

Summary
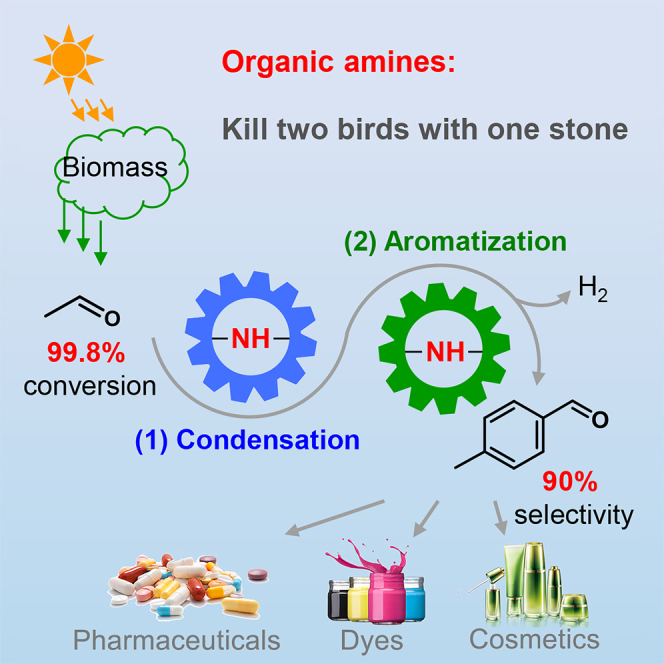
p-Methyl benzaldehyde (p-MBA) is a class of key chemical intermediates of pharmaceuticals. Conventional industrial processes for p-MBA production involve the consecutive photochlorination, amination, and acid hydrolysis of petroleum-derived p-xylene, while producing vast pollutants and waste water. Herein, we report a direct, green route for selective synthesis of p-MBA from acetaldehyde using a diphenyl prolinol trimethylsilyl ether catalyst. The optimized p-MBA selectivity is up to 90% at an acetaldehyde conversion as high as 99.8%. Intermediate structure and 18O-isotope data revealed that the conversion of acetaldehyde to p-methylcyclohexadienal intermediates proceeds in an enamine-iminium intermediate mechanism. Then, controlled experiments and D-isotope results indicated that the dehydrogenation of p-methylcyclohexadienal to p-MBA and H2 is catalyzed by the same amines through an iminium intermediate. This is an example that metal-free amines catalyze the dehydrogenation (releasing H2), rather than using metals or stoichiometric oxidants.
Subject areas: Chemistry, Catalysis, Organic chemistry, Organic synthesis
Graphical abstract
Highlights
-
•
A direct route to produce p-methyl benzaldehyde from biomass-derived acetaldehyde
-
•
Revealing the reaction kinetics and mechanism under reaction conditions
-
•
An example of an organic amine-catalyzed dehydrogenation-aromatization reaction
Introduction
p-Methyl benzaldehyde (p-MBA) is one of the primary precursors in fine chemicals manufacturing, e.g., pharmaceuticals (Yu et al., 2010; Chen et al., 2019). Conventionally, it was produced via a successive photochlorination, amination, and acid hydrolysis of petroleum-derived p-xylenes (Scheme 1A, Angyal, 2004; Schoch et al., 1982). However, this reaction co-produces vast pollutants and waste acid water (Schoch et al., 1982), which run counter to the current green development policy. Therefore, this situation becomes a strong incentive to develop new routes for the energy-efficient and green production of p-MBA.
Scheme 1.

Two routes for synthesis of p-MBA
(A) A conventional industrial route.
(B and C) A biomass-derived acetaldehyde route. o-MBA was reported as a major product using heterogeneous catalysts. p-MBA was synthesized using amines in this work.
Construction of value-added products with aromatic rings from low-molecular weight molecules is a long-standing research topic (Jiao et al., 2016; Li et al., 2019; Cheng et al., 2017; Tempelman et al., 2015; Moteki et al., 2016; Hulea, 2018). Extensive researches have reported the production of methyl benzaldehyde from biomass-derived acetaldehyde over MgO and Ca10(OH)2(PO4)6 (Wang et al., 2019; Lusardi et al., 2020; Zhang et al., 2016; Moteki et al., 2017). However, the selectivity of methyl benzaldehydes (MBA) in proceeding was no exceeding 30% (Scheme 1B), among which, p-MBA was less than 3% (Moteki et al., 2016; Wang et al., 2019), far from meeting the industrial production requirements. This insufficient selectivity was attributed to the complex network of competing reactions in acetaldehyde cascade processes, resulting in broad product distributions (Moteki et al., 2016; Lusardi et al., 2020).
Organic amines, such as tetrahydropyrrole (THP) and its derivatives, could catalyze enals to yield p-methyl benzaldehyde derivatives (Hong et al., 2006, 2007; Song et al., 2012), although the reaction intermediates and mechanism have not been investigated. Furthermore, the system that can directly catalyze the self-condensation of acetaldehyde and then, aromatize enal intermediates to para-aromatic aldehydes has not been reported yet. Here, we report a direct, green route for the selective synthesis of p-MBA from acetaldehyde using a diphenyl prolinol trimethylsilyl ether catalyst (Scheme 1C). The optimized p-MBA selectivity reaches 90% at conversion of 99.8%, yielding a formation rate of 0.33 mmolp-MBA mmolcat.−1 h−1, which is about six times of the-state-of-the-art report. This article will focus on the investigation of reaction intermediates, route, and mechanism to well understand this coupling-aromatization reaction and in turn, to make an optimization of the reaction rate. Controlled experiments, in situ spectroscopy, and 18O/D-isotope studies revealed that acetaldehyde undergoes an enamine-iminium intermediate mechanism and then an organic amine-catalyzed dehydrogenation mechanism to form p-MBA finally. This is an example that metal-free amines catalyze the dehydrogenation-aromatization (releasing H2), rather than using metals or stoichiometric oxidants. The striking p-MBA selectivity shows the charming merit of homogeneous catalysts in terms of fine chemicals manufacturing.
Results and discussion
Catalytic performance
Figure 1A illustrates the reaction scheme of acetaldehyde to p-MBA and the structure of catalysts used. Figures 1B and 1C and Table S1 summarize the catalytic activity of acetaldehyde condensation and aromatization to p-MBA with several organic amines. As shown, the product distributions differ and change with the type of catalysts, acid additives, and solvents. Notably, among C8 aromatic oxygenates, p-MBA is the major product. Reaction in the presence of tetrahydropyrrole (denoted as THP) or 3-pyrrolidinol (denoted as THP-OH, considering it as the derivative from THP) generates p-MBA and p-methylcyclohexadienal (p-MCA, Figure 1B), accompanying with undesired o-methyl benzaldehyde (o-MBA) and massive 2,4,6-octatrienal (C8 enals) as co-products (see Table S1), i.e., showing broad product distributions. With L-proline (denoted as THP-COOH), the acetaldehyde is almost completely converted, but 2-butenal is the major product with trace amounts of p-MBA (less than 0.7%, see Table S1). 2-(Diphenylmethyl) pyrrolidine (denoted as THP-DPh), grafted with large steric hindrance groups, slightly improves the selectivity of para-products (p-MBA + p-MCA, 62.6%) compared to those of Py and Py-OH. An encouraging result comes from the reaction in the presence of diphenyl prolinol trimethylsilyl ether (denoted as THP-DPh-OTMS), where p-MBA and p-MCA are formed in a selectivity of 82.5%, whereas the yield of C8 enals is dramatically reduced to 2.6% (see Table S1). In addition, the enhancement of para-products selectivity from THP to THP-DPh-OTMS (Figure 1B) can be explained by the steric hindrance inducing the preferential formation of intermediates with specific configuration (Mukherjee et al., 2007; Schmid et al., 2010; Zhang et al., 2012).
