

Keywords: calcium, cancer, diabetes, genetics, metabolism
Abstract
In the mid-1980s, the identification of serine and threonine residues on nuclear and cytoplasmic proteins modified by a N-acetylglucosamine moiety (O-GlcNAc) via an O-linkage overturned the widely held assumption that glycosylation only occurred in the endoplasmic reticulum, Golgi apparatus, and secretory pathways. In contrast to traditional glycosylation, the O-GlcNAc modification does not lead to complex, branched glycan structures and is rapidly cycled on and off proteins by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), respectively. Since its discovery, O-GlcNAcylation has been shown to contribute to numerous cellular functions, including signaling, protein localization and stability, transcription, chromatin remodeling, mitochondrial function, and cell survival. Dysregulation in O-GlcNAc cycling has been implicated in the progression of a wide range of diseases, such as diabetes, diabetic complications, cancer, cardiovascular, and neurodegenerative diseases. This review will outline our current understanding of the processes involved in regulating O-GlcNAc turnover, the role of O-GlcNAcylation in regulating cellular physiology, and how dysregulation in O-GlcNAc cycling contributes to pathophysiological processes.
CLINICAL HIGHLIGHTS.
The modification of proteins by sugars is one of the most common posttranslational modifications of proteins. Such modifications were believed to occur only on extracellular and secreted proteins and to consist of large branching structures comprising different sugar molecules. In the mid-1980s, a new modification was identified which consisted of a single N-acetylglucosamine moiety (O-GlcNAc) attached to serine and threonine residues of nuclear and cytoplasmic proteins. Since its discovery, O-GlcNAc modification of proteins has been shown to affect numerous cellular functions, and changes in O-GlcNAc levels have been implicated in a wide variety of diseases. The goal of this review is to summarize our current knowledge of O-GlcNAc biology and its contribution to normal physiology and disease.
1. INTRODUCTION
1.1. Brief History of O-GlcNAc
The modification of proteins by carbohydrates, otherwise known as protein glycosylation, is the most common posttranslational modification of proteins and occurs in all cells and organisms (1). In the early 1900s, there was considerable speculation that carbohydrates were important parts of the structure of proteins, but the technology was lacking to provide definitive evidence. It was not until the 1960s and 1970s that a better understanding of the structure and function of these complex glycans on proteins started to emerge. Through the mid-1980s, the consensus was that protein glycosylation was restricted to extracellular proteins that originated in the endoplasmic reticulum (ER), Golgi apparatus, and secretory pathway (1). However, in 1984, Torres and Hart (2) designed a study to characterize terminal N-acetylglucosamine (GlcNAc) residues on the surface of lymphocytes. Unexpectedly, they demonstrated that the majority of these terminal residues were localized inside the cell, and that rather than part of extended glycan structures, they existed as a single O-linked GlcNAc monosaccharide.
Two years later, Holt and Hart (3) characterized the cellular distribution of O-GlcNAc-modified proteins in rat liver cells, demonstrating that while they were found in nearly every cellular compartment, they were particularly enriched in the cytoplasm and nucleus. In 1987, a monoclonal antibody to rat liver nuclear pore complex (clone RL2), appeared to primarily recognize O-linked O-GlcNAc groups (4). Hanover et al. (5) also reported nuclear pore proteins were modified by O-GlcNAc; however, the functional consequence of this modification was unclear at that time. During the same period, Holt et al. (6) identified both serine (Ser) and threonine (Thr) residues as the primary residues modified by O-GlcNAc. The fact that O-GlcNAcylated proteins were especially enriched in the nucleus raised the possibility that the modification could be involved in protein transport into the nucleus; however, the observation that cytoskeletal proteins in erythrocytes, which lack a nucleus, were modified by O-GlcNAc suggested other functions for this modification (7). In 1992, the protein responsible for adding O-GlcNAc to proteins, O-GlcNAc transferase (OGT) was purified (8), but it was not until 1997 that the gene encoding OGT was identified, revealing a glycosyltransferase that was unrelated to any other previously known glycosyltransferases (9).
In contrast to traditional protein glycosylation, it was quickly established that O-GlcNAc modifications occurred rapidly and reversibly (10), suggesting the existence of an N-acetyl-glucosaminidase(s) responsible for its removal from proteins. Dong et al. (11) purified an O-GlcNAcase that was distinctly different from lysosomal hexosaminidases, in that it was localized in the cytosol and was optimally active at a neutral, rather than acidic, pH. In 2001, O-GlcNAcase was cloned and recognized to be identical to a previously known hexosaminidase C of unknown function, which specifically cleaved O-GlcNAc but not O-linked N-acetylgalactosamine (O-GalNAc) from glycopeptides (12). O-GlcNAcase was subsequently shown to have an identical sequence to a previously identified protein from meningioma patients called meningioma expressed antigen 5 (MGEA5) (13).
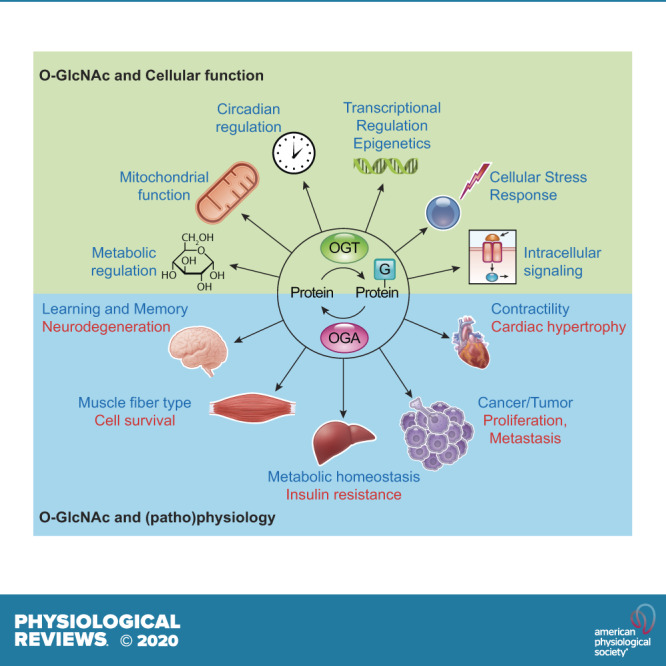
Over the decades since its discovery, O-GlcNAc modified proteins have been identified in all metazoans, some bacteria, protozoa, and viruses, but to date, not in yeast (14). Recent studies have suggested that in yeast, the addition of O-linked mannose (O-Man) on nuclear and cytoplasmic proteins might play a similar role to O-GlcNAc (15). In support for the necessity of O-GlcNAc signaling in mammals, germline deletion of OGT in mice has been shown to be embryonically lethal (16), and deletion of OGA results in perinatal mortality in mice (17). Furthermore, mammalian OGT and OGA are ubiquitously expressed, and proteins of every functional class have been shown to be subject to O-GlcNAcylation (FIGURE 1A) (18). Since its discovery, O-GlcNAcylation has been shown to contribute to numerous cellular functions, including signaling, protein localization and stability, transcription, chromatin remodeling, mitochondrial function, and cell survival (FIGURE 1B) (14). Given its diverse roles, it is not surprising that dysregulation in O-GlcNAcylation has been implicated in a wide range of pathophysiological processes, such as diabetes, diabetic complications, cancer, cardiovascular, and neurodegenerative diseases (19–24).
FIGURE 1.

A: O-linked N-acetylglucosamine acylated (O-GlcNAcylated) proteins belong to many different classes of proteins responsible for regulating diverse cellular processes. Some of the largest classes of proteins include those in regulating metabolism, transcription, and translation, as well as structural proteins. B: O-GlcNAcylated proteins are present in numerous cellular compartments, including the nucleus, cytosol, and mitochondria. Cytosolic domains of membrane proteins are also O-GlcNAcylated, as well as proteins involved in autophagy and proteosomal degradation of proteins, chaperone proteins, vesicle proteins, and numerous cytosolic proteins and enzymes. This figure is based, in part, on information presented in Chapter 19, The O-GlcNAc Modification, Essentials in Glycobiology, 3rd ed. (14). ER, endoplasmic reticulum.
1.2. Differences between O-GlcNAc and Traditional Glycosylation
Until the paradigm-changing study by Torres and Hart (2), protein glycosylation was thought to be limited to extracellular and excreted proteins. These proteins are processed via the ER-Golgi pathway, which contains a large number of glycosyltransferases that are responsible for creating N- and O-linked glycan structures. N-glycans are attached to proteins as asparagine residues via an N-glycosidic bond; whereas, O-glycans are attached to Ser or Thr residues. These proteins are subject to processing and maturation by numerous glycosyltransferases, leading to stable elongated and branched structures comprising a number of different monosaccharides. Glycosyltransferases are estimated to account for at least 2% of the human genome (25). The importance of the tight regulation of this process is highlighted by the fact that mutations in genes related to glycosylation are associated with more than 100 human genetic diseases, which are frequently associated with intellectual disabilities, as well as abnormalities in most organ systems (25).
Key distinguishing features of the O-GlcNAc modification are that 1) with few exceptions, it occurs primarily on nuclear and cytoplasmic proteins; 2) it consists of a single monosaccharide; 3) it is dynamic and rapidly reversible; 4) it is catalyzed by a single unique O-GlcNAc transferase, and 5) it is removed by a glycohydrolase that is specific for the removal of O-GlcNAc. It is of note that only very recently have mutations in the OGT gene been linked to human disease, such as intellectual disability (26–29). How mutations of OGT lead to disease and what specific functions are perturbed remain to be determined. In light of the growing recognition of the importance of O-GlcNAc-modified proteins in regulating cellular homeostasis—and that it is likely as abundant a modification as phosphorylation—it may seem surprising that it had not been identified earlier. One reason for this is that proteins modified by O-GlcNAc do not exhibit changes in mobility during gel electrophoresis, because of their low molecular weight and because unlike O-phosphate, it is uncharged. Moreover, despite its abundance, it exhibits low stoichiometry, estimated to be 5–10% of a specific site. In addition, the presence of hexosaminidases can remove O-GlcNAc from proteins unless they are specifically inhibited.
1.3. Identification of O-GlcNAcylated Proteins
The most widely used approach to detect O-GlcNAcylated proteins are pan-specific O-GlcNAc antibodies, the most commonly being RL2 and CTD110.6, but there are also a number of other commercially available O-GlcNAc antibodies as listed in TABLE 1 These can be used to provide a semiquantitative assessment of overall changes in O-GlcNAc levels via immunoblot or distribution of O-GlcNAc via immunohistochemistry in tissues and cells (FIGURE 2). The limitations of these antibodies include the fact that they have epitope specificity, and their selectivity for high- versus low-abundance proteins. Furthermore, a continuing limitation in the study of O-GlcNAcylated proteins is the lack of commercially available site-specific O-GlcNAc antibodies, although several have been developed for specific studies (79–82). Because of the lack of site-specific antibodies to determine whether a protein of interest is an O-GlcNAc target, the simplest approach is to immunoprecipitate the protein of interest followed by an O-GlcNAc immunoblot; however, because of differences in epitope specificities, a negative result with a single antibody does not preclude the possibility that a protein is modified. Moreover, because of the possibility of cross-reactivity with other sugars, if a positive result is obtained, a number of additional control experiments should be considered, such as preincubation with free GlcNAc to outcompete the antibody. Additionally, their cross-reactivity with N-linked modifications varies by detection conditions (83). A number of methodological reviews on different approaches for studying O-GlcNAcylated proteins have been published that provide valuable technical details (44,45, 84,85).
Table 1.
Tools for use in studying O-GlcNAc levels in cells and tissues
| Antibodies* | Characteristics and Limitations | Commercial Availability |
|---|---|---|
| CTD110.6 (IgM) (30) | Monoclonal antibody raised against RNA polymerase II subunit 1C terminal domain. Reportedly less dependent on protein structure than other antibodies, thereby, recognizing more proteins. Relatively low-binding affinity, therefore, biased toward more highly abundant proteins. | Multiple sources |
| HGAC39 (IgG) (31–33) | Monoclonal antibody raised against streptococcal group A carbohydrate (GAC) demonstrated to recognize O-GlcNAcylated proteins. | No |
| HGAC85 (IgG) (33) | Monoclonal antibody raised against streptococcal group A carbohydrate (GAC) demonstrated to recognize O-GlcNAcylated proteins. Not as widely used as CTD110.6 or RL2. Some of the most recent studies have used it in ChIP-chip assays. | Multiple sources |
| MY95 (IgG) (34,35) | Originally generated as an antinuclear pore complex antibody, subsequently shown to recognize O-GlcNAc modification. | No |
| RL2 (IgG) (4) | Because of epitope specificity, having been raised against nuclear pore protein, it recognizes only a subset of O-GlcNAc proteins. Relatively low binding affinity; therefore, it is biased toward more highly abundant proteins. This is the case for all pan-O-GlcNAc antibodies. | Multiple sources |
| 1F5.D6(14) (IgG) (36) | Appears to recognize a wider range of O-GlcNAcylated proteins, including those below 50 kDa that are not usually identified by RL2 or CTD110.6. | Millipore |
| 9D1.E4(10) (IgG) (36) | Appears to recognize a wider range of O-GlcNAcylated proteins, including those below 50 kDa, which are not usually identified by RL2 or CTD110.6. | Millipore |
| 18B10.C7(3) (IgG) (36) | Appears to recognize a wider range of O-GlcNAcylated proteins, including those below 50 kDa, which are not usually identified by RL2 or CTD110.6. | Millipore |
| Other Identification Methods | Characteristics and Limitations | Commercial Availability |
|---|---|---|
| Agrocybe aegerita GlcNAc-specific lectin (AANL) (37) | AANL, also reported as AAL2, is a useful tool for enrichment and identification of O-GlcNAcylated proteins and peptides. | Not known |
| Click-IT O-GlcNAc Enzymatic Labeling System (38–40) | Chemoenzymatic labeling of proteins resulting in O-GlcNAc residues being replaced by azido-modified galactose (GalNAz), followed by chemoselective ligation azide. Resulting proteins can be detected via Western blot using a using a variety of alkyne-modified chemical probes. This approach is not epitope specific, which has advantages over pan-O-GlcNAc antibodies; however, more sample processing steps are required. | Multiple sources |
| Galactosyl transferase/ [3H]-galactose (41) | Results in incorporation of 3H into terminal GlcNAc residues on proteins allowing for detection by autoradiography. Can be very time intensive because of low sensitivity of 3H. | Multiple sources |
| Wheat germ agglutinin (WGA) and succinylated WGA (sWGA) (41) | Both WGA and sWGA can be used for immunoblotting. WGA identifies all terminal GlcNAc residues, as well as sialic acid. sWGA reduces affinity for sialic acid. Lack of specific for O-GlcNAc requires careful interpretation. | Multiple sources |
| Enrichment Strategies | Characteristics and Limitations | Commercial Availability |
|---|---|---|
| β-elimination followed by Michael addition of dithiothreitol (BEMAD) (42) | BEMAD relies on the β-elimination of phosphate or O-GlcNAc under basic conditions followed by Michael addition using dithiothreitol or a biotin-thiol probe. | |
| Chemi-enzymatic labeling (43) | Using the same approach described for BEMAD, it replaces O-GlcNAc with GalNAz, combined with a biotin- or streptavidin-cleavable linker can be used to enrich O-GlcNAcylated peptides. A range of linkers have been developed that have different properties. | |
| Immunoprecipitation (36, 44) | The most widely used antibodies, RL2 and CTD110.6, do not perform well for immunopurification. Although not as widely used, the Millipore antibodies listed above have been reported to be effective for IP. | |
| WGA-agarose (45) | Because of the lack of specificity for O-GlcNAc, other glycoproteins will be copurified. This can be minimized by pretreatment to remove the N- and O-linked glycans. | Multiple sources |
| GFAT Inhibitors | Characteristics and Limitations | Commercial Availability |
|---|---|---|
| Azaserine (46) | Glutamine analog used to decrease HBP flux by inhibiting GFAT; however, it lacks specificity as it can inhibit other pathways that utilize glutamine (47). In the absence of any other small molecule GFAT inhibitors, it continues to be widely used, but interpretation of results needs to be cautious. | Multiple sources |
| 6-diazo-5-oxo-L-norleucine (DON) (46) | Glutamine analog used to decrease HBP flux by inhibiting GFAT; however, it lacks specificity as it can inhibit other pathways that utilize glutamine (48). In the absence of any other small molecule GFAT inhibitors, it continues to be widely used, but interpretation of results needs to be cautious. | Multiple sources |
| OGA Inhibitors | Characteristics and Limitations | Commercial Availability |
|---|---|---|
| α-GlcNAc thiolsulfonate (49) | OGA inhibitor exhibiting selectivity of short OGA compared to long OGA. | No |
| Gluco-nagstatin (50,51) | Based on natural product, nagstatin, which is a potent inhibitor of β-hexosaminidase. Inhibits OGA but is more effective for of β-hexosaminidase. | No |
| GlcNAcstatins (50, 52,53) | Family of OGA inhibitors, with high degree of potency and selectivity. | Abmole Bioscience, Inc. |
| NAG-thiazoline (50, 54) | Inhibitor of OGA with much greater selectivity over other hexosaminidases than PUGNAc. | No |
| NButGT (50, 55,56) | OGA inhibitor based on NAG-thiazoline scaffold. Less potent that NAG-thiazoline but even more selective over other hexosaminidases. | No |
| PUGNAc (57) | Widely used cell permeable OGA inhibitor, but also inhibits hexosaminidase A and B inhibitor. Limited aqueous solubility; usually dissolved in DMSO prior to dilution in aqueous solutions for biological studies. | Multiple sources |
| Streptozotocin (50, 54, 58,59) | A GlcNAc analog initially thought to inhibit OGA. Has major off target effects and more recent studies have reported that it does not inhibit OGA. Not recommended for use. | Multiple sources |
| Thiamet-G (50, 60) | Derivative of NButGT, with greater stability, water soluble, and orally available. | Multiple sources |
| OGT Inhibitors | Characteristics and Limitations | Commercial Availability |
|---|---|---|
| Alloxan (61,62) | A weak OGT inhibitor with numerous off target effects. Requires millimolar concentrations to lower cellular O-GlcNAc levels. Not recommended for use. | Multiple sources |
| Benzyl-2-acetamido-2-deoxy-α-d-galactopyranoside (BADGP)(63,64) | Although BADGP decreases O-GlcNAc levels in millimolar range it lacks specificity for OGT. Inhibits other O-Glycosyltransferases. Not recommended for use. | Sigma |
| L01 (63) | Effective in reducing cellular O-GlcNAc levels in 10–100-µM range. | No |
| OSMI-1 (65–68) | Reduces O-GlcNAc levels in cells at ∼50 µM. Shown to be specific for OGT compared with other glycosyltranserases. | Sigma |
| OSMI-3, OSMI-4 (69) | On the basis of OSMI-1 scaffold, but with greater potency. Reduces cellular O-GlcNAc levels in ∼5-µM range. | ProbeChem |
| ST045849 (TT04) (63, 66, 70–73) | Identified via high throughput screen. Specificity for OGT compared to other glycosyltranserases unclear. | TimTech |
| ST060266 (70, 74) | Identified via high-throughput screen. Specificity for OGT compared to other glycosyltranserases unclear. | TimTech |
| ST078925 (70, 72, 75) | Identified via high-throughput screen. Specificity for OGT compared to other glycosyltranserases unclear. | TimTech |
| UDP-5SGlcNAc; (61, 76) | Effective in vitro inhibitor of OGT but lacks cell permeability. | No |
| 2‐deoxy‐2‐N‐hexanamide‐5‐thio‐d‐glucopyranoside (5SGlcNHex); (77) | Shown to be effective in lowering tissue O-GlcNAc levels following in vivo administration. | No |
| 4-methoxyphenyl 6-acetyl-2-oxo-2,3-dihydro-1,3-benzoxazole-3-carboxylate (78) | A cell permeable, irreversible inhibitor of OGT. | No |
| 5-thioglucosamine (5SGlcNAc) (76) | The peracetylated form of 5SGlcNAc crosses the cell membrane and converts to UDP-5SGlcNAc, which binds to the active site of OGT. Reduces O-GlcNAc levels in cells in the 10-100-µM range. | No |
AAL2, Agrocybe aegerita GlcNAc-specific lectin; CTD, COOH-terminal domain; GFAT, d-fructose-6-phosphate amidotransferase; GlcNAc, N-acetylglucosamine moiety; HBP, hexosamine biosynthesis pathway; IP, immunoprecipitation; NAG, N-acetylglucosamine; O-GlcNAc, O-linked N-acetylglucosamine; OGT, O-GlcNAc transferase; RL2, rat liver nuclear pore complex; UDP, uridine diphosphate-azido-modified galactose.
FIGURE 2.
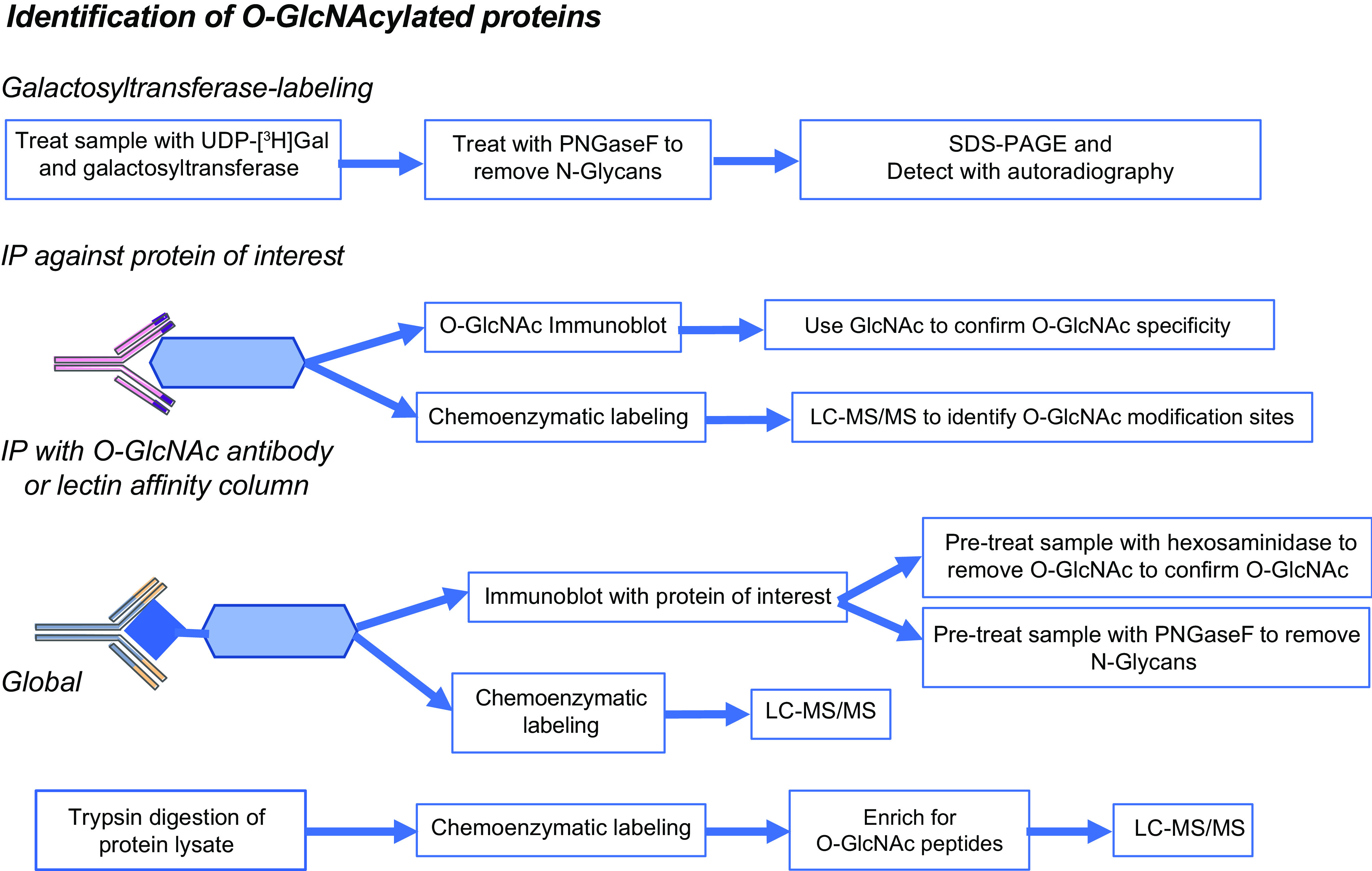
Overview of different approaches for the identification of O-linked N-acetylglucosamine acylated (O-GlcNAcylated) proteins, including galactosyltransferase labeling; immunopurification and chemoenzymatic labeling were combined with LC-MS/MS. Further details are found in TABLE 1. IP, immunoprecipitation; UDP, uridine diphosphate-azido-modified galactose.
Another challenge in the identification and characterization of O-GlcNAcylated proteins is that O-GlcNAc-modified peptides are frequently not detected using traditional collision-induced dissociation mass spectrometry (MS), as O-GlcNAc is very labile and is usually lost during collision-induced fragmentation. The development of electron transfer dissociation (ETD) MS techniques (86,87) significantly enhanced the identification of O-GlcNAc modification sites, since ETD does not usually result in the loss of the O-GlcNAc moiety from the peptide; however, the problems of ion suppression and low stoichiometry remain. To overcome these limitations, it is necessary to enrich the sample for O-GlcNAcylated peptides (88). One approach is to use a traditional immunoprecipitation with a single anti-O-GlcNAc antibody, as described in several studies (36, 89). On the other hand, because of the limitations of epitope specificity, there is a concern that only a subset of O-GlcNAc proteins will be identified. One way to overcome that limitation is to use a combination of antibodies; for example, Lee et al. (90) developed a G5-lectibody resin column that consisted of four different O-GlcNAc antibodies and the lectin wheat germ agglutinin (WGA) (FIGURE 2). Another enrichment approach is chemoenzymatic labeling (38, 44, 91), which involves using uridine diphosphate-azido-modified galactose (UDP-GalNAz) and a mutant galactosyltransferase to tag O-GlcNAc moieties with a reactive azide group. This is followed by the addition of a biotin group attached to a cleavable linker, using a copper-based azide-alkene cycloaddition (84). The resulting modified proteins or peptides can then be affinity purified using an avidin column. A number of different linkers have been described, including a UV-cleavable linker (92,93) and, more recently, a hydrazine-sensitive linker (94). A key advantage of the latter approach is the resulting tag at the O-GlcNAc site allows for more efficient fragmentation. There are several different versions of this technique, and refinements continue to be developed focused on improving the efficiency of the release of proteins/peptides from the avidin column, as well as simplifying the underlying chemistry (43, 95,96).
Compared with most other PTMs, our understanding of how O-GlcNAcylation regulates protein function and cellular physiology remains limited, although our knowledge is rapidly growing. The development of increasingly selective and specific small-molecule inhibitors of OGT and OGA, as summarized in TABLE 1, have helped advance our knowledge of the role of O-GlcNAc in cellular function. These new tools have helped stimulate research in O-GlcNAc biology, as illustrated in FIGURE 3. About 20 years ago, there were less than 20 articles published per year, but this has been steadily increasing, and in 2018, it reached ∼225 (or more than the first 20 years combined). As this becomes a more widely recognized area of biology, it is likely to grow more rapidly, given the increasing appreciation of its importance in regulating key physiological processes combined with its contributions to the development of diverse pathologies. Several thousands of proteins have been identified as O-GlcNAc targets (44), and the number continues to increase, as new techniques are developed. Many new O-GlcNAc sites are identified via high-throughput MS; consequently, the biological function of the modification is often not known. In TABLE 2, we provide a list of some key proteins, divided via cellular location or function, which are validated O-GlcNAc targets in which the specific modification site has been identified. In TABLE 3, the physiological effects of gain or loss of function of OGT and OGA in mammals are summarized.
FIGURE 3.
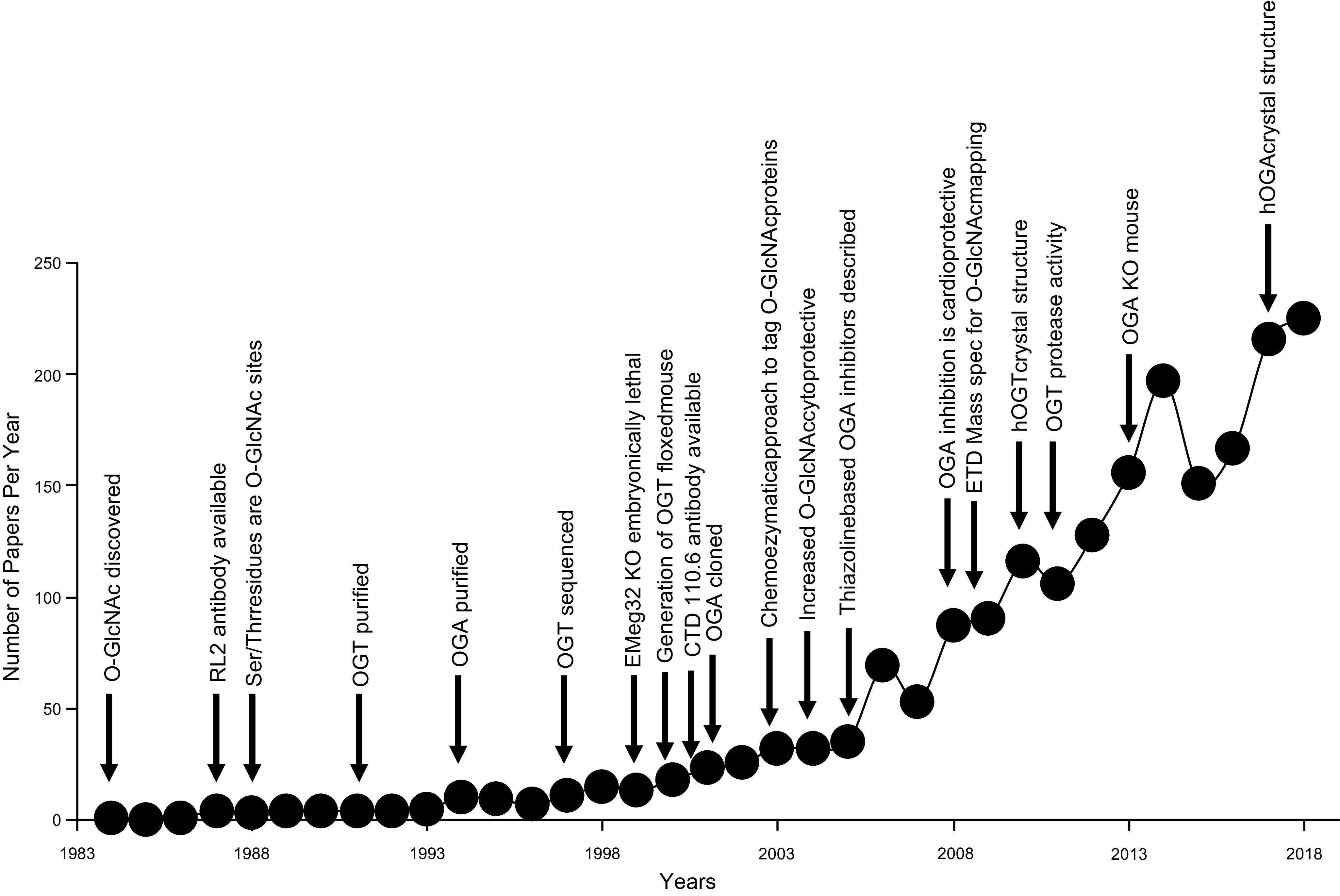
Number of O-linked N-acetylglucosamine acylated (O-GlcNAc) publications by year annotated by key events in O-GlcNAc biology from its initial discovery. Relevant citations are all included in the main text. CTD, COOH-terminal domain; ETD, electron dissociation transfer hOGA, human O-GlcNAcase; hOGT, human O-GlcNAc transferase; KO, knockout; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; RL2, rat liver nuclear pore complex.
Table 2.
List of selected O-GlcNAcylated proteins and their modification sites
| Transcription Factors and Transcriptional Regulators | ||||
|---|---|---|---|---|
| Protein Symbol | O-GlcNAc Modification Sites* | Effects on Protein Function | Citations | |
| C/EBPβ | Ser-180, Ser-181 | Regulates both the phosphorylation and DNA binding activity of C/EBPβ. | (97) | |
| cMyc | Thr-58 | Reduces phosphorylation of Ser-62 and Thr-58 and increases protein stability. | (98,99) | |
| CREB | Ser-40 | Represses both basal and activity-dependent transcription. | (100) | |
| ERα | Ser-10, Thr-50, Thr-575 | May regulate protein turnover. | (101,102) | |
| ERβ | Ser-16 | May regulate protein turnover. | (103) | |
| ERRγ | Ser-317, Ser-319 | Enhances receptor activity. | (104) | |
| FOXO1 | Thr-317, Ser-550, Thr-648, Ser-654 | Increases target gene transcription. | (105) | |
| FXR | Ser-62 | Enhances FXR gene expression and protein stability. | (106) | |
| KEAP1 | Ser-104 | O-GlcNAc is required for the efficient ubiquitination and degradation of Nrf2. | (107) | |
| Sp1 | Ser-491, Ser-612, Thr-640, Ser-641, Ser-698, Ser-702 | Inhibits transcriptional activation. | (108,109) | |
| LXRα/β | Ser-49 | Increases transactivation. | (110–112) | |
| Oct4 | Thr-116, Thr-225, Ser-236, Ser-288/889/890, Ser-335, Ser-349, Thr-351, Thr-352, Ser-355, Ser-359 | O-GlcNAc increases transcriptional activity. | (113) | |
| P27 | Ser-2 | Suppresses protein interactions. | (114) | |
| p53 | Ser-139 | Reduces phosphorylation and stabilizes protein. | (115) | |
| Per2 | Ser-566, Ser-580, Ser-653, Ser-662, Ser-668, Ser-671, Thr-734, Thr-965, Ser-983, Thr-1180 | O-GlcNAc increases its suppressor activity. | (116) | |
| PGC-1α | Ser-334, Ser-333 | Enhances stability and upregulated downstream genes. | (117,118) | |
| PPARγ | Thr-54 | Reduces its transcriptional activity. | (119) | |
| SIRT1 | Ser-549 | O-GlcNAcylation increases deacetylase activity, promotes cytoprotection under stress. | (120) | |
|
Insulin Signaling and Other Metabolic Proteins
| ||||
|---|---|---|---|---|
| Protein Symbol | O-GlcNAc Modification Sites* | Effects on Protein Function | Citations | |
| Akt1/2 | Ser-126, Ser-129, Thr-305, Thr-308, Thr-312, Ser-473 | Decreases phosphorylation and activity. | (121–126) | |
| CRTC2 | Ser-70, Ser-171 | Increased O-GlcNAc leads to nuclear translocation and promotes gluconeogenesis. | (127) | |
| GAPDH | Thr-227 | Increases nuclear translocation. | (128) | |
| GFAT1 | Ser-243 | AMPK phosphorylation of Ser-243 reduced GFAT activity. | (129,130) | |
| GSK3β | Ser-9, Thr-38, Thr-39, Thr-43 | Decreased activity. | (125, 131–133) | |
| G6PDH | Ser-84 | Increases activity. | (134) | |
| IRS1 | Ser-914, Ser-1009, Ser-1036, Ser-1041 | Attenuates insulin-mediated phosphorylation of IRS1. | (135,136) | |
| PDH (E1) | Ser-13, Ser-15, Ser-134, Ser-232 | Higher O-GlcNAc levels associated with greater activity. | (137,138) | |
| PDH (E2) | Ser-7, Ser-15, Ser-239, Ser-411 | Remains to be determined. | (137–139) | |
| PDK2 | Ser-110 | Remains to be determined. | (138) | |
| PFK1 | Ser-529 | O-GlcNAc levels increased in hypoxia and decreases activity. | (140) | |
| PKM2 | Thr-405, Ser-406 | Reduces activity and increases nuclear translocation. | (141) | |
|
Contractile and Cytoskeletal Proteins
| ||||
|---|---|---|---|---|
| Protein Symbol | O-GlcNAc Modification Sites* | Effects on Protein Function | Citations | |
| αB-crystallin | Thr-170 | Regulates stress induced translocation. | (142,143) | |
| β-Actin (Skeletal Muscle) | Ser-52, Ser-155, Ser-199, Ser-232, Ser-323, Ser-368 | O-GlcNAcylation of Ser-199 may regulate elongation of actin filaments. | (144–147) | |
| Keratin18 | Ser-29, Ser-30, Ser-48 | Increased O-GlcNAc promotes cell survival by activating Akt. | (148,149) | |
| Myosin heavy chain(Skeletal Muscle) | Ser-1097, Ser-1299, Ser-1708, Ser-1920 | Reduced myosin Ca2+ sensitivity. | (145,146) | |
| Myosin heavy 6 (Cardiac) | Thr-35, Thr-60, Ser-172, Ser-179, Ser-196 Ser-392, Ser-622, Ser-626, Ser-644, Ser-645, Ser-749. Ser-880, Ser-1038, Ser-1148, Ser-1189, Ser-1200, Ser-1308, Ser-1336, Ser-1470, Ser-1597, Thr-1600, Thr-1606, Ser-1638, Thr-1697, Ser-1711, Ser-1777, Ser-1838, Ser-1916 | Reduced myosin Ca2+ sensitivity. | (147, 150) | |
| MYL1 | Ser-45, Thr-93, Thr-164 | Remains to be determined. | (147, 150) | |
| MYL2 | Ser-15 | Remains to be determined. | (147) | |
| Synapsin I | Ser-55, Thr-56, Thr-87, Ser-436, Ser-516, Thr-524, Thr-562, and Ser-576 | Regulation of synaptogenesis.O-GlcNAc of Thr-87 regulates localization of synapsin I. | (151,152) | |
| Tau | Ser-208, Ser-238, Ser-400, Ser-692 | Loss of O-GlcNAc induces hyperphosphorylation. | (153–155) | |
| TnI | Ser-150 | Remains to be determined. | (147) | |
| TnT | Ser-190 | Increased O-GlcNAc reduces Ser208 phosphorylation. | (156) | |
| Tropomyosin α1 | Ser-87, Ser-123, Ser-186, Ser-206 | Remains to be determined. | (150) | |
| Oxidative Phosphorylation and Other Mitochondrial Proteins | ||||
|---|---|---|---|---|
| Protein Symbol | O-GlcNAc Modification Sites* | Effects on Protein Function | Citations | |
| ATP5A | Ser-76, Thr-432 | Decreased activity. | (138, 157,158) | |
| ATP5B | Ser-106, Ser-128 | Decreased activity. | (138) | |
| DRP1 | Thr-585, Thr-586 | Associated with increased mitochondrial fragmentation and decreased mitochondrial membrane potential. | (159) | |
| Milton | Ser-447, Ser-829, Ser-830, Ser-938 | Attenuates mitochondrial motility. | (160) | |
| NDUFA9 | Ser-156, Ser-230 | Impaired complex activity. | (138, 161) | |
| Prohibitin | Ser-121 | Decreases phosphorylation. | (138,139, 162) | |
| VDAC1 | Thr-2, Ser-240, Ser-260 | Increased O-GlcNAc attenuates mitochondrial Ca2+ uptake. | (138, 163) | |
| VDAC2 | Ser-2 | Increased O-GlcNAc contributes to mitochondrial dysfunction and apoptosis. | (139, 164) | |
|
Kinases, Phosphatases, and Other Signaling Molecules
| ||||
|---|---|---|---|---|
| Protein Symbol | O-GlcNAc Modification Sites* | Effects on Protein Function | Citations | |
| CaMKII | Ser-279, Ser-280 | Activates enzyme.Increases NOX2 ROS production | (165)(166) | |
| CaMKIV | Thr-57, Ser-58, Ser-137, Ser-189, Ser-344, Ser-345, Ser-356 | Activates enzyme. | (167) | |
| CDK5 | Ser-46, Thr-245, Thr-246, and Ser-247 | Decreases activity. | (168) | |
| CK2α | Ser-347 | Attenuates interaction with Pin1 and facilitates proteasomal degradation.Reduces CKII phosphorylation decreasing its stability. May also affect substrate selectivity. | (131, 169,170) | |
| GRASP55 | Ser-389, Ser-390,Thr-403, Thr-404,Thr-413 | O-GlcNAcylation attenuates autophagy. | (171) | |
| IKKβ | Ser-733 | Increases activity. | (172) | |
| PKC-α, β,δ, ε, θ, ζ | Numerous (see citations for details) | Function not well defined and could be isoform specific. In some cases, likely competes with phosphorylation and decreases enzyme activity. | (173–175) | |
| PTP1B | Ser-104, Ser-201, Ser-386 | Lower O-GlcNAc levels leads to lower phosphatase activity. | (176) | |
| RACK1 | Ser-122 | Promotes stability and interaction with PKCβII. | (177,178) | |
| RIPK3 | Thr-467 | Prevents dimerization and limits inflammation. | (179) | |
| SNAP29 | Ser-2, Ser-61, Thr-130, Ser-153 | O-GlcNAcylation attenuates autophagy. | (180) | |
| ULK1,2 | Thr-613, Thr-635, Thr-726, Thr-754, | O-GlcNAc increases AMPK-mediated phosphorylation of ULK, leading to increased activity. | (92, 181,182) | |
| YAP1 | Ser-109, Thr-241 | Ser-109 O-GlcNAc Disrupts interaction with upstream kinase LATS1. Thr-241 O-GlcNAc increases activity by improving stability. | (183,184) | |
| Miscellaneous | ||||
| Protein Symbol | O-GlcNAc Modification Sites | Effects on Protein Function | Citations | |
| α-synuclein | Thr-64, Thr-72, Thr-75, Thr-81, Ser-87 | Increased O-GlcNAcylation associated with decrease in aggregation; however, this has primarily been identified in in vitro studies. | (185) | |
| β-catenin | Ser-23, Thr-40, Thr-41, Thr-112 | Promotes its recruitment to the plasma membrane and its binding to E-cadherin and regulates stability. | (186,187) | |
| CREB-binding protein | Ser-147, Ser-2360 | Remains to be determined. | (188) | |
| eIF4A | Ser-322, Ser-323 | Regulates formation of translation initiation complex. | (189) | |
| eNOS | Ser-1177 | Impairs activity. | (190) | |
| FNIP1 | Ser-938 | Attenuates phosphorylation and increases proteasomal degradation. | (191) | |
| HCF-1 | Thr-490, Ser-569, Ser-620, Ser-622, Ser-623, Thr-625, Ser-685, Thr-726, Thr-779 Thr-801, Thr-861, Thr-1143, Thr-1273, Thr-1335, Thr-1743, Thr-1238, Thr-1241 | O-GlcNAcylation is a signal for its proteolytic processing | (188, 192) | |
| HSP90 | Ser-434, Ser-452, Ser-461 | Remains to be determined | (193) | |
| PLB | Ser-16 | Inhibits phosphorylation. | (194) | |
| TAB1 | Ser-395 | Required for full activation of TAK1 | (81) | |
| TAB2 | Ser-166, Ser-350, Ser-354, Thr-456 | May control the activity of IL-1 signaling pathway | (95, 188) | |
| TAB3 | Ser-408 | Mediates cell migration via activation of NF-κB, | (195) | |
The proteins included in this table were selected primarily on the basis of their inclusion within the main body of the article. In addition to the citations listed in Table 2, other resources that were used included PhosphoSite Plus, www.phosphosite.org (196), Dias et al. (197), Ma et al. (138,139). CKII, casein kinase II; FXR, farnesoid X receptor; GFAT, l-glutamine: d-fructose-6-phosphate amidotransferase; IRS1, insulin receptor substrate 1; LATS1, large tumor suppressor kinase 1; NOX2, NADPH phagocyte oxidase isoform; Nrf2, nuclear factor E2–related factor-2; ROS, reactive oxygen species; O-GlcNAc, O-linked N-acetylglucosamine; TAK1, transforming growth factor-β activated kinase 1; ULK, Unc-51-like autophagy activating kinase 1.
