

Abstract
Chronic traumatic encephalopathy (CTE) is a neurodegenerative disease associated with exposure to repetitive head impacts, such as those from contact sports. The pathognomonic lesion for CTE is the perivascular accumulation of hyper-phosphorylated tau in neurons and other cell process at the depths of sulci. CTE cannot be diagnosed during life at this time, limiting research on risk factors, mechanisms, epidemiology, and treatment. There is an urgent need for in vivo biomarkers that can accurately detect CTE and differentiate it from other neurological disorders. Neuroimaging is an integral component of the clinical evaluation of neurodegenerative diseases and will likely aid in diagnosing CTE during life. In this qualitative review, we present the current evidence on neuroimaging biomarkers for CTE with a focus on molecular, structural, and functional modalities routinely used as part of a dementia evaluation. Supporting imaging-pathological correlation studies are also presented. We targeted neuroimaging studies of living participants at high risk for CTE (e.g., aging former elite American football players, fighters). We conclude that an optimal tau PET radiotracer with high affinity for the 3R/4R neurofibrillary tangles in CTE has not yet been identified. Amyloid PET scans have tended to be negative. Converging structural and functional imaging evidence together with neuropathological evidence show frontotemporal and medial temporal lobe neurodegeneration, and increased likelihood for a cavum septum pellucidum. The literature offers promising neuroimaging biomarker targets of CTE, but it is limited by cross-sectional studies of small samples where the presence of underlying CTE is unknown. Imaging-pathological correlation studies will be important for the development and validation of neuroimaging biomarkers of CTE.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13311-021-01028-3.
Key Words: Chronic traumatic encephalopathy, Biomarkers, Neuroimaging, Atrophy, MRI, Traumatic brain injury, Neurodegenerative disease
Introduction
Contact sport participation, military combat, and physical violence are associated with exposure to repetitive head impacts (RHI) [1]. These impacts can result in mild traumatic brain injuries (TBIs), including symptomatic concussions and the more frequent asymptomatic sub-concussions [2–7]. Exposure to RHI has emerged as a public health concern because of its association with the neurodegenerative disease chronic traumatic encephalopathy (CTE) [8–14]. CTE has been neuropathologically studied in boxers since the late 1970s [15] with clinical syndromes reported earlier (e.g., “Punch Drunk” in 1928, “Dementia Pugilistica” in 1937) [16, 17]. Evidence of abnormal hyper-phosphorylated tau (p-tau) deposition was reported in a deceased former National Football League (NFL) for the first time in 2005 [18]. Neuropathological diagnostic criteria for CTE were published in 2016, and the pathognomonic lesion of CTE includes the perivascular deposition of p-tau in neurons, with or without astrocytes, at the depths of the cerebral sulci [12, 19, 20]. CTE has been neuropathologically diagnosed in hundreds of deceased American football players [12, 14] and various other contact sport athletes and military veterans [11, 12, 18, 21–28]. Risk for CTE has been shown to be a function of duration of exposure to RHI [13], with a prominent role for sub-concussions as opposed to concussion [12, 29].
CTE cannot be diagnosed during life at this time, limiting research on risk factors, mechanisms, epidemiology, and treatment. Multiple sets of criteria to diagnose CTE in life have been proposed [30, 31], including provisional clinical research diagnostic criteria known as traumatic encephalopathy syndrome (TES), proposed in 2014 [32] and revised in 2021 [33]. Based on retrospective interviews with informants of brain donors, the clinical presentation of CTE has been reported to include a constellation of cognitive, behavior, and mood symptoms and, in some instances, parkinsonism and other motor symptoms, often with progression to dementia [11, 12, 14, 25, 34, 35]. The reported clinical presentations of CTE are heterogeneous [32]. The etiology of the diverse symptoms is unknown and likely multifactorial [8, 11, 34, 36–43]. The ill-defined clinical presentation of CTE is one reason for the current inability to accurately detect and diagnose CTE during life.
CTE also cannot be diagnosed in life at this time due to the lack of validated in vivo biomarkers that can accurately detect and differentiate CTE from other neurological disorders, such as Alzheimer’s disease (AD) and frontotemporal dementia (FTD). Developing in vivo biomarkers of CTE will be instrumental for accurate disease detection and diagnosis, monitoring of disease progression, evaluating efficacy of therapeutic interventions, and studying disease mechanisms and symptom etiology. There have been advances in biomarker development for CTE in the past 5 years, particularly as the neuropathology of CTE has become increasingly well-defined. Similar to biomarker frameworks of other neurodegenerative diseases (e.g., A/T/N in AD) [44], in vivo biomarkers for CTE will likely include those that are specific and non-specific to CTE. Specific biomarkers would directly inform on the presence of underlying CTE p-tau pathology in the central nervous system (CNS). Non-specific biomarkers (e.g., patterns of atrophy) would be useful to support a diagnosis of CTE when combined with other risk factors, clinical and biomarker data points, as well as to track disease progression and to facilitate study of disease mechanisms, etiology, and pathogenesis.
Analysis of proteins in the cerebrospinal fluid and blood may be viable biomarker approaches in CTE [45–48]. Neuroimaging is an integral component of the clinical evaluation of neurodegenerative diseases and is used as part of large-scale multi-center clinical trials of disease-modifying therapies. Neuroimaging is dynamic and can provide insight into the structure, function, and molecular make-up of the CNS and can characterize disease stages. Neuroimaging will likely help to facilitate a “probable CTE” diagnosis during life. In this qualitative review, we present the current evidence on neuroimaging biomarkers for CTE with a focus on modalities routinely used in an academic memory disorders clinic for differential diagnoses of neurodegenerative diseases. Because it is necessary to have a clear understanding of the neuropathology of CTE to inform on neuroimaging targets, we begin with an overview of the microscopic and gross pathological features of CTE. Then, molecular, structural, and functional neuroimaging biomarker findings in living participants at high risk for CTE are presented. Existing imaging-pathological correlation studies are reviewed. We conclude with our opinions of current clinical and research implications and propose directions for future research. While this review has a clinical framework, it is important to note that validated neuroimaging biomarkers of CTE do not yet exist and are not yet ready for integration into the clinic to support an in vivo diagnosis of CTE.
Methods
This review focuses on neuroimaging techniques routinely used as part of dementia evaluations for differential diagnoses of neurodegenerative diseases. These include T1-weighted MRI, fluid attenuated inversion recovery (FLAIR), susceptibility weighted imaging (SWI)/T2*-weighted gradient-recalled echo (GRE), 18F fluorodeoxyglucose (FDG)-PET, single-photon emission computerized tomography (SPECT) imaging, and amyloid and tau PET imaging. These techniques are approved by the Food and Drug Administration (FDA), and selection of neuroimaging methods was also guided by the National Institute on Aging-Alzheimer’s Association (NIA-AA) Research Framework for defining the underlying pathophysiological processes of AD [44]. Note that FDG-PET, SPECT, and amyloid and tau PET are not necessarily routine, but requested for complex patients where symptom etiology is uncertain and there is a need for differential diagnosis to guide treatment planning.
Because CTE cannot be diagnosed during life and validated clinical criteria do not exist, recruitment of individuals with clinically diagnosed CTE is not currently possible. CTE has been shown to be strongly associated with exposure to RHI from American football [13]. CTE has also historically been neuropathologically described in boxers (e.g., “dementia pugilistica”) [16, 17]. RHI in these populations are frequent and homogenous. For these reasons, clinical research has drawn on aging former elite American football players and fighters to study and draw inferences about the clinical presentation and neuroimaging biomarkers of CTE. Although it is unclear if findings from these populations generalize to the broader population, it provides some level of certainty of underlying CTE pathology that is critical for biomarker development. Risk for CTE in other populations is not yet well-defined, such as in former soccer and ice hockey players and military veterans. These populations are less suitable for CTE biomarker development at this time due to the uncertainty of underlying CTE pathology. This review thus focuses on in vivo MRI data from former aging elite American football players and fighters. The Professional Fighters Brain Health Study (PFBHS) has resulted in major contributions to the field on the effects of RHI from fighting sports on the CNS [49–52]. Many of those studies have been among active fighters (or active + retired fighters with smaller representation of retired), and we discuss findings that reported separately on aging retired fighters.
Although there are numerous “case only” studies that are descriptive or report on exposure-MRI relationships [53–58], we place emphasis on “case-control” studies given biomarker sensitivity and specificity are key criterion for biomarker development. This methodology does not apply to the review of clinical-pathological correlation or autoradiography studies.
Studies that include samples of participants actively being or had acutely been exposed to RHI (e.g., young active college American football players) and the late effects of a single traumatic brain injury (TBI) were not included in this review. CTE is a neurodegenerative disease associated with age [20]. While the pathophysiological processes of CTE are likely initiated during active exposure to RHI, widespread neuropathology and clinical symptoms often manifest years after exposure to RHI has ended. Studies that use young active otherwise healthy contact sport athletes are unlikely to inform on biomarkers of CTE. Although a single TBI has been associated with increased risk for dementia [59–63] and an accumulation of various neurodegenerative disease proteins and other pathologies [64–66], there is insufficient evidence to suggest that single or multiple TBIs are associated with the diagnostic entity of CTE.
Our literature review was conducted using key words and associated permutations in PubMed, such as “repetitive head impacts,” “sub-concussion,” “contact sport athletes,” “American football,” “fighters,” “boxers,” “chronic traumatic encephalopathy,” “neuroimaging,” “biomarkers,” “PET,” “atrophy,” “brain,” “magnetic resonance imaging,” “molecular imaging,” “neurodegeneration,” “dementia,” “tau,” “FTP,” “FDDNP,” “PET radiotracers,” and other search terms. References of research articles were used for identifying relevant studies for this review. Because of the overall limited evidence on this topic, we did not restrict the articles to a specific timeframe.
Neuropathology of CTE
Microscopic Pathology
On the microscopic level, neurofibrillary tangle (NFT) pathology—intracellular aggregates of p-tau protein—are the hallmark of CTE and were initially described by Hof and colleagues [67]. Inclusions form in both neurons and glial cells. NFTs in CTE are irregularly/unevenly distributed with a tendency to form around blood vessels and at the depths of the sulci [68]. Dr. Ann McKee at the Veteran Affairs (VA)-Boston University (BU)-Concussion Legacy Foundation (CLF) brain bank has characterized the neuropathology of CTE through the National Institutes of Health (NIH)-funded Understanding Neurologic Injury in Traumatic Encephalopathy (UNITE) study [14, 69]. In 2015, a NINDS and National Institute of Biomedical Imaging and Bioengineering sponsored conference was convened to evaluate proposed neuropathological diagnostic criteria for CTE [12, 19]. A panel of seven neuropathologists with expertise in neurodegenerative tauopathies evaluated 25 selected cases of represented tauopathies. The panel defined the pathognomonic lesion of CTE as “an accumulation of abnormal hyperphosphorylated tau (p-tau) in neurons and astroglia distributed around small blood vessels at the depths of cortical sulci and in an irregular pattern.” Since these neuropathological criteria were published, it has been clarified that the perivascular lesions of CTE must be neuronal, consisting of NFTs and disordered neurites in the variable presence or absence of p-tau immunoreactive astrocytes [20]. P-tau immunoreactive astrocytes in CTE lesions are associated with advancing age [70].
CTE is a mixed three and four microtubule binding domain repeat (3R/4R) tauopathy. Cryo-electron microscopy studies showed differences in the molecular structure of tau filaments in CTE from AD [71], and from Pick’s disease [72] and corticobasal degeneration [73]. CTE tau isoforms may change with disease severity, including becoming 3R predominant with disease progression [70]. Astrocytic tau lesions tend to be 4R, whereas neuronal lesions are 3R and 4R [70]. Unlike AD, Aβ deposits in CTE are not an early feature, not diagnostic, and tend to accumulate with advancing age [36]. When present, Aβ plaques also tend to be diffuse and not neuritic [14, 36]. TAR DNA-binding protein 43 (TDP-43) neuronal and glial inclusions are also found in some CTE cases, most commonly in the cortex and subcortical white matter, in association with severe p-tau pathology [12, 37].