Figure 1.

Reaction scheme and catalytic performance
(A) Reaction scheme of CH3CHO to p-MBA.
(B) Effect of catalyst structure on product distributions.
(C) Effect of acidity strength (pKa) on selectivity of para-products. Reaction conditions: CH3CHO (1 mmol), solvent (3 mL), catalyst (0.1 mmol), acidic additives (0.3 mmol), 20°C for 2 h under Ar. ∗ More amines were added and the reaction was kept at 60°C for 2 h (see supplemental text for more details).
Furthermore, we found that organic amines can catalyze the conversion of p-MCA to yield p-MBA (the details were as below). Thus, more THP-DPh-OTMS was added to the reaction mixture (i.e., a “one-pot, two-step” process), which was then subjected to 60°C for more time. Surprisingly, a 90% selectivity of p-MBA was obtained with a yield of 90% and a formation rate of 0.33 mmolp-MBA mmolcat.−1 h−1, which is about six times of the report (using 2-butenal as reactants, Hong et al., 2006; Song et al., 2012). Notably, the ratio of p-MBA to o-MBA is up to 30 (see Table S1), which is more than two orders of magnitude higher than the literature (Moteki et al., 2016; Wang et al., 2019).
Next, the effect of acidic additives on the product distributions was examined. Figure 1C shows acidic groups and their strength playing a determinant role to the selective formation of para-products. To clarify the role of acidic groups, the −OH group of p-NO2-PhOH was replaced as −COOH. However, the total selectivity of p-MBA and p-MCA is dramatically reduced to 4.4%. To elucidate the effect of acidity, −H, −CH3, and −COOH substituents were selected to replace the −NO2 group of p-NO2-PhOH, respectively. Correspondingly, only moderate selectivity towards p-MBA and p-MCA was detected, i.e., 33.7, 23.2, and 33.3%. Interestingly, it is noted that there is a volcano-type dependence of the selectivity of p-MBA and p-MCA on the acid strength (pKa), i.e., 23.2% (p-CH3-PhOH, pKa = 10.2) < 33.7% (PhOH, pKa = 9.9) < 82.5% (p-NO2-PhOH, pKa = 7.15), and then 33.3% (p-COOH-PhOH, pKa = 4.6) > 4.4% (p-NO2-PhCOOH, pKa = 3.4). The strong acidic group possibly reacts or interacts with the amine group, thus preventing multistep C-C coupling, as demonstrated by the high selectivity of 2-butenal (see Table S1). Thus, the synergy between THP-DPh-OTMS and acidic additives boosts the conversion of acetaldehyde to para-aromatic aldehyde (Lokesh et al., 2019).
Screening solvents on the product distributions showed that the selectivity of p-MBA and p-MCA increases in the following order: dimethylsulfoxide (DMSO) ≈ dimethylformamide (DMF) < CH3CN < C2H5OH < CHCl3 (see Table S1). This order is almost in parallel to the solvent polarity (εr), i.e., 7.2 (DMSO) > 6.4 (DMF) > 6.2 (CH3CN) > 4.3 (C2H5OH) ≈ 4.4 (CHCl3). Therefore, the solvent with relative weak polarity favors the condensation and subsequent aromatization of acetaldehyde.
Reaction route
To understand the reasons for the selective p-MBA formation from acetaldehyde, the reaction route was studied by tracking the reaction progress. To distinguish the primary from the secondary products, the effect of reaction time on the product distributions was investigated (Anbarasan et al., 2012). Product yields and acetaldehyde conversion were plotted as a function of reaction time (Figure 2A). The steep initial rise in 2-butenal yield with acetaldehyde conversion indicates that the primary product is derived directly from acetaldehyde aldol-condensation (Wang et al., 2019). In a few minutes, the 2-butenal yield reaches a maximum and then it is converted to C8 oxygenates, such as p-MCA and p-MBA, via a secondary reaction. Furthermore, p-MCA and p-MBA become the major products at reaction time over 10 min. The high initial slope of p-MCA implies that it is the precursor for p-MBA formation. In addition, C8 enals and o-MBA were also simultaneously detected as secondary products but at much lower formation rates. This result suggests that the conversion of 2-butenal to p-MCA/p-MBA and to C8 enals/o-MBA possibly undergoes parallel reaction channels. Thus, acetaldehyde undergoes organic amines-catalyzed condensation, forming the 2-butenal intermediate (Equation 1), which yields p-MCA and p-MBA through cycloaddition and aromatization in sequence.
| (Equation 1) |
Figure 2.

Reaction route
(A) Dependence of conversion and product yields on the reaction time (1 mmol CH3CHO, 3 mL CHCl3, 0.05 mmol THP-DPh-OTMS, 0.3 mmol p-NO2-PhOH, 20°C).
(B) IR difference spectra. The green line in Figure 2B was obtained at 180 min (45 μmol CH3CHO, 0.5 μmol THP-DPh-OTMS, 1.2 μmol p-NO2-PhOH, CHCl3 as solvent, 20°C).
(C) Intensity changes of IR peaks in Figure 2B with reaction time.
(D) Evolution of IR peaks using THP as a catalyst with reaction time (see Figure S1). Star: CH3CHO, diamond: 2-butenal, hollow cycle: p-MCA, square: p-MBA, hollow up triangle: o-MBA, hollow down triangle: C8 enals.
In situ infrared spectroscopy (IR) was conducted to confirm whether 2-butenal and other intermediates were formed (see Figure S1). Figure 2B shows the corresponding IR difference spectra, which were obtained by subtracting a reference spectrum taken soon after the mixture of CH3CHO and catalysts from the real-time spectrum (Chen et al., 2007). An obvious negative peak at 1724 cm−1, assigned to the C=O stretching vibration [ν(C=O)] of acetaldehyde, was detected. Positive peaks at 1,686, 1,676, 1,642, 1,607, 1,603, 1,593, and 1,577 cm−1 were detected concomitantly. Peaks at 1,686 and 1,642 cm−1, assigned to the ν(C=O) and ν(C=C) modes of 2-butenal (see Figure S1), respectively, became prominent as the reaction proceeded. A series of IR peaks at 1,676, 1,642, 1,603, and 1,593 cm−1 are characteristic frequencies of enals with two C=C bonds (Aboaly et al., 1985), indicating the formation of p-MCA in view of the catalytic data shown in Figure 2A. Characteristic peaks at 1,607 and 1,577 cm−1 are attributed to the ν(C=C) modes of aromatic aldehydes (see Figure S1). Although the C=O stretching vibration peak of aromatic aldehydes (at ∼1,686 cm−1) is overlapped with that of 2-butenal, the characteristic ν(C=C) frequencies at 1,607 and 1,577 cm−1 indicate the formation of p-MBA. The peaks at 1724, 1,686, 1,676, and 1,607 cm−1 were used to track the concentration change of acetaldehyde, 2-butenal, p-MCA, and p-MBA, respectively. Figure 2C shows the varying tendency of the reactant and products with time. Difference in the initial slope of these curves indicates acetaldehyde yielding p-MBA via 2-butenal and p-MCA intermediates (Equation 2).
| (Equation 2) |
CH3CHO-IR experiments using THP as catalysts were also conducted (Figures 2D and S1). In contrast, although products p-MCA (frequencies at 1,676, 1,642, 1,603, and 1,593 cm−1) are shown, characteristic ν(C=C) peaks of C8 enals at 1,635, 1,617, and 1,604 cm−1 (Aboaly et al., 1985), also appear and increase with time. These IR data support the mechanism that 2-butenal derived from acetaldehyde condensation is the intermediate to yield p-MCA and p-MBA, and prove that the steric hindrance group, i.e., -DPh-OTMS, contributes to the formation of target products.