Table 3.
Physiological roles of OGT and OGA in mammals
| Genotype | Phenotype | Citations |
|---|---|---|
| Human | ||
| Ogt human mutation L254F or E1974H | Intellectual disability. | (26,27, 29, 198) |
| Rat | ||
| Oga Goto-Kakizaki (GK) rat mutation | Diabetes. | (199) |
| Systemic inhibition of OGA by thiamet G in wildtype rat | Impaired novel place recognition. | (200) |
| Mouse | ||
| AgRP-cre::Ogtf/f | Ogt ablation in AgRP neurons. Obese due to lack of browning of white fat. | (201) |
| CaMKII-cre::Ogtf/f | Constitutive Ogt ablation in forebrain neurons. Loss of body and brain weight and significant neurodegeneration almost as early as birth, decreased lifespan. | (202) |
| CaMKII-creER::Ogtf/f | Inducible Ogt ablation in forebrain neurons. Obese due to overeating after acute deletion of Ogt. | (203) |
| γ-F-crystallin-rtTA:: dnOGA | OGA overexpression in the eye. Premature cataracts. | (204) |
| MCK-rtTA::dnOGA | OGA overexpression in the skeletal muscle. Skeletal muscle atrophy. | (205) |
| MMTV-cre::Ogaf/f | Constitutive Oga ablation in mammary gland. Obesity, in females. | (17) |
| MHC-cre::Ogtf/f | Constitutive Ogt ablation in cardiomyocytes. Postnatal lethality, dilated hearts, and signs of heart failure. | (206) |
| Oga -/- | Germline knockout. Perinatal lethality. | (17, 207) |
| Oga +/- | Germline heterozygote. Lean not due to overeating or higher energy expenditure, associated with higher RER. | (207) |
| Ogt -/- | Germline knockout. Embryonic stem cell lethality. | (16, 208) |
| Systemic inhibition of OGA by thiamet G in Aβ and Tau overexpression models | Decrease pathology. | (60, 209) |
AgRP, Agouti-related protein.
In the following sections, we have provided an overview of our current understanding of the regulation of O-GlcNAcylation, its role in regulating cellular physiology, as well as its potential role in pathophysiology of diseases, including diabetes, cardiovascular disease, and cancer.
2. REGULATION OF O-GlcNACYLATION
2.1. Hexosamine Biosynthesis Pathway
Uridine diphosphate-GlcNAc (UDP-GlcNAc) is the common substrate for all amino sugars involved in the synthesis of glycoproteins and proteoglycans. As the end product of the hexosamine biosynthesis pathway (HBP), UDP-GlcNAc integrates multiple metabolic pathways and has long been considered an important nutrient signaling pathway partially regulated by substrate availability (FIGURE 4) (210–212). l-glutamine: d-fructose-6-phosphate transaminase (GFPT, EC 2.6.1.16), often referred to as l-glutamine: d-fructose-6-phosphate amidotransferase (GFAT), is the rate limiting enzyme of the HBP, catalyzing the transfer of the amide group from glutamine to fructose 6-phosphate, leading to the synthesis of glucosamine 6-phosphate (213). In mice and humans, there are two different isoforms of GFAT (GFAT1, GFAT2) encoded by two different genes, located on different chromosomes, and each isoform has markedly different tissue distribution (213, 214). In tissue from the central nervous system, GFAT2 expression predominates over GFAT1, whereas, in most other tissues, GFAT1 is more highly expressed (214). A splice variant of GFAT1, known as GFAT1Alt, has been identified, and it appears to be predominantly expressed in skeletal muscle and exhibits a higher Km for fructose-6-phosphate and a lower Ki for UDP-GlcNAc than those for GFAT1 (213, 215). In mammals, GFAT exists as a tetramer, and its activity is highly dependent on the availability of both glutamine and glucose (213). Both glucosamine-6-phosphate and UDP-GlcNAc are potent allosteric inhibitors of mammalian GFAT (FIGURE 5) (214). Up to 20 different phosphorylation sites have been identified in GFAT; however, the responsible kinases and function(s) of the majority of them are unknown. GFAT1 and GFAT2 are regulated by cAMP-dependent protein kinase (PKA) phosphorylation on Ser-205 and Ser-235, respectively (213, 216,217); both AMPK and calcium/calmodulin-dependent kinase (CaMK) II phosphorylate GFAT1 at Ser-243 (FIGURE 5) (129, 218). There are contradictory reports on the effects of phosphorylation on GFAT activity, which may be due to isoform-specific differences. In the heart, a number of studies indicate that AMPK phosphorylation of GFAT1 reduces its activity (219); however, it remains to be determined whether this is also the case in other cells and tissues. GFAT is also transcriptionally regulated (FIGURE 5) by specificity protein 1 (Sp1) (220) and activating transcription factor 4 (ATF4) (221). Multiple GFAT1 missense mutations leading to loss of activity and reduced O-GlcNAc levels have been linked to neuromuscular transmission defects (222). Despite reports of several putative GFAT inhibitors (213, 223, 224), the regulation of HBP flux via small-molecule inhibitors of GFAT has been limited to the glutamine analogs azaserine and 6-diazo-5-oxo-l-norleucine (DON), which are limited because of their lack of specificity (TABLE 1).
FIGURE 4.

Schematic of UDP-GlcNAc and O-GlcNAc synthesis. Glucose enters the cell via the glucose transporter system where it is rapidly phosphorylated by hexokinase (HK) and converted to fructose-6-phosphate by phosphoglucoseisomerase (PGI). Fructose-6-phosphate is subsequently metabolized to glucosamine-6-phosphate by l-Glutamine: d-fructose-6-phosphate amidotransferase (GFAT), which requires glutamine. Glucosamine-6-phosphate is converted to N-acetylglucosamine-6-phosphate by glucosamine 6-phosphate N-acetyltransferase (Emeg32), utilizing acetyl-CoA. Phosphoacetylglucosamine mutase (Agm1) converts N-acetylglucosamine-6-phosphate to N-acetylglucosamine-1-phosphate. The synthesis of uridine-diphosphate-N-acetylglucosamine (UDP-GlcNAc) is catalyzed by UDP-N-acetylglucosamine pyrophosphorylase (Uap1), which consumes uridine triphosphate (UTP). UDP-GlcNAc is the substrate for (O-GlcNAc transferase (OGT) leading to the formation of O-linked β-N-acetylglucosamine (O-GlcNAc)-modified proteins. β-N-acetylglucosaminidase (OGA) catalyzes the removal of O-GlcNAc from the proteins. GlcNAc can reenter the HBP via two salvage pathways: 1) via N-acetylglucosamine kinase (NAGK) to generate N-acetylglucosamine 1-phosphate and 2) involving the conversion by N-acetylgalactosamine kinase (GALK2) of N-acetyl-galactosamine to N-acetylgalactosamine 1-phosphate and UDP-N-acetylgalactosamine, with subsequent conversion by an epimerase to UDP-GlcNAc. Glucosamine, which enters the cell via the glucose transport system and can be phosphorylated by hexokinase (HK) to form glucosamine 6-phosphate thereby bypassing GFAT. The kinases that have been identified as regulating GFAT, OGT, and OGA are indicated; additional details may be found in the text (see FIGURE 6 and TABLE 3). AMPK, AMPK-activated protein kinase; CaMKII, calcium/calmodulin (Ca2+/CaM) dependent protein kinase II; CHK1, checkpoint kinase-1; CK2, casein kinase 2; EPI, epimerase; GalNAc, glucosamine fructose-6-phosphate amidotransferase; GFAT, glucosamine fructose-6-phosphate amidotransferase; GlcNAc, N-acetylglucosamine; GlcNAc-1P, N-acetylglucosamine-1-phosphate; GSK3β, glycogen synthase kinase 3b; HEX, hexokinase; IRS1, insulin receptor substrate-1; PPi, pyrophosphate; UDP-GalNAc, uridine diphosphate N-acetylgalactosamine.
FIGURE 5.

Regulation of glucosamine fructose-6-phosphate amidotransferase (GFAT). GFAT activity is regulated at several levels including substrate availability, feedback inhibition by uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) and glucosamine-6-phosphate (GlcN-6-P), as well as post-translational modifications by phosphorylation, acetylation, succinylation, and ubiquitination. The only kinases that have been identified as phosphorylating GFAT are PKA, AMPK, and calcium/calmodulin (Ca2+/CaM) dependent protein kinase II (CaMKII). Sp1, ATF4, and XBP1s have been shown to regulate GFAT at the transcriptional level. The database phosphosite.org and the citations contained therein were used to identify known posttranslational modification sites of GFAT (196). ATF4, activating transcription factor 4; Lys, lysine; Sp1, specificity protein 1; XBP1s, X-box binding protein 1.
Glucosamine 6-phosphate N-acetyltransferase (GNPNAT, GNA1), known as Emeg32 in mice, uses acetyl-CoA to convert glucosamine 6-phosphate to N-acetylglucosamine 6-phosphate (225), which is subsequently isomerized by phosphoglucomutase (PGM) to N-acetylglucosamine-l-phosphate. The final step in the HBP is conversion of N-acetylglucosamine-1-phosphate to UDP-GlcNAc, which is catalyzed by UDP-N-acetylglucosamine pyrophosphorylase (UAP1), also known as UDP-N-acetylhexosamine pyrophosphorylase (226). In addition to its de novo synthesis, UDP-GlcNAc can also be generated by two salvage pathways (FIGURE 4) (227). In one pathway, GlcNAc is phosphorylated by N-acetylglucosamine kinase to generate N-acetylglucosamine 6-phosphate. A second pathway involves the conversion of N-acetyl-galactosamine to N-acetylgalactosamine-1-phosphate and UDP-N-acetylgalactosamine, with subsequent conversion by an epimerase to UDP-GlcNAc (FIGURE 4). The relative contribution of the salvage pathways to total UDP-GlcNAc synthesis is not known. That deletion of GNPNAT gene is embryonically lethal (228) suggests that de novo synthesis is likely the predominant pathway. On the other hand, while deletion of EMeg32 substantially decreased UDP-GlcNAc levels, they were not eliminated. Further, the loss of EMeg32 did not lead to major changes in N- and O-glycosylation, whereas, O-GlcNAc levels were markedly suppressed, suggesting that salvage pathways were sufficient to maintain ER- and Golgi-mediated glycosylation, but not O-GlcNAcylation (228). Alternatively, under conditions in which UDP-GlcNAc levels are limiting, N- and O-glycosylation are prioritized over O-GlcNAcylation.
It is frequently stated that 2–5% of glucose entering a cell is metabolized via GFAT and the HBP. The origin of this statement is from a study using cultured adipocytes in which metabolic flux through glycolysis and the HBP was not directly measured, and the fraction of glucose metabolized by the HBP was inferred from other measurements (46). Moreover, energy metabolism, including glycolysis, varies widely from quiescent cells in culture to high-energy demands of the heart and brain. Consequently, the relative flux of glucose via glycolysis and HBP will also vary widely. There are very few studies that have directly measured the flux of glucose via the HBP, relative to other glucose-utilizing pathways, using radio- or stable-isotope techniques, either in cell culture or intact organs. In 2017, Gibb et al. (229), using 13C6-glucose labeling in cultured neonatal cardiomyocytes, concluded that glucose is metabolized more by the HBP than the pentose phosphate pathway, suggesting that glucose utilization via the HBP could be considerably greater than previous estimates; however, this was under the low-energy demands of cell culture, as well as being in neonatal cardiomyocytes which would have different energetic demands from those in an adult heart. Olsen et al. (230) have recently developed an LC/MS technique using 13C-labeled substrates to measure the rate of glucose metabolism via the HBP at the same time as glycolytic flux. In the isolated perfused working heart, an HBP flux of ∼2.5 nmol/g protein/min was measured, which represented 0.003–0.006% of the glycolytic flux. This is several orders of magnitude lower than earlier estimates, most likely because of the high metabolic rate of the heart. Moreover, when increasing glucose concentration from 5 to 25 mM, changes in HBP flux relative to glycolysis occurred as a result of changes in glycolytic, not HBP, flux rates. This illustrates the limitation of evaluating HBP flux as a fraction of glycolysis, as well as the assumption that HBP flux will be similar across biological systems with widely varying metabolic demands. While, much remains to be understood about of the regulation of HBP, the implementation of new techniques will enable direct measures of HBP flux in diverse biological systems.
2.2. O-GlcNAc Transferase
O-GlcNAc transferase (OGT; uridine diphospho-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase; EC 2.4.1.255) is a soluble glycosyltransferase primarily located in the cytoplasm and nucleus, which is responsible for using UDP-GlcNAc to modify proteins with O-GlcNAc (211, 231–234). OGT is located on the X-chromosome and encodes a multidomain protein containing multiple tetratriopeptide repeats (TPRs) in the NH2-terminal domain and two catalytic domains in the COOH-terminal region (FIGURE 6A) (211, 231–234). OGT is highly conserved, across all metazoans, and with the exception of zebrafish (237, 238), a single gene encodes OGT. Alternative splicing results in three mammalian isoforms of OGT, which differ primarily by the number of TPRs. A form of OGT, epidermal growth factor domain-specific OGT (EOGT), has also been identified in the ER, but it shares little homology with other forms of OGT. In contrast to OGT, it can also elongate O-GlcNAc into complex glycans (239), as reviewed in detail elsewhere (240, 241).
FIGURE 6.
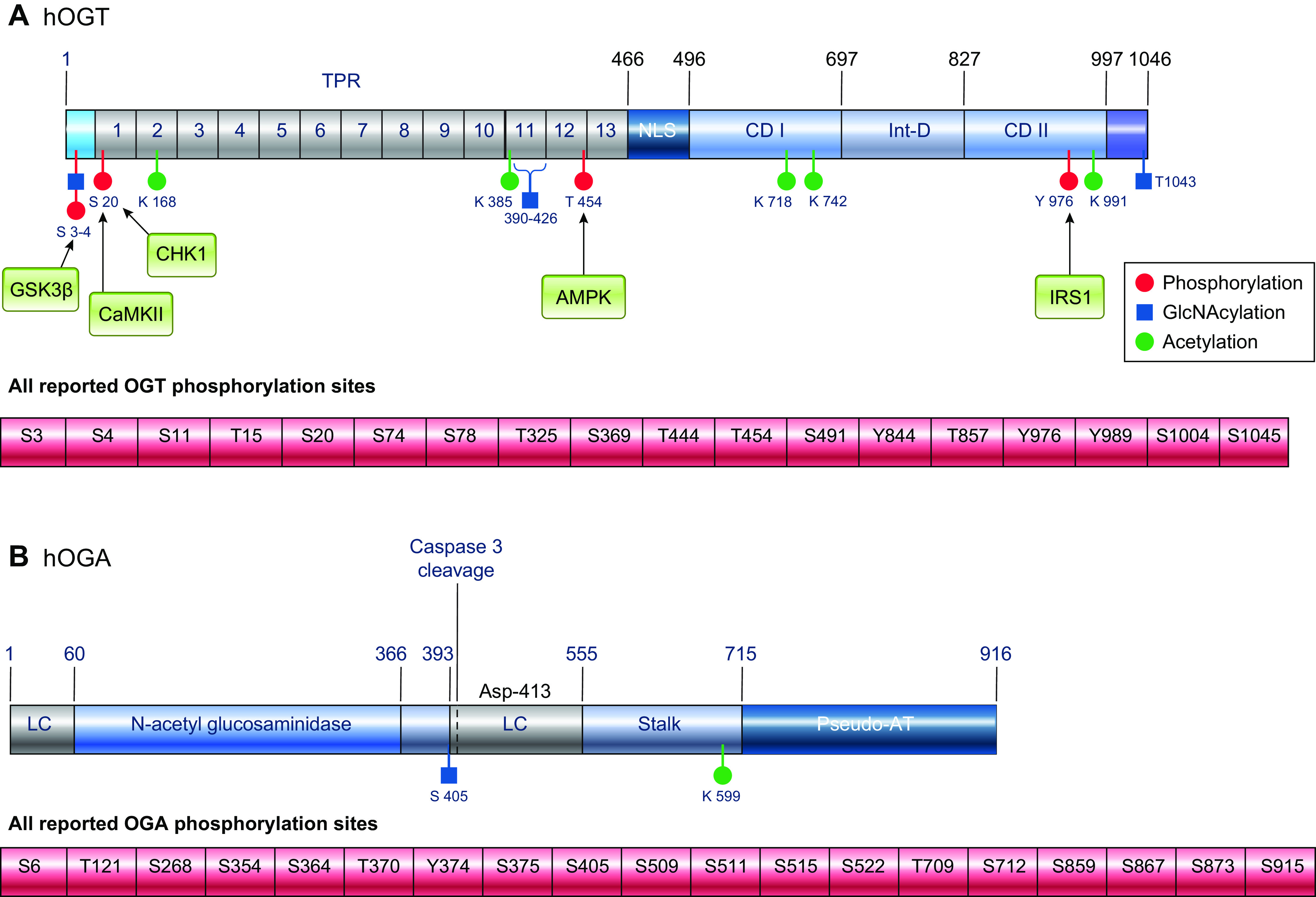
Structure and posttranslational modifications of human OGT (A) and OGA (B). Phosphorylation sites with identified kinases are shown as red circles, and all known phosphorylation sites are listed below the figures. O-GlcNAc sites are shown as blue squares, and acetylation sites are shown as green circles. AT, acetyl transferase; CaMKII, calcium/calmodulin (Ca2+/CaM) dependent protein kinase II; CD, catalytic domain; CHK1, checkpoint kinase-1; GSK3β, glycogen synthase kinase 3β; hOGT, human O-GlcNAc transferase; hOGA, huma O-GlcNAcase; Int-D, intervening domain; IRS1, insulin receptor substrate-1; LC, low complexity region; NLS, nuclear localization sequence; TPR, tetratricopeptide repeats. The databases https://www.phosphosite.org and http://www.phosphonet.ca/, and the citations contained therein were used to for known phosphorylation sites and acetylation modification sites(196). Citations for sites where kinases have been identified are included in the text; additional resources include Lundby, et al. (235), Levine and Walker (232) and Roth et al. (236). Other PTMs not shown include ubiquitination and sumoylation.
The full length or nucleocytoplasmic OGT (ncOGT; 110 kDa) and short OGT (sOGT; 78 kDa) have up to 13.5 and 4 TPRs, respectively; the precise number of TPRs is species specific (232). The mitochondrial OGT (mOGT; 90 kDa) has a mitochondrial targeting sequence at the NH2 terminus of the protein and 9 TPRs. All three OGT isoforms have the same two catalytic domains, as well as a putative phosphatidylinositol (3,4,5)-trisphosphate (PIP3)-binding domain (242); however, studies of the crystal structure of human OGT-UDP-peptide complex were unable to confirm the presence of the PIP3 domain (90). OGT is a member of the GT-B glycosyltransferase superfamily, and it is the only glycosyl transferase to have a ∼120 amino acid sequence in the middle of the catalytic domain, known as the intervening domain (Int-D) (232, 234, 243). The function of this region of the protein remains unclear, although it contains a large number of basic residues, suggesting contacts with negatively charged partners, which may affect cellular localization or protein-protein interactions (232, 243).
The catalytic domain of OGT represents less than half of the total molecular weight of the protein with the remainder of the protein primarily consisting of the TPRs (234). TPRs comprise 34 amino acid motifs clustered in repeats that form α-helical superspirals and have been proposed to play a key role in determining OGT interactions with target substrate proteins (234, 244). The importance of the TPR domain in regulating OGT substrate selectivity was demonstrated by the observation that modification of only two aspartate residues in the TPR substantially changed OGT substrate preference (245). Although most OGT substrates require the presence of the TPR domains for O-GlcNAcylation to occur, some proteins only interact with the catalytic region (131, 232, 234). OGT exists as a homodimer (243, 244, 246), and the TPR regions are also required for dimerization; however, in vitro disruption of this interaction did not change OGT activity (247). The function of OGT dimerization is unclear; however, it could serve to stabilize interactions with specific substrates. For example, mutations in the TPR 6–7 region prevent OGT dimerization, as well as reduce O-GlcNAc levels of nuclear pore glycoprotein p62 (Nup62) (244).
A definitive consensus sequence for O-GlcNAcylation has not been identified; consequently, although our knowledge of OGT structure and its molecular interactions with substrates has improved, our knowledge of the regulation of OGT function and the mechanisms underlying its substrate specificity remains limited (231). Reports demonstrating that interactions between UDP-GlcNAc and the peptide play a role in orientation of the peptide relative to the active site suggest that the active site contributes to substrate selection (248). Moreover, preference by OGT for Ser/Thr residues that have prolines and branched-chain amino acids in close proximity, resulting in an extended peptide orientation, also suggests that the active site imposes some degree of sequence constraint and, thus, substrate selection (249). However, perhaps the most widely accepted view is that OGT substrate selection is largely determined by binding to the TPR domains. This is supported by the fact that the TPR domains are essential for O-GlcNAcylation of most proteins, as well as the fact that specific substrates interact with specific TPR regions. For example, in brain tissue, ataxin-10 (Atx-10) binds to TPRs 6–8 (246), whereas mSIN3A, ten-eleven translocation (TET) 2/3, and trafficking kinesin protein 1 (TRAK1) all require TPRs 1–6 (232). Moreover, mutations of just two aspartate residues in the TPR domain changed OGT activity as well as target specificity (245). Structural studies have revealed a hinge region around TPRs 12–13, which could influence the access of protein substrates to the active site of OGT (232, 234). There is also evidence that under some conditions, sOGT can act as a negative regulator of ncOGT (233, 246). The importance of the TPR domain is reflected in the observations that missense mutations in this region result in decreased OGT function and neurodevelopmental abnormalities (29, 250).
OGT activity and substrate recognition can also be regulated by phosphorylation of OGT on Ser, Thr, and tyrosine (Tyr) residues (233). Almost 20 different phosphorylation sites have been reported on OGT, many identified by large-scale proteomic studies (FIGURE 6A). Although the function of many of these sites remains unknown, as do the kinase(s) responsible for their phosphorylation, a few have been characterized. For example, insulin increases Tyr phosphorylation of OGT via activation of the insulin receptor (IR), resulting in increased OGT activity (251). Glycogen synthase kinase (GSK)-3β phosphorylates OGT on Ser-3/4, leading to increased OGT activity (116), and AMPK phosphorylates Thr-444, resulting in changes in subcellular localization and substrate binding targets (252). In addition, OGT is phosphorylated on Ser-20 by checkpoint kinase 1 (Chk1), leading to stabilization of OGT, which is required for cytokinesis (253). Ser-20 on OGT is also a target for CaMKII, which increases its activity (181). O-GlcNAcylation of Ser-389, located in the TPR domain regulates OGT nuclear localization (254). Serines 3 and 4 on OGT are also sites of O-GlcNAcylation (116); however, the function of this modification remains to be determined. Recent work that has focused on some of these phosphorylation sites has identified that in sOGT, mutation of either Thr-12 or Ser-56 to an alanine significantly altered substrate binding by over 500 proteins (255). OGT is also acetylated on multiple residues (FIGURE 6A) (235). While the effects of this modification are not known, the fact that two of the sites occur within one of the catalytic domains suggests they could regulate OGT activity in some manner.
Targeting of specific proteins by OGT can also occur via its interaction with adaptor or scaffold proteins, which recruit substrates on OGT. For example, in the liver during fasting host cell factor 1 (HCF1) targets OGT to peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), where increased O-GlcNAcylation increases PGC-1α stability and upregulates gluconeogenic genes (118). Other such interactions include p38 MAPK, which recruits OGT to neurofilament-heavy polypeptide (NFH) (256), REV-ERBα, which prevents OGT degradation (65), and OGA, which increases interactions between OGT and pyruvate kinase isoform M2 (PKM2) (257). The potential for an OGT-OGA interaction raises the possibility of an “O-GlcNAczyme” complex, which would be consistent with rapid and reversible changes in O-GlcNAcylation (258). Insulin treatment results in translocation of OGT from the nucleus to the cytosol and plasma membrane, and changes in nutrient availability also lead to redistribution of OGT between the nucleus and cytosol (242, 251). The three OGT isoforms also exhibit differences in their subcellular localization, and along with differences in the TPR regions between the isoforms, different proteome subsets can result; however, this has only been demonstrated with in vitro studies (169).
In addition to its role as a glycosyl transferase, OGT also exhibits protease activity, although, to date, only one proteolytic substrate, the transcriptional coregulator HCF1, has been identified (259, 260). To fully function as a coregulator HCF1 is required to undergo proteolytic cleavage; however, the mechanism by which this occurred remained elusive until 2011 when two reports demonstrated that OGT played a key role in this process (259, 260). These studies demonstrated that both UDP-GlcNAc and OGT were required for HCF1 proteolysis, consistent with the notion that O-glycosylation of HCF1 was necessary. It was initially thought that OGT had a specific protease active site; however, subsequent studies demonstrated that the COOH-terminal of HCF1 binds to the TPR domain of OGT, and the cleavage region in the glycosyltransferase active site was similar to that for regular glycosylation substrates (261). Moreover, these studies also showed that UDP-5SGlcNAc, which binds to the OGT-active site in the same manner as UDP-GlcNAc, but is resistant to glycosyltransferase, inhibited HCF1 proteolysis. The role of OGT mediated cleavage of HCF1 remains unclear; however, it could represent a link between cellular metabolism and the regulation of cell cycle, which is supported by reports that the OGT/HCF1 complex itself is an important regulator of glucose metabolism (118).
2.3. O-GlcNAcase
O-GlcNAcase (OGA, EC 3.2.1.169) is a hexosaminidase that was first characterized in 1994 by Dong and Hart (11) following purification from rat spleen; it has also been referred to as NCOAT, nuclear cytoplasmic O-GlcNAcase and acetyltransferase (262), GCA (263), MEA5, and MGEA5 (264). Dong and Hart (11) demonstrated that it was distinct from lysosomal hexosaminidases by virtue of its subcellular localization to the cytosol and nucleus, its neutral pH optimum, and its selectivity for GlcNAc over GalNAc. Subsequent sequence identification and cloning demonstrated that OGA was identical to a previously identified protein, MGEA5, found in meningioma patients (264). The human OGA gene is located on chromosome 10 and encodes a protein with an NH2-terminal glycosyl hydrolase domain and a COOH-terminal domain that demonstrates sequence homology to histone acetyl transferase (AT) proteins (FIGURE 6B). The glycosyl hydrolase domain has two low-complexity or disordered regions [i.e., repeats of single amino acids or short amino acid motifs (265)], on either side. The larger low-complexity region is followed by the stalk domain, which connects to the COOH-terminal domain (231, 266). Between the catalytic domain and the COOH-terminal region is a noncanonical caspase cleavage site (267). It has been reported that OGA exhibited AT activity (199, 268); however, this has not been replicated by others (269). Recent structural studies found that human OGA (hOGA) lacks the residues necessary for binding to acetyl-CoA and described this region as a “catalytically incompetent pseudo-AT domain” (270). Alternative splicing results in the generation of a short OGA (sOGA), which lacks the pseudo-AT region and has a different 15-amino acid COOH-terminal region (271). sOGA has lower hexosaminidase activity than hOGA in vitro (56, 272) and appears primarily localized to the surface of lipid droplets (273), although its specific function remains to be elucidated.
For many years, full-length hOGA proved resistant to crystallization, consequently, most of the initial structural and mechanistic information were derived from bacterial homologs of OGA (274). In 2017, three independent studies reported the crystal structure of hOGA using different, catalytically functional, truncated versions of the protein (236, 275, 276). A key and unexpected finding in all three studies was that hOGA formed an obligate homodimer in which the stalk domain of one monomer is positioned over the catalytic site of the other monomer (266). One consequence of this arrangement is the creation of a substrate binding site, comprising conserved hydrophobic residues, which supports the notion that the dimer is the active form of OGA. Further structural analysis suggests OGA may preferentially remove O-GlcNAc from certain sites, suggesting that OGA may be an equal partner with OGT in regulating O-GlcNAc turnover (277). Moreover, it has also been reported that a number of specific residues on OGA contribute to its interactions with different peptide substrates, which has implications for differential regulation of O-GlcNAcylation on different proteins (278,279). While the active site of OGA is now better characterized than ever before, other regions, including the low-complexity region and the pseudo-AT domain remain poorly understood (266).
Similar to OGT, OGA is also subject to both phosphorylation and O-GlcNAcylation. At least 20 different Ser, Thr, and Tyr phosphorylation sites have been mapped by MS in both the glycosyl hydrolase and pseudo-HAT domains (FIGURE 6B); however, the effects of these modifications on OGA activity have not been determined. Interestingly, the Ser-405 O-GlcNAcylation site on OGA, is in the central low-complexity region, which is also the region where OGA interacts with OGT (266). Thus, O-GlcNAcylation of this residue plays a role in the regulation of the interaction between OGT and OGA. OGA is also acetylated in the stalk domain at Lys-599 (235).
2.4. Maintenance of O-GlcNAc Homeostasis
Changes in cellular O-GlcNAc levels occur in response to a diverse array of physiological and pathological stimuli, and dysregulation of O-GlcNAc homeostasis has been linked to a wide array of diseases, as discussed in detail later. Deletion of the OGT gene is embryonically lethal (208), and in cell culture, mouse embryonic fibroblasts (MEFs) die around 4–5 days after OGT is knocked out (280). Although OGA knockout mice survive to birth, the majority die before weaning (17). Chronic increases in O-GlcNAc levels induced by overexpression of a dominant negative OGA (dnOGA) in a tissue-specific manner results in apoptosis in skeletal muscle (205) and altered metabolism in the heart (281). Therefore, it is evident that maintenance of O-GlcNAc homeostasis is essential for the normal physiological function of cells and tissues. This has led to the concept that there is an optimal range of O-GlcNAcylation that supports normal physiological processes and that outside that range, either too high or too low, cellular dysfunction results. Consequently, it has been proposed that to keep within this optimal range, OGT and OGA work together to form a “buffering” system that can respond to moderate changes in O-GlcNAcylation (257).
The prevailing wisdom for many years was that the primary mechanism regulating cellular O-GlcNAc levels was nutrient availability, so that under conditions of nutrient excess, such as hyperglycemia, O-GlcNAc levels increased, and if glucose levels dropped O-GlcNAc levels would decrease. This concept is consistent with the fact that glucosamine, which bypasses GFAT, leads to uncontrolled synthesis of UDP-GlcNAc and increased O-GlcNAc levels (24, 271, 282, 283). This also enables glucosamine to be used pharmacologically to increase UDP-GlcNAc synthesis and O-GlcNAc levels. Acute changes in glucose availability either by increasing exogenous glucose or via insulin-stimulated increases in glucose uptake can have little or no effect on cellular O-GlcNAc levels (284). This is also consistent with the report that a five-fold increase in glucose had no effect on HBP flux in the isolated perfused heart (230). However, such responses likely vary greatly, depending on cell type and duration of treatments (285). In addition, OGT activity is very sensitive to UDP-GlcNAc concentrations, exhibiting multiple apparent Km values for UDP-GlcNAc under varying UDP-GlcNAc concentrations (247). Consequently, changes in UDP-GlcNAc concentrations can directly influence global as well as regional OGT activities and O-GlcNAc levels.
However, while nutrient availability as a regulatory mechanism is a valuable concept, it does not address the mechanisms that underlie widely differing rates of changes in O-GlcNAcylation that occur in response to different stimuli. For example, depolarization of neuroblastoma cells dramatically increased OGT activity, reaching a peak within 1 min, leading to increased O-GlcNAc levels (286), and stimulation of human neutrophils leads to an approximately five fold increase in O-GlcNAc within 30 s, returning to normal levels over the next 5–10 min (287). On the other hand, stress, such as ischemia (288) or heat shock occur over periods of minutes to hours, induced increases in O-GlcNAc levels (289). Conversely, in certain disease states, such as cancer (21, 290), diabetes (283, 290), or cardiac hypertrophy (291,292), O-GlcNAc levels are chronically elevated, and the mechanisms that lead to this resetting of steady-state O-GlcNAc levels outside of the normal range remains poorly understood (293). As discussed above, GFAT, OGT, and OGA are all modified by phosphorylation, indicating that these posttranslational modifications may contribute to the dynamic regulation of O-GlcNAcylation.
Given that HBP flux and O-GlcNAc levels are modulated, in part, by nutrient availability, it is perhaps not surprising that O-GlcNAc levels can also be regulated by nutrient-regulating hormones. Insulin is probably the most studied of these, and it has been shown that insulin treatment recruits OGT from the nucleus to the plasma membrane, where it is phosphorylated by the IR (242, 251, 294). This phosphorylation increases OGT activity and leads to O-GlcNAcylation of insulin receptor substrate-1 (IRS1) and AKT, resulting in attenuation of insulin signaling. Treatment of HepG2 cells with the adipokine leptin resulted in an approximately twofold increase in overall O-GlcNAc levels within 15 min with parallel increases in GFAT protein levels (295). However, the mechanism by which leptin stimulated GFAT expression and O-GlcNAc levels was not identified. Ghrelin, a hormone that is released in response to fasting, increases O-GlcNAc levels in hypothalamic appetite-stimulating agouti-related protein (AgRP) neurons, leading to their increased firing rate (201). In the liver, glucagon, which increases in response to fasting, increased OGT phosphorylation and O-GlcNAc levels in a CaMKII-dependent manner (181).
In addition to nutrient-dependent hormones, G protein-coupled receptor agonists have also been shown to increase cellular O-GlcNAc levels. For example, activation of the endothelin (ET)A receptor with ET-1 resulted in a time-dependent increase in O-GlcNAc levels in vascular cells, and the subsequent downstream signaling by ET-1 was dependent on the increase in O-GlcNAc levels (296, 297). Phenylephrine (PE), primarily an α-receptor agonist, also increased O-GlcNAc levels in cardiomyocytes, and the increase in O-GlcNAc was required for subsequent activation of PE-mediated signaling pathways (298, 299). One explanation for PE-dependent increases in O-GlcNAc levels was an increase in GFAT expression (298, 299). Another study suggested that the increase was mediated by the Ca2+ dependent CaMKII/calcineurin pathway (298).
The transcriptional regulation of OGT and OGA has been understudied; however, there are a number of reports indicating that they regulate each other and that O-GlcNAcylation itself might contribute to their transcriptional regulation (257), further discussed in Sect. IIIA and IIIB. For example, in OGT knockout MEFs the time course of loss of OGT was paralleled by a decrease in OGA (280). Increasing O-GlcNAc levels via OGA inhibition demonstrated that OGA transcription was O-GlcNAc dependent (300). Other studies have reported that low O-GlcNAc levels contribute to increased OGT transcription (68). The strongest evidence to date of mutual regulation of OGT and OGA was by Qian et al. (301), who reported that overexpression of OGA resulted in increased OGT transcription, whereas knockdown OGA with siRNA significantly decreased OGT levels. Using promoter luciferase reporters for both OGT and OGA, they clearly demonstrated reciprocal transcriptional regulation (301). They also showed that OGA cooperated with p300m, a histone acetyltransferase and the transcription factor CCAAT/enhancer-binding protein-β (C/EBP-β) to promote OGT transcription (301). E2F transcription factor 1 (E2F1), which contributes to the activation of many genes, was found to be a repressor of both OGT and OGA (302). It has been shown that E2F1 activity itself might be regulated by O-GlcNAcylation, illustrating another mechanism by which O-GlcNAc can regulate the transcription of OGT and OGA (303). The OGT promoter contains a TATA box, which likely facilitates OGT transcription, whereas the OGA transcription is not dependent on a TATA box (302). In macrophages, Cullin 3 (CUL3), an E3 ubiquitin ligase, has been reported to downregulate OGT expression in a nuclear factor E2-related factor-2 (Nrf2)-dependent manner (304).
Another potential mechanism for maintaining O-GlcNAc homeostasis is alternative splicing of both OGT and OGA, resulting in intron retention, which is regulated by changes in O-GlcNAc levels (68, 305). Consequently, when O-GlcNAc levels are high, nuclear retention of OGT due to intron retention increases, whereas low-O-GlcNAc levels decrease this process; thereby, decreasing or increasing OGT protein, respectively (68, 305). Conversely, low O-GlcNAc levels increase nuclear retention of OGA (305). It is noteworthy that these changes occur relatively rapidly and, as such, represent a potentially important mechanism in O-GlcNAc-mediated regulation of O-GlcNAc homeostasis (305). A number of microRNAs (miRs), including miR-101, 200a/b, 423-5p, 501-3p, 539, and 619-3p, have also been shown to regulate O-GlcNAc levels by targeting OGT (306–309) or OGA (310). Consequently, although our knowledge of the transcriptional regulation of OGT and OGA is improving, the physiological role of these pathways remains to be determined.
One of the more puzzling aspects of O-GlcNAc homeostasis is that glucose deprivation leads to a marked increase in overall cellular O-GlcNAc levels. This was first reported in HepG2 cells where it was observed that 12 h following the removal of glucose, there was approximately eight-fold increase in O-GlcNAc levels, which was accompanied by a ∼40% decrease in UDP-GlcNAc levels, suggesting that increased HBP flux was not contributing to the elevated O-GlcNAcylation (311). A later study came to a similar conclusion, as the addition of 50–100 µM glucosamine blocked the O-GlcNAc increase, resulting from glucose deprivation (312). Contrary to this finding was a report that glycogen degradation triggered by glucose deprivation provided the substrate for O-GlcNAcylation (313). Moreover, ATF4, a regulator of the unfolded protein response (UPR), increased GFAT1 expression in response to glucose deprivation, and ATF4 inhibition or knockdown prevented the increase in O-GlcNAc and GFAT1 (221). They also suggested that ATF4, along with another UPR-related protein X-box binding protein 1 (XBP1), mediated the steady-state levels of GFAT1 (221). This is consistent with an earlier report demonstrating that stress-induced increases in O-GlcNAc were mediated via XBP1 increases in GFAT1 protein levels (314). Glucose deprivation increased mRNA levels of both OGT and OGA; however, this was not associated with an increase in the levels of either protein (312). Interestingly, extracellular Ca2+ was required for this increase in O-GlcNAc levels, and inhibition of CaMKII blunted the response to glucose deprivation (312).
Historically, nutrient availability was considered to be the primary regulator of cellular O-GlcNAc homeostasis; however, it is increasingly evident that it is only one of many factors. Our knowledge of transcriptional regulation is growing, but it remains limited, and the role of phosphorylation in regulating GFAT and OGT activity is improving; however, the role of phosphorylation in regulating OGA activity is underexplored. Several lines of evidence suggest that Ca2+-mediated activation of CaMKII contributes to O-GlcNAc homeostasis, which would be consistent with rapid agonist-induced increases in O-GlcNAc being independent of nutrient availability or transcriptional regulation of GFAT1, OGT, and OGA.
2.5. Cross Talk Between O-GlcNAcylation and Phosphorylation
As the dynamic nature of O-GlcNAcylation was recognized, there was speculation that it might play a regulatory role in protein function. Once it was recognized that O-GlcNAc modified Ser and Thr residues, which are also potential sites of phosphorylation, there was increasing consideration about possible interactions between O-GlcNAc and phosphorylation on proteins (211, 293). One concept that gained early popularity was the possibility of reciprocity between O-GlcNAcylation and phosphorylation. In other words, that a specific residue on a protein could be modified by either O-GlcNAc or phosphorylation became more widely known as the “Ying-Yang” hypothesis (315). There are, indeed, several proteins that support this concept, such as cMyc (Thr-58) (98), estrogen receptor (ER)-β (Ser-16), and endothelial nitric oxide synthase (eNOS) (Ser-1177) (190). Alternatively, modification of adjacent sites can negatively interact with each other, for example, histone deacetylase 4 (HDAC4) is O-GlcNAcylated at Ser-642, and this blocks CaMKII-mediated phosphorylation of Ser-632 (316). Consequently, it is becoming increasingly clear that the cross talk between O-GlcNAc and phosphorylation is much more complex than first thought (FIGURE 7). A high-throughput proteomic analysis estimated that in only 8% of O-GlcNAcylated proteins was the same residue modified by phosphorylation (182). Some proteins are modified by both O-GlcNAc and phosphate, but not at the same sites; for example, increases in phosphorylation of Thr-200 of CaMKIV decreases overall O-GlcNAcylation at several sites, including Ser-189, and conversely, prevention of Thr-200 phosphorylation increased CaMKIV O-GlcNAcylation (167). In myosin light chain 1, the O-GlcNAc-modified sites are Thr-93 and Thr-164, whereas the phosphorylation sites are at Thr-69 and Ser-200 (147).
FIGURE 7.
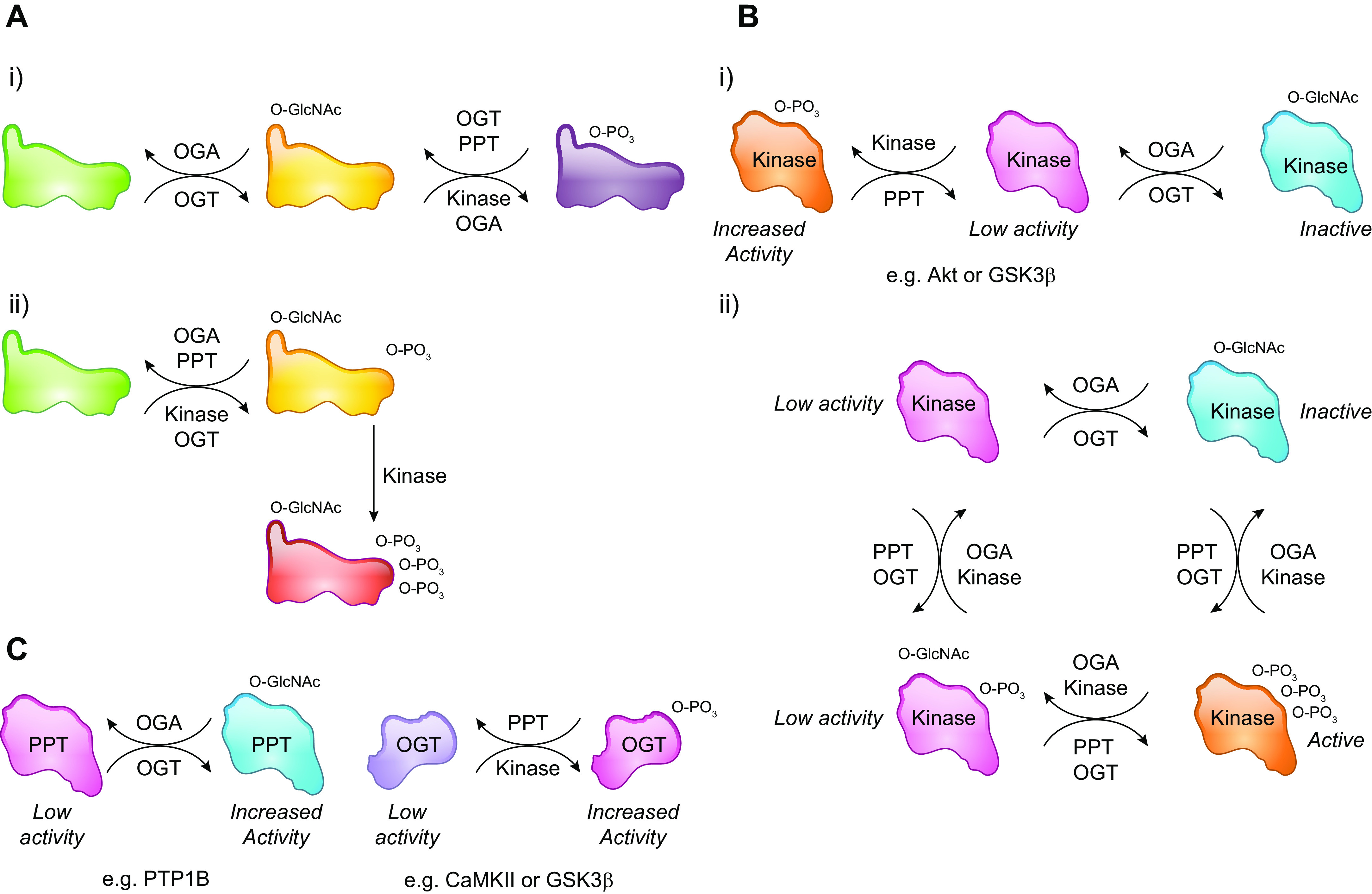
Extensive crosstalk between phosphorylation and O-GlcNAcylation. A, i: example of phosphorylation and O-GlcNAcylation of the same or adjacent sites that prevent simultaneous modifications. and A, ii: modification by both phosphorylation and O-GlcNAcylation, as well as O-GlcNAcylation enhancing phosphorylation via for example increased binding to adaptor proteins. B, i: example of a kinase that is activated when phosphorylated and inactivated by O-GlcNAcylation. B, ii: a kinase that can be modified by both O-GlcNAc and phosphorylation involving complex interactions between OGT/OGA and kinases/phosphatases. C: O-GlcNAcylation also regulates phosphorylation via modification of phosphatases, and OGT activity is directly regulated by kinase phosphorylation. GSK3β, glycogen synthase kinase 3β; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; PPT, protein phosphatase.