In 2013, McKee et al. [176] proposed a staging system to characterize the severity of p-tau, the McKee CTE staging scheme [12, 20]. This scheme includes four pathological stages, ranging from stage I (mild) to stage IV (severe) (Fig. 1). In stage I CTE, there are 1 or 2 isolated foci of p-tau NFTs and dot-like neurites around small blood vessels at the depths of the sulci, especially in the frontal cortex. In stage II, 3 + CTE lesions are found in multiple cortical regions, the lesions are larger, and NFTs are present in adjacent cortices. In stage III, larger perivascular patches of p-tau NFTs, astrocytes, and dotlike and threadlike neurites are found at the sulcal depths. Diffusely distributed NFTs are found in the hippocampus, entorhinal and perirhinal cortices, and amygdala. There is more brainstem p-tau pathology. In stage IV, perivascular p-tau lesions and NFTs are distributed throughout the cerebral cortex. Perivascular p-tau lesions and NFTs extend to the diencephalon, and brainstem and NFTs are in the cerebellar dentate nucleus, basis pontis, and spinal cord. The McKee CTE staging scheme was recently shown to be associated with semi-quantitative and quantitative scales of p-tau severity and density throughout the brain. Higher CTE stage was strongly linked to increasing age, exposure to RHI (defined by years of American football), and increased odds for dementia [20].
Fig. 1.
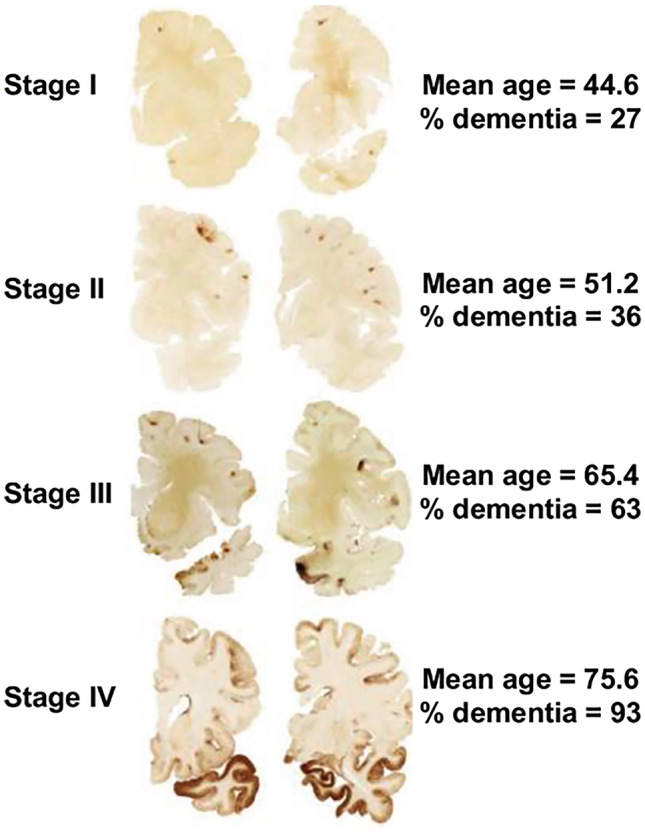
Pathological staging of p-tau in chronic traumatic encephalopathy. The figure shows regional p-tau accumulation (brown) based on the McKee CTE staging scheme. The photo is
adapted from [176] and the data shown are based on Alosco et al. [20]
Gross Pathology
Gross features of CTE include progressive cerebral, medial temporal lobe (MTL), and anterior diencephalic atrophy [12]. There is mild lateral ventricle enlargement in stage I CTE and mild enlargement of the frontal horn of the lateral ventricles and/or the third ventricle in stage II CTE. Most brains in stage III show cerebral atrophy with dilation of the lateral and third ventricles. Frontal and temporal lobe atrophy are most prominent. There is marked MTL atrophy by stage III CTE [12, 19]. Atrophy of the mammillary bodies and thalamus are observed, often beginning in stage III. By stage IV, atrophy is widespread. Other common gross features include a cavum septum pellucidum (CSP) and corpus callosum (CC) thinning. There is pallor of the locus coeruleus and substantia nigra, beginning in stage II and progressing as the disease advances.
Neuroimaging Biomarkers of CTE
Next, we review structural, molecular, and functional neuroimaging patterns in participants at high risk for CTE. We begin with a description of nuclear medicine findings, followed by a review of structural MRI patterns. The section concludes with an overview of advanced research MRI sequences. Note that although PET imaging is not routine due to high costs and lack of insurance coverage, amyloid (i.e., 18F-Florbetaben, 18F-Florbetapir, 18F-Flutemetamol) and tau (i.e.,18F-Flortaucipir, FTP) PET imaging are approved by the FDA for the detection of Aβ and p-tau AD fibrils of AD, respectively. PET imaging is frequently used in clinical trials as an eligibility criterion and/or endpoint [74–76] and has important implications for clinical management and prognosis [77].
Nuclear Medicine Imaging
Tau PET Imaging
PET imaging is a nuclear medicine functional imaging technique that detects gamma rays emitted from radiolabeled tracers injected into the body. Radiotracers that bind to p-tau in the CNS have been developed and hold promise for the detection of underlying CTE pathology. An overview of the current evidence is presented in Table 1. FDDNP was the first radiotracer to be injected in vivo in patients with AD that showed binding to tau pathology [78–80]. The tau PET tracer FDDNP has been examined in small samples of participants at risk for CTE [81–83]. In 2013, Small et al. examined FDDNP binding in five former NFL players (ages 45 to 73 years) with mood and cognitive symptoms compared with five unimpaired individuals who were similar in age, education, race, body mass index (BMI), and dementia family history [82]. The former NFL players had increased FDDNP binding in the amygdala, caudate, putamen, thalamus, subthalamus, midbrain, and cerebellar white matter. There were no group effects in frontal, parietal, posterior cingulate, or medial and lateral temporal regions. In a follow-up study, Barrio et al. compared FDDNP binding between 14 symptomatic former professional American football players (ages 40 to 86 years, 12 with mild cognitive impairment), 28 cognitively intact controls, and 24 participants with AD dementia [81]. Compared with the cognitively intact controls, the former professional American football players had greater binding in the amygdala, MTL, dorsal and ventral midbrain, hypothalamus, thalamus, pons, striatum, frontal lobe, and anterior cingulate gyrus. There were no effects for the parietal lobe, posterior cingulate gyrus, lateral temporal lobe, or occipital lobe. The strongest effects were for the dorsal midbrain and amygdala. The former professional American football players additionally had higher FDDNP values in the amygdala, dorsal and ventral midbrain, hypothalamus, pons, and the striatum compared with AD dementia. However, there were no group effects for any of the other regions (listed above). That author group also reported increased FDDNP binding in military personnel in a somewhat similar pattern, a population not of focus for this review [83]. In sum, these studies showed relatively diffuse uptake patterns that are not entirely consistent with the neuroanatomical distribution of p-tau pathology in CTE. Omalu et al. also conducted an FDDNP PET-pathological correlation study of a deceased former elite American football player [84]. FDDNP tau PET imaging was done 52 months prior to death. FDDNP binding was correlated with p-tau deposition at autopsy. However, FDDNP DVR values were highest in brain regions such as the striatum, thalamus, and midbrain, and there was lower cortical involvement—a pattern not typical of CTE [84]. Interest in FDDNP has since waned because it binds diffusely, lacks specificity for tau, and binds to Aβ and other proteins [85, 86], and there has been poor reproducibility of results [87].
Table 1.
Summary of tau PET studies in participants at risk for CTE. See “Methods” for study selection criterion. This review focuses on in vivo neuroimaging data from former aging elite American football players and fighters, as clinical research has drawn on aging former elite American football players and fighters to study and draw inferences about the clinical presentation and neuroimaging biomarkers of CTE. Although there have been case only studies on tau PET, we placed emphasis on “case-control” studies given biomarker sensitivity and specificity are key criterion for biomarker development
| Study | Tau tracer | Study type | Sample | Summary of findings | Amyloid status |
|---|---|---|---|---|---|
| Small et al. [82] | FDDNP | Case-control |
- 5 symptomatic former National Football League (NFL) players (45–73 years) - 5 male cognitively normal controls similar in age, education, race, body mass index, dementia family history |
Former NFL players had higher FDDNP DVR values in the caudate, thalamus, subthalamus, midbrain, cerebellar white matter, and amygdala. No cortical effects reported | Amyloid PET not acquired |
| Barrio et al. [81] | FDDNP | Case-control |
- 14 symptomatic former professional American football players (40–86 years), 12 with mild cognitive impairment - 24 Alzheimer’s disease dementia - 28 cognitively intact controls |
Increased FDDNP binding for various cortical and subcortical regions in former professional football players compared with controls. Compared with AD, elevated binding in the amygdala and subcortical brain regions; MTL binding intensity was comparable. Strongest effects observed for the amygdala (compared with both groups) and the dorsal midbrain (compared with controls) | Amyloid PET not acquired |
| Omalu et al. [84] | FDDNP | Imaging-pathological correlation | 59-year-old symptomatic former NFL player who had an antemortem FDDNP-PET scan 52 months prior to death | FDDNP DVR values highest in striatum, thalamus, amygdala and midbrain, with lower cortical involvement. Significant correlation between FDDNP DVRs and p-tau pathology | Amyloid PET not acquired. Diffuse and neuritic Aβ plaques present at autopsy |
| Stern et al. [92] | FTP | Case-control |
- 26 former NFL players (40–69 years) - 31 same-age asymptomatic men without a history of RHI or TBI |
Mean FTP SUVRs higher in former NFL players in the bilateral superior frontal, medial temporal lobe, and left parietal regions. Overlapping individual-level FTP measurements between former NFL players and controls | 25/26 former football players had negative florbetapir PET. No significant difference in mean SUVR between former football players and controls. |
| Lesman-Segev et al. [93] | FTP | Case-control |
- 11 participants who met criteria for TES (30–70 years) - 67 age- and education-matched amyloid negative participants (male and female) without RHI - 22 amyloid-positive age- and education matched participants with MCI (n = 9) or dementia (n = 13) due to AD (male and female) without RHI |
Mildly elevated FTP retention in the frontal-temporal and medial temporal regions in TES participants. Individual-level overlap with the amyloid negative participants and tracer retention was lower than observed in similarly impaired participants with AD | 9/11 TES participants had negative PiB PET |
| Mantyh et al. [94] | FTP | Imaging-pathological correlation | Symptomatic former NFL player who died at 72 and had an antemortem FTP PET at age 68 | Participant had stage IV CTE. Modest correlation between FTP SUVR and tau pathology that did not reach statistical significance. While there was regional correspondence between SUVR and tau pathology, there were regions with high SUVR values that had low p-tau burden | Negative PiB PET and no Aβ plaques at autopsy |
| Marquie et al. [95] | FTP | Autoradiography |
- 5 male donors with pathologically confirmed CTE: one CTE stage II-III, three CTE stage III, one stage IV (25–58 years) - 9 males and females with pathologically confirmed AD (66–96 years) - 6 age- and sex-matched participants without pathological diagnoses |
Negligible binding across cortical regions with tau aggregates in CTE tissue samples. Off-target binding to leptomeningeal melanocytes. No correlation between FTP autoradiographic regional binding and tau measures in CTE tissue. Strong tracer binding to tau aggregates in the AD tissue samples | No Aβ plaques in CTE tissue |
| Aguero et al. [98] | MK-6240 | Autoradiography |
- 5 male donors with pathologically confirmed CTE: one stage II-III, three stage III, one stage IV (25–65 years) - 14 male and female donors with AD (60–97 years) - 9 donors with FTLD-tau (male and female, 61–80 years) - 3 donors with Lewy body disease - 1 donor with multiple systemic atrophy - 4 donors with FTLD-TDP-43 - 2 donors with hemorrhage - 1 donor with diffuse cerebral amyloid angiopathy - 1 unspecified histopathology - 3 donors free of pathology (73–101 years) |
Strong binding to tau aggregates in the hippocampal formation, entorhinal cortex, frontal, temporal, parietal, occipital cortices in AD tissue. No detectable binding in tissue from any of the non-AD cases including CTE. Off-target binding in substantia nigra, brain metastatic melanoma, parenchymal hemorrhages, retinal pigment epithelium and extracutaneous meningeal melanocytes | No Aβ plaques in CTE tissue |
| Krishnadas et al. [103] | MK-6240 | Case (n = 1)-control |
-1 63 year-old male former Australian rules footballer, CDR = 0.5 -99 cognitively normal amyloid and tau negative controls -57 amyloid and tau positive MCI and AD dementia participants |
Former Australian rules footballer had high tracer retention in frontotemporal and medial temporal regions compared with the controls. Only frontotemporal retention was higher than the MCI participants, whereas the MCI participants had higher medial temporal lobe and temporo-parietal retention. Tracer retention in all cortical regions was lower than AD dementia | Former Australian footballer had moderate burden of Aβ on amyloid (18F-NAV4694) PET |
18F-FTP (FTP) is a first generation tau tracer that has high affinity to the PHF of mixed 3R/4R p-tau present in AD NFTs particularly in late stages [88, 89]. FTP is approved by the FDA to detect p-tau in patients who have cognitive impairment due to AD (https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212123s000lbl.pdf). Unlike FDDNP, FTP has improved specificity and selectivity to p-tau over fibrillar deposits of Aβ [88, 89]. Two separate case studies examined FTP binding in a 71-year-old and a 39-year-old former NFL player [90, 91]. However, we only identified two case-control studies on FTP at the time of the writing of this review. FTP retention was compared between 26 symptomatic former NFL players (ages 40–69) and 31 same-age asymptomatic men without any history of TBI [92]. The former NFL players were required to have at least 2 years of NFL play and a minimum of 12 total years of American football play. At the group level, FTP retention was higher in the former NFL players in the bilateral superior frontal, MTL, and left parietal regions, after p-value adjustment for multiple comparisons. However, variation was seen across participants and there were overlapping individual-level FTP measurements between the former NFL players and controls. The level of FTP uptake in the three brain regions was associated with the number of years playing football. FTP levels were not associated with cognitive and neuropsychiatric test scores.
