

Keywords: hypoxia, immune therapy, metabolism, molecular targets, tumor microenvironment
Abstract
The host immune system shapes the fate of tumor progression. Hence, manipulating patients’ immune system to activate host immune responses against cancer pathogenesis is a promising strategy to develop effective therapeutic interventions for metastatic and drug-resistant cancers. Understanding the dynamic mechanisms within the tumor microenvironment (TME) that contribute to heterogeneity and metabolic plasticity is essential to enhance the patients’ responsiveness to immune targeted therapies. Riera-Domingo et al. (Riera-Domingo C, Audige A, Granja S, Cheng WC, Ho PC, Baltazar F, Stockmann C, Mazzone, M. Physiol Rev 100: 1–102, 2020) describe the immune landscape within the TME and highlight the significance of metabolic and hypoxic signatures that impact immune function and response to immunotherapy strategies. Current literature in this field confirms that targeting tumor metabolism and the acidic microenvironment commonly associated with tumors may present viable strategies to modulate the host immune system in favor of response to immune targeted therapies. However, development of better tools to understand tumor-immune interactions and identify mechanisms driving nonresponders, more innovative clinical trial design, and new therapies will need to be identified to move the field forward. Personalized immune therapies incorporating metabolic and microbiome-based gene signatures to influence the therapeutic response and novel methods to generate immunologically “hot” tumors are at the forefront of immunotherapy currently. The combination of these approaches with clinically approved immunotherapies will be valuable moving forward.
CLINICAL HIGHLIGHTS.
Defining aspects of the tumor microenvironment, including cellular metabolism, immune infiltration, hypoxia, and acidosis, could direct patient-specific therapeutic interventions.
Determination of the immunological landscape, i.e., the dampened inflammatory response within immunologically “cold” tumors or the antitumor inflammatory response within immunologically “hot” tumors, could impact clinical decisions, since these distinct immune phenotypes influence the composition of the tumor microenvironment.
1. INTRODUCTION
Cancer remains a leading cause of morbidity and mortality worldwide despite treatment advances. Exploiting the patient’s own immune system to fight malignancy has emerged as a promising treatment option, yet patient response rates to existing immune targeted therapies vary widely (1). Currently, four main immunotherapeutic strategies are utilized for the treatment of cancer, including adoptive cell transfer, cancer vaccines, targeting of specific immune cell subsets, and checkpoint blockade (2, 3). However, mechanisms of immune surveillance and access of immune cells to malignant tissues can affect response rates. Therefore, recent studies with immune target therapies have focused on combinatorial approaches, perioperative use, predicting response, toxicity management, and use in special populations/new tumor entities (4).
Questions remain: How can we increase patient responses to immunotherapy? Why are some patients nonresponders? When is the best time to introduce immunotherapy? An elegant review by Riera-Domingo et al. recently published in Physiological Reviews (5) thoroughly describes the role of the immune system in tumor progression and how the tumor microenvironment (TME), specifically metabolic signatures and hypoxia within the TME, impacts immune function and subsequent immune targeted therapies. Additionally, the authors provide a comprehensive list of recent and ongoing clinical trials involving checkpoint blockade therapies, detailing tumor type, phase of the trials, and study outcomes. Since this review, new therapeutic strategies have been identified, including targeting acidosis within the TME, altering the microbiome, new procedures to alter tryptophan metabolism, methods to generate immunologically “hot” tumors, and novel combinatorial strategies. Each of these strategies is discussed in detail here.
2. THE TUMOR MICROENVIRONMENT: METABOLISM AND HYPOXIA
The complex interplay between cellular cross talk, matrix interactions, and the biochemical environment within a tumor impacts the metabolic phenotype and differentiation/polarization of immune cells. The concerted actions of various immune subsets promote or suppress tumor growth. Immune cells in the TME can have dual roles, either promoting or inhibiting tumor growth, in response to surrounding microenvironmental and external signals. As shown in FIGURE 1, immune cells that promote tumor growth include myeloid-derived suppressor cells (MDSCs), regulatory T (Treg)/Th2 cells, N2 neutrophil populations, M2-like macrophages, and stromal fibroblasts. In contrast, M1-like macrophages, N1 neutrophils, dendritic cells, natural killer (NK) cells, B cells, and effector T (Teff)/Th1/cytotoxic T cells within the TME have an antitumor effect. The presence of these cell populations varies within the TME and is highly dependent on extracellular cues within the TME. Riera-Domingo et al. (5) nicely detailed the interconnected factors that influence the tumor-associated immune response, including the hypoxic microenvironment and metabolic profile of the tumor, and emphasized how immune cell phenotype and positioning can be altered in response to these stimuli.
FIGURE 1.
