

Summary
Metabotropic glutamate receptors (mGluRs) are dimeric G-protein-coupled receptors activated by the main excitatory neurotransmitter, L-glutamate. mGluR activation by agonists binding in the venus flytrap domain is regulated by positive (PAM) or negative (NAM) allosteric modulators binding to the 7-transmembrane domain (7TM). We report the cryo-electron microscopy structures of fully inactive and intermediate-active conformations of mGlu5 receptor bound to an antagonist and a NAM or an agonist and a PAM, respectively, as well as the crystal structure of the 7TM bound to a photoswitchable NAM. The agonist induces a large movement between the subunits, bringing the 7TMs together and stabilizing a 7TM conformation structurally similar to the inactive state. Using functional approaches, we demonstrate that the PAM stabilizes a 7TM active conformation independent of the conformational changes induced by agonists, representing an alternative mode of mGlu activation. These findings provide a structural basis for different mGluR activation modes.
Keywords: G-protein-coupled receptors, signal transduction, glutamate, metabotropic glutamate receptor 5, allosteric modulators, cryo-EM, X-ray crystallography, photochromic ligands
Graphical abstract
Highlights
-
•
Cryo-EM analysis of thermostabilized mGlu5 receptor bound to inhibitors or activators
-
•
X-ray structure of trans-Alloswitch-1 bound to thermostable mGlu5 7TMs
-
•
Photopharmacology provides insight into allosteric regulation of mGlu5 7TMs
-
•
Multiple conformations of mGlu5 receptor activate G protein
Nasrallah et al. present cryo-EM structures of thermostabilized mGlu5 dimer bound to inhibitors and activators as well as an X-ray structure of mGlu5 7TM-bound photoswitchable ligand alloswitch-1. The structural and functional analyses reveal different modes of mGLu5 receptor activation and provide a structural basis for mGlu receptor activation mechanism.
Introduction
L-glutamate is the major excitatory neurotransmitter in the adult mammalian brain. This neurotransmitter is sensed by different receptors including the ionotropic glutamate receptors that are ligand-gated ion channels and the metabotropic glutamate (mGlu) receptors that belong to class C G-protein-coupled receptors (GPCRs). Eight subtypes of the mGlu receptors, mGlu1 to mGlu8, are expressed in the central nervous system (CNS) where they play a major physiological role in the precise tuning of synaptic activity and plasticity (Gregory and Goudet, 2021). Dysfunctions of mGlu receptors are associated with neurological disorders and psychiatric diseases. Among the mGlu receptors family, the mGlu5 subtype is a post-synaptic receptor, which is strongly expressed in the cortex, hippocampus, and striatum of the mammalian brain. It is an attractive therapeutic target for several diseases including anxiety, depression, Parkinson disease, schizophrenia, or fragile X syndrome (Gregory and Goudet, 2021).
Class C GPCRs are obligatory dimers whereby oligomerization is fundamental to their function as illustrated by the dimeric metabotropic glutamate receptors (mGlu) and the heterodimeric gamma amino-butyric acid receptor (GABAB) (Pin and Bettler, 2016). For mGlu receptors, each monomer possesses a large extracellular domain (ECD) that mediates dimerization by forming an intermolecular disulphide bridge. The ECD is composed of the venus flytrap domain (VFT), which contains the orthosteric binding site for glutamate, and a cysteine-rich domain (CRD) that links the VFT to the 7-transmembrane domain (7TM)-activating G proteins. The multidomain architecture of the mGlu dimers makes them highly flexible receptors that can adopt multiple conformations. Understanding how ligand binding in the VFT controls the 7TM conformation that is 100 Å away to activate the G protein is a long-standing question in the field of mGlu receptors. Structural characterization of the mGlu receptor conformations, either bound to inhibitors or activators that regulate the mGlu receptor activation, is crucial for understanding the physiology of receptors and for designing molecules with therapeutic potential.
The VFT is composed of two lobes (lobe I and lobe II) that form the binding site for L-glutamate (Kunishima et al., 2000). Agonist binding stabilizes the closed state (c) and induces the re-orientation of the VFT dimer corresponding to an active (A) state, while antagonist binding stabilizes the open (o) and inactive resting (R) state of the dimer (Pin and Bettler, 2016). Recently, cryoelectron microscopy (cryo-EM) structures of the full-length human mGlu5 receptor in the ligand-free (apo) and agonist-bound conformations described the molecular reorganization occurring between the VFTs, CRDs, and 7TMs upon agonist-induced receptor activation (Koehl et al., 2019). These studies revealed that agonist binding stabilizes a conformation of the VFT dimer that is propagated through the rigid CRDs to the ECL2 of the 7TMs, leading to intermolecular interactions mediated by the top of TM6 from each protomer. It is expected that such large molecular reorganization of the mGlu5 quaternary structure also induces intramolecular conformational changes in the 7TMs to promote the intracellular G-protein coupling and activation, in the vicinity to the allosteric binding site. In addition, mGlu5 apo inactive state is characterized by the absence of molecular interaction between the 7TMs, whereas the heterodimeric class C GABAB receptor inactive conformation is stabilized by a molecular interface established between the 7TMs (Mao et al., 2020; Park et al., 2020; Shaye et al., 2020), illustrating the diversity of conformations of class C GCPRs. It remains to be understood how the diverse mGlu5 receptor conformations can lead to receptor signaling.
L-glutamate dependent signal transduction of mGlu receptor can be allosterically potentiated or inhibited by the binding of synthetic positive or negative allosteric modulators (PAM and NAM, respectively) to the 7TMs (Stansley and Conn, 2019), and allosteric modulators are of great interest for their therapeutic potential in treating mGlu-associated brain disorders. In some cases, PAMs, called ago-PAM, display intrinsic agonist activity and can activate the receptor in absence of orthosteric agonist. High-resolution X-ray structures of mGlu5 7TM domain bound to a NAM provided the molecular basis of the allosteric binding sites (Christopher et al., 2015, 2019; Doré et al., 2014). However, the binding mode of PAMs is still unclear, as is the 7TM conformation stabilized by such molecules that can directly activate isolated mGlu 7TMs (El Moustaine et al., 2012; Goudet et al., 2004). Thus, it is important to understand the conformational changes occurring at the 7TM upon orthosteric agonist activation and by allosteric modulation.
Here, we report multiple structures of a fully functional thermostabilized human mGlu5 receptor determined by cryo-EM and of the 7TM bound to a photoswitchable ligand by X-ray crystallography. Cryo-EM structures were determined of the full-length mGlu5 receptor in a fully inactive state and an intermediate-active state. The fully inactive state structure was obtained with the receptor bound to the orthosteric antagonist LY341495 and the NAM MPEP, while the ligand-activated structure contains the orthosteric agonist quisqualate and the ago-PAM VU0424465. In addition, a crystal structure of the thermostable mGlu5 7TM domain alone was determined with a photochromic NAM alloswitch-1. Together with pharmacological analysis of the receptor conformations and the photo-pharmacological characterization of multiple receptor mutants, the structures provide insights into the mGlu5 receptor activation mechanisms.
Results
Thermostabilization of the full-length human mGlu5 receptor
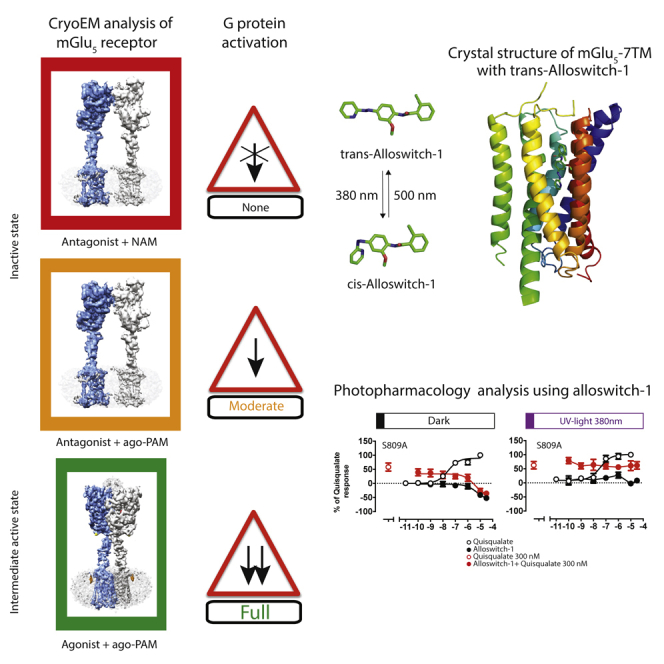
We previously described the expression and purification of a truncated version (mGlu5-Δ856) of the human mGlu5 receptor (Nasrallah et al., 2018). The yield of this receptor was low, and, more importantly, the receptor was rather unstable in the detergent dodecyl-β-maltoside (DDM), more commonly used for structural characterization of GPCRs. To improve both the yield and stability, the MPEP-bound conformation of the full-length mGlu5 receptor was thermostabilized by introducing five mutations (T742A5.42, S753A5.53, T777A6.42, I799A7.29, A813L7.43) in the 7TM domain of the C-terminally truncated mGlu5 receptor (mGlu5-Δ856) (Figure 1A; Table S1). Throughout the manuscript, residues of the 7TM are named using an adapted version of the Ballesteros numbering scheme previously developed for class C GPCRs (Ballesteros and Weinstein, 1995; Pin et al., 2003). Thermostabilizing mutants were identified by combining a radioligand-binding assay to the alanine-scanning mutant library as previously described (Magnani et al., 2016). We screened 288 single mutants of the mGlu5 7TM domain, and 11 mutants were first selected based on their improved thermostability compared to the mGlu5-Δ856. The best single thermostable single mutant A813L localized in TM7 (mGlu5-1M) displays a Tm of 28°C, compared to 20.7οC for the WT receptor (Figure 1; Table S1). A813L was then combined with four other thermostabilizing mutants, T742 (TM5), S753A (TM5), T777A (TM6), and I799A (TM7), based on their additive properties to give the construct named mGlu5-5M (Figure 1; Table S1). Among the five thermostabilizing mutations selected here, two of them, T742A and S753A, were previously identified and selected for stabilizing and solving the structure of the mGlu5 7TM domain bound to mavoglurant (Doré et al., 2014). The other three mutants stabilizing the receptor were identified in this screen. This construct mGlu5-5M displays an improved thermal stability of ∼20°C in MNG3-CHS (apparent Tm = 41.42°C) and DDM-CHS (apparent Tm = 40.22°C) when bound to [3H]-MPEP (Figures 1B and S1; Table S2). mGlu5-5M retains its full capacity to activate G protein compared to the wild-type (WT) receptor (Figure 1C). The thermostabilized mGlu5-5M receptor was expressed and purified to homogeneity from a 2L culture volume for structural analysis by using cryo-EM (Figures S2A and S2B).
Figure 1.

Thermostabilization of the full-length human mGlu5 receptor dimer, mGlu5-5M
(A) Snake plot of the transmembrane domain and C terminus of the human mGlu5 receptor depicting the important residues mutated to improve the thermostability of the receptor. Eleven thermostabilizing mutants in mGlu5 7TM domain were identified (circled in yellow), among which five (yellow lined circle and filled in green) were combined to thermostabilize the NAM MPEP-bound conformation of the human mGlu5 receptor dimer.