A case series from the University of California, San Francisco (UCSF), compared FTP uptake between 11 cognitively/behaviorally impaired participants who met the 2014 TES criteria [1] (primarily former American football players), 67 age- and education-matched amyloid-negative participants (male and female) without a known history of exposure to RHI, and 22 amyloid-positive age- and education matched participants with MCI (n = 9) or dementia (n = 13) due to AD (male and female) who also had no known history of RHI exposure [93]. At the individual level, six TES participants showed elevated cortical FTP binding in a frontotemporal pattern (there was diffuse cortical binding in 2 participants). Global mean cortical binding intensity varied and values for three cases fell in the range of the control group. Two TES participants showed non-specific diffuse cortical binding and the other three participants had no binding; for all of these participants, global mean cortical binding fell in the control range. MTL binding intensity values were slightly higher in the TES participants but overlapped with the controls. MTL binding intensity was higher in AD. Frequency maps showed 60% of the TES participants had mildly elevated FTP binding, predominantly in the bilateral frontotemporal cortex and in the medial and lateral parietal lobes; when restricted to amyloid negative TES participants, frontotemporal effects became more confined and there was reduction in binding in parietal regions. The control group showed minimal abnormal voxels, whereas the MCI due to AD group had a diffuse pattern of abnormal voxels with most showing elevated binding in the temporoparietal regions and variable frontal involvement. Dementia due to AD participants had the most diffuse binding, and all had temporoparietal and frontal abnormalities. Voxel-wise analyses showed that the participants with TES had mildly elevated FTP in the left inferior-posterior temporal lobe, in addition to the inferior frontal lobes among the amyloid negative TES participants. There were no group effects between TES and MCI and AD dementia participants, and binding was higher in many of the cortical regions in the AD group. Overall, findings from that study showed individual-level overlap with the amyloid negative participants and tracer retention was lower than observed in participants with AD. The authors concluded that FTP is unlikely to be sensitive to the early detection of CTE. Although not a case-control study in the traditional sense, Vasilevskaya et al. showed higher mean FTP SUVR in former professional and semi-professional contact sport athletes (mostly former American football players) with the APOE e4 allele (n = 13) compared with those without this allele (n = 25) [58]. No effect was found for MAPT diplotypes.
A recent PET-to-autopsy study from UCSF found no statistically significant correlation between regional FTP binding and tau pathology (as measured by immunohistochemistry) in a deceased former NFL player who had stage IV CTE [94]. The time from PET scan to death was 52 months. An autoradiography study examined FTP binding in pathologically confirmed CTE tissue samples [95]. The study included five cases of CTE (n = 1 stage I, n = 3 stage III, n = 1 stage IV). The hippocampus, superior temporal cortex, superior frontal cortex, inferior parietal cortex, and occipital cortex were sampled. The cases had low or no AD neuropathological changes. Autoradiography detected overall minimal FTP binding with the exception of off-target binding to the choroid plexus and the meninges in two of the five cases. Research has shown that FTP has off-binding to the chorioid plexus, particularly in African Americans, and may specifically impact interpretation of binding in certain brain regions (e.g., hippocampus) [96]. In another FTP PET to autopsy study, FTP was sensitive to the detection of advanced Braak stage tau and did not reliably detect and differentiate non-AD NFT pathology [97]. Although CTE was not directly examined in that study, it shows the selectivity of FTP to tau NFTs in AD.
18F-MK-6240 is a second generation PHF-tau tracer developed for AD that shows less off-target (i.e., non-tau related) binding than FTP [98–102]. It is an experimental tau radiotracer not yet approved by the FDA for clinical use. MK-6240 binding mimics Braak neuropathological staging of AD [100, 102]. MK-6240 has a distinct chemical structure that may lead to unique binding affinities across tauopathies. MK-6240 may be more suitable for detecting focal tau deposits seen in early stages of CTE because of low background signal. Krishnadas et al. [103] conducted an in vivo case study on MK-6240 in a 63-year-old former Australian rule football player (who also had a thalamic stroke at age 50) with a Clinical Dementia Rating scale score of 0.5 [103]. The study included 99 cognitively normal amyloid and tau PET negative participants and 37 amyloid and tau PET positive MCI (n = 16) and dementia (n = 21) due to AD participants from the Australian Imaging Biomarkers and Lifestyle study as comparison groups. The comparison groups were 9–10 years older than the participant, on average (ranging from 66 to 76.4 years old). Regions of interest included dorsolateral prefrontal cortex (DLFPC), MTL (entorhinal cortex, hippocampus, parahippocampus, and amygdala), temporoparietal (inferior temporal, fusiform, supramarginal and angular gyri, posterior cingulate, precuneus, superior and inferior parietal, and lateral occipital), and a frontal composite (DLPFC, ventrolateral prefrontal, orbitofrontal, gyrus rectus, anterior cingulate). SUVR was higher in the Australian rule football player for all regions compared with the amyloid and tau negative participants, including > 4 standard deviations higher for the DLPFC. DLPFC SUVR was 0.70 standard deviations higher in the participant relative to the MCI group, whereas the MCI group had higher MTL and temporoparietal SUVR. The AD dementia group had the highest SUVR across all regions. Voxel-wise analysis showed that the participant had high tracer retention in the bilateral superior frontal gyri, particularly in the DLPFC and MTL. Compared with the amyloid and tau negative group, there was mild uptake in the right supramarginal, right orbitofrontal, and bilateral temporal gyri. The former Austrialia rule football player also had higher retention in the bilateral superior frontal regions compared with the MCI group. Conversely, MCI participants had higher MTL and temporoparietal retention. The amyloid and tau positive AD dementia group had greater retention in all cortical regions. MK-6240 binding patterns from that preliminary study are consistent with the neuroanatomical distribution of CTE p-tau pathology.
Despite the above case study, Aguero et al. [98] conducted an autoradiography study that included five cases with autopsy-confirmed tissue of CTE (n = 1 CTE stage II/III, n = 3 CTE stage III, n = 1 CTE stage IV). AD, FTLD-tau, FTLD-TDP-43, LBD, and control tissue samples were also included. Detectable binding was only observed in AD tissues and not in any of the other non-AD tauopathies. There was no detectable binding of MK-6240 in the CTE cases or in the FTLD cases.
Amyloid PET Imaging
Given Aβ deposition is not a consistent feature of CTE, FDA-approved PET ligands that bind to neuritic Aβ (e.g., [(18)F]-Florbetapir) have not been a biomarker target of CTE. It has instead been used to rule out AD. In the aforementioned FTP study of 26 symptomatic former NFL players (ages 40–69) and 31 same-age asymptomatic men without any history of TBI, all but one of the former NFL players had a negative amyloid (florbetapir) PET scan [92]. There were no statistically significant group differences between the former NFL players and the controls in mean cortical cerebellar florbetapir SUVRs. In the UCSF FTP study of 11 TES participants, two were amyloid ([11C]PIB) positive [93]. The two FTP case studies of former NFL players (ages 39 and 71) were also amyloid (florbetapir) negative [90, 91]. The Australian rule football player had moderate levels of Aβ plaques on amyloid (18F-NAV4694) PET imaging.
18F-Fluorodeoxyglucose PET
FDG-PET measures metabolic rates of glucose in the brain and hypometabolism reflects neurodegeneration. AD is characterized by hypometabolism in temporoparietal regions, posterior cingulate cortex, and the MTL with frontal lobe involvement as the disease advances [104–108]. Behavioral variant frontotemporal dementia (bvFTD) includes hypometabolism of bilateral temporal, dorsolateral, and medial prefrontal regions [109]. In the aforementioned UCSF study of 11 participants with TES (primarily former American football players) and 67 amyloid-negative participants [93], six and 30 of the TES participants and amyloid-negative participants had FDG-PET, respectively. Individual SUVR images showed that TES participants had greater frontotemporal hypometabolism with heterogeneous levels of parietal lobe involvement. Voxel-wise analyses showed that the TES participants had decreased metabolism in the frontotemporal regions with involvement of the parietal lobe (i.e., precuneus, supramarginal) but to a much lesser extent. Previous research showed frontal hypometabolism on FDG-PET in five retired professional boxers (mean age = 46.8) compared with four age-matched controls [110]. Hypometabolism was found in the bilateral dorsolateral prefrontal cortices and right middle orbitofrontal cortex. In another FDG-PET study of 19 boxers and 7 controls, boxers had reduced FDG uptake in the frontal lobes bilaterally, posterior cingulate cortex, parieto-occipital lobe, and the cerebellum [111].
SPECT Imaging
A SPECT scan is a nuclear medicine technique that uses a radiotracer to track patterns of cerebral blood flow. SPECT patterns of bilateral temporoparietal hypoperfusion support a diagnosis of AD [112–115]. The clinical application of SPECT is somewhat debated due to uncertain value above and beyond MRI. Amen et al. [116] examined brain SPECT imaging in 100 active and former NFL players and a comparison group of 20 cognitively normal men without a history of TBI. Although the breakdown of active versus retired was not clear, the mean age was 57.27. The NFL players had global reductions in cerebral blood flow on SPECT, including of the prefrontal, temporal, parietal, and occipital lobes, as well as the anterior and posterior cingulate gyrus and the cerebellum. In a subsequent study by Amen et al. [117], among 161 active and former NFL players (mean age = 52) and 124 cognitively normal men and women (mean age = 44), the NFL players had reduced cerebral blood flow on SPECT across 36 brain regions. Brain regions that most accurately discriminated the NFL players from the controls included the anterior superior temporal lobes, rolandic operculum, insula, superior temporal lobes, precuneus, and cerebellar vermis. These effects remained similar when accounting for age, sex, and psychiatric co-morbidities. Although these studies showed evidence of cerebral blood flow reductions, the regional patterns were diffuse and non-specific.
DaTscan is a form of SPECT imaging that involves injection of a radiotracer (i.e., phenyltropane) that binds to nigrostriatal dopaminergic transporters. It is FDA approved to differentiate Parkinson’s disease or parkinsonism from essential tremor, but can differentiate Lewy body dementia and AD [118–120]. To our knowledge, no studies to date have examined DaTscans in former American football players or fighters. Given nigral degeneration is common in CTE and parkinsonism has been reported as a late effect of RHI exposure, particularly from boxing [16, 17, 32, 34, 121], DaTscan may be a valuable imaging modality and warrants study.