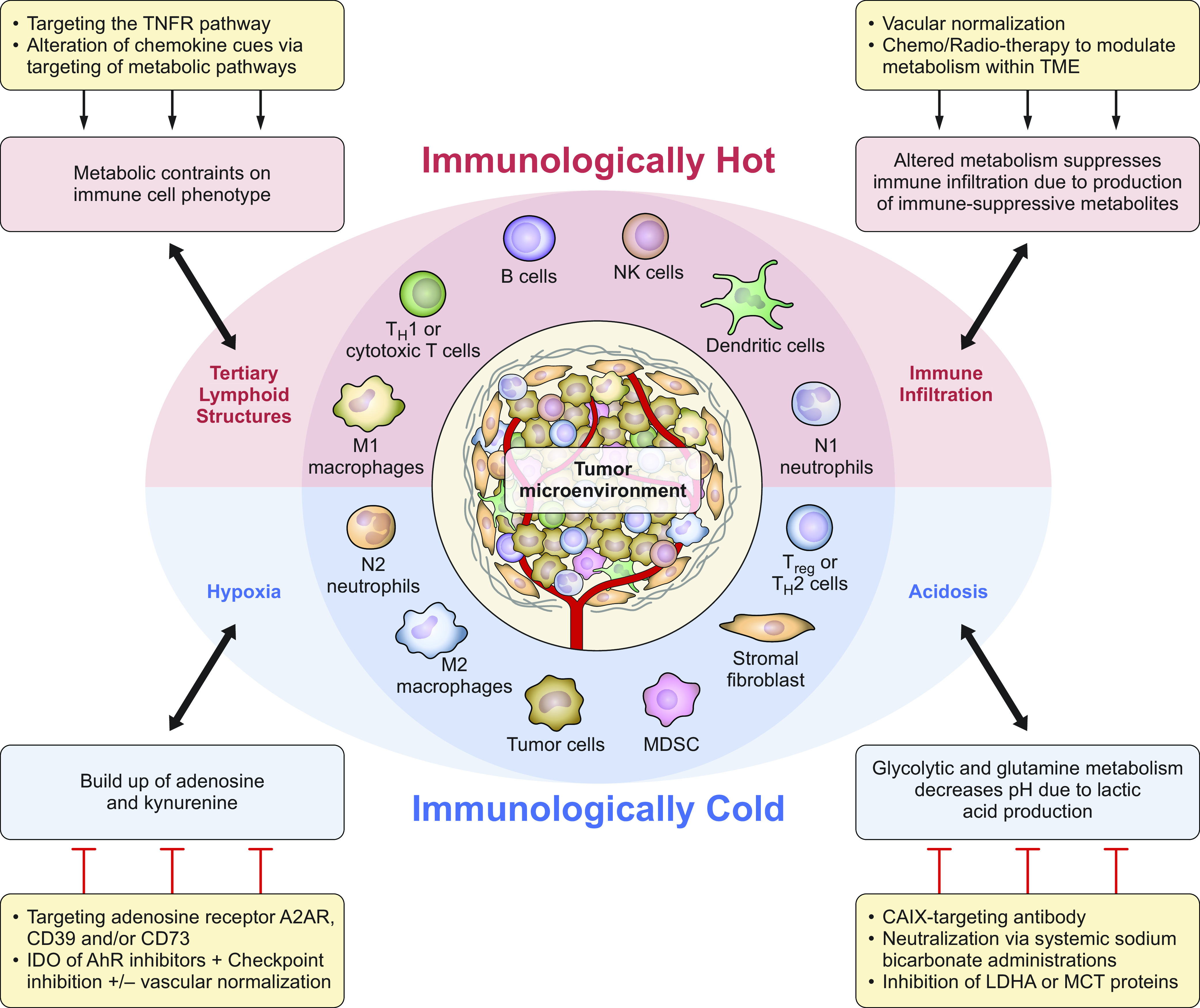
The tumor microenvironment (TME): tipping the scale to promote inflammatory reactions. Within the TME immune cell populations either are tolerant of tumor growth and exhibit a dampened inflammatory response (associated with immunologically cold tumors, within the blue half-circle) or promote an antitumor inflammatory response (associated with immunologically hot tumors, within the red half-circle). Factors within this microenvironment, including acidosis, hypoxia, infiltration of circulating immune cells, and formation of tertiary lymphoid structures, can also tip the scale and impact immune cell polarization/activation. Furthermore, various metabolic aspects within the TME influence these factors and subsequent immune cell presence and function. In addition to current therapies, targeting these cues within the TME, as indicated, may be useful in tipping the balance to promote the antitumor immune response. AhR, aryl hydrocarbon receptor; CAIX, carbonic anhydrase IX; IDO, indoleamine 2,3-dioxygenase; LDHA, lactate dehydrogenase A; MCT, monocarboxylate transporter; MDSC, myeloid-derived suppressor cell; NK, natural killer; Treg, regulatory T cell.
As reviewed by Riera-Domingo et al., solid tumors have a dynamic oxygen supply, with some regions of the tumor having reduced oxygen, or hypoxia, which promotes tumor progression and diminishes the response to therapy. The effects of hypoxia on interactions among cells of the immune system are not completely understood, as studies have focused on specific immune cell subsets, but many changes may occur simultaneously in tumor cells with altered oxygen availability, and this interplay could be important in predicting response to immune targeted therapies. The concept of solid tumors functioning as nutrient traps has also been suggested, and such a trap could compromise the immune response through effects on circulating immune cell populations and subsequent immune cell infiltration. Moreover, as Riera-Domingo et al. point out, metabolic processes utilized by immune cells, for example, tumor-associated macrophages (TAMs), alter immune responses by convergence of signaling cascades and epigenetic programs, further complicating metabolic targeting (5).