The intricate relationship between O-GlcNAcylation and phosphorylation was demonstrated by a study in which GSK3β was inhibited and O-GlcNAcylation levels of specific proteins were quantified. Of the 45 O-GlcNAcylated proteins identified, 10 exhibited an increase in O-GlcNAcylation, whereas 19 showed a decrease (317). In another study in which O-GlcNAc levels were increased by inhibition of OGA, increased phosphorylation was observed on 148 sites and lower phosphorylation was observed at 280 sites (318). A comparison between wild-type and OGT-null cells identified 232 phosphosites that were upregulated and 133 phosphosites that were downregulated 36 h after OGT deletion (319). Interestingly O-GlcNAc modifications have been reported to occur in clusters (182), a phenomenon that is also observed with phosphorylation (320). Therefore, it is evident that the cross talk between these two posttranslational modifications is complex, and it is currently not possible to predict a priori how they will interact on individual proteins. However, specific motifs have been identified, which when phosphorylated, strongly inhibit O-GlcNAcylation and vice versa; whereas, sites that are phosphorylated by proline-directed kinases do not appear to be subject to O-GlcNAcylation (321).
Recent studies have demonstrated that OGT and OGA form complexes with both kinases and phosphatases (322–324) demonstrating that changes in O-GlcNAcylation and phosphorylation could be occurring simultaneously on a target protein (FIGURE 7A). There is also a growing list of kinases that have been shown to be O-GlcNAcylated and that this modification directly affects their function (see TABLE 2 and FIGURE 7B). An analysis of glycoproteomic data sets reported that more than 100 kinases contain identified O-GlcNAcylation sites, emphasizing the importance of O-GlcNAc in regulating kinase function (325). A study of synaptic proteins found that kinases were more frequently a target for O-GlcNAcylation than other proteins; however, the specific modification sites were frequently outside the catalytic domain of the kinases in question (182). Thus, protein O-GlcNAcylation can alter phosphorylation either via direct modification of phosphorylated proteins, but also by regulation of kinases that are responsible for phosphorylation. Although there is less known about O-GlcNAcylation of phosphatases, protein tyrosine phosphatase 1B (PTP1B) has been shown to be O-GlcNAcylated at Ser-104, Ser-201, and Ser-386, resulting in increased enzymatic activity (176).
One specific example of the complex regulation between kinases and O-GlcNAcylation is CaMKIV, which is also known to activate OGT (286) (FIGURE 7C). Activation/deactivation of CaMKIV is rapid and tightly regulated beginning with displacement of protein phosphatase 2α (PP2A) and subsequent phosphorylation of Thr-200 by CaMKK; subsequent inactivation, involves the reassociation with PP2A and reduction in phosphorylation. Dias et al. (167) found that CaMKIV contained at least five O-GlcNAc modification sites and that during its activation, O-GlcNAc levels rapidly decreased as the interaction with OGA increased. Following inactivation, CaMKIV O-GlcNAc levels returned to normal, suggesting that OGT was recruited to CaMKIV; however, a direct interaction was not identified (167). Mutation of Thr-200 to alanine, thereby, preventing phosphorylation, resulted in an increase in CaMKIV O-GlcNAcylation; conversely, mutation to glutamate to mimic phosphorylation led to lower O-GlcNAc levels, demonstrating a direct interaction between Thr-200 and O-GlcNAcylation. Of the five O-GlcNAc sites identified, three Ser-189, Thr-57, and Ser-58, when mutated to alanine to prevent O-GlcNAcylation, markedly reduced Thr-200 phosphorylation. The double-mutant T57A/S58A resulted in no measurable kinase activity (167). Conversely, the S189A mutant, not only dramatically reduced O-GlcNAc levels, but also increased basal kinase activity. O-GlcNAcylation of CaMKII has also been shown to increase its kinase activity (165).
AMPK is another key example of a kinase that both regulates O-GlcNAc levels, as well as being regulated by O-GlcNAc itself, as reviewed in detail (219). As discussed earlier, GFAT is phosphorylated on Ser-243 by AMPK, resulting in decreased GFAT activity and lower O-GlcNAc levels (130, 218). Moreover, activation of AMPK leads to increased GFAT phosphorylation and decreased O-GlcNAc levels (299). AMPK also targets OGT by phosphorylating Thr-444; however, rather than alter its activity, this phosphorylation leads to changes in OGT cellular localization and substrate specificity (252). Interestingly, although OGA has not been shown to be a target for AMPK, deletion of the AMPKα2 isoform results in lower OGA protein levels and increased O-GlcNAc levels, without changes in either OGT or GFAT (299). Bullen et al. (252), demonstrated that all α- and γ-subunits of AMPK are O-GlcNAcylated and further that activation of AMPK increased O-GlcNAcylation of the γ1-subunit. They also reported that inhibition of OGA attenuated physiological and pharmacological activation of AMPK.
Although Ser and Thr residues are often the focus of discussions of cross talk between O-GlcNAcylation and phosphorylation, it is also clear that Tyr phosphorylation and O-GlcNAcylation also influence each other (326). An analysis of a small set of O-GlcNAcylated proteins concluded that >60% of them were also Tyr phosphorylated (326). The potential importance of interactions between O-GlcNAc and Tyr phosphorylation was first recognized, when it was demonstrated that insulin stimulates Tyr phosphorylation of OGT via the IR, thereby increasing OGT activity (251). A subsequent study revealed that increased O-GlcNAc levels reduced Tyr phosphorylation of IRS-1 (327). Studies on prohibitin, reported that O-GlcNAcylation attenuated its Tyr phosphorylation (328). Using peptides that contained residues that could be both Tyr phosphorylated and O-GlcNAcylated, Ande et al. (328) showed that while O-GlcNAcylation reduced Tyr phosphorylation, an increase in Tyr phosphorylation actually enhanced O-GlcNAcylation. Another study using peptide microarrays concluded that Tyr phosphorylation may have a greater effect on the regulation of O-GlcNAcylation than O-GlcNAcylation on phosphorylation (329).
Here, the focus has been on interactions between phosphorylation and O-GlcNAcylation; however, O-GlcNAcylation also interacts with other PTMs, although these are less widely studied (257). For example, both OGT and OGA have been found to be ubiquitinated, and O-GlcNAc has been shown to stabilize proteins by inhibiting their ubiquitination (330, 331). RelA/p65, a member of the nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) family of transcription factors, is both O-GlcNAcylated (Thr-305, Ser-319, Ser-337, Thr-352, and Ser-374) and acetylated (lysine-310). Acetylation is required for full transcriptional activity of RelA/p65, and O-GlcNAcylation at Thr-305 and Thr-315 promotes its acetylation at lysine-310 (332, 333). In addition, both OGT and OGA are also acetylated, although the effect of this modification on their function is not known (FIGURE 6) (235). Of note, HDAC4 has been shown to be modified by O-GlcNAc on Ser-642, providing further evidence of direct interactions between O-GlcNA cylation and acetylation (316). As further discussed in the next section, cross talk between acetylation and O-GlcNAcylation has also been implicated in epigenetic regulation (334).
3. O-GlcNACYLATION AND CELLULAR FUNCTION
In the prior sections, we discussed the discovery of O-GlcNAcylation, its unique place in glycobiology, and the numerous pathways that impact its regulation. In this section, we will focus on the cellular functions of regulation by O-GlcNAcylation, including modulation of gene expression at both the levels of transcription, as well as via epigenetic mechanisms, followed by specific examples of how O-GlcNAcylation impacts cellular signaling (e.g., insulin and calcium), metabolism (e.g., mitochondria), and survival (e.g., autophagy).
3.1. Transcription
Gene expression is controlled at a number of levels (FIGURE 8). First, at the level of DNA sequence are response elements that recruit transcriptional regulators that include numerous families of transcription factors. However, the regulation of gene expression is intricately controlled beyond proper recruitment of transcriptional machinery to the promoter regions of genes that contain these response elements. In particular, the direct modification of transcription factors and the transcriptional machinery can be either inhibitory or activating in altering RNA levels. One of the first molecular functions to show biological relevance of O-GlcNAc-mediated regulation is modification of these transcription factors (210). The early studies started with an initial focus on O-GlcNAcylation of the ubiquitously expressed Sp1, a zinc finger transcription factor that binds GC-rich motifs of many promoters (FIGURE 8A, I). Work by Jackson and Tjian (335) found that increased O-GlcNAcylation on Sp1 from either Drosophila or human cells increased its transcriptional activity. The mechanism of this transcriptional activation was identified by Kudlow and colleagues (336, 337), who reported that the O-GlcNAc modification protected Sp1 from proteasomal degradation. In turn, this allowed O-GlcNAc modification of Sp1 to act as a nutritional checkpoint for times of inadequate nutrients, hypoglycosylation, and resource sparing through decreased transcription (336). Subsequent studies on the mechanisms of O-GlcNAc action on transcriptional regulation also found that within this same factor, Sp1 could be O-GlcNAcylated at its activation domain to suppress transcriptional activity (109), suggesting multiple and opposing roles for this single modification depending on context. These mechanistic insights provided important clues as to how diabetes, for example, could disrupt cellular processes of signaling via decreased Sp1 transcriptional activation. In the context of diabetes, O-GlcNAc modification of Sp1 was further shown to modify transcriptional activity because of a dynamic interplay with its phosphorylation status, which further alters Sp1 subcellular compartmentalization and activity (338) and may impact transcriptional regulation of mitochondrial function (339). Additionally, Sp1 O-GlcNAcylation may simply interfere with its interaction with other transcription factors, such as Elf-1 (340), NF-Y (341), Oct1 (342), as well as Sp3 and Sp4 (343), highlighting the diverse set of mechanisms by which O-GlcNAcylation of transcription factors mediate gene expression.
FIGURE 8.

O-linked β-N-acetylglucosamine (O-GlcNAc) regulation of gene expression and epigenetics. A: examples of transcriptional regulation mediated by O-GlcNAc with additional details and references highlighted in the text. These examples include both inhibitory and activating roles of O-GlcNAc (blue square with G). A, I: for the transcription factor Sp1, one O-GlcNAc modification blocks an activation site to inhibit transcription, while a different O-GlcNAc site inhibits proteasomal degradation increasing transcription. A, II: a separate set of examples of this dual role of O-GlcNAc-mediated regulation is at ChREBP, which, under normal glucose levels, leads to O-GlcNAc modification (blue square) to enhance 14-3-3 binding decreasing transcriptional activity, while under high glucose levels, additional posttranslational regulation by phosphorylation (red circle with P) leads to a different O-GlcNAc modification and augments activity. A, III: multiple roles are shown for both direct O-GlcNAc modification of RNA polymerase II (RNAP II), as well as the contribution of uridine diphosphate-azido-modified galactose (UDP)-GlcNAc hydrolysis to enhance transcriptional activity. A, IV: multiple roles are shown for different O-GlcNAc modifications to either inhibit ubiquitination (gray square with U) to increase complex stability or other modifications that may impact nuclear cytoplasmic import/export of cargo (e.g., RNA). B: examples of epigenetic regulation mediated by O-GlcNAc with additional details and references highlighted in the text. B, I: highlights the multiple direct (i.e., O-GlcNAc of histone proteins) and indirect (i.e. interaction and modification of other histone modifiers), such as EZH2 to impact histone methylation (black square with M) and SIN3a to impact histone acetylation (purple square with A). B, II: highlights the interaction of O-GlcNAc transferase (OGT) with the ten-eleven translocation (TET) enzymes impacting DNA 5 hydroxymethylation (5hmC) which, in turn, can lead to DNA demethylation. B, III: a few examples are highlighted from the text of noncoding RNA [ncRNA; e.g., long-noncoding RNA (lncRNA) and microRNA (miR)] regulation of O-GlcNAc enzymes.
Although much of the early work was done on O-GlcNAc modification of Sp1, a number of subsequent studies have identified a continuously growing list of transcription factors with a direct O-GlcNAc modification and functional consequence. This includes additional key transcriptional regulators of insulin signaling and metabolism, such as brain and muscle ARNT-like 1 (BMAL1) (344), carbohydrate-responsive element-binding protein (ChREBP) (345), Forkhead Box O1 (FOXO1) (105, 346), liver X receptor (LXR)α (111, 347), PGC-1α (117), and peroxisome proliferator-activated receptors γ (PPARγ) (119), as well as a growing number of transcription factors involved in cancer: GLI (348), HIC1 (349), LXRα/β (350), and NF-κB (333), and many others (TABLE 2). In the case of ChREBP (FIGURE 8A, II), O-GlcNAcylation can again have both activating, as well as inhibitory roles (345). In the context of high glucose conditions, parallel phosphorylation enhances O-GlcNAc levels to maintain transcriptional activity, which was consistent with earlier findings showing higher hepatic ChREBP O-GlcNAcylation and transcriptional activity in diabetic mice (351). However, under normal glucose conditions ChREBP is O-GlcNAc modified at a different Ser residue, which increases its interaction with other factors, such as 14-3-3, resulting in nuclear exclusion and decreased transcriptional activity (345). These findings demonstrate the dynamic, and sometimes opposing, roles that O-GlcNAcylation can have on transcriptional regulation. Although not comprehensive, these examples, and numerous others, solidify our knowledge that modification of transcription factors by O-GlcNAcylation is responsible for changing transcriptional activity through altered DNA binding, localization, stability, and interaction with other coregulators (352).
The second level of transcriptional regulation is on the transcriptional machinery itself. Although the earliest studies were focused on the modification of the transcription factors, it was also recognized by the early 1990s that RNA polymerase II (RNAP II) is a direct target of protein O-GlcNAcylation at its COOH-terminal domain (CTD) (353). Subsequently, this observation was expanded into the idea that RNAP II posttranslational regulation occurred as mutually exclusive states of phosphorylation and O-GlcNAcylation to establish different functional states of this transcriptional regulator (354) (FIGURE 8A, III). Later, Ranuncolo et al. (355), defined the functional significance of this O-GlcNAc cycling on RNAP II as a critical regulatory circuit of the assembly of the transcriptional preinitiation complex (PIC), a mechanism that was further extended to suggest that the hydrolysis of UDP-GlcNAc may actually serve as a high-energy donor to facilitate PIC formation and the elongation step (356,357). Owing to the repetitive nature of the CTD domain of RNAP II, the O-GlcNAcylation of RNAP II additionally allows for a series of highly heterogeneous glycoforms, providing the potential to regulate transcription in response to fluctuating cellular conditions (358). This mechanism has subsequently been suggested to perform a nutrient-sensing role to buffer transcriptional activity to match environmental metabolic demands, as well as developmental milestones (32).
Other transcriptional machinery shown to be modified by O-GlcNAcylation include TATA-binding proteins (TBP) (359), topoisomerase I (Topo I) (360), and nucleoporins (NUPs) of the nuclear pore complex (NPC) (361) (FIGURE 8A, IV). Although the roles of O-GlcNAcylation on TBP or Topo I are generally understudied, the presence of O-GlcNAc on NUPs is extensive and has been widely studied (5,6, 362). In the case of TBP, it has been suggested that O-GlcNAc directs the cycling of this protein on and off promoter regions during the regulation of transcription. While the regulation of Topo I by O-GlcNAc appears to directly mediate DNA relaxation and, therefore, transcriptional accessibility to genes. On the other hand, modification of the NPC by O-GlcNAcylation has growing evidence to support a direct role in regulation of nucleocytoplasmic transport (363). Furthermore, the relatively high steady-state levels of O-GlcNAcylation on components of the NPC appear to preserve the integrity of the complex by preventing its ubiquitination and degradation (364). Extending beyond this role in trafficking, components of both UDP-GlcNAc synthesis and protein O-GlcNAcylation appear to involve the NPC toward regulation of speckle and paraspeckle formation, suggesting additional roles in gene expression (365), as it relates to chromatin structure.
3.2. Epigenetics and Chromatin Remodeling
Epigenetics is defined as the changes that occur above the genome to alter transcriptional regulation in a manner responsive to other events (366); in this way, it is a logical continuation of the O-GlcNAc-mediated control of gene expression described above. More specifically, epigenetics encompasses three areas (FIGURE 8B): 1) histone modifications (e.g., O-GlcNAcylation and acetylation), 2) DNA modifications (e.g., 5-mC and 5-hmC), and 3) noncoding RNA (e.g., miR, lncRNA). Direct O-GlcNAc modification of histones combined with the cross talk between epigenetics and the machinery involved in O-GlcNAcylation (e.g., OGT, OGA) have revealed this to be another important level of O-GlcNAc regulation as discussed here.
Sakabe and colleagues (367) were the first to demonstrate that the histone proteins themselves were directly O-GlcNAcylated. Despite some evidence questioning the relative abundance of direct O-GlcNAc modification of histones in mammalian cells (368), a robust and growing number of studies have mapped histone O-GlcNAcylation (80, 369–373). The role of this histone modification is still being determined, with recent evidence suggesting that it mediates cell cycle progression by suppressing histone H3 phosphorylation (369, 373). Alternatively, O-GlcNAcylation of histone H2B appears to facilitate ubiquitin ligase binding, and further histone modification in a mechanism proposed to initiate transcriptional activation (80). Additional sites of O-GlcNAcylation have been identified on histone H2B with unknown consequences (370). While still other histone variants have been identified with O-GlcNAcylation sites, including histone H2A (371,372), a modification that is in parallel with H2A phosphorylation and inverse to H2A acetylation. Clearly, more work in this area needs to be completed as O-GlcNAcylation has been identified on all four core histone proteins (374).
Another interesting component of the influence O-GlcNAcylation has on histones, is its interaction with other posttranslational mechanisms of histone modification (FIGURE 8B, I). Specifically, it was shown that OGT directly interacts with the histone deacetylase and transcriptional corepressor, SIN3a (375). This and other observations directly connected histone O-GlcNAcylation with acetylation, an observation that was subsequently shown to be regulated by both pathological conditions, as well as physiological stresses such as exercise (376). A second potential link between these two posttranslational modifications came from the identification of a putative histone acetyltransferase activity in OGA (199), an observation that has subsequently come under debate (270). However, the link between these two modifications continues to be explored with a more recent study finding that OGT interacts with the histone acetyltransferase nonspecific lethal (NSL) to alter histone H4 acetylation through complex stabilization (377). The influence of O-GlcNAc on other posttranslational modifications continues to expand and provide novel insights into transcriptional regulation with its interaction with protein methylation. Specifically, OGT can O-GlcNAcylate the histone methyltransferase enhancer of Zeste homolog 2 [EZH2, (378)], which, in turn, regulates gene expression involved in skeletal muscle insulin sensitivity (379), tumor suppression (380), as well as neuronal memory formation (381). It is clear that we are only at the beginning of uncovering the cross-talk between O-GlcNAcylation and the histone code.
DNA modification by 5-methylcytosine was one of the first epigenetic modifications recognized in the early 50s (382), with direct enzymatic regulation identified by the early 60s (383). We now have a relatively robust understanding of the addition of methylation to CpG sites within the genome (384); however, DNA demethylation is more diverse and less well understood (385). In 2009, two articles were published on the discovery and characterization of TET, methylcytosine-dioxygenase, enzymes responsible for active DNA hydroxymethylation (5hmC) and demethylation (386, 387). Subsequently, it was shown that OGT interacted with TET protein providing a connection between DNA modifications and O-GlcNAcylation (388–391) (FIGURE 8B, II). The role by which this interaction influences gene expression varies by the context and TET family member. For example, TET2 interacts with OGT to mediate O-GlcNAcylation of histone H2B (388). However, when OGT interacts with TET1, DNA 5hmC appears to increase (391). Interestingly, these interactions can be part of a larger complex, including TET2/TET3/OGT and HCF1, further bringing in regulation of histone methylation to mediate transcriptional activation (389). OGT, in turn, can O-GlcNAc modify each of the three TET enzymes, reducing phosphorylation, which could alter their activity. Therefore, this interaction between O-GlcNAc and DNA modifications appears to be an important additional area of study that is beginning to provide important insight linking a number of epigenetic mechanisms.
The last, and least explored, area connecting O-GlcNAcylation and epigenetics is that of noncoding RNA (ncRNA) (FIGURE 8B, III). Although ncRNA was described for decades as a having a passive role as a messenger between DNA and protein, this idea has been proven incorrect, as ncRNAs are now well known to play active roles in transcriptional regulation (392). A unique mechanism by which O-GlcNAc machinery is directly regulated in an epigenetic manner is through long noncoding RNA (lncRNA). The first example of this relates to the location of the OGT gene on the X chromosome. Specifically, the XIST lncRNA is localized to the inactive X chromosome (Xi), and its presence regulates OGT levels in females but not males (393). As mentioned above, it was recently shown that a novel splice variant of OGT, nuclear OGT retained-intron (OGT-RI), may function as a nuclear ncRNA to regulate O-GlcNAc homeostasis (68). In addition to regulating O-GlcNAcylation, lncRNA can also be regulated by O-GlcNAcylation. For example, it was shown that under high-glucose conditions O-GlcNAcylation of p65 can activate the lncRNA for hyaluronan synthase 2 (HAS2), a naturally occurring antisense transcript (HAS2-AS1), leading to increased HAS2 to regulate hyaluronan synthesis (394). This mechanism provides an additional example by which O-GlcNAcylation can regulate, as well as be regulated by ncRNA.
At the other end of the spectrum, miRs have also been implicated in the regulation of O-GlcNAcylation. One of the first examples of this was in a study of failing heart in which Methusamy et al. (310) identified miR-539 as being induced. This miR induction, in turn, suppressed OGA protein levels, resulting in higher total protein O-GlcNAcylation. However, in hepatocarcinoma cells, miR-24-1 was shown to be lower in those cell lines with higher metastasis potential (395). The authors showed that this partly occurs though binding of miR-24-1 to the 3'-UTR of the transcript for OGT, which regulates levels of O-GlcNAcylation and stability of the oncoprotein c-Myc. In contrast, high glucose decreases levels of miR-200a and miR-200b in endothelial cells, resulting in less binding of these miRs to OGT mRNA and increased OGT protein levels associated with increased O-GlcNAcylation (308). Further, treating diabetic mice (db/db) with miR-200a and miR-200b mimics leads to decreased OGT levels that could reduce O-GlcNAcylation and inflammation. A final example of the link between miR and O-GlcNAc came from a screen that identified miR-501-3p for its potential to reduce OGT protein levels, decrease infectivity of hepatitis C virus, and decrease liver disease progression and development of cancer (306).
3.3. Insulin Signaling
In the late 1980s, it was established that insulin resistance induced by different interventions was associated with a decrease in translocation of the insulin-sensitive glucose transporter, GLUT4. In cultured adipocytes, it was shown that neither elevated glucose nor insulin alone could reduce insulin sensitivity; however, together, they resulted in a marked decrease in maximal insulin response (46). Additional studies demonstrated that glutamine was required for the development of decreased insulin sensitivity, and subsequently, Marshall et al. (46) demonstrated that increased flux through the HBP was a factor in this process. Prolonged treatment with glucosamine in the presence of insulin, decreased basal and insulin-stimulated glucose uptake, and reduced plasma membrane GLUT4 levels, providing further evidence that products from the HBP regulated insulin signaling (396). This was supported by Patti et al. (397), who found that glucosamine infusion in rats resulted in impaired insulin-stimulated glucose uptake and glycogen synthesis in skeletal muscle. In the same study, they showed that this was associated with a glucosamine-dependent increase in O-GlcNAc levels of IRS1/2. Moreover, overexpression of GFAT in skeletal muscle and adipocytes resulted in peripheral insulin resistance (398). Together, these studies provided the first indications that insulin signaling may be regulated via an HBP-mediated increase in O-GlcNAcylation.
Increasing overall O-GlcNAc levels in 3T3-L1 adipocytes, by inhibiting OGA with PUGNAc, resulted in impaired insulin-stimulated glucose uptake, with no changes in insulin-mediated increase in IR-β or IRS2 phosphorylation (133). On the other hand, insulin-induced increases of AKT and GSK3β phosphorylation were attenuated following PUGNAc treatment. While AKT and GSK3β were not found to be O-GlcNAcylated, IRS1 and β-catenin were both modified in a PUGNAc-dependent manner (133). Subsequent studies have shown that AKT is also a target for O-GlcNAcylation and this attenuates its function (121, 126). Several studies have questioned the role of O-GlcNAcylation in the development of insulin resistance. For example, lowering O-GlcNAc levels via overexpression of OGA or knockdown of OGT did not mitigate hyperglycemia-induced insulin resistance in adipocytes (399). In addition, the use of more specific OGA inhibitors than PUGNAc, such as NBuGt and 6-Ac-Cas (599, 600), failed to recapitulate earlier observations of insulin resistance seen with PUGNAc, although they did confirm that IRS-1 was a target for O-GlcNAcylation (600). Buse et al. (399) concluded that increased O-GlcNAcylation was just one of many factors involved in insulin resistance and was not necessarily required. On the other hand, in the setting of normoglycemia, a reduction of O-GlcNAc in the liver in vivo via overexpression of O-GlcNAcase significantly increased AKT activity (400).
Although the precise role of increased O-GlcNAc in contributing to cellular insulin resistance remains unclear, there is consensus over the fact that insulin treatment stimulates Tyr phosphorylation of OGT (FIGURE 9A) (242, 251). Insulin stimulation of adipocytes resulted in marked increased in Tyr phosphorylation of OGT, resulting in an increase in OGT activity and an association between OGT and IR. Insulin treatment also resulted in translocation of OGT from the nucleus to the cytoplasm (251). Another study reported that insulin triggered the translocation of OGT from the nucleus to the plasma membrane, which was facilitated by PIP3 binding to OGT (242). As a result, it was proposed that OGT contained a PIP3-binding domain; however, structural studies have not been able to confirm the presence of such a domain (90). The recruitment of OGT to the plasma membrane, resulted in increased OGT phosphorylation and activity, with subsequent increases in O-GlcNAcylation of IRS1, AKT, and other downstream targets of insulin signaling (242). PTP1B, has long been considered to be a major mechanism in attenuating insulin signaling through the dephosphorylation of Tyr residues in the activation loop of the insulin receptor (405). Interestingly, PTP1B has been reported to be O-GlcNAcylated, leading an increase in enzyme activity, thereby potentially contributing to a reduction in insulin signaling (FIGURE 9A). These findings suggest that OGT translocation and O-GlcNAcylation of target proteins are part of a feedback mechanism in which sustained activation of insulin signaling is suppressed in an insulin-dependent manner.
FIGURE 9.
Examples of O-linked β-N-acetylglucosamine (O-GlcNAc) regulation of cellular signaling pathways. A: Insulin signaling. On initiation, in response to insulin, there is autophosphorylation of the insulin receptor (IR) and subsequent phosphorylation and activation of Akt, FOXO and GSK3β and their downstream signaling pathways. On termination, insulin triggers rapid translocation of O-GlcNAc transferase (OGT) from the nucleus to the plasma membrane, where it is phosphorylated by the IR, which increases OGT activity, leading to the subsequent O-GlcNAcylation of insulin receptor substrate 1 (IRS1), AKT, FOXO, and GSK3β, thereby attenuating activity at multiple steps in the insulin signaling cascade. The protein tyrosine phosphatase 1B (PTP1B) is responsible for decreasing phosphorylation of IR and attenuating insulin signaling. It is also O-GlcNAcylated, which increases its activity contributing to a further reduction in insulin signaling. It has been reported that translocation of OGT to the plasma membrane is via binding to PIP3 (242), which is generated in response to activation of insulin signaling; however, to date, structural studies have not revealed a phosphatidylinositol-3-phosphate (PIP3)-binding motif in OGT(90). B: Calcium signaling. Intracellular Ca2+ increases in response to diverse number of agonists, either as a result of Ca2+ release from ER/SR alone or via activation of plasma membrane Ca2+ channels as a result of ER/SR Ca2+ release (store-operated Ca2+ entry, SOCE). This increase in Ca2+ leads to activation of calmodulin (CaM), resulting in the phosphorylation and activation of both CaMKII and IV, which are responsible for regulation of numerous cellular processes. CaMKII also phosphorylates OGT, increasing its activity, and in a feedback manner, OGT O-GlcNAcylates CaMKII/IV reduces their activities. Ca2+-CaM also activates the calcium-dependent phosphatase calcineurin, which is responsible for regulating diverse cellular processes, and it has been reported that increased OGT expression and O-GlcNAcylation is sufficient to activate calcineurin-mediated transcription pathways (298). Influx of extracellular Ca2+ has been shown to contribute to the stress-induced increases in O-GlcNAc levels (312). Therefore, it is possible, that Ca2+-CaM and/or calcineurin regulate OGT (or OGA) activities; however, this has yet to be demonstrated experimentally. Multiple contractile proteins are O-GlcNAc modified (TABLE 2), and the increases in O-GlcNAc levels that can occur in diseases, such as diabetes, reduces Ca2+ sensitivity of some contractile proteins (147, 150). Phospholamban (PLB) and SERCA are responsible, in part, for the reuptake of Ca2+ into the sarcoplasmic reticulum (SR), contributing to muscle relaxation. Increases in O-GlcNAc levels decrease the activities of both PLB and SERCA directly or indirectly, which could be a contributing factor to impaired myocardial relaxation (i.e., diastolic function) that occurs with diabetes. The endoplasmic reticulum (ER)/SR transmembrane protein STIM1, which plays a key role in regulating SOCE, is also a target for O-GlcNAcylation and increases in O-GlcNAc levels impairs its function and attenuates SOCE (401). Increasing O-GlcNAc levels attenuates mitochondrial Ca2+ overload, although the precise mechanisms are unclear(163, 402). CaMKII phosphorylation of the mitochondrial Ca2+ uniporter (MCU) potentiates mitochondrial Ca2+ overload (403); therefore, as O-GlcNAcylation of CaMKII decreases its activity, this may represent a potential protective mechanism. However, others have questioned the role of CaMKII in the regulation of mitochondrial Ca2+ uptake by the MCU (404).
One limitation of many studies examining the role of O-GlcNAc in insulin signaling is that they have been mostly in cultured adipocytes; consequently, it may differ in other insulin-sensitive cells and tissues. Nevertheless, there is no doubt that key elements of the insulin signaling pathway, including IRS1/2, PDK1, AKT, and GSK3β (See TABLE 2) are all targets for O-GlcNAcylation and that in all cases, increasing O-GlcNAcylation suppresses their activities (290).
3.4. Calcium Signaling
There is growing recognition of reciprocal regulation between Ca2+ signaling and protein O-GlcNAcylation, perhaps best exemplified by the regulation of CaMKIV activity via O-GlcNAcylation and OGT activity regulated by CaMKIV-mediated phosphorylation (FIGURE 9B) (167). Depolarization of neuroblastoma cells resulted in a rapid increase in OGT activity and O-GlcNAc levels that was shown to be mediated by CaMKIV phosphorylation of OGT (286). An early indication of the potential for Ca2+/O-GlcNAc cross talk was the identification of seven O-GlcNAc modification sites on synapsin I that were clustered around its regulatory phosphorylation sites, and O-GlcNAcylation of these sites reduced the affinity of CaMKII for synapsin I (151). In the heart, CaMKII has also been shown to be O-GlcNAcylated on Ser-279, and this results in autonomous activation of CaMKII, which, in the setting of diabetes, was linked to increased arrhythmias (165). In liver, similar to the observations in neuroblastoma cells, CaMKII phosphorylated OGT, increasing O-GlcNAc levels, which subsequently activated autophagy (181). Given the diverse array of cellular functions that are regulated by the CaMK family of proteins (406), it is likely that many more links with O-GlcNAcylation remain to be discovered.
The nuclear factor of activated T cells (NFAT) family of transcription factors, are widely distributed and contribute to the regulation of numerous processes, including the immune system, cardiac and skeletal muscle, and brain. Ca2+-dependent activation of calmodulin, activates the phosphatase calcineurin which rapidly dephosphorylates NFAT, resulting in its nuclear translocation and activation. In neonatal cardiomyocytes NFAT translocation is initiated by hypertrophic agonists, such as angiotensin II (ANG II) or PE, and hyperglycemia was found to inhibit this translocation in an HBP-dependent manner (407). A subsequent study demonstrated that increasing O-GlcNAc levels attenuated the ANG II-induced increase in cytosolic Ca2+ (408). More recently, it was reported that activation of O-GlcNAc signaling was required for NFAT translocation in cardiomyocytes, and this could be blocked with the calcineurin inhibitor cyclosporin A (298). Thus, O-GlcNAcylation may have regulatory roles in both the initiation of NFAT signaling, as well as its inhibition (FIGURE 9B). Another protein that has been established as a key player in Ca2+-dependent activation of NFAT, particularly in the immune system, is stromal interacting molecule 1 (STIM1), via its role in regulating the store operated calcium entry pathway (SOCE) (409). STIM1 is positively and negatively regulated by phosphorylation, and increases in O-GlcNAc increased basal phosphorylation, but attenuated activation-dependent increases in phosphorylation (401). STIM1, is a highly conserved protein, considered to be a core component of mammalian Ca2+ signaling (409); consequently, its functional regulation by O-GlcNAc could have far reaching consequences.
In skeletal and cardiac muscle, Ca2+ also plays a central role in contraction of the myofilaments, via Ca2+ binding to key proteins, as well as regulating Ca2+ release and uptake by the ER/SR (FIGURE 9B) (410). Skeletal muscle myosin was the first contractile protein that was shown to be O-GlcNAcylated, with all isoforms being modified (411). Subsequent studies identified actin and myosin light chain (MLC) 1,2 as O-GlcNAc targets and found that in skinned muscle fibers, acute increases in O-GlcNAc levels reduced Ca2+ sensitivity, suggesting a possible role for O-GlcNAc in regulating skeletal muscle contractility (146). Myofilament proteins from cardiac muscle were also found to be O-GlcNAcylated, and in addition to those identified in skeletal muscle, troponin I (TnI) was modified at Ser-150, which is a phosphorylation site that regulates Ca2+ sensitivity (147). Pharmacological increases in O-GlcNAc levels reduced Ca2+ sensitivity, consistent with the earlier skeletal muscle study (146).
Uptake of cytosolic Ca2+ into the ER/SR is an important mechanism for regulating Ca2+ signaling, as well as muscle contraction, and this is controlled by sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) and its inhibitor phospholamban (PLB). Phosphorylation of PLB attenuates its inhibitory effect, thereby, facilitating more rapid uptake of Ca2+ into the ER/SR by SERCA. PLB is a target for O-GlcNAcylation, and Ser-16 was found to be the most likely target (194), although to date, this has not been confirmed by MS. Increased PLB O-GlcNAc levels, either by inhibition of OGA or under conditions of hyperglycemia, reduced PKA-mediated phosphorylation of PLB Ser-16 (194); conversely, knockdown of OGT significantly increased PLB phosphorylation (FIGURE 9B). Increased O-GlcNAc levels, were associated with lower SERCA activity and a greater association between SERCA and PLB (194). It was concluded that the increase in PLB O-GlcNAcylation could be a factor in the slower reuptake of Ca2+ by the ER/SR in the heart that occurs with diabetes. SERCA expression in the heart, has been shown to be decreased under conditions of increased O-GlcNAc levels, and this was attributed to an increase in O-GlcNAcylation of the transcription factor Sp1 (263). While Yokoe et al. (194), reported that SERCA was not an O-GlcNAc target, others have shown that it is O-GlcNAcylated (412, 413), although the functional consequence of this modification remains to be identified.
Another important contributor to cellular Ca2+ signaling is the inositol 1,4,5-trisphosphate (InsP3)-receptor, which is localized to the ER/SR membrane and is activated by InsP3 generated in response to a variety of extracellular stimuli. In C2C12 myotubes, increased O-GlcNAc levels attenuated bradykinin-induced production of IP3 and associated InsP3R-mediated Ca2+ release, which was associated with O-GlcNAcylation of PLC-β1 (414). The InsP3 receptor type 1 (InsP3R-1) itself is O-GlcNAcylated under basal conditions and this could be increased following treatment with the OGA inhibitor PUGNAc (415). Moreover, decreases in basal O-GlcNAc levels resulted in a marked increase in channel opening probability, and conversely, increasing O-GlcNAc levels reduced channel opening probability. These findings demonstrated a potential role for O-GlcNAc in regulating InsP3R-1 under physiological conditions (FIGURE 9B) (415). A subsequent study from the same group found that InsP3R-2 was not O-GlcNAcylated, and its function was unaltered by global changes in O-GlcNAc levels (416). Interestingly, however, they found that InsP3R-3 was O-GlcNAcylated, but that changes in its O-GlcNAc levels had the opposite effect to those observed with InsP3R-1. InsP3R is ubiquitously expressed, although the different isoforms exhibit tissue-specific differences in their function (417); consequently, there is a clear need for a better understanding of the role of O-GlcNAc in their regulation.
It is clear that O-GlcNAcylation provides a key link between nutrient and Ca2+ signaling contributing to the regulation of the majority of key Ca2+ signalling pathways (FIGURE 9B). What is also becoming increasingly evident is that O-GlcNAc levels are regulated in a Ca2+-dependent manner, as exemplified by CaMKII/IV-mediated phosphorylation of OGT, resulting in increased activity and high O-GlcNAc levels (181, 286). Stress induced increases in cellular O-GlcNAc levels have also been shown to be dependent on extracellular Ca2+ and activation of CaMKII (312). The role of Ca2+ in regulating GFAT activity is poorly understood, even though it can be phosphorylated by CaMKII, and to date, the role of Ca2+ in regulating OGA activity is unknown.
3.5. Metabolism and Mitochondrial Function
As discussed in sect. 2.1, substrate availability plays a key role in regulating HBP flux and O-GlcNAcylation; however, what is less well known is that O-GlcNAcylation contributes to the regulation of metabolism at multiple levels. For example, almost every enzyme in glycolysis has been shown to be O-GlcNAc modified from GLUT4 to pyruvate dehydrogenase (FIGURE 10A) (418). In addition, O-GlcNAcylation of glucose-6-phosphate dehydrogenase regulates pentose phosphate pathway activity, and GSK3β O-GlcNAcylation regulates glycogen synthesis. In the heart, increased flux through the HBP has been linked to greater fatty acid oxidation, potentially via O-GlcNAcylation of the fatty acid transporter, CD36 (419,420). Similarly, in adipocytes, activation of the HBP and increasing O-GlcNAc levels also stimulated fatty acid oxidation (421). Interestingly, a splice variant of OGA, sOGA, is associated with lipid droplets, further supporting a connection between O-GlcNAcylation and the regulation of lipid metabolism (273). In addition to direct regulation of metabolic fluxes, O-GlcNAcylation also contributes to the transcriptional regulation of metabolism via O-GlcNAcylation of multiple transcription factors, including PGC-1α, FoxO3, and CREB (100, 105, 117, 598).
FIGURE 10.

Metabolism and mitochondrial O-GlcNAcylation. A: majority of enzymes in glycolysis and glucose metabolism are targets for O-GlcNAcylation (418). ALD, aldolase; ENO, enolase; GAPDH, glyceraldehyde-6-phosphate dehydrogenase; GFAT, glutamine fructose-6phosphate amidotransferase; GP, glycogen phosphorylase; G6PDH, glucose-6-phosphate dehydrogenase; GS, glycogen synthase; HEX, hexokinase; LDH, lactate dehydrogenase; PFK, phosphofructokinase; PDC, pyruvate dehydrogenase complex; PGI, phosphoglucoisomerase; PGK, phosphoglycerate kinase; PGM phosphoglucomutase; PK, pyruvate kinase; UDP-GP, UDP-glucose pyrophosphorylase. B: wide range of different types of mitochondrial proteins that are O-GlcNAcylated. C: map of the O-GlcNAc modification sites on proteins that are central to the regulation of energy metabolism and mitochondrial function. Data largely based on a number of recent proteomic studies (138,139). Blue squares denote O-GlcNAc, whereas numbers inside blue squares indicate number of O-GlcNAc sites on the protein.
The identification of a splice variant of OGT, with a mitochondrial targeting sequence (422), suggested that mitochondrial proteins may be targets for O-GlcNAcylation. However, early reports indicated that there was little, if any, O-GlcNAcylation in mitochondria. It was proposed that the lack of mitochondrial O-GlcNAc was due to the lack of substrate, as there was no clear mechanism for UDP-GlcNAc transport into the mitochondria (423). Nevertheless, a number of studies suggested that alterations in cellular O-GlcNAc levels could alter mitochondrial function and particularly their response to stress. For example, increasing O-GlcNAc levels in cardiomyocytes with PUGNAc attenuated the loss of mitochondrial membrane potential induced by oxidative stress, and this was associated with O-GlcNAcylation of voltage-dependent anion channel (VDAC)1 (163). Both glucosamine treatment and OGT overexpression increased O-GlcNAc levels and reduced ischemia-reperfusion-induced injury in isolated cardiomyocytes (402). The same treatments attenuated hydrogen peroxide-induced mitochondrial membrane potential and enhanced recruitment of the anti-apoptotic protein BCL2 to the mitochondria (402). Subsequent studies suggested BCL2 itself might be a target for O-GlcNAcylation (424).
Although acute activation of O-GlcNAc levels protected mitochondria from oxidative stress, increases in O-GlcNAcylation associated with hyperglycemia were found to have adverse effects on mitochondrial function. For example, in pancreatic β-cells, hyperglycemia increased O-GlcNAc levels on the chaperone heat shock protein (HSP) 60, interfering with its binding to the proapoptotic factor BCL2 Associated X, Apoptosis Regulator (BAX), resulting in BAX translocation to the mitochondria and subsequent cytochrome-c release (425). Hyperglycemia resulted in impaired activity of mitochondrial complexes in neonatal cardiomyocytes, which could be reversed by overexpression of OGA. High glucose levels also increased O-GlcNAcylation on subunits of mitochondrial complexes, which was reduced following overexpression of OGA (161). Hyperglycemia also increased O-GlcNAcylation of dynamin-related protein 1 (DRP1), increasing mitochondrial fragmentation and decreasing mitochondrial membrane potential (159). In neurons, hyperglycemia impaired mitochondrial motility in an O-GlcNAc-dependent manner, due to O-GlcNAcylation of MILTON1 (also known as TRAK1), a trafficking protein that is essential for mitochondrial movement (160). 8-oxoguanine DNA glycosylase (Ogg1), is involved in mitochondrial DNA repaired and is O-GlcNAcylated in response to hyperglycemia, decreasing its activity and possibly contributing to increased mtDNA damage (426).
