

Summary
Age is the major risk factor in most carcinomas, yet little is known about how proteomes change with age in any human epithelium. We present comprehensive proteomes comprised of >9,000 total proteins and >15,000 phosphopeptides from normal primary human mammary epithelia at lineage resolution from ten women ranging in age from 19 to 68 years. Data were quality controlled and results were biologically validated with cell-based assays. Age-dependent protein signatures were identified using differential expression analyses and weighted protein co-expression network analyses. Upregulation of basal markers in luminal cells, including KRT14 and AXL, were a prominent consequence of aging. PEAK1 was identified as an age-dependent signaling kinase in luminal cells, which revealed a potential age-dependent vulnerability for targeted ablation. Correlation analyses between transcriptome and proteome revealed age-associated loss of proteostasis regulation. Age-dependent proteome changes in the breast epithelium identified heretofore unknown potential therapeutic targets for reducing breast cancer susceptibility.
Subject areas: Developmental biology, Complex system biology, Proteomics
Graphical abstract
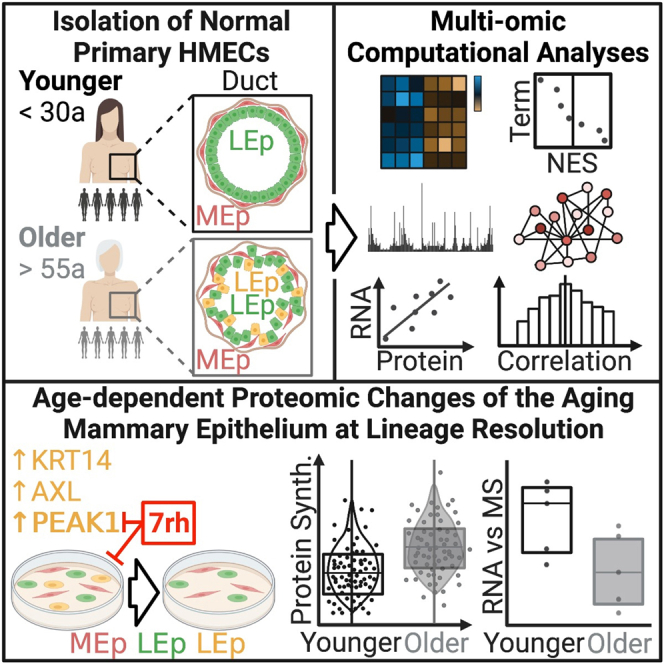
Highlights
-
•
Age-dependent proteomic profiling of HMECs at lineage resolution
-
•
Loss of proteostasis is unique to the luminal epithelial lineage with age
-
•
Identification of co-regulated proteins with age in luminal cells including PEAK1
-
•
PEAK1 inhibition selectively ablates older luminal cells – cancer cell of origin
Developmental biology; Complex system biology; Proteomics
Introduction
Aging is a major risk factor for most diseases in humans, and it is the greatest risk factor for solid tissue carcinomas. Aging is thought to increase susceptibility to cancer initiation, but the underlying reasons for this are unclear (Marusyk and DeGregori, 2008). A majority of effort spent characterizing effects of age in various human tissues have utilized transcriptome and genome-wide analyses of whole tissues (Berchtold et al., 2008; de Vries et al., 2017; Glass et al., 2013; Haakensen et al., 2011). There is poor correspondence between transcriptome and proteome (Kelmer Sacramento et al., 2020; Wang et al., 2019), and proteomic approaches to investigate effects from aging in human tissues have been limited to whole tissues, obscuring lineage-specific changes (Johnson et al., 2020).
We are using breast as a model for understanding how aging increases susceptibility to cancer because cell lineages are well-defined with biomarkers, and surgically discarded normal tissue is obtainable from cosmetic procedures. Age is the greatest risk factor for breast cancer as over 75% of new breast cancer diagnoses are made in women aged 50 years or older (Benz, 2008). Whole-breast tissue analyses identified a number of directional changes in gene expression (Lee and Lee, 2017; Song et al., 2017). However, aging is associated with significant shifts in proportions of epithelial and stromal cells so whole tissue signals mask age-dependent intrinsic changes to the different cell types (Garbe et al., 2012). Proteomic examination of breast-related tissue has been mostly limited to breast cancer cell lines, which revealed pathways that are active in different cancer subtypes but teaches nothing about aging or cancer susceptibility (Kalocsay et al., 2020; Lawrence et al., 2015). Indeed, there is a paucity of proteomic resources for understanding the effects of aging in breast, or in any other human tissue. We performed proteomics on purified populations of the two principle epithelial lineages in breast: the contractile and tumor suppressive myoepithelial cells, and the secretory luminal epithelial cells, which are thought to be the cancer cells of origin for the luminal subtypes of breast cancer that are most age-related (Prat and Perou, 2011).
Here, we examined age-dependent changes in the proteomes and phosphoproteomes of human mammary luminal epithelial and myoepithelial cells from 10 women who were aged 19–68 years at the time of surgery. We identified differentially expressed proteins (DEPs), enriched gene sets, weighted protein correlation network modules that change with age, and examined the age-dependent loss of matched mRNA-protein expression patterns. Previously identified age-dependent increases in expression of basal genes within luminal cells with age were verified proteomically and extended to include the receptor tyrosine kinase AXL and other proteins that control epithelial plasticity and epithelial to mesenchymal transitions. Heretofore unrecognized changes in the DDR1/PEAK1 pathway in luminal cells were identified as a potential age-dependent vulnerability, and we performed proof of principle drug-targeted ablation of older luminal epithelial cells. Correlation analyses of genome-wide transcriptome and proteome data with subsequent biological validation showed age-dependent loss of proteostasis in luminal cells. Our data presents previously unexplored opportunities to understand the cellular aging processes at the proteomic and phosphopeptide levels in two epithelial lineages from a normal human tissue.
