

Abstract
Aberrant expression of the zinc finger protein (ZIC) family has been extensively reported to contribute to progression and metastasis in multiple human cancers. However, the functional roles and underlying mechanisms of ZIC2 in non-small cell lung cancer (NSCLC) are largely unknown. In this study, ZIC2 expression was evaluated using qRT-PCR, western blot, and immunohistochemistry, respectively. Animal experiments in vivo and functional assays in vitro were performed to investigate the role of ZIC2 in NSCLC. Luciferase assays and chromatin immunoprecipitation (ChIP) were carried out to explore the underlying target involved in the roles of ZIC2 in NSCLC. Here, we reported that ZIC2 was upregulated in NSCLC tissues, and high expression of ZIC2 predicted worse overall and progression-free survival of NSCLC patients. Silencing ZIC2 repressed tumorigenesis and reduced the anoikis resistance of NSCLC cells. Mechanical investigation further revealed that silencing ZIC2 transcriptionally inhibited Src expression and inactivated steroid receptor coactivator/focal adhesion kinase signaling, which further attenuated the anoikis resistance of NSCLC cells. Importantly, our results showed that the number of circulating tumor cells (CTCs) was positively correlated with ZIC2 expression in NSCLC patients. Collectively, our findings unravel a novel mechanism implicating ZIC2 in NSCLC, which will facilitate the development of anti-tumor strategies in NSCLC.
Keywords: ZIC2, anoikis resistance, CTCs, Src/FAK signaling, NSCLC
Graphical abstract
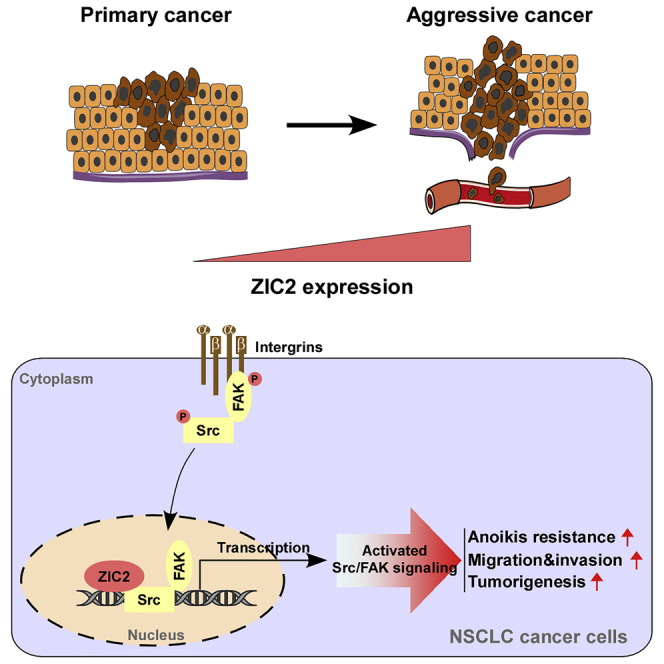
We pinpointed the oncogenic role of ZIC2 in NSCLC. Silencing ZIC2 abrogates tumorigenesis and anoikis resistance of non-small cell lung carcinoma cells (NSCLCs) by transcriptionally inhibiting Src/FAK signaling. Summarily, ZIC2 may hold the potential biomarker for early diagnosis and anti-tumor strategy in NSCLC.
Introduction
Lung cancer is the second-most common cancer diagnosed and the leading cause of cancer-related mortality in 2021, accounting for ∼12% of all new diagnoses; ranks next only to breast cancer; and accounts for one-quarter of all cancer deaths.1 According to its pathological characteristics, lung cancer is mainly divided into two histological types: small cell lung cancer (SCLC) and non-SCLC (NSCLC), where NSCLC primarily consists of lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC). Although substantial progress has been made with improvements in therapeutic strategies against NSCLC, the 5-year overall survival (OS) rate does not exceed 20% in NSCLC patients.2 Thus, it is of great necessity to identify potential biomarkers and therapeutic targets for NSCLC, which will be beneficial to improve the survival period and prognosis of NSCLC patients.
Aberrant expression or dysregulation of transcription factors has been extensively implicated in the pathogenesis of a variety of human cancers.3 Among the versatile transcription factor families, ZIC (zinc finger protein) is a kind of highly conserved protein with cysteine 2/histidine 2 motifs and exerts its function as a crucial transcription factor in multiple cellular and biological processes, such as embryo development, skeletal patterning, cell morphogenesis, myogenesis, and neurogenesis.4,5 Recently, accumulating momentum has implicated the zinc finger transcription factor ZIC2 in the pathogenesis and progression of multiple human cancer types. ZIC2 has been widely reported to be overexpressed in several human cancer types,6, 7, 8, 9, 10, 11, 12, 13 and overexpression of ZIC2 promoted cancer progression and metastasis by various mechanisms, suggesting that ZIC2 functions as an oncogene in the context of cancers. However, ZIC2 was found to be downregulated in breast cancer and pediatric medulloblastoma.14,15 These findings suggested that ZIC2 may play an oncogenic or tumor-suppressive role in different types of cancer. Furthermore, ZIC2 has been demonstrated to be upregulated in LUAD,16,17 as well as in SCLC,18 and high levels of ZIC2 predicted poor OS in patients with LUAD.16 However, the clinical significance and functional role of ZIC2 in NSCLC, especially in LUSC, remain to be further elucidated.
The extracellular environment, especially the extracellular matrix (ECM), provides adhesive support and connection among cells, which initiate signal transduction to promote cell survival and growth.19 Loss of this adhesive support or adhesion-mediated signaling leads to programmed cell death, referred to as anoikis.20 Anoikis resistance is a critical characteristic of cancer cells, which develop the ability of anchorage-independent growth for local dissemination and distant colonization.21 Cancer cells exploit multiple mechanisms to promote their resistance to anoikis, including epithelial-mesenchymal transition (EMT),22,23 deregulation of integrin,24 and constitutive activation of pro-survival pathways, such as phosphatidylinositol 3-kinase/protein kinase B (PI3K)/AKT signaling,25 signal transducer and activator of transcription 3 (STAT3) signaling,21 and steroid receptor coactivator/focal adhesion kinase (Src/FAK) signaling.26,27 Among these, the pro-anoikis resistance role of Src/FAK signaling in cancer seizes great attention, and activation of Src/FAK signaling has been demonstrated to protect cancer cells from anoikis.28,29 FAK is a broadly expressed non-receptor tyrosine kinase that elicit signals from integrins and plays an important role in cell adhesion, survival, and proliferation.30 Src has been found to interact with FAK by facilitating the phosphorylation (p) of FAK at Y397, leading to the activation of Src/FAK signaling.28,31 However, the underlying mechanisms responsible for anoikis resistance in NSCLC cells under the regulation of Src/FAK signaling remain unclear.