Macrostructural MRI
Anatomical T1-Weighted MRI
Figure 2 shows an exemplary antemortem MRI of an 82-year-old brain donor with autopsy confirmed stage IV CTE and who had no other neurodegenerative disease diagnoses. Macrostructural features shown in Fig. 2 are used to summarize our literature review findings.
Fig. 2.

Antemortem structural MRI macrostructural features of a brain donor with autopsy-confirmed CTE. This is an 85-year-old, White, male brain donor with autopsy-confirmed CTE who is from the Veteran Affairs (VA)-Boston University (BU)-Concussion Legacy Foundation (CLF) brain bank and part of the UNITE study. He was an elite American football player. The MRI was obtained via medical record requests and was completed during life as part of clinical care when he was 82 years old. He had a Functional Activities Questionnaire (FAQ) score of 19/30, and a panel of clinicians from the UNITE study determined he had dementia prior to death. Cause of death was cardiovascular disease, and the donor had a history of hypertension and diabetes. The brain donor was neuropathologically diagnosed with stage IV CTE and had no other neurodegenerative disease diagnoses. The MRI shows (a) global atrophy most prominent for the frontal and temporal lobes on axial T1 as well as a posterior cavum septum pellucidum, (b) bilateral medial temporal lobe atrophy on coronal T1, and (c) and moderate microvascular disease on FLAIR
Atrophy
Atrophy patterns on MRI are used to support diagnosis and monitoring of neurodegenerative diseases, like AD and FTD [44]. Atrophy rates serve as outcomes for large-scale multi-center clinical trials of disease-modifying therapies [122] and might have similar usefulness in clinical trials for post-traumatic neurodegeneration [123]. In vivo MRI studies have begun to discern structural MRI signatures in participants at risk for CTE using automated (e.g., Freesurfer) and/or a combination of automated and semi-automated imaging analysis pipelines to generate quantitative volumetric indices. Among the participants with TES and age- and education-matched amyloid-negative participants without known history of exposure to RHI from the UCSF study, voxel-wise analyses showed lower volume of the lateral and medial frontal cortices, insula, and anterior temporal lobes in the TES participants [93]. When compared against a normative MRI dataset, some, but not all, TES participants had cortical thinning and subcortical gray matter volume loss, with volume loss greatest for the hippocampus. In a study focused on frontotemporal regions comparing 19 former Canadian Football League players (mean age = 50, age range = 30–74 years) and age- and education-matched healthy men without a history of concussion, reduced thickness of the left anterior temporal lobe but not the orbitofrontal cortex was reported [124]. Another study compared MRI-derived prefrontal and temporal lobe cortical thickness values between 11 former NCAA Division I American football players (mean age = 27, age range = 24–32, scanned an average of four years after football participation ended) and 10 demographically similar track-and-field athletes. The former football players showed reduced cortical thickness of the left frontal pole, right superior frontal gyrus, superior temporal gyrus, left inferior temporal gyrus, right middle and superior temporal gyri, and bilaterally over the banks of the superior temporal sulcus [125]. The hippocampus was not examined, but there were no group differences for the parahippocampal gyrus or other aspects of the frontal and temporal lobes.
One MRI volumetric study examined limbic system structures among 86 symptomatic former NFL players (mean age = 54.86) and 22 same-age asymptomatic men without a history of RHI or TBI [126]. Findings showed that the former NFL players had smaller bilateral volumes of the amygdala, hippocampus, and the cingulate gyrus compared with the same age men without a history of RHI or TBI. Another study compared volume loss across 14 (left and right) regions of interest (hippocampus, amygdala, total gray matter, brain parenchymal volume, parahippocampal cortex, entorhinal cortex, temporal pole, supramarginal gyrus) in 9 former NFL players (all > 55 years old) and 9 age-matched cognitively normal controls [127]. The former NFL players had greater volume loss of the right hippocampus after correction for multiple comparisons. There were no effects for other regions of interest with multiple correction adjustment. Decreased volume of the hippocampus has similarly been reported in other small samples of aging symptomatic former NFL players [128]. Interestingly, Misquitta et al. [129] found an age-exposure interaction with a greater effect of age on left and right hippocampal volumes in 53 former Canadian Football League players (mean age = 55.6) compared with 25 age and education matched males without a history of concussion, and compared with 321 age-matched males from the Cambridge Centre for Aging and Neuroscience. In a sample of 34 retired fighters from the PFBHS (mean age = 47) and 62 age-matched individuals without a history of TBI, the retired fighters had lower volumes for the hippocampus, amygdala, and thalamus [130]. Effects were not observed for the caudate, putamen, or total brain volume.
There are sparse longitudinal MRI studies of people exposed to RHI. Bernick et al. examined longitudinal change in brain volume in 173 active and retired professional fighters from the PFBHS compared with people without RHI exposure history [131]. The sample included 23 retired boxers, 50 active boxers, 100 active mixed martial art fighters, and 31 cognitively normal individuals with no RHI history. Participants completed annual visits, and all had at least two MRIs with a mean follow-up of approximately 2–3 years, ranging from 1 to 4 years. The retired boxers showed an accelerated decline in the left and right amygdala and right hippocampus.
The above findings should be considered in the context of existing null studies. In a study devoted to glial cell activation and white matter integrity, Coughlin et al. presented supplementary material that compared 14 NFL players (4 active, 10 retired, ages 24–39 years) and 16 age, sex, education, and BMI-matched controls on volumes of the hippocampus, amygdala, temporal pole, entorhinal cortex, parahippocampal cortex, and supramarginal gyrus [132]. Group effects were not observed. These group comparisons were not the objective of the manuscript, and the sample’s young age may explain the null volumetric effects. Zivadinov et al. [133] compared 21 former NFL and National Hockey League (NHL) players (mean age = 56) with 21 age-matched non-contact sport athletes across a range of MRI indices. The authors did not find group differences in global or regional brain volumes. However, the authors did not conduct lobar regional analyses and it is unclear if the former NHL players may have suppressed the effects. Authors of another study claimed minimal evidence of chronic brain injury in sample of 45 former NFL players, including atrophy in only 2 of the 45 former NFL players [54]. However, the study was without a comparison group and was predominantly descriptive. Bang et al. did not find structural MRI differences in five retired professional boxers (mean age = 46.8) compared with four age-matched controls [110]. An additional study compared a priori volumetric regions of interest between 20 former high school football players who had two or more mild TBIs (ages 40–65 years) and 20 age, education, premorbid intellect, and symptom-matched former high school football players who denied a lifetime history of TBI [134]. Regions examined included total intracranial volume, total gray matter, total white matter, bilateral anterior cingulate cortex, bilateral hippocampi, and lateral ventricles. No group effects were found. The meaning of those null effects is not clear, given all participants played American football. Other studies have similarly compared neuroimaging indices between former university-level contact sport athletes with and without a concussion history, showing a range of effects with inferences being drawn on concussion as opposed to RHI exposure [135, 136].
CC Thinning
There is limited in vivo case-control evidence that report on the CC in aging participants at high risk for CTE. In the Bernick et al. longitudinal PFBHS study that included 23 retired boxers, the boxers showed accelerated shrinkage of the central CC compared with their control group (per Table 2 of that manuscript) [131]. In another sample of 34 retired fighters from the PFBHS, retired fighters had lower CC volume relative to 62 age-matched individuals without a history of TBI [130]. A diffusion tensor imaging (DTI) study compared CC integrity between 34 former NFL players (ages 41–79; both with and without symptoms) and 85 age, premorbid intellect, and education matched asymptomatic people without a history of concussion or elite football play. The symptomatic former NFL players (n = 14) had reduced fractional anisotropy (FA) of the CC relative to the matched control group (n = 14) [137]. No such effects were found for the asymptomatic former NFL players.
CSP
A CSP is not specific to CTE and is a frequent MRI finding in the general adult population[138]. In autopsy-confirmed CTE, there are frequent abnormalities of the septum pellucidum [12, 14, 19]. The in vivo MRI literature shows similar findings [93, 130, 139, 140]. In the UCSF study of 11 TES participants, 8 had a CSP ranging in width between 3 and 11 mm and between 1.5 and 4.5 mm in length [93]. In 39 retired fighters from the PFBHS and 63 age-matched individuals without a history of TBI, 77% and 51% of the retired fighters had a CSP and cavum vergae, respectively, compared with 17% and 0% of the age-matched comparison group (p < 0.01) [130]. Length of the CSP and cavum vergae was longer in the retired fighters. Similar findings were reported in a sample of 72 symptomatic former NFL players (mean age = 54.53) and 14 same age asymptomatic men without a history of RHI or TBI [139]. Specifically, 92% of the former NFL players and 57% of the same age men without RHI/TBI had a CSP (p < 0.01). CSP length was greater in the former NFL players. Other research in 17 symptomatic former professional American football players (mean age = 54.6) also found higher rates of a CSP compared with 17 age-/sex-matched individuals without a history of TBI (94% vs 18%) [140]. CSP was of a higher grade (an ordinal measure of width) and longer in length in the former professional football players.
FLAIR MRI
FLAIR MRI is used to visually assess the presence and severity of white matter hyperintensities (WMH). FLAIR WMH are of a presumed vascular etiology and accompany aging and vascular risk factors (e.g., hypertension, diabetes) [141–144]. As part of a dementia evaluation, FLAIR WMH are typically used to discern contributions from cerebral small vessel disease to cognitive or neuropsychiatric impairments and support diagnoses of vascular cognitive disorders (e.g., vascular dementia) [145]. Former elite contact sport athletes might be at increased risk for FLAIR WMH due to increased prevalence of vascular risk factors in this population [146–151]. FLAIR WMH are non-specific and may also reflect a range of white matter neuropathologies related to exposure to RHI, such as white matter rarefaction [8], neuroinflammation [132, 152], axonal loss, demyelination, and gliosis. Hart et al. [137] found greater total (mean diff. = 5.75) and deep (mean diff = 0.79) FLAIR WMH in 10 cognitively impaired former NFL players (mean age = 66.6) compared with 20 age-, estimated-IQ, and education-matched asymptomatic participants without a history of concussion or participation in college or professional football. There was not a statistically significant group effect for periventricular WMH. Among 86 symptomatic former NFL players (mean age = 54.86) and 23 same age asymptomatic men without a history of RHI or TBI, the former NFL players had increased volume of T1-weighted WM hypointensities from Freesurfer controlling for age and total brain volume [153]. The groups had nearly identical vascular risk scores (based on a modified Framingham Stroke Risk Profile score). A limitation of that study was the use of T1-weighted scans to estimate WM lesion volume as opposed to FLAIR. Zivadinov et al. [133] found no differences in total number or volume of WM signal abnormalities between 21 former NFL and NHL players (mean age = 56) with 21 age-matched non-contact sport athletes. White matter lesions were estimated using semi-automated methods on T2/PD/FLAIR images.
SWI/T2*-weighted GRE
SWI/T2*-weighted GRE sequences are routinely obtained to assess for the presence of cerebral microbleeds. Microbleeds can provide evidence for an underlying cerebrovascular disease etiology of cognitive and neuropsychiatric decline and inform on the presence and diagnosis of cerebral amyloid angiopathy (CAA) [154]. Microbleeds have been shown to be a neurological sequelae of TBI [155] and active RHI [50, 156, 157] and may have implications in CTE, especially given the links between exposure to RHI and microvascular pathology [8, 26, 38]. In the neuroimaging studies in participants at high risk for CTE, cerebral microbleeds have not typically been the study focus. In the data that exists, cerebral microbleeds have been infrequently reported [57, 133, 137]. This finding is consistent with minimal presence of microbleeds observed in neuropathological studies of deceased American football players [8].
