insertional disruption of the bilayer, as measured by circular dichroism (fig. S13, C and D). This solubilizing activity was required for bacterial restriction inside human cells, as shown by mutating four Phe residues to Ser (4F-S) on the hydrophobic face of AH-2 and -3 that structural homology modeling predicted were critical to positive curvature induction and membranolytic activity (Fig. 5F and fig. S14, A and B). The APOL3 4F-S mutant exhibited decreased liposome solvation and bacterial killing in vitro and could not restore IFN-γ–induced immunity when reintroduced into ΔAPOL3 human cells, despite continuing to traffic to bacteria (Fig. 5, G and H).
Finally, we used high-energy native mass spectrometry (nativeMS) (27, 28) to study APOL3 structural dynamics during membrane solubilization. In aqueous solution, rAPOL3 existed as a partly disordered “open” monomeric species with exposed surface area accumulating many positive charges (+16 to +10) during ionization. Upon engaging liposomes, rAPOL3 underwent a marked shift, adopting a tightly folded “closed” conformer of lower charge state (+7 to +5) associated with multiple lipid adducts, confirming lipoprotein particle assembly (Fig. 6A). This same conformational change occurred with live bacteria. Here, open rAPOL3 monomers converted to closed monomers and dimers (44% of soluble rAPOL3) upon killing bacteria and were only evident at a sufficiently high mass spectral collisional activation (HCD) energy to trigger collapse of the nanodisc and release the rAPOL3 scaffold (Fig. 6B) (29). EM analysis of recovered rAPOL3 from these same fractions confirmed the transition from lipid-free to discoidal lipoprotein form (Fig. 6C). Thus, APOL3 undergoes marked structural changes to extract lipids to form discoidal bacterial-human hybrid lipoprotein complexes during killing.
Fig. 6. APOL3 extracts bacterial lipid to form lipoproteins during killing.
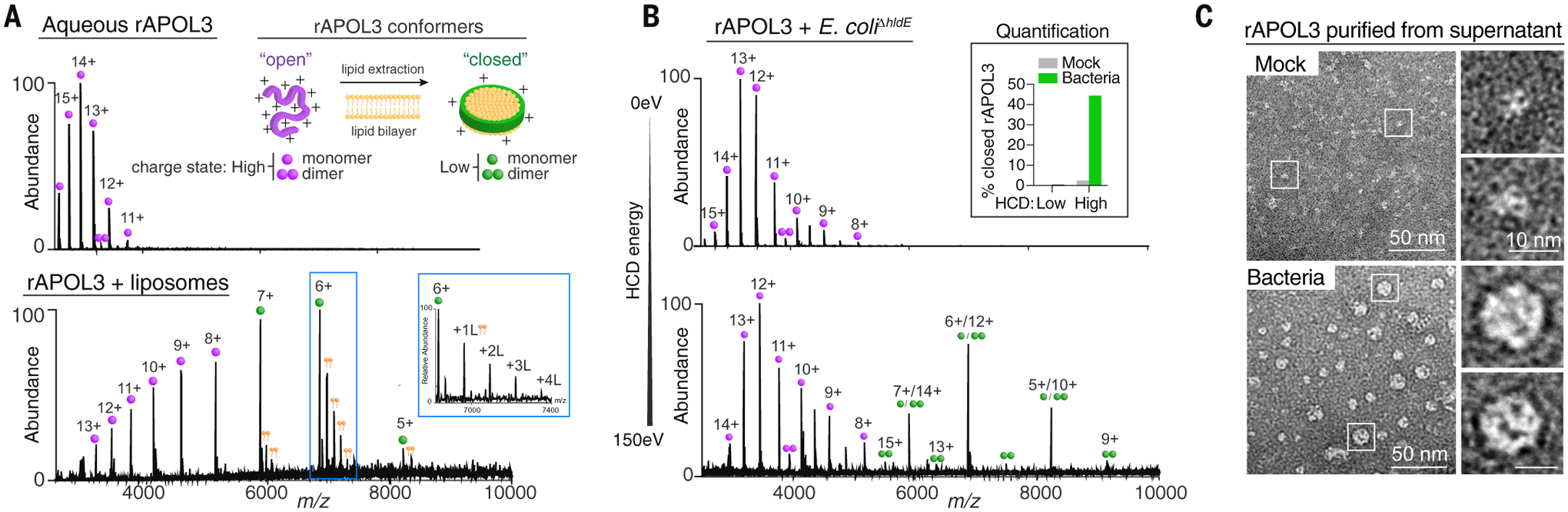
(A) Conformational analysis by native mass spectrometry (nativeMS) of rAPOL3 in ammonium acetate buffer (aqueous) or after incubation with DMPC/DMPG liposomes for 30 min. Inset shows that satellite peaks correspond to successive lipid (L) adducts. Schematic indicates the two observed charge states: lipid-free “open” or lipid-bound “closed” monomers (single circle) or dimers (doublet). (B) NativeMS spectra of soluble rAPOL3 after incubating with live E. coliΔhldE for 1 hour. Collisional activation energy (HCD) was set to 0 eV (top) or 150 eV (bottom). Inset shows nativeMS quantification of “closed” APOL3 conformers before (mock) and after treatment of bacteria and analyzed at the indicated HCD energy. (C) rAPOL3 was incubated with live E. coliΔhldE as in (B), purified from the supernatant by Ni-NTA pull-down and analyzed by negative-stain electron microscopy. Data from [(A) to (C)] are representative of three independent experiments.
Discussion
The double membranes surrounding Gram-negative bacteria make them exceptionally difficult to kill. Consequently, combination therapies that use OM-permeabilizing agents to facilitate passage of larger, more effective antibiotics have emerged as a promising treatment option (30–32). Our results show that humans have evolved an analogous strategy for cellular self-defense, with the natural detergent-like effector APOL3 being given access to the bacterial IM by other synergizing ISGs, including GBP1, that help to permeabilize the OM. Once inside the bacterium, APOL3 exerts broad-spectrum membranolytic activity through solubilization of the IM into discoidal lipoprotein complexes. This mode of killing appears to differ from that of canonical extracellular antimicrobial proteins (AMPs), which tend to form proteinaceous pores or induce local membrane dysfunction (33). Thus, APOL3 may have arisen as a host adaptation to support intracellular killing specifically, given that both the ionic strength and the divalent cation concentration of the human cytosol are incompatible with the activity of many AMPs (34).
Our biochemical studies found that APOL3 targets anionic lipids that are highly enriched in bacterial membranes (35). In contrast, cholesterol, which is present exclusively in eukaryotic membranes, inhibits APOL3 membranolytic activity. This selectivity may aid discrimination between self and non-self lipid structures, as seen for the cytolytic T cell antimicrobial protein granulysin, which similarly targets cholesterol-poor anionic membranes (36–38). Notably, organelles such as mitochondria also contain some of the same anionic lipids found in bacteria (cardiolipin, PG), whereas endoplasmic reticulum membranes exhibit naturally low cholesterol content (39). Thus, additional host factors probably help to restrain or localize APOL3 activity in order to prevent damage to cellular host structures. In this regard, an APOL3 loss-of-function variant shows signs of recent positive selection in Africans (40), which suggests that its potent membranolytic properties could be detrimental in certain modern human populations if spurious activation contributes to pro-inflammatory disease. Future studies may identify other APOL3 mutations in susceptible individuals and may determine whether they come with a fitness cost.
Examination of immune or microbial stimuli eliciting APOL3 expression found that type II IFN was the major trigger versus type I IFN (IFN-α, IFN-β), tumor necrosis factor (TNF)–α, IL-1β, or LPS as a Toll-like receptor 4 ligand. Hence, it principally operates as part of the IFN-γ–inducible defense program in human cells (2). This program enlists other IFN-γ–inducible defense factors, including the GBP family, that have emerged as central orchestra-tors of cell-autonomous immunity to intracellular bacterial pathogens (19–21). Cooperation between APOL3 and GBP1 was evident in loss-of-function, gain-of-function, and cell-free experimental systems. Notably, however, convergence of these proteins on the surface of Gram-negative bacteria yielded bifurcating outcomes. GBP1-mediated damage to the bacterial OM not only allowed APOL3 access to the IM for eventual killing but also activated human caspase-4 and IL-18 (18, 19). In contrast, APOL3 did not trigger the noncanonical caspase-4 inflammasome pathway, instead conferring protection through direct bactericidal activity. These data reveal distinct functional roles for these two IFN-γ–induced defense proteins within the “interferome” signature (2).