(B) Thermal stability curves for the best mutant and intermediate double, triple, quadruple, and quintuple mutants thermostabilized mGlu5-5M receptor (the details of the thermal stability are presented in Table S1). Two out of five mutations (T742A and S753A) of the mGlu5-5M are overlapping with the previously thermostabilized mGlu5 7TM described by Doré et al. (2014).
(C) Dose-response curve of the thermostabilized mGlu5-5M receptor in a signaling assay with a panel of ligands shows the conserved properties of the mutant mGlu5-5M to activate Gq protein. The pharmacological profile of the mGlu5-5M is similar to the profile of the mGlu5 WT receptor. The mGlu5-Δ856 and mGlu5-5M receptors are dose-dependently activated with similar potencies by the orthosteric agonists glutamate (pEC50 are 4.33 ± 0.07 [n = 4] and 4.12 ± 0.08 [n = 4], respectively) and quisqualate (pEC50 are 7.05 ± 0.15 [n = 5] and 6.69 ± 0.08 [n = 5], respectively) and by the ago-PAM VU0424465 (pEC50 are 7.54 ± 0.20 [n = 5] and 7.55 ± 0.15 [n = 5], respectively). In addition, the activation of mGlu5 WT and mGlu5-5M induced by quisqualate (1 μM) is similarly inhibited by the NAM MPEP (pIC50 are 7.64 ± 0.11 [n = 4] and 8.66 ± 0.34 [n = 3], respectively) in a dose-dependent manner. Average values from at least three independent experiments are plotted, and the error bar is represented as ±SEM.
We have further characterized the stability of the receptor in different detergents and evaluated the possible contribution of the ECD. The stabilized receptor, mGlu5-5M, displays stability in a shorter-chain detergent such as decyl β-D-maltopyranoside (DM) for which we could not previously measure the Tm on the WT mGlu5-Δ856 (Figure S1A; Nasrallah et al., 2018). We were also able to measure the stability in more denaturing short-chain detergents such as nonyl glucoside (NG), and C8E4 in which mGlu5-5M remains properly folded but less stable when compared to DDM or MNG3.
The ECD is composed of the VFT and the CRD that interacts with residues from the ECL2. This interaction was previously shown to be important for mediating signal transduction (Koehl et al., 2019). CRD-ECL2 interaction might also contribute to the thermostability of the full-length receptor. We measured the thermostability of the 7TM-5M alone in several different detergents similar to the full-length receptors. The thermostability of the 7TM-5M is dramatically impaired in MNG3 but also for all other detergents when compared to mGlu5-5M (Figures S1A and S1B; Table S2). This observation supports the contribution of the ECD not only for mediating the signal transduction but for maintaining the 7TM domain structural integrity required to its function.
Cryo-EM structures of the thermostabilized full-length human mGlu5 receptor
The cryo-EM structures of the mGlu5-5M receptor bound to the orthosteric antagonist LY341495 and the NAM MPEP was determined to an overall resolution of 4.0 Å (Figure 2A; Figure S3; Table S3), and the agonist quisqualate and the high-affinity ago-PAM VU0424465 was determined to an overall resolution of 3.8 Å (Figure 2D; Figure S4; Table S3). In both cryo-EM maps, the ECDs, in particular, the VFTs are better resolved, but the CRDs and 7TMs display lower resolution (Figures S3–S5).
Figure 2.

Cryo-EM structures of thermostabilized full-length mGlu5 receptor
(A and B) The cryo-EM map (sharpened with a B factor of −82 Å2) and the model of the receptor bound to the orthosteric antagonist LY341495 and NAM MPEP.
(C) Density for LY341495 and the surrounding residues in the VFT. The EM map sharpened with a B factor of −164 Å2 at 7 σ was used to make the figure with Pymol.
(D and E) The cryo-EM map (sharpened with a B factor of −61 Å2) and the model of the receptor bound to orthosteric agonist quisqualate and PAM VU0424465.
(F) Density for quisqualate and surrounding residues. The EM map sharpened with a B factor of −122 Å2 at 10 σ was used to make the figure with Pymol.
One of the monomers of the receptor in (A) and (D) is colored in blue and the other in gray. The rough position of NAM and PAM in the 7TM of the cryo EM maps are marked but not modelled. The fuzzy density at the top of the ECD at the dimer interface has not been modeled. Orthosteric ligands are represented in red (in both map and model). In the model, the VFTs are colored in green, CRDs in blue, and 7TMs in magenta. The density for cholesterol hemisuccinate in (D) is highlighted in orange and also shown in stick representation in (E).
In the antagonist-bound EM map, we observe an extra density at the hinge of lobe I and lobe II that is modeled as LY341495 in each VFT (Figures 2A and 2C). Interactions between LY341495 and the receptor are mediated by both lobe I (70% of the residue contacts) and lobe II (30% of the residue contacts) (Figure 3A; Table S4). Ser152 and Thr175 in lobe I establish hydrogen bonds with the amino and carboxylic group substituted on carbon-2 of LY341495. Similar hydrogen-bond networks are mediated by homologous residues in the mGlu1, mGlu3, and mGlu7 VFT X-ray structures in complex with LY341495, although Ser152 is not engaged in mGlu3 (Table S4; Figure S6). In lobe II, the direct interaction of LY341495 with the side chain of Tyr223 stabilizes an open conformation of lobe II relative to lobe I that is even larger than observed in the apo state (Figures 3B and 3C). Positioning of LY341495 in the apo structure reveals clashes with Ser173, Ala174, Thr175, and Tyr223 (Figure S7). Simultaneous binding of an antagonist in the orthosteric binding site and MPEP in the 7TMs abolishes G-protein signaling and basal activity of mGlu5. This structure represents therefore the fully inactive R conformation of the receptor (Figures 2A and 2B).
Figure 3.

LY341495-bound inactive conformation of the mGlu5 receptor
(A) Ligand binding site of the LY341495.
(B and C) Comparison of molecular interactions between orthosteric antagonist LY341495 and Y223 from lobe I in the structure. The position of Y223 in the apo inactive state (cyan; PDB 6N52) clashes with LY341495 if it adopted the same position seen in the mGlu5 structure.
(D) Compared to apo state (cyan; PDB 6N52), quisqualate induces the VFT closure (green), while LY341495 induces further opening (blue). In both cases, conformational changes are transmitted through the rigid CRDs to the 7TMs.
(E and F) Repositioning of 7TMs from LY341495- and quisqualate-bound conformation compared to apo inactive conformation. The depicted values (Δ) are the difference in distances between pairs of Cα atoms for the antagonist-bound and the apo states (E) and the antagonist- and agonist-bound states (F). Superposition of the mGlu structures was performed using residues R26 to V192 and S331 to D461 from lobe I of the LY341495-bound, quisqualate-bound, and the apo state (PDB:6N52) of mGlu5 receptor.
When we performed 3D classification of the agonist-bound receptor, a sub-population of receptors was found in the inactive state, and this is most likely due to the loss of quisqualate (Figure S4F). Due to the lower resolution of the map, analysis of this class was not pursued further. Overall, the quisqualate-bound map is better resolved than the antagonist-bound receptor (Figure S5) and is similar to the previously published mGlu5 receptor structure bound to a nanobody, quisqualate, and the ago-PAM CDPPB (Figures 2D–2F; Koehl et al., 2019). At the orthosteric binding site, the agonist quisqualate is found to interact with a similar set of residues in lobe I as the antagonist LY341495, including Y64, W100, Ser151, Ser152, Ser173, Ala174, and Thr175 (Table S4). However, the planar structure of quisqualate allows stabilization of the closed conformation of the VFTs, whereas LY341495, a larger molecule, prevents the VFTs closure, leading to a stable open state.
The resolution of the 7TMs is lower than that of the VFTs, and the model derived from the X-ray structure including the side chains was placed in the density and due to lower resolution we refrained from making any extensive structural comparisons but perform only global analysis. Superposition of the agonist-bound and apo state structures with LY341495-bound state structure illustrates the different dispositions of the 7TMs with respect to the lobe II movement. The movement of lobe II translates to the CRDs, which induces the repositioning of the 7TM domains (Figure 3D). The 7TMs of the LY341495-bound state are ∼25 Å apart (measured between Cα of Leu7385.38 from each 7TM residues) compared to 21 Å apart in the apo state (ΔLY341495- Δapo = 4.1 Å) (Figures 3E and 3F). There are no molecular contacts between the 7TMs of the mGlu5 receptor dimer in the fully inactive conformation (Figures 2A and 2B) similar to the previously described apo state (Koehl et al., 2019). This is different from the molecular interfaces between the 7TMs suggested for mGlu2 inactive conformation (Xue et al., 2015) and reported for the heterodimeric class C GABAB receptor (Mao et al., 2020; Papasergi-Scott et al., 2020; Park et al., 2020; Shaye et al., 2020) and may explain differences in the signaling properties of the various class C receptors. In its inactive state, each of the 7TM of the mGlu5 receptor protomers is an isolated entity.
In the intermediate-active conformation, VFTs, CRDs, and 7TMs form a compact mGlu5 receptor dimer, which stabilizes 7TM active interface via TM6-TM6 molecular contacts with Ile7916.56 separated by only 6.7 Å, whereas the same residues are 41.8 and 43.3 Å apart in the LY341495-bound state and apo state, respectively (Figures 3E and 3F), as measured between the Cα atoms. This large movement of the 7TMs is very likely mediated by the ECL2 of the 7TM that interacts with the CRD and with the linker connecting TM1 to the CRD (Koehl et al., 2019). This interaction was previously shown to be important for the quisqualate-bound conformation to activate G proteins, highlighting its critical role for translating VFT conformational changes to the 7TM (Koehl et al., 2019).
The protein preparation of agonist-bound mGlu5-5M also contains the ago-PAM VU0424465, which is thought to bind to the 7TMs. There is a strong density adjacent to residues Ser8097.39, Trp7856.50, and Pro6553.40 and is likely to be VU0424465 as this is part of the same allosteric binding pocket where NAMs bind (Christopher et al., 2015, 2019; Doré et al., 2014; Figure S5). The resolution obtained is insufficient to model and resolve the molecular details of PAM binding as the pose of the ligand cannot be assigned unambiguously. An additional density on the lipid-facing surface of TM1 at its extracellular side is modeled as cholesteryl hemisuccinate (CHS), which is present throughout the purification (Figure 2D). The modeled CHS molecule occupies a similar position to two of the oleic acid molecules modeled in the X-ray structure of mGluR5 7TM (Doré et al., 2014). Cholesterol is known to bind to the lipid-exposed surface of receptors and for stabilizing the receptor fold. This observation suggests that the rigidity of the cholesterol molecule may also contribute in stabilizing the interaction between TM1, 2, and 3. In the absence of the G protein to stabilize the full active conformation (Venkatakrishnan et al., 2013), this structure is likely to represent an intermediate-active state.