Furthermore, activated immune cells have increased energy demands much like tumor cells, with protumor immune populations generally being more resistant to metabolic stress. Therefore, these cell populations are more likely to be present within the TME. This complicates metabolism-targeted therapies, as interfering with tumor metabolic pathways will often impact other cell populations within the TME. Since immune cell fate is closely linked to metabolic programming, metabolic alterations may promote immune cell tolerance. Deregulation of energy metabolism plays a pivotal role in inhibiting the antitumor immune response and tumor progression and metastasis. There is growing evidence that the TME supports inappropriate metabolic reprograming, which dampens T cell function and impacts the immune response and tumor progression. Specifically, tumor-infiltrating lymphocytes (TILs) are at a metabolic disadvantage within the TME, as tumor cells impede T cell access to the nutrients needed for activation and generate increased levels of lactate. This lactate accumulation leads to acidification of the TME, reduces T cell activation, and leads to polarization to the Treg phenotype. Lactate accumulation also impacts macrophage polarization and represses the actions of NK cells (6), further restricting the antitumor response. Additionally, the immunosuppressive TME lacks carbon sources that are critical for T cell function, leading to increased inhibitory signals, such as checkpoint proteins (6).
Hypoxia and alterations in metabolism lead to acidosis, waste product accumulation, and nutrient competition generating a hostile TME, which includes deleterious effects on antitumor immune surveillance. This microenvironment has emerged as a major axis of tumor immunosuppression due, in part, to anergy and exhaustion of TILs [reviewed by Kouidhi et al. (6)]. Other metabolic alterations also occur in the TME, including upregulated tumor cell production of the enzyme indoleamine 2,3-dioxygenase (IDO) that breaks down tryptophan to produce l-kynurenine (Kyn), which promotes Treg polarization and reduces the proliferation of Teff cells, thereby blocking the T cell antitumor response (7). Furthermore, the metabolic changes associated with malignancy lead to the accumulation of metabolites that may modulate epigenetic factors, including enzymes involved in DNA and histone modifications. These metabolic intermediates consequently lead to changes in gene expression (8).
3. LOOKING FORWARD: POTENTIAL IMMUNE TARGETS TO EXPLORE
Moving forward, targeting tumor metabolism and the hypoxic microenvironment may be viable strategies to mitigate the adverse effect of the TME on the antitumor immune response (as detailed in FIGURE 1) and thereby improve the response to immune targeted therapies such as checkpoint inhibitors. Immune checkpoint inhibitor therapy involves blocking the specific interactions between T cells and tumor cells or protumor immune cells that limit Teff cell activation and contribute to T cell exhaustion. The two checkpoint pathways that have been successfully targeted for cancer immunotherapy involve interactions 1) between cytotoxic T lymphocyte-associated protein 4 (CTLA4, expressed on the surface of T cells) and CD80 or CD86 (expressed on the surface of professional antigen-presenting cells) and 2) between programmed death receptor 1 (PD-1, expressed on the surface of T cells) and programmed death ligand 1 (PD-L1, expressed on the surface of tumor and other immune cells) (9). Blocking these immune-inhibitory pathways is most efficacious in tumors with productive T cell infiltration, such as melanomas and lung cancers. It is presumed that other immune targeted therapies would similarly be most effective in tumors with brisk T cell infiltrates. However, many tumors, colloquially known as “cold” tumors, including many pancreatic and prostate tumors, are not well infiltrated by immune cells. Therefore, much recent research has focused on turning immunologically “cold” tumors “hot” by increasing immune cell infiltration and activity, or as Riera-Domingo et al. phrase it, “altering immune cell recruitment and positioning.” Methods proposed to achieve this goal include vascular normalization, promotion of tertiary lymphoid structures within the TME, drug-loaded nanoparticle treatment to increase neo-antigen burden, oncolytic virotherapy/intratumoral vaccine injection, TGF blockade, TNFR2 antagonism, inhibition of hypoxia-associated pathways (including carbonic anhydrases), and radiotherapy. These strategies impact immune cell trafficking, hypoxia, and the immunologic repertoire retained within the TME. Some particularly interesting strategies have been designed to target proangiogenic cytokines, including a bispecific antibody that targets vascular endothelial growth factor A (VEGF-A) and angiopoietin-2 (ANGPT2). This strategy is being investigated in combination with checkpoint inhibitors as well as agonistic anti-CD40 antibodies, which increase the function of professional antigen-presenting cells (10, 11). Overall, such approaches have shown a role for immune cells as effectors of antiangiogenic therapies and the cooperative benefits of reducing the leaky tumor vasculature/normalizing the vasculature while promoting proinflammatory polarization of macrophages and increased dendritic cell activation and T cell effector functions. Additionally, strategies focused on avoiding immune evasion or promoting effective immune recognition of cancer cells by altering hypoxia-induced immune checkpoint protein expression and/or blocking additional checkpoint pathways (e.g., CD47) may be of significant therapeutic value.