Our results elucidate the role of human APOL3 as a potent bactericidal agent deployed within nonimmune cells to combat cytosolic pathogens. Our findings reinforce the growing appreciation for the contributions made by cells outside of the hematopoietic compartment toward IFN-γ–induced host resistance (4, 5, 41). Our work also reveals the involvement of lipoproteins in intracellular killing in humans, adding to immune functions described earlier for insects that use serum lipoproteins as a form of systemic, extracellular defense (42, 43). Thus, although human APOL3 is considered a young gene that evolved relatively recently (~33 million years ago) (10), membrane solubilization may itself be an ancient bactericidal mechanism that appears to have been harnessed for sterilizing intracellular immunity in primates.
Materials and methods
Plasmids, antibodies, and reagents
CRISPR deletions were generated using pX459. Complementary DNAs (cDNAs) for the human APOL genes were amplified from HeLa cells and verified by sequencing. APOL3 isoform 2 was chosen, as this is considered the most commonly expressed isoform (15). cDNAs were inserted into the retroviral plasmid pMSCV-puro with an N-terminal hemagglutinin (HA) tag or N-terminal EGFP for stable expression in complementation and certain imaging experiments. For doxycycline-inducible expression, APOL3 or GBP1 cDNA was cloned into MCS1 or MCS2, respectively, of pCW57-MCS1–2A-MCS2 (Addgene) and transductants obtained by selection in puromycin. For live and high-resolution imaging, pmNeonGreen-C1 or pCMV-3XFLAG encoding APOL3 was used. N-terminal tags were used for all experiments. For recombinant protein expression, cDNA was inserted into a modified pET28a vector containing an N-terminal 6×-His tag followed by a precision protease cleavage site. Truncation mutants were generated by PCR with overlapping primers flanking deletion sites. Point mutations were inserted using a single mutated primer and Phusion polymerase (NEB). The ΔAH variant of APOL3 was obtained as a Geneblock from IDT. For qPCR, RNA was isolated using an RNeasy Mini Kit (Qiagen) and converted to cDNA using PrimeScript RT master mix (Takara). Amplification was done using PowerUp SYBR Green master mix (ThermoFisher) on a QuantStudio-5 Real Time PCR system with gene-specific primers and analyzed using the 2−ΔΔCt method with GAPDH as the housekeeping gene. For labeling of bacteria, pFPV25.1 (Addgene) encoding EGFP, mCherry, RFP, or mScarlet was used. The dual transcriptional reporter pFcCGi encoding a constitutive mCherry and PBAD-GFP has been described previously (44, 45) and was obtained from Addgene. To generate the minD reporter plasmid, minD was amplified from Stm 1344 genomic DNA using the primers 5′-atggcacgcattattgttgttacttcgggtaaaggg-3′ and 5′-ttatcctccgaacaggcgtttgaggaaacctttcttc-3′ and cloned into pmNeonGreen (mnGFP)-C1 using XhoI and EcoRI. The entire mnGFP-minD fusion protein was cloned into the PBAD-GFP position (XbaI and SphiI) of pFcCGi, creating pFcCmNmDi. To free the red channel for certain microscopy experiments, mCherry was removed by overlapping PCR, creating pFmNmDi. Plasmids were transformed into electrocompetent Stm and selected with carbenicillin (100 μg/ml). To induce expression of mnGFP-minD, overnight Stm was subcultured 1/33 and grown for 3.5 hours in the presence of carbenicillin (50 μg/ml) and 0.2% l-(+)-arabinose (Sigma) before infection. To express IM-anchored GFP, the TorA signal sequence (ssTorA) of Stm was ligated onto the N terminus of EGFP in pFPV25.1 using overlapping primers. This sequence directs properly folded EGFP through the twin arginine transporter pathway and then serves as a peripheral membrane anchor on the periplasmic face of the IM (46).
Antibodies used were anti-Flag M2 (Sigma), anti-HA (16B12 Biolegend), anti-APOL3 (ab154869), anti-Salmonella O Group B antiserum (BD), anti–β-actin (ab6276), anti-Lamp1 (PA1–654A, ThermoFisher), anti-p62 (bd 610832), anti-Galectin 8 (sc-377133), anti-GBP1 (sc-53857), anti-COX2 (sc-1747), anti-Mx2 (sc-271527), anti–IL-18 (PM014; MBL), anti-IFITM3 (Proteintech; 11714–1-AP), and anti–Caspase-4 (clone 4B9; Enzo). All lipids were purchased from Avanti Polar Lipids Inc. Fluorescein-labeled dextrans were from Sigma Aldrich. TbCl3+ and dipicolinic acid (DPA) were from Biotium. Calcein was from Life Technologies. l-Leucyl-l-leucine methyl ester (LLOMe) hydrochloride was from Cayman Chemicals. Zombie-UV was from Biolegend.
Bacterial strains
Bacterial strains were kindly provided by the following groups: Salmonella enterica serovar Typhimurium (Stm) strain 1344 and injectosome deficient StmΔinvA::pR1203 (J. Galan); Listeria monocytogenes 140203S (H. Agaisse); Shigella flexneri strain M90T (F. Randow); Stm 1344ΔcpxR:FRT (J. Vogel) (47); E. coli DH5αΔhldE (S. Gray-Owen) (48) Stm UK-1 wild-type, Δwzy, ΔwaaL, ΔwaaJ, ΔwaaI, and ΔwaaG (R. Curtiss III) (49). Burkholderia thailandensis strain 700388 and S. enterica serovar Typhi strain 700931 were purchased from ATCC.
Bacterial infections
For Stm infections, overnight bacterial cultures were diluted 1:33 in fresh LB, grown for 3 hours before being washed once in PBS, and used to infect HeLa cells at 80% confluence with an MOI of 5 unless otherwise indicated. Plates were centrifuged for 10 min at 1000g and incubated for 30 min at 37°C to allow invasion. Extracellular bacteria were killed by replacing media with fresh DMEM containing gentamicin (100 μg/ml) for 30 min. Cells were washed three times and incubated with gentamicin (20 μg/ml) for the duration. To enumerate live bacteria, cells were lysed in PBS + 0.5% Triton X-100 and serial dilutions plated on LB agar. To estimate bacterial load based on GFP intensity, StmGFP was used and cells were trypsinized and fixed in 4% PFA (Santa Cruz) for 15 min. After washing, cells were resuspended in PBS and GFP fluorescence determined on a FACSAria (BD). Analysis was done using Flowjo. To release bacteria from vacuoles, LLOMe (600 μM) and Z-VAD-FMK (20 μM) were added after 2 hours and incubated for the duration. For Shigella infections, overnight cultures were diluted 1:100 in tryptic soy broth (BD) and growth to OD600 = 0.5 before infecting HeLa cells at 80% confluence at MOI of 50. Cells were then processed as for Stm infections. For S. Typhi infections, overnight cultures were diluted 1:20 in LB + 0.3 M NaCl, grown to OD600 = 1.0 and processed as per Stm. For B. thailandensis infections, overnight cultures were diluted 1:10 and grown to OD600 = 1.0 (5 × 108 cfu/ml). Bacteria were washed once in PBS and added to cells at MOI of 200, centrifuged at 1000g for 10 min, and left for 1 hour at 37°C. Cells were rinsed and incubated for 24 hours in complete medium containing kanamycin (1 mg/ml). For L. monocytogenes infections, bacteria were grown overnight at 30°C in brain heart infusion broth (BHI; BD) and adjusted to OD600 = 1.0. Bacteria were washed and used to infect HeLa cells at MOI of 50 following the protocol outlined for Shigella.