Mechanism of acetaldehyde condensation and aromatization
Identification of active sites
To reveal the reaction mechanism, we designed controlled experiments to confirm the active sites. Figure 3A shows the catalytic activity of THP-DPh-OTMS with its N group grafted to a −CH3 group (Tang et al., 2004) and that of THP-DPh-OTMS treated with HCl. As shown, the introduction of a –CH3 group led to 2-butenal as the only product, i.e., neither p-MCA nor p-MBA detected. When THP-DPh-OTMS hydrochloride was used, the major product was 2-butenal, accompanying small amounts of p-MCA and p-MBA. Both catalysts showed much lower reaction rate than that of THP-DPh-OTMS. Therefore, the basic R–NH–R’ amine group of THP-DPh-OTMS can indeed be involved in the selective conversion of acetaldehyde to p-MCA and p-MBA.
Figure 3.

Reaction mechanism of acetaldehyde condensation and cycloaddition
(A) Comparison of catalytic performance with THP-DPh-OTMS, THP(–CH3)-DPh-OTMS, and (THP-DPh-OTMS)H+Cl− (Reaction conditions: 1 mmol CH3CHO, 3 mL CHCl3, 0.1 mmol catalyst, 0.3 mmol p-NO2-PhOH, 20°C for 2 h, Ar atmosphere).
(B) A TOF-MS spectrum of the reaction liquid after the mixture of acetaldehyde and THP-DPh-OTMS/p-NO2-PhOH.
(C and D) Effect of p-NO2-PhOH amounts on conversion and product distributions (Reaction conditions: 1 mmol CH3CHO, 3 mL CHCl3, 0.1 mmol THP-DPh-OTMS, 20°C for 2 h, Ar atmosphere). Star: CH3CHO, diamond: 2-butenal, hollow cycle: p-MCA, square: p-MBA, hollow up triangle: o-MBA, hollow down triangle: C8 enals.
Reaction mechanism of acetaldehyde condensation and cycloaddition
Up to now, several mechanisms were proposed in organic amine-catalyzed condensation and/or cycloaddition reaction (List et al., 2000; Northrup and MacMillan, 2002; Moyano and Rios, 2011; Erkkilä et al., 2007; Dalko Dr and Moisan, 2004). To clarify the possible intermediates, we tried to track them using H-NMR technique. However, the change of in situ H-NMR spectra with reaction time hardly provided clear evidence for intermediates formation in this case and typically only showed mixtures of reactants and products along with the THP-DPh-OTMS/p-NO2-PhOH, even though a high ratio of catalyst to reactant was used (see Figures S2–S5). This result was consistent with the report of Schnitzer et al. in an addition reaction using similar organic-amines as catalysts (Schnitzer and Wennemers, 2020). Therefore, we used a Time Of Flight Mass Spectrometer (TOF-MS) to detect the possible intermediate in the liquid phase as soon as the mixture of acetaldehyde and THP-DPh-OTMS/p-NO2-PhOH. MS data revealed that in addition to the signal of THP-DPh-OTMS {m/z = 326.1863, [M + H]+, ESI+}, the peaks at 431.2567 and 430.2489 were also detected (Figure 3B), which can be assigned to the diene iminium and trienamine, respectively, derived from the successive condensation and cycloaddition reaction between amines and 2-butenal (Scheme 2).
Scheme 2.

Proposed enamine/iminium catalytic cycle
The detected fragment ions of 18O/16O-incorporated aldehydes were marked in red/black.
To confirm the possible enamine-iminium mechanism, an 18O-isotope experiment was conducted. If the condensation and aromatization of acetaldehyde takes place in the presence of 18O-enriched water, incorporation of 18O at the carbonyl group of reactants and products would be expected (List et al., 2004) because of the final hydrolysis step in the iminium catalysis cycle (Scheme 2). Thus, prior to this experiment, the reaction order of H2O was determined by adding water (0–4 mmol) into the reaction system. The reaction rate and product distributions almost remain constant (see Figure S6), indicating a zero-order dependence on H2O under the controlled conditions. Then we detected the product distributions in the presence of 2 mmol 18O-enriched water (97 mol%, Aladdin). Scheme 2 and Figure S7 showed that 18O-contained reactants and products are observed.
Please note that the formation of iminium ions and their tautomerization to enamine species, see Scheme 2, need the assistance of acidic additives via a proton transfer process (List et al., 2000). Figure 1C showed that the functional group type and strength of acidic additives greatly influence the product distributions. Furthermore, Figures 3C and 3D showed that the selectivity of products dramatically changes with the variation in the amount of p-NO2-PhOH. Without acidic additives, neither p-MBA nor p-MCA was detected, but a 96.3% selectivity of 2-butenal. Other minor products were C6 enals (0.3%), C8 enals (2.4%), and o-MBA (1%). Interestingly, the p-MBA and p-MCA became the dominant products, when 0.15 mmol acidic additive was added (i.e., np-NO2-PhOH/ncat. = 1.5 mmol/mmol). Meanwhile, a dramatically declined selectivity of 2-butenal and an increased conversion were obtained. In the absence of THP-DPh-OTMS, no obvious reaction occurred. Therefore, the result of 18O-isotope studies and the dependence of products selectivity on acid amounts support that both the aldol condensation of acetaldehyde to 2-butenal and the following cycloaddition of 2-butenal to p-MCA undergo an enamine-iminium intermediate mechanism.
In addition, in Figure 3D, p-MCA and p-MBA were the major products, accompanying with less C6/C8 enals and their derivatives o-MBA (<5% in selectivity). This result suggests that the cycloaddition and subsequent aromatization of 2-butenal are thermodynamically more favorable than its chain growth (forming long-chain enals), as verified by the much lower Gibbs energy of the former reaction, i.e., −110 vs. −10 kJ/mol.
It is proven that 2-butenal is in a trans-configuration based on the in situ H-NMR data (see Figure S5). To reveal its cycloaddition course, the activation behavior of trans-2-butenal was investigated. We firstly adopted n-butyraldehyde as reactants to examine product distributions. Figure S8 showed that only aliphatic aldol-condensation products were detected from C4 saturated aldehydes. Branched chains at α-C sites of the dimer products convincingly indicate that the nucleophilic attack by the α-C of the aldehydes at the carbonyl C of others takes place. Next, when trans-2-butenal was used as reactants, aromatic aldehydes were obtained as main products. However, when the α-C site of 2-butenal was grafted with a methyl substituent, neither aldol dimers nor aromatic aldehydes were detected. This can be explained by the −CH3 at the α-C site of 2-methyl-2-butenal hindering the nucleophilic attack by this site, because this site is tetra-coordination without α-H. Therefore, these controlled experiments illustrated that the activation of the α-C site of trans-dienamines (or trans-2-butenal) results in the selective formation of p-MCA (Scheme 3).
Scheme 3.

Proposed reaction mechanism for the selective p-MCA formation from trans-2-butenal
The activation of the α-C site (marked in blue) of trans-2-butenal is crucial for the selective synthesis of p-MBA.
Note that, on a heterogeneous CaO catalyst, trans-2-butenal was preferentially deprotonated at the γ-C position because of allylic stabilization of the resulting anion (Wang et al., 2019). Nucleophilic attack by the γ-C of the anion at the carbonyl C of trans-2-butenal formed 2,4,6-octatrienal, which underwent cyclization and dehydrogenation to form o-MBA as the major product (Scheme 1B). Therefore, it is crucial to control the activation of the α-C site of trans-2-butenal for the selective synthesis of p-MBA.