Riera-Domingo et al. (5) also raised the idea of repolarizing or rewiring immune cell subsets, including TAMs, dendritic cells, NK cells, and T cells, by modulating metabolic pathways as a possible treatment strategy. Ongoing preclinical studies will continue to provide insight into how the TME regulates tumor-immune interactions and how these aspects can be targeted therapeutically. For example, it was recently reported that tumor cells prompt the efflux of membrane cholesterol from TAMs, which in turn converts these cells to an immune-suppressive/tumor-promoting phenotype. Targeting the cholesterol efflux abrogated reprogramming and reduced tumor progression in a model of ovarian cancer, and thus could be a new means of immunotherapy (12). Cell metabolism is an attractive target to restore antitumor immunity, but the overlapping metabolic requirements of tumor and immune cells must be considered. Li et al. (13) point out that the similar metabolic requirements of tumor cells and immune cells might preclude synergistic effects of combining metabolic therapies and immunotherapies unless specific metabolic pathways that are differentially essential to cancer cells and immune cells (specifically those modulated by tumor cells to evade immunosurveillance) are targeted. For example, targeting tumor glucose metabolism has been shown to impair T cell metabolism as well, and tumor cell-specific strategies to inhibit this metabolic pathway have not been extensively developed so far.
Targeting specific metabolic alterations or metabolic pathways shared by tumor cells and tumor-promoting immune populations in the TME may be a more straightforward therapeutic strategy. Cancer cells have an altered metabolic profile to meet their energy demands. Whereas normal cells rely on oxidation of pyruvate in mitochondria to generate energy, cancer cells utilize glycolysis and lactic acid fermentation, regardless of oxygen availability; this is known as the Warburg effect (14, 15). Cancer-specific aspects of this pathway may be targeted. One recent example involves the enzyme pyruvate kinase (PK) M2, which functions during the final step of glycolysis, converting phosphoenolpyruvate into pyruvate, and is thus essential for aerobic glycolysis. This enzyme is often highly expressed by tumor cells but is not as effective as PKM1 in the production of pyruvate, supporting the Warburg effect. Blocking PKM2 activity reduces the expression of PD-L1 on tumor and myeloid cells in a murine colon cancer model (13, 16). Therefore, modulation of PKM2 might synergize with PD-1/PD-L1 immune checkpoint inhibitors to reduce metabolic stress and immunosuppression within the TME by reducing aerobic glycolysis and PD-L1 expression in tumor cells. Targeting amino acid catabolism in combination with checkpoint inhibition is another approach that is currently in clinical trials (6). In addition, inhibitors of glutamine metabolism may have therapeutic efficacy, as suggested by Riera-Domingo et al., via impairment of tumor metabolism, promotion of M1 macrophage polarization, and modulation of available glutamine in the TME to fuel NK cells, but the clinical applicability of this target is currently unknown (17). Targeting metabolism of specific, aggressive tumor cell subsets (e.g., tumor-initiating cells or cancer stem cells) may be another viable strategy to target cancer-specific metabolic pathways. This strategy will probably be a more efficacious method for eliminating tumor recurrence, as long as antitumor immune cells do not also rely on the same metabolic processes. Wang et al. (18) recently showed that tumor-initiating cells have elevated methionine cycle activity, which can be targeted to block the tumor-forming ability of this cell population. Another recent preclinical study has shown an essential role of the stemness gene LIN28B in maintaining cancer stem cell glycolysis metabolism. Although blocking the LIN28B/MYC/miR-34a-5p signaling pathway with a LIN28B-specific inhibitor substantially inhibited tumor growth and metastasis in a murine model of breast cancer, the impact of this targeting strategy on immune cell populations is yet to be demonstrated (19). Carefully designed studies must also ensure that immune cell metabolic changes induced by this type of therapy do not promote tumor growth. Theoretically, targeting both tumor and T cell metabolism could regulate immunity to improve the response to immunotherapy. Evidence suggests that checkpoint inhibition may modify T cell metabolism, as checkpoint inhibition impacts the metabolic profile of the TME to favor T cell activation and inhibit cancer cells. Compounding on this metabolic switch with additional therapies may further promote antitumor immunity.