Cell culture and transfection
HeLa (CCL2) and 293T cells were purchased from ATCC. Cells were grown in DMEM supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. Autophagy-deficient Penta-KO and Hexa-KO cell lines have been described previously (50, 51). HUVECs from a single donor were obtained from LONZA (CC-2517) and maintained in EBM Basal Medium with growth factors and used prior to passage 10. Primary intestinal epithelial cells (CC-2931) and intestinal myofibroblasts (CC-2902) were from LONZA and maintained in SmGM Medium with growth factors. Epithelial cells were maintained at 33°C as per manufacturer’s instructions and thawed directly from frozen into 96-well plates and used on days 5 to 7. All cells were maintained in antibiotic-free media. Lentiviral (LentiCrisprV2) or retroviral (pMSCV-puro) transductions were done by incubating dilutions of 0.45 μm–filtered supernatants from transfected 293T cells with polybrene (8 μg/ml) for 24 hours. For selection of stable transductants, puromycin (1 μg/ml) was included. For transient transfections, TransIT-LT1 (MIRUS) was used according to manufacturer’s instructions. To minimize toxicity in microscopy experiments, 200 ng of DNA was transfected per 24-well cover slip. To generate stable gene knockouts, sgRNAs were cloned into pX459 (Addgene) per established protocols. 2–4 sgRNAs (table S2) targeting each gene (200 ng total DNA) were transfected in 24-well plates for 24 hours, followed by selection with puromycin (1 μg/ml) for 48 hours. Surviving cells were expanded into media lacking puromycin for 48 hours, then subjected to limiting dilution to obtain single colonies. Colonies were screened first by PCR, then by Western blot, and when appropriate the genotype of each positive clone was determined by Sanger sequencing. For siRNA knockdown, ON-TARGETplus Human APOL3 siRNA smartpool (Dharmacon) or nontargeting control were transfected (20 nM) with Dharmafect 1 transfection reagent for 48 hours as per manufacturer’s instructions. When required, HeLa cells were stimulated with IFN-γ (500 U/ml) and primary human cells stimulated with IFN-γ (50 U/ml) for 18 hours.
Genome-wide screen
LentiCrisprV2 pooled library (GeCKO v2) was a gift from F. Zhang (Addgene #1000000048) (52). 25 × 106 HeLa cells were transduced on four separate days and processed as individual biological replicates (N = 4) throughout the experiment. Transductants were selected with puromycin (1 μg/ml) for 48 hours, then allowed to rest without selection for an additional 48 hours. Surviving cells were split into two groups (±IFN-γ) and seeded into 6-well plates at 80% confluency. After 24 hours, IFN-γ (500 U/ml; R&D systems) was added for an additional 18 hours. Late log StmGFP was added to each well at MOI of 20 and incubated for 6 hours as described for infections. Cells were trypsinized and fixed in 4% PFA for 15 min and analyzed within 48 hours on a FACSAria (BD). Uninfected cells were gated out based on comparisons with cells only control, and infected cells gated into two groups, high GFP (HR) or low GFP (SR), based on the maximal difference obtained between the IFN-γ–treated and untreated control groups processed in parallel. For each of the four biological replicates, an average of 1 × 106 and 5 × 106 cells were collected for the HR and SR gates, respectively. After sorting, each group was pelleted and DNA extracted using the PicoPure DNA Extraction Kit (Applied Biosciences). sgRNA sequences were amplified using Herculese II Phusion DNA polymerase (Agilent) and amplicons purified from an 8% polyacrylamide Tris/Borate/EDTA (TBE) gel. Amplicons were sequenced using an Illumina HiSeq2500 (20 million reads per sample) and analyzed using the MAGeCK algorithm (53). Gene-level enrichment scores (P value) for sgRNAs enriched in the HR versus the SR populations were determined for both the IFN-γ–treated and untreated groups (table S1).
RNA sequencing
HeLa cells were stimulated with IFN-γ (500 U/ml) for 18 hours or were left untreated. Infections with Stm were performed on individual triplicates at MOI 5. After 5 hours, cells were washed three times in sterile prewarmed DMEM, lysed in 300 μl of RLT buffer, and processed via RNeasy (Qiagen) kits per the manufacturer’s protocol. RNA was checked for quality using a denaturing MOPS gel and Nanodrop. 10 μg of sample RNA was annealed to oligo-dT beads followed by first- and second-strand cDNA synthesis (Illumina). cDNA was then pair-end–barcoded with Illumina Universal Adapters and sequenced on a HiSeq4000 sequencer. Data acquired were bin-sorted and de-barcoded through a Sickle-Schythe Pipe. FASTA files were then aligned to human reference genome HsGRCh37 by Hisat and Tophat2, yielding 95% alignment. Annotated genes were quantitated via CufflinksV2 and subsequent data were processed for visual display through CummeRbund, ggplots, and ggplot2 in R.
Microscopy
HeLa cells were seeded on 12-mm high-performance cover glass #1.5h (Thorlabs). For live imaging, cells were seeded on four-well chambers with #1.5 high-performance cover glass (Cellvis). Cells were seeded 48 hours prior to imaging to reach 80% confluency on the day of infection and treated with IFN-γ (500 U/ml) where required for 18 to 24 hours prior to imaging. To image bacterial infections, bacteria were added to cells as described for infections at an MOI of 20. Images were analyzed on a DeltaVision OMX SR microscopy system (GE Healthcare) or a laser scanning confocal model SP8 (Leica). For analysis of relative positions of APOL3 and LPS, HA-APOL3 was detected with anti-HA, followed by anti-mouse Alexa-fluor 488 (ThermoFisher) and LPS detected with anti-Stm LPS and Alexa-fluor 568 or 647 anti-rabbit antibody. APOL3-positive bacteria were identified and the intensity profiles for LPS and APOL3 signal were determined from linescans drawn on a single plane slices of 2-μm z-stacks [following alignment and structure illumination microscopy (SIM) reconstruction]. Images were analyzed using Fiji. Bacteria with APOL3 signal intensity of >50% of max inside the LPS layer were quantified at 45 min or 150 min post-infection in the presence or absence of IFN-γ. For analysis of the bacterial response to arabinose post-infection, overnight Stm pFcCGi were subcultured 1/33 in LB containing carbenicillin (50 μg/ml) for 3 hours and used to infect HeLa cells in 24-well plates, transfected 24 hours earlier with 200 ng pCMV-3XFLAG-APOL3 in the presence or absence of IFN-γ, at an MOI of 20 for 15 min. Extracellular bacteria were killed with gentamicin (100 μg/ml) for 30 min, and after three washes, replaced with fresh media containing gentamicin (10 μg/ml) and 0.4% l-(+)-arabinose for an additional 2 hours prior to fixation with 4% PFA (Santa Cruz). 50 APOL3-positive or -negative bacteria were selected at random from micrographs in IFN-γ–treated or nontreated conditions and the maximum intensities of mCherry and GFP signals for each bacterium were determined using Fiji. For live high-content imaging, 5 × 104 cells (primary intestinal epithelium; InEpC and HeLa) or 1 × 104 primary intestinal fibroblasts (InMyoFib) were seeded into black 96-well clear-bottom plates 7 days (InEpC), 72 hours (InMyoFib), or 48 hours (HeLa) prior to imaging. When required, 48 hours prior to imaging, cells were transfected with control or APOL3-targeting siRNA for 30 hours, then treated with IFN-γ (50 U/ml) [primary cells, or HeLa cells (500 U/ml)]. Infections were done with StmmScarlett or StmGFP at an MOI of 20 as described above with the following modifications for InEpC: Bacteria were incubated for 60 min after centrifugation and 60 min after addition of gentamicin (100 μg/ml) rather than the usual 30 min. Cells were imaged live at 1-hour intervals while incubating at 33°C (InEpC) or 37°C (InMyoFib, HeLa) and 5% CO2 on an ImageXpress Pico Automated Imaging System (Molecular Devices) at 10× magnification. Analysis was done in an unbiased manner using CellReporterXpress with the preconfigured “Endocytosis” protocol template modified to identify the following intracellular Stm populations: Based on preliminary experiments in HeLa cells, SR Stm foci were defined as objects between 1 and 10 μm and HR Stm foci defined as objects between 10 and 35 μm. A threshold intensity of 60 units above background was set to exclude nonspecific fluorescence. Where required, Sytox Orange was included at 2 μM for the duration and dead cells defined as objects 2 to 10 μm. Data was normalized to total number of cells from parallel wells stained with Hoechst 33342 (5 μg/ml; Invitrogen). For in vitro imaging, cytosol-enriched Stm pFmNmDi or pFPV25.1-ssTorA-GFP bacteria were harvested from infected cells using Triton X-100 as described for bacterial killing assays, washed three times, and resuspended in Buffer A [50 mM MES pH 6.0, 100 mM potassium gluconate (KGl)] containing 5 μM 568-labeled rAPOL3. For minD imaging, bacteria were immediately imaged live on 1.5% agarose pads. To image ssTorA-GFP and simultaneously monitor membrane integrity, Zombie-UV (1/200) was added for 5 min, bacteria were washed once, resuspended in PBS, and imaged live on 1.5% agarose pads.