In this study, we found that ZIC2 expression was elevated in both LUAD and LUSC tissues, especially in LUSC tissues, which predicted poor OS and short progression-free survival (PFS) in NSCLC patients. Loss-of-function assays demonstrated that silencing ZIC2 repressed the tumorigenesis of NSCLC cells in vivo and attenuated the migratory, invasive ability and anoikis resistance of NSCLC cells in vitro. Mechanically, silencing ZIC2 transcriptionally inhibited Src expression and inactivated steroid receptor coactivator/focal adhesion kinase signaling, which further attenuated the anoikis resistance of NSCLC cells. Importantly, our results demonstrated that the number of circulating tumor cells (CTCs) with the ability to withstand anoikis in NSCLC patients with high levels of ZIC2 expression was dramatically higher than that in those with low levels of ZIC2 expression. Therefore, our results uncover a novel mechanism by which ZIC2 promotes tumorigenesis and anoikis resistance of NSCLC cells.
Results
ZIC2 is upregulated in NSCLC tissues
To determine the expression levels of ZIC2 in NSCLC, ZIC2 expression was first analyzed in the lung cancer dataset from The Cancer Genome Atlas (TCGA: https://tcga-data.nci.nih.gov/tcga/). As shown in Figure 1A, ZIC2 expression was dramatically upregulated in lung cancer tissues compared with that in the adjacent normal tissues (ANTs). Further analysis revealed that ZIC2 was differentially elevated in LUAD and LUSC tissues (Figure 1B). Similarly, we found that ZIC2 was upregulated in both LUAD and LUSC tissues in our previously integrative data profile of lung cancer based on the Affymetrix U133 Plus 2.0 microarray (AE-meta)32 (Figure 1C). Then, we further examined ZIC2 expression in 10 paired NSCLC tissues, including 6 LUADs and 4 LUSCs, by qRT-PCR and western blot, and the results showed that messenger RNA (mRNA) and protein levels of ZIC2 were elevated in 5/6 LUAD tissues and 3/4 LUSC tissues compared with those in the corresponding ANTs (Figures 1D and 1E). ZIC2 expression was further examined in the normal lung bronchial epithelial cell line BEAS-2, as well as in multiple lung cancer cell lines. As shown in Figures 1F and 1G, ZIC2 expression was differentially upregulated in lung cancer cells compared with that in BEAS-2. These results suggested that high expression of ZIC2 may be implicated in the pathogenesis of NSCLC.
Figure 1.

ZIC2 is upregulated in NSCLC
(A) Comparison of ZIC2 expression between NSCLC tissues and the matched adjacent normal tissues (ANTs) in The Cancer Genome Atlas (TCGA). (B) Expression level of ZIC2 in 109 ANT, 514 lung adenocarcinoma (LUAD), and 496 lung squamous cell carcinoma (LUSC) tissues in TCGA. Each bar represents the mean ± SD. (C) Expression level of ZIC2 in 340 ANT, 1109 LUAD, and 414 LUSC tissues in AE-meta. Each bar represents the mean ± SD. (D) ZIC2 mRNA expression in paired NSCLC tissues compared with their matched ANT patient samples. Transcript levels were normalized to GAPDH expression. Each bar represents the mean ± SD of three independent experiments. ∗ p < 0.05. (E) ZIC2 protein expression in paired NSCLC tissues compared with their matched ANT patient samples. α-tubulin served as the loading control. (F) ZIC2 mRNA expression in normal lung epithelial cell lines and NSCLC cell lines. Transcript levels were normalized to GAPDH expression. Each bar represents the mean ± SD of three independent experiments. ∗ p < 0.05. (G) ZIC2 protein expression in normal lung epithelial cell lines and NSCLC cell lines. α-tubulin served as the loading control.
High levels of ZIC2 predict poor prognosis and early progression
To investigate the clinical significance of ZIC2 in NSCLC, we further examined ZIC2 expression in a larger number of clinical NSCLC samples, including 168 LUADs and 93 LUSCs, by immunohistochemistry (IHC). As shown in Figures 2A−2C, ZIC2 was mainly expressed in the nucleus of cancer cells, and its expression levels were markedly upregulated in both LUAD and LUSC tissues compared with that in benign lung lesion tissues. Kaplan-Meier (KM) survival analysis showed that NSCLC patients with high levels of ZIC2 expression displayed poorer OS and PFS than those with low levels of ZIC2 expression (Figures 2D and 2E). We then explored the clinical correlation of ZIC2 expression with OS and PFS in two independent lung cancer datasets from AE-meta and Kaplan-Meier Plotter and found that high levels of ZIC2 expression predicted poor OS and short PFS of NSCLC patients compared with patients with low levels of ZIC2 expression (Figures 2F−2I), which further supported the findings from our clinical samples. Furthermore, the expression of ZIC2 was gradually increased with T classification, M classification, and the advancement of clinical stage, respectively (Table 1). Therefore, these findings indicated that high levels of ZIC2 predict a poor prognosis and early disease progression in NSCLC patients.
Figure 2.

High levels of ZIC2 predict poor prognosis and early progression in NSCLC patients
(A) Representative images of ZIC2 immunostaining of 43 benign lung lesion tissues, 168 LUAD tissues, and 93 LUSC tissues. Scale bars, upper panels, 200 μm; lower panels, 50 μm. (B) The number of lung tissues stratified by the staining index of ZIC2. (C) Staining index of ZIC2 in different histologic types of NSCLC. Error bar represents the 10th−90th percentile. (D and E) Kaplan-Meier survival curve of overall survival (OS) and progression-free survival (PFS) of NSCLC patients with low ZIC2 expression versus high ZIC2 expression. (F and G) Kaplan-Meier survival curve of OS and PFS in NSCLC patients with low ZIC2 expression versus high ZIC2 expression from AE-meta. (H and I) Kaplan-Meier Plotter of OS and PFS in NSCLC patients with low ZIC2 expression versus high ZIC2 expression. p value was determined by log-rank test. HR, hazard ratio; 95% CI, 95% confidence interval.
Table 1.
The relationship between ZIC2 expression level and clinical pathological characteristics in 261 patients with NSCLC
| Parameters | Number of cases | ZIC2 IHCa expression |
p values | |
|---|---|---|---|---|
| Low (n = 121) | High (n = 140) | |||
| Gender | ||||
| Female | 107 | 46 | 61 | 0.363 |
| Male | 154 | 75 | 79 | |
| Age | ||||
| <60 | 68 | 38 | 30 | 0.067 |
| ≥60 | 193 | 83 | 110 | |
| Grade | ||||
| G1−G2 | 156 | 73 | 83 | 0.864 |
| G3 | 105 | 48 | 57 | |
| T classification | ||||
| T1−2 | 198 | 102 | 96 | 0.002b |
| T3−4 | 59 | 17 | 42 | |
| N classification | ||||
| N0 | 141 | 70 | 71 | 0.344 |
| N1−3 | 110 | 48 | 62 | |
| M classification | ||||
| M0 | 243 | 117 | 126 | 0.033b |
| M1 | 18 | 4 | 14 | |
| Stage | ||||
| I−II | 181 | 93 | 88 | 0.014b |
| III−IV | 80 | 28 | 52 | |
IHC, immunohistochemistry.
p < 0.05.