Results
Deep proteomic and phosphopeptide quantification of the mammary epithelium
To investigate age-dependent changes in the mammary epithelium, we utilized tandem mass tag mass spectrometry (TMT-MS) to quantify the proteomic and phosphopeptide landscape of normal human mammary epithelial cells (HMECs) from 5 younger (age <30 years) and 5 older (age >50 years) women (Figure 1A, Table S1). Lineages were FACS enriched prior to TMT-MS into luminal epithelial (LEp, CD133+/CD271-) and myoepithelial (MEp, CD133-/CD271+) cells (Figure S1). The two different lineages from the same specimen were examined in tandem in separate runs. The proteomic analyses yielded >9,000 proteins (at 5% spectral mapping false discovery rate [FDR]) for both LEp and MEp lineages, the majority (>84%) were detected in both data sets (Figure 1B). Samples were analyzed as all LEp cells in tandem or all MEp cells in tandem enabling within lineage comparisons but not between lineage comparisons. The phosphopeptide analyses detected 9,799 and 15,713 distinct phosphopeptides for the LEp and MEp data sets, respectively (Figure 1B). A global overview of the LEp and MEp MS data sets demonstrated that the largest number of proteins observed was for proteins and peptides categorized as transcription factors or mitochondria associated proteins (Figures 1C–1F). Our previous work highlighted KRT14 and KRT19 as exemplar genes and proteins that show age-dependent changes in LEp cells (Garbe et al., 2012; Pelissier Vatter et al., 2018), and we also detected an increase in KRT14 and a decrease of KRT19 expression with age in these MS data (Figure 1G). Additionally, we demonstrated examples of age-dependent expression of ZNF542P and AVIL in the MEp cells as a characterization of the changing proteomic composition of the mammary epithelium (Figure 1H). These data provided a high-level overview of the produced datasets that vastly enhanced a proteome and phosphopeptide wide understanding of the aging mammary epithelium.
Figure 1.

Proteomic and phosphopeptide lineage specific profiling of primary mammary epithelial cells
(A) Flowchart of experimental workflow.
(B) Venn diagram of quantified proteins and phosphopeptides across datasets.
(C–F) Overview of proteins' and phosphopeptides' log2 fold changes (old vs young) in selected functional protein classes (young: from women <30 years (n = 5), old: >50 years old (n = 5)) Filtered by category members using curated publicly available datasets (Key Resource Table). Phosphopeptide data is shown on peptide level and filtered by masterprotein.
(G) KRT14 and KRT19 expression as a function of age in luminal epithelial (LEp) cells.
(H) ZNF542P and AVIL expression as a function of age in myoepithelial (MEp) cells.
Age-dependent changes are more pronounced in luminal epithelial cells
To assess the contribution of age to the proteomic and phosphopeptide landscape we conducted nonlinear dimension reduction analyses using t-distributed stochastic neighbor embedding (tSNE) (Figure 2). A robust age group-related separation was observed in the tSNE analyses for LEp cells along with a larger 95% confidence ellipsoid indicative of increased expression variance in LEp cells with age (Figures 2A and 2C, Video S1). Notably, MEp cells were inseparable by age and the 95% confidence ellipsoids demonstrated a greater overlap for both proteomic and phosphopeptide data, indicative that effects of aging were less pronounced in these cells (Figures 2B and 2D, Video S1). These analyses show that the age-dependent changes appear more prominent in LEp cells compared to MEp cells.
Figure 2.

Dimension reduction analyses of datasets
(A–D) tSNE plots with 95% confidence ellipsoid of HMEC datasets by age (young: from women <30 years (n = 5), old: > 50 years old (n = 5)) for proteins in (A) LEp, (B) MEp, and phosphopeptides (p-peptides) for (C) LEp, (D) MEp.
Protein level data for LEp and MEp cells and phosphopeptide level data for LEp and MEp cells (from left to right)
Aging signatures of the mammary epithelium
Differential expression analyses were conducted to identify proteins that are age-dependent and we detected 155 DEPs in LEp cells and 234 in MEp cells (cutoff of adjusted p value <0.05, only the 100 most significant proteins shown in Figures 3A and 3B, Table S1). In the LEp cells, 114 DEPs were upregulated, including KRT14, KRT10, KRT15, and AXL, whereas only 41 DEPs were downregulated. For MEp cells we detected that 197 of the 234 DEPs were downregulated and 37 were upregulated. Differential expression analyses also were performed for phosphopeptides (DEpP) in LEp cells (Figures 3C and Table S1) and MEp cells (Figures 3D and Table S1) and identified 77 DEpPs in LEp cells with 61 upregulated and 16 downregulated peptides. We identified 365 DEpPs for MEp cells; 53 were upregulated and 312 were downregulated.
Figure 3.

Differential expression analyses between younger and older HMECs by lineage
(A–D) Heatmaps of the top 100 differentially expressed proteins for LEp (A), MEp (B) and phosphopeptide (p-peptide) level (Z score scale). Differentially expressed phosphopeptides for LEp (C) and MEp (D).
(E and F) Gene set enrichment analyses of protein samples for (E) LEp and (F) MEp cells.
(G and H) Phosphorylation site-specific signature enrichment analysis for (G) LEp and (H) MEp cells.
Gene set enrichment analyses (GSEAs) were conducted for the proteome data sets using a ranked gene list based on the product of log2 fold change and the inverse of the p value to identify hallmark signatures enriched with age for LEp cells (Figure 3E) and MEp cells (Figure 3F). In LEp cells we identified 12 significantly enriched hallmark terms (FDR <0.05). Three gene sets were enriched in older LEp cells, including reduced KRAS signaling, expression of E2F targets, and G2M checkpoint. In younger LEp cells, we identified 9 gene sets enriched including IFN-alpha and -gamma, allograft rejection, IL2 STAT2 signaling, cholesterol homeostasis, fatty acid metabolism, peroxisome, and hypoxia (Figure 3E). Of the 15 significantly enriched gene sets for MEp cells, 10 gene sets are enriched in older MEp cells including oxidative phosphorylation, peroxisome, cholesterol homeostasis, bile acid metabolism, fatty acid metabolism, adipogenesis, xenobiotic metabolism, complement, protein secretion, and late estrogen response. In younger MEp cells, 5 gene sets were found to be enriched including G2M checkpoint, targets of the transcription factor E2F, spermatogenesis, MYC targets version 2, and mitotic spindle (Figure 3F). For the phosphopeptides, a phosphorylation site centric enrichment was performed utilizing PTM signature enrichment analysis (PTM-SEA). In LEp cells, 7 significantly enriched signatures (FDR <0.05) were identified including kinase activity associated with cell cycle control (CHEK1, CDK2, CDK11, CSNK2A1, and PRKACA) (Figure 3G). In the phosphoproteome of MEp cells, 16 significant (FDR <0.05) signatures were found to be enriched. These also contained multiple signatures of kinases associated with cell cycle control (CDK1, CSNK2A1, CDK2, PRKCA, AURKB, and CHEK1), and pathway activation of key signaling cascades (AKT1, ERK1, EGFR1, and GSK2B) (Figure 3H). For both LEp and MEp cells, these PTM-SEA results are consistent with the protein centric GSEA analysis and increase the understanding of signaling events in the mammary epithelium. Collectively, these analyses show that aging-dependent changes are highly lineage specific. Moreover, for certain gene sets (including cholesterol homeostasis, E2F targets, and G2M checkpoints) the age-dependent regulation was inverse when comparing MEp cells to LEp cells.