Purification of recombinant proteins
APOL proteins:
Overnight cultures of BL21 (DE3) pLysS harboring the APOL3 or APOL1 expression plasmid (pET28a-6XHis-PP) were grown in LB containing kanamycin (50 μg/ml) and chloramphenicol (20 μg/ml). Cultures were grown to OD600 = 0.7 in media without chloramphenicol and induced with 1 mM IPTG for 4 hours at 37°C. Cell pellets were lysed in 50 mM Tris pH 8.0, 5 mM EDTA and lysozyme (100 μg/ml; Sigma) with sonication. The presence of lysozyme had no effect on the quality or activity of the recovered protein but did increase yield. Insoluble material was pelleted at 20,000g and washed with lysis buffer containing 0.5 M NaCl. Pellets were solubilized in 6 M guanidine hydrochloride, 50 mM potassium phosphate pH 8.0, 1 mM TCEP, 10 mM imidazole for 1 hour at room temperature with gentle sonication and clarified by centrifugation at 40,000g for 30 min. Solubilized protein was affinity purified using Ni-NTA beads (Qiagen) and dialyzed extensively (>4 buffer changes of at least 1000 fold v/v) over 24 hours into 20 mM acetic acid. For certain experiments, the His-tag fusion protein was digested with 3C protease (Genscript) in 50 mM MES pH 6.0 overnight at 4°C. To remove the tag, undigested protein and the protease, the reaction and all insoluble precipitate was solubilized in 6 M guanidine hydrochloride, 50 mM potassium phosphate pH 8.0, 1 mM TCEP, 10 mM imidazole and incubated overnight with fresh Ni-NTA beads. Flow through was collected and dialyzed extensively into 20 mM acetic acid. The absence of the His-tag was confirmed using Western blot.
To refold the ΔAH variant, protein was first dialyzed against 20 mM acetic acid, 250 mM arginine hydrochloride (Sigma) for 6 hours before dialysis into 20 mM acetic acid. All purified proteins were concentrated (>10 mg/ml) and flash-frozen in liquid nitrogen and stored at −80°C. Proteins were thawed once, as activity decreased upon each freeze/thaw. rHis-APOL3 and rHis-cleaved APOL3 exhibited almost identical biological activity in both killing assays and liposome leakage/solubilization assays. However, rHis-APOL3 demonstrated increased stability at higher pH, maintained stability at a higher concentration, and was thus purified to greater yield. Therefore, the His-tagged protein was used when required. rHis-APOL1 was used for bactericidal and liposome solubilization assays. Preliminary experiments revealed that rAPOL3 protein stability was compromised in high concentrations (>0.1 M) of traditional chloride salts such as NaCl and KCl. Therefore, a gluconate salt of potassium (KGl, the most common ion in the cytosol) was included in reaction buffers, unless otherwise indicated, to maintain a near-physiological salt concentration. To prepare fluorescently labeled protein, 750 μg of protein was mixed with 333 μM AFDye 568 maleimide (Fluoroprobes) in a 300-μl reaction volume. pH was adjusted to pH 7.0 with 1 M HEPES pH 7.4 and reaction incubated for 2 hours at room temperature. During this time, ~75% of rAPOL3 protein precipitated. Precipitated protein was collected by centrifugation, dissolved in 6 M guanidine hydrochloride, 50 mM potassium phosphate pH 8.0 and dialyzed extensively (10 kDa MWCO) into 20 mM acetic acid to both refold the protein and remove unincorporated dye.
Guanylate binding protein 1 (GBP1):
The coding sequence of human GBP1 (hGBP1) was cloned into a customized vector to generate pCMV-His10-Halo-HRV-mRFP-TEV-hGBP1. HEK293f suspension cells (a gift from J. Rothman) was maintained at a concentration of 0.4 × 106 to 4 × 106 cells/ml in Expi293 expression medium (ThermoFisher Scientific). 24 hours prior to transfection, cells were seeded at a concentration of 1.2 × 106 cells/ml. For transfection, cells were harvested and resuspended in fresh medium at a concentration of 2.5 × 106 cells/ml. Cells were transfected by adding pCMV-His10-Halo-HRV-mRFP-TEV-hGBP1 to a final concentration of 1 μg/ml in media containing PEI at a concentration of 5 μg/ml. 24 hours after transfection, cells were diluted 1:1 (v/v) with fresh medium containing 4 mM valproic acid and cultured for an additional 2 days. 2 × 109 cells were harvested via centrifugation (500g, 10 min), washed once in cold PBS, resuspended in lysis buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 1 mM MgCl2, 10% glycerol, 0.5% CHAPS, 1 mM TCEP) and lysed via sonication. Cells were cleared at 35,000g for 1 hour at 4°C. Supernatant was collected and incubated with 1 ml bed volume of HaloLink resin (Promega) at 4°C overnight with gentle rotation. The resin was sequentially washed twice (10 min each) with wash buffer 1 (50 mM HEPES, pH 7.5, 500 mM NaCl, 1 mM MgCl2, 10% glycerol, 0.5% CHAPS), wash buffer 2 (50 mM HEPES, pH 7.5, 1 M NaCl, 10% glycerol) followed by wash buffer 1. To elute bound proteins, Halo resin was resuspended in lysis buffer and digested with homemade GST-HRV-His protease overnight at 4°C with gentle rotation. Resin was pelleted and the HRV protease was removed from the supernatant via Ni-NTA beads by affinity chromatography (Qiagen). Flow-through was collected, concentrated, and further purified and buffer-exchanged via size exclusion chromatography (Superdex 200 Increase; GE Healthcare) equilibrated with storage buffer [20 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 1 mM TCEP]. Fractions were analyzed by SDS-PAGE, pooled, concentrated, and flash-frozen in liquid nitrogen before storing at −80°C.
Recovery of APOL3 lipoprotein complexes:
To isolate rAPOL3-lipoprotein complexes from bacteria, overnight E. coliΔhldE was subcultured 1/20 and grown to OD600 = 0.5. 10 ml of bacteria were washed and resuspended in Buffer A containing 20 μM rHis-APOL3 for 2 hours at 30°C and insoluble material removed by centrifugation. The pH of the supernatant was adjusted to pH 7.2 with NaOH, and both NaCl and imidazole were added to 200 mM and 10 mM final concentrations, respectively. The solution was incubated with Ni-NTA beads at room temperature for 1 hour, washed 5 times with 10 mM Tris pH 7.2, 200 mM NaCl, and 20 mM imidazole, and eluted with 400 mM imidazole in the same buffer. Eluates were loaded directly onto glow-discharged copper grids and examined by negative-stain electron microscopy.