Silencing ZIC2 inhibits tumorigenesis in vivo and metastasis in vitro of NSCLC cells
To determine the effect of ZIC2 on tumorigenesis of NSCLC cells in vivo, we first constructed ZIC2-stably downexpressing cell lines by endogenously knocking down ZIC2 via lentivirus infection in A549 and NCI-H520 cells, which expressed the highest levels of ZIC2 among all lung cancer cell lines (Figure 3A), where ZIC2-sh#2 of NCI-H520 cells presented higher silencing efficiency and was chosen for further analysis in vivo. Then, mice were randomly divided into two groups (n = 6/group), and the NCI-H520 vector or ZIC2-sh#2 cells were injected into the mice via tail veins. 6 weeks after cell inoculation, we found that silencing ZIC2 reduced the nodular growth of NCI-H520 cells in the lung and prolonged the cumulative survival of the mice (Figures 3B−3D). Interestingly, our results revealed that silencing ZIC2 dramatically reduced the number of CTCs extracted from the mice compared with the vector mice groups (Figure 3E). Moreover, with the use of a wound-healing assay, we demonstrated that silencing ZIC2 significantly inhibited the migratory activity of NSCLC cells compared with that of control cells (Figure 4A). Similarly, Transwell assays revealed that ZIC2 downregulation abrogated the migratory and invasive ability of NSCLC cells (Figures 4B and 4C). Collectively, these results demonstrated that silencing ZIC2 inhibits the metastasis of NSCLC cells in vivo and in vitro.
Figure 3.

Silencing ZIC2 inhibits tumorigenesis of lung cancer cells in vivo
(A) ZIC2 expression in endogenous ZIC2-knockdown A549 and NCI-H520 cell lines. Transcript levels were normalized to GAPDH expression. α-tubulin served as the loading control. Each bar represents the mean ± SD of three independent experiments. ∗ p < 0.05. (B) Metastatic lung tumor nests (upper panel, red arrow) and lung metastatic tumor tissues (lower panel) in the indicated mice group. Scale bars, 200 μm. (C) The numbers of tumor nodules in the lung section of each group. (D) Kaplan-Meier survival curves of the mice after tail-vein injection of the indicated lung cancer cells. (E) The number of CTCs in blood samples of the indicated mice group. Each bar represents the median values ± quartile values.
Figure 4.

Silencing ZIC2 inhibits metastasis of lung cancer cells in vitro
(A and B) The effect of ZIC2 on migration of lung cancer cells was assessed by wound-healing assay (A) and Transwell migration assay (B). (C) The effect of ZIC2 on invasion of lung cancer cells was assessed by Transwell invasion assay. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05.
Silencing ZIC2 abrogates anoikis resistance in NSCLC cells
To further determine the functional roles of ZIC2 downregulation in inhibiting the tumorigenic ability of NSCLC cells in vitro, Cell Counting Kit 8 (CCK-8) and colony-formation assays were first performed. As shown in Figures 5A−5C, neither the cell growth nor the colony-forming capability of NSCLC cells was affected by silencing ZIC2. This finding suggested that silencing ZIC2 inhibits the growth of NSCLC cells in vivo independent of cell proliferation. The above-mentioned in vivo results showed that silencing ZIC2 reduced the number of CTCs that could survive under suspension conditions, namely, anoikis resistance. Several lines of evidence have reported that anoikis resistance plays a pivotal role in the metastasis of cancer cells, including lung colonization and subsequent outgrowth.33 Therefore, the effects of silencing ZIC2 on anoikis resistance in NSCLC cells was further investigated. As shown in Figures 5D−5F, silencing ZIC2 enhanced the expression levels of cleaved caspase-3 and caspase-9 and the anoikis ratio of NSCLC cells. Mitochondrial potential assay demonstrated that silencing ZIC2 reduced the mitochondrial potential of NSCLC cells (Figure 5G). Therefore, our findings supported the hypothesis that silencing ZIC2 inhibits the tumorigenic ability of NSCLC cells by blocking anoikis resistance.
Figure 5.

Silencing ZIC2 abrogates anoikis resistance in lung cancer cells in vitro
(A−C) The effect of ZIC2 on proliferation of lung cancer cells was assessed by CCK-8 assay (A and B) and colony-formation assay (C). Each bar represents the mean value ± SD of three independent experiments. n.s, no significance. (D and E) The effect of ZIC2 on the activities of caspase-3 (D) and caspase-9 (E) was detected by the cleaved forms of these two proteins. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05. (F) The effect of ZIC2 on apoptosis of lung cancer cells was assessed under suspension growth conditions. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05. (G) The effect of ZIC2 on mitochondrial membrane potential of lung cancer cells under suspension growth condition. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05.
Silencing ZIC2 represses Src/FAK signaling via transcriptionally inhibiting Src
Numerous studies have reported that unrestrained activation of Src/FAK signaling plays a crucial role in maintaining anoikis resistance in cancer cells.26,34 Thus, we further investigated whether Src/FAK signaling is implicated in the functional role of ZIC2 in NSCLC. Western blot analysis showed that silencing ZIC2 reduced the p-Src and p-FAK levels in NSCLC cells but did not affect the total expression of FAK (Figure 6A). Notably, we found that silencing ZIC2 dramatically inhibited the total mRNA and protein expression of Src in NSCLC cells (Figures 6A and 6B). This finding suggested that ZIC2 regulates Src expression at the transcriptional level, which was further supported by the finding that a decrease in the Src promoter luciferase activity was observed upon downregulation of ZIC2 in NSCLC cells (Figure 6C). A chromatin immunoprecipitation (ChIP) assay indicated that ZIC2 could bind to the P2 and P7 binding sites in the promoter region of Src in NSCLC cells (Figure 6D). These results demonstrated that ZIC2 transcriptionally regulates Src expression, which further activates Src/FAK signaling in NSCLC cells.
Figure 6.

Silencing ZIC2 inhibits Src/FAK signaling by transcriptionally inhibiting Src
(A) Effect of ZIC2 on p-Src, Src, p-FAK, and FAK expression in the indicated lung cancer cells. α-tubulin served as the loading control. (B) Effect of ZIC2 on the mRNA expression of SRC and FAK. Transcript levels were normalized to GAPDH expression. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05. (C) Luciferase reporter activity of the Src promoter in ZIC2-downexpressing lung cancer cells. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05. (D) Upper panel: schematic illustration of histone 3 lysine 27 acetylation (H3K27Ac) enrichment and RT-PCR fragments of the human SRC promoter using the UCSC genome browser. Lower panel: the binding sites of ZIC2 in the promoter region of Src in lung cancer cells. Each bar represents the mean values ± SD of three independent experiments. ∗ p < 0.05.
Activation of Src/FAK signaling is essential for ZIC2-induced anoikis resistance
To further determine the functional role of Src/FAK signaling in mediating the effect of ZIC2 on anoikis resistance, constitutively active Src (Y529F)35 and FAK (Y397E)36 were applied to ZIC2-silenced NSCLC cells. As shown in Figures 7A and 7B, the expression levels of cleaved caspase-3 and caspase-9 were dramatically inhibited by Src (Y529F)35 and FAK (Y397E) in ZIC2-silenced NSCLC cells. Consistently, either Src (Y529F) or FAK (Y397E) reduced the anoikis ratio in ZIC2-silenced NSCLC cells (Figure 7C). Src (Y529F) or FAK (Y397E) reversed the inhibitory effect of ZIC2 downregulation on the mitochondrial potential in NSCLC cells (Figure 7D). These findings indicated that silencing ZIC2 inhibits anoikis resistance in NSCLC cells by inactivating Src/FAK signaling.
Figure 7.