Identification of age-dependent co-expression modules and targetable factors
Weighted correlation network analyses (WGCNAs) were performed to identify groups of proteins with expression profiles that correlate with age. A co-expression network based on the protein and phosphopeptide expression profiles was constructed. A soft threshold power β of 18 was determined in all datasets to reach scale free independence. Eighteen co-expression modules (fast cluster algorithm with a minimum module size of 100 proteins) were identified in the LEp proteome with 4 of the modules significantly (adjusted p < 0.1) correlated with chronological age (Figures 4A and 4B). Modules “grey60” (rbicor = 0.92, adjusted p = 0.003) and “lightcyan” (rbicor = −0.75, adjusted p = 0.06) were positively correlated with age, whereas modules “lightgreen” (rbicor = −0.75, adjusted p = 0.06) and “midnightblue” (rbicor = −0.84, adjusted p = 0.02) were negatively correlated with age. The “grey60” module contained 111 proteins (Figure 4C), whereas module “midnightblue” contained 121 proteins (Table S1, Data S1 gene set). The expression patterns in these modules were homogeneous per age group as indicated by eigengene expression plots (Figures 4C–4F). The most interconnected proteins of the “grey60” (positively correlated with age) and “midnightblue” (negatively correlated with age) modules are shown in network plots (Figures 4G and 4H). KRT14 was identified as one of the most interconnected proteins within the “grey60” module, and other key signaling proteins such as PEAK1, IPPK, and CDK13 were also identified to be positively correlated with age (Figure 4G). Among the key negatively correlated proteins in the “midnightblue” module were KRT19, ALDH1A3, UBA6, and AVIL (Figure 4H).
Figure 4.

Weighted correlation network analyses for protein expression in LEp cells
(A) Protein dendrogram acquired by average linkage hierarchical clustering. Module assignment determined through dynamic tree cut and indicated by color row.
(B) Module eigengene correlation with age. Heatmap color refer to the biweight midcorrelation and the Benjamini-Hochberg adjusted p value is given in parenthesis.
(C–F) Heatmaps of modules significantly correlated with age (Z score scale) and barplots of eigengene expression of samples within the modules.
(G and H) Network plots of the most correlated modules (G) “grey60” and (H) “midnightblue”. Modules are colored by the protein expression log2 fold change and the size of the nodes are relative to the number of connections. The network only displays connections if the topological overlap is above a threshold of 0.08. Nodes with less than 3 connections were removed from plot.
Importantly, the identification of key known age-dependent proteins such as KRT14 and KRT19 within the modules serve as validation of the biological relevance of the analyses. Additionally, we validated PEAK1 as an additional central kinase positively correlated with age in our LEp cells. PEAK1 is heretofore unexamined in the context of the aging breast epithelium and is a factor established downstream of discoidin domain receptor 1 (DDR1), which is involved in pro-tumorigenic signaling in other carcinoma model systems (Aguilera et al., 2017; Hur et al., 2017; Saby et al., 2018). PEAK1 is a downstream effector of DDR1 in pancreatic cancer (Aguilera et al., 2014, 2017) and we thought it remarkable to identify its age-dependent expression here. Utilizing the human protein atlas we independently confirmed the protein expression of PEAK1 in the breast epithelium (Figure 5A) (Uhlen et al., 2017). RNAseq data from breast, also from the human protein atlas, indicated a significant increase of PEAK1 expression with age (p = 0.0018) (Figure 5C). As a biological validation of the WGCNA analysis we examined total PEAK1 protein expression levels as a function of age in two exemplar primary HMEC strains and showed an increased expression with age via western blot (Figure 5C). By utilizing the DDR1-specific drug 7rh, which was previously utilized to target DDR1 signaling in pancreatic cancer (Aguilera et al., 2017), phosphorylation of both DDR1 and PEAK1 were reduced in HMECs from older women (Figure 5D). Dose-response analyses assessed cell viability at different concentrations of 7rh. Sensitivity to 7rh significantly increased in HMECs with age (2-way ANOVA p < 1e-4, Figure 5E). To further elucidate if 7rh targets aged LEps specifically, we treated HMECs with increasing concentrations (0.5 μM and 2 μM of 7rh) for 48h. LEp and MEp populations were then analyzed using flow cytometry, and we identified a strong bias toward LEp ablation in the older HMEC strains (Figure 5F).
Figure 5.

Targeting PEAK1 expression through DDR1 inhibition
(A) PEAK1 DAB staining in breast specimen accessed via human protein atlas. Scale bar 100 μM.
(B) RNAseq data of PEAK1 expression in breast tissue by age accessed through human protein atlas. Boxes of box whisker plot represent 25–75 percentile ranges, vertical lines represent 1.5 × inter quartile range, and horizontal bars represent medians.
(C) Western blot of PEAK1 expression in younger (SubjectID 163, age 27) and older (SubjectID 237, age 66).
(D) Western blot of phospho-PEAK1 and phospho-DDR1 after inhibition of DDR1 mediated signaling through 24h treatment with 1 μM 7rh.
(E) Cell viability assay (cell titer glo) after 72h treatment with 7rh. Data are represented as mean ± SD.
(F) Change of LEp population size in percent in 2 younger and 2 older HMEC specimens after 48h of 7rh treatment.
Continuing the WGCNA analyses to interrogate the phosphopeptides, ten modules were identified for the LEp data set including the “brown” module (rbicor = 0.67, P = 0.03) which included 298 phosphopeptides significantly correlated with age (Figures 6A–6C, Table S1). We generated a co-expression network plot that depicts up and downregulated proteins (Figure 6D) including cell cycle related peptides, a YAP pathway-associated transcription factor (TEAD3), multiple RAB peptides, and key pathway kinases (MAPK2K7 and ILK). These results are consistent with PTM-SEA (Figure 3G). To identify a potential master kinase associated with the peptide targets in the “brown” module (Figure 6D), we conducted kinase enrichment analysis KEA2 (Lachmann and Ma'ayan, 2009) and identified a significant enrichment for targets of PDPK1, PDK1, and PDK3 (Figure 6E). No co-expression modules significantly correlated with age in the MEp datasets, indicating a reduced effect of aging on co-expression networks. Thus, WGCNA analyses identified key protein correlation networks for LEp cells that were associated with age and we presented data that experimentally confirmed the findings and targetability.
Figure 6.

Weighted correlation network analyses for phosphopeptide expression in LEp cells
(A) Protein dendrogram acquired by average linkage hierarchical clustering. Module assignment determined through dynamic tree cut and indicated by color row.
(B) Module eigengene correlation with age. Heatmap colors refer to the biweight midcorrelation and the p value is given in parenthesis.
(C) Heatmap of brown module (Z score scale) and barplots of eigengene expression of samples.