Bacterial killing assays
To isolate bacteria from different cellular compartments, ΔAPOL3 HeLa cells with or without IFN-γ (500 U/ml, 18 hours) were infected with Stm at MOI of 20 as described for infections. After 45 min, cells were either lysed in Buffer A containing 0.5% Triton X-100 (vacuolar population) or treated with 1 mM LLOMe (Sigma) in the presence of cell death inhibitor Z-VAD-FMK (R&D) for 2 hours before lysis (cytosolic). Bacteria were then mixed with rAPOL3 diluted in 20 mM acetic acid for 3 hours and enumerated by colony counting after serial dilution. To induce transient permeabilization of the OM by EDTA, overnight bacterial cultures were grown to OD600 = 0.5 in LB containing 2 mM CaCl2 and 2 mM MgCl2 and immediately washed twice in 0.1 M Tris pH 8.0, 0.75 M sucrose. Pellets were resuspended in 350 μl of the same buffer and 700 μl of 1 mM EDTA in H2O was added for 20 min. 50 μl of 0.5 M MgCl2 was added on ice for 5 min and bacteria pelleted at 4°C. Bacteria were resuspended in Buffer A, and rAPOL3 diluted in 10 mM acetic acid was added to the indicated concentration and incubated for 3 hours at 30°C. The final dialysate from rAPOL3 purifications was used as the negative control. Bacteria were enumerated by serial dilution on LB agar. To induce transient OM permeabilization by other means, mid–log-phase bacteria were washed and resuspended in Buffer A supplemented with the indicated concentration of polymyxin B nanopeptide (PMBN; Sigma), poly-l-lysine hydrobromide (avg. mw 20,000 Da; PKLB20, Alamanda Polymers), or human platelet factor IV 18 (C18G; Eurogentec). To induce hypotonic stress, bacteria were resuspended in Buffer A with 20 mM KCl substituted for 100 mM KGl. Bacteria were incubated for 30 min before washing and addition of rAPOL3 in Buffer A. Bacteria incubated in LB or Buffer A alone served as the nonpermeabilized control. After initial washes, a sample of bacteria was plated on LB agar prior to addition of any OM permeabilizing agent to define the input. For LD50 assays, Stm were incubated for 3 hours with twofold serial dilutions of rAPOL3, recombinant mouse RegIIIβ (R&D), or human β-defensin-2 (Biolegend) in either Buffer A or low salt (20 mM KCl) buffer. The minimum concentration required to kill >50% of Stm was determined. For LPS-truncated mutants, bacteria were grown to OD600 = 0.4 and 0.5 ml washed once in Buffer A, resuspended to 0.5 ml in Buffer A with 150 mM KGl and incubated with 10 μM rAPOL3 or dialysate at 30°C for 3 hours with shaking at 250 rpm. Bacteria were enumerated by serial dilution and colony counting. For treatment with hGBP1, bacteria were incubated for 1 hour with 5 μM hGBP1 in 50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl2, with or without 2 mM GTP. Bacteria were then pelleted and resuspended in Buffer A containing rAPOL3 and incubated at 37°C for 1 hour prior to plating.
Bacterial membrane and cytotoxicity assays
Permeability of the OM was determined using the fluorescent dye NPN (Sigma) uptake assay. Stm were prepared as described for the killing assay, treated with permeabilizing agent at the indicated concentration for 15 min, and resuspended in Buffer A containing 10 μM NPN. rAPOL3 or dialysate was added and incubated for 15 min. Fluorescence (Ft15) was recorded by SpectraMax i3X plate reader; λEx = 350 nm and λEm = 420 nm. NPN uptake after 15 min was calculated as (t) (%) = (Ft15 – Ft0) × 100/ (Ft100 – Ft0), where the fluorescence from untreated Stm was defined as Ft0 and in the presence of 5 mM EDTA and lysozyme (10 μg/ml) as Ft100. Permeability of the IM was determined by Sytox orange (ThermoFisher) or propidium iodide (PI; Sigma) uptake. Stm were prepared as described for the killing assay and treated with 10 μM rAPOL3 or dialysate in Buffer A for 15 min (static) or for the indicated time (time course). PI was included at 50 μM and fluorescence measured using a SpectraMax i3X plate reader; λEx = 535 nm and λEm = 620 nm. PI uptake at each time point was calculated as (t) (%) = (Ft – Ft0) × 100/(Ft100 – Ft0). Fluorescence from untreated Stm was defined as Ft0 and in the presence of polymyxin B (25 μg/ml) and 0.2% SDS as Ft100. Sytox orange was included at 0.2 μM and fluorescence determined as described for PI uptake with λEx = 545 nm and λEm = 570 nm. IM potential of 5 × 107 Stm was determined using the BacLight bacterial membrane potential kit (ThermoFisher) following the manufacturer’s protocol. Stm were treated as described for the killing assay, then incubated with 5 μM CCCP, 10 μM rAPOL3 wild-type, ΔAH mutant, or equal volume of dialysate for 1.5 hours before addition of DiOC2(3) for 30 min. Samples were analyzed on a FACSAria (BD). The ratio of red to green fluorescence provides an indication of membrane potential and was calculated for each treatment by dividing the mean fluorescence intensity (MFI) for the red channel (FL-2) by the MFI for the green channel (FL-1) after gating bacteria by forward and side scatter. Bacterial ATP content was determined using BacTiter-Glo microbial cell viability assay (Promega) following the manufacturer’s protocol. To measure membrane fluidity, mid-log E. coliΔhldE were washed and resuspended in PBS containing 0.2% glucose and 10 μM Laurdan (Cayman Chemicals) and incubated for 10 min. Bacteria were washed 3× with PBS containing 0.2% glucose and 1% dimethylformamide (DMF). 500 nM rAPOL3 was added and fluorescence at two wavelengths recorded over time using a SpectraMax i3X plate reader: (i) λEx = 350 nm and λEm = 460 nm; (ii) λEx = 350 nm and λEm = 500 nm. Background fluorescence was subtracted from each value and generalized polarization calculated as GP = (I460 – I500)/(I460 + I500) where I is the normalized intensity at each λEm wavelength. For treatment with rGBP1, bacteria were incubated for 1 hour with 5 μM rGBP1 (or mock) in 50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl2, 2 mM GTP. Bacteria were then pelleted and resuspended in Buffer A supplemented with 10 μM NPN and 0.2 μM Sytox orange containing 5 μM rAPOL3 (or mock) for 1 hour before fluorescence at two wavelengths was simultaneously measured using the SpectraMax i3X plate reader as above. Background fluorescence of NPN and Sytox orange in buffer alone was subtracted from each value. ATP content was then determined as described above.
Liposome preparation
Phospholipids were dissolved in chloroform and mixed in a glass vial. Solvent was evaporated under nitrogen and dried overnight in a vacuum. For liposome leakage assays, lipid film was hydrated in Buffer A. For liposome solubilization and nativeMS experiments, lipids were hydrated with 20 mM ammonium acetate. Lipids were solubilized with continual vortexing followed by five freeze/thaw cycles. Liposomes were generated by extrusion through a 0.1-μm polycarbonate filter (Avanti Polar Lipids Inc.) 30 times using a mini-extruder device (Avanti Polar Lipids Inc.). To generate calcein-encapsulated liposomes, lipid was hydrated in 50 mM MES pH 6.0, 20 mM potassium gluconate, and 80 mM calcein. For Tb3+-encapsulated liposomes, lipids were hydrated with 50 mM MES pH 6.0, 35 mM KGl, 50 mM sodium citrate, and 15 mM TbCl3. Non-encapsulated calcein or Tb3+ was removed using Illustra Microspin G50 columns (GE Healthcare). For dextran liposomes, hydration was done with Buffer A and indicated FITC-Dextran (2 mg/ml). Nonincorporated dextran was removed by buffer exchange with a centrifugal filter device (Amicon Ultra-15 100K MWCO, Millipore). All liposomes were used within 24 hours.