Activation of Src/FAK signaling is essential for ZIC2-induced anoikis resistance
(A and B) Effect of Src (Y529F) or FAK (Y397E) on the activities of caspase-3 (A) and caspase-9 (B) in the indicated lung cancer cells. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05. (C) Src (Y529F) or FAK (Y397E) reversed the induction effects of ZIC2 silencing on apoptosis analyzed under suspension growth conditions. Each bar represents the mean value ± SD of three independent experiments. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05. (D) Src (Y529F) or FAK (Y397E) reversed the inhibitory effects of ZIC2 silencing on the mitochondrial membrane potential under suspension growth conditions. Each bar represents the mean value ± SD of three independent experiments. ∗ p < 0.05.
Correlation of ZIC2 expression with the CTC count in clinical NSCLC samples
Since CTCs could withstand anoikis triggered by detaching from the ECM for subsequent successful colonization,37 the clinical correlation of ZIC2 expression with the number of CTCs in NSCLC patients was further analyzed. Immunofluorescence staining of CD45 and 4′,6-diamidino-2-phenylindole (DAPI) and fluorescence in situ hybridization (FISH) with the centromere of chromosome 8 (CEP8) was utilized to identify CTCs in NSCLC patients. CD45−/CEP8 > 2 cells were defined as CTCs, whereas those with CD45+/CEP8 = 2 were defined as white blood cells (Figure 8A). In line with the observations in vivo, we found that the number of CTCs in NSCLC patients with low levels of ZIC2 was fewer than that in those with high levels of ZIC2 (Figure 8B), whereas the number of CTC-white blood cell (WBC) clusters between them has no significant difference (Figure 8C). Our results indicated that ZIC2 expression levels are positively correlated with the number of CTCs in NSCLC patients.
Figure 8.

ZIC2 expression is positively correlated with the number of CTCs in NSCLC patients
(A) Identification of CTCs in NSCLC patients. DAPI, blue; CD45, red; CEP8, orange. Scale bars, 10 μm. (B) Number of CTCs in ZIC2-low (n = 32) and ZIC2-high (n = 33) expression NSCLC patients. Error bar represents the 10th−90th percentile. (C) Number of CTC-WBC clusters in ZIC2-low (n = 32) and ZIC2-high (n = 33) expression NSCLC patients. Error bar represents the 10th−90th percentile.
Discussion
The primary findings of the current study provide insights into the clinical significance and biological function of ZIC2 in NSCLC. In this manuscript, our results revealed that ZIC2 expression was elevated in NSCLC tissues, and high levels of ZIC2 predicted shorter overall and PFS in NSCLC patients. Loss-of-function assays demonstrated that silencing ZIC2 repressed tumorigenesis of NSCLC cells in vivo and attenuated anoikis resistance in NSCLC cells in vitro. Our results further indicated that silencing ZIC2 transcriptionally inhibited Src expression and inactivated Src/FAK signaling, which further attenuated the anoikis resistance of NSCLC cells. Therefore, our findings pinpoint the oncogenic role of ZIC2 in NSCLC.
Numerous studies have demonstrated that ZIC2 was upregulated in multiple human cancer types, including pancreatic cancer,6 meningioma,7 nasopharyngeal carcinoma,8 oral squamous cell carcinoma,9 hepatocellular carcinoma,10,11 cervical cancer,12 and epithelial ovarian tumor,13 and overexpression of ZIC2 promoted cancer progression and metastasis by varying mechanisms as an oncogene. However, Liu and colleagues14 have reported that ZIC2 was downregulated in breast cancer, and ectopic expression of ZIC2 repressed cell proliferation and the colony-formation ability in vitro and tumor growth in vivo. Furthermore, a decrease in ZIC2 expression was reported in pediatric medulloblastoma.15 These findings suggested that the oncogenic or tumor-suppressive role of ZIC2 largely depends on cancer type. Furthermore, ZIC2 has been widely reported to be upregulated in LUAD16,17 and SCLC,18 and its high expression was associated with poor OS in patients with LUAD.16 Consistently, our results in the current study demonstrated that ZIC2 was upregulated in both LUAD and LUSC tissues. High expression of ZIC2 was positively correlated with poor OS and PFS in NSCLC patients. Our results further demonstrated that silencing ZIC2 attenuated the tumorigenic ability of NSCLC cells in vivo and the anoikis resistance of NSCLC cells in vitro. In addition, silencing ZIC2 attenuated the anoikis resistance of NSCLC cells dependent on transcriptional repression of Src expression, leading to inactivation of Src/FAK signaling. Collectively, our findings uncover a novel mechanism by which ZIC2 promotes tumorigenesis and anoikis resistance in NSCLC.
As one of the originally identified members, ZIC2 belongs to a superfamily of zinc finger transcription factors and exerts its function as a transcription factor by virtue of its nomenclature.38 A study from Lu et al.11 has reported that ZIC2 directly bound to the P21-activated kinase 4 (PAK4) promoter and transcriptionally upregulated PAK4 expression, leading to sustained activation of the rapidly accelerated fibrosarcoma/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway, which further promoted tumor growth and metastasis in hepatocellular carcinoma; in addition, ZIC2 acted as an upstream transcription factor to recruit the nuclear remodeling factor (NURF) complex to the octamer-Binding Protein 4 (OCT4) promoter, thereby increasing OCT4 expression at the transcriptional level.10 Interestingly, ZIC2 has also been demonstrated to function as a transcriptional repressor in osteosarcoma and breast cancer by transcriptionally inhibiting SH2 domain-containing inositol 5'-phosphatase 2 (SHIP2) and STAT3 expression, respectively.14,39 However, the transcriptional regulatory role of ZIC2 in NSCLC has not yet been elucidated. In the current study, our results clarified for the first time, to the best of our knowledge, the transcriptional activating role of ZIC2 in NSCLC, where we found that silencing ZIC2 dramatically reduced the promoter luciferase activity of Src. Specifically, our results further demonstrated that ZIC2 could bind to the P2 and P7 binding sites in the promoter region of Src in NSCLC cells. Therefore, our results elucidate the transcriptional regulatory role of ZIC2 in NSCLC.
It has been extensively documented that ZIC2 may be used as a potential biomarker for the early diagnosis of cancer and the prediction of prognosis of patients with cancer. ZIC2 was found to be upregulated in hepatocellular carcinoma, which predicted worse overall and disease-free survival of hepatocellular carcinoma patients. Importantly, multivariate analyses showed that ZIC2 could serve as an independent indicator of a poor outcome of hepatocellular carcinoma patients.11 Moreover, high levels of ZIC2 were positively correlated with poor OS of oral squamous cell carcinoma patients compared with those with low levels of ZIC2.9 In the context of lung cancer, overexpression of ZIC2 predicted poor OS in patients with LUAD.16 Notably, antibodies against ZIC2 were present in approximately 30% of SCLC patients but were not identified in healthy controls.40 However, the potential of ZIC2 serving as a biomarker to predict the prognosis in NSCLC remains unknown. In this study, our results revealed that ZIC2 was upregulated in both LUAD and LUSC tissues, which predicted poor OS and short PFS in our clinical NSCLC patients, as well as in two independent lung cancer datasets from AE-meta and Kaplan-Meier Plotter, respectively, which was consistent with the previously reported finding.16 Therefore, our results further suggested that ZIC2 may hold the potential applicable value as a biomarker to predict prognosis in NSCLC patients. However, a more solid conclusion needs to be warranted in a large series of clinical studies in the following work.
