

Abstract
The SARS-CoV-2 papain-like protease (PLpro) is a target for antiviral drug development. It is essential for processing viral polyproteins for replication and functions in host immune evasion by cleaving ubiquitin (Ub) and ubiquitin-like protein (Ubl) conjugates. While highly conserved, SARS-CoV-2 and SARS-CoV PLpro have contrasting Ub/Ubl substrate preferences. Using a combination of structural analyses and functional assays, we identify a molecular sensor within the S1 Ub-binding site of PLpro that serves as a key determinant of substrate specificity. Variations within the S1 sensor specifically alter cleavage of Ub substrates but not of the Ubl interferon-stimulated gene 15 protein (ISG15). Significantly, a variant of concern associated with immune evasion carries a mutation in the S1 sensor that enhances PLpro activity on Ub substrates. Collectively, our data identify the S1 sensor region as a potential hotspot of variability that could alter host antiviral immune responses to newly emerging SARS-CoV-2 lineages.
Keywords: papain-like protease, PLpro, COVID-19, coronavirus, cysteine protease, deubiquitinase, ubiquitin, ISG15, DUB, SARS-CoV-2, SARS-CoV-1, K48-linked Ub
Graphical abstract
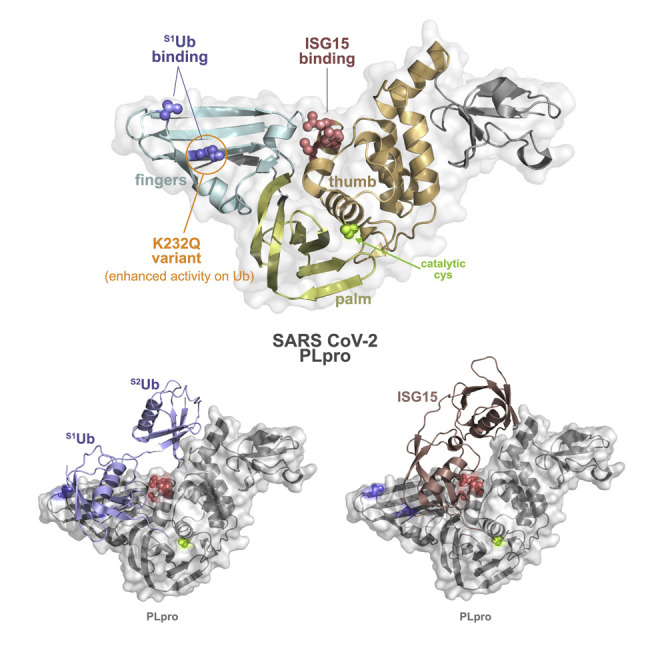
Patchett et al. use structural and biochemical methods to examine differential substrate preference between SARS-CoV and SARS-CoV-2 PLpro enzymes, pinpointing variant residues that determine substrate specificity. This study highlights the importance of studying PLpro variants in emerging SARS-CoV-2 lineages, as one such variant alters enzyme function.
Introduction
Following its initial emergence in Wuhan, China, severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2), the virus that causes coronavirus disease 2019 (COVID-19), has spread worldwide, with >100 million confirmed cases and >4 million COVID-19-related deaths (as of July 2021; https://www.who.int/). The COVID-19 pandemic marks the third emergence of a zoonotic coronavirus capable of causing severe disease in humans in the past 2 decades, with the SARS-CoV outbreak beginning in 2002 and the appearance of Middle East respiratory syndrome (MERS-CoV) a decade later. SARS-CoV-2 (SCoV-2) is highly similar to SARS-CoV (SCoV-1) in genome sequence and in disease physiology. However, SCoV-2 has a lower case-fatality rate and increased infectivity, leading to rapid and widespread transmission (Lu et al., 2020; Zhou et al., 2020). Although the recent approval and emergency deployment of SCoV-2 vaccines has helped curtail incidence, new variants of the virus have emerged that may be able to evade immune responses triggered by vaccines, and there remains an urgent need for antiviral therapeutics to combat COVID-19 in vulnerable populations.
Coronaviral proteases are attractive drug targets as their activity is essential for viral replication. Upon host cell entry, coronaviruses translate large polyproteins (pp1a and pp1ab) that encode the non-structural proteins (NSPSs) that direct viral genome replication (Ziebuhr, 2005). Processing of the polyproteins by viral proteases is required to release mature and functional NSPS. Papain-like protease (PLpro), a domain within nsp3, recognizes the P4–P1 consensus sequence LXGG located between nsp1/2, nsp2/3, and nsp3/4 (Harcourt et al., 2004; Han et al., 2005; Barretto et al., 2005). PLpro-catalyzed cleavage of the peptide bond after the P1 glycine releases nsp1, nsp2, and nsp3 and is required for viral replication. In addition to viral polyprotein processing, PLpro can recognize and hydrolyze isopeptide bonds following the LXGG motif at the C terminus of cellular ubiquitin (Ub) and the ubiquitin-like protein (Ubl) ISG15 (interferon-induced gene 15) (Lindner et al., 2005; Barretto et al., 2005; Lindner et al., 2007). As both Ub and ISG15 modifications play important roles in the immune response to viral infection (Davis and Gack, 2015; Perng and Lenschow, 2018; Dzimianski et al., 2019), their PLpro-catalyzed deconjugation has been shown to modulate the innate immune response to viral infection and affect viral pathogenesis (Devaraj et al., 2007; Frieman et al., 2009; Clementz et al., 2010; Bailey-Elkin et al., 2014; Mielech et al., 2015; Ratia et al., 2014; Sun et al., 2012). Consequently, targeting PLpro would impede viral replication and may also promote host immune function. Thus, the structural and molecular characterizations of SCoV-2 PLpro will be important to facilitate drug design and targeting.
Previous work on SCoV-1 PLpro demonstrates an “open hand” architecture characteristic of deubiquitylating enzymes (DUBs) (Ratia et al., 2006). However, whereas most DUBs cleave polyubiquitin by binding to di-Ub across the active site, recognizing a single Ub moiety via an enzymatic S1 site (Mevissen and Komander, 2017), PLpro binds Ub/Ubl substrates at 2 distinct sites (S1 and S2), providing specificity for K48-polyubiquitin and for the structurally similar ISG15 (Chou et al., 2014; Ratia et al., 2014; Békés et al., 2015, 2016). SCoV-2 PLpro amino acid sequence is 83% identical to SCoV-1 (Figure S1A), and structural studies demonstrate high similarity in overall structure and substrate binding (Shin et al., 2020; Rut et al., 2020; Klemm et al., 2020). Our lab and others have shown that SCoV-2 PLpro proteolytic activity is similar to SCoV-1 as both enzymes are active on ISG15 as well as Ub substrates, and both enzymes prefer K48- over K63-linked Ub chains (Rut et al., 2020, Shin et al., 2020; Klemm et al., 2020; Freitas et al., 2020). Despite these similarities, the enzymes differ in substrate preference. While SCoV-1 PLpro preferentially cleaves K48-Ub chains (Lindner et al., 2007; Ratia et al., 2014; Békés et al., 2015, 2016), SCoV-2 PLpro preferentially cleaves ISG15 substrates (Rut et al., 2020; Shin et al., 2020; Klemm et al., 2020; Freitas et al., 2020), and the molecular determinants underlying this difference remain unclear. As differences in substrate preference are likely to influence how PLpro modulates the immune response during coronaviral infection, it is critical to understand the molecular determinants that dictate differences in enzymatic activity between SCoV-1 and SCoV-2 PLpro.
Here, we sought to determine the molecular basis underlying ISG15 and Ub substrate preferences of SCoV-1 and SCoV-2 PLpro. Using structural and biochemical methods, we pinpointed variant residues in the S1 Ub-binding site responsible for differential substrate preference. Notably, currently circulating SCoV-2 variants of concern (VOCs) carry mutations in PLpro, including a K232Q substitution in the S1 Ub sensor region. We demonstrate that K232Q confers enhanced K48-Ub chain cleavage and is likely to affect the landscape of K48-linked ubiquitin modifications in infected cells, validating the importance of studies that characterize PLpro activity and substrate specificity for therapeutic considerations.