Structure-function insight from the thermostabilized mGlu5 7TM domain bound to the photochromic ligand alloswitch-1
The cryo-EM structures described above reveal the structural determinants of the mGlu5-5M receptor dimer in a fully inactive and agonist-bound intermediate-active state. The lower resolution of the 7TM regions precluded molecular insights into NAM and PAM binding. Thus, it is essential to understand the binding mode of allosteric modulators as they can control specific conformations of the 7TMs. More importantly, such compounds offer the possibility to develop photoswitchable molecules to control mGlu receptor activity with light (Berizzi and Goudet, 2020; Goudet et al., 2018; Hüll et al., 2018) as illustrated by recent studies that described the use of the photoswitchable ligand, alloswitch-1, that is selective for the mGlu5 receptor (Pittolo et al., 2014).
Alloswitch-1 is an azobenzene-containing compound that acts as a NAM with robust inverse agonist activity in its active, trans configuration (pIC50 = 7.07 ± 0.17; Table S5) and can rapidly and reversibly isomerise with exposure to UV light (380 nm; trans-to-cis) into its less active cis-configuration. The potency of the cis-isomer is significantly reduced (pIC50 = 5.58 ± 0.24; Table S6), consistent with previous investigations (Ricart-Ortega et al., 2020) and notably due to the lower affinity of the cis isomer. Alloswitch-1’s photoswitchable properties make it a powerful tool for investigating the role of 7TM residues in mGlu5 receptor allosteric modulation. We determined the crystal structure of the thermostabilized mGlu5 7TM, mGlu5-StaR (569-836)-T4L (Doré et al., 2014), bound to the photoswitchable NAM alloswitch-1 in its trans configuration (Pittolo et al., 2014) (Figure 4; Figures S2 and S8). The mGlu5-StaR(569-836)-T4L comprises 6 thermostabilizing mutations in the 7TM (Figure 1A), a T4L fusion protein at the intracellular tip of TM5 and TM6 in the intracellular loop 3 (ICL3), and this construct was previously used for determining the X-ray structures of the receptor with NAMs (Doré et al., 2014).
Figure 4.

High-resolution X-ray structure of the alloswitch-1-bound mGlu5 7TM
(A) The binding site of alloswitch-1 trans-isomer (yellow) in the thermostabilized mGlu5 7TM. The 2Fo-Fc map of the alloswitch-1 and key residues contoured at 1.5 σ.
(B and C) Chlorophenyl moiety of the photoactivable NAM fits into a hydrophobic sub-pocket formed by residues from TM5 and TM6 that also involves outward motion of F788 away from the core of the receptor.
(C and D) The phenylazopyridine binds to the NAM binding site that has been previously described and forms hydrogen bonds with S809 and Y659. S809 is a key residue that also binds to the pyridine ring. (D) Structural water molecule and W785 further stabilizes the alloswitch-1 trans-isomer and receptor inactive conformation.
(E and F) Photopharmacology of alloswitch-1 for the S809A and W785A receptor mutants in dark and photo-illuminated conditions. Dose responses represent the average measurement of at least three independent experiments performed in triplicate, and the error bar is represented as ±SEM.
The 2.5 Å structure of mGlu5-StaR(569-836)-T4L shows that alloswitch-1 binds between TM2, 3, 5, 6, and 7 and overlaps with the previously characterized NAM binding site (Figure 4A; Figure S8; Table S7; Christopher et al., 2015, 2019; Doré et al., 2014; Gregory and Conn, 2015; Gregory et al., 2013). The chlorophenyl ring binds high up in the 7TM domain between TM5 and 6, engaging in hydrophobic interactions with Trp7856.50, Val7896.54, Tyr7926.57, Phe7936.58, Val7405.40, and Leu7445.44 (Figure 4B; Figure S8B). The hydrophobic pocket is created by the repositioning of the Phe7886.53, which is tilted away from the receptor core compared to previously reported structures of NAM bound to thermostabilized mGlu5 StaR (Christopher et al., 2015, 2019; Doré et al., 2014; Figure 4C). The phenylazopyridine moiety of alloswitch-1 overlaps mostly with the M-MPEP NAM binding pocket and is stabilized by hydrophobic contacts by residues Ser6543.39, Gly6242.45, Ile6252.46, Pro6553.40, Val8067.36, and Ala8107.40 (Figure S8). Tyr6593.44 and Ser8097.39 complete the pocket and hydrogen bond to the N1 of pyridine ring (Figures 4C and 4D). Ser8097.39 makes an additional hydrogen bond with N2 from the phenylazopyridine moiety. Several of these residues, including Tyr6593.44, Trp7856.50, and Ser8097.39 have been identified as important in mediating the effects of positive and negative allosteric modulators (Gregory et al., 2013, 2014; Hellyer et al., 2020; Mølck et al., 2014; Sengmany et al., 2020). The structure with alloswitch-1 serves as a template for mutational and biochemical analysis and to further investigate the role of the allosteric binding site residues in stabilizing the mGlu5 inactive state.
The UV-light-induced conversion of trans-alloswitch-1 to the cis-isomer implies the displacement of the azobenzene moiety in the allosteric binding site results in the likely loss of affinity (Ricart-Ortega et al., 2020). Thus, mutation of the key residue Ser8097.39 strongly reduces the pIC50 of alloswitch-1 (Δ pIC50 = 1.5 log units; pIC50 = 5.58 ± 0.24), suggesting its strong contribution to alloswitch-1’s ability to inhibit receptor activity (Figure 4E; Table S5). However, in the S809A mutant, when the alloswitch-1 isomerization from trans-to-cis is triggered with UV illumination, there is a complete loss in cis-alloswitch-1 inhibitory potency (Figures 4E and 4F; Table S6). This observation has two important implications. First, Ser8097.39 is a key residue for stabilizing the NAM-dependent inactive conformation of the receptor (Christopher et al., 2015; Doré et al., 2014; Gregory et al., 2013, 2014). Second, the binding pocket exploited by the phenylazopyridine moiety is key for mediating alloswitch-1 negative allosteric effect. Tyr6593.44 also binds to the pyridine nitrogen and similar to Ser8097.39, Tyr659A mutation reduces the NAM activity of both trans- and cis-alloswitch-1 (Tables S5 and S6; Figures S9–S11). The inactive conformation of mGlu5 7TM is further stabilized by Thr7816.46. When this residue is mutated to alanine, alloswitch-1 has significantly reduced inhibitory potency for the receptor in dark or illuminated conditions. The hydroxyl of Tyr6593.44, the main chain carbonyl of Ser8097.39, and Thr7816.46 coordinate a water molecule and together stabilize the TM3, TM6, and TM7 helices (Figure 4D). Thus, the phenylazopyridine moiety of trans-alloswitch-1, the hydrogen bond network formed by residues Tyr6593.44, Ser8097.39, Thr7816.46, previously identified in mGlu5 NAM-bound conformation (Christopher et al., 2015, 2019; Doré et al., 2014), and the alloswitch-1 hydrophobic interactions in TM2, 3, 5, 6, and 7 all mediate alloswitch-1’s inhibitory effects and stabilize the 7TM inactive conformation.
A last component of the inactivation mechanism is the residue Trp7856.50, equivalent to the toggle switch Trp6.48 in class A GPCRs (Kobilka, 2007). Mutating this residue was previously reported to turn some NAMs into PAMs (Gregory et al., 2014). Here, we found that Trp7856.50 makes a strong interaction with the anisole ring of allsowitch-1 and also engages Ser8097.39 through a hydrogen bond (Figure 4D). Replacing the tryptophan residue to alanine (W785A) switches the activity of the NAM alloswitch-1 to a PAM (Figure 4E; Figure S12). Triggering alloswitch-1 trans-to-cis photoisomerization impairs this PAM effect and Gq activation, suggesting that the trans azobenzene binding into the conserved NAM pocket is also important for the PAM activity of the W785A mutant (Figures 4E and 4F; Figure S12). The Trp7856.50 side chain is likely to restrict the movement of TM6 in the WT receptor. Conformational changes in the 7TM and contribution of TM6, which represents the molecular signature of class A and B active conformation, remain to be fully characterized for mGlu receptors. The Trp7856.50 residue might play a role in controlling the motion of TM6 and its conformation in the 7TM dimer interface. A recent structure of GABAB receptor bound to Gi protein shows only a small displacement of the TM6 compared to the inactive state (Mao et al., 2020). The structure-function analysis presented here also supports only subtle conformational differences between the inactive and active conformations of the mGlu5 7TM.
7TM signaling from the mGlu5 orthosteric antagonist-bound state
From the present data and those recently reported (Koehl et al., 2019), two key molecular interactions explain how the VFT conformational change can lead to the activation of at least one 7TM. One is the interaction between the ECL2 and the CRD and its linker that connects to the first transmembrane domain. ECL2 and the CRD form a rigid bridge between the ECD and the 7TM. The change in orientation relative to the membrane plane of the CRD resulting from the reorientation of the VFTs is important for stabilizing the active conformation. Consistent with this interaction, mutation in the apical region of ECL2 largely affects the ability of the activated VFTs to transduce the signal to the 7TMs (Koehl et al., 2019). The second important interaction is defined by the molecular contact between the TM6s of both 7TMs in the active state, allowing stabilization of at least one 7TM in its active state, to finally lead to G-protein activation. The role of this second interaction is functionally supported by the large constitutive activity of a dimeric receptor in which the TM6 are crosslinked (Koehl et al., 2019) but also by the role of the W7856.50 in mediating alloswitch-1’s allosteric effect (Figure 4E), highlighting the contribution of TM6 in stabilizing 7TM active conformation.
Building on this, we further examined whether the individual 7TM from mGlu5 receptor dimer can induce G-protein signaling upon PAM activation, independent of VFT/CRD orientation relative to the plane of the membrane and the 7TM dimer interface. The ago-PAM VU0424465 can activate mGlu5 (Rook et al., 2013), and here we show that this response can only be partially inhibited by saturating concentrations of the competitive antagonist LY341495 that prevents VFT closure (Figure 5A; Table S8). Previously, it has been reported that the open VFTs can still move to reach the active orientation, as illustrated by the crystal structure of the mGlu1 VFT dimer with bound LY341495 (PDB: 3KS9), we cannot exclude the possibility of ago-PAM VU0424465 inducing the active (A) state of the VFT dimer. However, this seems unlikely as the VFT sensor based on the FRET signal between N-terminally fused snap-tags that directly reports the relative orientation of the VFTs (Doumazane et al., 2013) did not reveal any decrease in FRET upon VU0424465 activation, in contrast to what is observed with quisqualate (Figure 5B). We then confirmed the ability of the mGlu5 receptor to signal despite its VFTs being in inactive resting R conformation, by measuring G-protein activation from membrane expressing the mGlu5 receptor, excluding the possibility for endogenous L-glutamate to interfere. As expected, VU0424465 triggers G-protein activation even though the VFTs are bound to LY341495 that stabilizes the VFTs inactive R state (Figures 5A and 5C). These data thus demonstrate that the ago-PAM VU0424465 can activate the 7TMs even if the VFTs are maintained in their inactive conformation, indicating that the orientation of CRD-ECL2 rigid linker required for transmitting the VFT conformational changes is not a prerequisite for 7TM activation upon PAM stimulation. In summary, the ago-PAM can bypass the large glutamate-dependent structural rearrangement that brings the 7TM closer and triggers signal transduction from the isolated 7TM in absence of glutamate.