In summary, our results demonstrate that ZIC2 promotes the tumorigenesis and anoikis resistance of NSCLC by transcriptionally inhibiting the Src/FAK signaling pathway. In-depth understanding of the underlying mechanism of ZIC2 in NSCLC will facilitate the early detection of patients with NSCLC.
Materials and methods
Cells and cell culture
Non-cancerous, immortalized human lung bronchial epithelial cell line BEAS-2B was purchased from Zhong Qiao Xin Zhou Biotechnology (Shanghai, China). Normal fetal lung fibroblast cell HFL1; human LUAD cell lines Calu-3, NCI-H1975, and NCI-H1395; LUSC line NCI-H520; and other NSCLC cell lines A549 and NCI-H1299 were purchased from Procell (Wuhan, China). LUSC lines NCI-H226 and SK-MES-1 were purchased from Shanghai Chinese Academy of Science Cell Bank (Shanghai, China). BEAS-2B was grown in Bronchial Epithelial Cell Growth Medium BulletKit (BEGM; Lonza, Basel, Switzerland). HFL1 was cultured in Ham’s F-12K medium (Gibco, Pittsburgh, PA, USA). Calu-3 and SK-MES-1 were cultured in Eagle’s minimum essential medium (MEM; Gibco). NCI-H1975, NCI-H1395, NCI-H1299, NCI-H520, and NCI-H226 were maintained in RPMI-1640 (Gibco). All cell lines, except for BEAS-2B, were supplemented with 10% fetal bovine serum (FBS), streptomycin (100 μg/mL), and penicillin G (100 U/mL; Gibco). All cell lines were authenticated by short tandem repeat (STR) profiling. Cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Patients and tissue specimens
The 6 fresh LUAD tissues, 4 fresh LUSC tissues, and paired adjacent non-cancerous lung tissues were obtained during surgery, and the clinicopathological features of the patients are summarized in Table S1. A total of 304 frozen section and archived lung samples, including 43 benign lung disease lesions, 168 LUAD tissues, and 93 LUSC tissues, were obtained during surgery or needle biopsy. The clinicopathological features of the 43 patients with benign lung disease are summarized in Table S2, and the 261 patients with NSCLC are summarized in Table S3. All tissues were collected from the Affiliated Jiangmen Hospital of Sun Yat-sen University (Guangdong, China) between January 2008 and December 2019. Patients were diagnosed based on clinical and pathological evidence, and the specimens were immediately snap frozen and stored in liquid nitrogen tanks or the 6.0-μm frozen section stored in a −86°C refrigerator. For the use of these clinical materials for research purposes, prior patients’ consents and approval from the Institutional Research Ethics Committee of the Affiliated Jiangmen Hospital of Sun Yat-sen University were obtained. The proportions of tumors versus non-tumors in hematoxylin and eosin (H&E)-stained tissue samples were evaluated independently by two professional pathologists. All clinical lung cancer tissue samples analyzed in this study with tumor proportions exceeding 75% were used for further analysis.
IHC
IHC analysis was performed to examine ZIC2 expression in 304 frozen section and archived lung samples. The IHC procedures and expression scoring were as described previously.32,41 Briefly, the slides of a frozen section were antigen retrieved in Tris/EDTA (TE; pH 9.0) buffer, 10 min by microwave heating, blocked by hydrogen peroxide and goat serum, respectively, and incubated overnight at 4°C in a humidified chamber with the anti-ZIC2 antibody (Invitrogen, Carlsbad, CA, USA) diluted 1:100 in Antibody Diluent (Abcam, Cambridge, MA, USA). After incubation, slides were washed in Tris-buffered saline (TBS)/0.05% Tween 20, incubated with biotin-conjugated secondary antibody (Proteintech, Wuhan, China) and peroxidase-conjugated streptavidin (Proteintech), 30 min at 37°C, respectively, stained by the 3,3′-diaminobenzidine (DAB) Enhanced Liquid Substrate System (Sigma-Aldrich, St. Louis, MO, USA). Staining index (SI) given by the two experienced independent investigators was averaged for further comparative evaluation of ZIC2 expression. Tumor cell proportion was scored as follows: 0 (no positive tumor cells), 1 (<10% positive tumor cells), 2 (10%–35% positive tumor cells), 3 (36%–70% positive tumor cells), and 4 (>70% positive tumor cells). Staining intensity was graded according to the following criteria: 0 (no staining), 1 (weak staining, light yellow), 2 (moderate staining, yellow brown), and 3 (strong staining, brown). SI was calculated as the product of staining intensity score and the proportion of positive tumor cells. Based on this method of assessment, ZIC2 expression in lung tumor samples was evaluated by the SI, with scores of 0, 1, 2, 3, 4, 6, 8, 9, or 12. SI score 4 was the median of all sample tissues’ SI. High and low expression of ZIC2 was stratified by the following criteria: the SI ≤ 4 was used to define tumors with low expression of ZIC2 and SI score of >4 as tumors with high expression of ZIC2. Images were collected under 10× and 40× objective magnification in human lung tissues using M8 Digital Microscopy (PreciPoint, Freising, Bavaria, Germany).
Plasmids and transfection
The short hairpin RNA (shRNA) for human ZIC2 was cloned into a U6-MCS-CBh-gcGFP-IRES-puromycin lentiviral vector (GV493; Genechem, Shanghai, China), and the list of primers used in clone reactions is presented in Table S4. The promoter of SRC was PCR amplified from genomic DNA and cloned into the luciferase reporter pGL4.10[luc2] vector (Promega, Madison, WI, USA). Constitutively activate Src (Y529F) and FAK (Y397E) plasmids were synthesized by Transheep (Shanghai, China). Transfection of plasmids was performed using the Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s instructions. Stable cell lines expressing ZIC2-sh#1 or ZIC2-sh#2 were generated by filtered lentivirus infection using HEK293T cells and selected with 0.5 mg/L puromycin (Sigma-Aldrich) for 10 days, as described previously.32
Western blot analysis
Western blot was performed according to a standard method, as previously described.41 Briefly, the cell lysates were loaded with 10% loading buffer (Beyotime, Shanghai, China) and heated for 5 min at 100°C. Equal quantities of denatured protein samples were resolved on 8%–16% SDS-polyacrylamide gels and then transferred onto Immobilon-PSQ polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). After blocking with 5% non-fat dry milk in TBS/0.05% Tween 20, the membranes were incubated with a specific primary antibody, followed by a horseradish peroxidase-conjugated secondary antibody (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were visualized using BeyoECL Plus reagents (Beyotime). Specific primary antibodies against ZIC2 (Abcam), p-Src (Invitrogen), Src (Proteintech), p-FAK (Invitrogen), and FAK (Proteintech) were applied, and α-tubulin was detected by anti-α-tubulin (Proteintech) as the loading control.