Combination of the detected enamine and iminium intermediates and the viewpoint of the α-C site activation of trans-2-butenal, the possible cycloaddition mechanism, is proposed. Before the aldol condensation to form cyclic products (Scheme 3), dienamine species possibly serve as Michael donors for the conjugate addition to form iminium ions, i.e., forming the first C-C bond for p-MCA (Hong et al., 2006). Next, the resulting Michael adduct possibly proceeds in hydrolysis, intramolecular condensation (Hammer et al., 2020), and dehydration in sequence to form trienamine and diene iminium intermediates, which can hydrolyze to form p-MCA.
Aromatization reaction of p-methylcyclohexadienal (p-MCA)
The aromatization of p-MCA to p-MBA is thermodynamically favorable (ΔG (25–60°C) = −50 kJ/mol) and its mechanism is further investigated. Figure 4A showed that the selectivity of p-MBA and p-MCA is almost independent of air, O2, or Ar, even though prolonging reaction time (under Air or Ar, see Table S2). In addition, although both degassed CHCl3 solvent and Ar were used, product distributions were the same as that of its counterpart (Figure 4A). A similar experiment result was reported using 2-butenal as a reactant (Hong et al., 2006, 2007), but the reason had not been revealed yet. There were two possible explanations for these findings: (1) the hydrogen species abstracted from p-MCA were captured by certain acceptors or (2) a dehydrogenation process takes place by releasing H2. Nitro groups in p-NO2-PhOH could be reduced to form corresponding hydroxylamine and amino units (Serna and Corma, 2015). To substantiate this hypothesis, PhOH and its derivatives (for example, with a −COOH or −CH3 group at the para site), were used as acidic additives, respectively, but p-MBA was still formed (3.2%–8.5% in selectivity, see Table S1). Please note that, when no acids were used (Figure 3D), o-MBA was detected in a selectivity of 1%. Furthermore, no obvious alcohols and enols, possibly formed from a selective transfer hydrogenation of aldehydes and/or enals (Shi et al., 2010), were detected. Based on these data of controlled experiments, the possibility of the first hypothesis could be ruled out.
Figure 4.

Aromatization mechanism of p-MCA to p-MBA
(A) Product distributions under different atmospheres.
(B) Detection of gaseous products by MS (CH3CHO and CD3CDO as reactants).
(C) HCl poisoning experiments. Hollow: with HCl. Star (green): CH3CHO, cycle (red): p-MCA, square (blue): p-MBA.
(D) Effect of the type of organic amines on the selectivity and formation rate of p-MBA (reaction conditions: 1 mmol CH3CHO, 3 mL CHCl3, 0.1 mmol THP-DPh-OTMS, 0.3 mmol p-NO2-PhOH, 20°C for 2 h; then amines were added and stirred for 1 or 2 h at 60°C under Ar).
(E) Effect of the amounts of acidic additives on the dehydrogenation rate of p-MCA (reaction conditions: 0.04 mmol p-MCA, 0.04 mmol amines, 3 mL CHCl3, 60°C for 1.5 h under Ar).
To verify the validity of the second hypothesis, we used MS to detect the gas products in a batch reactor. Interestingly, in line with prediction, H2 was quantitatively obtained in a yield of ∼0.05 mmol/2 h (Figure 4B). Furthermore, CD3CDO (chemical purity, Aladdin) were employed as reactants, and HD/D2 were qualitatively detected (Figure 4B), which were originated from the dehydrogenation of D-substituted and H/D-substituted p-MCA (see Figure S9). Therefore, the direct dehydrogenation of p-MCA to p-MBA and H2 takes place indeed. This can be explained by the fact that the carbonyl and vinyl groups in cyclic p-MCA possibly induce the acidity (pKa) enhancement of their α/γ-C−H sites (Bordwell pKa Table, 2020), suggesting the easy C−H activation and dissociation.
The nucleophilic N center in the NHx group of organic amines activates the C-H bond of aldehydes and then catalyzes the subsequent condensation (Moyano and Rios, 2011; Erkkilä et al., 2007). Thus, we thought that the basic R-NH-R′ group of THP-DPh-OTMS possibly catalyzes the dehydrogenation of p-MCA via an H-abstract process. Therefore, we used HCl to poison the R-NH-R′ group (forming a hydrochloride) to verify this hypothesis. The poisoning experiment was performed by adding HCl (nHCl/nCat. = 1 mmol/mmol) into the mixture after reaction 2 h under normal conditions. The product distributions were then tracked with reaction time. Figure 4C showed the selectivity of p-MBA and p-MCA remaining almost constant, i.e., 24% and 58%, respectively, in 60 h after HCl addition. In comparison, controlled experiments without HCl addition showed that p-MBA selectivity slowly increases to 60% from 24% in 80 h, whereas that of p-MCA gradually decreases.
Furthermore, we found that the selectivity of p-MBA increased, though in different degree, when more primary-, secondary-, and tertiary-amines [such as H2N-(CH2)6-NH2, NH(C2H5)2/THP-DPh-OTMS, and N(C2H5)3] were added (Figure 4D). With the aid of secondary amines, the selectivity of p-MBA is about 2 times higher than those of other amines. For example, a 90% selectivity of p-MBA and a formation rate of 0.33 mmolp-MBA mmolNHx−1 h−1 were obtained, when more THP-DPh-OTMS (0.1 mmol) was added (i.e., a “one-pot, two-step’’ process) and the reaction was conducted at 60°C for 2 h. In comparison, the selectivity of p-MBA under H2N-(CH2)6-NH2 and N(C2H5)3 is 47.1% and 40.3%, respectively. When 0.2 mmol THP-DPh-OTMS was added into the reaction liquid at the beginning of the reaction (conducted at 60°C), however, 62% selectivity towards p-MBA was obtained with a carbon balance of as low as 58.7% (see Table S3, Entry 16). It indicates that acetaldehyde more easily undergoes over-polymerization to form high molecular weight products at high reaction temperatures. Therefore, this reaction was carried out in a “one-pot, two-steps” process, i.e., the conversion of CH3CHO to p-MCA at 20°C and then, the aromatization of p-MCA to p-MBA at 60°C. In this case, the carbon balance remains to ∼90% (see Table S3). Note that under these conditions, the molar ratio of p-MBA formed to catalyst is about 1.2–1.5, which further suggests that the amines catalyze the dehydrogenation of p-MCA to p-MBA and H2.
Next, cyclohexane was used to separate the p-MCA from the p-NO2-PhOH and THP-DPh-OTMS, which offers an unmissable chance to directly use it as a reactant to investigate the dehydrogenation mechanism. Figure 4E showed the dehydrogenation rate of p-MCA using THP-DPh-OTMS with or without acidic additives. p-MBA is the only product, i.e., a 100% selectivity. Amazingly, the reaction rate increases from 0.05 to 0.64 mmolp-MCA mmolCat.−1 h−1 when varying the ratio of n(p-NO2-PhOH)/n(THP-DPh-OTMS) from 0 to 3 (mmol/mmol). No reaction occurred with only acidic additives. This drastic difference suggests that the existence of acids, i.e., H-proton, possibly changed the dehydrogenative intermediates and reaction mechanism. However, a poor dehydrogenation rate was detected using acidic additives and H2N-(CH2)6-NH2 or N(C2H5)3 (Figure 4E). Please note that, iminium species are easily formed from aldehydes under the co-existence of acids and secondary amines (Scheme 2). These results guide us to presume that the p-MCA first converts to diene iminium, and then undergoes an amine-catalyzed dehydrogenation reaction to form p-MBA finally (Scheme 4). During this process, abstracting the first H from the acidic δ(γ)-C sites (pKa) should be crucial. This pKa in aldehydes is about 25 because of the activation effect by C=O (Bordwell pKa Table, 2020), thus possibly showing a low C-H activation barrier, about −6 kJ/mol (Liu et al., 2015). Compared to C=O, the electron-deficient C=N+ group in iminium will further decrease the pKa (Kitamura et al., 2014). This may explain the increased reaction rate of p-MCA when acids co-existed. In comparison, the electron-rich C-N group in enamine shows a negative effect for the C-H activation, which will not be discussed in the following sections.