Liposome binding, leakage, and turbidity assays
To measure liposome binding, indicated liposomes (2.5 mM lipid) were incubated for 20 min with recombinant APOL3 (rAPOL3; 1 μM protein such that liposomes were not completely dissolved) for 20 min in Buffer A. Samples were centrifuged for 1 hour at 120,000g in a Beckman Optima XE-100 Ultracentrifuge at 4°C. Supernatant (S) was collected and the pellet (P) washed twice with 700 μl of incubation buffer and resuspended in the same volume as supernatant. Samples were analyzed by SDS page followed by Coomassie blue staining. To measure leakage, liposomes of the indicated composition (500 μM lipid) were mixed with rAPOL3 (500 nM or the indicated concentration) in Buffer A. To measure Tb3+ efflux, 15 μM DPA was included in the buffer. The excitation and emission wavelengths were: λEx = 495 nm and λEm = 525 nm for calcein, λEx = 270 nm and λEm = 490 nm for Tb3+/DPA chelates, and λEx = 495 nm and λEm = 520 nm for FITC-Dextran. Fluorescence prior to addition of protein was treated as Ft0. 5 μl of APOL3 diluted in 10 mM acetic acid was added after ~1 min, and fluorescence recorded continuously (at 10- to 15-s intervals) using a SpectraMax i3X plate reader (Molecular Devices). 5 μl of final dialysate (20 mM acetic acid) was used as a mock treatment. 10 μl of 1% Triton X-100 was added to achieve complete dye release and the average of the top three fluorescence values defined as Ft100. The percentage of dye efflux at each time point was calculated as (t) (%) = (Ft – Ft0) × 100/(Ft100 – Ft0). To measure FITC-dextran efflux, liposome-protein mixtures were incubated for 20 min at room temperature and released FITC-dextran was collected in the flowthrough following centrifugation through a centrifugal filter device. Supernatant fluorescence from untreated liposomes was defined as Ft0 and in the presence of 0.1% Triton X-100 as Ft100. To measure liposome turbidity, DMPG or DMPC liposomes (2 mM lipid) were generated in 20 mM ammonium acetate and mixed with 40 μM rAPOL3 (50:1 lipid:protein ratio) in 20 mM ammonium acetate at the indicated temperature and absorbance at 400 nm determined. For the temperature transition, liposome-APOL3 mixtures were incubated at 37°C for 2 min before being transferred to room temperature for the duration.
Giant unilamellar vesicle (GUV) assays
79 nmol of DOPC, 20 nmol of DOPG, and 1 nmol of Cy5-labeled DOPC were mixed in 50 μl of 3:1 chloroform:methanol and spotted onto two indium tin oxide (ITO)–coated slides and evaporated under vacuum for 2 hours. ITO slides were sandwiched between PFTE spacers to create a GUV chamber and filled with swelling buffer (50 mM MES pH 6.0, 195 mM sucrose) and sealed with lipid-free modeling clay. Electroformation was conducted by applying a sinusoidal alternating voltage (10 Hz) increasing from 0.02 to 1.2 V over 50 min and holding this voltage for 120 min. Vesicles were removed and subjected to buffer exchange by adding 100 μl to 1 ml 100 mM MES pH 6.0, 150 mM KGl containing 50 μM Dylight 488 free acid (ThermoFisher) and mixed gently by inversion. After 30 min incubation at room temperature, vesicles were collected from the bottom of the tube and added to BSA-coated 20 mM glass-bottom dishes. 300 nM 568-labeled rAPOL3 was added to the well, mixed by pipetting, and imaged using a Nikon TiE inverted spinning disc confocal microscope or Nikon TE2000 microscope.
Circular dichroism
Spectra were taken of 3 μM rAPOL3 proteins in 10 mM MES pH 6.0, 20 mM KGl, 1 mM CaCl2 using a Chirascan circular dichroism spectrometer (Applied Photophysics). To determine the lipid-associated spectra, 3 μM rAPOL3 proteins were mixed with 3 mM PC/PG liposomes in the same buffer for 20 min and insoluble material removed by centrifugation. The amount of soluble protein remaining was determined and adjusted accordingly so that the lipid-bound and lipid-free concentrations were equivalent. Baseline spectra for buffer or liposome alone were used as the blank. CAPITO software (54) was used to estimate the secondary structure based on the observed spectra. Protein content was determined by BCA assay (ThermoFisher).
Electron microscopy
Negative-stain electron microscopy:
To visualize the effect of rAPOL3 addition to liposomes, liposomes containing 75:25 DMPC/DMPG (2 mM total lipid) were generated in 20 mM ammonium acetate and mixed with 40 μM rAPOL3 at 37°C for 5 min. The reaction was transferred to room temperature for an additional 30 min, then diluted 1/20 before loading onto glow-discharged copper coated EM grids (EMS, cat#CF400-Cu-50) and stained with 2% uranyl formate for 1 min. Grids were examined in JEOL1400 plus electron microscope with acceleration voltage of 80 kV. To visualize rAPOL3 on bacteria, log-phase E. coliΔhldE or StmΔwaaL was incubated with 10 μM, 5 μM, or 2 μM rHis-APOL3 in 100 μl Buffer A for 5 min at room temperature. Bacteria were pelleted and blocked by resuspension in 360 μl 20 mM Tris pH 7.4, 10 mM imidazole, 200 mM NaCl containing 1.5% skim milk for 5 min at room temperature. 40 μl of 5 nm Ni-NTA nanogold beads (nanoprobes) was added for 10 min at room temperature and washed three times in 20 mM Tris pH 7.4, 20 mM imidazole, 200 mM NaCl before loading directly onto glow-discharged copper grids. Bacteria were treated with dialysate alone and processed in parallel to assess nonspecific binding of beads to bacteria.
Cryo–immunoelectron microscopy:
HeLa cells were transduced with pMSCV-EGFP-APOL3 for 7 days, then infected with Stm as above before fixation/rehydration steps and immunogold labeling with anti-GFP antibodies as described (19).
Negative-stain EM particle averaging:
More than 400 meshed copper grids coated with carbon film (EMS, cat #CF400-Cu-50) were glow-discharged (PELCO easiGlow Glow Discharge Cleaning System) for 20 s to increase hydrophilicity of the carbon surface. 5-μl droplets containing rAPOL3 and liposomes were transferred to the glow-discharged EM grid and incubated for 1 min. Droplet samples were blotted with filter paper (Whatman qualitative filter paper, Grade 1), quickly washed with 5 μl 2% uranyl formate solution and stained on the grid by the same solution for 1 min. Residual staining solution was again removed by filter paper. Negative-staining EM grids were transferred to a JEOL1400plus electron microscope and images taken under an 80-kV electron gun at 40,000× magnification, and captured by a Hamamatsu ORCA HR camera, resulting in 0.234 nm/pixel. Five subframes were automatically taken at near in-focus plane and merged into a single image stack. All images were then preprocessed by e2proc2d.py function from eman2 packages for generating the images with MRC format (http://blake.bcm.edu/emanwiki/EMAN2). MRC format images were imported into Relion3 package for complete processing from 2D classification to 3D reconstruction (55). 2705 nanodisc-like particles were manually picked from 14 micrographs without processing motion correction and contrast transfer function correction. Two repeats on 2D classification were performed to remove nonspecific particles (56). Ten conformational classes representing 2071 particles from a final 2D classification were used to generate an initial model and 3D classification. Because no significant structural variation arose from 3D classification, 3D refinement was performed to obtain an averaged nanodisc structure.
Cryo-EM sample preparation:
5 mM DMPC/DMPG (3:1) liposomes were mixed with 100 μM rAPOL3 in 20 mM ammonium acetate for 5 min at 37°C, then transferred to 22°C for 1 hour and any insoluble material removed by centrifugation at 16,000g for 10 min. 4 μl was added to a C-Flat EM grid (C-Flat, 300 mesh CF-2/2–3Cu-50) that had been glow-discharged for 20 s (PELCO easiGlow Glow Discharge Cleaning System). Samples were incubated for 5 s and blotted in a Vitrobot Mark IV (Thermo Fisher Scientific). Blotting conditions are as follows: 5 s of blotting time, a blotting force of 8 at 90% humidity. The grid was subsequently plunged into liquid ethane and then transferred to liquid nitrogen for sample screening.