(D) Network plots of the most correlated modules. Modules are colored by the proteins log2 fold change and the size of the nodes are relative to the number of connections. The network only displays connections if the topological overlap is above a threshold of 0.2. Nodes with less than 3 connections were removed.
(E) Kinase enrichment analyses (KEA2) based on peptides identified in WGCNA “brown” module.
Correlation of transcriptome and proteome
We correlated RNAseq-derived transcriptomes with proteomes (Figures 7A and 7B) and demonstrated an observed median correlation of 0.19 (rbicor = 0.19) for LEp cells and 0.04 (rbicor = 0.04) for MEp cells. The correlation of keratins and ribosomal transcripts and proteins was greater in LEp cells than in MEp cells, whereas protein transport genes and proteins showed a lower correlation in LEp cells than in MEp cells. Using the gene-protein correlation pairs, we investigated differential correlation changes by age (Figures 7C and 7D). For LEp cells four transcript-protein pairs that changed the direction of correlation with age were identified. A positive correlation in younger cells that changed to a negative correlation in older cells was observed for ACAT1 (mitochondria associated), PRKACB (serine/threonine protein kinase), and TTC39C (unknown function). A negative correlation in younger strains and strong positive correlation with age was identified for NOP16 (ribosomal protein), which suggested a possible change of the ribosomal complex assembly machinery. In MEp cells, five differentially correlated transcript/protein pairs were detected. POLR2M (RNA Pol II subunit M), PPP2CB (protein phosphatase), and PEX6 (peroxisomal protein import) were highly positively correlated in younger strains and anticorrelated with age. Two proteins/transcripts that gained correlation with age were PPCS (metabolomic protein) and ANKMY1 (ankyrin repeat and MYND domain containing 1).
Figure 7.

Correlation between protein and RNA expression
(A and B) Histogram of the biweight midcorrelation distribution between protein and RNA expression for (A) LEp and (B) MEp with ridgeplot depicting distribution of selected gene groups curated by HGNC.
(C and D) Differentially correlated transcripts-proteins pairs by age for LEp cells and (D) MEp cells.
(E and F) Box and whisker plots of protein-transcript biweight midcorrelation by age and lineage (Wilcoxon one sided signed rank test F) Protein synthesis quantification using OPP-AF594 with quantification of nuclear integrated density per cell for 2 strains (scale bar 50 μm).
(G and H) Change in protein expression stoichiometry of (G) ribosomal and (H) proteasomal proteins by age and lineage). Boxes of box whisker plots represent 25–75 percentile ranges, vertical lines represent 1.5 × inter quartile range, and horizontal bars represent medians.
We further investigated the correlation between gene expression and protein levels with age (Figure 7E). Although MEp cells did not show loss of correlation, there was a significant age-dependent loss of correlation between the transcriptome and proteome in LEp cells (Wilcoxon test one-sided, p = 0.048). Since proteostasis is considered a hallmark of aging (Kaushik and Cuervo, 2015; Lopez-Otin et al., 2013), it was investigated whether the loss of correlation was attributable to an alteration of net protein production. These experiments were performed with 2 randomly selected cell strains. A significant increase in protein production was observed in an older HMEC strain (age 66 years) as compared with a younger one (age 27 years) (Figure 7F) utilizing an OPP-AF594-based protein synthesis assay. To further investigate the dysregulation of the translational machinery, we evaluated stoichiometric changes of the proteasomal and ribosomal subunits. Janssens et al. (2015) showed that loss of stoichiometry (uncoordinated change of expression) can alter the assembly of multiprotein complexes, which ultimately alters protein homeostasis (Kelmer Sacramento et al., 2020). Using the HUGO gene group annotation for the ribosomal subunits and proteasome associated proteins we determined a potential loss of stoichiometry for both LEp cells and Mep cells in an age-dependent manner (Figures 7G and 7H, results not significant). These data link the loss of the mRNA-protein correlation with age and dysregulation of the proteostasis machinery.
Discussion
Age is the most important risk factor for developing cancer. Thus, there is an urgent need to investigate the biology and mechanisms underlying this increased susceptibility in order to develop prevention and screening modalities. Here we present the first comprehensive and in-depth characterization of the proteomic and phosphopeptide changes of the normal aging mammary epithelium at lineage resolution. We present differentially expressed proteins and phosphopeptides and have identified key co-expression modules that correlate with the chronological age of the tissue donor. We also utilized matched RNAseq data to investigate the age-dependent decoupling between transcriptome-proteome correlations. Current proteomic studies that investigate human aging predominantly focus on blood samples generally lacking epithelial tissue (Johnson et al., 2020; Ubaida-Mohien et al., 2019). Moreover, proteomic profiling of breast-derived cells involve the use of abnormal and cancer cell lines (Kalocsay et al., 2020; Lawrence et al., 2015). Here we present a data resource for gaining insight into expression and phosphorylation of over 9,000 proteins in normal primary breast epithelia at lineage and age resolution.
We identified a number of age-dependent protein and phosphopeptide changes that could be leveraged as aging biomarkers or targets in future studies of aging in the breast. The most prominent age-dependent changes were identified in LEp cells, consistent with our previous findings, although TMT-MS was sensitive enough to also identify putative changes in MEps. Previous studies by our group characterized changes in human mammary epithelia with age and one of the most striking changes between cells from young compared with older HMECs is keratin expression in luminal cells (Garbe et al., 2012; Pelissier Vatter et al., 2018). KRT14 is a lineage-specific marker for MEp cells in young women, but LEp cells of older women acquire expression of this intermediate filament. KRT14 downregulation in cancer cell lines showed reduced proliferation, tumorigenicity, and reduced activation of the AKT pathway and presents a potential target to reduce cancer susceptibility with age (Alam et al., 2011). Here we have detected a significant increase of KRT14 expression, as well as KRT10 and KRT15, in our LEp data set and detected KRT14 in the WGCNA module that is most correlated with age. Given that the genes for these keratins, in addition to KRT19, are proximally located to one another on chromosome 17 suggests that they may be co-regulated in an age-dependent manner. Our data were consistent with scRNA sequencing from aging mammary pads of 7 inbred mice (Li et al., 2020). Utilizing the age-dependent genes from Li et al. (2020), we identified 160 homologs in our human TMT-MS data. Of those 160 proteins, 89 changed in the same direction with age in both data sets suggesting there is some conservation (Table S2). Taken together, our analyses of TMT-MS data for known changes validate the approach we have taken, giving confidence for future interrogation of other targets presented here.