Advanced Research Neuroimaging Sequences
Studies that have used advance research neuroimaging techniques among individuals at high risk for CTE have shown increased inflammation on translocator protein 18 kDa (TSPO) PET imaging [127, 132]; greater WM injury on DTI, particularly of frontotemporal tracks [125, 132, 136, 137, 158]; activation and connectivity deficits on functional MRI [159–161]; neurochemical alterations on MR spectroscopy global and regional cerebral blood flow reductions on arterial spin labeling MRI [117, 137]; blood-brain barrier disruption on dynamic contrast-enhanced (DCE) MRI [162]; and electrophysiological changes on EEG [116, 163]. DTI has been extensively studied in the setting of exposure to RHI, given diffuse axonal injury is cardinal pathology of TBI [64, 164]. Ex vivo DTI of CTE tissue has also shown an association between fractional anisotropy and axonal disruption in white matter regions that co-localized to areas with CTE p-tau pathology [165]. DTI involves extended scan acquisition time, post-processing can be complex, and the clinical meaning of DTI-derived indices (e.g., fractional anisotropy) is not clear, particularly in the setting of neurodegenerative diseases. It is for these reasons and lack of FDA approval for indications in AD/ADRD that DTI, along with the other aforementioned sequences, are not routine for dementia evaluations. Research neuroimaging sequences have and will continue to inform on disease pathogenesis, mechanisms, and co-morbid neuropathologies.
Discussion
Here, we presented the current evidence on promising neuroimaging biomarkers of CTE, focusing on neuroimaging modalities routinely used as part of a dementia evaluation. We make the following conclusions: (1) an optimal tau radiotracer that has high affinity for the 3R/4R PHF of NFTs in CTE has not yet been identified, (2) negative amyloid PET has been a consistent findining in symptomatic former elite American football players, (3) anatomical MRI and FDG-PET studies of participants at risk for CTE show patterns of frontotemporal and MTL neurodegeneration, (4) former elite American football players have increased burden of FLAIR WMH, but cerebral microbleeds are less frequent, (5) case-control evidence shows that people with substantial exposure to RHI are at increased odds for having a CSP, and (6) the evidence on neuroimaging biomarkers of CTE is limited by cross-sectional studies of small samples.
Identification of a tau PET radiotracer with affinity to CTE p-tau is of high priority, as it would enable disease detection and facilitate clinical trials of treatment andprevention. Although the CTE tau isoform has similarities to other tauopathies (e.g., AD), cryo-electron microscope studies show differences in the tau molecularstructure[71, 72]. FDDNP is not a targeted tracer due to off-target and non-specific binding. The current evidence on FTP for the detection of CTE p-tau pathology isinconclusive. Although mean group differences in FTP binding have been observed between participants at risk for CTE and same age men without a history of TBI [92, 93], there has not been clear separation at the individual level and tracer retention has been lower than expected. Autoradiography studies also show minimal binding of FTP to post-mortem CTE tissue. Larger studies on FTP in former NFL players, former college football players, and asymptomatic men without RHI/TBI are currently underway (i.e., DIAGNOSE CTE Research Project) to better understand the useulness of FTP as a biomarker for CTE. Second-generation radiotracers may have better binding properties for CTE p-tau, e.g., MK-6240 or PI-2620. Members of our team are collaborating with UCSF on a study known as the “Focused Imaging for the Neurodegenerative Disease CTE” (FIND-CTE) (PI: Rabinovici, MPI: Alosco). The goal of FIND-CTE is to identify neuroimaging biomarkers of CTE with a focus on tau PET.
Even if a tracer binds to p-tau isoforms in multiple tauopathies (e.g., AD, FTD, CTE), tau PET could inform on the distribution of p-tau to facilitate differential diagnosis. Tau PET binding in CTE would be expected to follow the progression and distribution of p-tau severity according to the McKee CTE staging scheme, which is distinct from the distribution and progression of p-tau in AD and FTD. Florbetapir PET can aid in differential diagnosis. A negative amyloid PET in an individual at risk for CTE (based on substantial exposure to RHI and clinical presentation) in combination with evidence of binding on tau PET would increase probability of underlying CTE. An elevated amyloid PET in a similar individual at risk for CTE does not rule out underlying CTE pathology; rather, it suggests that there may be co-morbid AD neuropathologic changes.
There is consistent MRI evidence of frontotemporal and MTL neurodegeneration in participants with substantial exposure to RHI. These patterns are similar to AD and bvFTD. A comprehensive review on the similarities and differences, including on molecular and structural neuroimaging, between CTE, AD, and bvFTD, was recently published [166]. CTE can be differentiated from AD by more severe patterns of frontotemporal atrophy and sparing of posterior brain regions especially in early disease stages. In CTE, the hippocampus is affected later in the disease course and with different hippocampal subfields disproportionately affected (CA2 and CA4) compared with AD. No neuroimaging studies have examined hippocampal subfields in participants at high risk for CTE. Differential diagnosis based on atrophy patterns between CTE and bvFTD is more challenging, as bvFTD is characterized by frontal and anterior temporal lobe atrophy [167–169]. The dorsolateral frontal cortex is an initial site of p-tau in CTE, and regional frontal lobe atrophy may assist with CTE versus bvFTD differential. Perhaps a ventral atrophy pattern involving the salience network, commonly seen in bvFTD [169, 170], may differentiate it from CTE. There are other non-specific MRI features that may be supportive of CTE. For example, thalamic atrophy and an enlarged third ventricle are observed in CTE and might help with disease differentiation. CSP is a common finding of CTE that may be related to exposure to RHI and that has been infrequently reported in other neurodegenerative tauopathies. Although to a lesser extent, thinning of the CC has also been a reported neuropathological feature of CTE. CC atrophy is a frequent MRI finding in AD and FTD [171–173] and may be a downstream effect of neurodegeneration [171]. In CTE, however, the CC white matter bundle might be susceptible to injury from the effects of RHI exposure [55] and TBI [174], with callosal atrophy worsening with advancing aging and neurodegeneration.
While the literature offers promising targets, validated neuroimaging biomarkers of CTE do not currently exist. Although PET imaging and structural MRI are commonly used tools in memory disorders clinics, they should primary be used to guide diagnosis of dementing illnesses for which they are indicated, rather than to make diagnostic determinations of CTE. Before these tools can be used for CTE diagnosis, additional studies are required. Prospective imaging-pathological correlation studies will be essential for biomarker development and validation in CTE. These studies are ongoing, but they require a sufficient number of participants to pass away to make inferences and can therefore take years to complete. Neuroimaging biomarkers also need to show a high level of sensitivity and specificity in distinguishing people at high risk for CTE from those with normal cognition and with other neurodegenerative diseases. Larger longitudinal studies among participants at high risk for CTE are much needed, as sample sizes have been quite small and nearly all studies have been cross-sectional. Documentation of change over time of both biomarkers (e.g., increased tau accumulation, worsening atrophy) and clinically meaningful signs and symptoms (e.g., neuropsychological test performance) is crucial for biomarker validation. Such longitudinal studies would also help to differentiate whether neuroimaging abnormalities in former contact sport athletes at risk for CTE are related to past stable damage from TBI or are from an underlying neurodegenerative disease process. As with other neurodegenerative diseases, CTE does not occur in isolation and CTE tau pathology is unlikely to be the sole cause of cognitive impairment. As opposed to focusing on isolated biomarkers, examination of multiple biomarkers or a biomarker panel may increase specificity and may help to identify multiple pathologies concurrently [175].
There are recruitment considerations in biomarker development for CTE. Unlike AD, validated clinical diagnostic criteria for CTE do not yet exist. A large, autopsy-based study assessing the validity of the 2014 TES research criteria suggests they rule out but do not rule in CTE pathology [177]. Updated criteria have recently been proposed but have not been validated. Although CTE has been neuropathologically diagnosed in various other contact sport athletes, military veterans, and other populations [11, 12, 18, 21–28], RHI exposure in these populations is quite heterogeneous and little is known about the extent or severity of RHI from these sources that is needed to confer risk for CTE. Our current knowledge of risk for CTE is based on studies in former elite football players and boxers, including published research that identified cutoffs for odds of CTE based on years of American football play [13]. In addition, CTE, like other neurodegenerative diseases, is strongly associated with age [20]. Recruitment for CTE biomarker development should target populations with the highest probability of meaningful CTE pathology, based on exposure and age.
Recruitment of an appropriate “control” group is also challenging in this setting, especially in terms of identifying individuals who have comparable physical, medical, socioeconomic, and psychosocial backgrounds as the former elite contact sport athletes. There are also multiple different ways a control group can be operationalized depending on the question being investigated. A control group can be defined based on the predictor (e.g., exposure to RHI) or on the outcome (e.g., CTE). Because CTE cannot accurately be diagnosed during life at this time, defining the control group based on CTE status in human subject studies is difficult. Therefore, most human subject studies have utilized a control group of individuals who have no history of exposure to RHI. Some studies have also required controls to be asymptomatic in addition to having no exposure to RHI. Selection of participants on both the predictor (exposure to RHI) and variables related to the biomarker outcome (symptoms) may result in biased effect estimates. This study design may be appropriate in certain circumstances, such as to examine the usefulness of a new/novel biomarker and to answer questions like, “Does this novel tau PET radiotracer bind to CTE p-tau pathology?” If that novel tau tracer cannot differentiate symptomatic individuals with substantial exposure to RHI from asymptomatic individuals without RHI exposure, it can be confidently concluded that the tau tracer is not a useful biomarker for CTE. Conversely, this study design would not be appropriate if the goal was to make inferences on the magnitude of group differences on the novel biomarker. Regardless, there should be clear operationalization of the control group that is determined by the scientific question(s) posed.
In summary, the current review provides insights into promising neuroimaging biomarkers of CTE. Tau PET tracers that have high affiniity to CTE p-tau have not yet been identified and converging imaging evidence shows frontotemporal and MTL neurodegenation in participants at high risk for CTE. Once validated, these biomarkers will likely help to support a “probable CTE” diagnosis in life. Disease detection and diagnosis is a critical next step in this field to counsel patients and families and so that treatment and prevention trials can begin.
Supplementary Information
Below is the link to the electronic supplementary material.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This work was supported by grant funding from the National Institute on Aging (R01AG061028, P30AG13846 supplement 0572063345-5), NINDS (U54NS115266, K23NS102399), and the DOD (W81XWH1810580).
Declarations
Disclaimer
Funders did not have a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
6/14/2021
The original version of this paper was updated. The heading “MRI-Pathological Correlation Evidence” needs to be removed.