Scheme 4.

Aromatization reaction route of p-MCA to p-MBA.
Combination of the catalytic data, in situ spectroscopic evidence, and isotope experiments described above, we propose a possible pathway for p-MBA synthesis from acetaldehyde using THP-DPh-OTMS as catalysts (Scheme 5). First, fast condensation converts acetaldehyde into trans-2-butenal, which then successively proceeds in Michael addition, aldol condensation (cycloaddition), and dehydrogenation to form para-site aromatic aldehydes, i.e., CH3CHO → trans-2-butenal → p-MCA → p-MBA. Among this course, the formed diene iminium species, derived from trans-2-butenal cycloaddition, proceed in a dehydrogenation process to form p-MBA and H2. This reaction follows an organic amine-catalyzed dehydrogenation mechanism via diene iminium intermediates. This process is different to the dehydrogenation-aromatization of N-heterocycles, in which potassium salt catalyzes the cleavage of N-H and C-H, thus resulting into H2 formation (Liu et al., 2019). In addition, the overall reaction rate remains constant when 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), a commonly used radical-trapping reagent (Tang, et al., 2021), was added in the reaction system, indicating that the conversion of acetaldehyde to p-MBA does not undergo a free radical route (see Figure S10). No transition metals were involved in this reaction (see ICP analysis in Method Details), thus excluding the possible existence of a redox mechanism. Therefore, the overall reaction proceeds in a mechanism of ionic reaction. THP-DPH-OTMS (with acidic additives) also catalyzes the self-condensation of 2-butenal followed by dehydrogenation to form o-MBA, but in small amounts.
Scheme 5.

Reaction pathways for aromatic aldehydes formation from acetaldehyde
All the reactants, intermediates, and products were detected except the one in the yellow panel.
Overall, this direct conversion of acetaldehyde to p-MBA is performed under mild reaction conditions with a remarkable yield. Further scale-up studies for p-MBA synthesis were carried out to demonstrate the utility of this method. For example, 440 mg CH3CHO (98%) was fed as reactants and, finally, 206.5 mg of p-MBA was obtained via an extraction-rotary evaporation technique (see Figure S11). In contrast, the commercial process for converting p-xylene to p-methyl benzaldehyde yields massive chlorides as by-products and produces waste strong acid water (Schoch et al., 1982). In addition, the inevitable chlorine residues in products further limits its application for pharmaceuticals. Therefore, this work represents a step change in developing a direct, green synthesis route for para-aromatic aldehydes and guides the design of more efficient heterogeneous catalysts for this reaction. More importantly, this report shows an experimental example of an organic amine-catalyzed dehydrogenation reaction, which is different to the reported metal-dehydrogenation and stoichiometric-dehydrogenation processes (Gnaim et al., 2021; Teskey et al., 2019; Mukaiyama et al., 2000).
The synthesis of aromatic aldehydes from low-carbon aliphatic aldehydes (n ≤ 6) was explored under the optimal conditions (Figure 5). When formaldehyde or acrolein were co-fed with acetaldehyde, a 10–12% selectivity of benzaldehyde was obtained, which was attributed to the complex network of competing reactions in these coupling-aromatization processes, leading to broad product distributions (Zhou et al., 2020). Interestingly, p-MBA could be selectively synthesized from acetaldehyde or 2-butenal. The butyraldehyde yielded the corresponding aldol-condensation product. Both C5 and C6 enals underwent the coupling-aromatization reaction yielding aromatic aldehydes with selectivity of over 94%.
Figure 5.

Selectivity of aromatic aldehydes from several aliphatic aldehydes (For [a], as shown in Scheme 3, the δ-C site of iminium intermediates involved in the cycloaddition step is indispensable; however, this site can’t be formed from CH2O or C3H4O, thus 1 mmol acetaldehyde was co-fed).
Conclusions
In this work, we developed a direct route for the selective synthesis of p-methyl benzaldehyde from acetaldehyde via condensation, cycloaddition, and dehydrogenation in consequence, using organic amines as catalysts. The optimized p-MBA selectivity reached 90% at an acetaldehyde conversion of 99.8%, leading to a product formation rate of 0.33 mmolp-MBA mmolcat.−1 h−1, which is about six times of the report. The basic R-NH-R′ group of THP-DPh-OTMS was proved to be the active site for acetaldehyde condensation, cycloaddition, and aromatization. Kinetic and in situ IR studies indicated that acetaldehyde undergoes aldol condensation to yield 2-butenal, which further forms p-MCA through cycloaddition and then to p-MBA via dehydrogenation. Intermediate structure and 18O-isotope data revealed that the conversion of acetaldehyde to p-MCA proceeds in an enamine-iminium intermediate mechanism. Then, the dehydrogenation of p-MCA to p-MBA was catalyzed by the same organic amine through an iminium intermediate. This is an experimental example that an organic amine catalyzes the dehydrogenation-aromatization reaction. Furthermore, this report proves that it is possible to selectively synthesize p-MBA from acetaldehyde by controlling the activation of the α-C site of 2-butenal intermediates.
Limitations of the study
We reported that the organic amine catalyzes the dehydrogenation of p-methylcyclohexadienal to p-methyl benzaldehyde with H2 releasing. This process was verified by controlled experiments and D-isotope results. However, during this dehydrogenation-aromatization process, the H-abstract, recombination, and H2 releasing were not clear yet. We preliminary tried to reveal these processes using DFT calculation, but failed, because it was a complex molecular system (involving 129 atoms with 381 parameters, including bond lengths, angels, and dihedrals). More rational models and feasible methods would be used to address these in future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals | ||
| Tetrahydropyrrole (THP) | Bidepharm | CAS: 123-75-1 |
| 3-pyrrolidinol (THP-OH) | ACMEC biochemical | CAS: 40499-83-0 |
| L-Proline (THP-COOH) | Bidepharm | CAS: 147-85-3 |
| 2-(Diphenylmethyl) pyrrolidine (THP-DPh) |
Daicel | CAS: 23627-61-4 |
| (S)-2-(diphenyl((trimethylsilyl)oxy)Methyl)pyrrolidine (THP-DPh-OTMS) | Bidepharm | CAS: 848821-58-9 |
| CHCl3 | Sinopharm | CAS: 67-66-3 |
| p-NO2-Ph-OH | Macklin | CAS: 100-02-7 |
| HCHO | Sinopharm | CAS: 50-00-0 |
| CH3CHO | Aladdin | CAS: 75-07-0 |
| CD3CDO (Chemical purity) | Aladdin | CAS: 1632-89-9 |
| Trans-C3H4O | Aladdin | CAS: 107-02-8 |
| Trans-C4H6O | Aladdin | CAS: 4170-30-3 |
| n-C4H8O | Aladdin | CAS: 123-72-8 |
| p-MBA | Aladdin | CAS: 104-87-0 |
| H218O | Aladdin | CAS: 14314-42-2 |
| Cyclopentanol | Aladdin | CAS: 96-41-3 |
| Trans-C5H8O (95%) | TCI | CAS: 1576-87-0 |
| Trans-C6H10O (98%) | D&B | CAS: 6728-26-3 |
| Software and algorithms | ||
| ChemDraw Professional 17.0 | PerkinElmer | https://www.perkinelmer.com/ |
| Origin Pro 9.0 | OriginLab | https://www.originlab.com/ |
| MathType 6.9 | WIRIS | https://www.wiris.com/en/mathtype/ |
| Other | ||
| 7890A GC/GC-MS (5975C) | Agilent Technologies | https://www.agilent.com/ |
| TOF-MS (6540 UHD Q-TOF) | Agilent Technologies | https://www.agilent.com/ |
| Avance 400 MHz NMR/HD 700 MHz NMR spectrometer | Bruker | https://www.bruker.com/ |
| ICP-OES (7300DV) | PerkinElmer | https://www.thermofisher.cn/ |
| NEXUS 470 FT-IR | Thermo Nicolet | https://www.thermofisher.cn/ |
| Mass spectrometer (DECRA) | Hiden Analytical | https://www.hidenanalytical.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Can Li (canli@dicp.ac.cn).