Modulating T cell metabolism during in vitro expansion for adoptive cell transfer therapy may improve its therapeutic efficacy and may be less complex and/or have less widespread impact on the metabolism of other cell types in the TME. Recent research has also focused on fatty acid metabolism, as cancer cells exhibit metabolic plasticity in fatty acid desaturation. Sapienate and its elongation products may influence cancer cell fatty acid and lipid signaling networks and could be targeted to block cancer cell proliferation (20). In a recent study published in Nature Immunology (21), Liu et al. showed that the use of a TLR9 agonist, CpG, alters carbon metabolism of macrophages to induce antitumor behavior. This therapy induces an altered metabolic state for de novo lipid biosynthesis, allowing macrophages to overcome the CD47 inhibitory signal on tumor cells. These findings suggest that carbon metabolism may be a potential therapeutic target to stimulate antitumor activity of macrophages (21). The impact of this therapeutic strategy on other immune cells is yet to be determined. However, a recently published study details the regulation of tumor ferroptosis by CD8+ T cells during immunotherapeutic intervention. The mechanisms of ferroptosis are not well defined, but this form of cell death relies on reactive oxygen species (ROS) and iron and occurs because of loss of plasma membrane permeability as a consequence of membrane lipid peroxidation. Although the exact molecular mechanisms underlying this process are yet to be understood, immunotherapy-activated CD8+ T cells were shown to enhance ferroptosis-specific lipid peroxidation in tumor cells. In turn, increased ferroptosis contributed to the antitumor efficacy of immunotherapy. This novel antitumor mechanism may also be a viable therapeutic approach to be employed clinically in combination with checkpoint blockade (22).
Strategies aimed at reducing immunomodulatory metabolites may also be effective. One such strategy includes altering the acidic microenvironment. Various studies have investigated the molecular pathways responsible for acid-base regulation and pH homeostasis and have found isoforms of carbonic anhydrase and anion exchangers, monocarboxylate transporters (MCTs), cotransporters, and Na+/H+ exchangers to be the most important pH regulators in tumor cells. Therapies targeting these pH regulators are currently at various stages of clinical development. Reversion of acidosis by administration of systemic buffers such as sodium bicarbonate hinders tumor growth in experimental models, and targeting tumor acidosis with this method increases the effectiveness of checkpoint inhibition (23). Administration of proton pump inhibitors (PPIs), including omeprazole and analogs, reduces the acidity within the TME. The prodrug forms are specifically activated by the low pH of the TME, increasing the tumor pH. PPIs enhance dendritic cell-based cancer vaccines and adoptive T cell transfer in murine tumor models and synergize with chemotherapeutics in the clinic (24). Blocking the tryptophan metabolism pathway is another method by which immunomodulatory metabolites could be reduced. Although previous results with IDO inhibitors in clinical trials have been unremarkable, likely because of pathway redundancies, new inhibitors that target multiple portions of the pathway are under development. One example, CMG017, is a dual IDO/tryptophan 2,3-dioxygenase (TDO) inhibitor aimed at maximal blockade of this pathway. Also, groups are focusing on inhibiting downstream targets of this pathway [including Kyn and the aryl hydrocarbon receptor (AhR)] in combination with checkpoint inhibition and other therapies (7). Accumulation of adenosine within the TME contributes to tumor progression and impairs Teff function. This buildup is mediated by increased PKA activation and impairment of the mammalian target of rapamycin complex 1 (mTORC1)-adenosine receptor (A2AR) interaction, as well as by CD73 (an ectoenzyme involved in adenosine production from AMP) and CD39 (an ectoenzyme involved in generating AMP from ATP). Each of these players may be targeted to block this signaling pathway (25). Additionally, tumor-derived lactate within the TME is known to alter immune function and serves as a fuel for many protumor immune cell populations. Blockage of LDH-A, a l-lactate dehydrogenase (LDH) isoform, was recently demonstrated to increase the number of NK and Teff cells in a murine model of melanoma, and these cell populations were shown to have augmented cytolytic activity. LDH blockade combined with anti-PD-1 therapy further reduced melanoma growth over that observed with PD-1 therapy alone (26). Better LDH inhibitors need to be developed for this strategy to gain traction because of low potency or off-target effects. One alternate approach would be to inhibit lactate transporters, such as monocarboxylate transporters (MCTs) in tumor cells, thereby blocking this source of fuel and subsequent immune evasion (26).
Potential new molecular targets are of great interest and could shift the immunotherapy landscape in the coming years. This includes the ability to reeducate the microbiome to enhance immunotherapeutic effectiveness. Recent reports have implicated gut microbiota in modulating patients’ response to checkpoint inhibition (1). Studies have shown that the gut microbiome affects the systemic immune system and that specific taxa of microbiota increase antitumor immunity and response to checkpoint inhibitors in mice and melanoma patients (27). Additionally, modulating gut bacteria, via probiotic supplement, microbiome-based metabolite therapy, depletion of unfavorable bacterial taxa via antibiotic treatment, or fecal microbiota transplant, may be a strategy to turn an immunologically cold tumor into a more immunologically active, or hot, tumor as well as reverse resistance to immunotherapy (28). Recently, short-chain fatty acids produced through bacterial fermentation of dietary fiber have been associated with resistance to CTLA-4-directed checkpoint blockade therapy and increased levels of Tregs (29). More thorough investigation is needed before these approaches are utilized clinically.