References
- 1.Montenigro PH, Alosco ML, Martin BM, et al. Cumulative Head Impact Exposure Predicts Later-Life Depression, Apathy, Executive Dysfunction, and Cognitive Impairment in Former High School and College Football Players. J Neurotrauma. 2017;34:328–340. doi: 10.1089/neu.2016.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morley WA. Environmental Subconcussive Injury, Axonal Injury, and Chronic Traumatic Encephalopathy. Front Neurol. 2018;9:166. doi: 10.3389/fneur.2018.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahrami N, Sharma D, Rosenthal S, et al. Subconcussive Head Impact Exposure and White Matter Tract Changes over a Single Season of Youth Football. Radiology. 2016;281:919–926. doi: 10.1148/radiol.2016160564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belanger HG, Vanderploeg RD, McAllister T. Subconcussive Blows to the Head: A Formative Review of Short-term Clinical Outcomes. J Head Trauma Rehabil. 2016;31:159–166. doi: 10.1097/HTR.0000000000000138. [DOI] [PubMed] [Google Scholar]
- 5.Broglio SP, Eckner JT, Paulson HL, Kutcher JS. Cognitive decline and aging: the role of concussive and subconcussive impacts. Exerc Sport Sci Rev. 2012;40:138–144. doi: 10.1097/JES.0b013e3182524273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gysland SM, Mihalik JP, Register-Mihalik JK, Trulock SC, Shields EW, Guskiewicz KM. The relationship between subconcussive impacts and concussion history on clinical measures of neurologic function in collegiate football players. Ann Biomed Eng. 2012;40:14–22. doi: 10.1007/s10439-011-0421-3. [DOI] [PubMed] [Google Scholar]
- 7.Martini D, Eckner J, Kutcher J, Broglio SP. Subconcussive head impact biomechanics: comparing differing offensive schemes. Med Sci Sports Exerc. 2013;45:755–761. doi: 10.1249/MSS.0b013e3182798758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alosco ML, Stein TD, Tripodis Y, et al. Association of White Matter Rarefaction, Arteriolosclerosis, and Tau With Dementia in Chronic Traumatic Encephalopathy JAMA Neurol 2019;76:1298. [DOI] [PMC free article] [PubMed]
- 9.Bieniek KF, Blessing MM, Heckman MG, et al. Association between contact sports participation and chronic traumatic encephalopathy: a retrospective cohort study. Brain Pathol. 2020;30:63–74. doi: 10.1111/bpa.12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. 2015;130:877–889. doi: 10.1007/s00401-015-1502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling H, Morris HR, Neal JW, et al. Mixed pathologies including chronic traumatic encephalopathy account for dementia in retired association football (soccer) players. Acta Neuropathol. 2017;133:337–352. doi: 10.1007/s00401-017-1680-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mez J, Daneshvar DH, Abdolmohammadi B, et al. Duration of American Football Play and Chronic Traumatic Encephalopathy. Ann Neurol. 2020;87:116–131. doi: 10.1002/ana.25611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mez J, Daneshvar DH, Kiernan PT, et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA. 2017;318:360–370. doi: 10.1001/jama.2017.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/S0033291700049588. [DOI] [PubMed] [Google Scholar]
- 16.Martland HS. Punch drunk. JAMA. 1928;91:1103–1107. doi: 10.1001/jama.1928.02700150029009. [DOI] [Google Scholar]
- 17.Millspaugh JA. Dementia pugilistica. US Naval Med. Bulletin. 1937;35:297–361. [Google Scholar]
- 18. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player Neurosurgery 2005;57:128-134. [DOI] [PubMed]
- 19.McKee AC, Cairns NJ, Dickson DW, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131:75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alosco ML, Cherry JD, Huber BR, et al. Characterizing tau deposition in chronic traumatic encephalopathy (CTE): utility of the McKee CTE staging scheme. Acta Neuropathol. 2020;140:495–512. doi: 10.1007/s00401-020-02197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckland ME, Sy J, Szentmariay I, et al. Chronic traumatic encephalopathy in two former Australian National Rugby League players. Acta Neuropathol Commun. 2019;7:97. doi: 10.1186/s40478-019-0751-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omalu B, Hammers JL, Bailes J, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus. 2011;31:E3. doi: 10.3171/2011.9.FOCUS11178. [DOI] [PubMed] [Google Scholar]
- 23.Omalu BI, Fitzsimmons RP, Hammers J, Bailes J. Chronic traumatic encephalopathy in a professional American wrestler. J Forensic Nurs. 2010;6:130–136. doi: 10.1111/j.1939-3938.2010.01078.x. [DOI] [PubMed] [Google Scholar]
- 24.Pearce AJ, Sy J, Lee M, et al. Chronic traumatic encephalopathy in a former Australian rules football player diagnosed with Alzheimer's disease. Acta Neuropathol Commun. 2020;8:23. doi: 10.1186/s40478-020-0895-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81:1122–1129. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tagge CA, Fisher AM, Minaeva OV, et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain. 2018;141:422–458. doi: 10.1093/brain/awx350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omalu BI, DeKosky ST, Hamilton RL, et al. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery 2006;59:1086–1092. [DOI] [PubMed]
- 28.Omalu BI, Hamilton RL, Kamboh MI, DeKosky ST, Bailes J. Chronic traumatic encephalopathy (CTE) in a National Football League Player: Case report and emerging medicolegal practice questions. J Forensic Nurs. 2010;6:40–46. doi: 10.1111/j.1939-3938.2009.01064.x. [DOI] [PubMed] [Google Scholar]
- 29.Stein TD, Alvarez VE, McKee AC. Concussion in Chronic Traumatic Encephalopathy. Curr Pain Headache Rep. 2015;19:47. doi: 10.1007/s11916-015-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jordan BD. The clinical spectrum of sport-related traumatic brain injury. Nat Rev Neurol. 2013;9:222–230. doi: 10.1038/nrneurol.2013.33. [DOI] [PubMed] [Google Scholar]
- 31.Victoroff J. Traumatic encephalopathy: review and provisional research diagnostic criteria. NeuroRehabilitation. 2013;32:211–224. doi: 10.3233/NRE-130839. [DOI] [PubMed] [Google Scholar]
- 32.Montenigro PH, Baugh CM, Daneshvar DH, et al. Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther. 2014;6:68. doi: 10.1186/s13195-014-0068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katz DI, Bernick C, Dodick DW, et al. NINDS Consensus Diagnostic Criteria for Traumatic Encephalopathy Syndrome. Neurology in press. https://pubmed.ncbi.nlm.nih.gov/33722990/ [DOI] [PMC free article] [PubMed]
- 34.Adams JW, Alvarez VE, Mez J, et al. Lewy Body Pathology and Chronic Traumatic Encephalopathy Associated With Contact Sports. J Neuropathol Exp Neurol. 2018;77:757–768. doi: 10.1093/jnen/nly065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alosco ML, Mez J, Tripodis Y, et al. Age of first exposure to tackle football and chronic traumatic encephalopathy. Ann Neurol. 2018;83:886–901. doi: 10.1002/ana.25245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein TD, Montenigro PH, Alvarez VE, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. 2015;130:21–34. doi: 10.1007/s00401-015-1435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Standring OJ, Friedberg J, Tripodis Y, et al. Contact sport participation and chronic traumatic encephalopathy are associated with altered severity and distribution of cerebral amyloid angiopathy. Acta Neuropathol. 2019;138:401–413. doi: 10.1007/s00401-019-02031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee EB, Kinch K, Johnson VE, Trojanowski JQ, Smith DH, Stewart W. Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol. 2019;138:389–399. doi: 10.1007/s00401-019-02030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stein TD, Alvarez VE, McKee AC. Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther. 2014;6:4. doi: 10.1186/alzrt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83:74–83. doi: 10.1002/ana.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchman AS, Yu L, Wilson RS, et al. Progressive parkinsonism in older adults is related to the burden of mixed brain pathologies. Neurology. 2019;92:e1821–e1830. doi: 10.1212/WNL.0000000000007315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu L, Wang T, Wilson RS, et al. Common age-related neuropathologies and yearly variability in cognition. Ann Clin Transl Neurol. 2019;6:2140–2149. doi: 10.1002/acn3.50857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jack CR, Jr, Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alosco ML, Tripodis Y, Fritts NG, et al. Cerebrospinal fluid tau, Abeta, and sTREM2 in Former National Football League Players: Modeling the relationship between repetitive head impacts, microglial activation, and neurodegeneration. Alzheimers Dement. 2018;14:1159–1170. doi: 10.1016/j.jalz.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cherry JD, Stein TD, Tripodis Y, et al. CCL11 is increased in the CNS in chronic traumatic encephalopathy but not in Alzheimer's disease. PLoS One. 2017;12:e0185541. doi: 10.1371/journal.pone.0185541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alosco ML, Tripodis Y, Jarnagin J, et al. Repetitive head impact exposure and later-life plasma total tau in former National Football League players. Alzheimers Dement (Amst) 2017;7:33–40. doi: 10.1016/j.dadm.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernick C, Zetterberg H, Shan G, Banks S, Blennow K. Longitudinal Performance of Plasma Neurofilament Light and Tau in Professional Fighters: The Professional Fighters Brain Health Study. J Neurotrauma. 2018;35:2351–2356. doi: 10.1089/neu.2017.5553. [DOI] [PubMed] [Google Scholar]
- 49.Bernick C, Banks SJ, Shin W, et al. Repeated head trauma is associated with smaller thalamic volumes and slower processing speed: the Professional Fighters' Brain Health Study. Br J Sports Med. 2015;49:1007–1011. doi: 10.1136/bjsports-2014-093877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JK, Wu J, Banks S, et al. Prevalence of Traumatic Findings on Routine MRI in a Large Cohort of Professional Fighters. AJNR Am J Neuroradiol. 2017;38:1303–1310. doi: 10.3174/ajnr.A5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mishra V, Sreenivasan K, Banks SJ, et al. Investigating structural and perfusion deficits due to repeated head trauma in active professional fighters. Neuroimage Clin. 2018;17:616–627. doi: 10.1016/j.nicl.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mishra VR, Zhuang X, Sreenivasan KR, et al. Multimodal MR Imaging Signatures of Cognitive Impairment in Active Professional Fighters. Radiology. 2017;285:555–567. doi: 10.1148/radiol.2017162403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuhn AW, Zuckerman SL, Solomon GS, Casson IR, Viano DC. Interrelationships Among Neuroimaging Biomarkers, Neuropsychological Test Data, and Symptom Reporting in a Cohort of Retired National Football League Players. Sports Health. 2017;9:30–40. doi: 10.1177/1941738116674006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Casson IR, Viano DC, Haacke EM, Kou Z, LeStrange DG. Is There Chronic Brain Damage in Retired NFL Players? Neuroradiology, Neuropsychology, and Neurology Examinations of 45 Retired Players. Sports Health. 2014;6:384–395. doi: 10.1177/1941738114540270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stamm JM, Koerte IK, Muehlmann M, et al. Age at First Exposure to Football Is Associated with Altered Corpus Callosum White Matter Microstructure in Former Professional Football Players. J Neurotrauma. 2015;32:1768–1776. doi: 10.1089/neu.2014.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alosco ML, Tripodis Y, Koerte IK, et al. Interactive Effects of Racial Identity and Repetitive Head Impacts on Cognitive Function, Structural MRI-Derived Volumetric Measures, and Cerebrospinal Fluid Tau and Abeta. Front Hum Neurosci. 2019;13:440. doi: 10.3389/fnhum.2019.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardner RC, Possin KL, Hess CP, et al. Evaluating and treating neurobehavioral symptoms in professional American football players: Lessons from a case series. Neurol Clin Pract. 2015;5:285–295. doi: 10.1212/CPJ.0000000000000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vasilevskaya A, Taghdiri F, Burke C, et al. Interaction of APOE4 alleles and PET tau imaging in former contact sport athletes. Neuroimage Clin. 2020;26:102212. doi: 10.1016/j.nicl.2020.102212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K. Association of Mild Traumatic Brain Injury With and Without Loss of Consciousness With Dementia in US Military Veterans. JAMA Neurol. 2018;75:1055–1061. doi: 10.1001/jamaneurol.2018.0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fann JR, Ribe AR, Pedersen HS, et al. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry. 2018;5:424–431. doi: 10.1016/S2215-0366(18)30065-8. [DOI] [PubMed] [Google Scholar]