In 2009, Hu et al. (161), identified O-GlcNAcylation on a number of mitochondrial oxidative phosphorylation complex proteins, which was associated with decreased complex I, II, and IV activity in mitochondrial of cardiomyocytes exposed to high glucose. Cao et al. (157) identified 11 O-GlcNAcylated proteins from rat liver mitochondria, which included enzymes in the TCA cycle, as well as those involved in ATP synthesis (FIGURE 10B). It remained unclear, however, as to how mitochondrial proteins could be modified by O-GlcNAc. In 2015, transport of 3H-UDP-GlcNAc into cardiac mitochondria was reported and the pyrimidine nucleotide carrier (PNC1) was identified as the UDP-GlcNAc mitochondrial transporter (427). Immunogold labeling and live cell imaging were also used to demonstrate the presence of OGA localized to the mitochondria, demonstrating for the first time that an active O-GlcNAc cycle was present in the mitochondria (427). Subsequent proteomics studies identified 86 mitochondrial proteins as O-GlcNAc targets that were involved in a diverse array of mitochondrial functions, including the TCA cycle, oxidative phosphorylation, fatty acid oxidation, and calcium regulation (FIGURE 10C) (138, 139). Others have been unable to detect mOGT isoform in cells or tissues, and ncOGT was reported to be the isoform responsible for O-GlcNAcylation of mitochondrial proteins (428). It has also been suggested that mOGT regulates mitochondrial stress responses, while ncOGT is responsible for the regulating mitochondrial bioenergetics (429). Thus, additional work remains to elucidate the fundamental mechanisms regulating O-GlcNAc cycling in the mitochondria, as well as understanding the physiological role of O-GlcNAcylation in regulating mitochondrial function.
3.6. Cell Survival/Autophagy
Pharmacological studies of OGT or OGA inhibition in INS-1 and βTC-6 cells have linked high O-GlcNAcylation with cell death and increased O-GlcNAcylation with reduced phosphorylation of Ser-473 on AKT1 as the potential mechanism (122). In human embryonic kidney 293 and HeLa cells, hyper-O-GlcNAcylation by overexpression of OGT induced apoptosis, which was associated with increased AKT O-GlcNAcylation and decreased AKT phosphorylation (124). Hyper-O-GlcNAcylation is also evident in response to cerebral ischemia (124). In a model of diabetic retinopathy, increased O-GlcNAcylation of NF-κB was associated with retinal ganglion cell death (430). Recent studies also have shown that O-GlcNAcylation on Ser-56 and Ser-57 of c-Fos is increased in 5XFAD mouse model of Alzheimer’s disease, as well as neuroblastoma cells exposed to amyloid beta (Aβ), this increased its stability and interaction with c-JUN, increased target gene Bim expression, and promoted cell death (431). Increased O-GlcNAc levels have also been reported to exacerbate acetaminophen-induced liver injury, whereas lower O-GlcNAc levels, resulting from liver-specific OGT knockout reduced the degree of injury (432). Increased cardiomyocyte apoptosis observed in a rat model of diabetes was linked to an overall increase in O-GlcNAc levels, as well as increased O-GlcNAcylation and reduced phosphorylation of the proapoptotic protein BCL2-associated agonist of cell death (or BAD) (433).
In contrast to the increase in O-GlcNAc levels, leading to cell death, Zachara et al. (289), demonstrated that exposure of cells to a variety of stressors induced an increase in O-GlcNAc that they showed was cytoprotective and represented an endogenous cell survival response. Subsequent studies revealed a link between the increase in O-GlcNAc and cell survival with the greater induction of heat shock protein expression via inactivation of G3K3β (280). A number of proteins involved in the DNA repair pathway have also been shown to be regulated either directly or indirectly by O-GlcNAcylation (89, 319, 434–436). O-GlcNAcylation is required for the normal regulation of DNA damage pathways, and OGT is recruited to sites of DNA damage (434). Additional studies found that pharmacologically increasing O-GlcNAc levels was cytoprotective, particularly in the setting of acute oxidative stress or ischemia-reperfusion (163, 402, 437). There have been several reports demonstrating a strong correlation between O-GlcNAc levels and tissue injury, with low O-GlcNAc levels associated with increased injury (437–439). The specific mechanisms by which the increase in O-GlcNAc is protective remains to be determined; however, there are indications that it attenuates Ca2+ overload, protects mitochondria against oxidative stress, and attenuates proinflammatory responses (163, 402, 440–443).
Autophagy is an important cell survival mechanism, and a role for O-GlcNAc in regulating autophagy is starting to emerge. Recent studies have shown Unc-51-like autophagy activating kinase 1 (ULK1) (444), mammalian target of rapamycin (mTOR) (445), hypoxia-inducible factor 1α (HIF1 α) (446), synaptosome-associated protein 29 (SNAP29), tubulin polymerization-promoting protein (TPPP), Golgi reassembly-stacking protein of 55 kDa (GRASP55) (180, 447–450), all of which play important roles in the regulation of autophagy, are modified by O-GlcNAcylation. Increased O-GlcNAcylation of SNAP29 and GRASP55 resulted in decreased autophagy (FIGURE 11) (180, 450). In C. elegans, an ogt mutation elevated autophagy during development and knockdown of OGT in mammalian cells also promoted autophagy (180). Reduced OGT was associated with increased formation of the SNARE complex, which includes SNAP29 and increased O-GlcNAcylation of SNAP29-inhibited autophagy at the step of autophagosome maturation (180). Impaired autophagic signaling in the diabetic heart was associated with increased O-GlcNAc levels and O-GlcNAcylation of BCL2 and Beclin 1 (424). Arsenic-induced inhibition of autophagic flux was shown to be mediated by O-GlcNAcylation of SNAP29, and this was prevented by reducing O-GlcNAc levels by knockdown of OGT (451). Transfection of SNAP29 containing an O-GlcNAc site mutant attenuated arsenic inhibition of autophagy, further substantiating the role of SNAP29 O-GlcNAcylation in regulating autophagy (451).
FIGURE 11.

O-linked β-N-acetylglucosamine (O-GlcNAc) regulation of cell survival and autophagy. A role of O-GlcNAc in cell survival is evidenced by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) knockout/knockdown and overexpression studies, although the identities of specific modified proteins in promoting cell death or survival are largely unclear. Among others, AKT, NF-κB, and c-Fos have been shown to be O-GlcNAcylated, and their O-GlcNAcylation is associated with cell death processes. In terms of pathology, Alzheimer’s disease-associated tau phosphorylation has been shown to be attenuated by enhancement of its O-GlcNAcylation. Although there are also studies demonstrating that increased O-GlcNAcylation can enhance proteotoxicity, conceivably by altering the degradation of O-GlcNAcylated forms of specific proteins. The involvement of O-GlcNAcylation in autophagy is best demonstrated in studies on SNAP29 and GRASP55, whose O-GlcNAcylation attenuates autophagic flux.
A mutagenesis study has shown that single mutations of GRASP55 at Ser-389, Ser-390, Thr-403, Thr-404, and Thr-413 decreased its O-GlcNAcylation, while a single mutation at Ser-380 increased its O-GlcNAcylation, whereas, mutating all Ser-389, Ser-390, Thr-403, Thr-404, and Thr-413 sites or these five sites in combination with Ser-380 both abolished its O-GlcNAcylation (FIGURE 11) (450). Notably, these sites are not the previously identified phosphorylation sites, and mutating known phosphorylation sites did not affect GRASP55 O-GlcNAcylation. During normal conditions, GRASP55 is O-GlcNAcylated, and in response to glucose deprivation, it is de-O-GlcNAcylated and interacts with LC2-II and lysosomal associated membrane protein 2 (or LAMP2), activating autophagy through enhancing autophagosome-lysosome fusion (450).
In neuroblastoma SH-SY5Y cells, O-GlcNAcylation increased autophagy, and this was linked to O-GlcNAcylation of ATG4B, which enhanced its proteolytic activity (452). However, the O-GlcNAcylation sites on ATG4B were not identified, and it remains to be determined whether ATG4 O-GlcNAcylation is necessary or sufficient for activation of autophagy (452). Interestingly, OGT interacted with ATG4B and downregulation of OGT decreased ATG4B. Pharmacological inhibition of OGA activated mTOR and inhibited autophagic flux in primary neurons (453), while in glia/primary neuron-mixed cultures, a similar intervention enhanced autophagic flux without affecting mTOR (454). In vivo treatment with the OGA inhibitor, Thiamet G, increased autophagic flux in the brain (454). Taken together, there is growing evidence that there is cross talk between autophagy and protein O-GlcNAcylation and that autophagic proteins are targets for O-GlcNAcylation; however, the precise role of O-GlcNAc in regulating autophagy remains unclear (FIGURE 11).
Conceivably, O-GlcNAcylation of a subset of proteins may also change their recognition and/or degradation by autophagy; therefore, as a result, inhibition or activation of autophagy will change the ratio of O-GlcNAcylated versus un-O-GlcNAcylated species of specific proteins. Additionally, activation or inhibition of autophagy may change the levels and the intracellular localization of OGA and OGT, or autophagy adaptor proteins that recognize specific autophagy substrates. Such an example has been demonstrated in breast cancer cells in which rapamycin treatment decreased O-GlcNAc levels by decreasing OGT level (455). These assumptions need to be critically examined by experiments to be substantiated. Specific effects of autophagy activation or inhibition at distinct steps on specific protein species and functional consequences need to be defined.
3.7. Circadian Clock
The circadian clock, a transcriptional mechanism that results in ∼24-h cycles of many physiological processes, has been identified in almost all cell types (456). In mammals, there is a central clock located in the suprachiasmatic nucleus (SCN), and there are peripheral clocks located in all other tissues and cells. The central clock is sensitive to light and regulates systemic rhythms in activity, body temperature, hormone levels, immune function, and blood pressure, whereas peripheral clocks regulate tissue-specific processes. Dyssynchrony between the SCN and external cues, as well as between the SCN and peripheral clocks, which occur as a result of shift work, jet lag, or poor sleep, have been linked to an increased risk of metabolic disease, including Type 2 diabetes, cardiovascular diseases, and cancer (457). A number of studies have demonstrated that there is complex cross talk between O-GlcNAc cycling and regulation of the circadian clock.
In the heart there were time-of-day-dependent changes in overall O-GlcNAc levels, as well as OGT and OGA protein levels that were not present when the cardiomyocyte circadian clock was deleted (344). In the same study, the authors showed that BMAL1, a circadian clock-regulating transcription factor, was also O-GlcNAcylated in the heart. It was also demonstrated that pharmacologically increasing O-GlcNAc levels resulted in a phase shift in the SCN clock (344). This was the first indication that not only were cellular O-GlcNAc levels regulated by the circadian clock, but also that acute changes in O-GlcNAcylation directly affected the circadian clock. Berthier et al. (65) found that REV-ERBα, a transcription factor that integrates circadian clock and energy metabolism, interacted with OGT preventing its degradation and, thus, increasing OGT activity, providing a potential mechanism by which the circadian clock can regulate cellular O-GlcNAc levels.
In Drosophila, upregulation of OGT increases the duration of circadian rhythms, whereas OGT knockdown shortened them (458). This was associated with changes in O-GlcNAcylation of the Drosophila PERIOD protein (dPER) and stabilization of dPER. Several studies further demonstrated a key role for O-GlcNAc in regulating the mammalian circadian clock (116, 330, 459). Central components of the circadian clock, including BMAL1, PER2, and CLOCK, are all targets for O-GlcNAcylation, and O-GlcNAc modification stabilized them, at least in part, by preventing their ubiquitination (330). There was also a consistent observation that reduced O-GlcNAc levels shortened, while increased O-GlcNAc lengthened, the duration of circadian rhythms. PER2 was shown to be O-GlcNAcylated in a region that regulates sleep phase in humans, competing with phosphorylation in the same region (116). Modulation of O-GlcNAc levels in the liver by either overexpression or knockout of OGT changed the diurnal rhythms of circulating glucose levels (330). Together, these studies demonstrate that OGT, OGA, and O-GlcNAcylation are key components of the circadian clock and, as such, contribute to the regulation of daily rhythms in physiological processes in central and peripheral clocks. Moreover, since dysregulation in O-GlcNAc cycling is associated with chronic diseases, this might also contribute to circadian dysregulation seen in response to stress and disease (460).
4. O-GlcNACYLATION IN HEALTH AND DISEASE
The wide-ranging effects of O-GlcNAcylation on protein and cellular function combined with its integration of nutrient availability and diverse signaling pathways make it ideally positioned to coordinate cellular response to physiological and pathological stimuli. In this section, we will summarize what is known about the role of O-GlcNAc involvement in normal physiology and in pathologies of diseases.
4.1. Development
In Xenopus and Drosophila, there are marked changes in the O-GlcNAc proteome during embryogenesis, including changes in the extent of O-GlcNAcylation on specific proteins, as well as the number of proteins that are O-GlcNAc targets (461, 462). Key regulators of embryonic development in Drosophila are O-GlcNAcylated, and disruption of O-GlcNAc cycling resulted in abnormal development (463, 464). Polycomb group (PcG) proteins play an important role in developmental regulation in Drosophila and mammals. In Drosophila, there is overlap between O-GlcNAc modification sites and PcG protein binding sites, indicating a role for OGT in regulating PcG gene silencing, further supporting a critical connection between O-GlcNAc cycling and developmental regulation (465).
In mice, the loss of EMeg32, which catalyzes the conversion of glucosamine-6-phosphate to glucosamine N-acetyl-6-phosphate, is embryonically lethal at day 7.5 (228). EMeg32 deletion, significantly decreased UDP-GlcNAc levels. This primarily leads to loss of O-GlcNAc protein modification rather than Golgi-mediated glycosylation, suggesting that O-GlcNAcylation plays an essential role in development. This is supported by the observation that OGT gene deletion is embryonically lethal at the blastocyst stage (∼day 4.5) and that this also results in embryonic stem cell death (208). To determine the effects of a loss of OGT in specific cell types O’Donnell et al. (16) used Cre-recombinase approaches to delete OGT in T cells, fibroblasts, and neurons. They found that loss of OGT resulted in T-cell apoptosis and growth arrest of fibroblasts. Selective ablation of OGT in neurons resulted in much smaller pups that did not survive beyond 10 days and was associated with marked increases in phosphorylation of tau (16). Cardiomyocyte-specific deletion of OGT (cmOGT KO) resulted in marked postnatal lethality, with only 12% surviving to weaning age. The surviving cmOGT KO mice exhibited signs of hypertrophy and heart failure characterized by dilation and accompanied by increased fibrosis and apoptosis (206). The constitutive KO of OGA, resulted in embryonic developmental delay and very high perinatal mortality, which was associated with mitotic defects (466). Keembiyehetty et al. (17) extended these observations, demonstrating that following OGA deletion, only 3% of pups survive to weaning, with the majority dying soon after birth. However, no structural defects were observed at either E16.5 or E18.5 in OGA KO animals, and surviving KO pups were almost indistinguishable from WT littermates. They observed profound hypoglycemia in KO mice, suggesting that perinatal lethality was likely a result of impaired glycogen metabolism. Collectively, these studies demonstrate the importance of normal O-GlcNAc cycling in developmental regulation.
Maternal stress, whether it is due to overnutrition or undernutrition, drug or alcohol consumption, or other factors, is known to increase the risk of neurodevelopmental and metabolic disorders (467). In a mouse model of early prenatal stress, placental OGT levels were significantly decreased; moreover, a placenta-specific reduction in OGT adversely affected early neurodevelopment, which was more pronounced in males compared to females (468,469). Severe hyperglycemia in rats, resulted in structural and functional changes in the placenta that was associated with increased O-GlcNAc levels in endothelial and trophoblast cells (470). In a model of maternal corticosterone exposure, Pantaleon et al. (471) demonstrated that there was an increase in OGT levels and O-GlcNAcylation of AKT in male, but not female, placentae. Exposure to corticosteroids in utero also resulted in depression-like behavior that was more pronounced in males, possibly due to an OGT-mediated effect on mitochondrial motility (472). One mechanism contributing to sex-specific differences in placental OGT could be due to increased resilience in females via increased OGT-mediated stabilization of EZH2 and higher levels of the epigenetic modification H3K27me3 (473). The observations of sex-specific differences in OGT and O-GlcNAc in the placenta provide a potential mechanism underlying sexual dimorphic responses to maternal stress.
An important consideration regarding the role of OGT and O-GlcNAc in developmental regulation is the fact that in all mammals, OGT is present on the X-chromosome: XqD in mice, and Xq13 in humans (208, 474). One consequence of this is that when maternally inherited, female heterozygous OGT KO is embryonically lethal, whereas when paternally inherited, mice are viable (475). Moreover, because of X-inactivation, female heterozygous OGT KO mice would be expected to see a reduction in OGT by ∼50%; however, this is not the case, suggesting a selection bias in early development against cells expressing the inactivated maternal allele (208). OGT expression in most tissues is equal between males and females, indicating that it is subject to silencing by X-inactivation of one of the two X-chromosomes in females (475). The fact that OGT is an X-linked gene raises the possibility that it could be a factor in sex differences in susceptibility to certain diseases. This could occur when a mutation occurs on one of X-chromosomes, which in females could be silenced, thereby, reducing, or even eliminating risks for certain diseases (FIGURE 8B, III). An example of this is reflected in the recent reports of several OGT mutations associated with X-linked intellectual disability (28,29, 476). In contrast, in two female twins, a wild-type OGT was silenced via X-inactivation, leading to expression of an OGT mutation in the catalytic domain, resulting in intellectual disability and developmental delays (28). Incomplete X-inactivation or X-reactivation could also be a factor in diseases that are known to predominantly affect women, including autoimmune diseases, such as lupus (475). Indeed, hypomethylation of X-linked genes, including OGT, was observed in women with active lupus compared to inactive disease or healthy women (477). The degree of overexpression of OGT mRNA and protein was directly related to the severity of disease, and both were higher in women with lupus compared to men with lupus, consistent with a contribution from the inactive X-chromosome (477). It has also been proposed that OGT reactivation could contribute to cancer progression in women (475).
4.2. Obesity and Diabetes
As discussed above, early studies linked increased HBP flux to the possible development of insulin resistance, and other reports demonstrated a close relationship between O-GlcNAcylation and insulin signaling. Consequently, excessive HBP flux and sustained increases in O-GlcNAc levels have been implicated in the development of metabolic disease. For example, overexpression of GFAT in skeletal muscle and adipose tissue resulted in systemic hyperinsulinemia and insulin resistance, and its overexpression in the liver caused obesity, dyslipidemia accompanied by insulin resistance, and in older animals, overt diabetes (478). A selective increase of GFAT in pancreatic β-cells resulted in hyperinsulinemia and insulin resistance (478). These findings support the concept that the HBP contributes to systemic metabolic regulation, but that under conditions of excess, it leads to metabolic dysfunction characteristic of insulin resistance and diabetes. Single-nucleotide polymorphisms in the GFAT2 gene are linked to Type 2 diabetes and increased risk of diabetic complications, such as diabetic nephropathy (479,480). In leukocytes, GFAT2 gene expression was found to be lower in prediabetic Caucasian subjects compared with controls, and this was even more pronounced in diabetic individuals; GFAT1 was not different between groups (481). How these changes related to GFAT1/2 protein levels or activities is not known.
OGT overexpression in the liver also leads to insulin resistance, supporting the idea that the effects of GFAT overexpression are most likely the result of increased O-GlcNAcylation (242). Additional support for a role of O-GlcNAc cycling in metabolic regulation comes from the observation that hepatic knockdown of OGT resulted in a decreased expression of gluconeogenic genes and improved glucose homeostasis in diabetic mice (118). Moreover, OGA polymorphism has been linked to Mexican Americans with increased risk for diabetes (482–484). Both OGT and OGA mRNA levels were significantly lower in diabetic individuals compared to both prediabetic and control subjects (481); it is unclear what factors are driving these changes or how they translate to changes in protein levels or activity. Muscle O-GlcNAc levels were significantly increased in patients with Type 2 diabetes compared with controls, regardless of insulin levels; however, there were no changes in protein levels of GFAT, OGT, or OGA (379). Skeletal muscle-specific OGT knockout increased overall energy expenditure, markedly increased insulin sensitivity, and reversed insulin resistance associated with a high-fat diet-induced insulin resistance (379, 485). Modest overexpression of OGT in skeletal muscle and adipose tissue resulted in insulin resistance and increased circulating leptin levels, despite having no effect on body weight, fat pad weight, or fasting blood glucose levels. O-GlcNAc signaling has also been implicated in regulating whole body metabolism in response to overnutrition by modulating macrophage proinflammatory activity (486). Collectively, these findings demonstrate the importance of O-GlcNAc cycling in multiple tissues in the regulation of whole body metabolic homeostasis.
OGT deletion specifically in pancreatic β-cells resulted in severe hyperglycemia, hypoinsulinemia, and β-cell apoptosis (487). Thioredoxin-interacting protein (TXNIP) is widely recognized as playing a key role in β-cell dysfunction and death, factors that underlie both Type 1 and Type 2 diabetes (488). Hyperglycemia increases O-GlcNAcylation of TXNIP in β-cell lines and in islets from diabetic rodents, which was associated with increased inflammatory processes (489). The dnOGA variant described above, which lacks OGA activity, was initially identified in the Goto-Kakizaki rat and is associated with a nonobese diabetic phenotype (199), as well as significantly increased O-GlcNAcylated proteins in the pancreas and islets (490). Increased O-GlcNAc levels have also been implicated in diabetic complications associated with many organ systems, diabetic cardiomyopathy, nephropathy, and retinopathy (20, 491–493).
Supporting the importance of brain protein O-GlcNAcylation in sensing nutrients and signaling satiety are the observations that forebrain neuron-specific OGT knockout results in overeating-dependent obesity (203). These data suggest that a lack of neuronal O-GlcNAcylation leads to excessive food intake, which can contribute to obesity. In further support of the importance for tight regulation of O-GlcNAcylation, OGT ablation in AgRP neurons, promotes white adipose tissue browning and protects mice against diet-induced obesity and insulin resistance (201). These data suggest that a lack of AgRP neuron O-GlcNAcylation sends signals that attenuate white adipose tissue browning. OGA deletion specifically in the brain resulted in a substantially elevated body fat, fatty liver, and metabolic dysfunction, including increased leptin, triglycerides, and insulin (494). These findings suggest that the hypothalamic-pituitary axis, which plays a key role in metabolic regulation in the brain, was adversely affected by the loss of OGA. Global OGA haploinsufficiency (Oga+/−) results in decreased body weight, decreased inguinal subcutaneous lipid content, with increased respiratory exchange ratio, suggesting increased glucose utilization, with no change in food intake or activity, yet also with higher energy expenditure (17). In response to a high-fat diet, only female Oga+/− mice demonstrated increased weight gain. On the other hand, it has been reported that Oga+/− mice were also resistant to high-fat diet-induced obesity, and they had enhanced differentiation of subcutaneous white adipose tissue-derived stromal vascular fraction cells into energy-consuming brown-like adipocytes (207). These observations indicate a complex regulation by OGT and OGA that involve multiple central and peripheral mechanisms.
4.3. Neurological Function and Diseases
It has been shown that cytosolic OGT activity is 10 times higher in brain compared to muscle, adipose, heart, and liver (495), suggesting significant involvement of O-GlcNAcylation in nervous system function. Using postsynaptic density preparation of mouse brain, Vosseller et al. (448) and Shapira et al. (496) identified ∼500 unique O-GlcNAc-modified proteins, including vesicle docking and fusion protein, Bassoon, and post-synaptic cytoskeletal structure protein, ankyrin G. Additional proteins involved in synaptic plasticity, synaptic vesicle trafficking, and axonal branching have been identified as O-GlcNAc targets (100, 152, 448, 497). Other examples of O-GlcNAcylation play a role in neuronal function, which include: 1) inhibition of cyclic-AMP response element binding protein (CREB) activity by O-GlcNAcylation of Ser-40 (100); 2) altered mitochondrial dynamics by O-GlcNAcylation of the motor adaptor protein Milton (160); and 3) altered hippocampal synaptic plasticity by GluA2 O-GlcNAcylation (200). It is estimated that ∼40% of neuronal proteins and 19% of synaptosome proteins are O-GlcNAcylated (182). It has also been shown that O-GlcNAcylated proteins, OGT, and OGA are abundant in widespread regions of the brain, especially the synapses and postsynaptic density preparations (100, 200, 448, 498–500). O-GlcNAcylated proteins have been found in multiple cell types in the brain, including pyramidal cells, GABAergic interneurons, and astrocytes in the hippocampus (200), as well as Purkinje cells in cerebellar cortex (501). These data are consistent with O-GlcNAc signaling playing an important role in neuronal function.
As discussed above, OGT mutations have been linked to intellectual disability (26,27, 29, 198). For example, Parkinsonian-dystonia (DYT3) has been mapped to the X chromosomal region that includes the Ogt locus (502), although whether the OGT mutation is causative for DYT3 has yet to be demonstrated. Interestingly, OGA is localized to chromosome 10(10q24), a region associated with late-onset Alzheimer’s disease (AD) (503). Protein O-GlcNAcylation was shown to be decreased in postmortem AD brains compared with controls, and this was associated with hyperphosphorylation of tau (504). Pharmacological (60, 505) or genetic (505) interventions that enhance O-GlcNAcylation have been shown to increase nonamyloidogenic α-secretase processing, resulting in increased levels of the neuroprotective sAPPα fragment and decreased Aβ secretion. Hyper-O-GlcNAcylation induced by an OGA inhibitor, decreased levels of Aβ40 and Aβ42 peptides in the brain, decreased plaque formation, and improved cognition in the 5xFAD amyloid-β mouse model (55). Pharmacological approaches to inhibit OGA have been used in tau overexpression models and were found to increase tau O-GlcNAcylation and decrease neurodegenerative phenotypes (60, 209). On the basis of these studies, clinical trials are currently ongoing to determine OGA inhibition in treatment of AD (506, 507); however, controversy remains as to whether lower O-GlcNAc levels contribute to AD. This was highlighted in a quantitative proteomics study, which found that in AD brains, 12 peptides exhibit ed decreased, while 119 peptides exhibited increased, O-GlcNAcylation compared with controls (508). Consequently, the long-term impact of globally increasing O-GlcNAc levels by inhibiting OGA as a treatment for AD remains unclear.
In contrast to increasing O-GlcNAc levels as a potential therapeutic approach for AD, other studies suggest that increased O-GlcNAc levels may lead to impaired neuronal function. For example, in a Caenorhabditis elegans model of neurodegeneration, the OGA inactive mutant, which is associated with increased O-GlcNAc levels, was shown to increase proteotoxicity (509). Hyper-O-GlcNAcylation also induces neuronal apoptosis in vitro, is associated with downregulation of AKT Ser-308 and Ser-473 phosphorylation, and correlates with greater tissue damage in an in vivo model of cerebral ischemia (124). Increased O-GlcNAcylation of NF-κB has also been found to be associated with retinal ganglion cell death in a model of diabetic retinopathy (430). There is also an increase in protein O-GlcNAcylation in postmortem brains of patients with Parkinson’s disease (PD) (453). α-Synuclein, a component of Lewy bodies that is involved in the pathogenesis of PD, is also O-GlcNAcylated, and this could affect α-synuclein degradation. Other O-GlcNAc-modified proteins associated with neurodegenerative diseases include tau and amyloid precursor protein (APP), involved in AD (93, 510), as well as superoxide dismutase (SOD)—a protein involved in amyotrophic lateral sclerosis (511)—and gigaxonin, mutations of which cause giant axonal neuropathy (512).
Since α-synuclein aggregation is the main component of Lewy bodies appearing in PD, as well as the nonamyloid component in AD (513–517), its modification by O-GlcNAcylation is drawing attention. In vivo proteomic studies have identified up to nine different O-GlcNAc modification sites, several within the region required for aggregation (i.e., residues 61–95) (185). Earlier studies demonstrated that α-synuclein O-GlcNAcylation at Thr-64 and Thr-72 in rat brain, and at Ser-87 in humans (92,93, 518) and in vitro O-GlcNAcylation at Thr-72 or Ser-87 decreased aggregation propensity and toxicity in cultured cells (519,520). Additional in vitro studies demonstrated that O-GlcNAcylation of Thr-72, Thr-75, or Thr-81 inhibited the nucleation step of aggregation, whereas O-GlcNAc modification of Ser-87 had little effect (185). Simultaneous O-GlcNAc modification of α-synuclein at three sites—Thr-72, Thr-75, and Thr-81—completely abrogated it aggregation (185). On the other hand, studies in primary cultured neurons found that increasing O-GlcNAc levels by inhibiting OGA increased the total levels of α-synuclein (453). It is possible that O-GlcNAcylation of α-synuclein may change its turnover or degradation; however, whether O-GlcNAcylated or un-O-GlcNAcylated α-synuclein is turned over more slowly remains unclear (453). Thus, although it is clear that α-synuclein is subject to O-GlcNAcylation, the impact of this modification on its aggregation and turnover remains to be understood. In light of the importance of α-synuclein in neurodegenerative diseases, a more comprehensive understanding of how O-GlcNAc levels impact α-synuclein homeostasis is important and would be a significant new finding at the intersection of glycobiology and neurodegenerative diseases.
As discussed in sect. 3, there is increasing evidence that O-GlcNAcylation is involved in regulating quality control of proteins and organelles through its effects on autophagy. Therefore, in addition to a direct effect on the accumulation and toxicity of proteins, such as α-synuclein, APP, tau, and SOD, their insufficient clearance due to impaired autophagy may also contribute to the pathogenesis associated with PD, AD, and dementia with Lewy bodies (DLB) (521–525). TPPP is a prime candidate linking O-GlcNAc to AD, DLB, and PD, because it has been found in Lewy bodies (526) and because it has been shown to promote α-synuclein secretion by inhibition of autophagosome-lysosome fusion (447). In primary neurons, OGA inhibition has been shown to attenuate autophagic flux (453), providing a further link between increased O-GlcNAc levels and neuronal dysfunction. Thus, the importance of O-GlcNAcylation in directly modulating toxic protein function and accumulation and indirectly modulating toxic protein accumulation by modulating autophagy will need to be further investigated.
In addition to O-GlcNAc’s roles in neurodegenerative diseases, there is evidence that acute changes in O-GlcNAc levels are involved in regulating learning and memory in vitro and in vivo via O-GlcNAcylation of the GluA2 subunit of the AMPAR receptor (200). Acute increases in O-GlcNAc levels decreased excitability of hippocampal neurons and attenuated excitatory synaptic transmission, which was also mediated by GluA2 containing AMPAR receptors (527). Increases in O-GlcNAc also decreased intrinsic neuronal excitability, suggesting that it could regulate overall excitation/inhibition balance and neuronal output (528). In Drosophila, loss of OGA activity altered locomotion and deficits in learning, suggesting that OGA and O-GlcNAc may play a conserved role in cognitive function (529). OGA+/− mice, which have chronically elevated O-GlcNAc levels, exhibited antidepressant-like behavior, which was associated with reduced inhibitory synaptic transmission in the medial prefrontal cortex, suggesting a possible link between altered O-GlcNAc levels in the brain and depression (530).
In the context of chronic sensorimotor perturbation, such as hindlimb unloading, overall O-GlcNAc levels were unchanged; however, O-GlcNAcylation of synapsin I in the cytosol was increased, suggesting a possible role for O-GlcNAc in regulating sensorimotor synaptic plasticity (531). OGT and O-GlcNAc have also been implicated in memory consolidation and fear conditioning via the epigenetic regulation of genes by EZH2 (43). In a C. elegans model of neuronal injury and regeneration, both reducing and increasing O-GlcNAc levels were found to enhance regeneration via two distinct mechanisms (532). Lower O-GlcNAc levels increased regeneration via an AKT-dependent increase in glycolysis, whereas the positive effects of higher O-GlcNAc levels were mediated by changes in mitochondrial function. The beneficial effect of acute increases in O-GlcNAc levels was also shown in seizure models, where pharmacological increase in O-GlcNAc attenuated hyperexcitability both in vitro and in vivo (533). Epilepsy was shown to decrease OGT and O-GlcNAc levels in human brains and in brains from a rat model of epilepsy; moreover, increasing O-GlcNAc levels by OGA inhibition decreased seizure duration, as well as epileptic spike events (534).
4.4. Cardiovascular Function and Diseases
The first studies of O-GlcNAcylation in the cardiovascular system identified the small heat shock protein αB-crystallin and the transcription factor Sp1 as O-GlcNAc targets in the heart and vascular smooth muscle cells, respectively (143, 336). Both studies emphasized the dynamic nature of this modification and its potential role in regulating the function of these proteins. Dong and Hart (11) reported that the heart exhibited measurable OGA activity, and a subsequent study showed that OGT activity was higher in the rat heart than many other tissues, including liver and adipose (535). However, GFAT activity, while measurable, was much lower in heart than most tissues (535). Brownlee and colleagues (190, 536) demonstrated in endothelial cells that hyperglycemia increased O-GlcNAc levels on Sp1 and eNOS and concluded that this could contribute to the endothelial dysfunction that occurs in diabetes.
In cardiomyocytes, hyperglycemia resulted in a prolonged Ca2+ transients, which was mimicked by increasing HBP flux with glucosamine or inhibition of OGA (263). Interestingly, overexpression of OGT had no effect under normal conditions and did not exacerbate the effects of hyperglycemia; however, OGA overexpression blunted the effects of hyperglycemia on Ca2+ transients (263). A subsequent study found that OGA overexpression in the diabetic heart improved cardiomyocyte contractility, and this was associated with a reduction in overall O-GlcNAc levels and the normalization of SERCA protein levels (537). Others have suggested that increased O-GlcNAcylation of PLB, a major regulator of SERCA activity, may contribute to impaired contractility that occurs with diabetes (194). Diabetes is associated with electrical abnormalities, including prolonged QT interval, which are associated with increased risk or cardiac arrhythmias (538). Increased O-GlcNAcylation of both CaMKII (165) and the voltage-gated sodium channel Nav1.5 (539) have been implicated as contributors to increased arrhythmogenesis in diabetes. Interestingly, O-GlcNAcylation of CaMKII has also been shown to be required for increased NOX2-mediated ROS production induced by hyperglycemia (166).
It is widely accepted that diabetes leads to a sustained increase in cardiac O-GlcNAcylation and that this sustained increase in cardiac O-GlcNAcylation has been implicated in the adverse effects of diabetes on the heart (540–542). This is commonly believed to be a direct consequence of increased availability of glucose; however, this has yet to be definitively confirmed. In addition to higher cardiac O-GlcNAc levels, 2 wk after Type 1 diabetes was induced, Hu et al. (537) observed increased mRNA and protein levels of both OGT and OGA, although surprisingly there was no change in activities of either enzyme. Another study reported biphasic changes in cardiac O-GlcNAc levels with increases at 2 wk after the initiation of diabetes, followed by a subsequent decrease to normal levels at 4 wk, with a second increase after 16 wk of diabetes (543). However, mRNA levels of OGT, OGA, and GFAT1 increased only in the 16-wk diabetic group, and only OGT protein levels were elevated at that time point. Thus, while diabetes is clearly associated with increased cardiac O-GlcNAc levels, the mechanisms underlying this response remain unclear.
The adverse effects of diabetes on the heart has also been linked to alterations in metabolism characterized by decreased carbohydrate oxidation and increased dependence on lipids for energy production. As discussed in sect. 3.3, multiple components involved in insulin signaling are O-GlcNAcylated, and increased O-GlcNAcylation is found to attenuate insulin signaling. This could be a contributing factor to the reduction in carbohydrate metabolism seen in diabetes; however, this has not yet been examined in the heart. The increase in lipid metabolism in rodent models of diabetes has been attributed, at least in part, to increased plasma membrane levels on the fatty acid transporter FAT/CD36 (544). In the isolated perfused heart, the addition of glucosamine, which acutely increases cardiac O-GlcNAc levels, also increases fatty acid oxidation and leads to an increase in plasma membrane levels of FAT/CD36 (419). FAT/CD36 was found to be O-GlcNAcylated and was associated with OGT (419). The link between O-GlcNAcylation, fatty acid oxidation, and FAT/CD36 translocation has been confirmed by others (545). It is also worth noting that the addition of physiological concentrations of glutamine increased fatty acid oxidation in the perfused heart, which occurred via activation of the HBP and an increase in plasma membrane FAT/CD36 levels (420). O-GlcNAcylation of E2 and E3 subunits of PDH in the heart, has been linked to an increase in PDH activity, which was attenuated under conditions of impaired branched-chain amino acid metabolism (137). Studies have also found a link between O-GlcNAcylation and cardiac ketone body metabolism, with increased O-GlcNAc levels associated with decreased ketone body oxidation (281, 546). A large number of enzymes in glycolysis are also O-GlcNAcylated (547), although the effect of this modification on regulating glucose metabolism in the heart is unknown. Consequently, O-GlcNAcylation may contribute to the regulation of cardiac metabolism under normal physiological conditions, as well as contribute to metabolic dysregulation in diabetes.
Impaired mitochondrial function is also a common feature in the heart in response to diabetes, and O-GlcNAcylation of mitochondrial proteins has been shown to reduce mitochondrial oxidative metabolism and contribute to increased mitochondrial fission (159, 161). Others have also reported alterations in mitochondrial O-GlcNAcylation in the heart in diabetes (427). It should be noted however, that whether O-GlcNAcylation mediates the effects of hyperglycemia on mitochondrial function has been questioned (548). While the O-GlcNAcome of cardiac mitochondria is diverse (138), our understanding of how it is regulated is limited. Mitochondrial uptake of UDP-GlcNAc in cardiomyocytes is regulated by PNC1, but it is not known whether transport is mediated by primarily by UDP-GlcNAc availability or whether it is determined by regulation of PNC1 activity by allosteric inhibitors or activators such as Ca2+ or by regulation of expression levels by insulin and IGF-1 (549). It also remains to be determined whether O-GlcNAc cycling rates in the mitochondria are similar or different than those in the nucleus or cytosol. The mechanisms by which O-GlcNAcylation is targeted to specific proteins and residues in the mitochondria are also unknown.
Cardiac O-GlcNAc levels are also increased in hypertrophy, heart failure, and aging, although the role of O-GlcNAcylation in the development of cardiac dysfunction has not been as extensively studied as in the context of diabetes. As noted above, in diabetes, it is assumed that O-GlcNAc levels in the heart increase because of high glucose levels increasing HBP flux. The mechanism leading to high O-GlcNAc levels in other pathological conditions is less clear, as there is no primary underlying metabolic abnormality. In a rat model of pressure overload-induced hypertrophy, in addition to an increase in O-GlcNAc levels, OGT, and OGA mRNA, and protein levels were significantly increased (291). Similar changes in O-GlcNAc, OGT, and OGA were found in biopsies from patients with aortic stenosis compared with samples from nonhypertrophied hearts (291). The increase in OGT could lead to the increase in O-GlcNAc, and the increase in OGA could be an adaptive response to try and lower O-GlcNAc levels; however, the cause and effect of these changes are unknown. Interestingly, acute hypertrophic stimuli increased cardiomyocyte O-GlcNAc levels, which was linked to increased expression of OGT protein, as well as increased GFAT phosphorylation and protein levels (298, 299). These studies demonstrated that the initial increase in O-GlcNAc levels was necessary and sufficient to activate hypertrophic signaling in cardiomyocytes. Further support for a role of the HBP and O-GlcNAcylation in regulating cardiac hypertrophy was provided by studies reporting that GFAT1 overexpression stimulates growth of neonatal cardiomyocytes in culture and accelerates cardiac remodeling in a model of pressure overload-induced hypertrophy; conversely, GFAT1 deletion blunted adverse remodeling (550). Preliminary reports also demonstrate that chronic overexpression of OGT in the heart leads to a dilated cardiomyopathy, in the absence of any additional stress (551).
In addition to its oncogenic role, cMyc also regulates cardiac hypertrophy, where it is reexpressed in response to hypertrophic stimuli, and its repression decreases the degree of hypertrophy (552). Inducible, cardiomyocyte overexpression of cMyc resulted in cardiac hypertrophy that was associated with an overall increase in cardiac O-GlcNAc levels, and cMyc KO attenuated the response to hypertrophy and reduced O-GlcNAc levels (553, 554). OGT deletion either before or after pressure overload hypertrophy resulted in worse outcomes, providing further support for O-GlcNAc being required for normal hypertrophic signaling (555). It was suggested that O-GlcNAcylation of PKA was a possible mechanism by which O-GlcNAc mediates the effects of hypertrophy. Interestingly, cardiac specific overexpression of LXRα was protective against pressure overload-induced hypertrophy, and this was associated with induction of cytoprotective mechanisms via O-GlcNAcylation of the transcription factors GATA4 and MEF2c (556).
Sustained exercise is also associated with cardiac remodeling and hypertrophy that is typically considered to be beneficial. Interestingly, in contrast to pathological hypertrophy, hypertrophy induced as a result of exercise swim-training reduced cardiac protein O-GlcNAcylation under normal conditions (557), as well as in the context of diabetes (558). This was associated with a decrease in OGT protein and OGT, OGA, and GFAT2 mRNA. A single 15-min bout of exercise resulted in a decrease in nuclear, but not cytosolic, cardiac O-GlcNAc levels, which was associated with a reduced interaction between OGT and the REST chromatic repressor (559). Surprisingly, treadmill exercise for 1 and 4 wk increased cardiac O-GlcNAc levels in hearts from db/db mice, independent of changes in OGT and OGA protein levels (376). The mechanism by which exercise affects O-GlcNAc levels is not known; however, in cultured neonatal cardiomyocytes, exogenous NAD+ resulted in a time- and dose-dependent decrease in O-GlcNAc levels that was not accompanied by changes in OGT or OGA protein levels (344).
Although considerable attention has been paid to the adverse effects of increased O-GlcNAc levels in chronic cardiac diseases, a number of studies have demonstrated that acute pharmacological increases in O-GlcNAc levels decrease cell and tissue injury and improve cardiac functional recovery (163, 402, 437, 442). Other studies have shown that decreases in O-GlcNAc levels are associated with increased sensitivity of cardiomyocytes to oxidative stress (438). It is also worth noting that well-established cardioprotective strategies, including ischemic preconditioning and glucose–insulin–potassium therapy, have also been shown to increase cardiac O-GlcNAc levels (560–563), although the extent to which this contributes to their protective effects is not known.
Protein O-GlcNAcylation has also been shown to regulate vascular function with sustained increases associated with impaired endothelial cell and vascular smooth muscle cell function, as seen with diabetes. Increased vascular O-GlcNAc levels have also been implicated in increased vascular reactivity, thereby, directly contributing to the development of hypertension. Of note, pharmacologically increasing O-GlcNAc levels in normal vascular smooth muscle also increases vascular reactivity (173, 564). ET-1, which contributes to acute and long-term changes in vascular function, also directly increases vascular O-GlcNAc levels in an ETA and ETB receptor-dependent fashion (173, 296). The effects of O-GlcNAc in regulating vascular reactivity appear to be mediated via the RhoA/Rho-kinase pathway (297); however, the direct link between O-GlcNAc and the RhoA/Rho-kinase pathway has yet to be identified. In addition to regulating vascular reactivity, acute increases in O-GlcNAc have been shown to be protective by reducing endoluminal injury and subsequent infiltration of inflammatory mediators (565, 566). Increasing O-GlcNAc levels also blunted the adverse effects of TNF-α on endothelial function via suppression of inducible nitric oxide synthase expression (567). Vascular dysfunction is also a hallmark of idiopathic pulmonary arterial hypertension, which has been associated with alterations in glucose metabolism. Barnes et al. (568) found that in tissues and cells from IPAH patients that GFAT, OGT, and O-GlcNAc levels were all increased and that knockdown of OGT attenuated the proliferative phenotype of pulmonary arterial smooth muscle cells.
4.5. Skeletal Muscle
Skeletal muscle is distinctly different from cardiac muscle in both its origin (569) and its regulation by O-GlcNAcylation (570). Within healthy muscle, there are higher levels of O-GlcNAcylation in proteins in the slow oxidative soleus muscle, compared to fast glycolytic extensor digitorum longus muscle (411). Furthermore, this difference in O-GlcNAcylation was lost in the soleus muscle following mechanical unloading and muscle wasting. These authors identified 14 O-GlcNAc-modified proteins involved in signal transduction, metabolism, and contraction. Over a decade later, using more sophisticated enrichment and MS techniques, this list was expanded to 342 O-GlcNAcylated skeletal muscle proteins (571). In contrast to the earlier study, they were also able to identify specific O-GlcNAc protein modification sites and solidified the role of O-GlcNAcylation in normal sarcomere cytoarchitecture.