4. REMAINING QUESTIONS
Important factors fueling the progress of immunotherapy have been outlined and include development of better tools to understand tumor-immune interactions, more innovative clinical trial design, and new therapies (30). Specific questions remain, including: How can we determine specific patient populations likely to respond to new therapies? How can we determine the appropriate timing of treatment for new immunotherapies? And which tumor types will be most likely to respond to metabolic targeting? The immune system is interconnected, and many therapies will impact multiple facets of the TME. Precise evaluation of the immunotherapeutic window of opportunity is critical to provide maximum benefit. Rational design of combination therapy will be key moving forward, and the impact of each monotherapy on the immune system will be a critical consideration to ensure that appropriate combinations and timing are being chosen. Leveraging current therapies in combination with new immune targeted therapies to increase efficacy and safety is the most logical way to move the field forward. However, it is important to consider that combination therapies can have antagonistic rather than synergistic effects (4). For example, combined checkpoint inhibitors have been associated with increases in immune-related adverse events, with differences seen based on the combination strategy (31). Current studies are investigating the perioperative use of immunotherapies as well; theoretically, this strategy may prime the immune system for tumor surveillance after tumor resection and may thereby provide clinical benefit to patient populations who may not otherwise respond well to immunotherapies (currently in clinical trials) (4). Recent studies detailing molecular links between cancer genotypes and metabolic dependence provide a basis for patient stratification in metabolism-targeted therapies and will inevitably be utilized during clinical trial design of combination therapies to include patient populations most likely to respond (32).
Many tumor-associated metabolic alterations described in the literature as potential therapeutic targets have only been evaluated in the context of the tumor cells themselves, such as stearoyl-CoA desaturase (33). Understanding how these alterations impact various cell populations, especially immune cell subsets within the TME, will be critical when moving forward with the development of new therapeutic strategies. Furthermore, cancer cells are metabolically plastic and can switch their metabolism phenotype between glycolysis and oxidative phosphorylation (OXPHOS). Thus targeting both glycolysis and OXPHOS may be necessary to suppress metabolic plasticity, as tumor cells can also acquire a hybrid metabolic phenotype. Other stromal cell populations [including cancer-associated fibroblasts (CAFs), adipocytes, and neurons] can be found within the TME. These cell populations are also influenced by hypoxia- and metabolism-targeted therapies, including those described here. The response of these cell subpopulations must be considered to develop more efficacious cancer therapies and achieve a clear understanding of how the TME is modulated by these interventions. CAFs have been described as a developing target of anticancer immunotherapy. These stromal cells are present within many tumor types and are involved in immune evasion and resistance to immune targeted therapies. CAFs can secrete cytokines, chemokines, and metabolites involved in inhibition of the antitumor immune response and the generation of an immunosuppressive TME, similar to the tumor cells themselves. Additionally, some CAF subsets can express the immune checkpoint protein PD-L1. CAF-targeted immunotherapies that block CAF function, deplete CAFs, or alter CAF activation have been suggested, although none of these strategies has made it to the clinic yet (34). Altering CAF activation by targeting the metabolic reprogramming of these stromal cells is particularly intriguing. CAFs acquire a catabolic phenotype in response to the tumor metabolic state. Some of the metabolic targets described for tumor cell inhibition, including blocking LDH-A, MCTs, and acidosis, may be effective (35). The interplay between these stromal cell populations and immune cells within the TME warrants more research.
Differences between tumor types are likely, and rationally designed clinical trials, with tumor biology taken into account, are needed to most appropriately test new therapeutic strategies. For example, in tumor types in which immunotherapy has been relatively successful, such as melanoma, the focus will be on finding therapeutic strategies for nonresponders and for those with acquired resistance. This subset of patients will likely benefit from combinatorial approaches that pair currently approved immune checkpoint inhibitors with targeted therapies that modulate other aspects within the TME, such as CD40, tryptophan metabolism, or acidosis (23,36). A triple therapy approach, combining checkpoint inhibitors with MEK and BRAF inhibitors, has also been suggested for the treatment of melanoma, as a survival benefit has been recently shown with this strategy in advanced melanoma cases (37). Immunotherapy treatment of melanoma has a long history, starting with high-dose interferon and high-dose interleukin 2 (IL-2) treatment, which are both associated with many adverse events. The approval of the first checkpoint inhibitor occurred in 2011, and this method of reversing immune suppression has been effective for the treatment of melanoma ever since (38). Recent trials combining checkpoint inhibitors with other immune targeted therapies have had mixed success. For example, the combination of pembrolizumab (anti-PD-1) with epacadostat (an IDO1 inhibitor) did not show benefit over pembrolizumab alone, suggesting that other combinations are necessary (39). Other tumor types that do not respond well to current immunotherapeutic approaches, like glioblastoma (GBM) and pancreatic cancer, will likely require more aggressive and/or modified tactics. GBM, for example, has been a continuous challenge, as this tumor type is incredibly heterogeneous. Data show that GBMs can be recognized by the immune system, yet these tumors have numerous methods to suppress antitumor immunity; therefore trials targeting only a single component of antitumor immunity have been unsuccessful. Recent data suggest that macrophage repolarization and combination immune checkpoint blockade may be beneficial within this tumor type (40). Other potential therapeutic targets discussed, such as extracellular acidosis and accumulation of immunomodulatory metabolites, are involved in GBM pathobiology, but the efficacy of targeting these aspects is currently unknown. Under these circumstances, personalized treatment approaches developed from genomics of nonresponders may be more effective.