Figure 5.

The 7TMs of mGlu5 receptor inactive conformation are decoupled from the ECD and can couple to G protein
(A) IP1 functional assays show the activation of the 7TM domain by the high-affinity PAM VU0424465 even in the presence of the saturating amounts of antagonist LY341495 in the VFT. Quisqualate displaces LY341495 and provides a full response compared to the PAM that displays moderate efficacy. One point measurement with saturating concentration of antagonist LY341495 and NAM MPEP was used as a negative control of IP1 production.
(B) Conformational analysis using a mGlu5 FRET sensor to define different states of the mGlu5 receptor in the membrane in the presence of different ligands.
(C) In vitro G-protein activation assay using a purified G protein and membrane-bound mGlu5 receptor.
(D) A mechanistic model of receptor conformation and G-protein activation. Dose responses represent the average measurement of at least three independent experiments performed in triplicate, and the error bar is represented as ±SEM.
Discussion
Here, we present a detailed analysis of the class C mGlu5 receptor conformations by determining the structures of the detergent-purified full-length mGlu5 receptor in fully inactive and the intermediate-active state.
The thermostabilized version of the receptor used here, mGlu5-5M, enabled the fully inactive structure of the receptor to be solved at 4 Å. We observed a slightly more open conformation of the VFTs, which is required for accommodating the antagonist LY341495 in the orthosteric binding site, compared to the apo state. The previously described inactive apo state conformation was obtained by reconstituting the receptor in nanodiscs to resolve the density within the 7TMs (Koehl et al., 2019). Both the structures of mGlu5 in detergent environment as described here and in nanodiscs by Koehl et al. (2019) reveal that the 7TMs are physically distant, and further stabilization and improvement in data collection will be required to obtain higher resolution maps of inactive states in particular the 7TMs. This lack of molecular interaction between the 7TMs in the inactive conformations of mGlu5 is not a general feature for class C GPCRs. The mGlu5, mGlu2, mGlu7, and GABAB receptor structures diverge in their inactive state. In GABAB and mGlu2 the dimer is stabilized by 7TM interactions (Du et al., 2021; Park et al., 2020), whereas the mGlu7 displays an inactive state comparable to mGlu5, in which the 7TMs are physically distant (Du et al., 2021). Such diversity in inactive state is likely to apply to some other mGlu receptors and other class C GPCRs.
Based on the functional analysis and biophysical characterization using the FRET-based VFTs sensor, we propose that this inactive conformation of the mGlu5 dimer may offer an alternative conformation from which intracellular G protein can be activated and may account for some signaling diversity. This is a highly relevant observation that needs to be considered when developing PAMs targeting mGlu5. The ago-PAM-dependent signaling mechanism proposed here might account for side effects reported for such ligands (Rook et al., 2013), which act independently of L-glutamate activation and do not require the receptor dimer to adopt the active conformation (Figure 5D). However, the level of ago-PAM induced biological response is lower compared to glutamate (Figure 5A).
The agonist-bound state (intermediate-active) of the thermostabilized mutant mGlu5-5M was determined without the need for stabilizing the VFT active state with nanobody as previously reported (Koehl et al., 2019). The structure presented here and the previously reported structure with nanobody bound to the VFT (PDB 6N52) are very similar, and both show the closure and the reorientation of the VFTs induced by quisqualate binding that triggers the rotation of the 7TMs stabilized by a molecular interaction at the extracellular tip of TM6. Additional density observed in the 7TM is likely to be associated with the PAM VU0424465 used in the preparation. However, a high-resolution structure of the 7TM domain will be required to fully characterize the binding mode of the PAMs.
Conformational understanding of the receptor dimer cannot be separated from the molecular mechanism of the 7TMs that directly activate G proteins (Pin and Bettler, 2016). We then solved the structure of the mGlu5 7TM bound to a photoswitchable NAM, alloswitch-1. Combined with alanine mutants of the alloswitch-1 binding site residues and photopharmacology, we show that the 7TM conformation is stabilized by a set of hydrophilic residues that links TM3, 5, 6, and 7 together and that directly contributes to mGlu5 allosteric modulation (Christopher et al., 2015; Doré et al., 2014). Differences distinguishing the inactive from the active 7TM conformation are very subtle, as illustrated here for Trp7856.50 mutant. This suggests that the energy barrier between active and inactive conformations is low. The energy barrier separating conformational states for class A and B receptors was previously suggested to directly justify receptor signaling properties (Hilger et al., 2020). The very low energy barrier for class C mGlu receptor might be the cue for L-glutamate to trigger G-protein activation from its binding to VFT and the subsequent stabilization of 7TM active conformation through the 7TM dimerization.
In summary, the functional and structural study presented here reveals two different possible modes of activation of the dimeric metabotropic glutamate receptor and provides information on the activation process of class C receptor dimers.
Resource availability
The X-ray model and structure factors have been deposited under PDB: 7P2L. The cryo-EM maps (the half maps as well as the B-factor sharpened maps used during model building) and models for agonist and antagonist have been deposited under codes EMDB: 31536, PDB: 7FD8 and EMDB: 31537, PDB: 7FD9, respectively.
STAR★Methods
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli cells DH5α | NEB | C2987 |
| E. coli cells DH10Bac | Invitrogen/ThermoFisher | 10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| pFastBac vector | Invitrogen/ThermoFisher | Cat#10712024 |
| BacMam pCMV-DEST vector | Invitrogen/ThermoFisher | A24223 |
| pCDNA3.1 vector | Invitrogen/ThermoFisher | Cat#V79020 |
| Tag-Lite buffer 5x | PerkinElmer/Cisbio | Cat#LABMED |
| SNAP-Lumi4-Tb | PerkinElmer/Cisbio | Cat#SSNPTBX |
| SNAP-red | PerkinElmer/Cisbio | Cat#SSNPREDF |
| IPOne Gq kit | PerkinElmer/Cisbio | Cat#62IPAPEC |
| Iodoacetamide | Sigma | Cat#I6125 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma | Cat#P7626 |
| cOmplete EDTA-free inhibitor cocktail | Sigma | Cat# 5056489001 |
| Lauryl Maltose Neopentyl (MNG) | Anatrace | Cat#NG310 |
| n-Dodecyl-beta-D-maltopyranoside (DDM) | Anatrace | Cat#D310LA |
| n-Decyl-beta-D-maltopyranoside (DM) | Anatrace | Cat#D322 |
| n-Nonyl-beta-D-Glucopyranoside (NG) | Anatrace | Cat#N324 |
| 1-O-(n-Octyl)-tetraethyleneglycol (C8E4) | Anatrace | Cat#T350 |
| Monoolein, 9.9 MAG | Molecular dimensions | Cat#MD2-67 |
| Cholesterol | Anatrace | Cat#CH200 |
| Cholesterol hemisuccinate, Tris Salt (CHS) | Anatrace | Cat#CH210 |
| HisTrap HP | GE Healthcare | Cat#17-5248-02 |
| ANTI-FLAG® M1 Agarose Affinity Gel | Sigma-Aldrich | Cat#A4596 |
| FLAG peptide | Covalab | https://www.covalab.com/peptide-synthesis |
| Superdex 200 increase 10/300 GL | GE Healthcare | Cat#28-9909-44 |
| EX-CELL420 Serum-Free Medium | Sigma-Aldrich | Cat#14420 |
| DMEM | Life Technologie | 41965062 |
| DMEM Glutamax | Life Technologie | 31966021 |
| Freestyle serum-Free Medium | Life Technologie | 12338018 |
| MPEP | Bio-techne | Cat#1212/10 |
| Tritiated MPEP | Isobio | ART 1209-250 μCi |
| VU0424465 | Nasrallah et al., 2018 | N/A |
| LY341495 disodium salt | Bio-techne | Cat#4062/1 |
| L-Quisqualic acid | Bio-techne | Cat#0188/10 |
| Polyethylenimine | Polyscience Europe | Cat#02371 |
| GTPγS | Roche | Cat#10220647001 |
| YM-254890 | Cayman Chemicals | Cat#29735 |
| GTPase-GloTM assay | Promega | Cat#V7681 |
| Experimental models: Cell lines | ||
| Insect cell line Sf9 | Life Technologies | Cat#11496015 |
| HEK293 human cells | ATCC | CRL-1573 |
| HEK293T human cells | ATCC | CRL-3216 |
| HEK293S GNTi− human cells | Reeves et al., 2002 | N/A |
| Recombinant DNA | ||
| pBacMam-pCMV-DEST mGlu5-5M | This study | N/A |
| pFastBac-mGlu5-5M | This study | N/A |
| pFastBac-7TM-mGlu5-5M | This study | N/A |
| pFastBac- mGlu5-StaR(569-836)-T4L | Doré et al., 2014 | N/A |
| pcDNA-SNAP-mGlu5 | Gift from CisBio | N/A |
| pcDNA-SNAP-mGlu5-Δ856 | This study | N/A |
| pcDNA-SNAP- mGlu5-5M | This study | N/A |
| pcDNA-SNAP- mGlu5-4M | This study | N/A |
| pcDNA-SNAP- mGlu5-3M | This study | N/A |
| pcDNA-SNAP- mGlu5-2M | This study | N/A |
| pcDNA-SNAP- mGlu5-1M | This study | N/A |
| pcDNA-SNAP-mGlu5-Δ856 single point mutants (I651A, P655A, S658A, Y659A, R668A, V740A, T742A, L744A, S753A, N747A, G748A, T777A, T781A, W785A, A787L, F788A, Y792A, G794A, I799A, M802A, S805A, V806A, S809A, A810L, A813L, A855L, A856L) | This study | N/A |
| Software and algorithms | ||
| PyMOL | Schrödinger | https://pymol.org/2/ |
| Prism v.6.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| XDS | https://xds.mr.mpg.de/ | VERSION Mar 15, 2019 BUILD = 20190806 |
| XDSCC12 | https://xds.mr.mpg.de/ | xdscc12 KD 2019-07-26 |
| XSCALE | https://xds.mr.mpg.de/html_doc/xscale_program.html | VERSION Jan 31, 2020 BUILD = 20200417 |
| CTFFIND4 | Rohou and Grigorieff, 2015 | CTFFIND4 |
| Relion | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page | Relion 3.1 |
| SerialEM | https://bio3d.colorado.edu/SerialEM/ | SerialEM 3.9 beta |
| Coot | https://www.ccp4.ac.uk/ | Coot 0.9.3 |
| ccpem | https://www.ccp4.ac.uk/ | CCPEM 1.5.0 |
| Phenix | https://phenix-online.org/ | Phenix 1.18.2-3874 |
| Refmac | http://www.ccp4.ac.uk | Refmac 5.8.0267 |
| Chimera | www.cgl.ucsf.edu/chimera | UCSF Chimera Version 1.13.1 |
| Topaz | http://cb.csail.mit.edu/cb/topaz/; Bepler et al., 2019 | topaz v0.2.5 |
| Deposited data | ||
| mGlu5 −5M bound to LY341495 and MPEP | This study | EMDB-31537, PDB – 7FD9 |
| mGlu5 −5M bound to quisqualate and VU0424465 | This study | EMBD-31536, PDB – 7FD8 |
| mGlu5 StaR(569-836)-T4L bound to alloswitch-1 | This study | PDB - 7P2L |
| Others | ||
| Quantifoil 0.6/1 um 300 mesh Au grids | https://www.quantifoil.com | N1-C11nAu30-01 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Guillaume Lebon (guillaume.lebon@igf.cnrs.fr).