Materials availability
This study did not generate new unique reagents.
Method details
Catalytic reaction test
All commercially available reagents were reagent grade and used without further purification unless otherwise specified. CHCl3 were further degassed before use for controlled experiments according to a standard freeze-pump-thaw method. Normally, reactions were carried out under Ar atmosphere in new, fire dried Schlenk tube (35 mL, Synthware). The amount of transition metals in amines was detected by ICP, and their content is negligible.
General procedure: a solution of acetaldehyde (1 mmol) in solvent (for example, CHCl3, 3 mL) was treated with p-nitrophenol (0.3 mmol) and catalysts (0.1 mmol). The reaction mixture was stirred in a tube at 20°C for a certain period. Cyclopentanol is co-added and used as an internal standard to calculate formation rate and carbon balance (Table S3). To accelerate the conversion of p-MCA to p-MBA, after the reaction carried out at 20°C for 2 h under normal conditions, more organic amines (0.1-0.45 mmol -NH- groups) were added and then the mixture was stirred for more time (1-20 h) at a certain temperature (20, 40, or 60°C). The reaction under Air, Ar, and O2 atmosphere was carried out using a batch reactor (75 mL). After reaction, a gas chromatograph (GC) with a flame ionization detector, equipped with a HP-5 column (30 m × 0.320 mm × 0.25 μm), was used to quantify the products. Their identities were confirmed by GC-MS analysis (Agilent 7890A GC, interfaced with 5975C MS, USA), and NMR spectrometer (1H spectra, BrukerAvance 400 NMR spectrometer). Time of Flight Mass Spectrometer (TOF-MS, Agilent, USA) was used to capture the possible reaction intermediates in the liquid mixture. Gaseous products (such as H2) were analyzed by a mass spectroscopy (Hiden Analytical). All conversion and selectivity were calculated on basis of moles of carbon in the reactants and products, as follows:
- Acetaldehyde conversion:
- Product selectivity:
The carbon number, FID peak area, and response factor of each product are designated Ni, Ai, and ƒi, respectively.
- Product yield:
Aromatization of p-MCA to p-MBA: Cyclohexane was used to separate p-methylcyclohexadienal from acidic additives and catalysts. Briefly, acetaldehyde reaction was firstly carried out at 20°C for 2 h under Ar. Then, the CHCl3 was removed by a rotary evaporator at 20°C. 25 mL (5 mL×5 times) cyclohexane was added to the yellow viscous residues to dissolve the methylcyclohexadienal intermediates. Please note that cyclohexane doesn't dissolve the p-NO2-PhOH and THP-DPh-OTMS. Yellowish products were obtained by removing the cyclohexane from the extraction liquid. Although methylcyclohexadienal was co-existed with methyl benzaldehyde, reaction rate can be calculated by using cyclopentanol as an internal standard.
Scale-up experiments: A solution of acetaldehyde (10 mmmol) in CHCl3 (30 mL) was treated with p-nitrophenol (3 mmol) and THP-DPh-OTMS (1 mmol). The reaction mixture was stirred in a glass reactor (100 mL) at 20°C for 2 h; then more THP-DPh-OTMS (1 mmol) was added and stirred for 2 h at 60°C under Ar. After reaction, a GC was used to quantify the products. Hexane was used to obtain the products in a similar method described above, i.e., an extraction-rotary evaporation technique.
Synthesis of several aromatic aldehydes: C1-C6 aliphatic aldehydes (CH2O, C2H4O, trans-C3H4O, trans-C4H6O, C4H8O, trans-C5H8O, and trans-C6H10O) were used to as reactants. A solution of aldehydes (1 mmmol C1-C3/0.5 mmol C4-C6 aldehydes) in CHCl3 (3 mL) was treated with p-nitrophenol (0.3 mmol) and THP-DPh-OTMS (0.1 mmol). The reaction mixture was stirred in a tube at 20°C for 2 h; then more THP-DPh-OTMS (0.1 mmol) were added and stirred for 2 h at 60°C under Ar. The product structure was analyzed and identified by GC-MS. Cyclopentanol is co-added and used as an internal standard to calculate formation rate and carbon balance (Table S3).
18O-isotope experiments: H218O (97 atom%) was supplied by Aladdin. Prior to adding acetaldehyde, 2 mmol H218O was firstly added into the mixture liquid of catalyst (0.1 mmol) and solvent (CHCl3, 3 mL). The reaction mixture was stirred in a vial at 20°C for 2 h. After reaction, a GC-MS was used to analyze the fragment peaks (m/z) of reactants and products.
D-isotope experiments: A solution of CD3CDO (4 mmmol) in CHCl3 (10 mL) was treated with p-nitrophenol (0.6 mmol) and THP-DPh-OTMS (0.2 mmol). The reaction mixture was stirred in a batch reactor at 20°C for 2 h; then more THP-DPh-OTMS (0.2 mmol) were added and stirred for 2 hr at 60°C under Ar. Aldehyde conversion and product selectivity were quantified by GC. The product structure was identified by GC-MS. Gaseous products (such as H2, HD, and D2) were qualitatively analyzed by a mass spectroscopy (Hiden Analytical DECRA). In comparison, experiments used CH3CHO as reactants were also conducted in a similar procedure. And the gaseous products were also analyzed.
In situ HCl poisoning experiments: A solution of acetaldehyde (1 mmol) in CHCl3 was treated with p-nitrophenol (0.3 mmol) and THP-DPh-OTMS (0.1 mmol). The reaction mixture was stirred in a vial at 20°C for 2 h under Ar. Then, 0.1 mmol HCl was added into the reaction mixture, which was continued to be treated at 20°C. The product distributions were detected with reaction time.
In situ IR experiments: CH3CHO-IR was carried out on a Thermo Nicolet spectrometer (NEXUS 470) equipped with a liquid reaction cell (Omni-Cell) and a deuterated triglycine sulfate (DTGS) detector. This liquid cell was equipped with a pair of CaF2 windows and sealed by Teflon, forming a cavity of about 120 μL. The reaction mixture was injected into the cell via one of the circular holes (2 mm in diameter) located at the CaF2 window (see Figure S1E). After the cell sealed, the reaction mixture was maintained at 20°C to collect the spectrum. The evolution of reactants and products was monitored by IR in a transmission mode by averaging 32 scans at a resolution of 4 cm-1. Standard IR spectra of acetaldehyde, 2-butenal, p-MBA, THP-DPh-OTMS, and p-NO2-PhOH in CHCl3 were also conducted using the same cell, respectively.