RNA extraction, reverse transcription, and qRT-PCR
Total RNA was extracted from cells using TRIzol (Invitrogen) according to the manufacturer’s instructions. mRNA was reverse transcribed from total mRNA using the HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China) according to the manufacturer’s protocol. qRT-PCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme) and quantified by ABI 7500 Fast System (Thermo Fisher Scientific). The primers are provided in Table S5 and were synthesized and purified by Biosune Biotechnology (Shanghai, China). Real-time PCR was performed according to the manufacturer’s protocol, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous control for mRNA. Relative fold expressions were calculated with the comparative threshold cycle (2−ΔΔCt) method, as described previously.32
Wound-healing assay
For the wound-healing assay, cells (5 × 105) were plated in 6-well plates, proliferating to 90% confluence. A wound was scratched into the cells using a sterile plastic tip and then washed by PBS. Then, cells were cultured in medium containing 2% FBS. Images of random fields were captured for analysis at the indicated times under a light microscope (Leica, Wetzlar, Germany).
Transwell assay
Cells (4 × 104) were plated on the upper compartment of the 24-well Transwell permeable chambers (Corning, Corning, NY, USA), chambers were coated without Matrigel for migration and with Matrigel (Corning) for invasion detection. The lower chamber of the Transwell was filled with complete media supplemented with 10% FBS as a chemoattractant. After incubation for 24 h, cells that migrated to the bottom side of the chamber were fixed with stationary solution (methanol:acetic acid = 3:1), stained with crystal violet, photographed, and quantified by counting in 5 random fields of view using a light microscope (Leica).
CCK-8 analysis and colony-formation assay
For CCK-8 analysis, cells (0.5 × 103) were seeded into 96-well plates and stained at the indicated time points with 100 μL CCK-8 (Dojindo, Japan) dye for 1 h at 37°C, followed by measuring the absorbance at 450 nm, with 650 nm used as the reference wavelength. For the colony-formation assay, cells (0.5−1.0 × 103) were plated into six-well plates and cultured for 7−10 days. Colonies were fixed for 15 min with 10% formaldehyde, stained with 1.0% crystal violet for 30 s, and washed with water.
Caspase-9 or caspase-3 activity assays
The activity of caspase-9 or caspase-3 was analyzed by spectrophotometry using the Caspase-9 Colorimetric Assay Kit or Caspase-3 Colorimetric Assay Kit (KeyGen, Nanjing, China), which was performed as previously described.42 The absorbance was measured at 405 nm, and bicinchoninic acid (BCA) protein quantitative analysis was used as the reference to normal for each experiment group.
Flow cytometric analysis
Cells were incubated on poly-hydroxyethylmethacrylate (HEMA; Sigma-Aldrich)-coated plates to prevent adhesion in suspension culture conditions for 48 h. Flow cytometric analysis of anoikis was performed using the Annexin V-FITC/PI (fluorescein isothiocyanate/propidium iodide) Apoptosis Detection Kit (KeyGen), as described in the protocol. Briefly, the cells were dissociated with trypsin and resuspended at 1 × 107 cells/mL in binding buffer with 500 μL/mL Annexin V-FITC and 500 μL/mL PI. The cells were subsequently incubated for 15 min at room temperature and then analyzed by a Gallios flow cytometer (Beckman Coulter, Brea, CA, USA). The inner mitochondrial membrane potential (ΔψM) of cells was detected by flow cytometry using a JC-1 Staining Kit (KeyGen) as described in the protocol. Briefly, the cells were dissociated with trypsin, resuspended at 1 × 107 cells/mL in assay buffer, and then incubated at 37°C for 15 min with 100 μL/mL JC-1. Before analysis by flow cytometry, the cells were washed twice with assay buffer and then filtered through a cell mesh. Flow cytometry data were analyzed using FlowJo VX software (Tree Star, San Carlos, CA, USA).
Dual-luciferase report experiments
Cells (5 × 104) were plated in 24-well plates, proliferating to 60%–80% confluence after 24 h of culture, and 0.2 μg of pGL4.10-promoter reporter plasmid and 0.2 mg of the Renilla luciferase expression vector thymidine kinase promoter-Renilla luciferase reporter plasmid were transfected into the cells using Lipofectamine 3000. The cells were harvested after 48 h of transfection, washed with PBS, and lysed with lysis buffer. The cell lysates were analyzed immediately using a Synergy 2 microplate system (BioTek, Winooski, VT, USA). Luciferase and Renilla luciferase were measured using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The luciferase activity of each lysate was normalized to Renilla luciferase activity. The relative transcriptional activity was converted into fold induction above the control group value.
ChIP
ChIP was performed as previously described.43 Cells (6 × 106) in a 100-mm culture dish were fixed with 1% formaldehyde to cross link the proteins to the DNA. The cell lysates were sonicated to shear the DNA to sizes of 300 to 1,000 bp. Equal aliquots of chromatin supernatants were incubated with 1 μg of anti-FLAG antibody (Cell Signaling Technology, Danvers, MA, USA) or an anti-immunoglobulin G (IgG) antibody (Cell Signaling Technology) overnight at 4°C with constant rotation. After reverse cross-linking of the protein/DNA complexes to free the DNA, real-time PCR was carried out. The primers used to detect the DNA fragments in the ChIP are provided in Table S6.
Animal study
The animal study was approved by the Ethical Committee of Xiangya Hospital, Central South University. At least 6 nude mice per group were used to ensure the adequate power, and mice were randomly allocated to the groups. For tail-vein injection, 1−2 × 106 cells in 100 μL PBS were injected into the lateral tail vein of BALB/c-nu mice (4−6 weeks old; 18−20 g). Mice were monitored twice weekly and were sacrificed by cervical dislocation depending on their survival time. Lungs of each group of mice were dissected and fixed with 4% paraformaldehyde and subjected to H&E staining. The tumor cell number was evaluated in 9 random fields (square millimeters) of the H&E tissues under 20× magnification using M8 Digital Microscopy (PreciPoint). To detect CTCs in the animal samples, 100 μL whole-blood samples were collected by tail-vein bleeding into EDTA-coated tubes 14 days after intravenous (i.v.) injection. GFP-positive tumor cells were selected by flow cytometry using GFP antibody (Cell Signaling Technology), and viable cells were counted using trypan blue exclusion assay.
Subtraction enrichment (SE) of blood samples for CTCs
For the clinical samples, enrichment of CTCs was performed using the Human Circulating Tumor Cell (hCTC) Subtraction Enrichment Kit (SHE-011; Cytelligen, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, 6 mL of peripheral blood was collected and centrifuged at 200 × g for 15 min. Sedimented blood cells were gently mixed with 3.5 mL hCTC buffer and subsequently loaded on a nonhematopoietic cell separation matrix, followed by centrifugation at 450 × g for 5 min. The sedimented red blood cells were removed. Supernatants containing the tumor cells were centrifuged and resuspended in hCTC buffer to 14 mL. Samples were then spun at 500 × g for 4 min at room temperature, followed by aspirating the supernatants down to 100 μL. The sedimented cells were gently resuspended and mixed with cell fixative (Cytelligen). The cell mixture was smeared on the formatted and coated CTC slides and then dried for subsequent immunostaining–fluorescence in situ hybridization (iFISH) processing.