Results
Structural comparison of inhibitor-bound SCoV-1 and SCoV-2 PLpro
To explore the difference in SCoV-1 and SCoV-2 PLpro substrate preference, we set out to determine whether there are differences in how each PLpro active site engages with substrates. To this end, we determined crystal structures of SCoV-1 PLpro in complex with our recently discovered covalent peptide inhibitors VIR250 and VIR251 (Rut et al., 2020). We previously determined structures of VIR250 and VIR251 in complex with SCoV-2 PLpro and reasoned that a direct comparison of SCoV-1 and SCoV-2 PLpro active sites bound to the same peptide substrates would provide an ideal opportunity to identify differences within the active sites. The structure of SCoV-1 PLpro in complex with VIR250 (Figure 1 A) (PDB: 7LFU) was determined by molecular replacement using the structure of VIR250-bound SCoV-2 PLpro (PDB: 6WUU) and was resolved to 2.29 Å with R/Rfree values of 0.224/0.265 (Table S1). This structure was used as the molecular replacement search model for the determination of the SCoV-1 PLpro structure in complex with VIR251 (Figure 1A) (PDB: 7LFV). The SCoV-1 PLpro/VIR251 structure was resolved to 2.23 Å and refined to R/Rfree values of 0.197/0.235 (Table S1). Overlay of the SCoV-1 PLpro-VIR250/VIR251 structures with the equivalent SCoV-2 structures shows that, except for a small rotation in the fingers domain, the 4 molecules are very similar, and electron density maps show that inhibitors are placed unambiguously in the models (Figure S1). Comparison of inhibitor-bound SCoV-1 PLpro structures to the corresponding SCoV-2 structures shows that organization of the active site is highly conserved (Figure 1B). Each inhibitor is composed of 4 amino acid residues and a vinylmethyl ester (VME) reactive group at position P1, as previously described (Rut et al., 2020) (Figures 1B and S1). P3-P1 positions of VIR250 and VIR251 are identical (Dap-Gly-GlyVME), while the P4 position comprises an Abu(Bth) in VIR250 and an hTyr residue in VIR251. As expected, VIR250/VIR251 bind to the catalytic pocket with a covalent bond between the P1 GlyVME and the catalytic cysteine Cys112 of SCoV-1 PLpro (Figure 1B). The P3-P1 residues engage in a nearly identical network of backbone-mediated interactions with the S3-S1 sites of the PLpro active site in SCoV-1 and SCoV-2 PLpro structures, including a fully conserved network of hydrogen bonds between the P3 Dap and Tyr265, Tyr269, and Gly272 of PLpro, P2 Gly, and Gly164 of PLpro, and P1 Gly with Gly272 of PLpro (Figure 1B).
Figure 1.

Structural comparison of SCoV-1 and SCoV-2 PLpro in complex with inhibitors
(A) Crystal structure of SCoV-1 PLpro-VIR250 complex (left) and SCoV-1 PLpro-VIR251 complex (right) with PLpro shown as illustration and VIR250 and VIR251 as spheres.
(B) Comparison of SCoV-1 and SCoV-2 catalytic sites in complex with VIR250 and VIR251. P4-P1 residues of VIR250/251 are indicated with boxes and arrows.
(C) Enlarged view of SCoV-1 residue Trp107 (W107) (left 2 panels) and SCoV-2 PLpro Trp106 (W106) (right 2 panels) and the P1 GlyVME of inhibitors.
(D) Enlarged view of SCoV-1 residue Tyr269 (Y269) (left panels) and SCoV-2 PLpro Tyr268 (Y268) (right panels) and the P4 Abu(Bth) of VIR250 or hTyr of VIR251.
(E) Enlarged view of the P4 aromatic rings of VIR250 and VIR251 in SCoV-1 and SCoV-2 PLpro-inhibitor complexes with additional SCoV-2 PLpro-inhibitor complexes aligned and indicated in yellow. Two partially overlapping P4 conformations are apparent.
The highly conserved manner in which SCoV-1 and SCoV-2 PLpro active sites engage VIR250 and VIR251 peptide inhibitors suggests that differences in substrate specificity are not the result of differences in the active site. However, we observed several notable differences in SCoV-1 and SCoV-2 PLpro complexes that highlight active site plasticity. The Trp107 side chain of PLpro (Trp106 in SCoV-2), which is proposed to stabilize the oxyanion hole to facilitate catalysis (Ratia et al., 2008, 2014), adopts different conformations in the 4 PLpro-inhibitor structures (Figures 1B and 1C). In the SCoV-2/VIR250 and VIR251 structures, the Trp106 side chain projects toward the thumb domain and participates in hydrogen bonds with the VIR251 VME group in SCoV-2/VIR251. In contrast, in SCoV-1 structures, the Trp107 side chain points down toward β strands in the palm domain, >6 Å away from VME (Figures 1B and 1C). Given the proximity of Trp106/107 to the S1′ site and P1′ position of a bona fide substrate, the observed differences in conformation suggest that the oxyanion hole may exist in various active and inactive conformations, depending on substrate binding status and step of reaction. A second region of variability is at the P4 site of inhibitors. In SCoV-2 structures, the P4 Abu(Bth) and hTyr side chains of VIR250 and VIR251 project into opposite parts of a broad and structurally variable PLpro S4 pocket (Figures 1B and 1D). Within the pocket, Tyr268 of SCoV-2 PLpro projects away from the P4 side chain, regardless of the different Abu(Bth) and hTyr conformations (Figures 1B and 1D). Similar to SCoV-2 structures, the P4 side chains of inhibitors project into opposite parts of the SCoV-1 PLpro S4 pocket (Figures 1B and 1D). However, Abu(Bth) from SCoV-1/VIR250 superimposes with hTyr from SCoV-2/VIR251 and hTyr from SCoV-1/VIR251 superimposes with Abu(Bth) from SCoV-2/VIR250 (Figures 1B and 1D). Furthermore, where the PLpro Tyr268 side chain projects away from the P4 inhibitor site in SCoV-2 structures, the corresponding Tyr269 side chain instead projects toward the P4 site in both SCoV-1 structures (Figure 1B and 1D). In the SCoV-1 PLpro/VIR251 structure, Tyr269 projects toward P4 hTyr and engages in direct contacts with hTyr and Pro248 and Pro249 of the S4 pocket. In the SCoV-1 PLpro/VIR250 structure, the Tyr269 side chain adopts a different rotamer but also projects toward P4 hTyr, engaging in direct contacts with Abu(Bth) and the VIR250 N terminus (Figures 1B and 1D).
In summary, structural variations at Trp107/106 and Tyr269/268 demonstrate the plasticity of PLpro active sites, and the conformational variability of the P4 side chain of inhibitors suggests a degree of flexibility in the substrate binding mode of PLpro. We previously hypothesized that the space occupied by different conformations of the P4 ring could be used in the refinement of inhibitors (Rut et al., 2020). Consistent with this, rings of inhibitors in recent PLpro structures (Gao et al., 2021; Fu et al., 2021; Osipiuk et al., 2021; Shan et al., 2021) partially overlap with the P4 ring conformations in our structures (Figure 1E), indicating that our results may facilitate the drug development of PLpro inhibitors.