Riera-Domingo et al. rightly stress that additional studies and technical advances are needed to better understand subset-specific metabolic dependencies in vivo. Importantly, the authors caution that the unique TME composition in various tumors likely varies from the environment studied in current models. Therefore, it is critical to further investigate how metabolic and immunomodulatory signals in the TME regulate immune cell function. The authors also emphasize that the overall effect of metabolic targeted therapies should be evaluated carefully because of the varied impact on different cell populations within the TME and the potentially strong impact of immune cell composition and degree of immune infiltrate in each tumor type.
GRANTS
J.S.D. is supported by NIH Grant R01 HL-128502. M.A. is supported by NIH Grant R01 ES-026219.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.F.G., J.S.D., and M.A. conceived and designed research; K.F.G. and J.S.D. drafted manuscript; K.F.G., J.S.D., C.A.E., and M.A. edited and revised manuscript; M.A. approved final version of manuscript.
REFERENCES
- 1.Sambi M, Bagheri L, Szewczuk MR. Current challenges in cancer immunotherapy: multimodal approaches to improve efficacy and patient response rates. J Oncol 2019: 4508794, 2019. doi: 10.1155/2019/4508794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3: 95ra73, 2011. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271: 1734–1736, 1996. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 4.Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, Schuebbe G, Renz BW, D'Haese JG, Schloesser H, Heinemann V, Subklewe M, Boeck S, Werner J, von Bergwelt-Baildon M. Advances in cancer immunotherapy 2019—latest trends. J Exp Clin Cancer Res 38: 268, 2019. doi: 10.1186/s13046-019-1266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riera-Domingo C, Audigé A, Granja S, Cheng WC, Ho PC, Baltazar F, Stockmann C, Mazzone M. Immunity, hypoxia, and metabolism-the menage a trois of cancer: implications for immunotherapy. Physiol Rev 100: 1–102, 2020. doi: 10.1152/physrev.00018.2019. [DOI] [PubMed] [Google Scholar]
- 6.Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front Immunol 9: 353, 2018. doi: 10.3389/fimmu.2018.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim C, Kim JH, Kim JS, Chon HJ, Kim JH. A novel dual inhibitor of IDO and TDO, CMG017, potently suppresses the kynurenine pathway and overcomes resistance to immune checkpoint inhibitors. J Clin Oncol 37: e14228, 2019. doi: 10.1200/JCO.2019.37.15_suppl.e14228. [DOI] [Google Scholar]
- 8.Lameirinhas A, Miranda-Gonçalves V, Henrique R, Jerónimo C. The complex interplay between metabolic reprogramming and epigenetic alterations in renal cell carcinoma. Genes (Basel) 10: 264, 2019. doi: 10.3390/genes10040264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaidi N, Jaffee EM. Immunotherapy transforms cancer treatment. J Clin Invest 129: 46–47, 2019. doi: 10.1172/JCI126046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kashyap AS, Schmittnaegel M, Rigamonti N, Pais-Ferreira D, Mueller P, Buchi M, Ooi CH, Kreuzaler M, Hirschmann P, Guichard A, Rieder N, Bill R, Herting F, Kienast Y, Dirnhofer S, Klein C, Hoves S, Ries CH, Corse E, De Palma M, Zippelius A. Optimized antiangiogenic reprogramming of the tumor microenvironment potentiates CD40 immunotherapy. Proc Natl Acad Sci USA 117: 541–551, 2020. doi: 10.1073/pnas.1902145116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmittnaegel M, Rigamonti N, Kadioglu E, Cassará A, Wyser Rmili C, Kiialainen A, Kienast Y, Mueller HJ, Ooi CH, Laoui D, De Palma M. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 9: eaak9670, 2017. doi: 10.1126/scitranslmed.aak9670. [DOI] [PubMed] [Google Scholar]
- 12.Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, Ulas T, Papantonopoulou O, Van Eck M, Auphan-Anezin N, Bebien M, Verthuy C, Vu Manh TP, Turner M, Dalod M, Schultze JL, Lawrence T. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab 29: 1376–1389.e4,2019. doi: 10.1016/j.cmet.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol 16: 425–441, 2019. doi: 10.1038/s41571-019-0203-7. [DOI] [PubMed] [Google Scholar]
- 14.Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med 45: e45, 2013. doi: 10.1038/emm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Xia Y, Lu Z. Metabolic features of cancer cells. Cancer Commun (Lond) 38: 65–65, 2018. doi: 10.1186/s40880-018-0335-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palsson-McDermott EM, Dyck L, Zasłona Z, Menon D, McGettrick AF, Mills KH, O’Neill LA. Pyruvate kinase M2 is required for the expression of the immune checkpoint pd-l1 in immune cells and tumors. Front Immunol 8: 1300, 2017. doi: 10.3389/fimmu.2017.01300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Sullivan D, Sanin DE, Pearce EJ, Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol 19: 324–335, 2019. doi: 10.1038/s41577-019-0140-9. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Yip LY, Lee JH, Wu Z, Chew HY, Chong PK, Teo CC, Ang HY, Peh KL, Yuan J, Ma S, Choo LS, Basri N, Jiang X, Yu Q, Hillmer AM, Lim WT, Lim TK, Takano A, Tan EH, Tan DS, Ho YS, Lim B, Tam WL. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med 25: 825–837, 2019. doi: 10.1038/s41591-019-0423-5. [DOI] [PubMed] [Google Scholar]