NMR investigation: to confirm the synthesis of p-MBA, products were separated from reaction mixtures by a method of column chromatography with EtOAc-light petroleum (volume ratio = 1:10) to obtain a light-yellow oil. Merck 60 silica gel was used for chromatography. 1H and 13C NMR spectra were obtained on an Avance 400 MHz NMR spectrometer. NMR peaks, assigned to p-MBA, were shown, as follows: 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 7.77 (d, J = 8.1 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 2.43 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 191.97, 145.54, 134.22, 129.84, 129.71, 21.87. THP-DPh-OTMS: 1H NMR (400 MHz, CDCl3) δ 7.58–7.52 (m, 2H), 7.47–7.41 (m, 2H), 7.41–7.27 (m, 7H), 4.15 (t, J = 7.4 Hz, 1H), 3.02–2.81 (m, 2H), 2.25 (s, 1H), 1.76–1.61 (m, 3H), 1.52–1.41 (m, 1H), 0.00 (s, 9H). In situ 1H spectra were recorded on a 700 MHz spectrometer (Bruker) at 20°C. 40 spectra were collected at an interval of 80s within about 1 h. Chemical shifts are given in ppm. Standard H-NMR spectra of acetaldehyde, 2-butenal, p-MBA, THP-DPh-OTMS, and p-NO2-PhOH in CHCl3 were also conducted as references. Reaction conditions were shown in Figure S4.
Methods for characterization
ICP analysis: Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES, ICPS-8100, Japan) was used to analyze the content of transition metals. Briefly, 90 mg THP-DPh-OTMS was dissolved into 3 mL aqua regia (Take care: this oxidation is a strong exothermic reaction!) to obtain a clear aqueous solution. Then, the content of Pd, Ir, and Pt (which are reported active for oxidative dehydrogenation, Gnaim, et al., 2021) was analyzed by ICP-OES. The amount was calculated on basis of the peak area. The amount of Pt is ∼0.01 ppm, and no Pd and Ir were detected. These results are within the uncertainty of the measurement; therefore, the metal amount is negligible.
Acknowledgments
This work was supported by the Youth Program of National Natural Science Foundation of China (No. 22002161). This work was also conducted by the Fundamental Research Center of Artificial Photosynthesis (FReCAP), financially supported by National Natural Science Foundation of China (NSFC) under grant No. 22088102. We thank Prof. Dr. Zhaochi Feng and Zhendong Feng (Dalian Institute of Chemical Physics, Chinese Academy of Sciences) for their help in in situ IR experiments. We also thank Dr. Guanna Li (Wageningen University & Research, Netherlands) for her help to make a preliminary attempt in theoretical calculation.
Author contributions
C. L. directed this work. C. L., Q. -N. W., and X. L. conceived this work. Q. -N. W. performed the catalytic experiments and proposed the dehydrogenation mechanism. X. L. prepared the (in situ) NMR experiments and investigation. K. W. participated in the discussion of dehydrogenation mechanism and gave some suggestions. Y. L. proposed the cycloaddition reaction from acetaldehyde. S. -M. L. discussed the results and offered some suggestionsabout the dehydrogenation mechanism. C. L., Q.-N. W., and X. L. wrote the article with edits and approval from all authors.
Declaration of interests
C. L., Q.-N. W., and X. L. applied for a China patent based on the technology described in this work at Dalian Institute of Chemical Physics, Chinese Academy of Sciences (Application number: 202011448529.1). The other authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103028.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper dose not report original code.
-
•
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.
References
- Aboaly M.M., Baron M.H., Favrot J., Belloc J., Revaul M. Vibrational analysis of (E,E)-2,4-hexadienal, (E,E,E)-2,4,6-octatrienal, and (E,E,E)-3-methyl-2,4,6- octatrienal. Can. J. Chem. 1985;63:1587–1593. [Google Scholar]
- Anbarasan P., Baer Z.C., Sreekumar S., Gross E., Binder J.B., Blanch H.W., Clark D.S., Toste F.D. Integration of chemical catalysis with extractive fermentation to produce fuels. Nature. 2012;491:235–239. doi: 10.1038/nature11594. [DOI] [PubMed] [Google Scholar]
- Angyal S.J. The Sommelet reaction. Org. React. 2004;8:197–217. [Google Scholar]
- Bordwell pKa Table, 2020, https://organicchemistrydata.org/hansreich/resources/pka/#kaacetyl.
- Chen T., Feng Z., Wu G., Shi J., Ma G., Ying P., Li C. Mechanistic studies of photocatalytic reaction of methanol for hydrogen production on Pt/TiO2 by in situ Fourier transform IR and time-resolved IR spectroscopy. J. Phys. Chem. C. 2007;111:8005–8014. [Google Scholar]
- Chen, H., Zhang, C., He, Y., Lin, X. (2019). Preparation of Procarbazine. CN Patent, 110156629.
- Cheng K., Zhou W., Kang J., He S., Shi S., Zhang Q., Pan Y., Wen W., Wang Y. Bifunctional catalysts for one-step conversion of syngas into aromatics with excellent selectivity and stability. Chem. 2017;3:334–347. [Google Scholar]
- Dalko Dr P.I., Moisan L. In the golden age of organocatalysis. Angew. Int. Ed. Chem. 2004;43:5138–5175. doi: 10.1002/anie.200400650. [DOI] [PubMed] [Google Scholar]
- Erkkilä A., Majander I., Pihko P.M. Iminium catalysis. Chem. Rev. 2007;107:5416–5470. doi: 10.1021/cr068388p. [DOI] [PubMed] [Google Scholar]
- Gnaim S., Vantourout J.C., Serpier F., Echeverria P.-G., Baran P.S. Carbonyl desaturation: where does catalysis stand? ACS Catal. 2021;11:883–892. [Google Scholar]
- Hammer N., Christensen M.L., Chen Y., Naharro D., Liu F., Jørgensen K.A., Houk K.N. An experimental stereoselective photochemical [1s, 3s]-sigmatropic silyl shift and the existence of silyl/allyl conical intersections. J. Am. Chem. Soc. 2020;142:6030–6035. doi: 10.1021/jacs.9b11579. [DOI] [PubMed] [Google Scholar]
- Hong B.-C., Wu M.-F., Tseng H.-C., Liao J.-H. Enantioselective organocatalytic formal [3 + 3]-cycloaddition of α,β-unsaturated aldehydes and application to the asymmetric synthesis of (-)-isopulegol hydrate and (−)-cubebaol. Org. Lett. 2006;8:2217–2220. doi: 10.1021/ol060486+. [DOI] [PubMed] [Google Scholar]
- Hong B.-C., Tseng H.-C., Chen S.-H. Synthesis of aromatic aldehydes by organocatalytic [4+2] and [3+3] cycloaddition of α, β-unsaturated aldehydes. Tetrahedron. 2007;63:2840–2850. [Google Scholar]
- Hulea V. Toward platform chemicals from bio-based ethylene: heterogeneous catalysts and processes. ACS Catal. 2018;8:3263–3279. [Google Scholar]
- Jiao F., Li J., Pan X., Xiao J., Li H., Ma H., Wei M., Pan Y., Zhou Z., Li M. Selective conversion of syngas to light olefins. Science. 2016;351:1065–1068. doi: 10.1126/science.aaf1835. [DOI] [PubMed] [Google Scholar]