The thumb domain drives specific interaction between PLpro and ISG15
To understand the apparent preference of SCoV-2 PLpro for ISG15 substrates, we overlaid a structure of SCoV-2 PLpro bound to ISG15 (Shin et al., 2020) with our previous structure of SCoV-1 PLpro bound to diUbK48 (Békés et al., 2016) and compared the ISG15 position with the predicted position of diUbK48 on SCoV-2 PLpro (Figure 2 A). The distal S2Ub and the N-terminal ISG15 domain share an interaction surface on PLpro, as PLpro residues interfacing with ISG15 also make predicted interactions with S2Ub. Similarly, the majority of PLpro residues interfacing with C-terminal ISG15 also interact with S1Ub. However, 3 residues along the α7 helix in the PLpro thumb domain make predicted interactions with ISG15, but not with Ub: Ser170, Tyr171, and Gln174 (Figure 2A). To determine whether these residues were important for interaction with ISG15, we generated a triple mutant, changing each residue to Ala. We compared this S1 site mutant (S170A, Y171A, Q174A) to an S2 mutant (F69S, E70K, H73G) and to catalytic-dead PLpro (C111S) as controls (Figure 2A). S2 mutant residues are conserved between SCoV-1 and SCoV-2 and were previously shown to mediate interaction between PLpro and K48-linked Ub chains, but these residues were less important for ISG15 interaction (Békés et al., 2015, 2016). As predicted, the S2 mutant was active on mono-Ub substrates (Figures 2B and 2C) but deficient in K48-linked Ub chain cleavage (Figure 2D). In contrast, the S1 mutant was active on mono-Ub (Figures 2B and 2C) and on K48-Ub chains, with only a minor reduction in tetra-UbK48 cleavage (Figure 2D). We next tested activity on ISG15-VS and found that the S1 mutant was deficient in ISG15-VS labeling (Figure 2E), while the S2 mutant was labeled by ISG15-VS (Figure 2F). Using a more sensitive and quantitative approach, we compared PLpro activity on the fluorogenic substrate ISG15-AMC. Consistently, we saw no difference between wild-type (WT) PLpro and the S2 mutant; however, activity on ISG15-AMC was significantly reduced in the S1 mutant (Figure 2G). Similar to what we previously found in our analysis of SCoV1-PLpro (Békés et al., 2015, 2016), this demonstrates that recognition of ISG15 by SCoV-2 PLpro is more dependent on interactions within S1 compared to S2. This also complements recent work by the Komander lab in which mutations in the SCoV-2 PLpro thumb domain (A156E and Y171R) reduced activity on ISG15 substrates (Klemm et al., 2020). These results map out an interaction surface on the PLpro thumb domain that is an important driver of ISG15 interaction.
Figure 2.

The thumb domain drives specific interaction between SCoV-2 PLpro and ISG15
(A) SCoV-1 PLpro-diUbK48 (PDB 5E6J) aligned with SCoV-2 PLpro-ISG15 (PDB 6YVA). Residues targeted for mutational analysis indicated by colored spheres.
(B) Ub-VS labeling of SCoV-2 WT and mutant PLpro.
(C) Initial velocity (Vi) of AMC release from Ub-AMC with the indicated PLpro enzymes. Error bars represent SDs (n = 3 independent experiments).
(D) Cleavage 3UbK48 chains by the indicated WT and mutant SCoV-2 PLpro.
(E and F) ISG15-VS labeling of the indicated WT and mutant PLpro enzymes.
(G) Michaelis-Menten kinetics comparing ISG15-AMC hydrolysis by the indicated PLpro enzymes. Error bars represent SDs (n = 3 independent experiments).
All of the experiments were repeated at least 3 times independently, with similar results.
Variations in the PLpro finger domain drive differential activity on Ub substrates
We next focused on the interface between PLpro and Ub, hypothesizing that the primary difference between SCoV-1 and SCoV-2 PLpro was in their activity on Ub substrates rather than an enhanced affinity of SCoV-2 for ISG15. This was based on several lines of evidence: (1) our lab and others have found substantially diminished K48-linked polyubiquitin cleavage in SCoV-2 compared to SCoV-1 PLpro (Rut et al., 2020; Shin et al., 2020) (Figure S2A). (2) In mono-Ub assays in which Ub binds only at the S1 site, SCoV-2 PLpro activity was also significantly diminished compared to SCoV-1 (Figures S2B and S2C), suggesting that the difference in activity stems from the S1 site. (3) We saw comparable SCoV-1 and SCoV-2 PLpro activity on ISG15 substrates (Figures S2D and S2E), and similar catalytic efficiencies on ISG15 have been reported for both enzymes (Shin et al., 2020). These results suggest that the predominant difference between PLpro enzymes is in their activity on Ub, in which cleavage of Ub substrates is diminished in SCoV-2 compared to SCoV-1. Thus, we set out to determine whether differences at the Ub-PLpro interface accounted for the differential substrate specificity.
We used our structure of SCoV-1 PLpro-diUbK48 to identify residues at the S1Ub and S2Ub interfaces (Figure 3 A) and compared these to the corresponding residues in SCoV-2 (Figure 3B). The S1Ub interface was conserved with the exception of 2 residues located in the PLpro finger domain: Val226 and Gln233 (Thr225 and Lys232 in SCoV-2) (Figure 3B). We overlaid SCoV-1 PLpro-diUbK48 on the SCoV-2 PLpro-ISG15 structure, and, while the finger region is not resolved in SCoV-2 PLpro, differential binding of S1Ub and the ISG15 C-terminal domain is apparent as Ub associates with the SCoV-1 finger domain and ISG15 is shifted toward the thumb domain (Figure 3C). Based on this, we reasoned that variants in the finger domain could specifically affect Ub association, explaining the dramatic difference in activity on Ub substrates but not on ISG15. To test this, we mutated SCoV-2 residues Thr225 and Lys232 to mimic the S1 finger region of SCoV-1 (SCoV2-T225V, K232Q). Separately, we mutated SCoV-2 residues Thr75 and Asp179 at the S2 interface to mimic the S2 site of SCoV-1 (SCoV2-T75L, D179E). We compared WT and mutant SCoV-2 PLpro activity on the S1 substrate Ub-VS. Strikingly, the S1 finger mutant (SCoV2-T225V, K232Q) dramatically improved labeling by Ub-VS compared to WT, while the S2 mutant (SCoV2-T75L, D179E) had no impact (Figure 3D). The change in activity was specific to Ub as there was no difference in ISG15-VS binding (Figure 3E). Next, we tested the opposite mutations in SCoV-1 PLpro to determine whether mutant SCoV-1 would behave like SCoV-2 PLpro. The SCoV-1 S1 finger mutant (SCoV1-V226T, Q233K) exhibited diminished Ub-VS labeling and the S2 mutant (SCoV1-L76T, E180D) had no impact (Figure 3F). To gain kinetic insight, we measured cleavage of the fluorogenic substrate Ub-AMC. Interestingly, SCoV2-T225V, K232Q improved Ub-AMC cleavage activity to be not only similar but also faster than SCoV-1 PLpro. The opposite effect was seen in SCoV-1 PLpro: S1 finger mutant activity was reduced below WT SCoV-2 (Figure 3G), implying that additional residues may participate in allosteric interactions that influence Ub association. We next tested cleavage of K48-linked polyubiquitin, which requires binding across S1-S2 sites. In both SCoV-1 and SCoV-2 PLpro assays, S2 mutations had no detectable impact on tetra-UbK48 cleavage (Figure 3H). Instead, and consistent with Ub-VS results, tetra-UbK48 cleavage was reduced in SCoV1-V226T,Q233K compared to WT PLpro and improved in SCoV2-T225V,K232Q compared to WT (Figure 3H). These results are quite striking, as it was recently shown that a T75L point mutation in the S2 site of SCoV-2 PLpro improved cleavage of diUbK48-AMC; however, not to the extent of cleavage by SCoV-1 PLpro (Shin et al., 2020). Likely, our gel-based cleavage assay was not sensitive enough to detect the relatively modest improvement conferred by T75L. In contrast, the improvement in Ub chain cleavage by the S1 mutant (T225V,K232Q) was dramatic and easily detected in our assays (Figure 3H). Thus, while the S2 site may contribute to differential substrate preference to a degree, the predominant driver of Ub substrate preference is the S1 site finger domain.
Figure 3.

Variations in the finger domain are responsible for differential activity on ubiquitin substrates
(A) Crystal structure of SCoV-1 PLpro-diUbK48 (PDB 5E6J) with the S1Ub-PLpro interface highlighted in green and the S2Ub interface highlighted in magenta.
(B) Sequence alignment of SCoV-1 and SCoV-2 PLpro with secondary structure shown above sequence. Conserved residues indicated as dots. Residues at S1 and S2 interfaces are indicated by green or magenta outlining, respectively. Variant residues are highlighted.
(C) SCoV-1 PLpro-diUbK48 aligned with SCoV-2 PLpro-ISG15 (PDB 6YVA), enlarged to compare binding of S1Ub (blue) and C-terminal Ubl domain of ISG15 (red). SCoV-1 PLpro residues V226 and Q233 are shown in stick representation (green).
(D) Ub-vinyl sulfone (Ub-VS) labeling of SCoV-2 PLpro mutants.
(E) ISG15-VS labeling of SCoV-2 PLpro mutants.
(F) Ub-VS labeling of SCoV-1 PLpro mutants.