Identification of CTCs using iFISH
iFISH was performed according to the manufacturer’s instructions (Cytelligen). Briefly, dried monolayer cells on the coated slides were rinsed and incubated with PBS at room temperature for 3 min, followed by hybridization with a Vysis CEP8 SpectrumOrange (Abbott Laboratories, Chicago, IL, USA) for 3 h using a S500 StatSpin ThermoBrite Slide Hybridization/Denaturation System (Abbott Molecular, Abbott Park, IL, USA). Samples were subsequently incubated with Alexa Fluor 488-conjugated monoclonal anti-CD45 antibodies (Cytelligen) at 1:200 dilution for 30 min in dark. Cell nuclei were stained with DAPI (Beyotime). Images of the identified tumor cells were automatically acquired using automated 3D CTC image scanning and analyses. CTCs were defined as DAPI+, CD45−, CEP8 > 2 signals.
Statistical analysis
For categorical data, the chi-square test was used to analyze the relationship between ZIC2 expression and the clinicopathological characteristics. For variables with a normal distribution, data are presented as the mean ± standard deviation (SD), and Student’s t test was used to determine statistical differences between two groups. One-way ANOVA was used to determine statistical differences among multiple groups. Continuous variables without normal distribution are presented as median and interquartile range. The significance of differences was assessed using a non-parametric test (Mann-Whitney U-test). Survival curves for both ZIC2-high and ZIC2-low patients were applied by the Kaplan-Meier method and compared by the log-rank test. In vitro experiments were repeated three times, and the data are presented as the mean ± SD. p < 0.05 was considered statistically significant. All analyses were performed using SPSS 23.0 software (IBM, Chicago, IL, USA), and diagrams were drawn using GraphPad Prism 8.0 software (GraphPad, San Diego, CA, USA).
Acknowledgments
This study was funded by grants from the Natural Science Foundation of Hunan Province (2018JJ2646 and 2019JJ50970), National Natural Science Foundation of China (81702917 and 81770045), Science and Technology Project of Guangdong Province (2019A1515011565), and Science and Technology Project of Jiangmen (2020020500150004120, 2019-252-2-1, 2019030102430012905, and 2018090106380023859).
Author contributions
Y.H. and K.S. conceived the project and drafted the manuscript. A.L. designed the study and analyzed the data. D.R., X.Z., and R.L. performed the IHC experiments and analyzed the images. W.L. and Q.C. performed the bioinformatics analysis. B.Y. and L.D. performed the animal experiments. H.X. contributed to the cell biology and molecular biology experiments. B.L. and L.R. organized the patients’ information.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.05.008.
Contributor Information
Yanming Huang, Email: huangyanming_jxy@163.com.
Ke Shi, Email: csushike@163.com.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Jacobsen M.M., Silverstein S.C., Quinn M., Waterston L.B., Thomas C.A., Benneyan J.C., Han P.K.J. Timeliness of access to lung cancer diagnosis and treatment: A scoping literature review. Lung Cancer. 2017;112:156–164. doi: 10.1016/j.lungcan.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Bhagwat A.S., Vakoc C.R. Targeting Transcription Factors in Cancer. Trends Cancer. 2015;1:53–65. doi: 10.1016/j.trecan.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houtmeyers R., Souopgui J., Tejpar S., Arkell R. The ZIC gene family encodes multi-functional proteins essential for patterning and morphogenesis. Cell. Mol. Life Sci. 2013;70:3791–3811. doi: 10.1007/s00018-013-1285-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chervenak A.P., Hakim I.S., Barald K.F. Spatiotemporal expression of Zic genes during vertebrate inner ear development. Dev. Dyn. 2013;242:897–908. doi: 10.1002/dvdy.23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inaguma S., Ito H., Riku M., Ikeda H., Kasai K. Addiction of pancreatic cancer cells to zinc-finger transcription factor ZIC2. Oncotarget. 2015;6:28257–28268. doi: 10.18632/oncotarget.4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aruga J., Nozaki Y., Hatayama M., Odaka Y.S., Yokota N. Expression of ZIC family genes in meningiomas and other brain tumors. BMC Cancer. 2010;10:79–88. doi: 10.1186/1471-2407-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yi W., Wang J., Yao Z., Kong Q., Zhang N., Mo W., Xu L., Li X. The expression status of ZIC2 as a prognostic marker for nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2018;11:4446–4460. [PMC free article] [PubMed] [Google Scholar]
- 9.Sakuma K., Kasamatsu A., Yamatoji M., Yamano Y., Fushimi K., Iyoda M., Ogoshi K., Shinozuka K., Ogawara K., Shiiba M. Expression status of Zic family member 2 as a prognostic marker for oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2010;136:553–559. doi: 10.1007/s00432-009-0689-y. [DOI] [PubMed] [Google Scholar]
- 10.Zhu P., Wang Y., He L., Huang G., Du Y., Zhang G., Yan X., Xia P., Ye B., Wang S. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J. Clin. Invest. 2015;125:3795–3808. doi: 10.1172/JCI81979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu S.X., Zhang C.Z., Luo R.Z., Wang C.H., Liu L.L., Fu J., Zhang L., Wang H., Xie D., Yun J.P. Zic2 promotes tumor growth and metastasis via PAK4 in hepatocellular carcinoma. Cancer Lett. 2017;402:71–80. doi: 10.1016/j.canlet.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 12.Chan D.W., Liu V.W., Leung L.Y., Yao K.M., Chan K.K., Cheung A.N., Ngan H.Y. Zic2 synergistically enhances Hedgehog signalling through nuclear retention of Gli1 in cervical cancer cells. J. Pathol. 2011;225:525–534. doi: 10.1002/path.2901. [DOI] [PubMed] [Google Scholar]
- 13.Marchini S., Poynor E., Barakat R.R., Clivio L., Cinquini M., Fruscio R., Porcu L., Bussani C., D’Incalci M., Erba E. The zinc finger gene ZIC2 has features of an oncogene and its overexpression correlates strongly with the clinical course of epithelial ovarian cancer. Clin. Cancer Res. 2012;18:4313–4324. doi: 10.1158/1078-0432.CCR-12-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z.H., Chen M.L., Zhang Q., Zhang Y., An X., Luo Y.L., Liu X.M., Liu S.X., Liu Q., Yang T. ZIC2 is downregulated and represses tumor growth via the regulation of STAT3 in breast cancer. Int. J. Cancer. 2020;147:505–518. doi: 10.1002/ijc.32922. [DOI] [PubMed] [Google Scholar]