Consistent with the earlier suggestion of a link between protein O-GlcNAcylation levels and the muscle fiber type, skeletal muscle-specific knockout of OGT (mOGT-KO) reduced O-GlcNAc levels, increased Myh7 fiber expression, and reduced oxidative phosphorylation gene expression (379). Interestingly, this mOGT-KO protected mice from high-fat diet-induced glucose and insulin intolerance through a proposed systemic signaling by IL-15 that affects adipose tissue and whole body metabolism. This later finding speaks to an interesting O-GlcNAc-mediated signaling from muscle to influence normal physiology. In support of this skeletal muscle signaling, a separate group, also using an mOGT-KO model, identified the loss of OGT as sufficient to activate AMPK to enhance glucose utilization (485), a link that was previously identified by in vitro studies (252). In contrast to the OGT knockout models, increasing total muscle protein O-GlcNAcylation by overexpression of a naturally occurring dominant-negative OGA leads to muscle atrophy and increased mortality (205). Consequently, it is clear that O-GlcNAcylation of skeletal muscle proteins is an important molecular signaling pathway that affects both muscle and the entire organism.
Beyond the role of O-GlcNAcylation for basal physiology and muscle signaling, external stimuli can further alter skeletal muscle protein O-GlcNAc levels. For example, exercise treadmill-training, but not acute exercise, is sufficient to increase protein O-GlcNAcylation (572). These changes in protein modification appear to be most robust on cytoplasmic proteins, further supporting a role in cellular signaling. Somewhat surprisingly, myocardial infarction-induced heart failure had no effect on skeletal muscle protein O-GlcNAcylation, despite decreases in levels of both OGT and OGA protein (572). However, the latter could be explained by the 6-wk duration of the study. Subsequent work showed that in high-fat diet-induced diabetes, skeletal muscle protein O-GlcNAcylation was increased, and using both in vitro and in vivo studies, Wang et al. (573) suggest that this is directly linked to transcriptional reprogramming of mitochondria by promoting PGC-1α O-GlcNAcylation and degradation. These studies support that stimuli, such as exercise, require intact O-GlcNAc signaling as part of the adaptations seen in skeletal muscle.
As discussed in sect. 3.3, increasing HBP flux decreased skeletal muscle insulin sensitivity in normoglycemic but not diabetic conditions (574), which was related to UDP-GlcNAc levels in skeletal muscle glucose transporter 4 (GLUT4)-containing vesicles (575). Somewhat paradoxically, either short-term (16 h) or long-term (8 mo) treatment of rodents with the OGA inhibitor NButGT, increased skeletal muscle total protein O-GlcNAcylation without significantly altering glucohomeostasis (576). Further evidence for this nutrient signaling to skeletal muscle protein O-GlcNAcylation came from a study examining the combination of glutathione depletion and acute exercise training. As mentioned above, long-term exercise altered skeletal muscle, but acute exercise alone was not sufficient to produce significant changes. However, when intracellular glutathione was depleted in rats by diethyl maleate treatment before treadmill running, OGT and total protein O-GlcNAc levels were increased (577). The authors provided additional information to suggest that the mechanism of this protein O-GlcNAcylation was driven by the cellular redox state, providing a potential explanation of the exercise-induced adaptation to cellular oxidative stress. Together, these studies suggest a unique mechanism by which the source and means by which flux through the O-GlcNAcylation pathway occurs dictate its physiological consequences.
4.6. Liver
The liver is responsible for regulating whole body energy homeostasis in a continuously changing metabolic environment. Following a meal, when blood glucose levels are typically in excess, the liver takes up glucose and stores it as glycogen, whereas when glucose falls below normal, glycogenolysis is activated, resulting in glucose export to the blood. Once glycogen stores are depleted, gluconeogenesis will be activated, resulting in glucose generation from noncarbohydrate sources and when gluconeogenesis becomes limiting, such as during a prolonged fast, the liver will activate ketogenesis, facilitating the conversion of fatty acids to ketone bodies. All of these pathways are tightly regulated at the transcriptional, translational, and posttranslational levels. Insulin plays a central role in regulating liver metabolism, and as discussed in sect. 3.3, most of the components of the insulin signaling pathway are O-GlcNAcylated, and this attenuates their activity; thus, this represents a mechanism for attenuating insulin signaling in response to sustained increases in insulin (578). However, hepatic overexpression of OGT resulted in insulin resistance and increased genes associated with gluconeogenesis while decreasing lipogenic gene expression (242). Changes in O-GlcNAc cycling in the liver either by OGT overexpression or knockdown altered gluconeogenic gene expression and circulating glucose levels (330). In the liver, glucokinase plays a central role in controlling glycogen synthesis and glycolysis, thereby contributing to the regulation of glucose homeostasis. Glucokinase has been shown to be a target for O-GlcNAcylation, and higher O-GlcNAc levels increase glucokinase protein levels, most likely by increasing its stability (579). This represents a potential mechanism for the short-term increase in glucokinase activity, in response to changes in glucose levels.
In response to fasting, glucagon activates gluconeogenesis, in part, by triggering the translocation of transducer of regulated CREB 2 (CRTC2) to the nucleus. In response to refeeding, circulating insulin is increased, and inhibits this program by increasing the degradation of CRTC2. In diabetes, high circulating levels of glucose and insulin resistance cause high levels of hepatic gluconeogenesis, further contributing to hyperglycemia. Both high glucose and glucosamine increased CRTC2 nuclear translocation, and this was associated with increased O-GlcNAcylation of CRTC2 at Ser-70 and Ser-171 (127), which are key phosphorylation sites that keep CRTC2 sequestered in the cytoplasm. Increased O-GlcNAcylation of PGC-1α protects it from degradation, thereby, also promoting gluconeogenesis (118). An OGT/HCF-1 complex is required to O-GlcNAcylate PGC-1α, and the knockdown of either OGT or HCF-1 improved glucose homeostasis in Type 2 diabetic db/db mice. Both the LXRα receptor and the liver transcription factor ChREBP play key roles in regulating hepatic glucose and lipid metabolism, and both have been shown to be O-GlcNAcylated, with the levels of O-GlcNAcylation increasing in response to hyperglycemia (347, 351). Both refeeding and diabetes lead to increased LXRα O-GlcNAcylation, which, in turn, activates sterol regulatory element-binding protein 1c (SREBP-1c), a key regulator of lipogenesis (347). The nuclear receptor REV-ERBα is also involved in regulating hepatic metabolism, and a REV-ERBα/OGT complex has been shown to control SREBP-1c transcription by stabilizing OGT levels and decreasing AKT phosphorylation (65). ChREBP also interacts with OGT, resulting in increased O-GlcNAcylation and leading to stabilization of ChREBP and increased transcriptional activity toward both glycolytic and lipogenic genes (351, 580). Collectively these findings demonstrate the important role of O-GlcNAcylation in regulating hepatic metabolism in physiological and pathological conditions.
4.7. Cancer
Aberrant energy metabolism is a well-established hallmark of all forms of cancer, typically characterized by a high demand for glucose. Glutamine is also an essential nutrient that facilitates the rapid growth that occurs in cancer cells (581). The high demand for glucose and glutamine is due, in part, to their contributions to energy production; however, glutamine also contributes to a wide range of biosynthetic pathways (582). Glucose and glutamine metabolism also converge at GFAT, the first step in the HBP, leading to UDP-GlcNAc synthesis (FIGURE 4). Studies have linked increased O-GlcNAc levels to the increased survival, progression, invasion, and metastasis of many cancers (21, 210, 583, 584). In addition to increased availability of substrates for the HBP, oncogenes such as KRAS and c-Myc have been shown to increase HBP flux and O-GlcNAcylation via upregulation of GFAT (583). Disease-free survival and overall survival outcomes are significantly worse in prostate and lung cancer patients with high OGT and O-GlcNAc levels (74, 564, 584, 585), and knockdown of OGT reduces tumor growth and cancer cell proliferation (583, 584).
As discussed in sect. 3.1, O-GlcNAcylation is widely recognized as playing an important role in transcriptional regulation, and this represents one mechanism underlying its involvement in the progression of cancer. Numerous oncogenes and tumor suppressor genes are targets for O-GlcNAcylation, including p53, cMyc, HIF1α, FoxM1, cyclin D1, and others (290, 583, 586). Almost invariably, increased O-GlcNAcylation increases their activation, either directly or indirectly, typically resulting in increased stability, translocation, and transcription (583, 584). For example, GSK3β-mediated phosphorylation at Thr-58 on cMyc is required for its degradation; thus, mutations of this residue prevent phosphorylation, reducing its degradation, converts cMyc into a potent oncogene (583, 584). O-GlcNAcylation of this same residue markedly increases cMyc stability, whereas OGT inhibition or knockdown decreases cMyc levels (66). UDP-N-acetylglucosamine pyrophosphorylase-1-like-1 (UAP1L1), which shares ∼60% homology with UAP1 (FIGURE 4), but lacks its enzymatic activity, is increased in hepatocellular carcinoma and is associated with poor prognosis (587). UAP1L1 appears to interact directly with OGT, thereby contributing to increased overall O-GlcNAc levels in hepatoma cells and increased O-GlcNAcylation of cMyc. A complex between unconventional prefoldin RPB5 interactor (URI), the protein phosphatase 1γ (PP1γ), and OGT also regulates cMyc levels by its O-GlcNAcylation, thereby contributing to its role in tumorigenesis (588). In some cases, O-GlcNAcylation of cMyc or HIF1α, contributes to the increases in glycolysis and glutamine metabolism, while in others, hyper-O-GlcNAcylation increases cell proliferation, cell survival, or invasion (583, 584). In breast cancer cell lines, hyperglycemia upregulates chemoresistance pathways via O-GlcNAcylation of Hedgehog transcription factors GLI1 and GLI2 (348), which could be a factor in the poorer outcomes of breast cancer patients that also have diabetes. Changes in methylation also contribute to cancer progression, and this can be directly affected by O-GlcNAcylation. For example, O-GlcNAcylation of EZH2 increases H3K27me3 forming a feedback loop that promotes metastasis (307). In human colon cancer stem cells, cross talk between O-GlcNAcylation and DNA methylation of the promoter region of MYBL1, a transcriptional activator, was shown to regulate tumor cell growth both in vitro and in vivo (589).
Increases in O-GlcNAcylation have also been associated with the metabolic reprogramming that occurs in cancer. This is due, in part, to transcriptional regulation of metabolism, but it can also occur via direct O-GlcNAcylation of metabolic enzymes. For example, PFK1, which catalyzes the conversion of fructose 1,6-bisphosphate from fructose-6-phosphate is subject to allosteric regulation at Ser-529 by fructose-2,6-bisphosphate (F-2,6-BP). O-GlcNAcylation of Ser-529 on PFK1 inhibits glycolysis, redirecting glucose into the pentose phosphate pathway (PPP), resulting in greater cell growth and increased resistance to oxidative stress. Conversely, blocking this modification decreased cell proliferation and tumor formation (140). In addition, glucose-6-phosphate dehydrogenase, the rate-limiting enzyme of the PPP is O-GlcNAcylated at Ser-84, increasing its activity, and further activating the PPP and increasing tumor growth (134). While most glycolytic enzymes have been shown to be O-GlcNAc targets (547), the functional consequences of many of these modifications are not known. GAPDH is O-GlcNAcylated at Thr-227, which blocks interactions between GAPDH monomers, and this has been linked to its increased nuclear translocation (128), where it has been reported to influence gene expression and telomere integrity (590). The roles of nonglycolytic functions of GAPDH in cancer remains to be elucidated. Pyruvate kinase M2 (PKM2), which plays a key role in metabolic reprogramming in cancer, is O-GlcNAcylated at Thr-405 and Ser-406, and this was shown to increase glucose utilization and glycolysis, whereas, blocking PMK2 O-GlcNAcylation attenuated tumor growth in vivo (141).
5. CONCLUSIONS
The initial discovery in 1984 of nuclear and cytosolic proteins modified by a single O-GlcNAc moiety (2) overturned widely held assumptions in glycobiology and initiated an entirely new area of research that encompasses diverse areas of cell biology, physiology, and pathophysiology. Many of the early studies examined the role of O-GlcNAc in regulating cellular physiology in the context of diabetes and hyperglycemia, conditions in which O-GlcNAc levels are chronically elevated on multiple proteins and in tissues that are adversely affected by diabetes. As our knowledge of the regulatory role of O-GlcNAc grew, it was recognized that O-GlcNAcylation also contributed to the complex coordination of cellular signaling under normal physiological conditions. The intricate relationship between O-GlcNAcylation and phosphorylation, as well as other posttranslational modifications, is slowly being unraveled. Although protein O-GlcNAc levels are undoubtedly regulated, in part, by nutrient availability, there is increasing recognition of multiple layers of regulation, including transcriptional regulation of GFAT, OGT, and OGA, as well as kinase-mediated regulation of these same proteins contributing to maintaining O-GlcNAc homeostasis. In addition, OGT binds to numerous partner proteins, which affects its stability, localization, and targets for O-GlcNAcylation. It is also increasingly clear that Ca2+ is another component of the O-GlcNAc regulatory network, although the specific nature of this relationship remains to be identified. Thus, although the concept of O-GlcNAc levels as a “rheostat”, reflecting nutrient availability (210, 257), is still applicable under some circumstances, it is only one of several mechanisms underlying the regulation of O-GlcNAcylation.
While research in O-GlcNAc biology and physiology continues to expand (FIGURE 3), challenges remain with regard to more rapid growth. As recently as 2019, more than 30 years after its discovery, Gerald Hart, the founder of O-GlcNAc biology, wrote that “Despite the efforts of many laboratories, this field is still in its infancy.” (210). One important limitation that has yet to be overcome is the commercial availability of site-specific O-GlcNAc antibodies, although a few such antibodies have been published for a few proteins (79–82). There have been great improvements in techniques to map O-GlcNAc modification sites using mass spectrometry. While their successful application often remains beyond the scope of many proteomic cores, efforts are continuing to improve these techniques (95). The pharmacological modulation of O-GlcNAc levels was hampered for many years by the absence of well-characterized, selective, high-affinity inhibitors of OGT and OGA. The development of the thiazoline family of OGA inhibitors by Vocadlo and colleagues in 2005 (54) substantially helped move the field forward. The subsequent development of Thiamet-G (60), a highly selective, high-affinity OGA inhibitor, which is orally available, crosses the blood-brain barrier, and rapidly increases cellular O-GlcNAc levels at concentrations of <1 µM, has been of particular value. Not until 2015 were OGT inhibitors developed with the necessary level of specificity and affinity; it is anticipated that the wider availability of these compounds will be of great value to the field (67, 69). Recent reports of other new OGT inhibitors that are effective in decreasing O-GlcNAc levels in vivo are also encouraging (77).
Conceptually, perhaps one of the biggest challenges is understanding how only a single OGT and single OGA can regulate thousands of different proteins, particularly given the lack of a clear consensus sequence. The comparison with phosphorylation is inevitable, where many hundreds of kinases and phosphatases are responsible for controlling the phosphorylation of specific proteins; thus, it can be difficult to see how OGT and OGA achieve their specificity. However, most kinases have multiple downstream targets and phosphorylation of individual targets are determined by the specific stimulus, cellular location, as well as other factors, including scaffolding proteins or other interacting proteins. AMPK, which regulates diverse cellular processes, is an excellent example, with 64 validated downstream targets and potentially many more (591, 592). The 14-3-3 protein family, which regulates diverse protein kinase signaling pathways by binding to specific phosphoserine/phosphothreonine motifs (593), is also an O-GlcNAc binding protein, with the O-GlcNAc binding site overlapping with its phosphorylation binding motif (594). This raises the possibility of 14-3-3 proteins playing an important role in regulating O-GlcNAc signaling cascades, as well as potentially integrating O-GlcNAc and phosphorylation signaling pathways.
It has been estimated that OGT could have as many as 800 interacting partners, a large number of which are linked to transcription and metabolism (118). Although less studied, OGA has been reported to have at least 90 binding partners, many of which change in response to stress (595). Although the biological significance of many of these interactions have yet to be characterized, these findings clearly demonstrate potential mechanisms for achieving selective targeting and removal of O-GlcNAc from proteins. There is undoubtedly considerable scope for studies to better understand the regulation and function of interacting partners of both OGT and OGA. Another important goal for future studies is understanding the temporal and spatial changes in cellular O-GlcNAc levels, this will include improving our knowledge of functional consequences of many of the PTMs on OGT and OGA. New technologies are being developed to better understand OGT and OGA substrate recognition, O-GlcNAc sensors targeted to specific intracellular targets, and nanobodies that can be used to direct OGT to specific protein targets (596, 597), will help improve our understanding of the role of O-GlcNAc in physiological regulation of cellular function.
The importance of O-GlcNAc modification of proteins in regulating cellular and organismal physiology was slow to emerge, due, at least in part, to the lack of basic tools However, as we have discussed here, protein O-GlcNAcylation is now recognized as playing an important role in the physiology of most organ systems, including specific processes, such as contractility in cardiac and skeletal muscle, gluconeogenesis in the liver, hippocampus-based learning and memory, vascular function, as well as universal processes, such as mitochondrial function, metabolic regulation, autophagy, calcium signaling, transcription, and epigenetics. Therefore, with the continued development of new pharmacological and molecular tools, the opportunity for future research into the fundamental physiological role of protein O-GlcNAcylation, as well as its contribution to the progression of numerous diseases, remains wide open.
GRANTS
This work is supported in part by National Institutes of Health Grants P30AG050886 (to J. Z.), R56AG060959, I01BX004251, and R01HL142216 (to J.Z. and J.C.C.), R21HL152354 (to J.C.C and A.R.W), and R01HL133011 (to A.R.W.).
ACKNOWLEDGMENTS
We would also like to thank our numerous colleagues at University of Alabama at Birmingham and other institutions for insightful and wide-ranging discussions on O-GlcNAc biology.
REFERENCES
- 1.Varki A, Kornfeld S. Historical background and overview. In: Essentials of Glycobiology (3rd ed.), edited by Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH.. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, NY, 2015. [PubMed] [Google Scholar]
- 2.Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem 259: 3308–3317, 1984. [PubMed] [Google Scholar]
- 3.Holt GD, Hart GW. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J Bioll Chem 261: 8049–8057, 1986. [PubMed] [Google Scholar]
- 4.Snow CM, Senior A, Gerace L. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. J Cell Biol 104: 1143–1156, 1987. doi: 10.1083/jcb.104.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanover JA, Cohen CK, Willingham MC, Park MK. O-linked N-acetylglucosamine is attached to proteins of the nuclear pore. Evidence for cytoplasmic and nucleoplasmic glycoproteins. J Biol Chem 262: 9887–9894, 1987. [PubMed] [Google Scholar]
- 6.Holt GD, Snow CM, Senior A, Haltiwanger RS, Gerace L, Hart GW. Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine. J Cell Biol 104: 1157–1164, 1987. doi: 10.1083/jcb.104.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holt GD, Haltiwanger RS, Torres CR, Hart GW. Erythrocytes contain cytoplasmic glycoproteins. O-linked GlcNAc on Band 4.1. J Biol Chem 262: 14,847–14,850, 1987. [PubMed] [Google Scholar]
- 8.Haltiwanger RS, Blomberg MA, Hart GW. Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide beta-N-acetylglucosaminyltransferase. J Biol Chem 267: 9005–9013, 1992. [PubMed] [Google Scholar]
- 9.Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem 272: 9308–9315, 1997. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- 10.Kearse KP, Hart GW. Lymphocyte activation induces rapid changes in nuclear and cytoplasmic glycoproteins. Proc Natl Acad Sci U S A 88: 1701–1705, 1991. doi: 10.1073/pnas.88.5.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J Biol Chem 269: 19,321–19,330, 1994. [PubMed] [Google Scholar]
- 12.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem 276: 9838–9845, 2001. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 13.Comtesse N, Maldener E, Meese E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a β-N-acetylglucosaminidase. Biochem Biophys Res Commun 283: 634–640, 2001. doi: 10.1006/bbrc.2001.4815. [DOI] [PubMed] [Google Scholar]
- 14.Zachara N, Akimoto Y, Hart GW. The O-GlcNAc Modification. In: Essentials of Glycobiology (3rd ed.), edited by Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH.. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, NY, 2015, p. 239–251. [Google Scholar]
- 15.Halim A, Larsen ISB, Neubert P, Joshi HJ, Petersen BL, Vakhrushev SY, Strahl S, Clausen H. Discovery of a nucleocytoplasmic O-mannose glycoproteome in yeast. Proc Natl Acad Sci U S A 112: 15648–15653, 2015. doi: 10.1073/pnas.1511743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Donnell N, Zachara NE, Hart GW, Marth JD. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. MCB 24: 1680–1690, 2004. doi: 10.1128/MCB.24.4.1680-1690.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA. Conditional knock-out reveals a requirement for O-Linked N-acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J Biol Chem 290: 7097–7113, 2015. doi: 10.1074/jbc.M114.617779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hart GW, Akimoto Y. The O-GlcNAc modification. In: Essentials of Glycobiology, edited by Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME.. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2009. [PubMed] [Google Scholar]
- 19.Akan I, Olivier-Van Stichelen S, Bond MR, Hanover JA. Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration. J Neurochem 144: 7–34, 2018. doi: 10.1111/jnc.14242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee PS, Lagerlöf O, Hart GW. Roles of O-GlcNAc in chronic diseases of aging. Mol Aspects Med 51: 1–15, 2016. doi: 10.1016/j.mam.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Hanover JA, Chen W, Bond MR. O-GlcNAc in cancer: An Oncometabolism-fueled vicious cycle. J Bioenerg Biomembr 50: 155–173, 2018. doi: 10.1007/s10863-018-9751-2. [DOI] [PubMed] [Google Scholar]
- 22.Nagel AK, Ball LE. Intracellular protein O-GlcNAc modification integrates nutrient status with transcriptional and metabolic regulation. Adv Cancer Res 126: 137–166, 2015. doi: 10.1016/bs.acr.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Peterson SB, Hart GW. New insights: A role for O-GlcNAcylation in diabetic complications. Crit Rev Biochem Mol Biol 51: 150–161, 2016. doi: 10.3109/10409238.2015.1135102. [DOI] [PubMed] [Google Scholar]
- 24.Vaidyanathan K, Wells L. Multiple tissue-specific roles for the O-GlcNAc post-translational modification in the induction of and complications arising from type II diabetes. J Biol Chem 289: 34466–34471, 2014. doi: 10.1074/jbc.R114.591560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 94: 161–175, 2014. doi: 10.1016/j.ajhg.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gundogdu M, Llabrés S, Gorelik A, Ferenbach AT, Zachariae U, van Aalten DMF. The O-GlcNAc transferase intellectual disability mutation L254F distorts the TPR helix. Cell Chem Biol 25: 513–518,2018. e514doi: 10.1016/j.chembiol.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niranjan TS, Skinner C, May M, Turner T, Rose R, Stevenson R, Schwartz CE, Wang T. Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PloS One 10: e0116454, 2015. doi: 10.1371/journal.pone.0116454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pravata VM, Muha V, Gundogdu M, Ferenbach AT, Kakade PS, Vandadi V, Wilmes AC, Borodkin VS, Joss S, Stavridis MP, van Aalten DMF. Catalytic deficiency of O-GlcNAc transferase leads to X-linked intellectual disability. Proc Natl Acad Sci U S A 116: 14,961–14,970, 2019. doi: 10.1073/pnas.1900065116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willems AP, Gundogdu M, Kempers MJE, Giltay JC, Pfundt R, Elferink M, Loza BF, Fuijkschot J, Ferenbach AT, van Gassen KLI, van Aalten DMF, Lefeber DJ. Mutations in N-acetylglucosamine (O-GlcNAc) transferase in patients with X-linked intellectual disability. J Biol Chem 292: 12621–12631, 2017. doi: 10.1074/jbc.M117.790097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW. Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anall Biochem 293: 169–177, 2001. doi: 10.1006/abio.2001.5132. [DOI] [PubMed] [Google Scholar]
- 31.Akan I, Love DC, Harwood KR, Bond MR, Hanover JA. Drosophila O-GlcNAcase deletion globally perturbs chromatin O-GlcNAcylation. J Biol Chem 291: 9906–9919, 2016. doi: 10.1074/jbc.M115.704783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krause MW, Love DC, Ghosh SK, Wang P, Yun S, Fukushige T, Hanover JA. Nutrient-driven O-GlcNAcylation at promoters impacts genome-wide RNA Pol II distribution. Front Endocrinol 9, 2018. doi: 10.3389/fendo.2018.00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner JR, Tartakoff AM, Greenspan NS. Cytologic assessment of nuclear and cytoplasmic O-linked N-acetylglucosamine distribution by using anti-streptococcal monoclonal antibodies. Proc Natl Acad Sci U S A 87: 5608–5612, 1990. doi: 10.1073/pnas.87.15.5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuoka Y, Matsuoka Y, Shibata S, Yasuhara N, Yoneda Y. Identification of Ewing's sarcoma gene product as a glycoprotein using a monoclonal antibody that recognizes an immunodeterminant containing O-linked N-acetylglucosamine moiety. Hybrid Hybridomics 21: 233–236, 2002. doi: 10.1089/153685902760213831. [DOI] [PubMed] [Google Scholar]
- 35.Matsuoka Y, Takagi M, Ban T, Miyazaki M, Yamamoto T, Kondo Y, Yoneda Y. Identification and characterization of nuclear pore subcomplexes in mitotic extract of human somatic cells. Biochem Biophys Res Commun 254: 417–423, 1999. doi: 10.1006/bbrc.1998.9953. [DOI] [PubMed] [Google Scholar]
- 36.Teo CF, Ingale S, Wolfert MA, Elsayed GA, Nöt LG, Chatham JC, Wells L, Boons GJ. Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nat Chem Biol 6: 338–343, 2010. doi: 10.1038/nchembio.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu W, Han G, Yin Y, Jiang S, Yu G, Yang Q, Yu W, Ye X, Su Y, Yang Y, Hart GW, Sun H. AANL (Agrocybe aegerita lectin 2) is a new facile tool to probe for O-GlcNAcylation. Glycobiology 28: 363–373, 2018. doi: 10.1093/glycob/cwy029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, Hsieh-Wilson LC. A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. J Am Chem Soc 125: 16,162–16,163, 2003. doi: 10.1021/ja038545r. [DOI] [PubMed] [Google Scholar]
- 39.Vocadlo DJ, Hang HC, Kim EJ, Hanover JA, Bertozzi CR. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc Natl Acad Sci U S A 100: 9116–9121, 2003. doi: 10.1073/pnas.1632821100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc 125: 3192–3193, 2003. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 41.Roquemore EP, Chou TY, Hart GW. Detection of O-linked N-acetylglucosamine (O-GlcNAc) on cytoplasmic and nuclear proteins. Methods Enzymol 230: 443–460, 1994. doi: 10.1016/0076-6879(94)30028-3. [DOI] [PubMed] [Google Scholar]
- 42.Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mapping Sites of O-GlcNAc Modification Using Affinity Tags for Serine and Threonine Post-translational Modifications. Mol Cell Proteomics 1: 791–804, 2002. doi: 10.1074/mcp.m200048-mcp200. [DOI] [PubMed] [Google Scholar]
- 43.Thompson JW, Griffin ME, Hsieh-Wilson LC. Methods for the detection, study, and dynamic profiling of O-GlcNAc glycosylation. Methods Enzymol 598: 101–135, 2018. doi: 10.1016/bs.mie.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma J, Hart G. O-GlcNAc profiling: From proteins to proteomes. Clin Proteom 11, 8, 2014. doi: 10.1186/1559-0275-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zachara NE, Vosseller K, Hart GW. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Protein Sci Chapter12: Unit12.18, 2011.. doi: 10.1002/0471140864.ps1208s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem 266: 4706–4712, 1991. [PubMed] [Google Scholar]
- 47.National Center for Biotechnology Information. PubChem Database. Azaserine C. https://pubchem.ncbi.nlm.nih.gov/compound/460129.
- 48.National Center for Biotechnology Information. PubChem Database. 6-Diazo-5-oxo-L-norleucine C. https://pubchem.ncbi.nlm.nih.gov/compound/9087.
- 49.Kim EJ, Amorelli B, Abdo M, Thomas CJ, Love DC, Knapp S, Hanover JA. Distinctive inhibition of O-GlcNAcase isoforms by an α-GlcNAc thiolsulfonate. J Am Chem Soc 129: 14854–14855, 2007. doi: 10.1021/ja076038u. [DOI] [PubMed] [Google Scholar]
- 50.Macauley MS, Vocadlo DJ. Increasing O-GlcNAc levels: An overview of small-molecule inhibitors of O-GlcNAcase. Biochim Biophys Acta 1800: 107–121, 2010. doi: 10.1016/j.bbagen.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 51.Shanmugasundaram B, Debowski AW, Dennis RJ, Davies GJ, Vocadlo DJ, Vasella A. Inhibition of O-GlcNAcase by a gluco-configured nagstatin and a PUGNAc-imidazole hybrid inhibitor. Chem Commun: 4372–4374, 2006. doi: 10.1039/B612154C. [DOI] [PubMed] [Google Scholar]
- 52.Dorfmueller HC, Borodkin VS, Schimpl M, Shepherd SM, Shpiro NA, van Aalten DM. GlcNAcstatin: a picomolar, selective O-GlcNAcase inhibitor that modulates intracellular O-glcNAcylation levels. J Am Chem Soc 128: 16,484–16,485, 2006. doi: 10.1021/ja066743n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dorfmueller HC, Borodkin VS, Schimpl M, van Aalten DM. GlcNAcstatins are nanomolar inhibitors of human O-GlcNAcase inducing cellular hyper-O-GlcNAcylation. Biochem Jl 420: 221–227, 2009. doi: 10.1042/BJ20090110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. O-GlcNAcase uses substrate-assisted catalysis: kinetic analysis and development of highly selective mechanism-inspired inhibitors. J Biol Chem 280: 25,313–25,322, 2005. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]
- 55.Kim C, Nam DW, Park SY, Song H, Hong HS, Boo JH, Jung ES, Kim Y, Baek JY, Kim KS, Cho JW, Mook-Jung I. O-linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiol Aging 34: 275–285, 2013. doi: 10.1016/j.neurobiolaging.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Macauley MS, Vocadlo DJ. Enzymatic characterization and inhibition of the nuclear variant of human O-GlcNAcase. Carbohydr Res 344: 1079–1084, 2009. doi: 10.1016/j.carres.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 57.Haltiwanger RS, Grove K, Philipsberg GA. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-β-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate. J Biol Chem 273: 3611–3617, 1998. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- 58.Konrad RJ, Mikolaenko I, Tolar JF, Liu K, Kudlow JE. The potential mechanism of the diabetogenic action of streptozotocin: inhibition of pancreatic beta-cell O-GlcNAc-selective N-acetyl-beta-D-glucosaminidase. Biochem J 356: 31–41, 2001. doi: 10.1042/bj3560031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roos MD, Xie W, Su K, Clark JA, Yang X, Chin E, Paterson AJ, Kudlow JE. Streptozotocin, an analog of N-acetylglucosamine, blocks the removal of O-GlcNAc from intracellular proteins. Proc Assoc Am Physicians 110: 422–432, 1998. [PubMed] [Google Scholar]
- 60.Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol 4: 483–490, 2008. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- 61.Dorfmueller HC, Borodkin VS, Blair DE, Pathak S, Navratilova I, van Aalten DM. Substrate and product analogues as human O-GlcNAc transferase inhibitors. Amino acids 40: 781–792, 2011. doi: 10.1007/s00726-010-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Konrad RJ, Zhang F, Hale JE, Knierman MD, Becker GW, Kudlow JE. Alloxan is an inhibitor of the enzyme O-linked N-acetylglucosamine transferase. Biochem Biophys Res Commun 293: 207–212, 2002. doi: 10.1016/S0006-291X(02)00200-0. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Ren Y, Cao Y, Huang H, Wu Q, Li W, Wu S, Zhang J. Discovery of a low toxicity O-GlcNAc transferase (OGT) inhibitor by structure-based virtual screening of natural products. Sci Rep 7: 12334, 2017. doi: 10.1038/s41598-017-12522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pantaleon M, Tan HY, Kafer GR, Kaye PL. Toxic effects of hyperglycemia are mediated by the hexosamine signaling pathway and O-linked glycosylation in early mouse embryos. Biol Reprod 82: 751–758, 2010. doi: 10.1095/biolreprod.109.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berthier A, Vinod M, Porez G, Steenackers A, Alexandre J, Yamakawa N, Gheeraert C, Ploton M, Maréchal X, Dubois-Chevalier J, Hovasse A, Schaeffer-Reiss C, Cianférani S, Rolando C, Bray F, Duez H, Eeckhoute J, Lefebvre T, Staels B, Lefebvre P. Combinatorial regulation of hepatic cytoplasmic signaling and nuclear transcriptional events by the OGT/REV-ERBα complex. Proc Natl Acad Sci USA 115: E11033–E11042, 2018. doi: 10.1073/pnas.1805397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Itkonen HM, Gorad SS, Duveau DY, Martin SE, Barkovskaya A, Bathen TF, Moestue SA, Mills IG. Inhibition of O-GlcNAc transferase activity reprograms prostate cancer cell metabolism. Oncotarget 7: 12,464–12,476, 2016. doi: 10.18632/oncotarget.7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. A small molecule that inhibits OGT activity in cells. ACS Chem Biol 10: 1392–1397, 2015. doi: 10.1021/acschembio.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park S-K, Zhou X, Pendleton KE, Hunter OV, Kohler JJ, O’Donnell KA, Conrad NK. A conserved splicing silencer dynamically regulates O-GlcNAc transferase intron retention and O-GlcNAc homeostasis. Cell Rep 20: 1088–1099, 2017. doi: 10.1016/j.celrep.2017.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin SES, Tan ZW, Itkonen HM, Duveau DY, Paulo JA, Janetzko J, Boutz PL, Törk L, Moss FA, Thomas CJ, Gygi SP, Lazarus MB, Walker S. Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors. J Am Chem Soc 140: 13542–13545, 2018. doi: 10.1021/jacs.8b07328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gross BJ, Kraybill BC, Walker S. Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc 127: 14588–14589, 2005. doi: 10.1021/ja0555217. [DOI] [PubMed] [Google Scholar]
- 71.Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. Journal of molecular and cellular cardiology 45: 313–325, 2008. doi: 10.1016/j.yjmcc.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu W, Zheng X, Wang J, Yang T, Dai W, Song S, Fang L, Wang Y, Gu J. O-GlcNAcylation on Rab3A attenuates its effects on mitochondrial oxidative phosphorylation and metastasis in hepatocellular carcinoma. Cell Death Dis 9: 970, 2018. doi: 10.1038/s41419-018-0961-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zafir A, Readnower R, Long BW, McCracken J, Aird A, Alvarez A, Cummins TD, Li Q, Hill BG, Bhatnagar A, Prabhu SD, Bolli R, Jones SP. Protein O-GlcNAcylation is a novel cytoprotective signal in cardiac stem cells. Stem Cells 31: 765–775, 2013. doi: 10.1002/stem.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem 287: 11070–11081, 2012. doi: 10.1074/jbc.M111.302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheng S, Mao Q, Dong Y, Ren J, Su L, Liu J, Liu Q, Zhou J, Ye X, Zheng S, Zhu N. GNB2L1 and its O-GlcNAcylation regulates metastasis via modulating epithelial-mesenchymal transition in the chemoresistance of gastric cancer. PloS One 12: e0182696, 2017. doi: 10.1371/journal.pone.0182696. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol 7: 174–181, 2011. doi: 10.1038/nchembio.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu TW, Zandberg WF, Gloster TM, Deng L, Murray KD, Shan X, Vocadlo DJ. Metabolic Inhibitors of O-GlcNAc Transferase That Act In Vivo Implicate Decreased O-GlcNAc Levels in Leptin-Mediated Nutrient Sensing. Angew Chem Int Ed 57: 7644–7648, 2018. doi: 10.1002/anie.201803254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang J, Lazarus MB, Pasquina L, Sliz P, Walker S. A neutral diphosphate mimic crosslinks the active site of human O-GlcNAc transferase. Nat Chem Biol 8: 72–77, 2012. doi: 10.1038/nchembio.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cameron A, Giacomozzi B, Joyce J, Gray A, Graham D, Ousson S, Neny M, Beher D, Carlson G, O'Moore J, Shearman M, Hering H. Generation and characterization of a rabbit monoclonal antibody site-specific for tau O-GlcNAcylated at serine 400. FEBS Lett 587: 3722–3728, 2013. doi: 10.1016/j.febslet.2013.09.042. [DOI] [PubMed] [Google Scholar]
- 80.Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, Roeder RG, Brown M, Kato S. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 480: 557–560, 2011. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pathak S, Borodkin VS, Albarbarawi O, Campbell DG, Ibrahim A, van Aalten DM. O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release. EMBO J 31: 1394–1404, 2012. doi: 10.1038/emboj.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shan H, Sun J, Shi M, Liu X, Shi Z, Yu W, Gu Y. Generation and characterization of a site-specific antibody for SIRT1 O-GlcNAcylated at serine 549. Glycobiology 28: 482–487, 2018. doi: 10.1093/glycob/cwy040. [DOI] [PubMed] [Google Scholar]
- 83.Reeves RA, Lee A, Henry R, Zachara NE. Characterization of the specificity of O-GlcNAc reactive antibodies under conditions of starvation and stress. Anal Biochem 457: 8–18, 2014. doi: 10.1016/j.ab.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Banerjee PS, Hart GW, Cho JW. Chemical approaches to study O-GlcNAcylation. Chem Soc Rev 42: 4345–4357, 2013. doi: 10.1039/C2CS35412H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cecioni S, Vocadlo DJ. Tools for probing and perturbing O-GlcNAc in cells and in vivo. Curr Opin Chem Biol 17: 719–728, 2013. doi: 10.1016/j.cbpa.2013.06.030. [DOI] [PubMed] [Google Scholar]
- 86.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta 1764: 1811–1822, 2006. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao P, Viner R, Teo CF, Boons GJ, Horn D, Wells L. Combining high-energy C-trap dissociation and electron transfer dissociation for protein O-GlcNAc modification site assignment. J Proteome Res 10: 4088–4104, 2011. doi: 10.1021/pr2002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ma J, Hart GW. Analysis of protein O-GlcNAcylation by mass spectrometry. Curr Protoc Protein Sci 87: 24 10 21–24 10 16, 2017. doi: 10.1002/cpps.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zachara NE, Molina H, Wong KY, Pandey A, Hart GW. The dynamic stress-induced “O-GlcNAc-ome” highlights functions for O-GlcNAc in regulating DNA damage/repair and other cellular pathways. Amino Acids 40: 793–808, 2011. doi: 10.1007/s00726-010-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee A, Miller D, Henry R, Paruchuri VDP, O’Meally RN, Boronina T, Cole RN, Zachara NE. Combined antibody/lectin enrichment identifies extensive changes in the O-GlcNAc sub-proteome upon oxidative stress. J Proteome Res 15: 4318–4336, 2016. doi: 10.1021/acs.jproteome.6b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim EJ. The utilities of chemical reactions and molecular tools for O-GlcNAc proteomic studies. Chembiochem 16: 1397–1409, 2015. doi: 10.1002/cbic.201500183. [DOI] [PubMed] [Google Scholar]
- 92.Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, Wang Z, Camp DG 2nd, Shabanowitz J, Stanley P, Hart GW, Hunt DF, Yang F, Smith RD. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc Natl Acad Sci U S A 109: 7280–7285, 2012. doi: 10.1073/pnas.1200425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Z, Udeshi ND, O'Malley M, Shabanowitz J, Hunt DF, Hart GW. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol Cell Proteomics 9: 153–160, 2010. doi: 10.1074/mcp.M900268-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Griffin ME, Jensen EH, Mason DE, Jenkins CL, Stone SE, Peters EC, Hsieh-Wilson LC. Comprehensive mapping of O-GlcNAc modification sites using a chemically cleavable tag. Mol Biosyst 12: 1756–1759, 2016. doi: 10.1039/C6MB00138F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ma J, Wang WH, Li Z, Shabanowitz J, Hunt DF, Hart GW. O-GlcNAc site mapping by using a combination of chemoenzymatic labeling, copper-free click chemistry, reductive cleavage, and electron-transfer dissociation mass spectrometry. Anal Chem 91: 2620–2625, 2019. doi: 10.1021/acs.analchem.8b05688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thompson JW, Sorum AW, Hsieh-Wilson LC. Deciphering the functions of O-GlcNAc glycosylation in the brain: the role of site-specific quantitative O-GlcNAcomics. Biochemistry 57: 4010–4018, 2018. doi: 10.1021/acs.biochem.8b00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li X, Molina H, Huang H, Zhang Y-y, Liu M, Qian S-W, Slawson C, Dias WB, Pandey A, Hart GW, Lane MD, Tang Q-Q. O-linked N-acetylglucosamine modification on CCAAT enhancer-binding protein β: role during adipocyte differentiation. J Biol Chem 284: 19,248–19,254, 2009. doi: 10.1074/jbc.M109.005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chou TY, Hart GW, Dang CV. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem 270: 18,961–18,965, 1995. doi: 10.1074/jbc.270.32.18961. [DOI] [PubMed] [Google Scholar]