(G) Michaelis-Menten kinetics comparing WT and mutant SCoV-1 and SCoV-2 PLpro on Ub-AMC. Error bars represent SDs (n = 3 independent experiments).
(H) Cleavage of K48-linked tetra-Ub chains (3UbK48) by SCoV-1 (left panel) and SCoV-2 (right panel) WT and mutant PLpro.
(I and J) Lysates prepared from HEK293T cells transiently transfected with empty vector or the indicated PLpro and analyzed by SDS-PAGE and western blotting.
(K) Electrostatic representation of SCoV-1 (top) and SCoV-2 (bottom) PLpro in complex with ubiquitin. Enlarged view (far left) shows the difference in electrostatic surface at SCoV-1 PLpro Gln233 and SCoV-2 PLpro Lys232. Open book views (right) highlight the neutral surface of Ub (Ala46-Gly47) and SCoV-1 Gln233/SCoV-2 Lys232 surfaces.
All of the experiments in (D)–(J) were repeated at least 3 times independently, with similar results.
To test the ability of PLpro to cleave intracellular K48-Ub chains, we transfected HEK293T cells with PLpro-FLAG constructs and analyzed cell lysates by western blotting for K48-specific Ub. Cleavage of K48-Ub was observed as a reduction in high-molecular-weight K48-Ub chains in cells expressing PLpro compared to empty vector (Figure 3I). Consistent with our in vitro cleavage analysis, K48-Ub chains were reduced significantly more in cells expressing SCoV-1 PLpro compared to SCoV-2 (Figure 3I). In cells with the SCoV-1 S1 finger mutant, the level of K48-Ub chains was greater than WT SCoV-1 and comparable to SCoV-2 PLpro (Figure 3I), indicating a reduced ability of the mutant to cleave Ub chains. In contrast, the opposite mutation of SCoV-2 PLpro improved cleavage, visualized by a strong reduction in K48-Ub chains compared to WT SCoV-2 (Figure 3J). These results identify variants in the finger domain of the SCoV-2 PLpro S1 site that are responsible for its diminished activity on Ub substrates compared to SCoV-1, further supporting the idea that the principal difference between SCoV-1 and SCoV-2 PLpro activity is driven by the S1 Ub-binding site.
Recently, a structure of SCoV-2 PLpro bound to mono-Ub in the S1 site showed that S1Ub does in fact associate with the finger domain of SCoV-2 PLpro (Klemm et al., 2020). The exact mechanism by which Val226/Thr225 and Gln233/Lys232 influence this interaction is not known. While Val226 and Thr225 differ in hydrophobicity, they do not directly interact with any hydrophobic residues on Ub. In contrast, differing electrostatic properties between Lys232 and Gln233 offer some mechanistic insight. Comparison of the SCoV-2-Ub structure with SCoV-1-Ub (Ratia et al., 2014) shows Lys232/Gln233 positioned to interact with a neutral surface of Ub (Ala46-Gly47) (Figure 3K). The relatively neutral surface of SCoV-1 Gln233 may be advantageous compared to the positively charged Lys232 on SCoV-2 PLpro.
The K232Q variant enhances SCoV-2 PLpro activity on Ub substrates
In late 2020, variants of SARS-CoV-2 emerged that pose an increased risk to global public health and have been characterized as VOCs. The current designated VOCs include Alpha (lineage B.1.1.7), Beta (lineage B.1.351), Gamma (lineage P.1), and, more recently, Delta (lineage B.1.617.2). While the focus has been on Spike protein mutations, lineage-defining amino acid substitutions are found in the PLpro domain of Alpha, Beta, and Gamma. Alpha, identified in the United Kingdom, and Beta, identified in South Africa, include PLpro substitutions A145D (Rambaut et al., 2020) and K92N (Tegally et al., 2020), respectively. A145 and K92 are located in close proximity in the PLpro structure on a distinct surface where Ub and ISG15 binding is not known to occur (Figure 4 A). Strikingly, Gamma, the Brazilian VOC associated with increased transmissibility and re-infection (Faria et al., 2021; Naveca et al., 2020; Garcia-Beltran et al., 2021), includes the K232Q mutation in the PLpro fingers domain (Figure 4A). Because the same mutation was included in our T225V,K232Q double mutant that enhanced activity on Ub substrates (Figure 3), we wanted to test whether K232Q alone could affect PLpro activity. We purified SCoV-2 PLpro mutants harboring each of the VOC-associated substitutions and tested their activity on Ub-VS. The K232Q mutant improved labeling by Ub-VS compared to WT, while K92N and A145D had no impact (Figure 4B). This was specific to Ub-VS, as there was no difference in ISG15-VS labeling (Figure 4C). To analyze K232Q more quantitatively, we compared its activity on Ub-AMC to WT SCoV-2 and SCoV-1 PLpro. K232Q enhanced Ub-AMC cleavage compared to WT SCoV-2, with activity similar to SCoV-1 (Figure 4D). We next tested VOC mutant activity on K48-Ub chains and found that while all of the mutants could cleave tetra-UbK48, cleavage by the K232Q mutant was enhanced (Figure 4E). We also tested cleavage of K48-Ub chains in HEK293T cells following transfection with PLpro expression constructs. Western blotting for K48-Ub demonstrated that like the previously analyzed double mutant (T225V,K232Q) the K232Q mutant significantly improved cleavage of K48-Ub chains (Figure 4F).
Figure 4.

The K232Q variant enhances SCoV-2 PLpro activity on Ub substrates
(A) SCoV-2 PLpro-Ub complex (PDB: 6XAA) with ISG15 from the aligned SCoV-2 PLpro-ISG15 complex (PDB: 6YVA). Residues mutated in variants of concern (VOCs) are shown in stick representation. Residue K232 (green) in the S1 Ub site is mutated in VOC Gamma (K232Q). K92 and A145 (cyan) are mutated in VOCs Alpha (A145D) and Beta (K92N).
(B) Ub-vinyl sulfone (Ub-VS) labeling of PLpro VOC mutants.
(C) ISG15-VS labeling of PLpro VOC mutants.
(D) Michaelis-Menten kinetics comparing K232Q PLpro mutant activity to WT SCoV-1 and SCoV-2 PLpro on Ub-AMC. Error bars represent SDs (n = 3 independent experiments).
(E) Cleavage of K48-linked tetra-Ub chains (3UbK48) by SCoV-2 PLpro VOC.
(F) Lysates prepared from HEK293T cells transiently transfected with empty vector or the indicated PLpro and analyzed by SDS-PAGE and western blotting.
(G) Schematic representation of mammalian expression constructs with molecular masses. Triangles indicate sites of PLpro-catalyzed cleavage.
(H) Processing of HA-nsp123-FLAG in HEK283T cells following transient transfection with indicated plasmids. WT processing is demonstrated by comparison to full-length catalytic-dead mutant (C/S) and HA-nsp1 and nsp3-FLAG (left panels). VOC mutants in HA-nsp123-FLAG compared to WT, S2 mutant (F69S, E70K, H73G) and empty vector (ev) (right panels). UbK48 chain cleavage analyzed by western blotting for K48-specific Ub (bottom panel).
All of the experiments in (B)–(H) were repeated at least 3 times independently, with similar results.