WGCNA analysis revealed the age-dependent expression of PEAK1 (pseudopodium enriched atypical kinase 1), a signaling kinase previously unassociated with aging. PEAK1 is a non-receptor tyrosine kinase ubiquitously expressed and dysregulated in several cancer models including the pancreas (Aguilera et al., 2014, 2017) and breast (Wang et al., 2010). Aguilera et al. (2014) established PEAK1 as a downstream target of DDR1 (discoidin domain receptor 1) that can be pharmacologically targeted with a small molecular inhibitor, 7rh (Aguilera et al., 2017; Gao et al., 2013). Utilizing HMECs from four individuals we showed sensitivity to 7rh in an age-dependent manner and confirmed decreased phosphorylation of PEAK1 after treatment. Based on these experiments with randomly selected and limited number of HMECs, we suggest a role for PEAK1 in the mammary epithelium that may be age-dependent, targetable, and could lead to breast cancer prevention intervention strategies that include prophylactic ablation of LEp cells or their precursors that exhibit increased PEAK1 expression and activity. Mature LEps are thought to be the cells of origin for the luminal subtype breast cancers (Prat and Perou, 2011), 80% of which are age-associated, and DDR1/PEAK1 represents a prospective heretofore unknown vulnerability.
Our findings uncover a loss of transcriptome-proteome quality control in an age-dependent manner that is most prominent in LEp cells. We observed an increased overall protein production and reduced correlation to the transcriptome. Loss of proteostasis control has previously been proposed as a hallmark of aging (Balch et al., 2008) and utilizing the approach proposed by Janssens et al. (2015) we established an increase of interquartile range of multiple protein complexes involved in proteostasis. Dysregulation of the stoichiometry of these complexes could contribute to the decoupling that has been extensively studied in aging models of yeast and killifish (Janssens et al., 2015; Kelmer Sacramento et al., 2020). We speculate that this decoupling may be the result, or the cause, of age-dependent increased transcriptional variance that has been reported in immune, pancreatic, and breast cells (Enge et al., 2017; Martinez-Jimenez et al., 2017). The loss of correlation between RNA and protein highlights the need for protein evidence-based investigations, for example pediatric cancer subtypes were identified based on only proteome data (Petralia et al., 2020).
The key to this study is the utilization of pre-stasis epithelial cells from reduction mammoplasty tissues to examine the aging process, a model system which we generated and is well established (Garbe et al., 2012, 2014; Labarge et al., 2013; Pelissier et al., 2014; Pelissier Vatter et al., 2018). In our present study, HMECs were isolated from 10 different reduction mammoplasties spanning an expansive age range from 19 to 68 years. LEp cells and MEp cells were isolated from finite primary cells that were not exposed to immortalization factors that do not possess gross genetic alterations, transformations, nor genomic instabilities (Stampfer et al., 2013). The HMECs show remarkable preservation of in vivo lineage- and age-dependent states at 4th passage with minimal transcriptomic adaptions to in vitro conditions (Miyano et al., 2017, Miyano et al., 2021). This is unlike cell lines, such as MCF10A or HMLER, which display high genetic instability and overexpress factors consistent with immortal transformation. The power of using systems like these is that, unlike primary tissues which can usually only support n-of-one analyses or functional assays, pre-stasis HMEC cultures enable one to follow-up and test predictions from the proteomics data in cell-based assays.
The resource we provide here is a remarkable data set that allows in-depth analyses of aging in normal human mammary epithelium. We observe large scale expression changes especially in the LEp subpopulation of the epithelium, which is the most likely culprit of most age-associated breast cancers. Ultimately, these data could lead to further interrogations that may uncover additional aging biomarkers, high risk identifiers, and therapeutic interventions to prevent or treat age-associated breast cancers.
Limitations of the study
MS analyses do not capture the entire proteome nor phosphoproteome but lack of detection might not correlate with lack of expression. All analyses have been performed at lineage resolution in bulk, which does not allow for single-cell resolution. Thus, we cannot evaluate whether an entire lineage or just a subpopulation of cells changed expression or phosphorylation of any given protein with age.
Human aging is a non-homogenous and highly individual process and the changes observed here might not fully represent the changes observed in a population. It is not possible to adjust for all clinical, genetic, and racial covariates. All analyses have been conducted on the basis of age only. The study relied on primary HMECs and we have not fully validated the results in vivo.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the method section lead contact, Mark LaBarge (mlabarge@coh.org).
Materials availability
This study did not generate new unique reagents. Cell strains are available upon request through the lead contact.
Experimental model and subject details
Cell culture
Primary HMECs at passage 4 were grown at 37°C in M87A medium containing cholera toxin and oxytocin at 0.5 ng/ml and 0.1 nM (Garbe et al., 2009). HMEC strains used in this study were 51L, 124, 163, 172L, 240L, 29, 112R, 117R, 191L, and 237 (see Table S1). Media was changed every 48h with the last media change at 24h prior to cell dissociation.
Methods details
Flow cytometry
Cells dissociated from primary HMEC strains (passage 4) were stained with anti-human CD271-PerCP/Cy5.5 (Biolegend #345122) and anti-human CD133-PE (Biolegend #372804) by following standard flow cytometry protocol. Cells were sorted by S3 Cell Sorter (Bio-Rad). After sorting, cells were washed three times with PBS, snap frozen, and stored at −80°C.
Mass spectrometry
Cell pellets were dissolved in 0.5 M triethylammonium bicarbonate (TEAB) (T7408, Sigma-Aldrich, St. Louis, MO, USA) and 0.05% sodium dodecyl sulphate (SDS) (71,736, 50 μL in 10mL water/TEAB solution, Sigma-Aldrich, St. Louis, MO, USA), and lysed using pulsed probe sonication (Misonix, Farmingdale, NY, USA). Lysates were centrifuged (16,000 g, 10 min, 4°C) and supernatants were transferred to fresh tubes. Each sample was measured for protein content using the PierceTM BCA protein assay kit-reducing agent compatible per manufacturer's instructions (23,250, Thermo Fisher Scientific, Waltham, MA, US). 100 ug of protein was used per sample, adjusted to the highest volume using lysis buffer (0.5M TEAB, 0.05% SDS). Proteins were then reduced [4 μL of 100 mM tris (2-carboxyethyl) phosphine (TCEP); 646547, Sigma-Aldrich, St. Louis, MO, USA], alkylated [2 μL of 100 mM S-methyl methanethiosulfonate (MMTS); 64,306, Sigma-Aldrich, St. Louis, MO, USA] and enzymatically proteolysed using trypsin/LysC (1:25 enzyme:protein ratio; V5072, Promega, Madison, WI, USA). Peptides from each sample were labelled using the ten-plex TMT reagent kit (90110, Thermo Fisher Scientific, Waltham, MA, US). Two ten-plex experiments were performed, one for MEp cells and one for LEp cells from ten subjects. Labelled peptides per ten-plex experiment were mixed and phospho-enrichment was performed using the high-selectTM SMOAC protocol per manufacturer’s instructions (A32992 and A32993, Thermo Fisher Scientific, Waltham, MA, US). The flow-through containing native peptides was offline fractionated using alkaline C4 reverse phase chromatography (Kromasil® C4 HPLC column, 100 Å pore size, 3.5 μm particle size, length x I.D 150 x 2.1 mm, K08670362, Sigma-Aldrich, St. Louis, MO, USA) and each collected fraction was analyzed using the Orbitrap Fusion mass spectrometry system (Thermo Fisher Scientific, Waltham, MA, US).