Materials availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed materials transfer agreement if there is a potential for commercial application.
Experimental model and subject details
SNAP-tagged mGlu5 receptor (SNAP-mGlu5-Δ856) and its receptor mutants were expressed in HEK293 (ATCC CRL1573) for conformational thermostabilization as well as G protein functional assay and HEK293T (ATCC CRL-3216) for VFT’s conformational FRET sensor experiments. Large scale expression for structural study were performed from purified mGlu5-5M receptor expressing in HEK293S cell line lacking N-acetylglucosaminyltransferase I (HEK293S GnTI(-)) (Reeves et al., 2002) using the pBACMAM system (Thermofisher). mGlu5-StaR (569-836)-T4L was expressed in Sf9 cells using the Bac-to-Bac® Baculovirus expression system (Thermofisher).
Method details
Conformational thermostabilization of full-length MPEP-bound mGlu5 receptor
7TM alanine mutant library of the of SNAP-tagged mGlu5 receptor (SNAP-mGlu5-Δ856) was generated using Quick-change strategy (Agilent technologies) and verified by sequencing (Eurofins Genomics). The thermostability assay was performed in a 96 well-plate format with HEK293 cells transiently transfected with the expression vector encoding the human WT truncated mGlu5 receptor (mGlu5-Δ856) and receptor mutants as previously described (Nasrallah et al., 2018). The SNAP-mGlu5-Δ856 mutants were transiently transfected using Lipofectamine 2000 in a 96-well-plate pre-coated with Poly-dL-Ornithine according to the manufacturer protocol. Briefly, DNA mixture of each mGlu5 receptor mutant at 40 ng, 50 ng of the glutamate transporter EAAC1 cDNA (to avoid any influence of glutamate in the assay medium released by the cells), and 60 ng of pRK6 to make up the final amount of DNA to 150 ng per well. The mGlu5 receptor mutants were incubated with 80 nM of the high-affinity tritiated NAM 2-Methyl-6-(phenylethynyl) pyridine ([3H]-MPEP) and solubilised for 1 hour at 4°C using a buffer containing 25 mM HEPES, pH 7.4, 400 mM NaCl and 0.8% LMNG, 0.05% CHS (w/v). For screening, solubilised single-point mutant receptors were incubated for 30 minutes at 4°C and 20°C and then left on ice for 5 minutes. The free ligands were separated from bound ligands as previously described (Magnani et al., 2016). [3H]-MPEP-bound receptor mutants were mixed with liquid scintillant and quantified using the MicroBeta counter (Perkin Elmer). Selected thermostable single-point mutants were characterized by performing a full thermostability curve and combined. The best thermal stability was obtained for mGlu5-5M. Data were fitted using the Boltzmann equation.
Expression and purification of the human full-length thermostabilized mGlu5 receptor bound to inhibitors and activators
The construct chosen for large scale expression contained an expression cassette with a GP64 peptide signal sequence from envelope surface glycoprotein of the Autographa californica nuclear polyhedrosis virus (AcNPV baculovirus), the flag (DYKDDDDK), 10xHis tag, and the precision protease recognition site (LEVLFQGP) at the N terminus, followed by the human mGlu5-5M gene truncated at amino acid 856 and that had one more mutant H350L for a nanobody to bind and N455A mutation to remove one glycosylation site. The construct was subcloned into the BacMam vector using PCR with primer pairs encoding restriction sites HindIII at the 5′ and NotI at the 3′ termini with subsequent ligation into the corresponding restriction sites present in the vector. High-titer recombinant baculovirus was obtained using the Bac-to-Bac Baculovirus expression system (Invitrogen) as described by the manufacturer. Briefly, Sf9 cells grown at 28°C in EX-CELL 420 medium (Sigma Aldrich) were infected with P1 human mGlu5-5M virus and then centrifuged 96 hours post-infection to generate the P2 virus used for the large-scale expression. HEK293S cell line lacking N-acetylglucosaminyltransferase I (HEK293S GnTI(-)) cells were grown at 37°C in humidified 5% CO2 incubator in Freestyle medium (Reeves et al., 2002) (Life technologies). HEK293S GnTI(-) cells were infected at a density of 2.5 x106 cells per mL with 6% of freshly produced P2 virus (60 mL per liter). Sodium butyrate was added at 10 mM, 24 hours post-infection. Cells were collected 96 hours post-infection and stored at −80°C. Cell membranes were disrupted by thawing frozen cell pellets in a lysis buffer containing 25 mM HEPES (pH 7.4), 10 mM MgCl2, 20 mM KCl, 1 mM PMSF (Sigma Aldrich), and the complete protease inhibitor cocktail tablet (Roche). The pellets were washed two times with the same lysis buffer and an additional washing with a high salt buffer containing 25 mM HEPES (pH 7.4), 10 mM MgCl2, 20 mM KCl and 1 M NaCl with the centrifugation step at 45 000 rpm (≈200 000g). Membranes were resuspended with the lysis buffer supplemented with 40% glycerol then stored at −80°C. Washed membranes were resuspended into a buffer containing 10−5 M MPEP, (Abcam), 10 mM iodoacetamide (Sigma), and the complete protease inhibitor cocktail tablets. The mixture was then incubated at room temperature (RT) for 1 hour. The membranes were then solubilised in a buffer containing 25 mM HEPES buffer (pH 7.4), 0.4 M NaCl, 10% Glycerol, 1% (w/v) n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace), and 0.2% (w/v) cholesteryl hemisuccinate (CHS, Anatrace) for 1.5 hours at 4°C, with shaking. The supernatant was then obtained by centrifugation at 45 000 rpm for 1 hour and supplemented with 10 mM imidazole (Sigma) before loading onto nickel (Ni2+) resin (HisTrap HP, GE Healthcare) at 0.3 mL/min, overnight at 4°C. The protein was then eluted with 25 mM HEPES (pH 7.4), 400 mM NaCl, 10 μM MPEP, 10 μM LY341495, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, and 250 mM imidazole. The eluted mGlu5-5M protein was further incubated onto a Flag affinity resin (Anti-Flag M1, Sigma) for 1 hour at 4°C then washed with 10 column volumes of wash buffer (25 mM HEPES (pH 7.4), 150 mM NaCl, 10 μM MPEP, 10 μM LY341495, 0.03 (w/v) DDM, 0.006% (w/v) CHS, and 2 mM calcium). The protein was eluted with a buffer containing 0.03% DDM (w/v), 0.006% CHS (w/v), 25 mM HEPES pH 7.4, 0.15 M NaCl, 10 μM MPEP, 10 μM LY341495, 0.2 mg/mL Flag peptide and 2 mM EDTA. The eluted fractions were then concentrated using a Vivaspin 20 centrifugal concentrator 100 kDa (Sartorius), centrifuged for 10 minutes at 80 000 rpm to eliminate aggregates then loaded on a size exclusion chromatography column (Superdex 200 Increase 10/300, GE Healthcare) with the SEC buffer containing 0.03% DDM (w/v), 0.006% CHS (w/v), 25 mM HEPES pH 7.4, 0.15 M NaCl, 10 μM MPEP and 10 μM LY341495. The same protocol was used to purify the quisqualate and the VU0424465-bound thermostabilized mGlu5 receptor. After size-exclusion chromatography, the protein peak was collected and concentrated using Vivaspin 6 centrifugal concentrator 100 kDa (Sartorius) for cryoEM analysis.
Detergent-solubilized receptor thermostability measurements
After 48 hours of baculovirus infection, Sf9 membranes expressing mGlu5-5M or the 7TM-mGlu5-5M were incubated for 1 hour at RT with the NAM [3H]-MPEP (ARC, inc) at 80 nM (≈five times the KD) in a buffer containing 25 mM HEPES pH 7.4 and 400 mM NaCl. Membranes were then solubilized for 1 hour at 4°C with 0.83% (w/v) of the following detergents: Lauryl maltose-neopentyl glycol (MNG3), Dodecyl Maltoside (DDM), Decyl Maltoside (DM), Nonyl Glucoside (NG), and 1-O-(n-Octyl)-tetraethyleneglycol (C8E4), respectively, each detergent was supplemented with 0.083% (w/v) CHS. Non-specific binding was determined for each detergent by competition binding in the presence of 200 nM MPEP (Abcam). Solubilized receptors (mGlu5-5M and 7TM-mGlu5-5M) were then incubated for 30 minutes at various temperatures ranging from 4°C to 55°C. The samples were then chilled at 4°C for 5 minutes and detergent solubilized ligand binding experiment performed as previously reported (Magnani et al., 2016). Free and bound radioligand molecules were separated by rapid filtration through Toyopearl resin (Tosoh Bioscience) packed in 96 filter plates (Millipore). After mixing with liquid scintillation solution, bound radioligands were quantified using a MicroBeta liquid scintillation counter (Perkin Elmer). Tm values were calculated using GraphPad Prism version 7.0 (GraphPad Software, San Diego, CA). Data are fit with a Boltzmann function. Tm values are expressed as the mean ± SEM from at least 3 independent experiments performed in duplicate ± SEM, except for C8E4 for the mGlu5-5M, Tm is expressed as mean value ± SD.
Grids preparation for cryoEM analysis
The antagonist and agonist bound mGlu5 in detergent micelles, at 2.5 mg/mL and 2.85 mg/mL respectively, were applied to a Quantifoil® Au 0.6/1 μm 300 mesh grids. Grids were rendered hydrophilic by glow-discharging for 1 min at 0.2 mBar and 40 mA. Grids were plunge-frozen using Vitrobot Mk IV (ThermoFisher) with the chamber equilibrated at 100% RH and 10°C. Grids were blotted with Whatman no. 1 filter paper for 3.5 s with a blot force of 10 and no waiting/drying time and plunge frozen in liquid ethane. The frozen grids were stored in liquid nitrogen until further use.