Cryo-EM data collection and image processing:
EM grids were loaded into a 200kV cryo–electron microscope (Thermo Scientific Glacios) equipped with a K2 Summit direct electron detector. Data were collected at 45,000 magnification, resulting in a physical pixel size of 0.896 angstroms (Å). The stage was adjusted such that focus ranged from 1 μm to 2 μm for data collection and the illumination area was set to 1 μm in diameter. Data were collected in superresolution movie mode with a 7-s exposure equaling 35 frames with a total electron dose of 50 e− Å−2. In total, 4648 stacks were collected in a 2-day session. Superresolution frames with a pixel size of 0.448 Å were treated with motion correction process by MotionCor2 (57). Parameters for processing drift correction are as follows: -Pathc 5 5 -PixSize 0.448 -Iter 30 -FtBin 2 -FmDose 1.43 -Bft 150 -Group 3. Each micrograph was initially screened manually to remove ice contamination or aggregates. During motion correction two types of images were generated: dose-weighted images and non–dose-weighted images. The non–dose-weighted images were used to estimate contrast transfer function (CTF) by Gctf (58). The CTF fitting of each micrograph was examined by manually checking the fitting accuracy of the Thon ring. The dose-weighted micrographs were imported into Relion for further particle picking and image processing. For particle picking, 1194 particles were manually picked from five representative micrographs to generate a template for the auto-picking process. In total, 502,901 particles were automatically picked from 4155 micrographs. These particles were extracted in a binning factor 4 for 2D classification. The initial 3D model was generated with the stochastic gradient descent algorithm in Relion. Multiple rounds of 3D classification were performed to screen homogeneous particles. The predominant class consisted of 236,364 particles and was used for a final 3D reconstruction with a 13.5-Å resolution based on gold-standard Fourier shell correlation criterion. The final 3D EM map was visualized and segmented by UCSF Chimera (59). The 3D reconstruction of APOL3 lipoprotein from the cryo-EM dataset was fitted into the EM density map from the negative-staining TEM dataset using the fit-in-map function of UCSF Chimera.
Native mass spectrometry (nativeMS)
DMPC/DMPG (75:25; 2 mM lipid) liposomes made in 20 mM ammonium acetate were treated with 40 μM APOL3 at 37°C for 5 min before transfer to 22°C for 1 hour. Incubation with 20 mM ammonium acetate without lipid served as the negative control. For analysis of bacterial-treated rAPOL3, overnight E. coliΔhldE was diluted 1/20 and grown to OD600 = 0.5. 0.5 ml was centrifuged and washed three times in 20 mM ammonium acetate and resuspended in 250 μl of 20 mM ammonium acetate buffer. rHis-APOL3 was added to 20 μM and incubated for 1 hour before insoluble material was pelleted and supernatant harvested and placed on ice to limit degradation by released bacterial pro-teases. Remaining protein content was estimated by protein gel. All samples were diluted to 5 μM and equilibrated to room temperature for 15 min prior to analysis. NativeMS was performed on a Q Exactive UHMR mass spectrometer (Thermo Fisher Scientific) using in-house nano ion-emitting capillaries. The ultrahigh vacuum was set at 5.65 × 10−10 mbar and capillary voltage 1.5 kV. Insource trapping and higher-energy collisional dissociation (HCD) were optimized for best-quality spectra. Relative quantitation was performed by combining the area under curves for each charge state.
In silico protein sequence analysis
Physiochemical properties of APOL3, APOL3-ΔAH, or APOE1 were calculated using Heliquest (60). Transmembrane domains and protein structure was predicted using Phyre2.0 (61). Hydrophobicity and charge were visualized by applying YRB lighting (62) in Pymol.
Experimental design and statistics
No sample size calculation or blinding was performed. For quantification of micrographs, sample size reflects both prior knowledge of variation and the maximum number of events that could be reasonably quantified. No data were excluded. Samples were randomly allocated into experimental groups and typically started with common pools of cells or bacteria. Data were analyzed by GraphPad Prism 8.0 software. Unless otherwise indicated, statistical significance was determined by t test (two-tailed) or one-way analysis of variance (ANOVA) (Dunnett’s multiple-comparison tests) or two-way ANOVA (multiple comparisons).
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Nikolaus, A. Tunaru, M. Braun, K. Nelson, M. Llaguno, and X. Liu for experimental advice and technical help.
Funding:
Supported by National Institute of Allergy and Infectious Diseases grants R01AI068041-14 and R01AI108834-07 (J.D.M.); National Institute of Neurological Disorders and Stroke grant R01NS113236 (E.K.); and National Health and Medical Research Council grants GNT1106471 and GNT1160315 and Australian Research Council grants FT1601100063 and DP200100347 (M.L.). R.G.G. is an HHMI Helen Hay Whitney Foundation Fellow. J.D.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: The authors declare there are no competing interests.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/373/6552/eabf8113/suppl/DC1
Figs. S1 to S14
Tables S1 and S2
Movies S1 to S11
Data and materials availability:
All data are available in the main text or the supplementary materials. The cryo-EM density map for the APOL3 lipoprotein nanodisc is available in the Electron Microscopy Databank (EMDB) with accession code EMD-24144.
REFERENCES AND NOTES
- 1.Randow F, MacMicking JD, James LC, Cellular self-defense: How cell-autonomous immunity protects against pathogens. Science 340, 701–706 (2013). doi: 10.1126/science.1233028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacMicking JD, Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol 12, 367–382 (2012). doi: 10.1038/nri3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schroder K, Hertzog PJ, Ravasi T, Hume DA, Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol 75, 163–189 (2004). doi: 10.1189/jlb.0603252 [DOI] [PubMed] [Google Scholar]
- 4.Zhang S-Y et al. , Inborn errors of interferon (IFN)-mediated immunity in humans: Insights into the respective roles of IFN-α/β, IFN-γ, and IFN-λ in host defense. Immunol. Rev 226, 29–40 (2008). doi: 10.1111/j.1600-065X.2008.00698.x [DOI] [PubMed] [Google Scholar]
- 5.Zhang S-Y et al. , Human inborn errors of immunity to infection affecting cells other than leukocytes: From the immune system to the whole organism. Curr. Opin. Immunol 59, 88–100 (2019). doi: 10.1016/j.coi.2019.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krausgruber T et al. , Structural cells are key regulators of organ-specific immune responses. Nature 583, 296–302 (2020). doi: 10.1038/s41586-020-2424-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knodler LA et al. , Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. U.S.A 107, 17733–17738 (2010). doi: 10.1073/pnas.1006098107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beuzón CR et al. , Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19, 3235–3249 (2000). doi: 10.1093/emboj/19.13.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen M et al. , Itaconate is an effector of a Rab GTPase cell-autonomous host defense pathway against Salmonella. Science 369, 450–455 (2020). doi: 10.1126/science.aaz1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith EE, Malik HS, The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res 19, 850–858 (2009). doi: 10.1101/gr.085647.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanhamme L et al. , Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 422, 83–87 (2003). doi: 10.1038/nature01461 [DOI] [PubMed] [Google Scholar]
- 12.Pérez-Morga D et al. , Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 309, 469–472 (2005). doi: 10.1126/science.1114566 [DOI] [PubMed] [Google Scholar]
- 13.Thurston TLM, Wandel MP, von Muhlinen N, Foeglein A, Randow F, Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 (2012). doi: 10.1038/nature10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strahl H, Hamoen LW, Membrane potential is important for bacterial cell division. Proc. Natl. Acad. Sci. U.S.A 107, 12281–12286 (2010). doi: 10.1073/pnas.1005485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontaine F et al. , APOLs with low pH dependence can kill all African trypanosomes. Nat. Microbiol 2, 1500–1506 (2017). doi: 10.1038/s41564-017-0034-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miki T, Hardt W-D, Outer membrane permeabilization is an essential step in the killing of gram-negative bacteria by the lectin RegIIIβ. PLOS ONE 8, e69901 (2013). doi: 10.1371/journal.pone.0069901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallo RL, Hooper LV, Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol 12, 503–516 (2012). doi: 10.1038/nri3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng YT et al. , The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol 183, 5909–5916 (2009). doi: 10.4049/jimmunol.0900441 [DOI] [PubMed] [Google Scholar]