As SARS-CoV-2 infection depends on the ability of PLpro to process the viral polyprotein and release mature nsp1, nsp2, and nsp3, we tested the ability of PLpro VOC mutants to “self-cleave” in the context of the polyprotein. To test this, we generated an expression construct consisting of SCoV-2 polyprotein sequence from nsp1 through nsp3 with an N-terminal hemagglutinin (HA) tag and C-terminal FLAG tag to track cleavage products (HA-nsp123-FLAG). HA-nsp1 and nsp3-FLAG constructs were used as controls to represent polyprotein cleavage products (Figure 4G). Constructs containing nsp3 were truncated upstream of the trans-membrane region and the final “LKGG” cleavage site. Following transient transfection in HEK293T cells, HA-nsp123-FLAG self-cleavage was evident by western blotting, as both HA-nsp1 and nsp3-FLAG cleavage products were detected in cells transfected with HA-nsp123-FLAG (Figure 4H, left panels). In contrast, the catalytic-dead mutant (C/S) migrated at a higher molecular weight in western blots against both FLAG and HA tags, consistent with unprocessed HA-nsp123-FLAG (Figure 4H). We next introduced point mutations into the HA-nsp123-FLAG construct to test the ability of the VOC mutants to self-cleave. We also included the S2 mutant (SCoV2-F69S, E70K, H73G) that disrupted K48-Ub chain cleavage. Similar to WT, HA-nsp1 and nsp3-FLAG cleavage products were detected in all of the mutants, with the exception of the catalytic-dead full-length control (Figure 4H, right panels), demonstrating that all mutants are active in the context of the polyprotein. The ability of the K232Q and S2 mutants to self-cleave also suggests that polyprotein cleavage may occur by a mechanism independent of the PLpro regions critical for Ub association. Importantly, the ability of PLpro mutants to affect activity on K48-linked Ub was retained in the context of the polyprotein. In cells expressing HA-nsp123-FLAG containing the VOC mutant K232Q, the level of K48-Ub chains was reduced compared to WT, indicating improved K48-chain cleavage (Figure 4H, bottom panel). In contrast, the level of K48-chains was increased in the S2 mutant, consistent with the ability of the S2 mutant to disrupt K48-Ub chain cleavage (Figure 4H, bottom panel). Based on this work, mutations in the SARS-CoV-2 PLpro domain have the potential to affect PLpro substrate preference during infection. Our work also suggests that the already-circulating K232Q variant may have an enhanced ability to target K48-linked polyubiquitin substrates, which could in turn influence the host immune response in infected cells.
Discussion
While the sequence identity of SCoV-1 and SCoV-2 PLpro active sites is strictly conserved, our direct structural comparison of SCoV-1 and SCoV-2 PLpro bound to the same set of inhibitors demonstrates active site plasticity and shows structural malleability of the oxyanion hole, which is noteworthy given its crucial role in promoting catalysis. Conformational variability in the P4 side chain of inhibitors VIR250/VIR251 also suggests a degree of flexibility in substrate binding. Alternating P4 conformations and corresponding changes at the S4 pocket were noted in our previous VIR250/VIR251 work (Rut et al., 2020), and the same pocket was later noted to be a hotspot for drug discovery as the inhibitor GRL0617 binds to this site (Fu et al., 2021). Overall, our structures provide insight into the ensemble of conformations that this region of PLpro can adopt, which will guide future drug design.
Despite the observed plasticity, SCoV-1 and SCoV-2 PLpro active sites engage VIR250/VIR251 in a highly conserved manner, and our previous activity profiling indicates virtually identical substrate specificity at the active site (Drag et al., 2008; Rut et al., 2020). However, it is clear that PLpro substrate specificity is not only defined by binding in the active site but also by various allosteric binding surfaces on PLpro. Our biochemical and mutational analyses confirm that the S1 binding site is critical for activity on both Ub and ISG15, but that the association of each substrate with PLpro is driven by distinct regions of the S1 site. Whereas mutation of the α7 helix in the thumb domain diminished activity on ISG15 substrates, mutations in the PLpro fingers domain specifically modulated activity on Ub substrates. We also demonstrated that although SCoV-2 PLpro structure is highly similar to SCoV-1, variant residues in the fingers domain cause differential substrate preference. Previous work by our lab and others demonstrated that unlike SCoV-1 PLpro, which has a preference for K48-Ub chains (Békés et al., 2015, 2016; Lindner et al., 2007; Ratia et al., 2014), the preferred substrate for SCoV-2 PLpro is ISG15 (Klemm et al., 2020; Shin et al., 2020; Rut et al., 2020; Freitas et al., 2020). Our work here is consistent with this, but mechanistically explains the difference in substrate preference. Rather than an enhanced affinity for ISG15, SCoV-2 PLpro exhibited significantly diminished activity on both K48-linked Ub and mono-Ub substrates compared to SCoV-1, and our mutational analysis demonstrated that subtle changes to the primary amino acid sequence in the PLpro finger domain could modulate PLpro activity on both mono-Ub and K48-linked polyUb substrates. Thus, the differences in K48-Ub deconjugation that have been observed between SCoV-1 and SCoV-2 PLpro can be explained by differences in the S1 Ub binding site.
Differences in PLpro substrate preference are intriguing, given the roles of K48-Ub and ISG15 modifications in the evasion of the host immune response. While differing cellular interactomes have been established for SCoV-1 and SCoV-2 PLpro (Shin et al., 2020), the identities of bona fide ubiquitin/ISG15-conjugated PLpro targets whose cleavage is essential for viral pathogenicity remain to be defined. It will be interesting to determine whether PLpro targets cellular substrates to dampen the host immune response or whether PLpro cleaves Ub/ISG15 from viral proteins as a mechanism to evade the immune response. Thus, it will be important to define the ubiquitylated and ISGylated proteome of cells during infection and to define how differences in PLpro substrate preference affect viral life cycle and pathogenesis.
Currently, four SCoV-2 VOCs are circulating around the globe, raising fears of increased transmissibility and re-infection. Evidence is rapidly growing that VOCs may be able to evade immune responses triggered by vaccines or by previous infection (Andreano et al., 2020; Cele et al., 2021; Xie et al., 2021; Wang et al., 2021; Naveca et al., 2020; Garcia-Beltran et al., 2021). While the focus of VOC research has been on mutations in the Spike protein, VOCs also harbor mutations in PLpro. The VOCs Alpha, Beta, and Gamma each possess a different lineage-defining amino acid substitution in PLpro. We showed that the K232Q mutation present in the Gamma variant enhances cleavage of Ub substrates, which will undoubtedly have important ramifications in how PLpro influences host immune responses during SARS-CoV-2 infection. While the A145D mutation found in the Alpha variant and K92N mutation in the Beta variant did not significantly affect PLpro activity on Ub or ISG15 substrates, both substitutions alter charge in the same region of PLpro (Figure 4A), making it likely that these variants are functionally significant. Although we found that A145D and K92N mutants could self-cleave in the context of the polyprotein in cells, it will be important to develop a more sensitive in vitro assay to determine whether these mutations or others cause changes in the kinetics of viral polyprotein cleavage. The fourth VOC, Delta, is not defined by any PLpro mutations; however, ∼20% of Delta variant sequences currently available contain a P77L mutation in PLpro (Gong et al., 2020; Zhao et al., 2020). Given the continuous evolution of SARS-CoV-2 and the possibility that vaccines may not be universally effective against variants or new coronaviruses that emerge in the future, it will be critical to study PLpro mutations that may continue to arise and to determine how variants affect the landscape of K48-Ub- and ISG15-modified substrates in cells during infection.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG (M2) | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Rabbit polyclonal anti-HA.11 Epitope Tag | Biolegend | Cat# 902301; RRID: AB_2565018 |