CryoEM data acquisition and image processing
Data were collected on Titan Krios transmission electron microscope (ThermoFisher) equipped with a BioQuantum energy filter and K2 direct electron detector both from Gatan and operating at 300 kV. Movies were recorded in EFTEM mode, with a 20eV slit width, a sampling rate of 0.89 Å/pixel and a total dose was ∼50 and ∼67 e-/Å2, fractionated into 32 and 48 frames for the antagonist and agonist bound FL mGlu5 respectively. SerialEM was used to correct astigmatism, perform coma-free alignment, and automated data collection using in-house data collection macros (Mastronarde, 2005). Active beam tilt compensation was turned on during the acquisition. Movies were collected with the defocus range −1.2 to −2.8 μm. Two movies were collected for each hole and a beam-image shift was used to collect a series of 3 × 3 holes for faster data collection, while stage shift was used to center the 3 × 3 template acquisition (Cheng et al., 2018). Note that the hole size of the Quantifoil® grids used was close to 1 μm rather than 0.6 and when collecting two images, the beam overlapped but not the imaging area of the detector. Movies were motion-corrected with dose-weighting in RELION 3.1 using Relion’s own implementation (Scheres, 2012). CTF estimation was performed with CTFFIND4 (Rohou and Grigorieff, 2015). Particles were auto-picked with TOPAZ (Bepler et al., 2019). The final extracted box size for the antagonist and agonist datasets were 300 and 384 pixels respectively. The 2D classification of particles was performed in RELION 3.1, binned (4x or 8x) particles were used for the classification steps. Several rounds of 2D classification were carried out to enrich good particles. An initial model was calculated using Stochastic Gradient Descent (SGD) independently for both datasets in RELION 3.1. After initial 3D refinement, particles were separated into 3D classes, using the initial 3D-refined volume as reference. Particles that were sorted into well-resolved classes were kept for further analysis. Particles were sorted into 18 different optics groups defined based on the 3 × 3 groups for image shift-based data collection. CTF Refinement and Bayesian polishing were performed in RELION 3.1 (Zivanov et al., 2018). The overall and local resolution of the maps were calculated using the post-process and local resolution option in Relion. The handedness of the antagonist map was corrected using relion_image_handler and invert_hand option at the end of processing.
CryoEM model building and refinement
The model of mGlu5 with quisqualate was built using the PDB 6N50/6N51 for the VFT and CRD and the 7TM from the X-ray structure with alloswitch-1 (including all the side chains) described below. The graphics program Coot was used for all model building (Emsley and Cowtan, 2004). The resolution of the EM map of mGlu5 with quisqualate is not uniform and the application of a single B-factor calculated by default during the post-processing (with a sharpening value of −122 Å2) resulted in the blurring of the CRD and 7TM regions that were difficult to interpret. We then used maps with multiple B-factor sharpening (−61 Å2 and −20 Å2) to place the CRD and 7TM. The use of multiple maps helped us in identifying the CHS molecule, whose density otherwise was not clear. Different pose of CHS in particular the succinic acid group could be fit but as the density around this region is poor, one orientation with least clashes has currently been modeled. In parallel, we also tried LocScale within ccpem (Burnley et al., 2017; Jakobi et al., 2017) and phenix density modification options for cryoEM (Terwilliger et al., 2020) to see if these maps can aid in model building. Although, the density modification option predicted an increase in resolution of 0.2-0.3 Å, visual inspection of the map showed no significant improvement in the CRD and 7TM but was used to check the tracing and some side-chain rotamers of the VFT where possible. This map and other maps sharpened with different B-factors reveal the possibility of extra NAG moiety and loop extension in the 7TM region but these have not been modeled. The built models were then refined with phenix in real space and Refmac with jelly body restrains (Afonine et al., 2018; Murshudov et al., 1997; Nicholls et al., 2018). For the modeling of antagonist bound mGlu5, 6N52 and the X-ray structure with alloswitch were used as templates (the model 3KS9 was used for comparison and to analyze the binding of antagonist). The model was manually inspected (one chain) with Coot using EM maps sharpened with −164 Å2 or −82 Å2 or −20 Å2, modified and refined with phenix in real space and Refmac with jelly body restrains. Both models were validated with Molprobity within phenix (Chen et al., 2010). Figures of molecular structures were generated with PyMOL (https://pymol.org/2/) and Chimera (Goddard et al., 2007).
Expression, purification, and crystallization of alloswitch-1-bound mGlu5-7TM
Thermostabilized (E579A, N667Y, I669A, G675M, T742A, and S753A) human 7TM domain of mGlu5 receptor, mGlu5-StaR (569-836)-T4L (a generous gift from Sosei Heptares) was expressed in Sf9 cells grown in EX-CELL® 420 serum-free medium (Sigma Aldrich) using the Bac-to-Bac® Baculovirus expression system (Thermofisher). Briefly, the construct was truncated at the Pro569 at the N terminus and at Ala836 at the C terminus. A T4-lysozyme was inserted between Lys678 and Lys679, a GP64 signal sequence, and a 10xHis tag were fused at the N- and C-terminal, respectively, as described previously (Doré et al., 2014). Cells were infected at a density of 3-4 × 106 cells per mL with recombinant baculovirus P2 particles. Cells were grown at 27°C, harvested 48 hours post-infection by centrifugation at 3000 x g for 15 min, and stored at −80°C until use. Insect cells membranes were disrupted by repeated washing and centrifugation with a hypotonic (25 mM HEPES pH 7.4, 10 mM MgCl2, 20 mM KCl, 5 mM EDTA) and hypertonic (25 mM HEPES pH 7.4, 10 mM MgCl2, 20 mM KCl, 1 M NaCl) buffers supplemented with complete protease inhibitor cocktail tablets (Roche). Membranes were stored in hypotonic solution supplemented with 40% of glycerol at −80°C until use. Washed membranes were incubated with continuous stirring in 25 mM HEPES pH 7.4, 400 mM NaCl, 10 mM iodoacetamide, 100 μM alloswitch-1 (provided by Llebaria A.) for 1 hour at RT and then solubilized by adding 1% DDM (w/v) with 0.2% CHS (w/v) for an additional 3 hours at 4°C. All the following purification steps were performed at 4°C. Insoluble materials were removed by ultracentrifugation at 45000 rpm for 1 hour and the filtered supernatant was loaded on a HisTrap HP column (GE Healthcare) on the ÄKTA Purifier system in the presence of 10 mM imidazole. The resin was washed with a gradient of 10 mM to 50 mM imidazole and resin-bound material was eluted in 25 mM HEPES pH 7.4, 250 mM NaCl, 10% glycerol, 0.05% DDM, 0.01% CHS, 250 mM imidazole, 10 μM alloswitch-1. Eluted fractions were concentrated to 250 μL using Vivaspin-6 centrifugal concentrator MWCO 100 kDa (Sartorius) and insoluble material was removed by ultracentrifugation at 80000 rpm for 10 minutes. Soluble receptor was further applied to a Superdex 200 increase 10/300 column (GE Healthcare) equilibrated in size exclusion chromatography (SEC) buffer (25 mM HEPES pH 7.4, 150 mM NaCl, 0.03% DDM, 0.006% CHS, 10 μM alloswitch-1). mGlu5-staR(569-836)-T4L receptor purity was estimated by Coomassie blue-stained SDS-PAGE gel analysis and protein concentration was determined using the bicinchoninic acid (BCA) assay kit (ThermoFisher Scientific). mGlu5-staR(569-836)-T4L concentration was adjusted to ∼25 mg/mL in SEC buffer. For crystallization, purified mGlu5-staR (569-836)-T4L was reconstituted in lipid cubic phase by mixing the protein with monoolein supplemented with 10% (w/w) cholesterol (Anatrace) using 100 μL micro-syringes (Art Robbins Instruments) in a final protein: lipid ratio of 2:3 (w/w) (Cherezov, 2011). Between 40-50 nL of bolus was dispensed on a Laminex 96-well glass base (Molecular Dimensions) and overlaid with 750 nL of precipitant solution using the crystal Gryphon dispenser robot (Art Robbins Instruments) and plates were sealed and stored at 20°C. Crystals appeared after a few hours and grew to a range size of 20-50 μm in a screen of 0.15-0.25 M ammonium phosphate dibasic, 22%–24% polyethylene glycol 400, either with 0.10 M 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.7-6.8 or 0.1 M HEPES pH 6.8 (MemGoldMeso screen, Molecular Dimensions). Crystals were harvested on a loop from crystallization plates and immediately flash-frozen in liquid nitrogen.
X-ray data collection, processing, and structure determination of mGlu5 7TM bound to alloswitch-1
Crystals were measured at the protein crystallography beamline X06SA-PXI in Swiss Light Source (SLS), Villigen, Switzerland. Data were collected with a 5 × 5 or 10 × 10 μm2 micro-focused X-ray beam of 12.39 keV (1 Å in wavelength) at 100 K using data acquisition software suites (DA+) (Wojdyla et al., 2018). Continues grid-scans (Wojdyla et al., 2016) were used to locate crystals in frozen LCP samples. The crystals were collected by an automated serial data collection protocol (CY+) as described previously with 0.1 s exposure time, 0.1° oscillation and 10° wedge from each crystal for data collection (Basu et al., 2019) with the EIGER 16M detector operated in continuous/shutterless data collection mode. Data were processed with XDS and scaled and merged with XSCALE (Kabsch, 2010a, 2010b). Data selection using XDSCC12 was applied to improve the merging dataset (Assmann et al., 2020). From the processing of multiple datasets, the final data was merged from 60 partial datasets to 2.54 Å resolution. The structure was determined using the molecular replacement (MR) method using PDB code 4OO9 without ligand as the search model with the program Phaser (McCoy et al., 2007). The structure was refined using Refmac and model building were completed manually using Coot (Emsley and Cowtan, 2004; Murshudov et al., 1997). The final structure has Rwork/Rfree to 0.23/0.28. Data collection and processing statistics are provided in Table S7. Figures of molecular structures were generated with PyMOL (https://pymol.org/2/) an 2D plot using LigPlot+ (Laskowski and Swindells, 2011).
mGlu5 VFT’s conformational FRET sensor
Membrane fractions for tr-FRET measurements and G protein activation were prepared from adherent HEK293T cells (ATCC CRL-3216, LGC Standards S.a.r.l., France). Cells were cultured in GIBCO DMEM, high glucose, GlutaMAX Supplement, pyruvate (Thermo Fischer Scientific, France) supplemented with 10% (v/v) fetal calf serum (FCS, Sigma-Aldrich, France) in 25 cm2 cell culture treated flasks (TPP Techno Plastic Products AG, Switzerland) to approximately 80% confluence and then transfected with polyethylenimine (PEI 25K, Polysciences Europe GmbH, Germany) at a DNA to PEI ratio (w/w) of 1:3, using 4 μg DNA (pCDNA-SNAP-mGlu5-5M and empty pRK6 as control) per flask. In brief, 10 mg/mL PEI stock solution in 1 M HCl was diluted in 20 mM MES at pH 5, 150 mM NaCl and incubated at RT for 25 minutes before sequential addition of 0.5 mL complete medium followed by an additional 4 mL. The flask’s culture medium was then replaced by the diluted transfection mix and protein expression proceeded for 48 hours at 37°C, 5% CO2. The medium was then exchanged with 3 mL GlutaMAX medium without FCS and incubated for another 2 hours. For TR-FRET measurements, the medium was supplemented with 100 nM SNAP-Lumi4-Tb and 60 nM SNAP-green (Cisbio Bioassays, Codolet, France). The cells were then washed 3-times with 5 mL DPBS (Thermo Fischer Scientific, France) and subsequently detached mechanically in DPBS. After collection of the cells at 500 g and RT for 5 minutes, the pellet was placed on ice and resuspended in cold hypotonic lysis buffer composed of 10 mM HEPES pH 7.4 with protease inhibitors (cOmplete, EDTA-free Protease Inhibitor Cocktail, Roche, France). The suspension was then frozen once at −80°C and after thawing on ice was passed 30-times through a 0.4 mm needle. The lysed cells were then centrifuged 2-times at 500 g and 4°C for 5 minutes and the supernatant recovered, aliquoted and membranes collected at 21000 g and 4°C for 30 minutes. tr-FRET measurements of N- terminally SNAP-tag labeled receptors were carried out in white 384 well plates (polystyrene, flat-bottom, small volume, medium-binding, Greiner Bio-One SAS, France) using a PHERAstar FS microplate reader (BMG Labtech, Germany). Measurements were carried out in Tris-Krebs buffer without Glucose (20 mM Tris-HCl pH7.4, 118 mM NaCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 4.7 mM KCl, 1.8 mM CaCl2) supplemented with 100 μM GTPγS (Roche) at RT and data analyzed as previously described (Scholler et al., 2017).