- 99.Kamemura K, Hayes BK, Comer FI, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J Biol Chem 277: 19,229–19,235, 2002. doi: 10.1074/jbc.M201729200. [DOI] [PubMed] [Google Scholar]
- 100.Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, Hsieh-Wilson LC. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat Chem Biol 8: 253–261, 2012. doi: 10.1038/nchembio.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cheng X, Hart GW. Glycosylation of the murine estrogen receptor-α. J Steroid Biochem Mol Biol 75: 147–158, 2000. doi: 10.1016/S0960-0760(00)00167-9. [DOI] [PubMed] [Google Scholar]
- 102.Jiang MS, Hart GW. A subpopulation of estrogen receptors are modified by O-linked N-acetylglucosamine. J Biol Chem 272: 2421–2428, 1997. doi: 10.1074/jbc.272.4.2421. [DOI] [PubMed] [Google Scholar]
- 103.Cheng X, Hart GW. Alternative O-glycosylation/O-phosphorylation of serine-16 in murine estrogen receptor beta: post-translational regulation of turnover and transactivation activity. J Biol Chem 276: 10,570–10,575, 2001. doi: 10.1074/jbc.M010411200. [DOI] [PubMed] [Google Scholar]
- 104.Misra J, Kim DK, Jung YS, Kim HB, Kim YH, Yoo EK, Kim BG, Kim S, Lee IK, Harris RA, Kim JS, Lee CH, Cho JW, Choi HS. O-GlcNAcylation of orphan nuclear receptor estrogen-related receptor γ promotes hepatic gluconeogenesis. Diabetes 65: 2835–2848, 2016. doi: 10.2337/db15-1523. [DOI] [PubMed] [Google Scholar]
- 105.Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, Puigserver P, Hart GW. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem 283: 16,283–16,292, 2008. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Berrabah W, Aumercier P, Gheeraert C, Dehondt H, Bouchaert E, Alexandre J, Ploton M, Mazuy C, Caron S, Tailleux A, Eeckhoute J, Lefebvre T, Staels B, Lefebvre P. Glucose sensing O-GlcNAcylation pathway regulates the nuclear bile acid receptor farnesoid X receptor (FXR). Hepatology 59: 2022–2033, 2014. doi: 10.1002/hep.26710. [DOI] [PubMed] [Google Scholar]
- 107.Chen PH, Smith TJ, Wu J, Siesser PF, Bisnett BJ, Khan F, Hogue M, Soderblom E, Tang F, Marks JR, Major MB, Swarts BM, Boyce M, Chi JT. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J 36: 2233–2250, 2017. doi: 10.15252/embj.201696113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kommaddi RP, Dickson KM, Barker PA. Stress-induced expression of the p75 neurotrophin receptor is regulated by O-GlcNAcylation of the Sp1 transcription factor. J Neurochem 116: 396–405, 2011. doi: 10.1111/j.1471-4159.2010.07120.x. [DOI] [PubMed] [Google Scholar]
- 109.Yang X, Su K, Roos MD, Chang Q, Paterson AJ, Kudlow JE. O-linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc Natl Acad Sci U S A 98: 6611–6616, 2001. doi: 10.1073/pnas.111099998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bindesbøll C, Fan Q, Nørgaard RC, MacPherson L, Ruan HB, Wu J, Pedersen TA, Steffensen KR, Yang X, Matthews J, Mandrup S, Nebb HI, Grønning-Wang LM. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J Lipid Res 56: 771–785, 2015. doi: 10.1194/jlr.M049130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fan Q, Moen A, Anonsen JH, Bindesbøll C, Sæther T, Carlson CR, Grønning-Wang LM. O-GlcNAc site-mapping of liver X receptor-α and O-GlcNAc transferase. Biochem Biophys Res Commun 499: 354–360, 2018. doi: 10.1016/j.bbrc.2018.03.164. [DOI] [PubMed] [Google Scholar]
- 112.Fan Q, Norgaard RC, Bindesbøll C, Lucas C, Dalen KT, Babaie E, Itkonen HM, Matthews J, Nebb HI, Grønning-Wang LM. LXRα regulates hepatic ChREBPα activity and lipogenesis upon glucose, but not fructose feeding in mice. Nutrients 9: 678, 2017. doi: 10.3390/nu9070678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Constable S, Lim JM, Vaidyanathan K, Wells L. O-GlcNAc transferase regulates transcriptional activity of human Oct4. Glycobiology 27: 927–937, 2017. doi: 10.1093/glycob/cwx055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Qiu H, Liu F, Tao T, Zhang D, Liu X, Zhu G, Xu Z, Ni R, Shen A. Modification of p27 with O-linked N-acetylglucosamine regulates cell proliferation in hepatocellular carcinoma. Mol Carcinog 56: 258–271, 2017. doi: 10.1002/mc.22490. [DOI] [PubMed] [Google Scholar]
- 115.Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, Cho JW. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol 8: 1074–1083, 2006. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- 116.Kaasik K, Kivimäe S, Allen JJ, Chalkley RJ, Huang Y, Baer K, Kissel H, Burlingame AL, Shokat KM, Ptáĉek LJ, Fu YH. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell metabolism 17: 291–302, 2013. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Housley MP, Udeshi ND, Rodgers JT, Shabanowitz J, Puigserver P, Hunt DF, Hart GW. A PGC-1α:O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J Biol Chem 284: 5148–5157, 2009. doi: 10.1074/jbc.M808890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, Zhao L, Bennett AM, Samuel VT, Wu J, Yates JR 3rd, Yang X. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1α stability. Cell Metab 16: 226–237, 2012. doi: 10.1016/j.cmet.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ji S, Park SY, Roth J, Kim HS, Cho JW. O-GlcNAc modification of PPARγ reduces its transcriptional activity. Biochem Biophys Res Commun 417: 1158–1163, 2012. doi: 10.1016/j.bbrc.2011.12.086. [DOI] [PubMed] [Google Scholar]
- 120.Han C, Gu Y, Shan H, Mi W, Sun J, Shi M, Zhang X, Lu X, Han F, Gong Q, Yu W. W. O-GlcNAcylation of SIRT1 enhances its deacetylase activity and promotes cytoprotection under stress. Nat Commun 8: 1491, 2017. doi: 10.1038/s41467-017-01654-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gandy JC, Rountree AE, Bijur GN. Akt1 is dynamically modified with O-GlcNAc following treatments with PUGNAc and insulin-like growth factor-1. FEBS Lett 580: 3051–3058, 2006. doi: 10.1016/j.febslet.2006.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kang ES, Han D, Park J, Kwak TK, Oh MA, Lee SA, Choi S, Park ZY, Kim Y, Lee JW. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic β cells. Exp Cell Res 314: 2238–2248, 2008. doi: 10.1016/j.yexcr.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 123.Park SY, Ryu J, Lee W. O-GlcNAc modification on IRS-1 and Akt2 by PUGNAc inhibits their phosphorylation and induces insulin resistance in rat primary adipocytes. Exp Mol Med 37: 220–229, 2005. doi: 10.1038/emm.2005.30. [DOI] [PubMed] [Google Scholar]
- 124.Shi J, Gu JH, Dai CL, Gu J, Jin X, Sun J, Iqbal K, Liu F, Gong CX. O-GlcNAcylation regulates ischemia-induced neuronal apoptosis through AKT signaling. Sci Rep 5: 14500, 2015. doi: 10.1038/srep14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shi J, Wu S, Dai CL, Li Y, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX. Diverse regulation of AKT and GSK-3β by O-GlcNAcylation in various types of cells. FEBS Lett 586: 2443–2450, 2012. doi: 10.1016/j.febslet.2012.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang S, Huang X, Sun D, Xin X, Pan Q, Peng S, Liang Z, Luo C, Yang Y, Jiang H, Huang M, Chai W, Ding J, Geng M. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PloS one 7: e37427, 2012. doi: 10.1371/journal.pone.0037427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dentin R, Hedrick S, Xie J, Yates J 3rd, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 319: 1402–1405, 2008. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- 128.Park J, Han D, Kim K, Kang Y, Kim Y. O-GlcNAcylation disrupts glyceraldehyde-3-phosphate dehydrogenase homo-tetramer formation and mediates its nuclear translocation. Biochim Biophys Acta 1794: 254–262, 2009. doi: 10.1016/j.bbapap.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 129.Li Y, Roux C, Lazereg S, LeCaer JP, Laprevote O, Badet B, Badet-Denisot MA. Identification of a novel serine phosphorylation site in human glutamine:fructose-6-phosphate amidotransferase isoform 1. Biochemistry 46: 13163–13169, 2007. doi: 10.1021/bi700694c. [DOI] [PubMed] [Google Scholar]
- 130.Zibrova D, Vandermoere F, Göransson O, Peggie M, Mariño KV, Knierim A, Spengler K, Weigert C, Viollet B, Morrice NA, Sakamoto K, Heller R. GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis. Biochem J 474: 983–1001, 2017. doi: 10.1042/BCJ20160980. [DOI] [PubMed] [Google Scholar]
- 131.Lubas WA, Hanover JA. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J Biol Chem 275: 10,983–10,988, 2000. doi: 10.1074/jbc.275.15.10983. [DOI] [PubMed] [Google Scholar]
- 132.Maury JJ, Ng D, Bi X, Bardor M, Choo AB. Multiple reaction monitoring mass spectrometry for the discovery and quantification of O-GlcNAc-modified proteins. Anal Chem 86: 395–402, 2014. doi: 10.1021/ac401821d. [DOI] [PubMed] [Google Scholar]
- 133.Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci 99: 5313–5318, 2002. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rao X, Duan X, Mao W, Li X, Li Z, Li Q, Zheng Z, Xu H, Chen M, Wang PG, Wang Y, Shen B, Yi W. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat Commun 6: 8468, 2015. doi: 10.1038/ncomms9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ball LE, Berkaw MN, Buse MG. Identification of the major site of O-linked β-N-acetylglucosamine modification in the C terminus of insulin receptor substrate-1. Mol Cell Proteomics 5: 313–323, 2006. doi: 10.1074/mcp.M500314-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Klein AL, Berkaw MN, Buse MG, Ball LE. O-linked N-acetylglucosamine modification of insulin receptor substrate-1 occurs in close proximity to multiple SH2 domain binding motifs. Mol Cell Proteomics 8: 2733–2745, 2009. doi: 10.1074/mcp.M900207-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li T, Zhang Z, KolwiczSC, Jr, Abell L, Roe ND, Kim M, Zhou B, Cao Y, Ritterhoff J, Gu H, Raftery D, Sun H, Tian R. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab 25: 374–385, 2017. doi: 10.1016/j.cmet.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ma J, Liu T, Wei AC, Banerjee P, O'Rourke B, Hart GW. O-GlcNAcomic profiling identifies widespread O-linked β-N-acetylglucosamine modification (O-GlcNAcylation) in oxidative phosphorylation system regulating cardiac mitochondrial function. J Biol Chem 290: 29141–29153, 2015. doi: 10.1074/jbc.M115.691741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ma J, Banerjee P, Whelan SA, Liu T, Wei AC, Ramirez-Correa G, McComb ME, Costello CE, O’Rourke B, Murphy A, Hart GW. Comparative proteomics reveals dysregulated mitochondrial O-GlcNAcylation in diabetic hearts. J Proteome Res 15: 2254–2264, 2016. doi: 10.1021/acs.jproteome.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337: 975–980, 2012. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang Y, Liu J, Jin X, Zhang D, Li D, Hao F, Feng Y, Gu S, Meng F, Tian M, Zheng Y, Xin L, Zhang X, Han X, Aravind L, Wei M. O-GlcNAcylation destabilizes the active tetrameric PKM2 to promote the Warburg effect. Proc Natl Acad Sci U S A 114: 13,732–13,737, 2017. doi: 10.1073/pnas.1704145115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Krishnamoorthy V, Donofrio AJ, Martin JL. O-GlcNAcylation of αB-crystallin regulates its stress-induced translocation and cytoprotection. Mol Cell Biochem 379: 59–68, 2013. doi: 10.1007/s11010-013-1627-5. [DOI] [PubMed] [Google Scholar]
- 143.Roquemore EP, Chevrier MR, Cotter RJ, Hart GW. Dynamic O-GlcNAcylation of the small heat shock protein α B-crystallin. Biochemistry 35: 3578–3586, 1996. doi: 10.1021/bi951918j. [DOI] [PubMed] [Google Scholar]
- 144.Akimoto Y, Yan K, Miura Y, Tsumoto H, Toda T, Fukutomi T, Sugahara D, Kudo A, Arai T, Chiba Y, Kaname S, Hart GW, Endo T, Kawakami H. O-GlcNAcylation and phosphorylation of β-actin serine199 in diabetic nephropathy. Am J Physiol Renal Physiol 317: F1359–F1374, 2019. doi: 10.1152/ajprenal.00566.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hédou J, Bastide B, Page A, Michalski JC, Morelle W. Mapping of O-linked β-N-acetylglucosamine modification sites in key contractile proteins of rat skeletal muscle. Proteomics 9: 2139–2148, 2009. doi: 10.1002/pmic.200800617. [DOI] [PubMed] [Google Scholar]
- 146.Hédou J, Cieniewski-Bernard C, Leroy Y, Michalski JC, Mounier Y, Bastide B. O-linked N-acetylglucosaminylation is involved in the Ca2+ activation properties of rat skeletal muscle. J Biol Chem 282: 10360–10369, 2007. doi: 10.1074/jbc.M606787200. [DOI] [PubMed] [Google Scholar]
- 147.Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, Vecoli C, Hart GW, Murphy AM. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res 103: 1354–1358, 2008. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ku NO, Omary MB. Identification and mutational analysis of the glycosylation sites of human keratin 18. J Biol Chem 270: 11820–11827, 1995. doi: 10.1074/jbc.270.20.11820. [DOI] [PubMed] [Google Scholar]
- 149.Ku NO, Toivola DM, Strnad P, Omary MB. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat Cell Biol 12: 876–885, 2010. doi: 10.1038/ncb2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ramirez-Correa G, Ma J, Slawson C, Zeidan Q, Lugo-Fagundo NS, Xu M, Shen X, Gao WD, Caceres V, Chakir K, DeVine L, Cole R, Marchionni L, Paolocci N, Hart GW, Murphy AM. Removal of abnormal myofilament O-GlcNAcylation restores Ca2+ sensitivity in diabetic cardiac muscle. Diabetes 64: 3573–3587, 2015. doi: 10.2337/db14-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rn C, Hart GW. Glycosylation sites flank phosphorylation sites on synapsin I: O-linked N-acetylglucosamine residues are localized within domains mediating synapsin I interactions. J Neurochem 73: 418–428, 1999. [DOI] [PubMed] [Google Scholar]
- 152.Skorobogatko Y, Landicho A, Chalkley RJ, Kossenkov AV, Gallo G, Vosseller K. O-linked β-N-acetylglucosamine (O-GlcNAc) site thr-87 regulates synapsin I localization to synapses and size of the reserve pool of synaptic vesicles. J Biol Chem 289: 3602–3612, 2014. doi: 10.1074/jbc.M113.512814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kang MJ, Kim C, Jeong H, Cho BK, Ryou AL, Hwang D, Mook-Jung I, Yi EC. Synapsin-1 and tau reciprocal O-GlcNAcylation and phosphorylation sites in mouse brain synaptosomes. Exp Mol Med 45: e29, 2013. doi: 10.1038/emm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Smet-Nocca C, Broncel M, Wieruszeski JM, Tokarski C, Hanoulle X, Leroy A, Landrieu I, Rolando C, Lippens G, Hackenberger CP. Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation. Mol Biosyst 7: 1420–1429, 2011. doi: 10.1039/c0mb00337a. [DOI] [PubMed] [Google Scholar]
- 155.Yuzwa SA, Cheung AH, Okon M, McIntosh LP, Vocadlo DJ. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J Mol Biol 426: 1736–1752, 2014. doi: 10.1016/j.jmb.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 156.Dubois-Deruy E, Belliard A, Mulder P, Bouvet M, Smet-Nocca C, Janel S, Lafont F, Beseme O, Amouyel P, Richard V, Pinet F. Interplay between troponin T phosphorylation and O-N-acetylglucosaminylation in ischaemic heart failure. Cardiovascular Res 107: 56–65, 2015. doi: 10.1093/cvr/cvv136. [DOI] [PubMed] [Google Scholar]
- 157.Cao W, Cao J, Huang J, Yao J, Yan G, Xu H, Yang P. Discovery and confirmation of O-GlcNAcylated proteins in rat liver mitochondria by combination of mass spectrometry and immunological methods. PloS One 8: e76399, 2013. doi: 10.1371/journal.pone.0076399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cha M-Y, Cho HJ, Kim C, Jung YO, Kang MJ, Murray ME, Hong HS, Choi Y-J, Choi H, Kim DK, Choi H, Kim J, Dickson DW, Song HK, Cho JW, Yi EC, Kim J, Jin SM, Mook-Jung I. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer's disease. Hum Mol Genet 24: 6492–6504, 2015. doi: 10.1093/hmg/ddv358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates J, Hoshijima M, Dillmann WH. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287: 30024–30034, 2012. doi: 10.1074/jbc.M112.390682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158: 54–68, 2014. doi: 10.1016/j.cell.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Hu Y, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem 284: 547–555, 2009. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gu Y, Ande SR, Mishra S. Altered O-GlcNAc modification and phosphorylation of mitochondrial proteins in myoblast cells exposed to high glucose. Arch Biochem Biophys 505: 98–104, 2011. doi: 10.1016/j.abb.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 163.Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, Hart GW, Marban E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 117: 1172–1182, 2008. doi: 10.1161/CIRCULATIONAHA.107.730515. [DOI] [PubMed] [Google Scholar]
- 164.Palaniappan KK, Hangauer MJ, Smith TJ, Smart BP, Pitcher AA, Cheng EH, Bertozzi CR, Boyce M. A chemical glycoproteomics platform reveals O-GlcNAcylation of mitochondrial voltage-dependent anion channel 2. Cell Rep 5: 546–552, 2013. doi: 10.1016/j.celrep.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502: 372–376, 2013. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lu S, Liao Z, Lu X, Katschinski DM, Mercola M, Chen J, Heller Brown J, Molkentin JD, Bossuyt J, Bers DM. Hyperglycemia acutely increases cytosolic reactive oxygen species via O-linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ Res 126: e80–e96, 2020. doi: 10.1161/CIRCRESAHA.119.316288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dias WB, Cheung WD, Wang Z, Hart GW. Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J Biol Chem 284: 21327–21337, 2009. doi: 10.1074/jbc.M109.007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ning X, Tao T, Shen J, Ji Y, Xie L, Wang H, Liu N, Xu X, Sun C, Zhang D, Shen A, Ke K. The O-GlcNAc modification of CDK5 involved in neuronal apoptosis following in vitro intracerebral hemorrhage. Cell Mol Neurobiol 37: 527–536, 2017. doi: 10.1007/s10571-016-0391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lazarus BD, Love DC, Hanover JA. Recombinant O-GlcNAc transferase isoforms: identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates. Glycobiology 16: 415–421, 2006. doi: 10.1093/glycob/cwj078. [DOI] [PubMed] [Google Scholar]
- 170.Tarrant MK, Rho HS, Xie Z, Jiang YL, Gross C, Culhane JC, Yan G, Qian J, Ichikawa Y, Matsuoka T, Zachara N, Etzkorn FA, Hart GW, Jeong JS, Blackshaw S, Zhu H, Cole PA. Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat Chem Biol 8: 262–269, 2012. doi: 10.1038/nchembio.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Brewer RA, Collins HE, Berry RD, Brahma MK, Tirado BA, Peliciari-Garcia RA, Stanley HL, Wende AR, Taegtmeyer H, Rajasekaran NS, Darley-Usmar V, Zhang J, Frank SJ, Chatham JC, Young ME. Temporal partitioning of adaptive responses of the murine heart to fasting. Life sciences 197: 30–39, 2018. doi: 10.1016/j.lfs.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of p53 enhances catalytic activity of IKKβ through O-linked β-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A 106: 3431–3436, 2009. doi: 10.1073/pnas.0813210106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lima VV, Giachini FR, Hardy DM, Webb RC, Tostes RC. O-GlcNAcylation: a novel pathway contributing to the effects of endothelin in the vasculature. Am J Physiol Regul Integr Comp Physiol 300: R236–R250, 2011. doi: 10.1152/ajpregu.00230.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Miura T, Kume M, Kawamura T, Yamamoto K, Hamakubo T, Nishihara S. O-GlcNAc on PKCζ inhibits the FGF4-PKCζ-MEK-ERK1/2 pathway via inhibition of PKCζ phosphorylation in mouse embryonic stem cells. Stem Cell Reports 10: 272–286, 2018. doi: 10.1016/j.stemcr.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Robles-Flores M, Melendez L, Garcia W, Mendoza-Hernandez G, Lam TT, Castaneda-Patlan C, Gonzalez-Aguilar H. Posttranslational modifications on protein kinase c isozymes. Effects of epinephrine and phorbol esters. Biochim Biophys Acta 1783: 695–712, 2008. doi: 10.1016/j.bbamcr.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 176.Zhao Y, Tang Z, Shen A, Tao T, Wan C, Zhu X, Huang J, Zhang W, Xia N, Wang S, Cui S, Zhang D. The role of PTP1B O-GlcNAcylation in hepatic insulin resistance. Int J Mol Sci 16: 22856–22869, 2015. doi: 10.3390/ijms160922856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Duan F, Wu H, Jia D, Wu W, Ren S, Wang L, Song S, Guo X, Liu F, Ruan Y, Gu J. O-GlcNAcylation of RACK1 promotes hepatocellular carcinogenesis. J Hepatol 68: 1191–1202, 2018. doi: 10.1016/j.jhep.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 178.Ohn T, Kedersha N, Hickman T, Tisdale S, Anderson P. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat Cell Biol 10: 1224–1231, 2008. doi: 10.1038/ncb1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, Kalil AC, Bhinderwala F, Powers R, Yin G, Herring LE, Asara JM, Lei YL, Yang X, Rodriguez DA, Yang M, Green DR, Singh PK, Wen H. O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity 50: 576–590,2019. e576doi: 10.1016/j.immuni.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovacs AL, Zhang Z, Feng D, Chen S, Zhang H. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol 16: 1215–1226, 2014. doi: 10.1038/ncb3066. [DOI] [PubMed] [Google Scholar]
- 181.Ruan H-B, Ma Y, Torres S, Zhang B, Feriod C, Heck RM, Qian K, Fu M, Li X, Nathanson MH, Bennett AM, Nie Y, Ehrlich BE, Yang X. Calcium-dependent O-GlcNAc signaling drives liver autophagy in adaptation to starvation. Genes Dev 31: 1655, 2017. doi: 10.1101/gad.305441.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics 11: 215–229, 2012. doi: 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Peng C, Zhu Y, Zhang W, Liao Q, Chen Y, Zhao X, Guo Q, Shen P, Zhen B, Qian X, Yang D, Zhang JS, Xiao D, Qin W, Pei H. Regulation of the Hippo-YAP pathway by glucose sensor O-GlcNAcylation. Mol Cell 68: 591–604,2017. doi: 10.1016/j.molcel.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 184.Zhang X, Qiao Y, Wu Q, Chen Y, Zou S, Liu X, Zhu G, Zhao Y, Chen Y, Yu Y, Pan Q, Wang J, Sun F. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat Commun 8: 15280, 2017. doi: 10.1038/ncomms15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Levine PM, Galesic A, Balana AT, Mahul-Mellier AL, Navarro MX, De Leon CA, Lashuel HA, Pratt MR. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc Natl Acad Sci U S A 116: 1511–1519, 2019. doi: 10.1073/pnas.1808845116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ha JR, Hao L, Venkateswaran G, Huang YH, Garcia E, Persad S. β-catenin is O-GlcNAc glycosylated at serine 23: implications for β-catenin's subcellular localization and transactivator function. Exp Cell Res 321: 153–166, 2014. doi: 10.1016/j.yexcr.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 187.Olivier-Van Stichelen S, Dehennaut V, Buzy A, Zachayus JL, Guinez C, Mir AM, El Yazidi-Belkoura I, Copin MC, Boureme D, Loyaux D, Ferrara P, Lefebvre T. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41. FASEB J 28: 3325–3338, 2014. doi: 10.1096/fj.13-243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nagel AK, Schilling M, Comte-Walters S, Berkaw MN, Ball LE. Identification of O-linked N-acetylglucosamine (O-GlcNAc)-modified osteoblast proteins by electron transfer dissociation tandem mass spectrometry reveals proteins critical for bone formation. Mol Cell Proteomics 12: 945–955, 2013. doi: 10.1074/mcp.M112.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Li X, Zhu Q, Shi X, Cheng Y, Li X, Xu H, Duan X, Hsieh-Wilson LC, Chu J, Pelletier J, Ni M, Zheng Z, Li S, Yi W. O-GlcNAcylation of core components of the translation initiation machinery regulates protein synthesis. Proc Natl Acad Sci U S A 116: 7857–7866, 2019. doi: 10.1073/pnas.1813026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest 108: 1341–1348, 2001. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sager RA, Woodford MR, Backe SJ, Makedon AM, Baker-Williams AJ, DiGregorio BT, Loiselle DR, Haystead TA, Zachara NE, Prodromou C, Bourboulia D, Schmidt LS, Linehan WM, Bratslavsky G, Mollapour M. Post-translational regulation of FNIP1 creates a rheostat for the molecular chaperone Hsp90. Cell Rep 26: 1344–1356.e5, 2019. doi: 10.1016/j.celrep.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Myers SA, Daou S, Affar e B, Burlingame A. Electron transfer dissociation (ETD): the mass spectrometric breakthrough essential for O-GlcNAc protein site assignments-a study of the O-GlcNAcylated protein host cell factor C1. Proteomics 13: 982–991, 2013. doi: 10.1002/pmic.201200332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Overath T, Kuckelkorn U, Henklein P, Strehl B, Bonar D, Kloss A, Siele D, Kloetzel PM, Janek K. Mapping of O-GlcNAc sites of 20 S proteasome subunits and Hsp90 by a novel biotin-cystamine tag. Mol Cell Proteomics 11: 467–477, 2012. doi: 10.1074/mcp.M111.015966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yokoe S, Asahi M, Takeda T, Otsu K, Taniguchi N, Miyoshi E, Suzuki K. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: implications for diabetic cardiomyopathy. Glycobiology 20: 1217–1226, 2010. doi: 10.1093/glycob/cwq071. [DOI] [PubMed] [Google Scholar]
- 195.Tao T, He Z, Shao Z, Lu H. TAB3 O-GlcNAcylation promotes metastasis of triple negative breast cancer. Oncotarget 7: 22,807–22,818, 2016. doi: 10.18632/oncotarget.8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–520, 2015. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dias WB, Cheung WD, Hart GW. O-GlcNAcylation of kinases. Biochem Biophys Res Commun 422: 224–228, 2012. doi: 10.1016/j.bbrc.2012.04.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bouazzi H, Lesca G, Trujillo C, Alwasiyah MK, Munnich A. Nonsyndromic X-linked intellectual deficiency in three brothers with a novel MED12 missense mutation [c.5922G>T (p.Glu1974His)]. Clin Case Rep 3: 604–609, 2015. doi: 10.1002/ccr3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Toleman C, Paterson AJ, Whisenhunt TR, Kudlow JE. Characterization of the histone acetyltransferase (HAT) domain of a bifunctional protein with activable O-GlcNAcase and HAT activities. J Biol Chem 279: 53,665–53,673, 2004. doi: 10.1074/jbc.M410406200. [DOI] [PubMed] [Google Scholar]
- 200.Taylor EW, Wang K, Nelson AR, Bredemann TM, Fraser KB, Clinton SM, Puckett R, Marchase RB, Chatham JC, McMahon LL. O-GlcNAcylation of AMPA receptor GluA2 is associated with a novel form of long-term depression at hippocampal synapses. J Neurosci 34: 10–21, 2014. doi: 10.1523/JNEUROSCI.4761-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ruan HB, Dietrich MO, Liu ZW, Zimmer MR, Li MD, Singh JP, Zhang K, Yin R, Wu J, Horvath TL, Yang X. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 159: 306–317, 2014. doi: 10.1016/j.cell.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci U S A 113: 15120–15125, 2016. doi: 10.1073/pnas.1606899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lagerlöf O, Slocomb JE, Hong I, Aponte Y, Blackshaw S, Hart GW, Huganir RL. The nutrient sensor OGT in PVN neurons regulates feeding. Science 351: 1293–1296, 2016. doi: 10.1126/science.aad5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wang K, Ho SR, Mao W, Huang P, Zhang F, Schwiebert EM, Kudlow JE, Paterson AJ. Increased O-GlcNAc causes disrupted lens fiber cell differentiation and cataracts. Biochem Biophys Res Commun 387: 70–76, 2009. doi: 10.1016/j.bbrc.2009.06.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Huang P, Ho SR, Wang K, Roessler BC, Zhang F, Hu Y, Bowe DB, Kudlow JE, Paterson AJ. Muscle-specific overexpression of NCOATGK, splice variant of O-GlcNAcase, induces skeletal muscle atrophy. Am J Physiol Cell Physiol 300: C456–C465, 2011. doi: 10.1152/ajpcell.00124.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Watson LJ, Long BW, DeMartino AM, Brittian KR, Readnower RD, Brainard RE, Cummins TD, Annamalai L, Hill BG, Jones SP. Cardiomyocyte Ogt is essential for postnatal viability. Am J Physiol Heart Circ Physiol 306: H142–153, 2014. doi: 10.1152/ajpheart.00438.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yang YR, Jang HJ, Choi SS, Lee YH, Lee GH, Seo YK, Choi JH, Park D, Koh A, Kim IS, Lee H, Ryu SH, Suh PG. Obesity resistance and increased energy expenditure by white adipose tissue browning in Oga(+/-) mice. Diabetologia 58: 2867–2876, 2015. doi: 10.1007/s00125-015-3736-z. [DOI] [PubMed] [Google Scholar]
- 208.Shafi R, Iyer SPN, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A 97: 5735–5739, 2000. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol 8: 393–399, 2012. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- 210.Hart GW. Nutrient regulation of signaling and transcription. J Biol Chem 294: 2211–2231, 2019. doi: 10.1074/jbc.AW119.003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80: 825–858, 2011. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zachara NE, Hart GW. O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim Biophys Acta 1673: 13–28, 2004. doi: 10.1016/j.bbagen.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 213.Milewski S. Glucosamine-6-phosphate synthase–the multi-facets enzyme. Biochim Biophys Acta 1597: 173–192, 2002. doi: 10.1016/S0167-4838(02)00318-7. [DOI] [PubMed] [Google Scholar]
- 214.Oki T, Yamazaki K, Kuromitsu J, Okada M, Tanaka I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics 57: 227–234, 1999. doi: 10.1006/geno.1999.5785. [DOI] [PubMed] [Google Scholar]
- 215.DeHaven JE, Robinson KA, Nelson BA, Buse MG. A novel variant of glutamine: fructose-6-phosphate amidotransferase-1 (GFAT1) mRNA is selectively expressed in striated muscle. Diabetes 50: 2419–2424, 2001. doi: 10.2337/diabetes.50.11.2419. [DOI] [PubMed] [Google Scholar]
- 216.Chang Q, Su K, Baker JR, Yang X, Paterson AJ, Kudlow JE. Phosphorylation of human glutamine:fructose-6-phosphate amidotransferase by cAMP-dependent protein kinase at serine 205 blocks the enzyme activity. J Biol Chem 275: 21981–21987, 2000. doi: 10.1074/jbc.M001049200. [DOI] [PubMed] [Google Scholar]
- 217.Hu Y, Riesland L, Paterson AJ, Kudlow JE. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J Biol Chem 279: 29988–29993, 2004. doi: 10.1074/jbc.M401547200. [DOI] [PubMed] [Google Scholar]
- 218.Eguchi S, Oshiro N, Miyamoto T, Yoshino K, Okamoto S, Ono T, Kikkawa U, Yonezawa K. AMP-activated protein kinase phosphorylates glutamine: fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells 14: 179–189, 2009. doi: 10.1111/j.1365-2443.2008.01260.x. [DOI] [PubMed] [Google Scholar]
- 219.Gelinas R, Dontaine J, Horman S, Beauloye C, Bultot L, Bertrand L. AMP-activated protein kinase and O-GlcNAcylation, two partners tightly connected to regulate key cellular processes. Front Endocrinol (Lausanne) 9: 519, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sayeski PP, Wang D, Su K, Han IO, Kudlow JE. Cloning and partial characterization of the mouse glutamine:fructose-6-phosphate amidotransferase (GFAT) gene promoter. Nucleic acids research 25: 1458–1466, 1997. doi: 10.1093/nar/25.7.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chaveroux C, Sarcinelli C, Barbet V, Belfeki S, Barthelaix A, Ferraro-Peyret C, Lebecque S, Renno T, Bruhat A, Fafournoux P, Manié SN. Nutrient shortage triggers the hexosamine biosynthetic pathway via the GCN2-ATF4 signalling pathway. Sci Rep 6: 27278, 2016. doi: 10.1038/srep27278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Senderek J, Müller JS, Dusl M, Strom TM, Guergueltcheva V, Diepolder I, Laval SH, Maxwell S, Cossins J, Krause S, Muelas N, Vilchez JJ, Colomer J, Jimenez Mallebrera C, Nascimento A, Nafissi S, Kariminejad A, Nilipour Y, Bozorgmehr B, Najmabadi H, Rodolico C, Sieb JP, Steinlein OK, Schlotter B, Schoser B, Kirschner J, Herrmann R, Voit T, Oldfors A, Lindbergh C, Urtizberea A, von der Hagen M, Hübner A, Palace J, Bushby K, Straub V, Beeson D, Abicht A, Lochmüller H. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet 88: 162–172, 2011. doi: 10.1016/j.ajhg.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Qian Y, Ahmad M, Chen S, Gillespie P, Le N, Mennona F, Mischke S, So SS, Wang H, Burghardt C, Tannu S, Conde-Knape K, Kochan J, Bolin D. Discovery of 1-arylcarbonyl-6,7-dimethoxyisoquinoline derivatives as glutamine fructose-6-phosphate amidotransferase (GFAT) inhibitors. Bioorg Med Chem Lett 21: 6264–6269, 2011. doi: 10.1016/j.bmcl.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 224.Vyas B, Silakari O, Bahia MS, Singh B. Glutamine: fructose-6-phosphate amidotransferase (GFAT): homology modelling and designing of new inhibitors using pharmacophore and docking based hierarchical virtual screening protocol. SAR QSAR Environ Res 24: 733–752, 2013. doi: 10.1080/1062936X.2013.797493. [DOI] [PubMed] [Google Scholar]
- 225.Boehmelt G, Fialka I, Brothers G, McGinley MD, Patterson SD, Mo R, Hui CC, Chung S, Huber LA, Mak TW, Iscove NN. Cloning and characterization of the murine glucosamine-6-phosphate acetyltransferase EMeg32. Differential expression and intracellular membrane association. J Biol Chem 275: 12,821–12,832, 2000. doi: 10.1074/jbc.275.17.12821. [DOI] [PubMed] [Google Scholar]
- 226.Mio T, Yabe T, Arisawa M, Yamada-Okabe H. The eukaryotic UDP-N-acetylglucosamine pyrophosphorylases. Gene cloning, protein expression, and catalytic mechanism. J Biol Chem 273: 14392–14397, 1998. doi: 10.1074/jbc.273.23.14392. [DOI] [PubMed] [Google Scholar]
- 227.Freeze HH, Hart GW, Schnaar RL. Glycosylation Precursors. In: Essentials of Glycobiology (3rd ed.), edited by Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH.. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, NY, 2015, p. 51–63. [Google Scholar]
- 228.Boehmelt G, Wakeham A, Elia A, Sasaki T, Plyte S, Potter J, Yang Y, Tsang E, Ruland J, Iscove NN, Dennis JW, Mak TW. Decreased UDP-GlcNAc levels abrogate proliferation control in EMeg32-deficient cells. EMBO J 19: 5092–5104, 2000. doi: 10.1093/emboj/19.19.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X, Bhatnagar A, Jones SP, Hill BG. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem J 474: 2785–2801, 2017. doi: 10.1042/BCJ20170474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Olson A, Bouchard B, Zhu WZ, Chatham JC, Des Rosiers C. First characterization of glucose flux through the hexosamine biosynthesis pathway (HBP) in ex vivo mouse. J Biol Chem 295: 2018–2033, 2020. doi: 10.1074/jbc.RA119.010565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Joiner CM, Li H, Jiang J, Walker S. Structural characterization of the O-GlcNAc cycling enzymes: insights into substrate recognition and catalytic mechanisms. Curr Opin Struct Biol 56: 97–106, 2019. doi: 10.1016/j.sbi.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Levine ZG, Walker S. The biochemistry of O-GlcNAc transferase: which functions make it essential in mammalian cells? Annu Rev Biochem 85: 631–657, 2016. doi: 10.1146/annurev-biochem-060713-035344. [DOI] [PubMed] [Google Scholar]
- 233.Nagel AK, Ball LE. O-GlcNAc transferase and O-GlcNAcase: achieving target substrate specificity. Amino Acids 46: 2305–2316, 2014. doi: 10.1007/s00726-014-1827-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Vocadlo DJ. O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation. Curr Opin Chem Biol 16: 488–497, 2012. doi: 10.1016/j.cbpa.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 235.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, Olsen JV. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2: 419–431, 2012. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Roth C, Chan S, Offen WA, Hemsworth GR, Willems LI, King DT, Varghese V, Britton R, Vocadlo DJ, Davies GJ. Structural and functional insight into human O-GlcNAcase. Nat Chem Biol 13: 610–612, 2017. doi: 10.1038/nchembio.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sohn KC, Do SI. Transcriptional regulation and O-GlcNAcylation activity of zebrafish OGT during embryogenesis. Biochem Biophys Res Commun 337: 256–263, 2005. doi: 10.1016/j.bbrc.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 238.Webster DM, Teo CF, Sun Y, Wloga D, Gay S, Klonowski KD, Wells L, Dougan ST. O-GlcNAc modifications regulate cell survival and epiboly during zebrafish development. BMC Dev Biol 9: 28, 2009. doi: 10.1186/1471-213X-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sakaidani Y, Nomura T, Matsuura A, Ito M, Suzuki E, Murakami K, Nadano D, Matsuda T, Furukawa K, Okajima T. O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat Commun 2, 2011. doi: 10.1038/ncomms1591. [DOI] [PubMed] [Google Scholar]
- 240.Ogawa M, Okajima T. Structure and function of extracellular O-GlcNAc. Curr Opin Struct Biol 56: 72–77, 2019. doi: 10.1016/j.sbi.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 241.Varshney S, Stanley P. EOGT and O-GlcNAc on secreted and membrane proteins. Biochem Soc Trans 45: 401–408, 2017. doi: 10.1042/BST20160165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, Kudlow JE, Michell RH, Olefsky JM, Field SJ, Evans RM. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 451: 964–969, 2008. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- 243.Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469: 564–567, 2011. doi: 10.1038/nature09638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jinek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti E. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha. Nat Struct Mol Biol 11: 1001–1007, 2004. doi: 10.1038/nsmb833. [DOI] [PubMed] [Google Scholar]
- 245.Joiner CM, Levine ZG, Aonbangkhen C, Woo CM, Walker S. Aspartate residues far from the active site drive O-GlcNAc transferase substrate selection. J Am Chem Soc 141: 12,974–12,978, 2019. doi: 10.1021/jacs.9b06061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.März P, Stetefeld J, Bendfeldt K, Nitsch C, Reinstein J, Shoeman RL, Dimitriades-Schmutz B, Schwager M, Leiser D, Özcan S, Otten U, Özbek S. Ataxin-10 interacts with O-linked β-N-acetylglucosamine transferase in the brain. J Biol Chem 281: 20263–20270, 2006. doi: 10.1074/jbc.M601563200. [DOI] [PubMed] [Google Scholar]
- 247.Kreppel LK, Hart GW. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J Biol Chem 274: 32015–32022, 1999. doi: 10.1074/jbc.274.45.32015. [DOI] [PubMed] [Google Scholar]
- 248.Pathak S, Alonso J, Schimpl M, Rafie K, Blair DE, Borodkin VS, Schüttelkopf AW, Albarbarawi O, van Aalten DMF. The active site of O-GlcNAc transferase imposes constraints on substrate sequence. Nat Struct Mol Biol 22: 744–750, 2015. doi: 10.1038/nsmb.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Chalkley RJ, Thalhammer A, Schoepfer R, Burlingame AL. Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides. Proc Natl Acad Sci U S A 106: 8894–8899, 2009. doi: 10.1073/pnas.0900288106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Vaidyanathan K, Niranjan T, Selvan N, Teo CF, May M, Patel S, Weatherly B, Skinner C, Opitz J, Carey J, Viskochil D, Gecz J, Shaw M, Peng Y, Alexov E, Wang T, Schwartz C, Wells L. Identification and characterization of a missense mutation in the O-linked β-N-acetylglucosamine (O-GlcNAc) transferase gene that segregates with X-linked intellectual disability. J Biol Chem 292: 8948–8963, 2017. doi: 10.1074/jbc.M116.771030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Whelan SA, Lane MD, Hart GW. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. J Biol Chem 283: 21411–21417, 2008. doi: 10.1074/jbc.M800677200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Bullen JW, Balsbaugh JL, Chanda D, Shabanowitz J, Hunt DF, Neumann D, Hart GW. Crosstalk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J Biol Chem 289: 10,592–10,606, 2014. doi: 10.1074/jbc.M113.523068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Li Z, Li X, Nai S, Geng Q, Liao J, Xu X, Li J. Checkpoint kinase 1-induced phosphorylation of O-linked β-N-acetylglucosamine transferase regulates the intermediate filament network during cytokinesis. J Biol Chem 292: 19,548–19,555, 2017. doi: 10.1074/jbc.M117.811646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Seo HG, Kim HB, Kang MJ, Ryum JH, Yi EC, Cho JW. Identification of the nuclear localisation signal of O-GlcNAc transferase and its nuclear import regulation. Sci Rep 6: 34614, 2016. doi: 10.1038/srep34614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Liu L, Li L, Ma C, Shi Y, Liu C, Xiao Z, Zhang Y, Tian F, Gao Y, Zhang J, Ying W, Wang PG, Zhang L. O-GlcNAcylation of Thr12/Ser56 in short form O-GlcNAc transferase (sOGT) regulates its substrate selectivity. J Biol Chem 294: 16,620–16,633, 2019. doi: 10.1074/jbc.RA119.009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Cheung WD, Hart GW. AMP-activated protein kinase and p38 MAPK activate O-GlcNAcylation of neuronal proteins during glucose deprivation. J Biol Chem 283: 13009–13020, 2008. doi: 10.1074/jbc.M801222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18: 452–465, 2017. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Whisenhunt TR, Yang X, Bowe DB, Paterson AJ, Van Tine BA, Kudlow JE. Disrupting the enzyme complex regulating O-GlcNAcylation blocks signaling and development. Glycobiology 16: 551–563, 2006. doi: 10.1093/glycob/cwj096. [DOI] [PubMed] [Google Scholar]
- 259.Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell 144: 376–388, 2011. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 260.Daou S, Mashtalir N, Hammond-Martel I, Pak H, Yu H, Sui G, Vogel JL, Kristie TM, Affar e. B. Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc Natl Acad Sci U S A 108: 2747–2752, 2011. doi: 10.1073/pnas.1013822108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 342: 1235–1239, 2013. doi: 10.1126/science.1243990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Toleman C, Paterson AJ, Kudlow JE. Location and characterization of the O-GlcNAcase active site. Biochim Biophys Acta 1760: 829–839, 2006. doi: 10.1016/j.bbagen.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 263.Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem 278: 44,230–44,237, 2003. doi: 10.1074/jbc.M303810200. [DOI] [PubMed] [Google Scholar]
- 264.Heckel D, Comtesse N, Brass N, Blin N, Zang KD, Meese E. Novel immunogenic antigen homologous to hyaluronidase in meningioma. Hum Mol Genet 7: 1859–1872, 1998. doi: 10.1093/hmg/7.12.1859. [DOI] [PubMed] [Google Scholar]