| Rabbit monoclonal anti-Ubiquitin, Lys48-Specific | Millipore | Cat# 05-1307; RRID: AB_1587578 |
| Mouse monoclonal anti-alpha-Tubulin | Millipore | Cat# CP06; RRID: AB_2617116 |
| Bacterial and virus strains | ||
| BL21(DE3) | New England Biolabs | Cat# C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| Ub-VS, Recombinant Human Ubiquitin Vinyl Sulfone | BostonBiochem | Cat# U-202 |
| Ub-AMC, Recombinant Human Ubiquitin AMC | BostonBiochem | Cat# U-550 |
| Tri-Ub Chains (K48-linked) | BostonBiochem | Cat# UC-215B |
| ISG15-VS, Recombinant Human ISG15 Vinyl Sulfone | BostonBiochem | Cat# UL-603 |
| ISG15-AMC, Recombinant Human ISG15 AMC | BostonBiochem | Cat# UL-553 |
| FuGENE6 Transfection Reagent | Promega | Cat# E2691 |
| VIR250 peptide inhibitor | Rut et al., 2020 | N/A |
| VIR251 peptide inhibitor | Rut et al., 2020 | N/A |
| Deposited data | ||
| SARS-CoV-1 PLpro/VIR250 | This study | PDB ID: 7LFU |
| SARS-CoV-1 PLpro/VIR251 | This study | PDB ID: 7LFV |
| SARS-CoV-2 PLpro/VIR250 | Rut et al., 2020 | PDB ID: 6WUU |
| SARS-CoV-2 PLpro/VIR251 | Rut et al., 2020 | PDB ID: 6WX4 |
| SARS-CoV-2 PLpro/Compound S43 | Shan et al., 2021 | PDB ID: 7D7T |
| SARS-CoV-2 PLpro/GRL0617 | Fu et al., 2021 | PDB ID: 7CJM |
| SARS-CoV-2 PLpro/GRL0617 | Gao et al., 2021 | PDB ID: 7CMD |
| SARS-CoV-2 PLpro/PLP_Snyder457 inhibitor | Osipiuk et al., 2021 | PDB ID: 7JIR |
| SCoV-1 PLpro-diUbK48 | Békés et al., 2016 | PDB ID: 5E6J |
| SCoV-2 PLpro-ISG15 | Shin et al., 2020 | PDB ID: 6YVA |
| SCoV-2 PLpro-Ub | Klemm et al., 2020 | PDB ID: 6XAA |
| SCoV-1 PLpro-Ub | Ratia et al., 2014 | PDB ID: 4MM3 |
| Experimental models: cell lines | ||
| HEK293T | ATCC | ATCC Cat# CRL-3216, RRID:CVCL_0063 |
| Oligonucleotides | ||
| 5′-AAGGTACCGAGAGCCTGGTGCCAGGCTTCAACG-3′ | This study | SPO166: N-term nsp1 |
| 5′-ACTCGAGTCATTTGTCATCATCGTCTTTATAG TCCCAAGGCACAGAGTTCACTGCTGCC-3′ |
This study | SPO167: C-term nsp3 (truncated) |
| 5′-CTTGGTACCATGGCCCCCACCAAGGTGACATTTGG-3′ | This study | SPO140: N-term nsp3 |
| 5′-CCTTGTAATCGGGCCCTCTAGACTCG-3′ | This study | SPO141: C-term nsp3 (truncated) |
| 5′-TTCTCGAGGTTCAGCTCTCTCATCAGCTCCCGGG −3′ | This study | SPO149: C-term nsp1 |
| Recombinant DNA | ||
| pET28_SCoV2_PLpro-6XHIS | Rut et al., 2020 | N/A |
| pET28_SCoV2_PLpro-C/S-6XHIS | This study | N/A |
| pET28_SCoV2_PLpro-S2-6XHIS (F69S, E70K, H73G) | This study | N/A |
| pET28_SCoV2_PLpro-S1-6XHIS (S170A, Y171A, Q174A) | This study | N/A |
| pET28_SCoV2_PLpro-T225V,K232Q-6XHIS | This study | N/A |
| pET28_SCoV2_PLpro-T75L,D179E-6XHIS | This study | N/A |
| pET28_SCoV2_PLpro-K232Q-6XHIS | This study | N/A |
| pET28_SCoV2_PLpro-K92N-6XHIS | This study | N/A |
| pET28_SCoV2_PLpro-A145D-6XHIS | This study | N/A |
| pET28_SCoV1_PLpro-6XHIS | Békés et al., 2015 | N/A |
| pET28_SCoV1_PLpro-V226T,Q233K-6XHIS | This study | N/A |
| pET28_SCoV1_PLpro-L76T,E180D-6XHIS | This study | N/A |
| pcDNA3.1-HA | A gift from Oskar Laur, Emory University | RRID: Addgene_128034 |
| pcDNA3_SCoV2_PLpro-FLAG | This study | N/A |
| pcDNA3_SCoV2_PLpro-T225V,K232Q-FLAG | This study | N/A |
| pcDNA3_SCoV2_PLpro-T75L,D179E-FLAG | This study | N/A |
| pcDNA3_SCoV2_PLpro-K232Q-FLAG | This study | N/A |
| pcDNA3_SCoV2_PLpro-K92N-FLAG | This study | N/A |
| pcDNA3_SCoV2_PLpro-A145D-FLAG | This study | N/A |
| pcDNA3_SCoV1_PLpro-FLAG | This study | N/A |
| pcDNA3_SCoV1_PLpro-V226T,Q233K-FLAG | This study | N/A |
| pcDNA3_SCoV2_HA-nsp1 | This study | N/A |
| pcDNA3_SCoV2_nsp3-FLAG | This study | N/A |
| pcDNA3_SCoV2_HA-nsp123-FLAG | This study | N/A |
| pcDNA3_SCoV2_HA-nsp123-C/S-FLAG | This study | N/A |
| pcDNA3_SCoV2_HA-nsp123-K232Q-FLAG | This study | N/A |
| pcDNA3_SCoV2_HA-nsp123-K92N-FLAG | This study | N/A |
| pcDNA3_SCoV2_HA-nsp123-A145D-FLAG | This study | N/A |
| pcDNA3_SCoV2_HA-nsp123-S2-FLAG (F69S, E70K, H73G) | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism (version 9) | GraphPad Software | RRID:SCR_002798; https://www.graphpad.com/ |
| HKL2000 (version v718) | Otwinowski and Minor, 1997 | N/A; https://hkl2000.hkl-xray.com |
| COOT (version 0.9) | Emsley and Cowtan, 2004 | RRID:SCR_014222; https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix.Refine (version 1.15.2-3472) | Adams et al., 2010 | RRID:SCR_016736; https://www.phenix-online.org/documentation/reference/refinement.html |
| PyMOL (version 2.0.6) | Schrödinger LLC | RRID:SCR_000305; https://pymol.org/2/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tony T. Huang, PhD (tony.huang@nyumc.org).
Materials availability
All unique/stable reagents generated in this study will be made available upon request.
Experimental model and subject details
Cell lines and culture
HEK293T cells (ATCC) were grown by standard cell culture technique in DMEM (GIBCO) with 10% FBS (Atlantic Biologicals), 1% penicillin/streptomycin (GIBCO), and 1% glutamine (GIBCO) at 37°C with 5% CO2. Transient plasmid transfections were performed using FuGene6 reagent (Promega) according to the manufacturer’s instructions. Cells were collected for analyses 24 hours after plasmid transfection.
BL21-DE3 E. coli cells were used for bacterial expression and purification of proteins, as described in Method details. Cells were cultured in LB medium at 37°C.
Method details
Plasmids
The sequence for SARS-CoV-1 PLpro was based on SARS-CoV-1 orf1ab (NC_004718.3) polyprotein amino acids 1541-1856 and the sequence for SARS-CoV-2 PLpro was based on SARS-CoV-2 orf1ab (NC_045512.2) polyprotein amino acids 1564-1879. For bacterial expression and purification, PLpro sequences were codon-optimized for E. coli expression, synthesized, and cloned into the NcoI and XhoI sites of pET-28a(+) for expression with a C-terminal 6xHIS tag (Genscript). For expression in human cells, PLpro sequences were codon-optimized for human expression, synthesized, and cloned into KpnI and XhoI sites of pcDNA3.1(+)-C-DYK for expression with a C-terminal FLAG tag (Genscript). All mutations were introduced by site-directed mutagenesis and verified by sequencing. The truncated SCoV-2 “nsp123” sequence (amino acids 1-2159 of the orf1ab polyprotein) was codon-optimized for human cell expression, synthesized, and cloned into KpnI and XhoI sites of pcDNA3.1(+)-C-DYK (Genscript). To incorporate an N-terminal HA-tag, the nsp123-FLAG sequence was amplified and cloned into KpnI and XhoI sites of pcDNA3.1-HA using the following oligos SPO166 and SPO167 (see Key resources table). Mutations were introduced by site-directed mutagenesis and verified by sequencing. The nsp3 sequence (amino acids 819-2159 of orf1ab polyprotein) was amplified using nsp123-FLAG plasmid as template with oligos SPO140 and SPO141 (see Key resources table). The nsp3 sequence was cloned into KpnI and XhoI sites of pcDNA3.1(+)-C-DYK to generate nsp3-FLAG plasmid. The HA-nsp1 plasmid was made by amplifying the nsp1 sequence (amino acids 1-180 of orf1ab polyprotein) from the HA-nsp123-FLAG plasmid using oligos SPO166 and SPO149 (see Key resources table). The nsp1 sequence was then cloned into KpnI and XhoI sites of pcDNA3.1-HA.