G protein turnover assay
The membranes were first incubated overnight at 4°C with the G protein inhibitor YM-254890 in the presence of 10 μM GTPγS, 5 mM MgCl2, and 150 mM NaCl. After extensive washing, a receptor-catalyzed GTP turnover assay was carried out using the GTPase-GloTM assay (Promega) as described (Strohman et al., 2019). Briefly, the receptor was incubated in the absence or presence of the ligands for 60 minutes at 20°C. The G protein (500 nM) was then added with a final concentration in GTP and GDP of 10 μM and 5 μM, respectively. The amount of GTP remaining after 15 minutes incubation at 20°C was finally assessed using the GTPase-Glo assay (Promega). The signal was normalized in each case to that in the absence of the receptor (100%).
Cell culture, transfections, and inositol phosphate one (IP1) accumulation assay
HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and were maintained at 37°C in a humidified atmosphere with 5% CO2. Cells were transiently transfected, using lipofectamine 2000 (Invitrogen), with the indicated mGlu5 construct. The mGlu5 constructs contained a Flag and SNAP-tag to enable cell surface expression measurement. The DNA mixture for transfection included 10 ng of the indicated mGlu5 mutants, 30 ng of the glutamate transporter EAAC1 cDNA (to reduce the influence of glutamate that may remain in the assay medium as released by the cells), and 110 ng of pRK6 to make up the final amount of DNA to 150 ng per well. Following 24 hours of transfection, HEK293 cells were incubated for 2 hours with glutamate-free DMEM GlutaMAX-I (Life Technologies) before the commencement of the IP1 accumulation assay, to reduce the extracellular concentration of glutamate. Wild-type and mutant SNAP-tagged mGlu5 expression were determined using the SNAP-tag labeled receptors and lumi4-Tb. In brief, cells were washed with 100 μL of glutamate-free DMEM GlutaMAX-I and then incubated for 1 hour at 37°C with 100 nM of SNAP-Lumi4-Tb in 50 μL of glutamate-free DMEM GlutaMAX-I. Cells were then washed twice with Tag-Lite buffer, and then 100 μL of Tag-Lite buffer was added to each well (Cisbio Bioassays). Lumi4-Tb fluorescence was then measured. The IP1 accumulation assay kit (Cisbio Bioassays, France) was used for the direct quantitative measurement of IP1 in HEK293 cells transiently transfected with mGlu5 constructs into black, clear-bottom 96-well culture plates (Greiner Bio-one). Cells were stimulated with various concentrations of orthosteric and/or allosteric compounds and were then incubated for 30 minutes at 37°C, 5% CO2 in either dark or 380 nm conditions. For 380 nm conditions, culture plates were placed above a 96-LED array plate (LEDA, Teleopto) connected to a LED array driver (LAD-1, Teleopto) with light pulsed for 50/50 ms on/off rather than continuous illumination to avoid overheating the cells and compromising the integrity of the assay as previously described (Ricart-Ortega et al., 2020). Irradiance was set to 0.12 mW/mm2 for 380 nm assay conditions. Cells were then lysed using the conjugate-lysis buffer mixed with the d2-labeled IP1 analog and the terbium cryptate-labeled anti-IP1 antibody according to the manufacturer’s instructions. After 1 hour incubation at RT, the HTRF measurement was performed after excitation at 337 nm with 50 μs delay, terbium cryptate fluorescence and tr-FRET signals were measured at 620 nm and 665 nm, respectively, using a PheraStar fluorimeter (BMG Labtech).
GraphPad Prism version 7 (San Diego, CA) was used for all other curve fitting and statistical analyses. For the empirical analysis of functional concentration-response data, agonist concentration-response curves, and the curves for the complete interaction of quisqualate with alloswitch-1 at mGlu5 WT or mutant constructs, under dark or 380 nm conditions, datasets were analyzed according to a standard logistic function to determine measures of potency (as negative logarithms; pEC50 or pIC50).
Quantification and statistical analysis
Graphs were analyzed using GraphPad Prism version 7.0 (GraphPad Software, San Diego, CA). A one-way ANOVA was used for multiple comparisons with a defined reference level, with a Dunnett’s post-test. Statistical significance was set as one, two, three and four stars to indicate p < 0.05, p < 0.01, p < 0.001 and p < 0.0001, respectively.
Acknowledgments
We would like to thank Joël Bockaert, Chris Tate, Philippe Marin, and Richard Henderson for careful reading and for their comments on the manuscript. We would like to thank SLS PSI XA06 microfocus beamline scientist (Villigen; Switzerland), MRC Laboratory Molecular Biology EM and computing core facilities (Cambridge, UK), and the platform of pharmacology Arpeges of the Institut de Génomique Fonctionnelle (Montpellier, France). This project was funded by the ATIP grant from CNRS. C.N. was funded by the Post-doctoral program from Montpellier University. A.E.B. was supported by the Labex EpiGenMed (program “Investissements d’avenir,” ANR-10-LABX-12-01), L.B. was supported by Ligue Contre le Cancer. J.F.-I., X.G.-S., J.C., and A.L. were supported by Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación and ERDF-FEDER European Fund (project CTQ2017-89222-R) and by the Catalan government (2017 SGR 1604). J.-L.B. was supported by ANR (ANR-17-CE11-0011). J.-P.P. was supported by the Fondation Recherche Médicale (FRM team: DEQ20170326522), grants from ANR (DynaMuR2, ANR-18-CE11-0004; Lanthslider, ANR-17-CE11-0046). G.C. and K.R.V. were supported by Medical Research Council grant MC-U105184322. K.R.V. acknowledges SERB, India for the Ramanujan Fellowship (RJN-094/2017), DBT B-Life grant DBT/PR12422/MED/31/287/2014, and the support of the Department of Atomic Energy, Government of India, Government of India, under Project Identification No. RTI4006. G.L. was supported by ANR grants AT2R-TRAAK Bioanalgesics (ANR-17-CE18-0001), DynaMuR2 (ANR-18-CE11-0004), SWITCH-ON (ANR-20-CE11-0019), and Fondation Recherche Médicale (ING20121226326).
Author contributions
C.N., G.C., and J.B. contributed equally to the work and share the co-first authorship. C.N. developed expression and purification for the full-length mGlu5-5M and mGlu5 7TMs and purified all protein samples for cryo-EM analysis. G.C. prepared cryo-EM grids and collected and processed cryo-EM data. J.B. developed the purification protocol for the thermostable mGlu5-StaR(569-836)-T4L bound to alloswitch-1, purified the thermostable 7TM bound to alloswitch-1, and produced crystals. L.B. was involved in mGlu5-StaR(569-836)-T4L expression and purification. K.R. generated the alanine-mutant library of the full-length mGlu5 receptor and performed conformational thermostabilization experiments. A.E.B. performed pharmacological and photopharmacology experiments on the mGlu5 receptor and receptor mutants. C.G. designed pharmacological and photopharmacological experiments and, together with A.E.B., analyzed the data. J.B., F.H., and G.L. collected X-ray diffraction data, and C.-Y.H. and G.L. performed X-ray data processing and model building. R.B.Q. performed VFT’s conformational FRET sensor experiments and data analysis. J.-L.B. performed G-protein turnover assay and analyzed the data. F.M., J.K., and A.D. performed pharmacological experiments on the WT mGlu5 receptor dimer. J.F.-I. synthesized the VU0424465, J.C. and X.G.-S. synthesized alloswitch-1, and A.L. designed and supervised compound synthesis. K.R.V. was involved in the initial screening of the receptor for cryo-EM and performed model building/refinement of full-length mGlu5-5M. J.K., C.G., J.-P.P., K.R.V., and G.L. interpreted the data. G.L. designed and supervised the project and wrote the manuscript with contributions from all authors.
Declaration of interests
The authors declare no competing interests.
Published: August 31, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109648.
Contributor Information
Kutti R. Vinothkumar, Email: vkumar@ncbs.res.in.
Guillaume Lebon, Email: guillaume.lebon@igf.cnrs.fr.
Supplemental information
Data and code availability
-
•
Adjective data reported in this paper will be shared by the lead contact upon request.