Unprocessed raw files were submitted to Proteome Discoverer 2.3.0.523 for target decoy search using Byonic. The UniProtKB homo sapiens database (release date Dec 2019) was utilized. The search allowed for up to two missed cleavages, a precursor mass tolerance of 10 ppm, a minimum peptide length of six and a maximum of two dynamic modifications of; oxidation (M), deamidation (N, Q), or phosphorylation (S, T, Y). Methylthio (C) and TMT (K, N-terminus) were set as static modifications. Standard FDR corrected p value cutoff at the peptide level was set to <0.05 for native proteins and to <0.01 for phosphopeptides (Billiard et al., 2018; Bouchal et al., 2015; Liu et al., 2016; Mournetas et al., 2021; Stopfer et al., 2020). Percent co-isolation excluding peptides from quantitation was set at 50.
Differential expression
Differential expression was determined utilizing multiple t-tests (1 per protein/phosphopeptide) with pooled standard deviation (Cohen, 1988) based on the log2 transformed abundance values. To correct for multiple comparisons the Benjamini et al., (2006) method was deployed and proteins were considered differentially expressed for FDRBKY < 0.05.
Gene set enrichment analyses
GSEA were conducted using the fgsea R package (Sergushichev, 2016). Proteome wide expression profiles were ranked using the product of the log2 fold change and inverse of the p value. Gene sets were accessed through MySigDB V7.0 (Subramanian et al., 2005) and PTM signature enrichment analysis signatures were retrieved via PTMsigDB v1.9.0 (Krug et al., 2019). Terms were considered significantly enriched if FDR <0.05.
Weighted correlation network analysis (WGCNA)
To perform weighted correlation network analysis the R package WGCNA (Langfelder and Horvath, 2008) utilizing the biweight midcorrelation was used. Scale independence and mean connectivity were then tested using a gradient method. Minimum scale independence of at least 0.80 was met for every dataset with soft threshold of 18. The modules were detected by hierarchical average linkage clustering analysis for the protein dendrogram of the topology overlap matrix. To identify age-dependent modules the module-age relationships were calculated using bicor function and significance was determined (FDR <0.1). Most interconnected proteins and phosphopeptide networks were illustrated using cytoscape 3.8.0 (Shannon et al., 2003).
RNA-protein correlation analysis
RNAseq data has been accessed from previously published dataset (GSE182338). Correlations between RNA and proteins were calculated using biweight midcorrelation. Differential correlation has been assessed using the fisher r-z-transformation based in the correlation coefficient. p value was calculated using two-sided t test and p value adjustments was performed using Benjamini et al., (2006) method. Changes were considered significant for FDRBKY < 0.1.
Protein synthesis assay
HMECs were cultured as described above on 4-well chamber slides. When cells reached subconfluence media was changed. Protein synthesis assay (Thermo Fisher # C10457) was performed according to manufacturer's instructions. Images were captured in the same imaging session using Nikon DS-Qi2 camera (3s exposure) on a Nikon Ti2 Microscope. Single cell level of nuclear fluorescence was quantified using Cell Profiler 3.1.5.
Cell viability assay
HMECs were plated in white walled 96-well plates at 1500 cells per well. After 24h cells were treated with increasing concentrations of 7rh for 72h. Cell viability was determined using CellTiter-Glo (Promega) and measured using Cytation 3 Cell Imaging Multi-Mode Reader (BioTek). Graphs were plotted and significance was assessed using Graphpad Prism.
Western blot
Sub-confluent HMECs were lysed, centrifugated at 13,000 rpm, and supernatend protein concentration was measured using BCA assay. Equal amounts of total protein were separated by SDS-PAGE and transferredonto PVDF membranes. Membranes were underwent blockade for 1 hour in 5% milk in TBS-T. Primary antibody was incubated overnight at 4°C. Membranes were incubated with corresponding HRP-conjugated secondary antibody for 2 hours. Bands were detected using the enhanced chemiluminescence reagent using Oddyssey Fc (Licor).
Quantification and statistical analysis
All performed tests were two-sided unless otherwise specified and calculated in R (V3.6.1) or Prism (Graphpad, V9).
Acknowledgments
We gratefully acknowledge Dr. Rolf A. Brekken for kindly providing 7rh, and our patient advocates Susan Samson and Sandy Preto for providing much needed context. Graphical abstract was created with BioRender.com. Funding: DOD CDMRP (BC141351 Era of Hope Scholar Award & BC181737), Hilton-Ludwig Foundation, and City of Hope Center for Cancer and Aging to ML; Margaret Early Memorial Research Trust, Pediatric Cancer Research Foundation to LDW; National Institutes of Health/National Cancer Institute, NIH/NCI) grants R01CA237602, U01CA244109, R33AG059206 to ML; and R01EB024989 to LLS and MAL; K08 CA201591 to LDW; NCI Cancer Metabolism Training Program Postdoctoral Fellowship T32CA221709 to RWS; American Cancer Society Postdoctoral Fellowship (131311-PF-18-188-01-TBG) to M.E.T; California Institute for Regenerative Medicine grant CLIN2-12153, Pediatric Cancer Research Foundation Translational Research Grant, and Margaret E. Early Medical Research Trust to LDW. Research reported in this publication included work performed in the Mass Spectrometry and Proteomics Core, Analytical Cytometry Core, and Integrative Genomics and Bioinformatics Core supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
Conceptualization, S.H., A.M., M.M., K.Y.A., L.D.W., and M.A.L; writing – original draft, S.H., K.Y.A., and M.A.L; writing – review & editing, S.H., A.M., M.M., R.W.S., K.Y.A., M.E.T., J.C.L., L.L.S., L.D.W., and M.A.L.; investigation, S.H., A.M., M.M., K.Y.A., R.W.S., and J.C.L; formal analysis, S.H. and A.M.; data curation, S.H. and A.M.; visualization, S.H.; supervision and funding acquisition, L.L.S., L.D.W., and M.A.L.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103026.
Contributor Information
Leo D. Wang, Email: lewang@coh.org.
Mark A. LaBarge, Email: mlabarge@coh.org.