Protein expression and purification
PLpro bacterial expression vectors were transformed in BL21-DE3 cells and grown in selective LB broth at 37°C with shaking. For crystallization, cells were grown in LB broth at 37°C with shaking until OD600 = 1.5; Isopropyl-β-D-thiogalactopyranoside (IPTG) (0.1 mM) and ZnSO4 (0.1 mM) were added to induce protein expression overnight at 18°C. For biochemical assays, cells were grown until OD600 = 0.5 and PLpro expression was induced with 0.5mM IPTG at 18°C overnight with shaking. Cell pellets were resuspended in lysis buffer [20 mM Tris-Cl (pH 8.0), 350 mM NaCl, 20 mM imidazole, and 2 mM β-mercaptoethanol] and lysed using sonication. Lysate was cleared by centrifugation at 4°C and passed onto Ni-NTA Agarose (QIAGEN) followed by washing with lysis buffer. The His-tagged PLpro was eluted in lysis buffer supplemented with 250 mM imidazole and was further purified using Superdex 75-pg size-exclusion columns (GE) equilibrated with 20 mM Tris-Cl (pH 8.0), 40 mM NaCl, and 2 mM dithiothreitol (DTT) (Figures S2F and S2G). The purified protein was then concentrated to ∼10 mg/ml and snap-frozen in liquid nitrogen for later use.
Crystallization
SARS-CoV-1 PLpro (3 μM) was reacted with 30 μM peptide inhibitor in 5 mM NaCl, 20 mM Tris-Cl (pH 8.0) at 37°C for 20 min. Protein was concentrated using a 30-kD cutoff Amicon Ultra Filter and desalted into 5 mM NaCl, 20 mM Tris-Cl (pH 8.0), and 10 mM DTT. Final protein concentration was 5 to 10 mg/ml. VIR250 complex crystals were grown by mixing 0.3 μL protein and 0.3 μL well solution containing 0.2 M sodium thiocyanate, and 20% w/v polyethylene glycol 3350 on a 96-well sitting plate at 18°C. VIR251 complex crystals were grown by mixing 0.3 μL protein sample with 0.3 μL well solution containing 0.2 M ammonium sulfate, 16% w/v polyethylene glycol 3350 on a 96-well sitting plate at 18°C. Crystals were cryo-protected by 25% (v/v) ethylene glycol and flash-frozen with liquid nitrogen.
Structure determination and refinement
A complete dataset was collected from the SCoV-1 PLpro/VIR250 crystals to 2.29 Å resolution at the Advanced Photon Source, NE-CAT beamline 24-IDE. Dataset was indexed, integrated, and scaled using HKL2000 (Otwinowski and Minor, 1997). Crystal belongs to space group I222 with unit cell dimensions a = 70.8, b = 90.4, c = 113.6. There is one SCoV-1 PLpro/VIR250 complexes per asymmetric unit. The structure was solved by molecular replacement using the program PHASER. The search model was SARS-CoV-2 PLpro (PDB: 6WUU). Apparent ligand density for both Fo-Fc and 2Fo-Fc maps was observed projecting off Cys111 after first round of refinement. Model and restraints for VIR250 was prepared using Phenix.Elbow. Model of SARS-CoV-1 PLpro/VIR250 was subjected to iterative rounds of refinement and rebuilding using PHENIX (Adams et al., 2010) and COOT (Emsley and Cowtan, 2004).
For SCoV-1 PLpro/VIR251 crystals, data were collected and processed as described above for VIR250 to a resolution of 2.23 Å. The crystal belongs to space group P6122 with unit cell dimensions a = b = 103.6, and c = 265.4. There are two SCoV-1 PLpro/VIR251 complexes per asymmetric unit. The structure was determined by molecular replacement with Phaser and the search model was SCoV-1 PLpro from the SCoV-1 PLpro/VIR250 structure described above. Structure with ligand was refined as described above for the VIR250 structure.
The final two models for PLpro-VIR250 and PLpro-VIR251 complexes have R/Rfree values of 0.224/0.265 and 0.197/0.235, respectively. The two structures also have excellent geometry as assessed using Molprobity: favored (96.4%), allowed (3.6%), and outliers (0%) for the PLpro/VIR250 structure and favored (96.6%), allowed (3.4%), and outliers (0%) for the PLpro/VIR251 structure.
Gel-based Ub chain cleavage assays and ABP labeling
Tetra-UbK48 chains (Boston Biochem) were cleaved in a reaction volume of 10 μL in reaction buffer [20 mM Tris-Cl (pH 7.5), 150 mM NaCl, and 5 mM DTT] with 1 μM tetra-UbK48 and 500 nM PLpro. Labeling with activity-based probes was performed in a reaction volume of 10 μL in reaction buffer with 250 nM PLpro and 1.5 μM Ub-Vinyl Sulfone (Ub-VS) or 500 nM ISG15-VS. Reactions were incubated at 37°C for 30 min or for the indicated time points and terminated with sample loading buffer (4X LDS, Invitrogen), and analyzed by SDS-PAGE (4%–12% bis-tris, NuPAGE) and SYPRO Ruby staining. Gels were imaged using an Azure Biosystems c500 imager.
Ubiquitin- and ISG15-AMC kinetics
To determine apparent kcat/KM for SCoV-1 and SCoV-2 PLpro, ubiquitin-AMC (amino-methyl-coumarine) and ISG15-AMC were prepared as 1∕2-fold serial dilutions (starting at 40 μM or 25 μM for Ub-AMC and 15 μM for ISG15-AMC) in reaction buffer [20 mM Tris-Cl (pH 7.5), 150 mM NaCl and 5 mM DTT]. PLpro was pre-incubated in reaction buffer for 5 min at room temp and used at a final concentration of 50 nM for Ub-AMC assays or 10 nM for ISG15-AMC assays. Cleavage was performed in 10 μL reaction volume at 30°C using a Tecan infinite 200Pro plate reader running Tecan i-control software, operated in fluorescence mode, in white, round-bottom 384-well plates. Free AMC fluorescence was monitored over time by excitation at 355 nm and emission at 460 nm. Initial linear cleavage rates (Vi) were fitted by the Michealis-Menten equation using Prism software (GraphPad) based on a free AMC standard curve. Comparisons of SCoV-2 PLpro and its mutants were performed as above, or at a single Ub-AMC concentration (5 μM) plotted as pM AMC/sec. All experiments were performed at least in triplicate; error bars indicate ± SD.
Western blotting
Western blots were performed with whole-cell extracts prepared in SDS sample buffer [0.1M Tris-Cl (pH 6.8), 2% (w/v) SDS and 12% (v/v) β-mercaptoethanol]. Protein extracts were separated on Nupage 4%–12% Bis-Tris or 3%–8% Tris-Acetate gels (Invitrogen) and transferred onto 0.45 μM PVDF membrane in Invitrogen Tris-Glycine transfer buffer. Membranes were blocked in 5% milk in TBST for 1 hr and incubated in primary antibody overnight at 4°C. Membranes were incubated with secondary antibodies in 5% milk TBST for 1 hr at room temperature, developed using ECL Prime Western Blotting Detection Reagent (Amersham), and imaged using an Azure Biosystems c500 imager. FLAG-tagged PLpro, nsp3, and nsp123 were detected using anti-FLAG M2 antibody (Sigma F1804). HA-tagged nsp1 and nsp123 were detected using anti-HA.11 antibody (Biolegend 902301). K48-linked Ub chains were detected by K48-linkage specific Ub antibody (Millipore 05-1307) and antibody against α-tubulin was used where indicated as a loading control (Cal Biochem CP06).
Quantification and statistical analysis
For qualitative gel-based assays, all reactions were repeated at least three times (biological replicates) and representative gel images are shown. Graphpad Prism software was used for plotting AMC-based assay data. Cleavage rates (Vi) are represented as the mean of three independent replicates and error bars indicate ± standard deviation of the mean, as indicated in each figure legend.
Acknowledgments
This work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the NIH (P30 GM124165). The Eiger 16M detector on the 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. This work is based on research conducted in the Structural Biology Core Facilities-X-ray Crystallography Core Laboratory, a part of the Institutional Research Cores at the University of Texas Health Science Center at San Antonio supported by the Office of the Vice President for Research and the Mays Cancer Center Drug Discovery and Structural Biology Shared Resource (NIH P30 CA054174). Research reported in this publication was supported by CPRIT RR200030 and NIH grants R01 GM115568 and R01 GM128731 (to S.K.O.) and R01 ES025166 and R35 GM139610 (to T.T.H.). S.P. is an American Cancer Society Postdoctoral Fellow (PF-18-235-01-RMC). This project was supported by the National Science Center grant 2015/17/N/ST5/03072 (Preludium 9) in Poland (to W.R.), UMO-2020/01/0/NZ1/00063 (to M.D.), and the “TEAM/2017-4/32” project, which is carried out within the TEAM program of the Foundation for Polish Science, cofinanced by the European Union under the European Regional Development Fund (to M.D.).