- 15.Pfister S., Schlaeger C., Mendrzyk F., Wittmann A., Benner A., Kulozik A., Scheurlen W., Radlwimmer B., Lichter P. Array-based profiling of reference-independent methylation status (aPRIMES) identifies frequent promoter methylation and consecutive downregulation of ZIC2 in pediatric medulloblastoma. Nucleic Acids Res. 2007;35:e51. doi: 10.1093/nar/gkm094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang H., Xu S., Chen C. A ten-gene signature-based risk assessment model predicts the prognosis of lung adenocarcinoma. BMC Cancer. 2020;20:782–792. doi: 10.1186/s12885-020-07235-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei-Hua W., Ning Z., Qian C., Dao-Wen J. ZIC2 promotes cancer stem cell traits via up-regulating OCT4 expression in lung adenocarcinoma cells. J. Cancer. 2020;11:6070–6080. doi: 10.7150/jca.44367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Güre A.O., Stockert E., Scanlan M.J., Keresztes R.S., Jäger D., Altorki N.K., Old L.J., Chen Y.T. Serological identification of embryonic neural proteins as highly immunogenic tumor antigens in small cell lung cancer. Proc. Natl. Acad. Sci. USA. 2000;97:4198–4203. doi: 10.1073/pnas.97.8.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins N.L., Reginato M.J., Paulus J.K., Sgroi D.C., Labaer J., Brugge J.S. G1/S cell cycle arrest provides anoikis resistance through Erk-mediated Bim suppression. Mol. Cell. Biol. 2005;25:5282–5291. doi: 10.1128/MCB.25.12.5282-5291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horowitz J.C., Rogers D.S., Sharma V., Vittal R., White E.S., Cui Z., Thannickal V.J. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell. Signal. 2007;19:761–771. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fofaria N.M., Srivastava S.K. Critical role of STAT3 in melanoma metastasis through anoikis resistance. Oncotarget. 2014;5:7051–7064. doi: 10.18632/oncotarget.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren D., Yang Q., Dai Y., Guo W., Du H., Song L., Peng X. Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-κB signaling pathway. Mol. Cancer. 2017;16:117–132. doi: 10.1186/s12943-017-0688-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaffer C.L., Weinberg R.A. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 24.Leng C., Zhang Z.G., Chen W.X., Luo H.P., Song J., Dong W., Zhu X.R., Chen X.P., Liang H.F., Zhang B.X. An integrin beta4-EGFR unit promotes hepatocellular carcinoma lung metastases by enhancing anchorage independence through activation of FAK-AKT pathway. Cancer Lett. 2016;376:188–196. doi: 10.1016/j.canlet.2016.03.023. [DOI] [PubMed] [Google Scholar]
- 25.Tang Y., Pan J., Huang S., Peng X., Zou X., Luo Y., Ren D., Zhang X., Li R., He P., Wa Q. Downregulation of miR-133a-3p promotes prostate cancer bone metastasis via activating PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2018;37:160–175. doi: 10.1186/s13046-018-0813-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mo C.F., Li J., Yang S.X., Guo H.J., Liu Y., Luo X.Y., Wang Y.T., Li M.H., Li J.Y., Zou Q. IQGAP1 promotes anoikis resistance and metastasis through Rac1-dependent ROS accumulation and activation of Src/FAK signalling in hepatocellular carcinoma. Br. J. Cancer. 2020;123:1154–1163. doi: 10.1038/s41416-020-0970-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li K., Zhao G., Ao J., Gong D., Zhang J., Chen Y., Li J., Huang L., Xiang R., Hu J. ZNF32 induces anoikis resistance through maintaining redox homeostasis and activating Src/FAK signaling in hepatocellular carcinoma. Cancer Lett. 2019;442:271–278. doi: 10.1016/j.canlet.2018.09.033. [DOI] [PubMed] [Google Scholar]
- 28.Sood A.K., Armaiz-Pena G.N., Halder J., Nick A.M., Stone R.L., Hu W., Carroll A.R., Spannuth W.A., Deavers M.T., Allen J.K. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Invest. 2010;120:1515–1523. doi: 10.1172/JCI40802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park E.K., Park M.J., Lee S.H., Li Y.C., Kim J., Lee J.S., Lee J.W., Ye S.K., Park J.W., Kim C.W. Cholesterol depletion induces anoikis-like apoptosis via FAK down-regulation and caveolae internalization. J. Pathol. 2009;218:337–349. doi: 10.1002/path.2531. [DOI] [PubMed] [Google Scholar]
- 30.Paoli P., Giannoni E., Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta. 2013;1833:3481–3498. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Subramani J., Ghosh M., Rahman M.M., Caromile L.A., Gerber C., Rezaul K., Han D.K., Shapiro L.H. Tyrosine phosphorylation of CD13 regulates inflammatory cell-cell adhesion and monocyte trafficking. J. Immunol. 2013;191:3905–3912. doi: 10.4049/jimmunol.1301348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J., Liu A., Wang Z., Wang B., Chai X., Lu W., Cao T., Li R., Wu M., Lu Z. LINC00173.v1 promotes angiogenesis and progression of lung squamous cell carcinoma by sponging miR-511-5p to regulate VEGFA expression. Mol. Cancer. 2020;19:98–116. doi: 10.1186/s12943-020-01217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin L., Chun J., Pan C., Kumar A., Zhang G., Ha Y., Li D., Alesi G.N., Kang Y., Zhou L. The PLAG1-GDH1 Axis Promotes Anoikis Resistance and Tumor Metastasis through CamKK2-AMPK Signaling in LKB1-Deficient Lung Cancer. Mol. Cell. 2018;69:87–99.e7. doi: 10.1016/j.molcel.2017.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Summy J.M., Gallick G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 35.Cai H., Smith D.A., Memarzadeh S., Lowell C.A., Cooper J.A., Witte O.N. Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc. Natl. Acad. Sci. USA. 2011;108:6579–6584. doi: 10.1073/pnas.1103904108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lechertier T., Hodivala-Dilke K. Focal adhesion kinase and tumour angiogenesis. J. Pathol. 2012;226:404–412. doi: 10.1002/path.3018. [DOI] [PubMed] [Google Scholar]
- 37.Lambert A.W., Pattabiraman D.R., Weinberg R.A. Emerging Biological Principles of Metastasis. Cell. 2017;168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyu Y., Nakano K., Davis R.R., Tepper C.G., Campbell M., Izumiya Y. ZIC2 Is Essential for Maintenance of Latency and Is a Target of an Immediate Early Protein during Kaposi’s Sarcoma-Associated Herpesvirus Lytic Reactivation. J. Virol. 2017;91:e00980-17. doi: 10.1128/JVI.00980-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Retraction. J. Cell. Biochem. 2021;122:145. doi: 10.1002/jcb.29851. [DOI] [PubMed] [Google Scholar]
- 40.Vural B., Chen L.C., Saip P., Chen Y.T., Ustuner Z., Gonen M., Simpson A.J., Old L.J., Ozbek U., Gure A.O. Frequency of SOX Group B (SOX1, 2, 3) and ZIC2 antibodies in Turkish patients with small cell lung carcinoma and their correlation with clinical parameters. Cancer. 2005;103:2575–2583. doi: 10.1002/cncr.21088. [DOI] [PubMed] [Google Scholar]
- 41.Chen J., Liu A., Lin Z., Wang B., Chai X., Chen S., Lu W., Zheng M., Cao T., Zhong M. Downregulation of the circadian rhythm regulator HLF promotes multiple-organ distant metastases in non-small cell lung cancer through PPAR/NF-κb signaling. Cancer Lett. 2020;482:56–71. doi: 10.1016/j.canlet.2020.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Ruan X., Liu A., Zhong M., Wei J., Zhang W., Rong Y., Liu W., Li M., Qing X., Chen G. Silencing LGR6 Attenuates Stemness and Chemoresistance via Inhibiting Wnt/β-Catenin Signaling in Ovarian Cancer. Mol. Ther. Oncolytics. 2019;14:94–106. doi: 10.1016/j.omto.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu A., Zhang X., Li R., Zheng M., Yang S., Dai L., Wu A., Hu C., Huang Y., Xie M., Chen Q. Overexpression of the SARS-CoV-2 receptor ACE2 is induced by cigarette smoke in bronchial and alveolar epithelia. J. Pathol. 2021;253:17–30. doi: 10.1002/path.5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.