- 19.Chen C, Bai L, Cao F, Wang S, He H, Song M, Chen H, Liu Y, Guo J, Si Q, Pan Y, Zhu R, Chuang TH, Xiang R, Luo Y. Targeting LIN28B reprograms tumor glucose metabolism and acidic microenvironment to suppress cancer stemness and metastasis. Oncogene 38: 4527–4539, 2019. doi: 10.1038/s41388-019-0735-4. [DOI] [PubMed] [Google Scholar]
- 20.Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, , et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 566: 403–406, 2019. doi: 10.1038/s41586-019-0904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M, O’Connor RS, Trefely S, Graham K, Snyder NW, Beatty GL. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated 'don't-eat-me' signal. Nat Immunol 20: 265–275, 2019. doi: 10.1038/s41590-018-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, Xia H, Zhou J, Li G, Li J, Li W, Wei S, Vatan L, Zhang H, Szeliga W, Gu W, Liu R, Lawrence TS, Lamb C, Tanno Y, Cieslik M, Stone E, Georgiou G, Chan TA, Chinnaiyan A, Zou W. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569: 270–274, 2019. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mulé JJ, Ibrahim-Hashim A, Gillies RJ. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res 76: 1381–1390, 2016. doi: 10.1158/0008-5472.CAN-15-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, Tuccitto A, Tagliabue E, Castelli C, Rivoltini L. Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol 43: 74–89, 2017. doi: 10.1016/j.semcancer.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, Coukos G. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol 10: 925–925, 2019. doi: 10.3389/fimmu.2019.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feichtinger RG, Lang R. Targeting L-lactate metabolism to overcome resistance to immune therapy of melanoma and other tumor entities. J Oncol 2019: 2084195, 2019. doi: 10.1155/2019/2084195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, , et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359: 97–103, 2018. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33: 570–580, 2018. doi: 10.1016/j.ccell.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coutzac C, Jouniaux JM, Paci A, Schmidt J, Mallardo D, Seck A, Asvatourian V, Cassard L, Saulnier P, Lacroix L, Woerther PL, Vozy A, Naigeon M, Nebot-Bral L, Desbois M, Simeone E, Mateus C, Boselli L, Grivel J, Soularue E, Lepage P, Carbonnel F, Ascierto PA, Robert C, Chaput N. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat Commun 11: 2168, 2020. doi: 10.1038/s41467-020-16079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brodsky AN. Cancer immunotherapy: the year in review and a look at the year ahead. In: Immune to Cancer: The CRI Blog. New York: Cancer Research Institute, 2019. [Google Scholar]
- 31.Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA, Lambotte O, Mariette X, Prat A, Suárez-Almazor ME. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers 6: 38, 2020. doi: 10.1038/s41572-020-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin N, Bi A, Lan X, Xu J, Wang X, Liu Y, Wang T, Tang S, Zeng H, Chen Z, Tan M, Ai J, Xie H, Zhang T, Liu D, Huang R, Song Y, Leung EL, Yao X, Ding J, Geng M, Lin SH, Huang M. Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat Commun 10: 2701, 2019. doi: 10.1038/s41467-019-10427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pisanu ME, Noto A, De Vitis C, Morrone S, Scognamiglio G, Botti G, Venuta F, Diso D, Jakopin Z, Padula F, Ricci A, Mariotta S, Giovagnoli MR, Giarnieri E, Amelio I, Agostini M, Melino G, Ciliberto G, Mancini R. Blockade of stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett 406: 93–104, 2017. doi: 10.1016/j.canlet.2017.07.027. [DOI] [PubMed] [Google Scholar]
- 34.Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, Yin R. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol 12: 86, 2019. doi: 10.1186/s13045-019-0770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett 380: 272–280, 2016. doi: 10.1016/j.canlet.2015.10.027. [DOI] [PubMed] [Google Scholar]
- 36.Weiss SA, Wolchok JD, Sznol M. Immunotherapy of melanoma: facts and hopes. Clin Cancer Res 25: 5191–5201, 2019. doi: 10.1158/1078-0432.CCR-18-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anonymous. Triple therapy better for advanced melanoma. Cancer Discov 10: OF7, 2020. doi: 10.1158/2159-8290.CD-NB2020-036. [DOI] [PubMed] [Google Scholar]
- 38.Onitilo AA, Wittig JA. Principles of immunotherapy in melanoma. Surg Clin North Am 100: 161–173, 2020. doi: 10.1016/j.suc.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, Arance A, Carlino MS, Grob JJ, Kim TM, Demidov L, Robert C, Larkin J, Anderson JR, Maleski J, Jones M, Diede SJ, Mitchell TC. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol 20: 1083–1097, 2019. doi: 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 40.Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 9: e52176, 2020. doi: 10.7554/eLife.52176. [DOI] [PMC free article] [PubMed] [Google Scholar]