- Kitamura M., Kawasaki F., Ogawa K., Nakanishi S., Tanaka H., Yamada K., Kunishima M. Role of linkers in tertiary amines that mediate or catalyze 1,3,5-triazine-based amide-forming reactions. J. Org. Chem. 2014;79:3709–3714. doi: 10.1021/jo500376m. [DOI] [PubMed] [Google Scholar]
- Li Z., Qu Y., Wang J., Li M., Miao S., Li C. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule. 2019;3:570–583. [Google Scholar]
- List B., Lerner R.A., Barbas C.F., III Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000;122:2395–2396. [Google Scholar]
- List B., Hoang L., Martin H.J. New mechanistic studies on the proline-catalyzed aldol reaction. PNAS. 2004;101:5839–5842. doi: 10.1073/pnas.0307979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Evans T.J., Cheng L., Nimlos M.R., Mukarakate C., Robichaud D.J., Assary R.S., Curtiss L.A. Catalytic upgrading of biomass-derived compounds via C−C coupling reactions: computational and experimental studies of acetaldehyde and furan reactions in HZSM-5. J. Phys. Chem. C. 2015;119:24025–24035. [Google Scholar]
- Liu T., Wu K., Wang L., Yu Z. Potassium tert-Butoxide-promoted acceptorless dehydrogenation of N-heterocycles. Adv. Synth. Catal. 2019;361:3958–3964. [Google Scholar]
- Lokesh N., Hioe J., Gramüller J., Gschwind M.R. Relaxation dispersion NMR to reveal fast dynamics in Brønsted acid catalysis: influence of sterics and h-bond strength on conformations and substrate hopping. J. Am. Chem. Soc. 2019;141:16398–16407. doi: 10.1021/jacs.9b07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusardi M., Struble T., Teixeira A.R., Jensen K.F. Identifying the roles of acid–base sites in formation pathways of tolualdehydes from acetaldehyde over MgO-based catalysts. Catal. Sci. Tech. 2020;10:536–548. [Google Scholar]
- Moteki T., Rowley A.T., Flaherty D.W. Self-terminated cascade reactions that produce methylbenzaldehydes from ethanol. ACS Catal. 2016;6:7278–7282. [Google Scholar]
- Moteki T., Rowley A.T., Bregante D.T., Flaherty D.W. Formation pathways toward 2- and 4-methylbenzaldehyde via sequential reactions from acetaldehyde over hydroxyapatite catalyst. ChemCatChem. 2017;9:1921–1929. [Google Scholar]
- Moyano A., Rios R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Chem. Rev. 2011;111:4703–4832. doi: 10.1021/cr100348t. [DOI] [PubMed] [Google Scholar]
- Mukaiyama T., Matsuo J.-I., Kitagawa H. A New and one-pot synthesis of α,β-unsaturated ketones by dehydrogenation of various ketones with N-tert-butyl phenylsulfinimidoyl chloride. Chem. Lett. 2000;29:1250–1251. [Google Scholar]
- Mukherjee S., Yang J.W., Hoffmann S., List Benjamin. Asymmetric enamine catalysis. Chem. Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- Northrup A.B., MacMillan D.W.C. The first general enantioselective catalytic Diels-Alder reaction with simple α, β-unsaturated ketones. J. Am. Chem. Soc. 2002;124:2458–2460. doi: 10.1021/ja017641u. [DOI] [PubMed] [Google Scholar]
- Schmid M.B., Zeitler K., Gschwind R.M. The elusive enamine intermediate in proline-catalyzed aldol reactions: NMR detection, formation pathway, and stabilization trends. Angew. Chem. Int. Ed. 2010;49:4997–5003. doi: 10.1002/anie.200906629. [DOI] [PubMed] [Google Scholar]
- Schnitzer T., Wennemers H. Deactivation of secondary amine catalysts via aldol reaction-amine catalysis under solvent-free conditions. J. Org. Chem. 2020;85:7633–7640. doi: 10.1021/acs.joc.0c00665. [DOI] [PubMed] [Google Scholar]
- Schoch, W., Kroener, M., Widder, R. (1982). Preparation of Aromatic Aldehydes by the Sommelet Reaction. US Patent, 4321412.
- Serna P., Corma A. Transforming nanometal nonselective particulates into chemoselective catalysts for hydrogenation of substituted nitrobenzenes. ACS Catal. 2015;5:7114–7121. [Google Scholar]
- Shi R., Wang F., Mu X., Ta N., Li Y., Huang X., Shen W. Transfer dehydrogenation of 1-octanol to 1-octanal over Cu/MgO catalyst: effect of Cu particle size. Chin. J. Catal. 2010;31:626–630. [Google Scholar]
- Song X., Zhang X., Zhang S., Li H., Wang W. Direct transformation of simple enals to 3,4-disubstituted benzaldehydes under mild reaction conditions via an organocatalytic regio- and chemoselective dimerization cascade. Chem. Eur. J. 2012;18:9770–9774. doi: 10.1002/chem.201201709. [DOI] [PubMed] [Google Scholar]
- Tang Z., Jiang F., Cui X., Gong L.-Z., Mi A.-Q., Jiang Y.-Z., Wu Y.-D. Enantioselective direct aldol reactions catalyzed by l-prolinamide derivatives. PNAS. 2004;101:5755–5760. doi: 10.1073/pnas.0307176101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J., Yu X., Wang Y., Yamamoto Y., Bao M. Interweaving visible-light and iron catalysis for nitrene formation and transformation with dioxazolones. Angew. Chem. Int. Ed. 2021;60:2–11. doi: 10.1002/anie.202016234. [DOI] [PubMed] [Google Scholar]
- Tempelman C.H.L., Zhu X., Hensen E.J.M. Activation of Mo/HZSM-5 for methane aromatization. Chin. J. Catal. 2015;36:829–837. [Google Scholar]
- Teskey C.J., Adler P., GonÅalves C.R., Maulide N. Chemoselective α,β-dehydrogenation of saturated amides. Angew. Chem. Int. Ed. 2019;58:447–451. doi: 10.1002/anie.201808794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.–N., Weng X.-F., Zhou B.-C., Lv S.-P., Miao S., Zhang D., Han Y., Scott S.L., Schüth F., Lu A.-H. Direct, selective production of aromatic alcohols from ethanol using a tailored bifunctional cobalt–hydroxyapatite catalyst. ACS Catal. 2019;9:7204–7216. [Google Scholar]
- Yu C., Zhao B., Zhao Y., Lin J. A new route for the synthesis of Ozagrel hydrochloride. Org. Prep. Proc. Int. 2010;42:183–185. [Google Scholar]
- Zhang B., Jiang Z., Zhou X., Lu S., Li J., Liu Y., Li C. The synthesis of chiral isotetronic acids with amphiphilic imidazole/pyrrolidine catalysts assembled in oil-in-water emulsion droplets. Angew. Chem. Int. Ed. 2012;124:13159–13162. doi: 10.1002/anie.201206438. [DOI] [PubMed] [Google Scholar]
- Zhang L., Pham T.N., Faria J., Santhanaraj D., Sooknoi T., Tan Q., Zhao Z., Resasco D.E. Synthesis of C4 and C8 chemicals from ethanol on MgO-incorporated faujasite catalysts with balanced confinement effects and basicity. ChemSusChem. 2016;9:736–748. doi: 10.1002/cssc.201501518. [DOI] [PubMed] [Google Scholar]
- Zhou B.-C., Wang Q.-N., Weng X.-F., He L., Li W.-C., Lu A.-H. Regulating aromatic alcohols distributions by cofeeding methanol with ethanol over cobalt-hydroxyapatite catalyst. ChemCatChem. 2020;12:2341–2347. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper dose not report original code.
-
•
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.