- 61.Lee YK, Hou SW, Lee CC, Hsu CY, Huang YS, Su YC. Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One. 2013;8:e62422. doi: 10.1371/journal.pone.0062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nordstrom A, Nordstrom P. Traumatic brain injury and the risk of dementia diagnosis: A nationwide cohort study. PLoS Med. 2018;15:e1002496. doi: 10.1371/journal.pmed.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plassman BL, Grafman J. Traumatic brain injury and late-life dementia. Handb Clin Neurol. 2015;128:711–722. doi: 10.1016/B978-0-444-63521-1.00044-3. [DOI] [PubMed] [Google Scholar]
- 64.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hof PR, Bouras C, Buee L, Delacourte A, Perl DP, Morrison JH. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer's disease cases. Acta Neuropathol. 1992;85:23–30. doi: 10.1007/BF00304630. [DOI] [PubMed] [Google Scholar]
- 68.Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 69.Mez J, Solomon TM, Daneshvar DH, et al. Assessing clinicopathological correlation in chronic traumatic encephalopathy: rationale and methods for the UNITE study. Alzheimers Res Ther. 2015;7:62. doi: 10.1186/s13195-015-0148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cherry JD, Kim SH, Stein TD, et al. Evolution of neuronal and glial tau isoforms in chronic traumatic encephalopathy. Brain Pathol. 2020;30:913–925. doi: 10.1111/bpa.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. 2019;568:420–423. doi: 10.1038/s41586-019-1026-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. 2018;561:137–140. doi: 10.1038/s41586-018-0454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration. Nature. 2020;580:283–287. doi: 10.1038/s41586-020-2043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsai RM, Boxer AL. Therapy and clinical trials in frontotemporal dementia: past, present, and future. J Neurochem. 2016;138(Suppl 1):211–221. doi: 10.1111/jnc.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cash DM, Rohrer JD, Ryan NS, Ourselin S, Fox NC. Imaging endpoints for clinical trials in Alzheimer's disease. Alzheimers Res Ther. 2014;6:87. doi: 10.1186/s13195-014-0087-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med 2014;6:228fs13. [DOI] [PMC free article] [PubMed]
- 77.Rabinovici GD, Gatsonis C, Apgar C, et al. Association of Amyloid Positron Emission Tomography With Subsequent Change in Clinical Management Among Medicare Beneficiaries With Mild Cognitive Impairment or Dementia. JAMA. 2019;321:1286–1294. doi: 10.1001/jama.2019.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Braskie MN, Klunder AD, Hayashi KM, et al. Plaque and tangle imaging and cognition in normal aging and Alzheimer's disease. Neurobiol Aging. 2010;31:1669–1678. doi: 10.1016/j.neurobiolaging.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shoghi-Jadid K, Small GW, Agdeppa ED, et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24–35. doi: 10.1097/00019442-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 80.Small GW, Kepe V, Ercoli LM, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 81.Barrio JR, Small GW, Wong KP, et al. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc Natl Acad Sci U S A. 2015;112:E2039–E2047. doi: 10.1073/pnas.1409952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Small GW, Kepe V, Siddarth P, et al. PET scanning of brain tau in retired national football league players: preliminary findings. Am J Geriatr Psychiatry. 2013;21:138–144. doi: 10.1016/j.jagp.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 83.Chen ST, Siddarth P, Merrill DA, et al. FDDNP-PET Tau Brain Protein Binding Patterns in Military Personnel with Suspected Chronic Traumatic Encephalopathy1. J Alzheimers Dis. 2018;65:79–88. doi: 10.3233/JAD-171152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Omalu B, Small GW, Bailes J, et al. Postmortem Autopsy-Confirmation of Antemortem [F-18]FDDNP-PET Scans in a Football Player With Chronic Traumatic Encephalopathy. Neurosurgery. 2018;82:237–246. doi: 10.1093/neuros/nyx536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Agdeppa ED, Kepe V, Liu J, et al. Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for beta-amyloid plaques in Alzheimer's disease. J Neurosci 2001;21:RC189. [DOI] [PMC free article] [PubMed]
- 86.Bresjanac M, Smid LM, Vovko TD, Petric A, Barrio JR, Popovic M. Molecular-imaging probe 2-(1-[6-[(2-fluoroethyl)(methyl) amino]-2-naphthyl]ethylidene) malononitrile labels prion plaques in vitro. J Neurosci. 2003;23:8029–8033. doi: 10.1523/JNEUROSCI.23-22-08029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ossenkoppele R, Tolboom N, Foster-Dingley JC, et al. Longitudinal imaging of Alzheimer pathology using [11C]PIB, [18F]FDDNP and [18F]FDG PET. Eur J Nucl Med Mol Imaging. 2012;39:990–1000. doi: 10.1007/s00259-012-2102-3. [DOI] [PubMed] [Google Scholar]
- 88.Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78:787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xia CF, Arteaga J, Chen G, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement. 2013;9:666–676. doi: 10.1016/j.jalz.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 90.Dickstein DL, Pullman MY, Fernandez C, et al. Cerebral [(18) F]T807/AV1451 retention pattern in clinically probable CTE resembles pathognomonic distribution of CTE tauopathy. Transl Psychiatry. 2016;6:e900. doi: 10.1038/tp.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mitsis EM, Riggio S, Kostakoglu L, et al. Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatry. 2014;4:e441. doi: 10.1038/tp.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stern RA, Adler CH, Chen K, et al. Tau Positron-Emission Tomography in Former National Football League Players. N Engl J Med. 2019;380:1716–1725. doi: 10.1056/NEJMoa1900757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lesman-Segev OH, La Joie R, Stephens ML, et al. Tau PET and multimodal brain imaging in patients at risk for chronic traumatic encephalopathy. Neuroimage Clin. 2019;24:102025. doi: 10.1016/j.nicl.2019.102025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mantyh WG, Spina S, Lee A, et al. Tau Positron Emission Tomographic Findings in a Former US Football Player With Pathologically Confirmed Chronic Traumatic Encephalopathy. JAMA Neurol. 2020;77:517–521. doi: 10.1001/jamaneurol.2019.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marquie M, Aguero C, Amaral AC, et al. [(18)F]-AV-1451 binding profile in chronic traumatic encephalopathy: a postmortem case series. Acta Neuropathol Commun. 2019;7:164. doi: 10.1186/s40478-019-0808-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee CM, Jacobs HIL, Marquie M, et al. 18F-Flortaucipir Binding in Choroid Plexus: Related to Race and Hippocampus Signal. J Alzheimers Dis. 2018;62:1691–1702. doi: 10.3233/JAD-170840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Soleimani-Meigooni DN, Iaccarino L, La Joie R, et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer's disease and other neurodegenerative diseases. Brain. 2020;143:3477–3494. doi: 10.1093/brain/awaa276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aguero C, Dhaynaut M, Normandin MD, et al. Autoradiography validation of novel tau PET tracer [F-18]-MK-6240 on human postmortem brain tissue. Acta Neuropathol Commun. 2019;7:37. doi: 10.1186/s40478-019-0686-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Betthauser TJ, Cody KA, Zammit MD, et al. In Vivo Characterization and Quantification of Neurofibrillary Tau PET Radioligand (18)F-MK-6240 in Humans from Alzheimer Disease Dementia to Young Controls. J Nucl Med. 2019;60:93–99. doi: 10.2967/jnumed.118.209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Betthauser TJ, Koscik RL, Jonaitis EM, et al. Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age. Brain. 2020;143:320–335. doi: 10.1093/brain/awz378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hostetler ED, Walji AM, Zeng Z, et al. Preclinical Characterization of 18F-MK-6240, a Promising PET Tracer for In Vivo Quantification of Human Neurofibrillary Tangles. J Nucl Med. 2016;57:1599–1606. doi: 10.2967/jnumed.115.171678. [DOI] [PubMed] [Google Scholar]
- 102.Pascoal TA, Shin M, Kang MS, et al. In vivo quantification of neurofibrillary tangles with [(18)F]MK-6240. Alzheimers Res Ther. 2018;10:74. doi: 10.1186/s13195-018-0402-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Krishnadas N, Dore V, Lamb F, et al. Case Report: (18)F-MK6240 Tau Positron Emission Tomography Pattern Resembling Chronic Traumatic Encephalopathy in a Retired Australian Rules Football Player. Front Neurol. 2020;11:598980. doi: 10.3389/fneur.2020.598980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim EJ, Cho SS, Jeong Y, et al. Glucose metabolism in early onset versus late onset Alzheimer's disease: an SPM analysis of 120 patients. Brain. 2005;128:1790–1801. doi: 10.1093/brain/awh539. [DOI] [PubMed] [Google Scholar]
- 105.Minoshima S, Frey KA, Kuhl DE. Cerebellar metabolic reduction in Alzheimer's disease and data normalization. J Nucl Med. 1998;39:374–376. [PubMed] [Google Scholar]
- 106.Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- 107.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging 2005;32:486–510. [DOI] [PubMed]
- 108.Silverman DH, Small GW, Chang CY, et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA. 2001;286:2120–2127. doi: 10.1001/jama.286.17.2120. [DOI] [PubMed] [Google Scholar]
- 109.Bejanin A, Tammewar G, Marx G, et al. Longitudinal structural and metabolic changes in frontotemporal dementia. Neurology. 2020;95:e140–e154. doi: 10.1212/WNL.0000000000009760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bang SA, Song YS, Moon BS, et al. Neuropsychological, Metabolic, and GABAA Receptor Studies in Subjects with Repetitive Traumatic Brain Injury. J Neurotrauma. 2016;33:1005–1014. doi: 10.1089/neu.2015.4051. [DOI] [PubMed] [Google Scholar]
- 111.Provenzano FA, Jordan B, Tikofsky RS, Saxena C, Van Heertum RL, Ichise M. F-18 FDG PET imaging of chronic traumatic brain injury in boxers: a statistical parametric analysis. Nucl Med Commun. 2010;31:952–957. doi: 10.1097/MNM.0b013e32833e37c4. [DOI] [PubMed] [Google Scholar]
- 112.Jagust W, Thisted R, Devous MD, Sr, et al. SPECT perfusion imaging in the diagnosis of Alzheimer's disease: a clinical-pathologic study. Neurology. 2001;56:950–956. doi: 10.1212/WNL.56.7.950. [DOI] [PubMed] [Google Scholar]
- 113.Jagust WJ, Johnson KA, Holman BL. SPECT perfusion imaging in the diagnosis of dementia. J Neuroimaging. 1995;5(Suppl 1):S45–52. doi: 10.1111/jon19955s1s45. [DOI] [PubMed] [Google Scholar]
- 114.Jagust WJ, Budinger TF, Reed BR. The diagnosis of dementia with single photon emission computed tomography. Arch Neurol. 1987;44:258–262. doi: 10.1001/archneur.1987.00520150014011. [DOI] [PubMed] [Google Scholar]
- 115.Rollin-Sillaire A, Bombois S, Deramecourt V, et al. Contribution of single photon emission computed tomography to the differential diagnosis of dementia in a memory clinic. J Alzheimers Dis. 2012;30:833–845. doi: 10.3233/JAD-2012-111067. [DOI] [PubMed] [Google Scholar]
- 116.Amen DG, Newberg A, Thatcher R, et al. Impact of playing American professional football on long-term brain function. J Neuropsychiatry Clin Neurosci. 2011;23:98–106. doi: 10.1176/appi.neuropsych.23.1.98. [DOI] [PubMed] [Google Scholar]
- 117.Amen DG, Willeumier K, Omalu B, Newberg A, Raghavendra C, Raji CA. Perfusion Neuroimaging Abnormalities Alone Distinguish National Football League Players from a Healthy Population. J Alzheimers Dis. 2016;53:237–241. doi: 10.3233/JAD-160207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Colloby SJ, Firbank MJ, Pakrasi S, et al. A comparison of 99mTc-exametazime and 123I-FP-CIT SPECT imaging in the differential diagnosis of Alzheimer's disease and dementia with Lewy bodies. Int Psychogeriatr. 2008;20:1124–1140. doi: 10.1017/S1041610208007709. [DOI] [PubMed] [Google Scholar]
- 119.O'Brien JT, Colloby S, Fenwick J, et al. Dopamine transporter loss visualized with FP-CIT SPECT in the differential diagnosis of dementia with Lewy bodies. Arch Neurol. 2004;61:919–925. doi: 10.1001/archneur.61.6.919. [DOI] [PubMed] [Google Scholar]
- 120.Vlaar AM, de Nijs T, Kessels AG, et al. Diagnostic value of 123I-ioflupane and 123I-iodobenzamide SPECT scans in 248 patients with parkinsonian syndromes. Eur Neurol. 2008;59:258–266. doi: 10.1159/000115640. [DOI] [PubMed] [Google Scholar]
- 121.Montenigro PH, Bernick C, Cantu RC. Clinical features of repetitive traumatic brain injury and chronic traumatic encephalopathy. Brain Pathol. 2015;25:304–317. doi: 10.1111/bpa.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Graham NS, Sharp DJ. Understanding neurodegeneration after traumatic brain injury: from mechanisms to clinical trials in dementia. J Neurol Neurosurg Psychiatry. 2019;90:1221–1233. doi: 10.1136/jnnp-2017-317557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Goswami R, Dufort P, Tartaglia MC, et al. Frontotemporal correlates of impulsivity and machine learning in retired professional athletes with a history of multiple concussions. Brain Struct Funct. 2016;221:1911–1925. doi: 10.1007/s00429-015-1012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Adler CM, DelBello MP, Weber W, et al. MRI Evidence of Neuropathic Changes in Former College Football Players. Clin J Sport Med. 2018;28:100–105. doi: 10.1097/JSM.0000000000000391. [DOI] [PubMed] [Google Scholar]