- 265.Toll-Riera M, Radó-Trilla N, Martys F, Albà MM. Role of low-complexity sequences in the formation of novel protein coding sequences. Mol Biol Evol 29: 883–886, 2012. doi: 10.1093/molbev/msr263. [DOI] [PubMed] [Google Scholar]
- 266.Kohler JJ. Carb cutting works better with a partner. Nat Struct Mol Biol 24: 433–435, 2017. doi: 10.1038/nsmb.3405. [DOI] [PubMed] [Google Scholar]
- 267.Butkinaree C, Cheung WD, Park S, Park K, Barber M, Hart GW. Characterization of β-N-acetylglucosaminidase cleavage by caspase-3 during apoptosis. J Biol Chem 283: 23,557–23,566, 2008. doi: 10.1074/jbc.M804116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Toleman CA, Paterson AJ, Kudlow JE. The histone acetyltransferase NCOAT contains a zinc finger-like motif involved in substrate recognition. J Biol Chem 281: 3918–3925, 2006. doi: 10.1074/jbc.M510485200. [DOI] [PubMed] [Google Scholar]
- 269.Campbell MP, Aoki-Kinoshita KF, Lisacek F, York WS, Packer NH. Glycoinformatics. In: Essentials of Glycobiology (3rd ed.), edited by Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH.. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2015, p. 667–679. [Google Scholar]
- 270.Rao FV, Schüttelkopf AW, Dorfmueller HC, Ferenbach AT, Navratilova I, van Aalten DM. Structure of a bacterial putative acetyltransferase defines the fold of the human O-GlcNAcase C-terminal domain. Open Biol 3: 130021, 2013. doi: 10.1098/rsob.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Bond MR, Hanover JA. A little sugar goes a long way: The cell biology of O-GlcNAc. J Cell Biol 208: 869–880, 2015. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kim EJ, Kang DO, Love DC, Hanover JA. Enzymatic characterization of O-GlcNAcase isoforms using a fluorogenic GlcNAc substrate. Carbohydr Res 341: 971–982, 2006. doi: 10.1016/j.carres.2006.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Keembiyehetty CN, Krzeslak A, Love DC, Hanover JA. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J Cell Science 124: 2851–2860, 2011. doi: 10.1242/jcs.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Alonso J, Schimpl M, van Aalten DM. O-GlcNAcase: promiscuous hexosaminidase or key regulator of O-GlcNAc signaling? J Biol Chem 289: 34433–34439, 2014. doi: 10.1074/jbc.R114.609198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Elsen NL, Patel SB, Ford RE, Hall DL, Hess F, Kandula H, Kornienko M, Reid J, Selnick H, Shipman JM, Sharma S, Lumb KJ, Soisson SM, Klein DJ. Insights into activity and inhibition from the crystal structure of human O-GlcNAcase. Nat Chem Biol 13: 613–615, 2017. doi: 10.1038/nchembio.2357. [DOI] [PubMed] [Google Scholar]
- 276.Li B, Li H, Lu L, Jiang J. Structures of human O-GlcNAcase and its complexes reveal a new substrate recognition mode. Nat Struct Mol Biol 24: 362–369, 2017. doi: 10.1038/nsmb.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Shen DL, Gloster TM, Yuzwa SA, Vocadlo DJ. Insights into O-linked N-acetylglucosamine ([0-9]O-GlcNAc) processing and dynamics through kinetic analysis of O-GlcNAc transferase and O-GlcNAcase activity on protein substrates. J Biol Chem 287: 15,395–15,408, 2012. doi: 10.1074/jbc.M111.310664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Martin JC, Fadda E, Ito K, Woods RJ. Defining the structural origin of the substrate sequence independence of O-GlcNAcase using a combination of molecular docking and dynamics simulation. Glycobiology 24: 85–96, 2014. doi: 10.1093/glycob/cwt094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Schimpl M, Schüttelkopf Alexander W, Borodkin VS, van Aalten Daan MF. Human OGA binds substrates in a conserved peptide recognition groove. Biochem J 432: 1–12, 2010. doi: 10.1042/BJ20101338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kazemi Z, Chang H, Haserodt S, McKen C, Zachara NE. O-linked β-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3β-dependent manner. J Biol Chem 285: 39096–39107, 2010. doi: 10.1074/jbc.M110.131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Brahma MK, Ha C‐M, Pepin ME, Mia S, Sun Z, Chatham JC, Habegger KM, Abel ED, Paterson AJ, Young ME, Wende AR. Increased glucose availability attenuates myocardial ketone body utilization. JAHA 9, 2020., In Pressdoi: 10.1161/JAHA.119.013039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Chatham JC, Not LG, Fulop N, Marchase RB. Hexosamine biosynthesis and protein O-glycosylation: the first line of defense against stress, ischemia, and trauma. Shock 29: 431–440, 2008. doi: 10.1097/SHK.0b013e3181598bad. [DOI] [PubMed] [Google Scholar]
- 283.Ma J, Hart GW. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev Proteomics 10: 365–380, 2013. doi: 10.1586/14789450.2013.820536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Balteau M, Tajeddine N, de Meester C, Ginion A, Des Rosiers C, Brady NR, Sommereyns C, Horman S, Vanoverschelde JL, Gailly P, Hue L, Bertrand L, Beauloye C. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res 92: 237–246, 2011. doi: 10.1093/cvr/cvr230. [DOI] [PubMed] [Google Scholar]
- 285.Walgren JL, Vincent TS, Schey KL, Buse MG. High glucose and insulin promote O-GlcNAc modification of proteins, including α-tubulin. Am J Physiol Endocrinol Metab 284: E424–E434, 2003. doi: 10.1152/ajpendo.00382.2002. [DOI] [PubMed] [Google Scholar]
- 286.Song M, Kim H-S, Park J-M, Kim S-H, Kim I-H, Ryu SH, Suh P-G. o-GlcNAc transferase is activated by CaMKIV-dependent phosphorylation under potassium chloride-induced depolarization in NG-108-15 cells. Cell Signal 20: 94–104, 2008. doi: 10.1016/j.cellsig.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 287.Kneass ZT, Marchase RB. Neutrophils exhibit rapid agonist-induced increases in protein-associated O-GlcNAc. J Biol Chem 279: 45,759–45,765, 2004. doi: 10.1074/jbc.M407911200. [DOI] [PubMed] [Google Scholar]
- 288.Fülöp N, Zhang Z, Marchase RB, Chatham JC. Glucosamine cardioprotection in perfused rat hearts associated with increased O-linked N-acetylglucosamine protein modification and altered p38 activation. Am J Physiol Heart Circ Physiol 292: H2227–H2236, 2007. doi: 10.1152/ajpheart.01091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zachara NE, O'Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem 279: 30,133–30,142, 2004. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- 290.Slawson C, Copeland RJ, Hart GW. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem Sci 35: 547–555, 2010. doi: 10.1016/j.tibs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Lunde IG, Aronsen JM, Kvaløy H, Qvigstad E, Sjaastad I, Tönnessen T, Christensen G, Grönning-Wang LM, Carlson CR. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol Genomics 44: 162–172, 2012. doi: 10.1152/physiolgenomics.00016.2011. [DOI] [PubMed] [Google Scholar]
- 292.Mailleux F, Gélinas R, Beauloye C, Horman S, Bertrand L. O-GlcNAcylation, enemy or ally during cardiac hypertrophy development? Biophys Biochim Acta 1862: 2232–2243, 2016. doi: 10.1016/j.bbadis.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 293.Butkinaree C, Park K, Hart GW. O-linked β-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta 1800: 96–106, 2010. doi: 10.1016/j.bbagen.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab 36: 423–435, 2010. doi: 10.1016/j.diabet.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 295.Zimmerman AD, Harris RB. In vivo and in vitro evidence that chronic activation of the hexosamine biosynthetic pathway interferes with leptin-dependent STAT3 phosphorylation. Am J Physiol Regul Integr Comp Physiol 308: R543–R555, 2015. doi: 10.1152/ajpregu.00347.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Lima VV, Giachini FR, Carneiro FS, Carneiro ZN, Saleh MA, Pollock DM, Fortes ZB, Carvalho MH, Ergul A, Webb RC, Tostes RC. O-GlcNAcylation contributes to augmented vascular reactivity induced by endothelin 1. Hypertension 55: 180–188, 2010. doi: 10.1161/HYPERTENSIONAHA.109.143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Lima VV, Giachini FR, Carneiro FS, Carvalho MH, Fortes ZB, Webb RC, Tostes RC. O-GlcNAcylation contributes to the vascular effects of ET-1 via activation of the RhoA/Rho-kinase pathway. Cardiovasc Res 89: 614–622, 2011. doi: 10.1093/cvr/cvq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol 302: H2122–H2130, 2012. doi: 10.1152/ajpheart.00775.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Gélinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois-Deruy E, Lauzier B, Gauthier C, Olson AK, Bouchard B, Des Rosiers C, Viollet B, Sakamoto K, Balligand JL, Vanoverschelde JL, Beauloye C, Horman S, Bertrand L. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun 9: 374, 2018. doi: 10.1038/s41467-017-02795-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Zhang Z, Tan EP, VandenHull NJ, Peterson KR, Slawson C. O-GlcNAcase expression is sensitive to changes in O-GlcNAc homeostasis. Front Endocrinol 5: 206, 2014. doi: 10.3389/fendo.2014.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Qian K, Wang S, Fu M, Zhou J, Singh JP, Li MD, Yang Y, Zhang K, Wu J, Nie Y, Ruan HB, Yang X. Transcriptional regulation of O-GlcNAc homeostasis is disrupted in pancreatic cancer. J Biol Chem 293: 13989–14000, 2018. doi: 10.1074/jbc.RA118.004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Muthusamy S, Hong KU, Dassanayaka S, Hamid T, Jones SP. E2F1 Transcription factor regulates O-linked N-acetylglucosamine (O-GlcNAc) transferase and O-GlcNAcase rexpression. J Biol Chem 290: 31013–31024, 2015. doi: 10.1074/jbc.M115.677534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Wells L, Slawson C, Hart GW. The E2F-1 associated retinoblastoma-susceptibility gene product is modified by O-GlcNAc. Amino Acids 40: 877–883, 2011. doi: 10.1007/s00726-010-0709-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, Herring LE, Asara JM, Singer JD, Wen H. Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J Exp Med 214: 1093–1109, 2017. doi: 10.1084/jem.20161105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Tan ZW, Fei G, Paulo JA, Bellaousov S, Martin SES, Duveau DY, Thomas CJ, Gygi SP, Boutz PL, Walker S. O-GlcNAc regulates gene expression by controlling detained intron splicing. Nucleic Acids Res 48: 5656–5669, 2020. doi: 10.1093/nar/gkaa263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Herzog K, Bandiera S, Pernot S, Fauvelle C, Jühling F, Weiss A, Bull A, Durand SC, Chane-Woon-Ming B, Pfeffer S, Mercey M, Lerat H, Meunier J-C, Raffelsberger W, Brino L, Baumert TF, Zeisel MB. Functional microRNA screen uncovers O-linked N-acetylglucosamine transferase as a host factor modulating hepatitis C virus morphogenesis and infectivity. Gut 69: 380–392, 2020. doi: 10.1136/gutjnl-2018-317423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Jiang M, Xu B, Li X, Shang Y, Chu Y, Wang W, Chen D, Wu N, Hu S, Zhang S, Li M, Wu K, Yang X, Liang J, Nie Y, Fan D. O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene 38: 301–316, 2019. doi: 10.1038/s41388-018-0435-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Lo W-Y, Yang W-K, Peng C-T, Pai W-Y, Wang H-J. MicroRNA-200a/200b modulate high glucose-induced endothelial inflammation by targeting O-linked N-acetylglucosamine transferase expression. Front Physiol 9: 355, 2018. doi: 10.3389/fphys.2018.00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Luo P, He T, Jiang R, Li G. MicroRNA-423-5p targets O-GlcNAc transferase to induce apoptosis in cardiomyocytes. Mol Med Rep 12: 1163–1168, 2015. doi: 10.3892/mmr.2015.3491. [DOI] [PubMed] [Google Scholar]
- 310.Muthusamy S, DeMartino AM, Watson LJ, Brittian KR, Zafir A, Dassanayaka S, Hong KU, Jones SP. MicroRNA-539 is up-regulated in failing heart, and suppresses O-GlcNAcase expression. J Biol Chem 289: 29,665–29,676, 2014. doi: 10.1074/jbc.M114.578682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Taylor RP, Geisler TS, Chambers JH, McClain DA. Up-regulation of O-GlcNAc transferase with glucose deprivation in HepG2 cells is mediated by decreased hexosamine pathway flux. J Biol Chem 284: 3425–3432, 2009. doi: 10.1074/jbc.M803198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Zou L, Zhu-Mauldin X, Marchase RB, Paterson AJ, Liu J, Yang Q, Chatham JC. Glucose deprivation-induced increase in protein O-GlcNAcylation in cardiomyocytes is calcium-dependent. J Biol Chem 287: 34,419–34,431, 2012. doi: 10.1074/jbc.M112.393207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Kang JG, Park SY, Ji S, Jang I, Park S, Kim HS, Kim SM, Yook JI, Park YI, Roth J, Cho JW. O-GlcNAc protein modification in cancer cells increases in response to glucose deprivation through glycogen degradation. J Biol Chem 284: 34,777–34,784, 2009. doi: 10.1074/jbc.M109.026351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, Lehrman MA, Rothermel BA, Lee AH, Lavandero S, Mammen PP, Ferdous A, Gillette TG, Scherer PE, Hill JA. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156: 1179–1192, 2014. doi: 10.1016/j.cell.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science 291: 2376–2378, 2001. doi: 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- 316.Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, Falcäo-Pires I, Leite-Moreira AF, Volk N, Gröne HJ, Müller OJ, Sickmann A, Katus HA, Backs J. O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation 140: 580–594, 2019. doi: 10.1161/CIRCULATIONAHA.117.031942. [DOI] [PubMed] [Google Scholar]
- 317.Wang Z, Pandey A, Hart GW. Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol Cell Proteomics 6: 1365–1379, 2007. doi: 10.1074/mcp.M600453-MCP200. [DOI] [PubMed] [Google Scholar]
- 318.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A 105: 13793–13798, 2008. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zhong J, Martinez M, Sengupta S, Lee A, Wu X, Chaerkady R, Chatterjee A, O’Meally RN, Cole RN, Pandey A, Zachara NE. Quantitative phosphoproteomics reveals crosstalk between phosphorylation and O-GlcNAc in the DNA damage response pathway. Proteomics 15: 591–607, 2015. doi: 10.1002/pmic.201400339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Yachie N, Saito R, Sugahara J, Tomita M, Ishihama Y. In silico analysis of phosphoproteome data suggests a rich-get-richer process of phosphosite accumulation over evolution. Mol Cell Proteomics 8: 1061–1071, 2009. doi: 10.1074/mcp.M800466-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Leney AC, El Atmioui D, Wu W, Ovaa H, Heck AJR. Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc Natl Acad Sci U S A 114: E7255–E7261, 2017. doi: 10.1073/pnas.1620529114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Hardivillé S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab 20: 208–213, 2014. doi: 10.1016/j.cmet.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Slawson C, Lakshmanan T, Knapp S, Hart GW. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol Biol Cell 19: 4130–4140, 2008. doi: 10.1091/mbc.e07-11-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Wells L, Kreppel LK, Comer FI, Wadzinski BE, Hart GW. O-GlcNAc transferase is in a functional complex with protein phosphatase 1 catalytic subunits. J Biol Chem 279: 38,466–38,470, 2004. doi: 10.1074/jbc.M406481200. [DOI] [PubMed] [Google Scholar]
- 325.Schwein PA, Woo CM. The O-GlcNAc modification on kinases. ACS Chem Biol 15: 602–617, 2020. doi: 10.1021/acschembio.9b01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Mishra S, Ande SR, Salter NW. O-GlcNAc modification: why so intimately associated with phosphorylation? Cell Commun Signal 9: 1, 2011. doi: 10.1186/1478-811X-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Whelan SA, Dias WB, Thiruneelakantapillai L, Lane MD, Hart GW. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-linked β-N-acetylglucosamine in 3T3-L1 adipocytes. J Biol Chem 285: 5204–5211, 2010. doi: 10.1074/jbc.M109.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Ande SR, Moulik S, Mishra S. Interaction between O-GlcNAc modification and tyrosine phosphorylation of prohibitin: implication for a novel binary switch. PloS One 4: e4586, 2009. doi: 10.1371/journal.pone.0004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Shi J, Tomašič T, Sharif S, Brouwer AJ, Anderluh M, Ruijtenbeek R, Pieters RJ. Peptide microarray analysis of the cross-talk between O-GlcNAcylation and tyrosine phosphorylation. FEBS Lett 591: 1872–1883, 2017. doi: 10.1002/1873-3468.12708. [DOI] [PubMed] [Google Scholar]
- 330.Li M-D, Ruan H-B, Hughes Michael E, Lee J-S, Singh Jay P, Jones Steven P, Nitabach Michael N, Yang XO. GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab 17: 303–310, 2013. doi: 10.1016/j.cmet.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Ruan HB, Nie Y, Yang X. Regulation of protein degradation by O-GlcNAcylation: crosstalk with ubiquitination. Mol Cell Proteomics 12: 3489–3497, 2013. doi: 10.1074/mcp.R113.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Allison DF, Wamsley JJ, Kumar M, Li D, Gray LG, Hart GW, Jones DR, Mayo MW. Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-κB acetylation and transcription. Proc Natl Acad Sci U S A 109: 16888–16893, 2012. doi: 10.1073/pnas.1208468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Ma Z, Chalkley RJ, Vosseller K. Hyper-O-GlcNAcylation activates nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling through interplay with phosphorylation and acetylation. J Biol Chem 292: 9150–9163, 2017. doi: 10.1074/jbc.M116.766568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Hayakawa K, Hirosawa M, Tabei Y, Arai D, Tanaka S, Murakami N, Yagi S, Shiota K. Epigenetic switching by the metabolism-sensing factors in the generation of orexin neurons from mouse embryonic stem cells. J Biol Chem 288: 17099–17110, 2013. doi: 10.1074/jbc.M113.455899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Jackson SP, Tjian R. O-glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell 55: 125–133, 1988. doi: 10.1016/0092-8674(88)90015-3. [DOI] [PubMed] [Google Scholar]
- 336.Han I, Kudlow JE. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol Cell Biol 17: 2550–2558, 1997. doi: 10.1128/MCB.17.5.2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115: 715–725, 2003. doi: 10.1016/S0092-8674(03)00974-7. [DOI] [PubMed] [Google Scholar]
- 338.Majumdar G, Wright J, Markowitz P, Martinez-Hernandez A, Raghow R, Solomon SS. Insulin stimulates and diabetes inhibits O-linked N-acetylglucosamine transferase and O-glycosylation of Sp1. Diabetes 53: 3184–3192, 2004. doi: 10.2337/diabetes.53.12.3184. [DOI] [PubMed] [Google Scholar]
- 339.Wende AR, Schell JC, Ha C-M, Pepin ME, Khalimonchuk O, Schwertz H, Pereira RO, Brahma MK, Tuinei J, Contreras-Ferrat A, Wang L, Andrizzi CA, Olsen CD, Bradley WE, Dell’Italia LJ, Dillmann WH, Litwin SE, Abel ED. Maintaining myocardial glucose utilization in diabetic cardiomyopathy accelerates mitochondrial dysfunction. Diabetes 69: 2094–2111, 2020. doi: 10.2337/db19-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Lim K, Chang HI. O-GlcNAc inhibits interaction between Sp1 and Elf-1 transcription factors. Biochem Biophys Res Commun 380: 569–574, 2009. doi: 10.1016/j.bbrc.2009.01.121. [DOI] [PubMed] [Google Scholar]
- 341.Lim K, Chang HI. O-GlcNAcylation of Sp1 interrupts Sp1 interaction with NF-Y. Biochem Biophys Res Commun 382: 593–597, 2009. doi: 10.1016/j.bbrc.2009.03.075. [DOI] [PubMed] [Google Scholar]
- 342.Lim K, Chang HI. O-GlcNAc modification of Sp1 inhibits the functional interaction between Sp1 and Oct1. FEBS Lett 583: 512–520, 2009. doi: 10.1016/j.febslet.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 343.Ha C, Lim K. O-GlcNAc modification of Sp3 and Sp4 transcription factors negatively regulates their transcriptional activities. Biochem Biophys Res Commun 467: 341–347, 2015. doi: 10.1016/j.bbrc.2015.09.155. [DOI] [PubMed] [Google Scholar]
- 344.Durgan DJ, Pat BM, Laczy B, Bradley JA, Tsai JY, Grenett MH, Ratcliffe WF, Brewer RA, Nagendran J, Villegas-Montoya C, Zou C, Zou L, Johnson RL Jr, Dyck JR, Bray MS, Gamble KL, Chatham JC, Young ME. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem 286: 44,606–44,619, 2011. doi: 10.1074/jbc.M111.278903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Yang AQ, Li D, Chi L, Ye XS. Validation, identification, and biological consequences of the site-specific O-GlcNAcylation dynamics of carbohydrate-responsive element-binding protein (ChREBP). Mol Cell Proteomics 16: 1233–1243, 2017. doi: 10.1074/mcp.M116.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Jeon JH, Suh HN, Kim MO, Ryu JM, Han HJ. Glucosamine-induced OGT activation mediates glucose production through cleaved Notch1 and FoxO1, which coordinately contributed to the regulation of maintenance of self-renewal in mouse embryonic stem cells. Stem Cells Dev 23: 2067–2079, 2014. doi: 10.1089/scd.2013.0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Anthonisen EH, Berven L, Holm S, Nygård M, Nebb HI, Grønning-Wang LM. Nuclear receptor liver X receptor is O-GlcNAc-modified in response to glucose. J Biol Chem 285: 1607–1615, 2010. doi: 10.1074/jbc.M109.082685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Das S, Bailey SK, Metge BJ, Hanna A, Hinshaw DC, Mota M, Forero-Torres A, Chatham JC, Samant RS, Shevde LA. O-GlcNAcylation of GLI transcription factors in hyperglycemic conditions augments Hedgehog activity. Lab Invest 99: 260–270, 2019. doi: 10.1038/s41374-018-0122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Lefebvre T, Pinte S, Guérardel C, Deltour S, Martin-Soudant N, Slomianny M-C, Michalski J-C, Leprince D. The tumor suppressor HIC1 (hypermethylated in cancer 1) is O-GlcNAc glycosylated. Eur J Biochem 271: 3843–3854, 2004. doi: 10.1111/j.1432-1033.2004.04316.x. [DOI] [PubMed] [Google Scholar]
- 350.Kim MJ, Choi MY, Lee DH, Roh GS, Kim HJ, Kang SS, Cho GJ, Kim YS, Choi WS. O-linked N-acetylglucosamine transferase enhances secretory clusterin expression via liver X receptors and sterol response element binding protein regulation in cervical cancer. Oncotarget 9: 4625–4636, 2018. doi: 10.18632/oncotarget.23588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, Moldes M, Burnol AF, Yang X, Lefebvre T, Girard J, Postic C. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60: 1399–1413, 2011. doi: 10.2337/db10-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Özcan S, Andrali SS, Cantrell JEL. Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta 1799: 353–364, 2010. doi: 10.1016/j.bbagrm.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Kelly WG, Dahmus ME, Hart GW. RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J Biol Chem 268: 10,416–10,424, 1993. [PubMed] [Google Scholar]
- 354.Comer FI, Hart GW. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry 40: 7845–7852, 2001. doi: 10.1021/bi0027480. [DOI] [PubMed] [Google Scholar]
- 355.Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA. Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. J Biol Chem 287: 23,549–23,561, 2012. doi: 10.1074/jbc.M111.330910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Lewis BA, Burlingame AL, Myers SA. Human RNA polymerase II promoter recruitment in vitro is regulated by O-linked N-acetylglucosaminyltransferase (OGT). J Biol Chem 291: 14056–14061, 2016. doi: 10.1074/jbc.M115.684365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Resto M, Kim BH, Fernandez AG, Abraham BJ, Zhao K, Lewis BA. O-GlcNAcase is an RNA polymerase II elongation factor coupled to pausing factors SPT5 and TIF1β. J Biol Chem 291: 22,703–22,713, 2016. doi: 10.1074/jbc.M116.751420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Lu L, Fan D, Hu C-W, Worth M, Ma Z-X, Jiang J. Distributive O-GlcNAcylation on the highly repetitive C-terminal domain of RNA polymerase II. Biochemistry 55: 1149–1158, 2016. doi: 10.1021/acs.biochem.5b01280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Hardivillé S, Han G, Ma J, Hu P, Banerjee PS, Hart GW. Dynamic interactions of TATA-box binding protein with promoters is regulated by O-GlcNAcylation. FASEB J 30: 617.611–617.611, 2016. [Google Scholar]
- 360.Noach N, Segev Y, Levi I, Segal S, Priel E. Modification of topoisomerase I activity by glucose and by O-Glcnacylation of the enzyme protein. Glycobiology 17: 1357–1364, 2007. doi: 10.1093/glycob/cwm105. [DOI] [PubMed] [Google Scholar]
- 361.Eustice M, Bond MR, Hanover JA. O-GlcNAc cycling and the regulation of nucleocytoplasmic dynamics. Biochem Soc Trans 45: 427–436, 2017. doi: 10.1042/BST20160171. [DOI] [PubMed] [Google Scholar]
- 362.Davis LI, Blobel G. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc Natl Acad Sci 84: 7552–7556, 1987. doi: 10.1073/pnas.84.21.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Li B, Kohler JJ. Glycosylation of the nuclear pore. Traffic 15: 347–361, 2014. doi: 10.1111/tra.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Zhu Y, Liu T-W, Madden Z, Yuzwa SA, Murray K, Cecioni S, Zachara N, Vocadlo DJ. Post-translational O-GlcNAcylation is essential for nuclear pore integrity and maintenance of the pore selectivity filter. J Mol Cell Biol 8: 2–16, 2016. doi: 10.1093/jmcb/mjv033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Sharif SR, Lee H, Islam MA, Seog DH, Moon IS. N-acetyl-D-glucosamine kinase is a component of nuclear speckles and paraspeckles. Mol Cells 38: 402–408, 2015. doi: 10.14348/molcells.2015.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Bird A. Perceptions of epigenetics. Nature 447: 396–398, 2007. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 367.Sakabe K, Wang Z, Hart GW. β-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci U S A 107: 19,915–19,920, 2010. doi: 10.1073/pnas.1009023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Gagnon J, Daou S, Zamorano N, Iannantuono NV, Hammond-Martel I, Mashtalir N, Bonneil E, Wurtele H, Thibault P, Affar EB. Undetectable histone O-GlcNAcylation in mammalian cells. Epigenetics 10: 677–691, 2015. doi: 10.1080/15592294.2015.1060387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Fong JJ, Nguyen BL, Bridger R, Medrano EE, Wells L, Pan S, Sifers RN. β-N-Acetylglucosamine (O-GlcNAc) is a novel regulator of mitosis-specific phosphorylations on histone H3. J Biol Chem 287: 12195–12203, 2012. doi: 10.1074/jbc.M111.315804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Hahne H, Kuster B. Discovery of O-GlcNAc-6-phosphate-modified proteins by re-analyzing large-scale phosphoproteomics data. Mol Cell Proteomics 11: 1063–1069, 2012. doi: 10.1074/mcp.M112.019760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Hayakawa K, Hirosawa M, Tani R, Yoneda C, Tanaka S, Shiota K. H2A O-GlcNAcylation at serine 40 functions genomic protection in association with acetylated H2AZ or γH2AX. Epigenet Chrom 10: 51, 2017. doi: 10.1186/s13072-017-0157-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Hirosawa M, Hayakawa K, Yoneda C, Arai D, Shiota H, Suzuki T, Tanaka S, Dohmae N, Shiota K. Novel O-GlcNAcylation on Ser40 of canonical H2A isoforms specific to viviparity. Sci Rep 6: 31785, 2016. doi: 10.1038/srep31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Zhang S, Roche K, Nasheuer HP, Lowndes NF. Modification of histones by sugar β-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. J Biol Chem 286: 37,483–37,495, 2011. doi: 10.1074/jbc.M111.284885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Arnaudo A, Garcia B. Proteomic characterization of novel histone post-translational modifications. Epigenet Chromat 6: 24, 2013. doi: 10.1186/1756-8935-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 110: 69–80, 2002. doi: 10.1016/S0092-8674(02)00810-3. [DOI] [PubMed] [Google Scholar]
- 376.Cox EJ, Marsh SA. Exercise and diabetes have opposite effects on the assembly and O-GlcNAc modification of the mSin3A/HDAC1/2 complex in the heart. Cardiovasc Diabetol 12: 101, 2013. doi: 10.1186/1475-2840-12-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Wu D, Zhao L, Feng Z, Yu C, Ding J, Wang L, Wang F, Liu D, Zhu H, Xing F, Conaway JW, Conaway RC, Cai Y, Jin J. O-Linked N-acetylglucosamine transferase 1 regulates global histone H4 acetylation via stabilization of the nonspecific lethal protein NSL3. J Biol Chem 292: 10014–10025, 2017. doi: 10.1074/jbc.M117.781401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Chu C-S, Lo P-W, Yeh Y-H, Hsu P-H, Peng S-H, Teng Y-C, Kang M-L, Wong C-H, Juan L-J. O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci 111: 1355–1360, 2014. doi: 10.1073/pnas.1323226111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Shi H, Munk A, Nielsen TS, Daughtry MR, Larsson L, Li S, Hoyer KF, Geisler HW, Sulek K, Kjøbsted R, Fisher T, Andersen MM, Shen Z, Hansen UK, England EM, Cheng Z, Højlund K, Wojtaszewski JFP, Yang X, Hulver MW, Helm RF, Treebak JT, Gerrard DE. Skeletal muscle O-GlcNAc transferase is important for muscle energy homeostasis and whole-body insulin sensitivity. Mol Metab 11: 160–177, 2018. doi: 10.1016/j.molmet.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Lo P-W, Shie J-J, Chen C-H, Wu C-Y, Hsu T-L, Wong C-H. O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc Natl Acad Sci U S A 115: 7302–7307, 2018. doi: 10.1073/pnas.1801850115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Butler AA, Sanchez RG, Jarome TJ, Webb WM, Lubin FD. O-GlcNAc and EZH2-mediated epigenetic regulation of gene expression during consolidation of fear memories. Learn Mem 26: 373–379, 2019. doi: 10.1101/lm.049023.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Wyatt GR, Cohen SS. A new pyrimidine base from bacteriophage nucleic acids. Nature 170: 1072–1073, 1952. doi: 10.1038/1701072a0. [DOI] [PubMed] [Google Scholar]
- 383.Gold M, Hurwitz J, Anders M. The enzymatic methylation of RNA and DNA. I. Biochem Biophys Res Commun 11: 107–114, 1963. doi: 10.1016/0006-291X(63)90075-5. [DOI] [PubMed] [Google Scholar]
- 384.Schmitz RJ, Lewis ZA, Goll MG. DNA methylation: shared and divergent features across eukaryotes. Trends Genet 35: 818–827, 2019. doi: 10.1016/j.tig.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Franchini D-M, Schmitz K-M, Petersen-Mahrt SK. 5-Methylcytosine DNA demethylation: more than losing a methyl group. Annu Rev Genet 46: 419–441, 2012. doi: 10.1146/annurev-genet-110711-155451. [DOI] [PubMed] [Google Scholar]
- 386.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324: 929–930, 2009. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935, 2009. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493: 561–564, 2013. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, Shih AH, Levine RL, Bernard O, Mercher T, Solary E, Urh M, Daniels DL, Fuks F. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 32: 645–655, 2013. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Shi F-T, Kim H, Lu W, He Q, Liu D, Goodell MA, Wan M, Songyang Z. Ten-eleven translocation 1 (Tet1) is regulated by O-Linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J Biol Chem 288: 20,776–20,784, 2013. doi: 10.1074/jbc.M113.460386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K, Pasini D. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell 49: 645–656, 2013. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 392.Wang J, Samuels DC, Zhao S, Xiang Y, Zhao YY, Guo Y. Current research on non-coding ribonucleic acid (RNA). Genes 8: 366–320, 2017. doi: 10.3390/genes8120366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Olivier-Van Stichelen S, Hanover JA. X-inactivation normalizes O-GlcNAc Transferase levels and generates an O-GlcNAc-depleted Barr body. Front Genet 5: 256, 2014. doi: 10.3389/fgene.2014.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Vigetti D, Deleonibus S, Moretto P, Bowen T, Fischer JW, Grandoch M, Oberhuber A, Love DC, Hanover JA, Cinquetti R, Karousou E, Viola M, D'Angelo ML, Hascall VC, De Luca G, Passi A. Natural antisense transcript for hyaluronan synthase 2 (HAS2-AS1) induces transcription of HAS2 via protein O-GlcNAcylation. J Biol Chem 289: 28,816–28,826, 2014. doi: 10.1074/jbc.M114.597401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Liu Y, Huang H, Liu M, Wu Q, Li W, Zhang J. MicroRNA-24-1 suppresses mouse hepatoma cell invasion and metastasis via directly targeting O-GlcNAc transferase. Biomed Pharmacother 91: 731–738, 2017. doi: 10.1016/j.biopha.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 396.Chen H, Ing BL, Robinson KA, Feagin AC, Buse MG, Quon MJ. Effects of overexpression of glutamine:fructose-6-phosphate amidotransferase (GFAT) and glucosamine treatment on translocation of GLUT4 in rat adipose cells. Mol Cell Endocrinol 135: 67–77, 1997. doi: 10.1016/S0303-7207(97)00191-3. [DOI] [PubMed] [Google Scholar]
- 397.Patti ME, Virkamaki A, Landaker EJ, Kahn CR, Yki-Jarvinen H. Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance of early postreceptor insulin signaling events in skeletal muscle. Diabetes 48: 1562–1571, 1999. doi: 10.2337/diabetes.48.8.1562. [DOI] [PubMed] [Google Scholar]
- 398.Hebert LF Jr, Daniels MC, Zhou J, Crook ED, Turner RL, Simmons ST, Neidigh JL, Zhu JS, Baron AD, McClain DA. Overexpression of glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J Clin Invest 98: 930–936, 1996. doi: 10.1172/JCI118876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Robinson KA, Ball LE, Buse MG. Reduction of O-GlcNAc protein modification does not prevent insulin resistance in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 292: E884–E890, 2007. doi: 10.1152/ajpendo.00569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Soesanto YA, Luo B, Jones D, Taylor R, Gabrielsen JS, Parker G, McClain DA. Regulation of Akt signaling by O-GlcNAc in euglycemia. Am J Physiol Endocrinol Metab 295: E974–E980, 2008. doi: 10.1152/ajpendo.90366.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB, Chatham JC. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J Biol Chem 287: 39094–39106, 2012. doi: 10.1074/jbc.M112.383778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am J Physiol Cell Physiol 294: C1509–C1520, 2008. doi: 10.1152/ajpcell.00456.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature 491: 269–273, 2012. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Nickel AG, Kohlhaas M, Bertero E, Wilhelm D, Wagner M, Sequeira V, Kreusser MM, Dewenter M, Kappl R, Hoth M, Dudek J, Backs J, Maack C. CaMKII does not control mitochondrial Ca2+ uptake in cardiac myocytes. J Physiol 598: 1361–1376, 2020. doi: 10.1113/JP276766. [DOI] [PubMed] [Google Scholar]
- 405.Asante-Appiah E, Kennedy BP. Protein tyrosine phosphatases: the quest for negative regulators of insulin action. Am J Physiol Endocrinol Metab 284: E663–E670, 2003. doi: 10.1152/ajpendo.00462.2002. [DOI] [PubMed] [Google Scholar]
- 406.Naz H, Islam A, Ahmad F, Hassan MI. Calcium/calmodulin-dependent protein kinase IV: A multifunctional enzyme and potential therapeutic target. Prog Biophys Mol Biol 121: 54–65, 2016. doi: 10.1016/j.pbiomolbio.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 407.Pang Y, Hunton DL, Bounelis P, Marchase RB. Hyperglycemia inhibits capacitative calcium entry and hypertrophy in neonatal cardiomyocytes. Diabetes 51: 3461–3467, 2002. doi: 10.2337/diabetes.51.12.3461. [DOI] [PubMed] [Google Scholar]
- 408.Nagy T, Champattanachai V, Marchase RB, Chatham JC. Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked N-acetylglucosamine. Am J Physiol Cell Physiol 290: C57–C65, 2006. doi: 10.1152/ajpcell.00263.2005. [DOI] [PubMed] [Google Scholar]
- 409.Hogan PG, Rao A. Store-operated calcium entry: mechanisms and modulation. Biochem Biophys Res Commun 460: 40–49, 2015. doi: 10.1016/j.bbrc.2015.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Cieniewski-Bernard C, Lambert M, Dupont E, Montel V, Stevens L, Bastide B. O-GlcNAcylation, contractile protein modifications and calcium affinity in skeletal muscle. Front Physiol 5: 421, 2014. doi: 10.3389/fphys.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Cieniewski-Bernard C, Bastide B, Lefebvre T, Lemoine J, Mounier Y, Michalski J-C. Identification of O-linked N-acetylglucosamine proteins in rat skeletal muscle using two-dimensional gel electrophoresis and mass spectrometry. Mol Cell Proteomics 3: 577–585, 2004. doi: 10.1074/mcp.M400024-MCP200. [DOI] [PubMed] [Google Scholar]
- 412.Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, Torres-Gonzalez M, Wang H, Ellrott I, Maya-Ramos L, Villarreal F, Dillmann WH. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol 303: R689–R699, 2012. doi: 10.1152/ajpregu.00548.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Johnsen VL, Belke DD, Hughey CC, Hittel DS, Hepple RT, Koch LG, Britton SL, Shearer J. Enhanced cardiac protein glycosylation (O-GlcNAc) of selected mitochondrial proteins in rats artificially selected for low running capacity. Physiol Genomics 45: 17–25, 2013. doi: 10.1152/physiolgenomics.00111.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Kim YH, Song M, Oh YS, Heo K, Choi JW, Park JM, Kim SH, Lim S, Kwon HM, Ryu SH, Suh PG. Inhibition of phospholipase C-β1-mediated signaling by O-GlcNAc modification. J Cell Physiol 207: 689–696, 2006. doi: 10.1002/jcp.20609. [DOI] [PubMed] [Google Scholar]
- 415.Rengifo J, Gibson CJ, Winkler E, Collin T, Ehrlich BE. Regulation of the inositol 1,4,5-trisphosphate receptor type I by O-GlcNAc glycosylation. J Neurosci 27: 13,813–13,821, 2007. doi: 10.1523/JNEUROSCI.2069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Bimboese P, Gibson CJ, Schmidt S, Xiang W, Ehrlich BE. Isoform-specific regulation of the inositol 1,4,5-trisphosphate receptor by O-linked glycosylation. J Biol Chem 286: 15,688–15,697, 2011. doi: 10.1074/jbc.M110.206482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Kerkhofs M, Seitaj B, Ivanova H, Monaco G, Bultynck G, Parys JB. Pathophysiological consequences of isoform-specific IP3 receptor mutations. Biochim Biophys Acta Mol Cell Res 1865: 1707–1717, 2018. doi: 10.1016/j.bbamcr.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 418.Bacigalupa ZA, Bhadiadra CH, Reginato MJ. O-GlcNAcylation: key regulator of glycolytic pathways. J Bioenerg Biomembr 50: 189–198, 2018. doi: 10.1007/s10863-018-9742-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Laczy B, Fulop N, Onay-Besikci A, Des Rosiers C, Chatham JC. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PloS One 6: e18417, 2011. doi: 10.1371/journal.pone.0018417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Lauzier B, Vaillant F, Merlen C, Gelinas R, Bouchard B, Rivard ME, Labarthe F, Dolinsky VW, Dyck JR, Allen BG, Chatham JC, Des Rosiers C. Metabolic effects of glutamine on the heart: anaplerosis versus the hexosamine biosynthetic pathway. J Mol Cell Cardiol 55: 92–100, 2013. doi: 10.1016/j.yjmcc.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Luo B, Parker GJ, Cooksey RC, Soesanto Y, Evans M, Jones D, McClain DA. Chronic hexosamine flux stimulates fatty acid oxidation by activating AMP-activated protein kinase in adipocytes. J Biol Chem 282: 7172–7180, 2007. doi: 10.1074/jbc.M607362200. [DOI] [PubMed] [Google Scholar]
- 422.Hanover JA, Yu S, Lubas WB, Shin SH, Ragano-Caracciola M, Kochran J, Love DC. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch Biochem Biophys 409: 287–297, 2003. doi: 10.1016/S0003-9861(02)00578-7. [DOI] [PubMed] [Google Scholar]
- 423.Love DC, Kochran J, Cathey RL, Shin SH, Hanover JA. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci 116: 647–654, 2003. doi: 10.1242/jcs.00246. [DOI] [PubMed] [Google Scholar]
- 424.Marsh SA, Powell PC, Dell'italia LJ, Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci 92: 648–656, 2013. doi: 10.1016/j.lfs.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Kim HS, Kim EM, Lee J, Yang WH, Park TY, Kim YM, Cho JW. Heat shock protein 60 modified with O-linked N-acetylglucosamine is involved in pancreatic β-cell death under hyperglycemic conditions. FEBS Lett 580: 2311–2316, 2006. doi: 10.1016/j.febslet.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 426.Cividini F, Scott BT, Dai A, Han W, Suarez J, Diaz-Juarez J, Diemer T, Casteel DE, Dillmann WH. O-GlcNAcylation of 8-oxoguanine DNA glycosylase (Ogg1) impairs oxidative mitochondrial DNA lesion repair in diabetic hearts. J Biol Chem 291: 26515–26528, 2016. doi: 10.1074/jbc.M116.754481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci USA 112: 6050–6055, 2015. doi: 10.1073/pnas.1424017112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Trapannone R, Mariappa D, Ferenbach AT, van Aalten DM. Nucleocytoplasmic human O-GlcNAc transferase is sufficient for O-GlcNAcylation of mitochondrial proteins. Biochem J 473: 1693–1702, 2016. doi: 10.1042/BCJ20160092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Sacoman JL, Dagda RY, Burnham-Marusich AR, Dagda RK, Berninsone PM. Mitochondrial O-GlcNAc transferase (mOGT) regulates mitochondrial structure, function, and survival in HeLa cells. J Biol Chem 292: 4499–4518, 2017. doi: 10.1074/jbc.M116.726752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Kim S-J, Yoo W-S, Choi M, Chung I, Yoo J-M, Choi W-S. Increased O-GlcNAcylation of NF-κB enhances retinal ganglion cell death in streptozotocin-induced diabetic retinopathy. Current Eye Res 41: 249–257, 2016. doi: 10.3109/02713683.2015.1006372. [DOI] [PubMed] [Google Scholar]
- 431.Choi H, Kim C, Song H, Cha M-Y, Cho HJ, Son SM, Kim HJ, Mook-Jung I. Amyloid β-induced elevation of O-GlcNAcylated c-Fos promotes neuronal cell death. Aging Cell 18: e12872, 2019. doi: 10.1111/acel.12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.McGreal SR, Bhushan B, Walesky C, McGill MR, Lebofsky M, Kandel SE, Winefield RD, Jaeschke H, Zachara NE, Zhang Z, Tan EP, Slawson C, Apte U. Modulation of O-GlcNAc levels in the liver impacts acetaminophen-induced liver injury by affecting protein adduct formation and glutathione synthesis. Toxicol Sci 162: 599–610, 2018. doi: 10.1093/toxsci/kfy002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Rajamani U, Joseph D, Roux S, Essop MF. The hexosamine biosynthetic pathway can mediate myocardial apoptosis in a rat model of diet-induced insulin resistance. Acta Physiol 202: 151–157, 2011. doi: 10.1111/j.1748-1716.2011.02275.x. [DOI] [PubMed] [Google Scholar]
- 434.Chen Q, Yu X. OGT restrains the expansion of DNA damage signaling. Nucleic Acids Res 44: 9266–9278, 2016. doi: 10.1093/nar/gkw663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Ma X, Liu H, Li J, Wang Y, Ding YH, Shen H, Yang Y, Sun C, Huang M, Tu Y, Liu Y, Zhao Y, Dong MQ, Xu P, Tang TS, Guo C. Polη O-GlcNAcylation governs genome integrity during translesion DNA synthesis. Nat Commun 8: 1941, 2017. doi: 10.1038/s41467-017-02164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Miura Y, Sakurai Y, Endo T. O-GlcNAc modification affects the ATM-mediated DNA damage response. Biochim Biophys Acta 1820: 1678–1685, 2012. doi: 10.1016/j.bbagen.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 437.Laczy B, Marsh SA, Brocks CA, Wittmann I, Chatham JC. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am J Physiol Heart Circ Physiol 299: H1715–H1727, 2010. doi: 10.1152/ajpheart.00337.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Ngoh GA, Facundo HT, Hamid T, Dillmann W, Zachara NE, Jones SP. Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to hypoxia/reoxygenation injury. Circ Res 104: 41–49, 2009. doi: 10.1161/CIRCRESAHA.108.189431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Not LG, Marchase RB, Fulop N, Brocks CA, Chatham JC. Glucosamine administration improves survival rate after severe hemorrhagic shock combined with trauma in rats. Shock 28: 345–352, 2007. [DOI] [PubMed] [Google Scholar]