Supplemental information
Data and code availability
The mass spectrometry proteomics data and analyses have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and are publically available with the dataset identifier PXD027540. The RNA-sequencing count data referenced in this study are deposited and publicly available at GEO: GSE182338. Original code and any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aguilera K.Y., Huang H., Du W., Hagopian M.M., Wang Z., Hinz S., Hwang T.H., Wang H., Fleming J.B., Castrillon D.H. Inhibition of discoidin domain receptor 1 reduces collagen-mediated tumorigenicity in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2017;16:2473–2485. doi: 10.1158/1535-7163.MCT-16-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera K.Y., Rivera L.B., Hur H., Carbon J.G., Toombs J.E., Goldstein C.D., Dellinger M.T., Castrillon D.H., Brekken R.A. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014;74:1032–1044. doi: 10.1158/0008-5472.CAN-13-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam H., Sehgal L., Kundu S.T., Dalal S.N., Vaidya M.M. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol. Biol. Cell. 2011;22:4068–4078. doi: 10.1091/mbc.E10-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch W.E., Morimoto R.I., Dillin A., Kelly J.W. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Krieger A.M., Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika. 2006;93:491–507. [Google Scholar]
- Benz C.C. Impact of aging on the biology of breast cancer. Crit. Rev. Oncol. Hematol. 2008;66:65–74. doi: 10.1016/j.critrevonc.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold N.C., Cribbs D.H., Coleman P.D., Rogers J., Head E., Kim R., Beach T., Miller C., Troncoso J., Trojanowski J.Q. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. U S A. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiard F., Karaliota S., Wang B., Stellas D., Serafimidis I., Manousopoulou A., Koutmani Y., Ninou E., Golubov J., DaNave A. Delta-like ligand-4-notch signaling inhibition regulates pancreatic islet function and insulin secretion. Cell Rep. 2018;22:895–904. doi: 10.1016/j.celrep.2017.12.076. [DOI] [PubMed] [Google Scholar]
- Bouchal P., Dvorakova M., Roumeliotis T., Bortlicek Z., Ihnatova I., Prochazkova I., Ho J.T., Maryas J., Imrichova H., Budinska E. Combined proteomics and transcriptomics identifies carboxypeptidase B1 and nuclear factor kappaB (NF-kappaB) associated proteins as putative biomarkers of metastasis in low grade breast cancer. Mol. Cell Proteomics. 2015;14:1814–1830. doi: 10.1074/mcp.M114.041335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. L. Erlbaum Associates; 1988. Statistical Power Analysis for the Behavioral Sciences. [Google Scholar]
- de Vries M., Faiz A., Woldhuis R.R., Postma D.S., de Jong T.V., Sin D.D., Bosse Y., Nickle D.C., Guryev V., Timens W. Lung tissue gene-expression signature for the ageing lung in COPD. Thorax. 2017 doi: 10.1136/thoraxjnl-2017-210074. [DOI] [PubMed] [Google Scholar]
- Enge M., Arda H.E., Mignardi M., Beausang J., Bottino R., Kim S.K., Quake S.R. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. 2017;171:321–330 e314. doi: 10.1016/j.cell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Duan L., Luo J., Zhang L., Lu X., Zhang Y., Zhang Z., Tu Z., Xu Y., Ren X. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2013;56:3281–3295. doi: 10.1021/jm301824k. [DOI] [PubMed] [Google Scholar]
- Garbe J.C., Bhattacharya S., Merchant B., Bassett E., Swisshelm K., Feiler H.S., Wyrobek A.J., Stampfer M.R. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009;69:7557–7568. doi: 10.1158/0008-5472.CAN-09-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe J.C., Pepin F., Pelissier F.A., Sputova K., Fridriksdottir A.J., Guo D.E., Villadsen R., Park M., Petersen O.W., Borowsky A.D. Accumulation of multipotent progenitors with a basal differentiation bias during aging of human mammary epithelia. Cancer Res. 2012;72:3687–3701. doi: 10.1158/0008-5472.CAN-12-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe J.C., Vrba L., Sputova K., Fuchs L., Novak P., Brothman A.R., Jackson M., Chin K., LaBarge M.A., Watts G. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell cycle. 2014;13:3423–3435. doi: 10.4161/15384101.2014.954456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D., Vinuela A., Davies M.N., Ramasamy A., Parts L., Knowles D., Brown A.A., Hedman A.K., Small K.S., Buil A. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013;14:R75. doi: 10.1186/gb-2013-14-7-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haakensen V.D., Lingjaerde O.C., Luders T., Riis M., Prat A., Troester M.A., Holmen M.M., Frantzen J.O., Romundstad L., Navjord D. Gene expression profiles of breast biopsies from healthy women identify a group with claudin-low features. BMC Med. Genomics. 2011;4:77. doi: 10.1186/1755-8794-4-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur H., Ham I.H., Lee D., Jin H., Aguilera K.Y., Oh H.J., Han S.U., Kwon J.E., Kim Y.B., Ding K. Discoidin domain receptor 1 activity drives an aggressive phenotype in gastric carcinoma. BMC Cancer. 2017;17:87. doi: 10.1186/s12885-017-3051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens G.E., Meinema A.C., Gonzalez J., Wolters J.C., Schmidt A., Guryev V., Bischoff R., Wit E.C., Veenhoff L.M., Heinemann M. Protein biogenesis machinery is a driver of replicative aging in yeast. Elife. 2015;4:e08527. doi: 10.7554/eLife.08527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A.A., Shokhirev M.N., Wyss-Coray T., Lehallier B. Systematic review and analysis of human proteomics aging studies unveils a novel proteomic aging clock and identifies key processes that change with age. Ageing Res. Rev. 2020;60:101070. doi: 10.1016/j.arr.2020.101070. [DOI] [PubMed] [Google Scholar]
- Kalocsay M., Berberich M.J., Everley R.A., Nariya M.K., Chung M., Gaudio B., Victor C., Bradshaw G.A., Hafner M., Sorger P.K. Data Descriptor: proteomic profiling across breast cancer cell lines and models. bioRxiv. 2020 doi: 10.1038/s41597-023-02355-0. 2020.2012.2015.422823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Cuervo A.M. Proteostasis and aging. Nat. Med. 2015;21:1406–1415. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- Kelmer Sacramento E., Kirkpatrick J.M., Mazzetto M., Baumgart M., Bartolome A., Di Sanzo S., Caterino C., Sanguanini M., Papaevgeniou N., Lefaki M. Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Mol. Syst. Biol. 2020;16:e9596. doi: 10.15252/msb.20209596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K., Mertins P., Zhang B., Hornbeck P., Raju R., Ahmad R., Szucs M., Mundt F., Forestier D., Jane-Valbuena J. A curated resource for phosphosite-specific signature analysis. Mol. Cell Proteomics. 2019;18:576–593. doi: 10.1074/mcp.TIR118.