Author contributions
Conceptualization, S.P., Z.L., S.K.O., and T.T.H.; methodology, S.P., Z.L., S.K.O., and T.T.H.; investigation, S.P., Z.L., and W.R.; resources, S.P., Z.L., W.R., M.B., and M.D.; formal analysis, S.P., Z.L., S.K.O., and T.T.H.; visualization, S.P. and Z.L.; writing – original draft, S.P.; writing – review & editing, S.P., Z.L., W.R., M.B., M.D., S.K.O., and T.T.H.
Declaration of interests
M.B. is an employee of Arvinas, Inc, which was not involved in this study. T.T.H. is a member of the advisory board for Cell Reports. The remaining authors declare no competing interests.
Published: September 28, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109754.
Supplemental information
Data and code availability
Structures from this study have been deposited in the RCSB (https://www.rcsb.org) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table. This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreano E., Piccini G., Licastro D., Casalino L., Johnson N.V., Paciello I., Monego S.D., Pantano E., Manganaro N., Manenti A., et al. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv. 2020 doi: 10.1101/2020.12.28.424451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Elkin B.A., Knaap R.C., Johnson G.G., Dalebout T.J., Ninaber D.K., van Kasteren P.B., Bredenbeek P.J., Snijder E.J., Kikkert M., Mark B.L. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014;289:34667–34682. doi: 10.1074/jbc.M114.609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Békés M., Rut W., Kasperkiewicz P., Mulder M.P., Ovaa H., Drag M., Lima C.D., Huang T.T. SARS hCoV papain-like protease is a unique Lys48 linkage-specific di-distributive deubiquitinating enzyme. Biochem. J. 2015;468:215–226. doi: 10.1042/BJ20141170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Békés M., van der Heden van Noort G.J., Ekkebus R., Ovaa H., Huang T.T., Lima C.D. Recognition of Lys48-Linked Di-ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease. Mol. Cell. 2016;62:572–585. doi: 10.1016/j.molcel.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cele S., Gazy I., Jackson L., Hwa S.-H., Tegally H., Lustig G., Giandhari J., Pillay S., Wilkinson E., Naidoo Y., et al. Escape of SARS-CoV-2 501Y.V2 variants from neutralization by convalescent plasma. Nature. 2021;593:142–146. doi: 10.1038/s41586-021-03471-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Lai H.Y., Chen H.Y., Cheng S.C., Cheng K.W., Chou Y.W. Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain-like protease. Acta Crystallogr. D Biol. Crystallogr. 2014;70:572–581. doi: 10.1107/S1399004713031040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.E., Gack M.U. Ubiquitination in the antiviral immune response. Virology. 2015;479-480:52–65. doi: 10.1016/j.virol.2015.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.T., Baker S.C., Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag M., Mikolajczyk J., Bekes M., Reyes-Turcu F.E., Ellman J.A., Wilkinson K.D., Salvesen G.S. Positional-scanning fluorigenic substrate libraries reveal unexpected specificity determinants of DUBs (deubiquitinating enzymes) Biochem. J. 2008;415:367–375. doi: 10.1042/BJ20080779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzimianski J.V., Scholte F.E.M., Bergeron É., Pegan S.D. ISG15: It’s Complicated. J. Mol. Biol. 2019;431:4203–4216. doi: 10.1016/j.jmb.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Faria N.R., Morales Claro I., Candido D., Moyses Franco L., Andrade P.S., Coletti T.M., Silva C.A., Sales F.C., Manuli E.R., Aguiar R.S. 2021. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings.https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 [Google Scholar]
- Freitas B.T., Durie I.A., Murray J., Longo J.E., Miller H.C., Crich D., Hogan R.J., Tripp R.A., Pegan S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020;6:2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Qin B., Chen P., Zhu K., Hou P., Wojdyla J.A., Wang M., Cui S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11:237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W.F., Lam E.C., St Denis K., Nitido A.D., Garcia Z.H., Hauser B.M., Feldman J., Pavlovic M.N., Gregory D.J., Poznansky M.C., et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184:2523. doi: 10.1016/j.cell.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z., Zhu J.W., Li C.P., Jiang S., Ma L.N., Tang B.X., Zou D., Chen M.L., Sun Y.B., Song S.H., et al. An online coronavirus analysis platform from the National Genomics Data Center. Zool. Res. 2020;41:705–708. doi: 10.24272/j.issn.2095-8137.2020.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.S., Chang G.G., Juo C.G., Lee H.J., Yeh S.H., Hsu J.T., Chen X. Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry. 2005;44:10349–10359. doi: 10.1021/bi0504761. [DOI] [PubMed] [Google Scholar]
- Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., Rota P.A., Baker S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P., Lu B.G., Kuchel N.W., Grohmann C., Shibata Y., et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39:e106275. doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Ménard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Lytvyn V., Qi H., Lachance P., Ziomek E., Ménard R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007;466:8–14. doi: 10.1016/j.abb.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen T.E.T., Komander D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017;86:159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- Mielech A.M., Deng X., Chen Y., Kindler E., Wheeler D.L., Mesecar A.D., Thiel V., Perlman S., Baker S.C. Murine coronavirus ubiquitin-like domain is important for papain-like protease stability and viral pathogenesis. J. Virol. 2015;89:4907–4917. doi: 10.1128/JVI.00338-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveca F., Da Costa C., Nascimento V., Souza V., Corado A., Nascimento F., Costa Á., Duarte D., Silva G., Mejía M. 2020. SARS-CoV-2 reinfection by the new Variant of Concern (VOC) P. 1 in Amazonas, Brazil.https://virological.org/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/596 [Google Scholar]
- Osipiuk J., Azizi S.A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Perng Y.C., Lenschow D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018;16:423–439. doi: 10.1038/s41579-018-0020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Loman N., Pybus O., Barclay W., Barrett J., Carabelli A., Connor T., Peacock T., Robertson D., Volz E. 2020. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations.https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 [Google Scholar]
- Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. USA. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Kilianski A., Baez-Santos Y.M., Baker S.C., Mesecar A. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10:e1004113. doi: 10.1371/journal.ppat.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., El Oualid F., Huang T.T., Bekes M., Drag M., Olsen S.K. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti-COVID-19 drug design. Sci. Adv. 2020;6:eabd4596. doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan H., Liu J., Shen J., Dai J., Xu G., Lu K., Han C., Wang Y., Xu X., Tong Y., et al. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021;28:855–865.e9. doi: 10.1016/j.chembiol.2021.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B., Clementz M.A., Banach B.S., Li K., Baker S.C., Chen Z. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS ONE. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. 2020 doi: 10.1101/2020.12.21.20248640. [DOI] [Google Scholar]
- Wang Z., Schmidt F., Weisblum Y., Muecksch F., Barnes C.O., Finkin S., Schaefer-Babajew D., Cipolla M., Gaebler C., Lieberman J.A., et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021;592:616–622. doi: 10.1038/s41586-021-03324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Zou J., Fontes-Garfias C.R., Xia H., Swanson K.A., Cutler M., Cooper D., Menachery V.D., Weaver S., Dormitzer P.R., Shi P.-Y. Neutralization of N501Y mutant SARS-CoV-2 by BNT162b2 vaccine-elicited sera. bioRxiv. 2021 doi: 10.1101/2021.01.07.425740. [DOI] [PubMed] [Google Scholar]
- Zhao W.M., Song S.H., Chen M.L., Zou D., Ma L.N., Ma Y.K., Li R.J., Hao L.L., Li C.P., Tian D.M., et al. The 2019 novel coronavirus resource. Yi Chuan. 2020;42:212–221. doi: 10.16288/j.yczz.20-030. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 2005;287:57–94. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structures from this study have been deposited in the RCSB (https://www.rcsb.org) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table. This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.