- 440.Fulop N, Marchase RB, Chatham JC. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res 73: 288–297, 2007. doi: 10.1016/j.cardiores.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Liu J, Pang Y, Chang T, Bounelis P, Chatham JC, Marchase RB. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol 40: 303–312, 2006. doi: 10.1016/j.yjmcc.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 442.Ngoh GA, Watson LJ, Facundo HT, Jones SP. Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids 40: 895–911, 2011. doi: 10.1007/s00726-010-0728-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Zou L, Yang S, Champattanachai V, Hu S, Chaudry IH, Marchase RB, Chatham JC. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-κB signaling. Am J Physiol Heart Circ Physiol 296: H515–H523, 2009. doi: 10.1152/ajpheart.01025.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Xing J, Liu H, Yang H, Chen R, Chen Y, Xu J. Upregulation of Unc-51-like kinase 1 by nitric oxide stabilizes SIRT1, independent of autophagy. PloS One 9: e116165, 2014. doi: 10.1371/journal.pone.0116165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Park S, Pak J, Jang I, Cho JW. Inhibition of mTOR affects protein stability of OGT. Biochem Biophys Res Commun 453: 208–212, 2014. doi: 10.1016/j.bbrc.2014.05.047. [DOI] [PubMed] [Google Scholar]
- 446.Li Y-N, Hu J-A, Wang H-M. Inhibition of HIF-1α; Affects Autophagy Mediated Glycosylation in Oral Squamous Cell Carcinoma Cells. Dis Markers, 2015: 9, 2015. doi: 10.1155/2015/239479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Ejlerskov P, Rasmussen I, Nielsen TT, Bergström AL, Tohyama Y, Jensen PH, Vilhardt F. Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem 288: 17,313–17,335, 2013. doi: 10.1074/jbc.M112.401174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Vosseller K, Trinidad JC, Chalkley RJ, Specht CG, Thalhammer A, Lynn AJ, Snedecor JO, Guan S, Medzihradszky KF, Maltby DA, Schoepfer R, Burlingame AL. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol Cell Proteomics 5: 923–934, 2006. doi: 10.1074/mcp.T500040-MCP200. [DOI] [PubMed] [Google Scholar]
- 449.Zhang J. Teaching the basics of autophagy and mitophagy to redox biologists–mechanisms and experimental approaches. Redox Biol 4: 242–259, 2015. doi: 10.1016/j.redox.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Zhang X, Wang L, Lak B, Li J, Jokitalo E, Wang Y. GRASP55 senses glucose deprivation through O-GlcNAcylation to promote autophagosome-lysosome fusion. Dev Cell 45: 245–261,2018. e246 doi: 10.1016/j.devcel.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Dodson M, Liu P, Jiang T, Ambrose AJ, Luo G, Rojo de la Vega M, Cholanians AB, Wong PK, Chapman E, Zhang DD. Increased O-GlcNAcylation of SNAP29 drives arsenic-induced autophagic dysfunction. Mol Cell Biol 38: e00595-00517, 2018. doi: 10.1128/MCB.00595-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Jo YK, Park NY, Park SJ, Kim B-G, Shin JH, Jo DS, Bae D-J, Suh Y-A, Chang JH, Lee EK, Kim S-Y, Kim JC, Cho D-H. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget 7: 57,186–57,196, 2016. doi: 10.18632/oncotarget.11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Wani WY, Ouyang X, Benavides GA, Redmann M, Cofield SS, Shacka JJ, Chatham JC, Darley-Usmar V, Zhang J. O-GlcNAc regulation of autophagy and α-synuclein homeostasis; implications for Parkinson’s disease. Mol Brain 10: 32, 2017. doi: 10.1186/s13041-017-0311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Zhu Y, Shan X, Safarpour F, Erro Go N, Li N, Shan A, Huang MC, Deen M, Holicek V, Ashmus R, Madden Z, Gorski S, Silverman MA, Vocadlo DJ. Pharmacological inhibition of O-GlcNAcase enhances autophagy in brain through an mTOR-independent pathway. ACS Chem Neurosci 9: 1366–1379, 2018. doi: 10.1021/acschemneuro.8b00015. [DOI] [PubMed] [Google Scholar]
- 455.Sodi VL, Khaku S, Krutilina R, Schwab LP, Vocadlo DJ, Seagroves TN, Reginato MJ. mTOR/MYC axis regulates O-GlcNAc transferase expression and O-GlcNAcylation in breast cancer. Mol Cancer Res 13: 923–933, 2015. doi: 10.1158/1541-7786.MCR-14-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Edery I. Circadian rhythms in a nutshell. Physioll Genomics 3: 59–74, 2000. doi: 10.1152/physiolgenomics.2000.3.2.59. [DOI] [PubMed] [Google Scholar]
- 457.Morris CJ, Yang JN, Scheer FA. The impact of the circadian timing system on cardiovascular and metabolic function. Prog Brain Res 199: 337–358, 2012. doi: 10.1016/B978-0-444-59427-3.00019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Kim EY, Jeong EH, Park S, Jeong HJ, Edery I, Cho JW. A role for O-GlcNAcylation in setting circadian clock speed. Genes Dev 26: 490–502, 2012. doi: 10.1101/gad.182378.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Ma YT, Luo H, Guan WJ, Zhang H, Chen C, Wang Z, Li JD. O-GlcNAcylation of BMAL1 regulates circadian rhythms in NIH3T3 fibroblasts. Biochem Biophys Res Commun 431: 382–387, 2013. doi: 10.1016/j.bbrc.2013.01.043. [DOI] [PubMed] [Google Scholar]
- 460.Hart GW. How sugar tunes your clock. Cell Metab 17: 155–156, 2013. doi: 10.1016/j.cmet.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Dehennaut V, Lefebvre T, Leroy Y, Vilain JP, Michalski JC, Bodart JF. Survey of O-GlcNAc level variations in Xenopus laevis from oogenesis to early development. Glycoconj J 26: 301–311, 2009. doi: 10.1007/s10719-008-9166-0. [DOI] [PubMed] [Google Scholar]
- 462.Mariappa D, Selvan N, Borodkin V, Alonso J, Ferenbach AT, Shepherd C, Navratilova IH, van Aalten DMF. A mutant O-GlcNAcase as a probe to reveal global dynamics of protein O-GlcNAcylation during Drosophila embryonic development. Biochem J 470: 255–262, 2015. doi: 10.1042/BJ20150610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Mariappa D, Ferenbach AT, van Aalten DMF. Effects of hypo-O-GlcNAcylation on Drosophila development. J Biol Chem 293: 7209–7221, 2018. doi: 10.1074/jbc.RA118.002580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Selvan N, Williamson R, Mariappa D, Campbell DG, Gourlay R, Ferenbach AT, Aristotelous T, Hopkins-Navratilova I, Trost M, van Aalten DMF. A mutant O-GlcNAcase enriches Drosophila developmental regulators. Nat Chem Biol 13: 882–887, 2017. doi: 10.1038/nchembio.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Sinclair DA, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, Honda BM. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc). Proc Natl Acad Sci U S A 106: 13427–13432, 2009. doi: 10.1073/pnas.0904638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Yang YR, Song M, Lee H, Jeon Y, Choi EJ, Jang HJ, Moon HY, Byun HY, Kim EK, Kim DH, Lee MN, Koh A, Ghim J, Choi JH, Lee-Kwon W, Kim KT, Ryu SH, Suh PG. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 11: 439–448, 2012. doi: 10.1111/j.1474-9726.2012.00801.x. [DOI] [PubMed] [Google Scholar]
- 467.Nugent BM, Bale TL. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol 39: 28–37, 2015. doi: 10.1016/j.yfrne.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Howerton CL, Bale TL. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc Natl Acad Sci U S A 111: 9639–9644, 2014. doi: 10.1073/pnas.1401203111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci U S A 110: 5169–5174, 2013. doi: 10.1073/pnas.1300065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Dela Justina V, Dos Passos RR Junior, Bressan AF, Tostes RC, Carneiro FS, Soares TS, Volpato GT, Lima VV, Martin SS, Giachini FR. O-linked N-acetyl-glucosamine deposition in placental proteins varies according to maternal glycemic levels. Life Sci 205: 18–25, 2018. doi: 10.1016/j.lfs.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 471.Pantaleon M, Steane SE, McMahon K, Cuffe JSM, Moritz KM. Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Sci Rep 7: 2017, 2017. doi: 10.1038/s41598-017-01666-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Liu W, Wang H, Xue X, Xia J, Liu J, Qi Z, Ji L. OGT-related mitochondrial motility is associated with sex differences and exercise effects in depression induced by prenatal exposure to glucocorticoids. J Affect Disord 226: 203–215, 2018. doi: 10.1016/j.jad.2017.09.053. [DOI] [PubMed] [Google Scholar]
- 473.Nugent BM, O’Donnell CM, Epperson CN, Bale TL. Placental H3K27me3 establishes female resilience to prenatal insults. Nat Commun 9: 2555, 2018. doi: 10.1038/s41467-018-04992-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Nolte D, Muller U. Human O-GlcNAc transferase (OGT): genomic structure, analysis of splice variants, fine mapping in Xq13.1. Mamm Genome 13: 62–64, 2002. doi: 10.1007/s00335-001-2108-9. [DOI] [PubMed] [Google Scholar]
- 475.Olivier-Van Stichelen S, Abramowitz LK, Hanover JA. X marks the spot: does it matter that O-GlcNAc transferase is an X-linked gene? Biochem Biophys Res Commun 453: 201–207, 2014. doi: 10.1016/j.bbrc.2014.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Selvan N, George S, Serajee FJ, Shaw M, Hobson L, Kalscheuer V, Prasad N, Levy SE, Taylor J, Aftimos S, Schwartz CE, Huq AM, Gecz J, Wells L. O-GlcNAc transferase missense mutations linked to X-linked intellectual disability deregulate genes involved in cell fate determination and signaling. J Biol Chem 293: 10,810–10,824, 2018. doi: 10.1074/jbc.RA118.002583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Hewagama A, Gorelik G, Patel D, Liyanarachchi P, McCune WJ, Somers E, Gonzalez-Rivera T, Michigan Lupus C, Strickland F, Richardson B. Overexpression of X-linked genes in T cells from women with lupus. J Autoimmun 41: 60–71, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.McClain DA. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J Diab Complic 16: 72–80, 2002. doi: 10.1016/S1056-8727(01)00188-X. [DOI] [PubMed] [Google Scholar]
- 479.Prasad P, Tiwari AK, Kumar KM, Ammini AC, Gupta A, Gupta R, Thelma BK. Association analysis of ADPRT1, AKR1B1, RAGE, GFPT2 and PAI-1 gene polymorphisms with chronic renal insufficiency among Asian Indians with type-2 diabetes. BMC Med Genet 11: 52, 2010. doi: 10.1186/1471-2350-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Zhang H, Jia Y, Cooper JJ, Hale T, Zhang Z, Elbein SC. Common variants in glutamine:fructose-6-phosphate amidotransferase 2 (GFPT2) gene are associated with type 2 diabetes, diabetic nephropathy, and increased GFPT2 mRNA levels. J Clin Endocrinol Metab 89: 748–755, 2004. doi: 10.1210/jc.2003-031286. [DOI] [PubMed] [Google Scholar]
- 481.Coomer M, Essop MF. Differential hexosamine biosynthetic pathway gene expression with type 2 diabetes. Mol Genet Metab Rep 1: 158–169, 2014. doi: 10.1016/j.ymgmr.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Cameron EA, Martinez-Marignac VL, Chan A, Valladares A, Simmonds LV, Wacher N, Kumate J, McKeigue P, Shriver MD, Kittles R, Cruz M, Parra EJ. MGEA5-14 polymorphism and type 2 diabetes in Mexico City. Am J Hum Biol 19: 593–596, 2007. doi: 10.1002/ajhb.20639. [DOI] [PubMed] [Google Scholar]
- 483.Farook VS, Bogardus C, Prochazka M. Analysis of MGEA5 on 10q24.1–q24.3 encoding the β-O-linked N-acetylglucosaminidase as a candidate gene for type 2 diabetes mellitus in Pima Indians. Molec Genet Metab 77: 189–193, 2002. doi: 10.1016/S1096-7192(02)00127-0. [DOI] [PubMed] [Google Scholar]
- 484.Lehman DM, Fu D-J, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, Hamlington J, Dyer TD, Arya R, Abboud H, Göring HHH, Duggirala R, Blangero J, Konrad RJ, Stern MP. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc–selective N-Acetyl-β-d glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes 54: 1214–1221, 2005. doi: 10.2337/diabetes.54.4.1214. [DOI] [PubMed] [Google Scholar]
- 485.Murata K, Morino K, Ida S, Ohashi N, Lemecha M, Park SY, Ishikado A, Kume S, Choi CS, Sekine O, Ugi S, Maegawa H. Lack of O-GlcNAcylation enhances exercise-dependent glucose utilization potentially through AMP-activated protein kinase activation in skeletal muscle. Biochem Biophys Res Commun 495: 2098–2104, 2018. doi: 10.1016/j.bbrc.2017.12.081. [DOI] [PubMed] [Google Scholar]
- 486.Yang Y, Li X, Luan HH, Zhang B, Zhang K, Nam JH, Li Z, Fu M, Munk A, Zhang D, Wang S, Liu Y, Albuquerque JP, Ong Q, Li R, Wang Q, Robert ME, Perry RJ, Chung D, Shulman GI, Yang X. OGT suppresses S6K1-mediated macrophage inflammation and metabolic disturbance. Proc Natl Acad Sci U S A, 117: 16616–16625, 2020. doi: 10.1073/pnas.1916121117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Ida S, Morino K, Sekine O, Ohashi N, Kume S, Chano T, Iwasaki K, Harada N, Inagaki N, Ugi S, Maegawa H. Diverse metabolic effects of O-GlcNAcylation in the pancreas but limited effects in insulin-sensitive organs in mice. Diabetologia 60: 1761–1769, 2017. doi: 10.1007/s00125-017-4327-y. [DOI] [PubMed] [Google Scholar]
- 488.Thielen L, Shalev A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr Opin Endocrinol Diabetes Obes 25: 75–80, 2018. doi: 10.1097/MED.0000000000000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Filhoulaud G, Benhamed F, Pagesy P, Bonner C, Fardini Y, Ilias A, Movassat J, Burnol AF, Guilmeau S, Kerr-Conte J, Pattou F, Issad T, Postic C. O-GlcNacylation links TxNIP to inflammasome activation in pancreatic β cells. Front Endocrinol (Lausanne) 10: 291, 2019. doi: 10.3389/fendo.2019.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Akimoto Y, Hart GW, Wells L, Vosseller K, Yamamoto K, Munetomo E, Ohara-Imaizumi M, Nishiwaki C, Nagamatsu S, Hirano H, Kawakami H. Elevation of the post-translational modification of proteins by O-linked N-acetylglucosamine leads to deterioration of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto-Kakizaki rats. Glycobiology 17: 127–140, 2007. doi: 10.1093/glycob/cwl067. [DOI] [PubMed] [Google Scholar]
- 491.Degrell P, Cseh J, Mohás M, Molnár GA, Pajor L, Chatham JC, Fülöp N, Wittmann I. Evidence of O-linked N-acetylglucosamine in diabetic nephropathy. Life Sci 84: 389–393, 2009. doi: 10.1016/j.lfs.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 492.Levi I, Segev Y, Priel E. Type 1 diabetes affects topoisomerase I activity and GlcNAcylation in rat organs: kidney, liver and pancreas. Glycobiology 22: 704–713, 2012. doi: 10.1093/glycob/cws008. [DOI] [PubMed] [Google Scholar]
- 493.McLarty JL, Marsh SA, Chatham JC. Post-translational protein modification by O-linked N-acetyl-glucosamine: its role in mediating the adverse effects of diabetes on the heart. Life Sci 92: 621–627, 2013. doi: 10.1016/j.lfs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Olivier-Van Stichelen S, Wang P, Comly M, Love DC, Hanover JA. Nutrient-driven O-linked N-acetylglucosamine (O-GlcNAc) cycling impacts neurodevelopmental timing and metabolism. J Biol Chem 292: 6076–6085, 2017. doi: 10.1074/jbc.M116.774042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Okuyama R, Marshall S. UDP-N-acetylglucosaminyl transferase (OGT) in brain tissue: temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J Neurochem 86: 1271–1280, 2003. doi: 10.1046/j.1471-4159.2003.01939.x. [DOI] [PubMed] [Google Scholar]
- 496.Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron 38: 237–252, 2003. doi: 10.1016/S0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- 497.Tallent MK, Varghis N, Skorobogatko Y, Hernandez-Cuebas L, Whelan K, Vocadlo DJ, Vosseller K. In vivo modulation of O-GlcNAc levels regulates hippocampal synaptic plasticity through interplay with phosphorylation. J Biol Chem 284: 174–181, 2009. doi: 10.1074/jbc.M807431200. [DOI] [PubMed] [Google Scholar]
- 498.Cole RN, Hart GW. Cytosolic O-glycosylation is abundant in nerve terminals. Journal of Neurochemistry 79: 1080–1089, 2008. doi: 10.1046/j.1471-4159.2001.00655.x. [DOI] [PubMed] [Google Scholar]
- 499.Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: Direct identification of O/-GlcNAc-modified proteins from the brain. Proceedings of the National Academy of Sciences of the United States of America 101: 13,132–13,137, 2004. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Liu Y, Li X, Yu Y, Shi J, Liang Z, Run X, Li Y, Dai C-L, Grundke-Iqbal I, Iqbal K, Liu F, Gong C-X. Developmental regulation of protein O-GlcNAcylation, O-GlcNAc transferase, and O-GlcNAcase in mammalian brain. PloS One 7: e43724, 2012. doi: 10.1371/journal.pone.0043724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Akimoto Y, Comer FI, Cole RN, Kudo A, Kawakami H, Hirano H, Hart GW. Localization of the O-GlcNAc transferase and O-GlcNAc-modified proteins in rat cerebellar cortex. Brain Res 966: 194–205, 2003. doi: 10.1016/S0006-8993(02)04158-6. [DOI] [PubMed] [Google Scholar]
- 502.Németh AH, Nolte D, Dunne E, Niemann S, Kostrzewa M, Peters U, Fraser E, Bochukova E, Butler R, Brown J, Cox RD, Levy ER, Ropers H-H, Monaco AP, Müller U. Refined linkage disequilibrium and physical mapping of the gene locus for X-linked dystonia–Parkinsonism (DYT3). Genomics 60: 320–329, 1999. doi: 10.1006/geno.1999.5929. [DOI] [PubMed] [Google Scholar]
- 503.Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RCP, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE. Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science 290: 2302–2303, 2000. doi: 10.1126/science.290.5500.2302. [DOI] [PubMed] [Google Scholar]
- 504.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong C-X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A 101: 10,804–10,809, 2004. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Borghgraef P, Menuet C, Theunis C, Louis JV, Devijver H, Maurin H, Smet-Nocca C, Lippens G, Hilaire G, Gijsen H, Moechars D, Van Leuven F. Increasing brain protein O-GlcNAc-ylation mitigates breathing defects and mortality of Tau.P301L mice. PloS One 8: e84442, 2013. doi: 10.1371/journal.pone.0084442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Medina M. An overview on the Clinical Development of Tau-Based Therapeutics. Int J Mol Sci 19, 2018. doi: 10.3390/ijms19041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Shoeibi A, Olfati N, Litvan I. Preclinical, phase I, and phase II investigational clinical trials for treatment of progressive supranuclear palsy. Expert Opin Investig Drugs 27: 349–361, 2018. doi: 10.1080/13543784.2018.1460356. [DOI] [PubMed] [Google Scholar]
- 508.Wang S, Yang F, Petyuk VA, Shukla AK, Monroe ME, Gritsenko MA, Rodland KD, Smith RD, Qian WJ, Gong CX, Liu T. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J Pathol 243: 78–88, 2017. doi: 10.1002/path.4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Wang P, Lazarus BD, Forsythe ME, Love DC, Krause MW, Hanover JA. O-GlcNAc cycling mutants modulate proteotoxicity in Caenorhabditis elegans models of human neurodegenerative diseases. Proc Natl Acad Sci U S A 109: 17,669–17,674, 2012. doi: 10.1073/pnas.1205748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Griffith LS, Mathes M, Schmitz B. β-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J Neurosci Res 41: 270–278, 1995. doi: 10.1002/jnr.490410214. [DOI] [PubMed] [Google Scholar]
- 511.Sprung R, Nandi A, Chen Y, Kim SC, Barma D, Falck JR, Zhao Y. Tagging-via-substrate strategy for probing O-GlcNAc modified proteins. J Proteome Res 4: 950–957, 2005. doi: 10.1021/pr050033j. [DOI] [PubMed] [Google Scholar]
- 512.Chen PH, Hu J, Wu J, Huynh DT, Smith TJ, Pan S, Bisnett BJ, Smith AB, Lu A, Condon BM, Chi JT, Boyce M. Gigaxonin glycosylation regulates intermediate filament turnover and may impact giant axonal neuropathy etiology or treatment. JCI Insight 5: 2020. doi: 10.1172/jci.insight.127751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron 79: 1044–1066, 2013. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Aβ, τ, and α-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 30: 7281–7289, 2010. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10: 378–384, 2006. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Kraybill ML, Larson EB, Tsuang DW, Teri L, McCormick WC, Bowen JD, Kukull WA, Leverenz JB, Cherrier MM. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 64: 2069–2073, 2005. doi: 10.1212/01.WNL.0000165987.89198.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Spencer B, Desplats PA, Overk CR, Valera-Martin E, Rissman RA, Wu C, Mante M, Adame A, Florio J, Rockenstein E, Masliah E. Reducing endogenous α-synuclein mitigates the degeneration of selective neuronal populations in an Alzheimer’s disease transgenic mouse model. J Neurosci 36: 7971–7984, 2016. doi: 10.1523/JNEUROSCI.0775-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Wang Z, Park K, Comer F, Hsieh-Wilson LC, Saudek CD, Hart GW. Site-specific GlcNAcylation of human erythrocyte proteins: potential biomarker(s) for diabetes. Diabetes 58: 309–317, 2009. doi: 10.2337/db08-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Lewis YE, Galesic A, Levine PM, De Leon CA, Lamiri N, Brennan CK, Pratt MR. O-GlcNAcylation of α-synuclein at serine 87 reduces aggregation without affecting membrane binding. ACS Chem Biol 12: 1020–1027, 2017. doi: 10.1021/acschembio.7b00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, Arnold DB, Langen R, Pratt MR. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nature Chem 7: 913–920, 2015. doi: 10.1038/nchem.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of α-synucleinopathy. PLoS One 5: e9313, 2010. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 522.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 19: 983–997, 2013. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 523.Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D, Liang Q, Crimmins S, Schneider L, Uchiyama Y, Iwatsubo T, Zhou Y, Peng L, Lu Y, Standaert DG, Walls KC, Shacka JJ, Roth KA, Zhang J. Lysosomal enzyme cathepsin D protects against α-synuclein aggregation and toxicity. Mol Brain 1: 17, 2008. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun 5: 5659, 2014. doi: 10.1038/ncomms6659. [DOI] [PubMed] [Google Scholar]
- 525.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. α-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278: 25009–25013, 2003. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 526.Oláh J, Tőkési N, Vincze O, Horváth I, Lehotzky A, Erdei A, Szájli E, Medzihradszky KF, Orosz F, Kovács GG, Ovádi J. Interaction of TPPP/p25 protein with glyceraldehyde-3-phosphate dehydrogenase and their co-localization in Lewy bodies. FEBS Lett 580: 5807–5814, 2006. doi: 10.1016/j.febslet.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 527.Hwang H, Rhim H. Acutely elevated O-GlcNAcylation suppresses hippocampal activity by modulating both intrinsic and synaptic excitability factors. Sci Rep 9: 7287, 2019. doi: 10.1038/s41598-019-43017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Stewart LT, Abiraman K, Chatham JC, McMahon LL. Increased O-GlcNAcylation rapidly decreases GABAAR currents in hippocampus but depresses neuronal output. Sci Rep 10: 7494, 2020. doi: 10.1038/s41598-020-63188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Muha V, Fenckova M, Ferenbach AT, Catinozzi M, Eidhof I, Storkebaum E, Schenck A, van Aalten DMF. O-GlcNAcase contributes to cognitive function in Drosophila. J Biol Chem 295: 8636–8646, 2020. doi: 10.1074/jbc.RA119.010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Cho Y, Hwang H, Rahman MA, Chung C, Rhim H. Elevated O-GlcNAcylation induces an antidepressant-like phenotype and decreased inhibitory transmission in medial prefrontal cortex. Sci Rep 10: 6924, 2020. doi: 10.1038/s41598-020-63819-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Fourneau J, Canu MH, Cieniewski-Bernard C, Bastide B, Dupont E. Synaptic protein changes after a chronic period of sensorimotor perturbation in adult rats: a potential role of phosphorylation/O-GlcNAcylation interplay. J Neurochem 147: 240–255, 2018. doi: 10.1111/jnc.14474. [DOI] [PubMed] [Google Scholar]
- 532.Taub DG, Awal MR, Gabel CV. O-GlcNAc signaling orchestrates the regenerative response to neuronal injury in Caenorhabditis elegans. Cell Rep 24: 1931–1938,2018. e1933doi: 10.1016/j.celrep.2018.07.078. [DOI] [PubMed] [Google Scholar]
- 533.Stewart LT, Khan AU, Wang K, Pizarro D, Pati S, Buckingham SC, Olsen ML, Chatham JC, McMahon LL. Acute increases in protein O-GlcNAcylation dampen epileptiform activity in hippocampus. J Neurosci 37: 8207–8215, 2017. doi: 10.1523/JNEUROSCI.0173-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Sanchez RG, Parrish RR, Rich M, Webb WM, Lockhart RM, Nakao K, Ianov L, Buckingham SC, Broadwater DR, Jenkins A, de Lanerolle NC, Cunningham M, Eid T, Riley K, Lubin FD. Human and rodent temporal lobe epilepsy is characterized by changes in O-GlcNAc homeostasis that can be reversed to dampen epileptiform activity. Neurobiol Dis 124: 531–543, 2019. doi: 10.1016/j.nbd.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Yki-Jarvinen H, Vogt C, Iozzo P, Pipek R, Daniels MC, Virkamaki A, Makimattila S, Mandarino L, DeFronzo RA, McClain D, Gottschalk WK. UDP-N-acetylglucosamine transferase and glutamine: fructose 6-phosphate amidotransferase activities in insulin-sensitive tissues. Diabetologia 40: 76–81, 1997. [DOI] [PubMed] [Google Scholar]
- 536.Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A 97: 12,222–12,226, 2000. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res 96: 1006–1013, 2005. doi: 10.1161/01.RES.0000165478.06813.58. [DOI] [PubMed] [Google Scholar]
- 538.Huxley RR, Filion KB, Konety S, Alonso A. Meta-analysis of cohort and case-control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol 108: 56–62, 2011. doi: 10.1016/j.amjcard.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Yu P, Hu L, Xie J, Chen S, Huang L, Xu Z, Liu X, Zhou Q, Yuan P, Yan X, Jin J, Shen Y, Zhu W, Fu L, Chen Q, Yu J, Hu J, Cao Q, Wan R, Hong KO. GlcNAcylation of cardiac Nav1.5 contributes to the development of arrhythmias in diabetic hearts. Int J Cardiol 260: 74–81, 2018. doi: 10.1016/j.ijcard.2018.02.099. [DOI] [PubMed] [Google Scholar]
- 540.Ducheix S, Magré J, Cariou B, Prieur X. Chronic O-GlcNAcylation and diabetic cardiomyopathy: the bitterness of glucose. Front Endocrinol 9, 2018. doi: 10.3389/fendo.2018.00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Marsh SA, Collins HE, Chatham JC. Protein O-GlcNAcylation and cardiovascular (patho)physiology. J Biol Chem 289: 34449–34456, 2014. doi: 10.1074/jbc.R114.585984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Qin CX, Sleaby R, Davidoff AJ, Bell JR, De Blasio MJ, Delbridge LM, Chatham JC, Ritchie RH. Insights into the role of maladaptive hexosamine biosynthesis and O-GlcNAcylation in development of diabetic cardiac complications. Pharmacol Res 116: 45–56, 2017. doi: 10.1016/j.phrs.2016.12.016. [DOI] [PubMed] [Google Scholar]
- 543.De Blasio MJ, Huynh N, Deo M, Dubrana LE, Walsh J, Willis A, Prakoso D, Kiriazis H, Donner DG, Chatham JC, Ritchie RH. Defining the progression of diabetic cardiomyopathy in a mouse model of Type 1 diabetes. Front Physiol 11: 124, 2020. doi: 10.3389/fphys.2020.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Coort SL, Bonen A, van der Vusse GJ, Glatz JF, Luiken JJ. Cardiac substrate uptake and metabolism in obesity and type-2 diabetes: role of sarcolemmal substrate transporters. Mol Cell Biochem 299: 5–18, 2007. doi: 10.1007/s11010-005-9030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Lauzier B, Merlen C, Vaillant F, McDuff J, Bouchard B, Beguin PC, Dolinsky VW, Foisy S, Villeneuve LR, Labarthe F, Dyck JR, Allen BG, Charron G, Des Rosiers C. Post-translational modifications, a key process in CD36 function: lessons from the spontaneously hypertensive rat heart. J Mol Cell Cardiol 51: 99–108, 2011. doi: 10.1016/j.yjmcc.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 546.Brahma MK, McCrory MA, Paterson AJ, Pepin ME, Young ME, Wende AR. Regulation of myocardial ketone oxidative proteins by increased O-GlcNAcylation. FASEB Journal 30: 1273–1271, 2016. doi: 10.1096/fasebj.30.1_supplement.1273.1. [DOI] [Google Scholar]
- 547.Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, Hsieh-Wilson LC. Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. J Am Chem Soc 130: 11576–11577, 2008. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Dassanayaka S, Readnower RD, Salabei JK, Long BW, Aird AL, Zheng YT, Muthusamy S, Facundo HT, Hill BG, Jones SP. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem J 467: 115–126, 2015. doi: 10.1042/BJ20141018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med 34: 465–484, 2013. doi: 10.1016/j.mam.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 550.Tran DH, May HI, Li Q, Luo X, Huang J, Zhang G, Niewold E, Wang X, Gillette TG, Deng Y, Wang ZV. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat Commun 11: 1771, 2020. doi: 10.1038/s41467-020-15640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Umapathi P, Mesubi O, Banerjee P, Zachara N, Wang Q, Granger J, Luczak E, Wu Y, Florea L, Talbot C, Hart GW, Anderson ME. Elevated O-Glcnacylation: an independent driver of cardiomyopathy. Circulation 140: A13696, 2019. [Google Scholar]
- 552.Zhong W, Mao S, Tobis S, Angelis E, Jordan MC, Roos KP, Fishbein MC, de Alborán IM, MacLellan WR. Hypertrophic growth in cardiac myocytes is mediated by Myc through a cyclin D2-dependent pathway. EMBO J 25: 3869–3879, 2006. doi: 10.1038/sj.emboj.7601252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Ledee D, Smith L, Bruce M, Kajimoto M, Isern N, Portman MA, Olson AK. c-Myc alters substrate utilization and O-GlcNAc protein posttranslational modifications without altering cardiac function during early aortic constriction. PloS One 10: e0135262, 2015. doi: 10.1371/journal.pone.0135262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Olson AK, Ledee D, Iwamoto K, Kajimoto M, O'Kelly Priddy C, Isern N, Portman MA. C-Myc induced compensated cardiac hypertrophy increases free fatty acid utilization for the citric acid cycle. J Mol Cell Cardiol 55: 156–164, 2013. doi: 10.1016/j.yjmcc.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Zhu WZ, El-Nachef D, Yang X, Ledee D, Olson AK. O-GlcNAc transferase promotes compensated cardiac function and protein kinase A O-GlcNAcylation during early and established pathological hypertrophy from pressure overload. J Am Heart Assoc 8: e011260, 2019. doi: 10.1161/JAHA.118.011260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Cannon MV, Silljé HH, Sijbesma JW, Vreeswijk‐Baudoin I, Ciapaite J, van der Sluis B, van Deursen J, Silva GJ, de Windt LJ, Gustafsson JÅ, van der Harst P, van Gilst WH, de Boer RA. Cardiac LXRα protects against pathological cardiac hypertrophy and dysfunction by enhancing glucose uptake and utilization. EMBO Mol Med 7: 1229–1243, 2015. doi: 10.15252/emmm.201404669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Belke DD. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J Appl Physiol 111: 157–162, 2011. doi: 10.1152/japplphysiol.00147.2011. [DOI] [PubMed] [Google Scholar]
- 558.Bennett CE, Johnsen VL, Shearer J, Belke DD. Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci 92: 657–663, 2013. doi: 10.1016/j.lfs.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 559.Medford HM, Porter K, Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Heart Circulatory Physiol 305: H114–H123, 2013. doi: 10.1152/ajpheart.00135.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Chun WJ, Nah DY, Bae JH, Chung JW, Lee H, Moon IS. Glucose-insulin-potassium solution protects ventricular myocytes of neonatal rat in an in vitro coverslip ischemia/reperfusion model. Korean Circ J 45: 234–241, 2015. doi: 10.4070/kcj.2015.45.3.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Howell NJ, Ashrafian H, Drury NE, Ranasinghe AM, Contractor H, Isackson H, Calvert M, Williams LK, Freemantle N, Quinn DW, Green D, Frenneaux M, Bonser RS, Mascaro JG, Graham TR, Rooney SJ, Wilson IC, Pagano D. Glucose-insulin-potassium reduces the incidence of low cardiac output episodes after aortic valve replacement for aortic stenosis in patients with left ventricular hypertrophy: results from the Hypertrophy, Insulin, Glucose, and Electrolytes (HINGE) trial. Circulation 123: 170–177, 2011. doi: 10.1161/CIRCULATIONAHA.110.945170. [DOI] [PubMed] [Google Scholar]
- 562.Jensen RV, Zachara NE, Nielsen PH, Kimose HH, Kristiansen SB, Bøtker HE. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc Res 97: 369–378, 2013. doi: 10.1093/cvr/cvs337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Vibjerg Jensen R, Johnsen J, Buus Kristiansen S, Zachara NE, Botker HE. Ischemic preconditioning increases myocardial O-GlcNAc glycosylation. Scand Cardiovasc J 47: 168–174, 2013. doi: 10.3109/14017431.2012.756984. [DOI] [PubMed] [Google Scholar]
- 564.Lin YC, Lin CH, Yeh YC, Ho HL, Wu YC, Chen MY, Chou TY. High O-linked N-acetylglucosamine transferase expression predicts poor survival in patients with early stage lung adenocarcinoma. Oncotarget 9: 31032–31044, 2018. doi: 10.18632/oncotarget.25772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Xing D, Feng W, Nöt LG, Miller AP, Zhang Y, Chen YF, Majid-Hassan E, Chatham JC, Oparil S. Increased protein O-GlcNAc modification inhibits inflammatory and neointimal responses to acute endoluminal arterial injury. Am J Physiol Heart Circ Physiol 295: H335–H342, 2008. doi: 10.1152/ajpheart.01259.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Xing D, Gong K, Feng W, Nozell SE, Chen YF, Chatham JC, Oparil S. O-GlcNAc modification of NF-κB p65 inhibits TNF-α-induced inflammatory mediator expression in rat aortic smooth muscle cells. PloS One 6: e24021, 2011. doi: 10.1371/journal.pone.0024021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Hilgers RH, Xing D, Gong K, Chen YF, Chatham JC, Oparil S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am J Physiol Heart Circ Physiol 303: H513–H522, 2012. doi: 10.1152/ajpheart.01175.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Barnes JW, Tian L, Heresi GA, Farver CF, Asosingh K, Comhair SA, Aulak KS, Dweik RA. O-linked β-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 131: 1260–1268, 2015. doi: 10.1161/CIRCULATIONAHA.114.013878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Costamagna D, Quattrocelli M, Duelen R, Sahakyan V, Perini I, Palazzolo G, Sampaolesi M. Fate choice of post-natal mesoderm progenitors: skeletal versus cardiac muscle plasticity. Cell Mol Life Sci 71: 615–627, 2014. doi: 10.1007/s00018-013-1445-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Lambert M, Claeyssen C, Bastide B, Cieniewski-Bernard C. O-GlcNAcylation as a regulator of the functional and structural properties of the sarcomere in skeletal muscle: An update review. Acta Physiol 228: e13301, 2019. doi: 10.1111/apha.13301. [DOI] [PubMed] [Google Scholar]
- 571.Deracinois B, Camoin L, Lambert M, Boyer J-B, Dupont E, Bastide B, Cieniewski-Bernard C. O-GlcNAcylation site mapping by (azide-alkyne) click chemistry and mass spectrometry following intensive fractionation of skeletal muscle cells proteins. J Proteomics 186: 83–97, 2018. doi: 10.1016/j.jprot.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 572.Hortemo KH, Lunde PK, Anonsen JH, Kvaløy H, Munkvik M, Rehn TA, Sjaastad I, Lunde IG, Aronsen JM, Sejersted OM. Exercise training increases protein O-GlcNAcylation in rat skeletal muscle. Physiol Rep 4: e12896, 2016. doi: 10.14814/phy2.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Wang X, Feng Z, Wang X, Yang L, Han S, Cao K, Xu J, Zhao L, Zhang Y, Liu J. O-GlcNAcase deficiency suppresses skeletal myogenesis and insulin sensitivity in mice through the modulation of mitochondrial homeostasis. Diabetologia 59: 1287–1296, 2016. doi: 10.1007/s00125-016-3919-2. [DOI] [PubMed] [Google Scholar]
- 574.Rossetti L, Hawkins M, Chen W, Gindi J, Barzilai N. In vivo glucosamine infusion induces insulin resistance in normoglycemic but not in hyperglycemic conscious rats. J Clin Invest 96: 132–140, 1995. doi: 10.1172/JCI118013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Hawkins M, Angelov I, Liu R, Barzilai N, Rossetti L. The tissue concentration of UDP-N-acetylglucosamine modulates the stimulatory effect of insulin on skeletal muscle glucose uptake. J Biol Chem 272: 4889–4895, 1997. doi: 10.1074/jbc.272.8.4889. [DOI] [PubMed] [Google Scholar]
- 576.Macauley MS, Shan X, Yuzwa SA, Gloster TM, Vocadlo DJ. Elevation of global O-GlcNAc in rodents using a selective O-GlcNAcase inhibitor does not cause insulin resistance or perturb glucohomeostasis. Chem Biol 17: 949–958, 2010. doi: 10.1016/j.chembiol.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Peternelj TT, Marsh SA, Strobel NA, Matsumoto A, Briskey D, Dalbo VJ, Tucker PS, Coombes JS. Glutathione depletion and acute exercise increase O-GlcNAc protein modification in rat skeletal muscle. Mol Cell Biochem 400: 265–275, 2015. doi: 10.1007/s11010-014-2283-0. [DOI] [PubMed] [Google Scholar]
- 578.Zhang K, Yin R, Yang X. O-GlcNAc: a bittersweet switch in liver. Front Endocrinol (Lausanne) 5: 221, 2014. doi: 10.3389/fendo.2014.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Baldini SF, Steenackers A, Olivier-Van Stichelen S, Mir AM, Mortuaire M, Lefebvre T, Guinez C. Glucokinase expression is regulated by glucose through O-GlcNAc glycosylation. Biochem Biophys Res Commun 478: 942–948, 2016. doi: 10.1016/j.bbrc.2016.08.056. [DOI] [PubMed] [Google Scholar]
- 580.Ido-Kitamura Y, Sasaki T, Kobayashi M, Kim HJ, Lee YS, Kikuchi O, Yokota-Hashimoto H, Iizuka K, Accili D, Kitamura T. Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of Chrebp O-glycosylation. PloS one 7: e47231, 2012. doi: 10.1371/journal.pone.0047231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123: 3678–3684, 2013. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16: 619–634, 2016. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Ma Z, Vosseller K. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem 289: 34,457–34,465, 2014. doi: 10.1074/jbc.R114.577718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Makwana V, Ryan P, Patel B, Dukie SA, Rudrawar S. Essential role of O-GlcNAcylation in stabilization of oncogenic factors. Biochim Biophys Acta 1863: 1302–1317, 2019. doi: 10.1016/j.bbagen.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 585.Gao J, Yang Y, Qiu R, Zhang K, Teng X, Liu R, Wang Y. Proteomic analysis of the OGT interactome: novel links to epithelial-mesenchymal transition and metastasis of cervical cancer. Carcinogenesis 39: 1222–1234, 2018. doi: 10.1093/carcin/bgy097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer 11: 678–684, 2011. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Lai CY, Liu H, Tin KX, Huang Y, Yeh KH, Peng HW, Chen HD, He JY, Chiang YJ, Liu CS, Weng SY, Tao MH, Yen JJ, Yang-Yen HF. Identification of UAP1L1 as a critical factor for protein O-GlcNAcylation and cell proliferation in human hepatoma cells. Oncogene 38: 317–331, 2019. doi: 10.1038/s41388-018-0442-6. [DOI] [PubMed] [Google Scholar]
- 588.Burén S, Gomes AL, Teijeiro A, Fawal MA, Yilmaz M, Tummala KS, Perez M, Rodriguez-Justo M, Campos-Olivas R, Megias D, Djouder N. Regulation of OGT by URI in response to glucose confers c-MYC-dependent survival mechanisms. Cancer Cell 30: 290–307, 2016. doi: 10.1016/j.ccell.2016.06.023. [DOI] [PubMed] [Google Scholar]
- 589.Guo H, Zhang B, Nairn AV, Nagy T, Moremen KW, Buckhaults P, Pierce M. O-linked N-acetylglucosamine (O-GlcNAc) expression levels epigenetically regulate colon cancer tumorigenesis by affecting the cancer stem cell compartment via modulating expression of transcriptional factor MYBL1. J Biol Chem 292: 4123–4137, 2017. doi: 10.1074/jbc.M116.763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J Cell Biochem 95: 45–52, 2005. doi: 10.1002/jcb.20399. [DOI] [PubMed] [Google Scholar]
- 591.Hardie DG, Schaffer BE, Brunet A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol 26: 190–201, 2016. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Marin TL, Gongol B, Martin M, King SJ, Smith L, Johnson DA, Subramaniam S, Chien S, Shyy JY. Identification of AMP-activated protein kinase targets by a consensus sequence search of the proteome. BMC Syst Biol 9: 13, 2015. doi: 10.1186/s12918-015-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Freeman AK, Morrison DK. 14-3-3 Proteins: diverse functions in cell proliferation and cancer progression. Seminars Cell Dev Biol 22: 681–687, 2011. doi: 10.1016/j.semcdb.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Toleman CA, Schumacher MA, Yu SH, Zeng W, Cox NJ, Smith TJ, Soderblom EJ, Wands AM, Kohler JJ, Boyce M. Structural basis of O-GlcNAc recognition by mammalian 14-3-3 proteins. Proc Natl Acad Sci U S A 115: 5956–5961, 2018. doi: 10.1073/pnas.1722437115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Groves JA, Maduka AO, O'Meally RN, Cole RN, Zachara NE. Fatty acid synthase inhibits the O-GlcNAcase during oxidative stress. J Biol Chem 292: 6493–6511, 2017. doi: 10.1074/jbc.M116.760785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Hu CW, Worth M, Li H, Jiang J. Chemical and biochemical strategies to explore the substrate recognition of O-GlcNAc-cycling enzymes. Chembiochem 20: 312–318, 2019. doi: 10.1002/cbic.201800481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Ramirez DH, Aonbangkhen C, Wu H-Y, Naftaly JA, Tang S, O’Meara TR, Woo CM. Engineering a proximity-directed O-GlcNAc transferase for selective protein O-GlcNAcylation in cells. ACS Chem Biol 15: 1059–1066, 2020. doi: 10.1021/acschembio.0c00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Jin N, Ma D, Gu J, Shi J, Xu X, Iqbal K, Gong CX, Liu F, Chu D. O-GlcNAcylation modulates PKA-CREB signaling in a manner specific to PKA catalytic subunit isoforms. Biochem Biophys Res Commun 497: 194–199, 2018. doi: 10.1016/j.bbrc.2018.02.053. ] [DOI] [PubMed] [Google Scholar]
- 599.Macauley MS, Bubb AK, Martinez-Fleites C, Davies GJ, Vocadlo DJ. Elevation of Global O-GlcNAc levels in 3T3-L1 adipocytes by selective inhibition of O-GlcNAcase does not induce insulin resistance. J Biol Chem 283: 34,687–34,695, 2008. doi: 10.1074/jbc.M804525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Macauley MS, He Y, Gloster TM, Stubbs KA, Davies GJ, Vocadlo DJ. Inhibition of O-GlcNAcase using a potent and cell-permeable inhibitor does not induce insulin resistance in 3T3-L1 adipocytes. Chem Biol 17: 937–948, 2010. doi: 10.1016/j.chembiol.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]