-
•
This manuscript does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann G.M., Wang M., Diederichs K. Making a difference in multi-data-set crystallography: simple and deterministic data-scaling/selection methods. Acta Crystallogr. D Struct. Biol. 2020;76:636–652. doi: 10.1107/S2059798320006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J.A., Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Basu S., Kaminski J.W., Panepucci E., Huang C.Y., Warshamanage R., Wang M., Wojdyla J.A. Automated data collection and real-time data analysis suite for serial synchrotron crystallography. J. Synchrotron Radiat. 2019;26:244–252. doi: 10.1107/S1600577518016570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T., Morin A., Rapp M., Brasch J., Shapiro L., Noble A.J., Berger B. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods. 2019;16:1153–1160. doi: 10.1038/s41592-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berizzi A.E., Goudet C. Strategies and considerations of G-protein-coupled receptor photopharmacology. Adv. Pharmacol. 2020;88:143–172. doi: 10.1016/bs.apha.2019.12.001. [DOI] [PubMed] [Google Scholar]
- Burnley T., Palmer C.M., Winn M. Recent developments in the CCP-EM software suite. Acta Crystallogr. D Struct. Biol. 2017;73:469–477. doi: 10.1107/S2059798317007859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A., Eng E.T., Alink L., Rice W.J., Jordan K.D., Kim L.Y., Potter C.S., Carragher B. High resolution single particle cryo-electron microscopy using beam-image shift. J. Struct. Biol. 2018;204:270–275. doi: 10.1016/j.jsb.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V. Lipidic cubic phase technologies for membrane protein structural studies. Curr. Opin. Struct. Biol. 2011;21:559–566. doi: 10.1016/j.sbi.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher J.A., Aves S.J., Bennett K.A., Doré A.S., Errey J.C., Jazayeri A., Marshall F.H., Okrasa K., Serrano-Vega M.J., Tehan B.G. Fragment and Structure-Based Drug Discovery for a Class C GPCR: Discovery of the mGlu5 Negative Allosteric Modulator HTL14242 (3-Chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile) J. Med. Chem. 2015;58:6653–6664. doi: 10.1021/acs.jmedchem.5b00892. [DOI] [PubMed] [Google Scholar]
- Christopher J.A., Orgován Z., Congreve M., Doré A.S., Errey J.C., Marshall F.H., Mason J.S., Okrasa K., Rucktooa P., Serrano-Vega M.J. Structure-Based Optimization Strategies for G Protein-Coupled Receptor (GPCR) Allosteric Modulators: A Case Study from Analyses of New Metabotropic Glutamate Receptor 5 (mGlu5) X-ray Structures. J. Med. Chem. 2019;62:207–222. doi: 10.1021/acs.jmedchem.7b01722. [DOI] [PubMed] [Google Scholar]
- Doré A.S., Okrasa K., Patel J.C., Serrano-Vega M., Bennett K., Cooke R.M., Errey J.C., Jazayeri A., Khan S., Tehan B. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- Doumazane E., Scholler P., Fabre L., Zwier J.M., Trinquet E., Pin J.-P., Rondard P. Illuminating the activation mechanisms and allosteric properties of metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA. 2013;110:E1416–E1425. doi: 10.1073/pnas.1215615110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Wang D., Fan H., Xu C., Tai L., Lin S., Han S., Tan Q., Wang X., Xu T. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature. 2021;594:589–593. doi: 10.1038/s41586-021-03641-w. [DOI] [PubMed] [Google Scholar]
- El Moustaine D., Granier S., Doumazane E., Scholler P., Rahmeh R., Bron P., Mouillac B., Banères J.-L., Rondard P., Pin J.-P. Distinct roles of metabotropic glutamate receptor dimerization in agonist activation and G-protein coupling. Proc. Natl. Acad. Sci. USA. 2012;109:16342–16347. doi: 10.1073/pnas.1205838109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Goddard T.D., Huang C.C., Ferrin T.E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007;157:281–287. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Goudet C., Gaven F., Kniazeff J., Vol C., Liu J., Cohen-Gonsaud M., Acher F., Prézeau L., Pin J.P. Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc. Natl. Acad. Sci. USA. 2004;101:378–383. doi: 10.1073/pnas.0304699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet C., Rovira X., Llebaria A. Shedding light on metabotropic glutamate receptors using optogenetics and photopharmacology. Curr. Opin. Pharmacol. 2018;38:8–15. doi: 10.1016/j.coph.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Gregory K.J., Conn P.J. Molecular Insights into Metabotropic Glutamate Receptor Allosteric Modulation. Mol. Pharmacol. 2015;88:188–202. doi: 10.1124/mol.114.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory K.J., Goudet C. International Union of Basic and Clinical Pharmacology. CXI. Pharmacology, Signaling, and Physiology of Metabotropic Glutamate Receptors. Pharmacol. Rev. 2021;73:521–569. doi: 10.1124/pr.119.019133. [DOI] [PubMed] [Google Scholar]
- Gregory K.J., Nguyen E.D., Reiff S.D., Squire E.F., Stauffer S.R., Lindsley C.W., Meiler J., Conn P.J. Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol. Pharmacol. 2013;83:991–1006. doi: 10.1124/mol.112.083949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory K.J., Nguyen E.D., Malosh C., Mendenhall J.L., Zic J.Z., Bates B.S., Noetzel M.J., Squire E.F., Turner E.M., Rook J.M. Identification of specific ligand-receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chem. Neurosci. 2014;5:282–295. doi: 10.1021/cn400225x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellyer S.D., Sengmany K., Keller A.N., Christopoulos A., Leach K., Gregory K.J. Probe dependence and biased potentiation of metabotropic glutamate receptor 5 is mediated by differential ligand interactions in the common allosteric binding site. Biochem. Pharmacol. 2020;177:114013. doi: 10.1016/j.bcp.2020.114013. [DOI] [PubMed] [Google Scholar]
- Hilger D., Kumar K.K., Hu H., Pedersen M.F., O’Brien E.S., Giehm L., Jennings C., Eskici G., Inoue A., Lerch M. Structural insights into differences in G protein activation by family A and family B GPCRs. Science. 2020;369:6503. doi: 10.1126/science.aba3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüll K., Morstein J., Trauner D. In Vivo Photopharmacology. Chem. Rev. 2018;118:10710–10747. doi: 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- Jakobi A.J., Wilmanns M., Sachse C. Model-based local density sharpening of cryo-EM maps. eLife. 2017;6:e27131. doi: 10.7554/eLife.27131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A., Hu H., Feng D., Sun B., Zhang Y., Robertson M.J., Chu M., Kobilka T.S., Laeremans T., Steyaert J. Structural insights into the activation of metabotropic glutamate receptors. Nature. 2019;566:79–84. doi: 10.1038/s41586-019-0881-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima N., Shimada Y., Tsuji Y., Sato T., Yamamoto M., Kumasaka T., Nakanishi S., Jingami H., Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Magnani F., Serrano-Vega M.J., Shibata Y., Abdul-Hussein S., Lebon G., Miller-Gallacher J., Singhal A., Strege A., Thomas J.A., Tate C.G. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 2016;11:1554–1571. doi: 10.1038/nprot.2016.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C., Shen C., Li C., Shen D.-D., Xu C., Zhang S., Zhou R., Shen Q., Chen L.-N., Jiang Z. Cryo-EM structures of inactive and active GABAB receptor. Cell Res. 2020;30:564–573. doi: 10.1038/s41422-020-0350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mølck C., Harpsøe K., Gloriam D.E., Mathiesen J.M., Nielsen S.M., Bräuner-Osborne H. mGluR5: exploration of orthosteric and allosteric ligand binding pockets and their applications to drug discovery. Neurochem. Res. 2014;39:1862–1875. doi: 10.1007/s11064-014-1248-8. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nasrallah C., Rottier K., Marcellin R., Compan V., Font J., Llebaria A., Pin J.-P., Banères J.-L., Lebon G. Direct coupling of detergent purified human mGlu5 receptor to the heterotrimeric G proteins Gq and Gs. Sci. Rep. 2018;8:4407. doi: 10.1038/s41598-018-22729-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls R.A., Tykac M., Kovalevskiy O., Murshudov G.N. Current approaches for the fitting and refinement of atomic models into cryo-EM maps using CCP-EM. Acta Crystallogr. D Struct. Biol. 2018;74:492–505. doi: 10.1107/S2059798318007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasergi-Scott M.M., Robertson M.J., Seven A.B., Panova O., Mathiesen J.M., Skiniotis G. Structures of metabotropic GABAB receptor. Nature. 2020;584:310–314. doi: 10.1038/s41586-020-2469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Fu Z., Frangaj A., Liu J., Mosyak L., Shen T., Slavkovich V.N., Ray K.M., Taura J., Cao B. Structure of human GABAB receptor in an inactive state. Nature. 2020;584:304–309. doi: 10.1038/s41586-020-2452-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin J.-P., Bettler B. Organization and functions of mGlu and GABAB receptor complexes. Nature. 2016;540:60–68. doi: 10.1038/nature20566. [DOI] [PubMed] [Google Scholar]
- Pin J.-P., Galvez T., Prézeau L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol. Ther. 2003;98:325–354. doi: 10.1016/s0163-7258(03)00038-x. [DOI] [PubMed] [Google Scholar]
- Pittolo S., Gómez-Santacana X., Eckelt K., Rovira X., Dalton J., Goudet C., Pin J.-P., Llobet A., Giraldo J., Llebaria A., Gorostiza P. An allosteric modulator to control endogenous G protein-coupled receptors with light. Nat. Chem. Biol. 2014;10:813–815. doi: 10.1038/nchembio.1612. [DOI] [PubMed] [Google Scholar]
- Reeves P.J., Callewaert N., Contreras R., Khorana H.G. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. USA. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricart-Ortega M., Berizzi A.E., Pereira V., Malhaire F., Catena J., Font J., Gómez-Santacana X., Muñoz L., Zussy C., Serra C. Mechanistic Insights into Light-Driven Allosteric Control of GPCR Biological Activity. ACS Pharmacol. Transl. Sci. 2020;3:883–895. doi: 10.1021/acsptsci.0c00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A., Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook J.M., Noetzel M.J., Pouliot W.A., Bridges T.M., Vinson P.N., Cho H.P., Zhou Y., Gogliotti R.D., Manka J.T., Gregory K.J. Unique signaling profiles of positive allosteric modulators of metabotropic glutamate receptor subtype 5 determine differences in in vivo activity. Biol. Psychiatry. 2013;73:501–509. doi: 10.1016/j.biopsych.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H.W. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 2012;415:406–418. doi: 10.1016/j.jmb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler P., Moreno-Delgado D., Lecat-Guillet N., Doumazane E., Monnier C., Charrier-Savournin F., Fabre L., Chouvet C., Soldevila S., Lamarque L. HTS-compatible FRET-based conformational sensors clarify membrane receptor activation. Nat. Chem. Biol. 2017;13:372–380. doi: 10.1038/nchembio.2286. [DOI] [PubMed] [Google Scholar]
- Sengmany K., Hellyer S.D., Christopoulos A., Lapinsky D.J., Leach K., Gregory K.J. Differential contribution of metabotropic glutamate receptor 5 common allosteric binding site residues to biased allosteric agonism. Biochem. Pharmacol. 2020;177:114011. doi: 10.1016/j.bcp.2020.114011. [DOI] [PubMed] [Google Scholar]
- Shaye H., Ishchenko A., Lam J.H., Han G.W., Xue L., Rondard P., Pin J.-P., Katritch V., Gati C., Cherezov V. Structural basis of the activation of a metabotropic GABA receptor. Nature. 2020;584:298–303. doi: 10.1038/s41586-020-2408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansley B.J., Conn P.J. Neuropharmacological Insight from Allosteric Modulation of mGlu Receptors. Trends Pharmacol. Sci. 2019;40:240–252. doi: 10.1016/j.tips.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohman M.J., Maeda S., Hilger D., Masureel M., Du Y., Kobilka B.K. Local membrane charge regulates β2 adrenergic receptor coupling to Gi3. Nat. Commun. 2019;10:2234. doi: 10.1038/s41467-019-10108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C., Sobolev O.V., Afonine P.V., Adams P.D., Read R.J. Density modification of cryo-EM maps. Acta Crystallogr. D Struct. Biol. 2020;76:912–925. doi: 10.1107/S205979832001061X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan A.J., Deupi X., Lebon G., Tate C.G., Schertler G.F., Babu M.M. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Wojdyla J.A., Kaminski J.W., Panepucci E., Ebner S., Wang X., Gabadinho J., Wang M. DA+ data acquisition and analysis software at the Swiss Light Source macromolecular crystallography beamlines. J. Synchrotron Radiat. 2018;25:293–303. doi: 10.1107/S1600577517014503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdyla J.A., Panepucci E., Martiel I., Ebner S., Huang C.Y., Caffrey M., Bunk O., Wang M. Fast two-dimensional grid and transmission X-ray microscopy scanning methods for visualizing and characterizing protein crystals. J. Appl. Cryst. 2016;49:944–952. doi: 10.1107/S1600576716006233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L., Rovira X., Scholler P., Zhao H., Liu J., Pin J.-P., Rondard P. Major ligand-induced rearrangement of the heptahelical domain interface in a GPCR dimer. Nat. Chem. Biol. 2015;11:134–140. doi: 10.1038/nchembio.1711. [DOI] [PubMed] [Google Scholar]
- Zivanov J., Nakane T., Forsberg B.O., Kimanius D., Hagen W.J., Lindahl E., Scheres S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7:e42166. doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Adjective data reported in this paper will be shared by the lead contact upon request.
-
•
This manuscript does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.