- 126.Lepage C, Muehlmann M, Tripodis Y, et al. Limbic system structure volumes and associated neurocognitive functioning in former NFL players. Brain Imaging Behav. 2019;13:725–734. doi: 10.1007/s11682-018-9895-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coughlin JM, Wang Y, Munro CA, et al. Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol Dis. 2015;74:58–65. doi: 10.1016/j.nbd.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Strain JF, Womack KB, Didehbani N, et al. Imaging Correlates of Memory and Concussion History in Retired National Football League Athletes. JAMA Neurol. 2015;72:773–780. doi: 10.1001/jamaneurol.2015.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Misquitta K, Dadar M, Tarazi A, et al. The relationship between brain atrophy and cognitive-behavioural symptoms in retired Canadian football players with multiple concussions. Neuroimage Clin. 2018;19:551–558. doi: 10.1016/j.nicl.2018.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee JK, Wu J, Bullen J, et al. Association of Cavum Septum Pellucidum and Cavum Vergae With Cognition, Mood, and Brain Volumes in Professional Fighters. JAMA Neurol. 2020;77:35–42. doi: 10.1001/jamaneurol.2019.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bernick C, Shan G, Zetterberg H, et al. Longitudinal change in regional brain volumes with exposure to repetitive head impacts. Neurology. 2020;94:e232–e240. doi: 10.1212/WNL.0000000000008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Coughlin JM, Wang Y, Minn I, et al. Imaging of Glial Cell Activation and White Matter Integrity in Brains of Active and Recently Retired National Football League Players. JAMA Neurol. 2017;74:67–74. doi: 10.1001/jamaneurol.2016.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zivadinov R, Polak P, Schweser F, et al. Multimodal Imaging of Retired Professional Contact Sport Athletes Does Not Provide Evidence of Structural and Functional Brain Damage. J Head Trauma Rehabil. 2018;33:E24–E32. doi: 10.1097/HTR.0000000000000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Terry DP, Miller LS. Repeated mild traumatic brain injuries is not associated with volumetric differences in former high school football players. Brain Imaging Behav. 2018;12:631–639. doi: 10.1007/s11682-017-9719-6. [DOI] [PubMed] [Google Scholar]
- 135.Tremblay S, De Beaumont L, Henry LC, et al. Sports concussions and aging: a neuroimaging investigation. Cereb Cortex. 2013;23:1159–1166. doi: 10.1093/cercor/bhs102. [DOI] [PubMed] [Google Scholar]
- 136.Tremblay S, Henry LC, Bedetti C, et al. Diffuse white matter tract abnormalities in clinically normal ageing retired athletes with a history of sports-related concussions. Brain. 2014;137:2997–3011. doi: 10.1093/brain/awu236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hart J, Jr, Kraut MA, Womack KB, et al. Neuroimaging of cognitive dysfunction and depression in aging retired National Football League players: a cross-sectional study. JAMA Neurol. 2013;70:326–335. doi: 10.1001/2013.jamaneurol.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Born CM, Meisenzahl EM, Frodl T, et al. The septum pellucidum and its variants. An MRI study. Eur Arch Psychiatry Clin Neurosci 2004;254:295–302. [DOI] [PubMed]
- 139.Koerte IK, Hufschmidt J, Muehlmann M, et al. Cavum Septi Pellucidi in Symptomatic Former Professional Football Players. J Neurotrauma. 2016;33:346–353. doi: 10.1089/neu.2015.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gardner RC, Hess CP, Brus-Ramer M, et al. Cavum Septum Pellucidum in Retired American Pro-Football Players. J Neurotrauma. 2016;33:157–161. doi: 10.1089/neu.2014.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Alosco ML, Sugarman MA, Besser LM, et al. A Clinicopathological Investigation of White Matter Hyperintensities and Alzheimer's Disease Neuropathology. J Alzheimers Dis. 2018;63:1347–1360. doi: 10.3233/JAD-180017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Arfanakis K, Evia AM, Leurgans SE, et al. Neuropathologic Correlates of White Matter Hyperintensities in a Community-Based Cohort of Older Adults. J Alzheimers Dis. 2020;73:333–345. doi: 10.3233/JAD-190687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.de Havenon A, Majersik JJ, Tirschwell DL, McNally JS, Stoddard G, Rost NS. Blood pressure, glycemic control, and white matter hyperintensity progression in type 2 diabetics. Neurology. 2019;92:e1168–e1175. doi: 10.1212/WNL.0000000000007093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shim YS, Yang DW, Roe CM, et al. Pathological correlates of white matter hyperintensities on magnetic resonance imaging. Dement Geriatr Cogn Disord. 2015;39:92–104. doi: 10.1159/000366411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822–838. doi: 10.1016/S1474-4422(13)70124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lehman EJ, Hein MJ, Baron SL, Gersic CM. Neurodegenerative causes of death among retired National Football League players. Neurology. 2012;79:1970–1974. doi: 10.1212/WNL.0b013e31826daf50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pokharel Y, Basra S, Lincoln AE, et al. Association of body mass index and waist circumference with subclinical atherosclerosis in retired NFL players. South Med J. 2014;107:633–639. doi: 10.14423/SMJ.0000000000000173. [DOI] [PubMed] [Google Scholar]
- 148.Tucker AM, Vogel RA, Lincoln AE, et al. Prevalence of cardiovascular disease risk factors among National Football League players. JAMA. 2009;301:2111–2119. doi: 10.1001/jama.2009.716. [DOI] [PubMed] [Google Scholar]
- 149.Willeumier K, Taylor DV, Amen DG. Elevated body mass in National Football League players linked to cognitive impairment and decreased prefrontal cortex and temporal pole activity. Transl Psychiatry. 2012;2:e68. doi: 10.1038/tp.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Grashow R, Weisskopf MG, Baggish A, et al. Premortem Chronic Traumatic Encephalopathy Diagnoses in Professional Football. Ann Neurol. 2020;88:106–112. doi: 10.1002/ana.25747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Roberts AL, Zafonte RD, Speizer FE, et al. Modifiable Risk Factors for Poor Cognitive Function in Former American-Style Football Players: Findings from the Harvard Football Players Health Study. J Neurotrauma. 2021;38:189–195. doi: 10.1089/neu.2020.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cherry JD, Tripodis Y, Alvarez VE, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2016;4:112. doi: 10.1186/s40478-016-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Alosco ML, Koerte IK, Tripodis Y, et al. White matter signal abnormalities in former National Football League players. Alzheimers Dement (Amst) 2018;10:56–65. doi: 10.1016/j.dadm.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537–539. doi: 10.1212/WNL.56.4.537. [DOI] [PubMed] [Google Scholar]
- 155.Griffin AD, Turtzo LC, Parikh GY, et al. Traumatic microbleeds suggest vascular injury and predict disability in traumatic brain injury. Brain. 2019;142:3550–3564. doi: 10.1093/brain/awz290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hasiloglu ZI, Albayram S, Selcuk H, et al. Cerebral microhemorrhages detected by susceptibility-weighted imaging in amateur boxers. AJNR Am J Neuroradiol. 2011;32:99–102. doi: 10.3174/ajnr.A2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shah BR, Holcomb JM, Davenport EM, et al. Prevalence and Incidence of Microhemorrhages in Adolescent Football Players. AJNR Am J Neuroradiol. 2020;41:1263–1268. doi: 10.3174/ajnr.A6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Multani N, Goswami R, Khodadadi M, et al. The association between white-matter tract abnormalities, and neuropsychiatric and cognitive symptoms in retired professional football players with multiple concussions. J Neurol. 2016;263:1332–1341. doi: 10.1007/s00415-016-8141-0. [DOI] [PubMed] [Google Scholar]
- 159.Hampshire A, MacDonald A, Owen AM. Hypoconnectivity and hyperfrontality in retired American football players. Sci Rep. 2013;3:2972. doi: 10.1038/srep02972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Terry DP, Adams TE, Ferrara MS, Miller LS. FMRI hypoactivation during verbal learning and memory in former high school football players with multiple concussions. Arch Clin Neuropsychol. 2015;30:341–355. doi: 10.1093/arclin/acv020. [DOI] [PubMed] [Google Scholar]
- 161.Ford JH, Giovanello KS, Guskiewicz KM. Episodic memory in former professional football players with a history of concussion: an event-related functional neuroimaging study. J Neurotrauma. 2013;30:1683–1701. doi: 10.1089/neu.2012.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Veksler R, Vazana U, Serlin Y, et al. Slow blood-to-brain transport underlies enduring barrier dysfunction in American football players. Brain 2020;143:1826-1842. https://pubmed.ncbi.nlm.nih.gov/30848432/ [DOI] [PMC free article] [PubMed]
- 163.De Beaumont L, Lassonde M, Leclerc S, Theoret H. Long-term and cumulative effects of sports concussion on motor cortex inhibition. Neurosurgery 2007;61:329–336. [DOI] [PubMed]
- 164.Giza CC, Hovda DA. The new neurometabolic cascade of concussion. Neurosurgery. 2014;75(Suppl 4):S24–33. doi: 10.1227/NEU.0000000000000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Holleran L, Kim JH, Gangolli M, et al. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol. 2017;133:367–380. doi: 10.1007/s00401-017-1686-x. [DOI] [PubMed] [Google Scholar]
- 166.Lesman-Segev OH, Edwards L, Rabinovici GD. Chronic Traumatic Encephalopathy: A Comparison with Alzheimer's Disease and Frontotemporal Dementia. Semin Neurol. 2020;40:394–410. doi: 10.1055/s-0040-1715134. [DOI] [PubMed] [Google Scholar]
- 167.Rosen HJ, Gorno-Tempini ML, Goldman WP, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208. doi: 10.1212/WNL.58.2.198. [DOI] [PubMed] [Google Scholar]
- 168.Schroeter ML, Raczka K, Neumann J, Yves von Cramon D. Towards a nosology for frontotemporal lobar degenerations-a meta-analysis involving 267 subjects. Neuroimage 2007;36:497–510. [DOI] [PubMed]
- 169.Seeley WW, Crawford R, Rascovsky K, et al. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol. 2008;65:249–255. doi: 10.1001/archneurol.2007.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Schroeter ML, Raczka K, Neumann J, von Cramon DY. Neural networks in frontotemporal dementia–a meta-analysis. Neurobiol Aging. 2008;29:418–426. doi: 10.1016/j.neurobiolaging.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 171.Hampel H, Teipel SJ, Alexander GE, et al. Corpus callosum atrophy is a possible indicator of region- and cell type-specific neuronal degeneration in Alzheimer disease: a magnetic resonance imaging analysis. Arch Neurol. 1998;55:193–198. doi: 10.1001/archneur.55.2.193. [DOI] [PubMed] [Google Scholar]
- 172.Teipel SJ, Bayer W, Alexander GE, et al. Progression of corpus callosum atrophy in Alzheimer disease. Arch Neurol. 2002;59:243–248. doi: 10.1001/archneur.59.2.243. [DOI] [PubMed] [Google Scholar]
- 173.Walterfang M, Luders E, Looi JC, et al. Shape analysis of the corpus callosum in Alzheimer's disease and frontotemporal lobar degeneration subtypes. J Alzheimers Dis. 2014;40:897–906. doi: 10.3233/JAD-131853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Arenth PM, Russell KC, Scanlon JM, Kessler LJ, Ricker JH. Corpus callosum integrity and neuropsychological performance after traumatic brain injury: a diffusion tensor imaging study. J Head Trauma Rehabil. 2014;29:E1–E10. doi: 10.1097/HTR.0b013e318289ede5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chetelat G, Arbizu J, Barthel H, et al. Amyloid-PET and (18)F-FDG-PET in the diagnostic investigation of Alzheimer's disease and other dementias. Lancet Neurol. 2020;19:951–962. doi: 10.1016/S1474-4422(20)30314-8. [DOI] [PubMed] [Google Scholar]
- 176.McKee AC, Alosco ML, Huber BR. Repetitive Head Impacts and Chronic Traumatic Encephalopathy. Neurosurg Clin N Am. 2016;27:529–535. doi: 10.1016/j.nec.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mez J, Alosco ML, Daneshvar DH, Saltiel N, Baucom Z, Abdolmohammadi B, Uretsky M, Nicks R, Martin B, Palmisano JN, Nowinski CJ, Montenigro P, Solomon TM, Mahar I, Cherry JD, Alvarez VE, Dwyer B, Goldstein LE, Katz DI, Cantu RC, Kowall NW, Tripodis Y, Huber BR, Stein TD, Stern RA, McKee AC. Validity of the 2014 traumatic encephalopathy syndrome criteria for CTE pathology. Alzheimer's & Dementia (in press) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.