- 19.Kim B-H et al. , A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–721 (2011). doi: 10.1126/science.1201711 [DOI] [PubMed] [Google Scholar]
- 20.Kim B-H et al. , Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat. Immunol 17, 481–489 (2016). doi: 10.1038/ni.3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shenoy AR et al. , GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–485 (2012). doi: 10.1126/science.1217141 [DOI] [PubMed] [Google Scholar]
- 22.Wandel MP et al. , Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat. Immunol 21, 880–891 (2020). doi: 10.1038/s41590-020-0697-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos JC et al. , Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat. Commun 11, 3276 (2020). doi: 10.1038/s41467-020-16889-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kutsch M et al. , Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. EMBO J 39, e104926 (2020). doi: 10.15252/embj.2020104926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayburt TH, Grinkova YV, Sligar SG, Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Lett 2, 853–856 (2002). doi: 10.1021/nl025623k [DOI] [Google Scholar]
- 26.Surewicz WK, Epand RM, Pownall HJ, Hui SW, Human apolipoprotein A-I forms thermally stable complexes with anionic but not with zwitterionic phospholipids. J. Biol. Chem 261, 16191–16197 (1986). doi: 10.1016/S0021-9258(18)66697-9 [DOI] [PubMed] [Google Scholar]
- 27.Gupta K et al. , Identifying key membrane protein lipid interactions using mass spectrometry. Nat. Protoc 13, 1106–1120 (2018). doi: 10.1038/nprot.2018.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta K et al. , The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 (2017). doi: 10.1038/nature20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keener JE et al. , Chemical additives enable native mass spectrometry measurement of membrane protein oligomeric state within intact nanodiscs. J. Am. Chem. Soc 141, 1054–1061 (2019). doi: 10.1021/jacs.8b11529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stokes JM et al. , Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol 2, 17028 (2017). doi: 10.1038/nmicrobiol.2017.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaara M, Polymyxins and Their Potential Next Generation as Therapeutic Antibiotics. Front. Microbiol 10, 1689 (2019). doi: 10.3389/fmicb.2019.01689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clairfeuille T et al. , Structure of the essential inner membrane lipopolysaccharide-PbgA complex. Nature 584, 479–483 (2020). doi: 10.1038/s41586-020-2597-x [DOI] [PubMed] [Google Scholar]
- 33.Brogden KA, Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol 3, 238–250 (2005). doi: 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- 34.Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ, Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov 19, 311–332 (2020). doi: 10.1038/s41573-019-0058-8 [DOI] [PubMed] [Google Scholar]
- 35.Sohlenkamp C, Geiger O, Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev 40, 133–159 (2016). doi: 10.1093/femsre/fuv008 [DOI] [PubMed] [Google Scholar]
- 36.Ernst WA et al. , Granulysin, a T cell product, kills bacteria by altering membrane permeability. J. Immunol 165, 7102–7108 (2000). doi: 10.4049/jimmunol.165.12.7102 [DOI] [PubMed] [Google Scholar]
- 37.Barman H et al. , Cholesterol in negatively charged lipid bilayers modulates the effect of the antimicrobial protein granulysin. J. Membr. Biol 212, 29–39 (2006). doi: 10.1007/s00232-006-0040-3 [DOI] [PubMed] [Google Scholar]
- 38.Stenger S et al. , An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 282, 121–125 (1998). doi: 10.1126/science.282.5386.121 [DOI] [PubMed] [Google Scholar]
- 39.van Meer G, Voelker DR, Feigenson GW, Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol 9, 112–124 (2008). doi: 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rausell A et al. , Common homozygosity for predicted loss-of-function variants reveals both redundant and advantageous effects of dispensable human genes. Proc. Natl. Acad. Sci. U.S.A 117, 13626–13636 (2020). doi: 10.1073/pnas.1917993117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaudet RG, Bradfield CJ, MacMicking JD, Evolution of Cell-Autonomous Effector Mechanisms in Macrophages versus Non-Immune Cells. Microbiol. Spectr 4, 10.1128/microbiolspec.MCHD-0050–2016 (2016). doi: 10.1128/microbiolspec.MCHD-0050-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamareddine L, Nakhleh J, Osta MA, Functional Interaction between Apolipophorins and Complement Regulate the Mosquito Immune Response to Systemic Infections. J. Innate Immun 8, 314–326 (2016). doi: 10.1159/000443883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zdybicka-Barabas A, Cytryńska M, Involvement of apolipophorin III in antibacterial defense of Galleria mellonella larvae. Comp. Biochem. Physiol. B 158, 90–98 (2011). doi: 10.1016/j.cbpb.2010.10.001 [DOI] [PubMed] [Google Scholar]
- 44.Figueira R, Watson KG, Holden DW, Helaine S, Identification of salmonella pathogenicity island-2 type III secretion system effectors involved in intramacrophage replication of S. enterica serovar typhimurium: Implications for rational vaccine design. mBio 4, e00065 (2013). doi: 10.1128/mBio.00065-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Helaine S et al. , Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343, 204–208 (2014). doi: 10.1126/science.1244705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karlsson AJ et al. , Engineering antibody fitness and function using membrane-anchored display of correctly folded proteins. J. Mol. Biol 416, 94–107 (2012). doi: 10.1016/j.jmb.2011.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chao Y, Vogel J, A 3′ UTR-Derived Small RNA Provides the Regulatory Noncoding Arm of the Inner Membrane Stress Response. Mol. Cell 61, 352–363 (2016). doi: 10.1016/j.molcel.2015.12.023 [DOI] [PubMed] [Google Scholar]
- 48.Gaudet RG et al. , Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science 348, 1251–1255 (2015). doi: 10.1126/science.aaa4921 [DOI] [PubMed] [Google Scholar]
- 49.Kong Q et al. , Effect of deletion of genes involved in lipopolysaccharide core and O-antigen synthesis on virulence and immunogenicity of Salmonella enterica serovar typhimurium. Infect. Immun 79, 4227–4239 (2011). doi: 10.1128/IAI.05398-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lazarou M et al. , The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015). doi: 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen TN et al. , Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol 215, 857–874 (2016). doi: 10.1083/jcb.201607039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shalem O et al. , Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). doi: 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li W et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). doi: 10.1186/s13059-014-0554-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiedemann C, Bellstedt P, Görlach M, CAPITO—A web server-based analysis and plotting tool for circular dichroism data. Bioinformatics 29, 1750–1757 (2013). doi: 10.1093/bioinformatics/btt278 [DOI] [PubMed] [Google Scholar]
- 55.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). doi: 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gallagher JR, Kim AJ, Gulati NM, Harris AK, Negative-Stain Transmission Electron Microscopy of Molecular Complexes for Image Analysis by 2D Class Averaging. Curr. Protoc. Microbiol 54, e90 (2019). doi: 10.1002/cpmc.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng SQ et al. , MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). doi: 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang K, Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12 (2016). doi: 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF et al. , UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). doi: 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 60.Gautier R, Douguet D, Antonny B, Drin G, HELIQUEST: A web server to screen sequences with specific α-helical properties. Bioinformatics 24, 2101–2102 (2008). doi: 10.1093/bioinformatics/btn392 [DOI] [PubMed] [Google Scholar]
- 61.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE, The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858 (2015). doi: 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hagemans D, van Belzen IAEM, Morán Luengo T, Rüdiger SGD, A script to highlight hydrophobicity and charge on protein surfaces. Front. Mol. Biosci 2, 56 (2015). doi: 10.3389/fmolb.2015.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. The cryo-EM density map for the APOL3 lipoprotein nanodisc is available in the Electron Microscopy Databank (EMDB) with accession code EMD-24144.