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarge M.A., Garbe J.C., Stampfer M.R. Processing of human reduction mammoplasty and mastectomy tissues for cell culture. J. Vis. Exp. 2013 doi: 10.3791/50011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A., Ma'ayan A. KEA: kinase enrichment analysis. Bioinformatics. 2009;25:684–686. doi: 10.1093/bioinformatics/btp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence R.T., Perez E.M., Hernandez D., Miller C.P., Haas K.M., Irie H.Y., Lee S.I., Blau C.A., Villen J. The proteomic landscape of triple-negative breast cancer. Cell Rep. 2015;11:630–644. doi: 10.1016/j.celrep.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G., Lee M. Classification of genes based on age-related differential expression in breast cancer. Genomics Inform. 2017;15:156–161. doi: 10.5808/GI.2017.15.4.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.M., Shapiro H., Tsiobikas C., Selfors L.M., Chen H., Rosenbluth J., Moore K., Gupta K.P., Gray G.K., Oren Y. Aging-associated alterations in mammary epithelia and stroma revealed by single-cell RNA sequencing. Cell Rep. 2020;33:108566. doi: 10.1016/j.celrep.2020.108566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Kenney J.W., Manousopoulou A., Johnston H.E., Kamei M., Woelk C.H., Xie J., Schwarzer M., Garbis S.D., Proud C.G. Quantitative non-canonical amino acid tagging (QuaNCAT) proteomics identifies distinct patterns of protein synthesis rapidly induced by hypertrophic agents in cardiomyocytes, revealing new aspects of metabolic remodeling. Mol. Cell Proteomics. 2016;15:3170–3189. doi: 10.1074/mcp.M115.054312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez C.P., Eling N., Chen H.C., Vallejos C.A., Kolodziejczyk A.A., Connor F., Stojic L., Rayner T.F., Stubbington M.J.T., Teichmann S.A. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science. 2017;355:1433–1436. doi: 10.1126/science.aah4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A., DeGregori J. Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochim. Biophys. Acta. 2008;1785:1–11. doi: 10.1016/j.bbcan.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyano M., Sayaman R.W., Shalabi S.F., Senapati P., Lopez J.C., Angarola B.L., Hinz S., Zirbes A., Anczukow O., Yee L.D. Breast specific molecular clocks comprised of ELF5 expression and promoter methylation identify individuals susceptible to cancer initiation. Cancer Prev. Res. (Phila) 2021;14(8):779. doi: 10.1158/1940-6207.CAPR-20-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyano M., Sayaman R.W., Stoiber M.H., Lin C.H., Stampfer M.R., Brown J.B., LaBarge M.A. Age-related gene expression in luminal epithelial cells is driven by a microenvironment made from myoepithelial cells. Aging (Albany NY) 2017;9:2026–2051. doi: 10.18632/aging.101298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mournetas V., Massourides E., Dupont J.B., Kornobis E., Polveche H., Jarrige M., Dorval A.R.L., Gosselin M.R.F., Manousopoulou A., Garbis S.D. Myogenesis modelled by human pluripotent stem cells: a multi-omic study of Duchenne myopathy early onset. J. Cachexia Sarcopenia Muscle. 2021;12:209–232. doi: 10.1002/jcsm.12665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelissier F.A., Garbe J.C., Ananthanarayanan B., Miyano M., Lin C., Jokela T., Kumar S., Stampfer M.R., Lorens J.B., LaBarge M.A. Age-related dysfunction in mechanotransduction impairs differentiation of human mammary epithelial progenitors. Cell Rep. 2014;7:1926–1939. doi: 10.1016/j.celrep.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelissier Vatter F.A., Schapiro D., Chang H., Borowsky A.D., Lee J.K., Parvin B., Stampfer M.R., LaBarge M.A., Bodenmiller B., Lorens J.B. High-dimensional phenotyping identifies age-emergent cells in human mammary epithelia. Cell Rep. 2018;23:1205–1219. doi: 10.1016/j.celrep.2018.03.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia F., Tignor N., Reva B., Koptyra M., Chowdhury S., Rykunov D., Krek A., Ma W., Zhu Y., Ji J. Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell. 2020;183:1962–1985 e1931. doi: 10.1016/j.cell.2020.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A., Perou C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saby C., Rammal H., Magnien K., Buache E., Brassart-Pasco S., Van-Gulick L., Jeannesson P., Maquoi E., Morjani H. Age-related modifications of type I collagen impair DDR1-induced apoptosis in non-invasive breast carcinoma cells. Cell Adh Migr. 2018;12:335–347. doi: 10.1080/19336918.2018.1472182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. 2016;060012 [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M.A., Brasky T.M., Weng D.Y., McElroy J.P., Marian C., Higgins M.J., Ambrosone C., Spear S.L., Llanos A.A., Kallakury B.V.S. Landscape of genome-wide age-related DNA methylation in breast tissue. Oncotarget. 2017;8:114648–114662. doi: 10.18632/oncotarget.22754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer M.R., LaBarge M.A., Garbe J.C. Cell and Molecular Biology of Breast Cancer. Springer; 2013. An integrated human mammary epithelial cell culture system for studying carcinogenesis and aging; pp. 323–361. [Google Scholar]
- Stopfer L.E., Mesfin J.M., Joughin B.A., Lauffenburger D.A., White F.M. Multiplexed relative and absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. Nat. Commun. 2020;11:2760. doi: 10.1038/s41467-020-16588-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubaida-Mohien C., Lyashkov A., Gonzalez-Freire M., Tharakan R., Shardell M., Moaddel R., Semba R.D., Chia C.W., Gorospe M., Sen R. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. Elife. 2019;8:e49874. doi: 10.7554/eLife.49874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M., Zhang C., Lee S., Sjostedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352) doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Wang D., Eraslan B., Wieland T., Hallstrom B., Hopf T., Zolg D.P., Zecha J., Asplund A., Li L.H., Meng C. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019;15:e8503. doi: 10.15252/msb.20188503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kelber J.A., Tran Cao H.S., Cantin G.T., Lin R., Wang W., Kaushal S., Bristow J.M., Edgington T.S., Hoffman R.M. Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression [corrected] Proc. Natl. Acad. Sci. U S A. 2010;107:10920–10925. doi: 10.1073/pnas.0914776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Protein level data for LEp and MEp cells and phosphopeptide level data for LEp and MEp cells (from left to right)
Data Availability Statement
The mass spectrometry proteomics data and analyses have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and are publically available with the dataset identifier PXD027540. The RNA-sequencing count data referenced in this study are deposited and publicly available at GEO: GSE182338. Original code and any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.